

ABSTRACT
Centromere structure and function are defined by the epigenetic modification of histones at centromeric and pericentromeric chromatin. The constitutive heterochromatin found at pericentromeric regions is highly enriched for H3K9me3 and H4K20me3. Although mis-expression of the methyltransferase enzymes that regulate these marks, Suv39 and Suv420, is common in disease, the consequences of such changes are not well understood. Our data show that increased centromere localization of Suv39 and Suv420 suppresses centromere transcription and compromises localization of the mitotic kinase Aurora B, decreasing microtubule dynamics and compromising chromosome alignment and segregation. We find that inhibition of Suv420 methyltransferase activity partially restores Aurora B localization to centromeres and that restoration of the Aurora B-containing chromosomal passenger complex to the centromere is sufficient to suppress mitotic errors that result when Suv420 and H4K20me3 is enriched at centromeres. Consistent with a role for Suv39 and Suv420 in negatively regulating Aurora B, high expression of these enzymes corresponds with increased sensitivity to Aurora kinase inhibition in human cancer cells, suggesting that increased H3K9 and H4K20 methylation may be an underappreciated source of chromosome mis-segregation in cancer.
This article has an associated First Person interview with the first author of the paper.
KEY WORDS: Centromere, Mitosis, Chromosome instability
Summary: Centromere enrichment of the methyltransferases Suv39 and Suv420 compromises localization of Aurora B kinase and corrupts mitotic chromosome segregation.
INTRODUCTION
Mitotic chromosome segregation is regulated, in part, through the composition and function of kinetochores, which are protein complexes that link spindle microtubules to chromatin (Thomas et al., 2017; Hinshaw and Harrison, 2018). Kinetochores are, in turn, assembled upon specialized domains of chromatin known as centromeres. Centromeric chromatin is composed of highly repetitive DNA sequences and is defined epigenetically by the presence of the centromere-specific histone 3 variant CENP-A (Hinshaw and Harrison, 2018; De Rop et al., 2012; Sharma et al., 2019; Ohzeki et al., 2019; Guse et al., 2011; Carroll and Straight, 2006). Flanking the CENP-A-containing centromeric chromatin are less ordered repeat sequences that make up the pericentromere. A defining characteristic of pericentromeric chromatin is its enrichment for repressive epigenetic marks, including di- and tri-methylation of lysine 9 on histone H3 and lysine 20 on histone H4 (H3K9me2/3 and H4K20me2/3, respectively), that are strikingly absent from the adjacent centromere (Fioriniello et al., 2020; Sullivan and Karpen, 2004). CENP-A chromatin is demonstrated to be a critical epigenetic marker upon which the kinetochore is built (De Rop et al., 2012; Barnhart et al., 2011); however, the function of the epigenetic marks at pericentromeric heterochromatin (PCH) domains remain less clear.
Although many studies have implicated changes in heterochromatin in the regulation of genome stability and cancer progression, most focus on the consequences of decreased H3K9me3 and H4K20me3 heterochromatin marks (reviewed in Janssen et al., 2018). Nevertheless, both increased expression of the methyltransferases responsible for placing these repressive marks and decreased expression of demethylases that remove these marks have been described in various cancer contexts (Janssen et al., 2018; Black et al., 2012; Zhou et al., 2019; Yokoyama et al., 2013). Functional studies in cancer cells indicate that increased levels of H3K9me3 and H4K20me3 contribute to increased motility and metastatic potential (Zhou et al., 2019; Yokoyama et al., 2013) and can limit therapeutic response (Cuellar et al., 2017; Guler et al., 2017).
Studies exploiting a human artificial chromosome (HAC) system have shown that expansion of heterochromatin via direct tethering of Suv39 to the centromere is sufficient to compromise segregation fidelity of the HAC (Ohzeki et al., 2012; Martins et al., 2016). Consistent with these observations, recent work has revealed that expression of Suv39 and Suv420, the methyltransferases responsible for H3K9me2/3 and H4K20me2/3, respectively, correlate with levels of aneuploidy across 10,000+ cancer samples from The Cancer Genome Atlas (TCGA) project (Taylor et al., 2018). Mitotic segregation errors underlie the generation of aneuploidy, and, together, these data suggest that misregulation of Suv39 and Suv420, and the epigenetic marks they regulate, may be functionally linked to the generation of aneuploidy in cancer.
Key to accurate chromosome segregation is the localization and function of the mitotic kinase Aurora B (Lampson et al., 2004; Liang et al., 2020; Gregan et al., 2011; Krenn and Musacchio, 2015). Aurora B, together with INCENP, Survivin (also known as BIRC5) and Borealin (also known as CDCA8), form the chromosomal passenger complex (CPC). Distributed along chromosome arms at mitotic entry, the CPC re-localizes first to the centromere and kinetochore and later, during anaphase, to the central spindle (Hindriksen et al., 2017). Concentration of the CPC at the centromere and kinetochore is believed to be crucial for its function in mitotic error correction as it places Aurora B in close proximity to its substrates on kinetochores (Welburn et al., 2010). Aurora B de-stabilizes microtubules by phosphorylating several key regulators of kinetochore–microtubule attachments to promote the removal of merotelic attachments (Cheeseman et al., 2006; Welburn et al., 2010; DeLuca et al., 2011; Cimini et al., 2006; DeLuca et al., 2006). The formation of merotelic attachments during early stages of mitosis are stochastic, and loss or functional inactivation of Aurora B kinase activity precludes their correction and corrupts mitotic fidelity (Abe et al., 2016; Broad et al., 2020; Hauf et al., 2003; Huang et al., 2018).
An intricate signaling network involving Haspin-dependent phosphorylation of H3T3 and BUB1-dependent phosphorylation of H2AT120 controls localization of the CPC to the inner centromere. These pathways represent two parallel modes of CPC regulation such that abrogation of either one results in dispersion of the CPC over chromatin and a reduction, but not loss, of CPC at the centromere (Hindriksen et al., 2017; Krenn and Musacchio, 2015; Carretero et al., 2013; Dai et al., 2006; Meppelink et al., 2015; Wang et al., 2010). It remains unclear whether other epigenetic marks at the centromere or pericentromere similarly impact the regulation of CPC localization and function.
RESULTS
High expression of Suv39 and Suv420 methyltransferases is prevalent in cancer
Expression data from the TCGA indicate that Suv39 and Suv420 are highly expressed in cancer contexts, with Suv39 isoforms h1 and h2 and Suv420 isoform h2 exhibiting increased average expression compared to corresponding normal tissue in 11 of 14 cancer subtypes for which paired tumor and normal data are available (Fig. 1A). These enzymes function to regulate constitutive heterochromatin at pericentromeres, and, in normal cells, H3K9me3 and H4K20me3 are enriched near centromeres. In a panel of high Suv420-expressing breast cancer cell lines, H4K20me3 was not dispersed uniformly throughout the genome and instead remained enriched near centromeres (Fig. S1), raising the possibility that Suv420 misexpression may preferentially compromise pericentromere or centromere function and perturb mitotic chromosome segregation. Indeed, recent work describes that high expression of Suv39 or Suv420 positively correlates with pan cancer analyses of increased aneuploidy (Taylor et al., 2018). This relationship is not an artifact of high Suv39 or Suv420 expression in a single cancer type that also happens to be highly aneuploid, but instead persists when samples are sorted by cancer subtype. We found that high expression of at least one Suv39 or Suv420 isoform demonstrated a significant positive correlation with aneuploidy in 12 out of 20 cancer contexts represented in the TCGA database (Fig. 1B; Table S1). Consistent with a correlation with aneuploidy, which is known to correspond with tumor aggressiveness and poor patient outcome (Pfau and Amon, 2012; Weaver et al., 2007; Davoli et al., 2013; Liu et al., 2016), high expression of a Suv39h1/h2 or Suv420h1/h2 isoform corresponded with a significant decrease in disease-free survival in four out of 20 cancer subtypes represented in the Gene Expression Omnibus (GEO), European Genome-phenome Archive (EGA) and TCGA databases (Table S2) (Nagy et al., 2018; Györffy et al., 2010).
Fig. 1.

High expression of Suv39 and Suv420 isoforms in cancer correlates with aneuploidy and reduced patient survival. (A) Gene expression values for SUV39H1, SUV39H2, SUV420H1 (also known as KMT5B) and SUV420H2 (also known as KMT5C) from TCGA, sorted by cancer subtype, indicate increased gene expression compared to normal tissue expression levels in multiple cancer subtypes. (B) Suv39 and Suv420 isoform expression exhibit a moderate, but highly significant, correlation with calculated aneuploidy score in several cancer subtypes. Red indicates significance at P<0.0025 using linear regression analysis (see also Table S1).
Depletion of the H3K9me3 demethylase KDM4A compromises mitotic fidelity
To define the consequences of increased H3K9 and H4K20 methylation on mitotic chromosome segregation, we used multiple independent approaches to increase the methylated state of H3K9 or H4K20 globally or specifically at the centromere in human telomerase reverse transcriptase (hTERT)-immortalized retinal pigment epithelial (RPE-1) cells. RPE-1 cells are diploid and genomically stable, exhibiting fewer than one segregation error every 100 cell divisions (Thompson and Compton, 2008). To enhance H3K9 methylation and monitor the impact on mitosis, we first utilized siRNA to deplete KDM4A, a demethylase responsible for removing di- and tri-methyl marks from H3K9 (Fig. S2A) (Berry and Janknecht, 2013). As expected, depletion of KDM4A led to a small but measurable increase in the global level of H3K9 methylation (Fig. S2D,E). Using time-lapse and fixed cell imaging approaches, we demonstrated that depletion of KDM4A compromises mitotic progression. RPE-1 cells expressing RFP-tagged histone 2B (RFP-H2B) to label chromatin were transfected with a non-targeting control (siControl) or KDM4A-targeting siRNA (siKDM4A) for 36 h and then monitored by live-cell fluorescence imaging (Fig. 2A). Images were captured every 5 min, and mitotic cells were tracked to determine the dynamics of chromosome alignment and the timing of anaphase onset. Cells lacking KDM4A exhibited a minor delay in metaphase chromosome alignment and anaphase onset (27.37±0.40 min versus 34.67±1.33 min, P=0.007; Fig. 2A). Consistent with live-cell imaging, immunofluorescence analysis of KDM4A-depleted cells revealed an enrichment in cells in which single chromosomes remain near spindle poles when metaphase alignment of the majority of chromosomes has already been achieved (Fig. 2B). Although live-cell imaging revealed that full metaphase alignment is eventually achieved prior to anaphase onset, nearly 25% of KDM4A-depleted anaphase cells exhibited lagging chromosomes (Fig. 2C). Similar results were seen when KDM4A mRNA was targeted for depletion with any one of four independent siRNA sequences (Fig. S2B, Table S3).
Fig. 2.
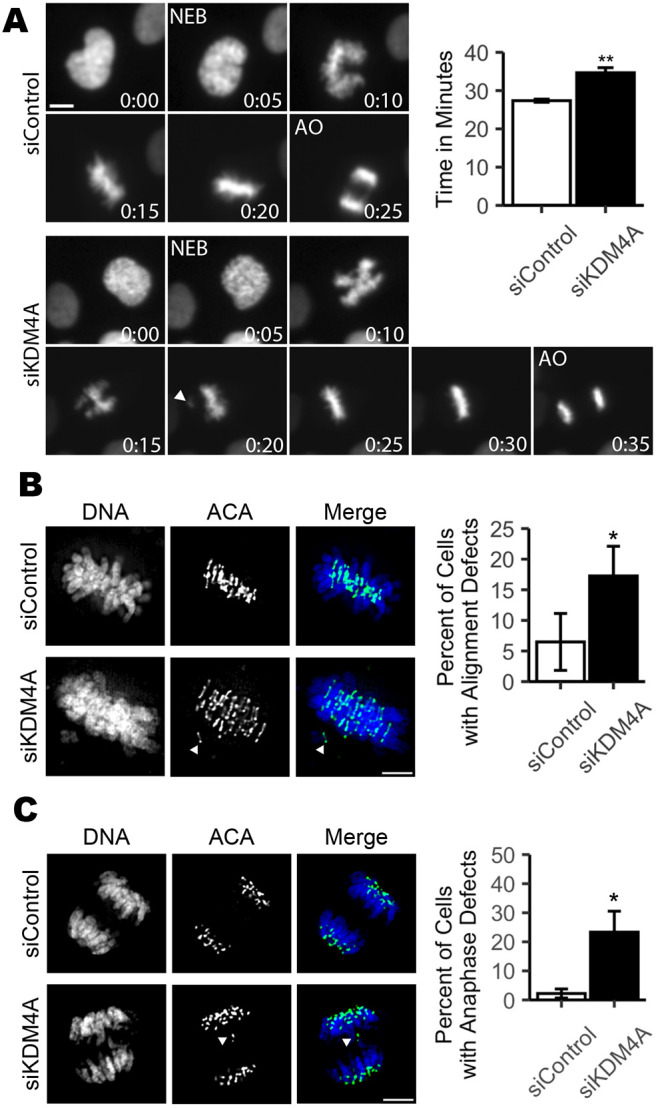
Depletion of KDM4A compromises mitotic fidelity. (A) Still images and quantification of average mitotic duration from time-lapse microscopy of RFP-H2B-expressing RPE-1 cells transfected with either non-targeting control (siControl) or KDM4A-targeting (siKDM4A) siRNAs show that cells lacking KDM4A exhibit delayed anaphase onset. Images were captured every 5 min. Number insets indicate time progression in minutes. Nuclear envelope breakdown (NEB) and anaphase onset (AO) are indicated. 50 mitotic cells were analyzed per condition for each of three biological replicates. (B) Images and quantification showing that KDM4A-depleted mitotic cells have an increase in late prometaphase/metaphase cells that retain one to three unaligned chromosomes. (C) Images and quantification showing that KDM4A-depleted anaphase cells exhibit an increase in lagging chromosomes during anaphase. For B and C, 30 metaphase or anaphase cells were analyzed per condition for each of three biological replicates. For all panels, white arrowheads indicate unaligned or lagging chromosomes. *P<0.05, **P<0.01 (paired Student's t-test). Error bars are ±s.d. and statistical analyses were performed between three biological replicates. Scale bars: 5 µm.
Centromere tethering of the histone methyltransferase Suv39 or Suv420 corrupts mitotic fidelity
As H3K9 methylation is not exclusive to the pericentromere, increased H3K9 methylation that follows KDM4A depletion is not restricted to centromeric heterochromatin or PCH (Fig. S2D,E) (Berry and Janknecht, 2013). Therefore, to test whether changes in centromere-specific H3K9 methylation levels are sufficient to cause mitotic errors, we engineered RPE-1 cells to inducibly express a GFP-tagged Suv39h1 protein fused to the DNA-binding domain of the centromere-localized protein CENP-B (cen-Suv39-GFP, Fig. S2C–E). The DNA-binding domain of CENP-B binds to a discrete sequence at the centromere (Pluta et al., 1992), targeting the cen-Suv39-GFP fusion protein to centromeres, where it promotes increased H3K9 methylation (Fig. S2D,E). Comparable to depletion of KDM4A, immunofluorescence imaging of cells following induced expression of cen-Suv39-GFP revealed an increase in metaphase cells with alignment defects and anaphase cells with lagging chromosomes that are not present in the absence of doxycycline-induced expression of the transgene (i.e. Mock vs Induced, Fig. 3A; Fig. S3B).
Fig. 3.

Increased H4K20me3 corresponds with decreased mitotic fidelity. (A) Images and quantification showing an increase in lagging chromosomes during anaphase following induced expression of cen-Suv39-GFP or cen-Suv420-GFP. White arrowheads indicate a lagging chromosome. Scale bar: 5 µm. (B) Images and quantification showing that induction of cen-Suv420-GFP promotes a substantial increase in centromere-localized H4K20me3 levels, whereas siKDM4A, cen-Suv39-GFP and (non-tethered) Suv420-GFP induce only moderate changes in H4K20me3 at centromeres. Cen-GFP does not alter centromere H4K20me3 levels. Insets are 1.5× enlargements of ACA-stained kinetochore pairs. Scale bar: 1 µm. (C) Images and quantification showing an increase in micronuclei formation following induction of cen-Suv39-GFP or cen-Suv420-GFP. Insets are 3× enlargements of individual micronuclei. Presence of an ACA-labeled kinetochore within each micronucleus indicates they likely form following mitotic segregation errors. Scale bar: 5 µm. Alignment and segregation errors were assessed in 30 cells per condition, per replicate. Intensity analyses were done on ten kinetochores for each of 30 cells per condition, per replicate. For micronuclei analyses, a minimum of 240 cells were scored per condition, per replicate. All statistical analyses were performed between three biological replicates. *P<0.05, **P<0.01; N.S., not significant. Error bars are ±s.d.
H3K9 methylation promotes the recruitment and binding of heterochromatin protein 1 (HP1). HP1, in turn, serves as a platform to recruit a number of factors to heterochromatin, including the H4K20 methyltransferase Suv420 (Janssen et al., 2018; Hahn et al., 2013; Schotta et al., 2004). Thus, establishment of H3K9me3 by Suv39 is indirectly implicated in deposition of H4K20me3. Consistent with this relationship, we found that centromere targeting of Suv39 (the H3K9me2/3 methyltransferase) enhances H4K20me3 levels at the centromere (Fig. 3B). To delineate the roles of H3K9 and H4K20 centromere methylation in the regulation of mitotic progression, we next expressed a GFP-tagged Suv420h2 protein fused to the DNA-binding domain of centromere protein CENP-B (cen-Suv420-GFP). Cen-Suv420-GFP localized to centromeres and was sufficient to promote a marked enrichment of H4K20me3 at centromeres without altering H4K20me3 levels along chromosome arms (Fig. 3B; Fig. S3C). As seen with KDM4A depletion and cen-Suv39-GFP expression, induced expression of cen-Suv420-GFP led to metaphase alignment defects (Fig. S3B) and anaphase segregation errors (Fig. 3A), suggesting that increased H4K20me3 at centromeres is sufficient to corrupt kinetochore regulation. Chromosomes that lag during anaphase can be excluded from the main nucleus when the nuclear envelope reforms, resulting in the formation of a separate micronucleus (Fenech et al., 2011). Consistent with the presence of chromosome segregation defects, cells expressing cen-Suv39-GFP or cen-Suv420-GFP also exhibited an increase in the number of interphase cells within the population that have centromere-positive micronuclei (Fig. 3C).
Centromere enrichment of Suv420 compromises mitotic error correction mechanisms
Formation of syntelic and merotelic microtubule attachments, where both kinetochores of a chromosome pair are bound by microtubules from the same spindle pole, occurs stochastically during mitosis. Error correction mechanisms to destabilize such mal-attachments persist throughout prometaphase and metaphase (Cimini et al., 2003). Delays in chromosome alignment to the metaphase plate, and the presence of lagging chromosomes during anaphase, can result from persistent merotelic attachments and may indicate that the number of merotelic attachments formed in mitosis overwhelm the error correction machinery (Cimini et al., 2003). Imbalances in error corrective mechanisms that result in merotelic attachments and segregation errors arise due to either an increased burden in the number of merotelic attachments formed, or from an underlying defect in the error correction machinery (Gregan et al., 2011). To determine whether cells with altered centromere methylation are predisposed to forming merotelic attachments or instead are deficient in correcting these mal-attachments, KDM4A-depleted cells or cells induced to express either cen-Suv39-GFP or cen-Suv420-GFP were exposed to the Eg5 inhibitor monastrol for 4 h (Fig. 4A). Inhibition of the mitotic kinesin Eg5 with the small-molecule inhibitor monastrol causes spindle poles to collapse, resulting in a monopolar spindle structure. These spindles cannot form amphitelic chromosome attachments and instead form syntelic or monotelic attachments (Kapoor et al., 2000). Upon washout of monastrol, a large portion of kinetochore attachments are converted to merotelic attachments (Kapoor et al., 2000; Lampson et al., 2004). Destabilization of mal-attached microtubules is a prerequisite for acquisition of proper kinetochore biorientation and chromosome alignment, such that the time needed to achieve complete metaphase alignment following monastrol washout is an indication of error correction efficiency. Immunofluorescence imaging demonstrated that, following monastrol washout, bipolar spindles form and the majority of control cells restore chromosome biorientation and achieve metaphase alignment within 40 min. In contrast, although spindle bipolarity was ultimately achieved similarly to control cells, KDM4A-depleted, cen-Suv39-GFP-expressing and cen-Suv420-GFP-expressing cells all exhibited a delayed progression to metaphase, with fewer than half of all cells able to achieve metaphase alignment within the same time frame as control cells (Fig. 4A). Taken together, these data indicate that the mitotic error correction machinery is compromised under conditions in which H3K9me3 or H4K20me3 is increased.
Fig. 4.

Mitotic error correction and Aurora B localization are compromised when centromere Suv420/H4K20 methylation levels are increased. (A) Images and quantification showing progression to full metaphase alignment following inhibition of Eg5 with monastrol and subsequent drug washout. Cells lacking KDM4A or expressing cen-Suv39-GFP or cen-Suv420-GFP are delayed in achieving metaphase alignment, indicating a deficiency in mitotic error correction capacity. (B) Images and quantification of Aurora B kinase localization at centromeres of nocodazole-treated prometaphase cells. Induced expression of cen-Suv39-GFP or cen-Suv420-GFP, but not cen-GFP, reduced Aurora B intensity at centromeres. Insets are 2× enlargements of single ACA-stained kinetochore pairs. Scale bars: 5 µm. Kinetochore analyses were done on ten kinetochores for each of 30 cells per condition, per replicate. Alignment was assessed in 50 cells per condition, per replicate. For all panels, statistical analyses were performed between three biological replicates. *P<0.05; N.S., not significant. Error bars are ±s.d..
Centromere localization of the CPC is sensitive to centromere enrichment of Suv39 or Suv420
Aurora B is a mitotic kinase that functions as a master regulator of error correction. Loss or functional inactivation of Aurora B kinase activity leads to persistent merotelic attachments and segregation errors (Lampson et al., 2004; Liang et al., 2020; Gregan et al., 2011; Krenn and Musacchio, 2015; Kallio et al., 2002; Hauf et al., 2003; Ditchfield et al., 2003). To determine whether changes in Aurora B, localization and/or function underlie observed defects in mitosis following manipulations that increase H3K9me3 and/or H4K20me3, we first performed quantitative immunofluorescence to measure Aurora B and INCENP (a component of the Aurora B-containing CPC complex) recruitment to centromeres following expression of either cen-Suv39-GFP or cen-Suv420-GFP, or depletion of KDM4A. By measuring pixel intensity of anti-Aurora B or anti-INCENP antibody staining across kinetochore pairs stained with anti-centromere antigen (ACA) in metaphase and/or nocodazole-arrested prometaphase cells, we identified and assessed Aurora B localization at centromeres and kinetochores. We found that the intensity of both Aurora B and INCENP at mitotic centromeres was significantly reduced following depletion of KDM4A or induced expression of either cen-Suv39-GFP or cen-Suv420-GFP (Fig. 4; Fig. S4). Aurora B, Borealin, INCENP and Survivin were all expressed at similar levels in cells with and without induction of cen-Suv39-GFP or cen-Suv420-GFP, suggesting that decreased staining intensity is not a consequence of changes in CPC expression (Fig. S4C). Importantly, expression of centromere-targeted GFP alone (cen-GFP) or non-targeted Suv420 (Suv420-GFP), conditions that do not enhance centromere enrichment of H4K20me3, did not perturb centromere levels of Aurora B (Fig. 4; Figs S3 and S4). Instead, reduction in centromere localization of CPC components was sensitive to Suv420 and/or H4K20me3 at centromeres, as treatment with A196, a specific small-molecule inhibitor of Suv420 methyltransferase activity (Bromberg et al., 2017), was sufficient to partially restore Aurora B localization to centromeres and to promote efficient chromosome alignment along the metaphase plate (Fig. 5).
Fig. 5.

Inhibition of Suv420 methyltransferase activity restores Aurora B localization and corrects metaphase alignment defects. (A,B) Treatment of cen-Suv420-GFP-expressing cells with 200 nM Suv420 inhibitor A196 reduces H4K20me3 levels and partially restored Aurora B localization to the centromere. (C) Chromosome alignment defects seen in cells expressing cen-Suv420-GFP are suppressed following concurrent treatment with A196. Insets are 2× enlargements of single ACA-stained kinetochore pairs. Scale bars: 5 µm. Kinetochore analyses were done on three kinetochores for each of 30 cells per condition, per replicate. Alignment was assessed in 30 cells per condition, per replicate. For all panels, statistical analyses were performed between three biological replicates. *P<0.05, **P<0.01.
Phosphorylation of Hec1 (also known as NDC80) (at serine 55) by Aurora B de-stabilizes kinetochore microtubule attachments to permit error correction. Consistent with compromised Aurora B localization, we found that centromeres in mitotic cells have decreased Hec1 phosphorylation, but not overall levels of Hec1, when cen-Suv39-GFP or cen-Suv420-GFP is expressed (Fig. 6A,B). A similar decrease was seen in phosphorylation levels of the Aurora B target CENP-A (at serine 7) at centromeres (Fig. S5A,B). These cells also exhibited a corresponding increase in stable microtubules that are resistant to cold-induced depolymerization (Fig. 6C). However, consistent with other conditions that specifically perturb Aurora B activity at centromeres, but not along chromosome arms (Wang et al., 2010), no change was seen in the phosphorylation of H3 (at serine 10), a substrate of Aurora B on chromosome arms (Fig. S5C).
Fig. 6.

Increased H4K20me3 reduces phosphorylation of Aurora B target Hec1 and increases microtubule stability. (A,B) Images and quantification showing that phosho (p)Hec1, but not total Hec1 levels at the centromere are decreased following cen-Suv39-GFP or cen-Suv420-GFP expression. Note that this anti-pHec1 antibody stains both centrosomes and kinetochores. Insets are 3× enlargements of single ACA-stained kinetochore pairs. Staining intensity between ACA peaks was measured for three kinetochore pairs in each of 30 metaphase cells per condition, in each of three biological replicates. (C) Images and quantification showing that induction of cen-Suv39-GFP or cen-Suv420-GFP expression increases the resistance of kinetochore microtubules to cold-induced depolymerization, indicating an increase in stability of kinetochore-microtubule attachments. Microtubule intensity was measured in each of 30 metaphase cells per condition, in each of three biological replicates. *P<0.05; N.S., not significant. Scale bars: 5 µm.
Aurora B localization to centromeres is influenced by several histone modifications, including phosphorylation of threonine 120 on histone 2A (H2A–T120p) by BUB1 and phosphorylation of threonine 3 on histone 3 (H3–T3p) by Haspin. Each of these histone phosphorylation events is independently sufficient to recruit Aurora B to centromeres (Liang et al., 2020; Broad et al., 2020; Hadders et al., 2020). However, we did not see reduction in either of these histone marks in contexts in which H4K20me3 has been directly or indirectly increased (Fig. S6A,B), suggesting that indirect changes in these known regulatory marks cannot explain decreased centromere CPC complex localization.
Localization of the CPC is also sensitive to centromere transcription. Centromere transcripts have been shown to bind the CPC and regulate both CPC centromere localization and Aurora B activation (Jambhekar et al., 2014; Blower, 2016; Ideue et al., 2014). To test whether centromere transcription is altered by centromere tethering of Suv39-GFP or Suv420-GFP, we measured levels of centromere transcripts in nocodazole-synchronized cells following 24 h of cen-Suv39-GFP or cen-Suv420-GFP expression. Although quantitative PCR (qPCR) analysis showed that expression of centromeric α satellite RNA is readily detected during mitosis, levels of each transcript were reduced by roughly half in mitotic cells expressing cen-Suv39-GFP or cen-Suv420-GFP (Fig. S6C). Consistent with the centromere tethering of these constructs, suppression of transcription appeared to be restricted to centromeres, as transcripts from non-centromere regions, such as housekeeping genes (GAPDH and actin) and CPC components remained unchanged in these samples (Fig. S4C). These data suggest that suppression of transcription underlies defects in Aurora B localization when centromere levels of Suv39/H3K9me3 and/or Suv420/H4K20me3 are increased.
Expression levels of Suv39 and Suv420 correspond with sensitivity to Aurora kinase inhibition in cancer cell lines
Decreased CPC localization and increased rates of chromosome segregation errors may render cells exquisitely sensitive to further inhibition of Aurora kinase activity. To test this possibility, cells with and without induction of cen-Suv39-GFP or cen-Suv420-GFP expression were treated with inhibitors targeting Aurora B kinase (Barasertib), the related Aurora A kinase (Alisertib) or the mitotic kinase MPS1 (MPS1IN1) and monitored for mitotic fidelity. Although control, cen-Suv39-GFP- and cen-Suv420-GFP-expressing cells were all similarly sensitive to MPS1 inhibition, we found that cells expressing cen-Suv39-GFP or cen-Suv420-GFP were susceptible to increased anaphase-lagging chromosomes following short-term treatment with low nanomolar concentrations of both Aurora kinase inhibitors, whereas control cells were not (Fig. 7A). We next tested whether mitotic defects in cen-Suv420-GFP-expressing cells could be suppressed by concurrently tethering the Aurora B-containing CPC to the centromere. Cen-Suv420-GFP-expressing cells were monitored for anaphase defects following expression of cen-INCENP-mCherry (Wang et al., 2011). Cells expressing cen-Suv420 exhibited a high rate of lagging chromosomes during anaphase (Figs. 3A and 7E). The frequency of these defects was reduced when the cen-INCENP-mCherry construct was expressed simultaneously. Importantly, the level of cen-Suv420-GFP at centromeres was not significantly altered following cen-INCENP-mCherry expression (Fig. 7B–D), suggesting that this rescue is not the result of a competition between INCENP and Suv420 for centromere binding.
Fig. 7.

Expression of Suv39 and Suv420 sensitizes cells to Aurora kinase inhibition. (A) Quantification of anaphase defects, showing that cells induced to express cen-Suv39 or cen-Suv420 exhibit an increase in anaphase-lagging chromosomes following low nanomolar concentrations of Aurora kinase inhibition, whereas their uninduced counterparts do not. Cells both with and without induction of cen-Suv39-GFP or cen-Suv420-GFP are similarly sensitive to inhibition of the mitotic kinase MPS1 (±cen-Suv39-GFP induction: P=0.00003, P=0.0003 and P=0.844 for Alisertib, Barasertib and MPS1IN1, respectively; ±cen-Suv420-GFP: P=0.003, P=0.004 and P=0.724 for Alisertib, Barasertib and MPS1IN1, respectively). Error bars are ±s.d. (B,C) Cen-INCENP-mCherry localizes to centromeres in cen-Suv420-GFP-expressing cells. Scale bar: 5 µm. (D) Expression of cen-INCENP-mCherry does not limit cen-Suv420-GFP localization to centromeres. GFP-staining intensity between ACA peaks was measured for three kinetochore pairs in each of 30 metaphase cells per condition, in each of three biological replicates. (E) Expression of cen-INCENP-mCherry reduces the frequency of lagging chromosomes seen in cen-Suv420-GFP expressing anaphase cells. A minimum of 30 anaphase cells were scored per condition, for each of three biological replicates. **P<0.01, ***P<0.001. (F) Quantification of area under the dose–response curve (AUC) for each drug in cancer cell lines from the TCGA sorted by top and bottom quartile of either Suv39 or Suv420 expression shows that cells with high expression of either Suv39 or Suv420 exhibit increased sensitivity to Aurora kinase inhibition. Each dot represents an individual cancer cell line tested. Open circles indicate the mean of IC50 AUCs for each condition. *P<0.00625, **P<0.00125, ***P<0.000125; N.S., not significant. Error bars are ±s.e.m.
To explore this relationship more broadly, we utilized drug sensitivity data described by the Wellcome Sanger Institute (Yang et al., 2013) and RNA-sequencing expression data from the Cancer Cell Line Encyclopedia that includes 1457 cancer cell lines (Ghandi et al., 2019). Cell lines were sorted based on Suv39 or Suv420 expression level and then drug sensitivity of the top and bottom quartiles were compared. We found that cell lines likely to have high H3K9 and/or H4K20 methylation states (owing to high expression of Suv39 or Suv420) exhibited increased sensitivity to five out of six inhibitors that preferentially target Aurora kinase activity (Alisertib, CD532, GSK1070916, Genentech-CPD-10 and ZM447439) (Fig. 7F). This relationship does not reflect a general sensitivity to mitotic poisons, as inhibition of the mitotic kinase MPS1 (MPS1IN1, TK3146) does not indicate a similar correlation with expression of these epigenetic regulators.
DISCUSSION
Here, we demonstrate that Aurora B recruitment to centromeres is sensitive to changes in the epigenetic regulation of PCH. We find that increased methylation of H3K9 and H4K20 corresponds with reduced Aurora B localization to centromeres and reduced phosphorylation of Aurora B substrates. Consistent with studies that directly perturb Aurora B function (Kallio et al., 2002; Hauf et al., 2003; Ditchfield et al., 2003), we show that such changes in centromere levels of the methyltransferases Suv39 or Suv420 are sufficient to stabilize kinetochore microtubule dynamics, limit merotelic error correction, increase lagging chromosomes during anaphase and promote whole-chromosome segregation errors.
Suv39 and Suv420 overexpression contributes to increased aneuploidy
Analyses presented here and previously reported by the Meyerson group (Taylor et al., 2018) indicate a moderate, but highly significant, correlation between degree of aneuploidy and the independent expression levels of Suv39h1, Suv39h2, Suv420h1 and Suv420h2. Consistent with previous reports, our experimental data indicate that these isoforms of Suv39 and Suv420 also share at least partially overlapping roles in heterochromatin regulation and mitotic fidelity (Tsang et al., 2010; O'Carroll et al., 2000). As such, we expect that misexpression of any one of these enzymes (Suv39h1, Suv39h2, Suv420h1 or Suv420h2) could be sufficient to promote segregation errors, and that the correlation of an individual enzyme with degree of aneuploidy would therefore be lower than if the function were served by a single enzyme. Although the correlation with aneuploidy for each enzyme is moderate, given the high significance of this relationship, we propose that these correlations are consistent with a model whereby high expression levels of a Suv39 or Suv420 isoform are sufficient to promote mitotic defects and contribute to the generation of aneuploidy. Our data do not argue against the possibility that Suv39 or Suv420 function outside regulation of PCH may compromise genome stability in cancer. However, our experimental data showing that centromere enrichment of Suv420 and/or H4K20me3 is sufficient to compromise mitotic fidelity, together with sustained pericentromere enrichment of H4K20me3 in cancer cells with high expression of Suv420, suggest that corruption of centromere regulation may be a contributing factor.
Short-term changes in centromere H3K9me3 and/or H4K20me3 corrupt mitotic fidelity without apparent defects in gross centromere structure
Work from other groups has described that a persistent increase in H3K9me3 over many cell cycles limits CENP-A deposition at the centromere on an HAC, thus compromising centromere maintenance (Ohzeki et al., 2012; Martins et al., 2016, 2020). However, although induction of cen-Suv39-GFP or cen-Suv420-GFP for 24 h (less than two cell cycles) is sufficient to cause dramatic changes in centromere levels of H3K9me3 and/or H4K20me3, it appears insufficient to compromise centromere formation or maintenance as we detect no change in overall levels of CENP-A at the centromere, or in the outer kinetochore protein Hec1 (Fig. 6; Fig. S5). Consistent with the sustained localization of critical centromere and outer kinetochore proteins, we find that cells expressing cen-Suv39-GFP or cen-Suv420-GFP remain competent to form stable end-on microtubule attachments that are sufficient to drive chromosome movement and segregation. However, we find that short-term increases in Suv39 and/or Suv420 at centromeres compromised CPC localization and reduced phosphorylation of Aurora B substrates, suggesting that spreading of the respective heterochromatin marks can impact centromere function in two distinct ways: first, by subtly moderating regulation of microtubule attachments, and then, more crudely, by preventing centromere formation and maintenance. As methylation levels likely increase with the duration of the experimental perturbation, whether these distinctions arise due to experimental differences in the total amount of H3K9/H4K20 methylation achieved at the centromere, or instead reflect a cumulative impact of persistent epigenetic deregulation over several cell cycles, remains unclear.
Both direct and indirect increases in H4K20me3 levels corrupt mitotic fidelity
In otherwise normal cells, depletion of KDM4A, or overexpression of Suv420 alone, promotes aberrant methylation throughout the genome; however, changes in H4K20me3 at the centromere are moderate (Fig. 3B) (Manning et al., 2014), suggesting that additional regulatory pathways may function to limit Suv420 enrichment or otherwise restrict H4K20me3 at centromeres. Nevertheless, this regulation of centromere/pericentromere Suv420 localization may not be as tightly controlled in cancer contexts, as we find that H4K20me3 remains enriched at centromeres in a panel of breast cancer cell lines that highly express Suv420 (Fig. S1). Furthermore, our study demonstrates that centromere targeting of Suv420 is sufficient to enhance H4K20me3 levels at centromeres without increasing H4K20me3 along chromosome arms. In this way, although we cannot rule out that H3K9me3 independently impairs Aurora B localization at centromeres, our data suggest that increased Suv420 and/or H4K20me3, independent of Suv39/H3K9me3, is sufficient to restrict Aurora B localization and compromise mitotic fidelity. Importantly, expression of centromere-targeted GFP, even at levels higher than that of cen-Suv39-GFP or cen-Suv420-GFP, is insufficient to alter centromere H4K20me3 or Aurora B localization (Figs. 3B and 4B). Taken together, these data suggest that Aurora B localization and mitotic fidelity are not generally perturbed by protein tethering to the centromere but are instead specifically sensitive to Suv420 and/or H4K20me3 levels.
Our data indicate that KDM4A depletion may disrupt Aurora B localization without a significant change in centromere H4K20me3 levels (Fig. 3). Unlike expression of the cen-targeted constructs for which the GFP tag allows us to confirm that each mitotic cell/centromere analyzed has overexpressed the enzyme, we do not have a similar report of KDM4A depletion at the single-cell level and cannot rule out the possibility that changes in methylation/Aurora B staining at centromeres may be impacted by variability of KDM4A depletion with the cell population. Nevertheless, our rescue experiments showing that inhibition of Suv420 methyltransferase activity (with A196) incompletely restores Aurora B localization (Fig. 5) are consistent with Aurora B localization being at least partially independent of H4K20me3 levels.
HP1 directly binds the Aurora B-containing CPC through INCENP, and in doing so enhances the enzymatic activity of Aurora B (Abe et al., 2016; Kang et al., 2011). In turn, Aurora B-dependent phosphorylation of H3S10 limits HP1 association with H3K9 methylation (Fischle et al., 2005; Hirota et al., 2005), such that the interaction between HP1 and Aurora B both positively (through functional regulation) and negatively (through reduction of HP1 recruitment) regulates Aurora B activity at the centromere. Suv420 is similarly recruited to the pericentromere through interactions with HP1, and our data raise the possibility that CPC interaction with HP1 is limited when Suv420 is bound. Consistent with this possibility, we find that centromere localization of the CPC is disrupted by centromere tethering of Suv420, but not overexpression of non-tethered Suv420. Interestingly, if true, this model would suggest that the role of Suv420 in regulating CPC localization were, in part, independent of its methyltransferase activity.
Disruption of heterochromatin boundaries impairs centromere cohesion, transcription and function
The constitutive heterochromatin marks H3K9me3 and H4K20me3 are enriched at pericentromeres but normally restricted from the core of the centromere (Sullivan and Karpen, 2004). This dynamic boundary between centromeric and PCH is defined by H3K9 methylation and its disruption impacts transcription of the underlying DNA (Lam et al., 2006). In our experiments, the Suv39 and Suv420 fusion constructs target methyltransferase activity to centromeres by exploiting the CENP-B DNA-binding domain. This binding domain recognizes a 17 bp motif at centromeres to which the constitutive centromere protein CENP-B localizes (Muro et al., 1992). In recruiting Suv39 and Suv420 to this domain, H3K9 and H4K20 methylation increases at ACA-stained centromeres, indicating that heterochromatin, and potentially heterochromatin-associated proteins, has spread from the pericentromere into the centromere. Consistent with spreading of heterochromatin, we find that levels of centromere and pericentromere transcripts are reduced following tethering of Suv39 or Suv420 to mitotic centromeres (Fig. S6C). In future studies, it will be important to determine whether CPC recruitment and centromere function is generally sensitive to increased Suv420 at centromere and pericentromere regions, or specifically sensitive to the spreading of the repressive H4K20me3 mark into the centromere, where it is normally absent.
Early in mitosis, Aurora B is localized along chromosome arms and must be re-localized to centromeres to ensure accurate chromosome segregation. Various regulatory mechanisms for the recruitment and retainment of Aurora B to centromeres have been identified, including positive regulation by phosphorylation of H3T3 and H2AT120 (Yamagishi et al., 2010; Kelly et al., 2010; Wang et al., 2010; Tsukahara et al., 2010). However, our data indicate that neither H3T3 phosphorylation nor H2AT120 phosphorylation are reduced when H4K20me3 is increased, making it unlikely that H3K9 or H4K20 methylation impinge on these regulatory mechanisms.
Instead, our data suggest that disruption of Aurora B localization following targeted recruitment of Suv39 or Suv420 to the centromere may arise due to misregulation of centromere cohesion and/or transcription. As with marks of heterochromatin, cohesin is enriched at pericentromeres (Eckert et al., 2007; Glynn et al., 2004; Weber et al., 2004). This distribution of cohesin is required for transcription of mitotic centromeres such that disruption of either pericentromere cohesin enrichment, or transcription itself, impairs Aurora B localization (Jambhekar et al., 2014; Kleyman et al., 2014; Perea-Resa et al., 2020). Work from our group and others has demonstrated that Suv420-dependent H4K20me3 enhances pericentromeric cohesin (Hahn et al., 2013; Manning et al., 2014; Bernard et al., 2001). This relationship would predict that Suv420-dependent methylation should promote centromere transcription and Aurora B localization. However, we find that following Suv39 or Suv420 tethering to centromeres, increased centromere H4K20me3 corresponds with reduced, not enhanced, transcript abundance and Aurora B localization at centromeres (Fig. S6C). Taken together, these data support a model whereby suppression of centromere transcription itself and/or the reduction in centromere transcripts resulting from spreading of H4K20me3 from the pericentromere into the centromere may be sufficient, irrespective of cohesin enrichment, to disrupt CPC localization.
Increased H4K20me3 contributes to chromosome instability
The aneuploid chromosome content that results from mitotic segregation errors contributes to infertility, and is a primary cause of non-viable embryos and birth defects (Baker et al., 2009; Choi et al., 2009; Gao et al., 2007; Heilig et al., 2010; Kuukasjarvi et al., 1997; McClelland et al., 2009; Nowell, 1976; Rajagopalan and Lengauer, 2004; Swanton et al., 2009; Sotillo et al., 2010; Cucco and Musio, 2016). Persistent underlying defects in chromosome segregation, termed chromosome instability (CIN), (Baker et al., 2009; Sotillo et al., 2007; Weaver et al., 2007; Hagstrom and Meyer, 2003) are prevalent in cancer contexts in which CIN promotes intratumor heterogeneity that in turn contributes to tumor evolution and drug resistance (Baker et al., 2009; Choi et al., 2009; Gao et al., 2007; Heilig et al., 2010; Kuukasjarvi et al., 1997; McClelland et al., 2009; Nowell, 1976; Rajagopalan and Lengauer, 2004; Swanton et al., 2009; Sotillo et al., 2010; Weaver et al., 2007). Merotelic kinetochore attachments are demonstrated to be the primary cause of segregation errors in human cells (Cimini et al., 2001, 2003; Ganem et al., 2009; Thompson and Compton, 2008), suggesting that corruption of the Aurora B error correction pathway may be prevalent in CIN. Nevertheless, although mutations in genes directly involved in spindle structure, chromosome segregation or mitotic checkpoints have been associated with a subset of cancers and hereditary disorders (Kim et al., 2012; Chung et al., 2012; Cahill et al., 1998, 1999; Cucco and Musio, 2016), these mutations do not explain CIN in the vast majority of contexts (Wang et al., 2004; Negrini et al., 2010; Rajagopalan and Lengauer, 2004).
Our data suggest that changes in centromere methylation may underlie CIN in some cancer contexts. Consistent with this model, our work and others demonstrate that depletion of H3K9 demethylase enzymes, or high expression of H3K9 or H4K20 methyltransferase enzymes compromise mitotic fidelity in experimental systems and/or correspond both with aneuploidy and poor outcome in human cancer patients (Fig. 1B; Table S1) (Janssen et al., 2018; Black et al., 2012; Kupershmit et al., 2014; Frescas et al., 2008). We find that centromere tethering of either Suv39 or Suv420 is sufficient to compromise Aurora B kinase localization at centromeres and results in reduced phosphorylation of Aurora B substrates that are critical for proper chromosome segregation. Hec1, a key Aurora B substrate that governs kinetochore microtubule stability and mitotic error correction, is also a substrate for the related Aurora A kinase (DeLuca, 2017). Consistent with this redundant regulation, our data indicate that, when centromere methylation is experimentally enhanced, or in cancer contexts in which Suv39 or Suv420 expression is high and H4K20me3 is therefore likely high, sensitivity to inhibition of both Aurora A and Aurora B kinase is increased (Fig. 7). The Aurora kinase family represent promising therapeutic targets that are actively being pursued in clinical and preclinical studies (Tang et al., 2017). Our results propose an intriguing possibility that levels of H4K20me3 (or expression of the enzymes that directly or indirectly promote this methylation state) may be predictive of CIN and may furthermore indicate sensitivity to molecular therapeutics that target Aurora B directly.
MATERIALS AND METHODS
Cell culture, siRNA and transgene expression
hTERT-RPE-1 (RPE-1) cells were grown in Dulbecco's modified Eagle medium (DMEM). Human breast cancer cell lines HCC1187, HCC202 and ZR-75-1 were grown in RPMI medium, SK-BR-3 grown in McCoy's 5a medium, and MCF7 grown in Eagle’s minimum essential medium. All cell culture media were supplemented with 10% fetal bovine serum and 1% penicillin+streptomycin, and cells were grown at 37°C with 5% CO2.
Depletion of KDM4A was carried out through transient transfection of one of four individual, or a SMARTpool of all four, ON-TARGETplus siRNA constructs (Horizon Inspired Cell Solutions) using RNAiMAX transfection reagent (Invitrogen) according to the manufacturer's instructions. Transfection with individual, or a SMARTpool of four, non-targeting siRNA sequences (Horizon Inspired Cell Solutions) was used as a negative control for KDM4A depletion. Knockdown efficiency was monitored by qPCR using KDM4A-specific primers. Expression of centromere-targeted, GFP-tagged cen-GFP, cen-Suv39-GFP and cen-Suv420-GFP fusion proteins, and non-targeted Suv420-GFP was achieved by cloning the respective cDNA into Addgene vector CENP-B DBD INCENP GFP (Addgene plasmid #45237) at NheI and BamH1. Inducible expression was achieved by cloning the GFP-tagged constructs into plvx-Tre3G-IRES (631362, Clonetech) at Not1 and NdeI restriction cut sites. Expression vectors were transiently or stably expressed using Lipofectamine 3000 transfection reagent, according to the manufacturer's instructions. Inducible expression of transgenes was achieved by the addition of 2 µg/ml doxycycline for 16–24 h. ‘Mock’ controls reflect the individual cell lines in the absence of doxycycline induction. Western blot analysis was used to confirm population-level expression of each construct. Immunofluorescence for GFP was used to confirm expression and centromere localization for all single-cell analyses performed. Sequences for all siRNA constructs and qPCR primers are presented in Table S3.
Metaphase spreads, fixation and staining for immunofluorescence imaging
Metaphase spreads were prepared as in Martins et al. (2016). Mitotic cells were collected by shake off following 3 h incubation in 100 ng/ml nocodazole, washed briefly in PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, KH2PO4) and incubated in 75 mM KCl for 12 min at 37°C. Cells were spun onto poly-L-lysine-coated chamber slides at 340 g for 5 min, then incubated in 37°C KCM buffer (120 mM KCl, 20 mM NaCl and 10 mM Tris-HCl pH 8.0, in 0.1% Triton X-100) for 10 min.
For analysis of kinetochore protein localization, untreated (metaphase analysis) or nocodazole-treated (prometaphase analysis) cells were prepared as in Kleyman et al. (2014). Cells were incubated in 3.5% paraformaldehyde for 15 min, quenched with 500 mM ammonium chloride in PBS for 10 min, incubated in ice-cold methanol for 5 min and washed briefly with PBS. For cen-INCENP-mCherry expression, cells were transiently transfected with Addgene plasmid #45233 36 h prior to doxycycline induction of cen-Suv420-GFP expression. For inhibition of Suv420 methyltransferase activity, cells were treated with 200 nM A196 concurrent with doxycycline induction of cen-Suv420-GFP expression.
For metaphase and anaphase analyses of chromosome alignment and segregation, cells were fixed in 4% paraformaldehyde for 20 min, washed briefly in PBS, post-extracted in PBS+0.5% Triton X-100 for 10 min, washed briefly in PBS, or alternatively fixed in ice-cold methanol for 10 min. For analysis of merotelic error correction capacity, cells were incubated in the presence of 100 µM monastrol for 4 h, washed twice for 5 min in drug-free growth medium at 37°C, and incubated in growth medium supplemented with 25 µM MG132 (to prevent anaphase progression) prior to fixation at indicated times. For inhibitor sensitivity assays, cells were treated with indicated concentrations of each drug for 12 h prior to fixation.
Chromosome spreads were stained with antibodies diluted in KCM+1% bovine serum albumin (BSA). All other fixation methods were followed by 30 min block in TBS-BSA (10 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% BSA)+0.1% Triton X-100 and incubation in primary then secondary antibodies in a humid chamber. Primary and secondary dilutions were made in TBS-BSA+0.1% Triton X-100. DNA was detected with 0.2 µg/ml 4′,6-diamidino-2-phenylindole (DAPI) added to secondary antibody incubations. Coverslips were mounted onto slides using Prolong Antifade Gold (Molecular Probes).
Antibodies
The following antibodies were used for immunofluorescence (IF) and/or western blot (WB) analyses: human anti-ACA (15-234, Antibodies Inc., 1:500); mouse anti-H3K9me2 (ab1220, Abcam, 1:1000); rabbit anti-H3K9me3 (ab176916, Abcam, 1:1000); rabbit anti-H3 (ab1791, Abcam, 1:1000); rabbit anti-H4K20me3 (ab9053, Abcam, 1:1000); rabbit anti-H4 (ab7311, Abcam, 1:1000); rabbit anti-H3S10p (04-817, Millipore, 1:1000); mouse anti-α-tubulin (dm1α, Sigma-Aldrich, 1:1000 for IF, 1:10,000 for WB); mouse anti-Aurora B (AIM-1, BD Biosciences, 1:1000); rabbit anti-H3T3p (97145, Cell Signaling Technology, 1:500); rabbit anti-H2AT120p (61196, Active Motif, 1:1000); rabbit anti-CENP-As7p (07-232, Upstate, 1:500); rabbit anti-CENP-A (2186, Cell Signaling Technology, 1:1000); rabbit anti-HEC1S55p (pA5-85846, Invitrogen, 1:500); mouse anti-HEC1 (9G3.23, Novus Biologicals, 1:1000); goat anti-GFP (ab6662, Abcam); rabbit anti-GFP (2956, Cell Signaling Technology, 1:2500); and rabbit anti-mCherry (PA5-34974, Invitrogen, 1:500).
Fluorescence microscopy, image analysis and statistics
Cells were captured with a Zyla sCMOS camera mounted on a Nikon Ti-E microscope with a 60× Plan Apo oil immersion objective for fixed immunofluorescence, or a 20× CFI Plan Fluor objective for live-cell imaging. All images within a single replicate were prepared and imaged in parallel, and images captured using the same exposure time. All single-cell or single-kinetochore measurements were normalized to the average intensity of the respective control condition and represented as fold change in fluorescence. For metaphase and anaphase cell analyses, a minimum of 30 mitotic cells were analyzed per condition for each of three biological replicates. In these analyses, lagging chromosomes are defined as single chromosomes (marked by ACA-stained kinetochore) that are located between the two masses of segregating chromosomes (anaphase plates) and at least as far from the next nearest kinetochore as the anaphase plate is wide (based on ACA staining). Significance between biological replicates (n=3) was determined using an unpaired two-tailed Student's t-test. To assess centromere specific protein levels, NIS Elements software was used to draw a line through ACA-stained centromere/kinetochore pairs. The intensity of antibody staining along the line was assessed for three kinetochore pairs per cell. To assess centromere enrichment, average pixel intensity of H4K20me3 staining was measured in ten uniform regions of interest (ROIs) defined at ACA-stained centromeres/kinetochores per cell and divided by the average H4K20me3 staining at ROIs defined on DAPI-stained chromatin. Analyses were performed for a minimum of 30 cells in each of three biological replicates. Significance between biological replicates (n=3) was determined using a two-tailed Student's t-test. Live-cell images were captured at ten coordinates per condition at 5-min intervals for 24 h for each of three biological replicates. Mitotic timing was recorded from NEB until anaphase onset (Mercadante et al., 2019) for a minimum of 50 cells per replicate, and significance between biological replicates (n=3) was determined using a two-tailed Student's t-test. All analyses were performed on unprocessed images. For publication images NIS Elements deconvolution software was used. All images within a single panel were cropped comparably using ImageJ software to allow for direct comparison.
qPCR analysis of centromere transcript abundance
RPE-1 cells carrying the inducible cen-Suv39-GFP or cen-Suv420-GFP construct were plated in cell culture flasks and induced, or not, with 2 µg/ml doxycycline for 24 h. Then, 100 ng/ml nocodazole was added to the growth medium for the last 4 h of induction. Mitotic cells were collected via manual shake off. RNA was isolated with Trizol and purified using Qiagen's RNAeasy columns. Purified RNA was quantitated by qPCR per the double delta Ct method that considers transcript changes relative to an internal control that is insensitive to the experimental manipulations (in this case GAPDH transcript), using transcript-specific primers (see Table S3), ABI 7500 and PowerUp SYBR Green Master Mix (Roche). Primers for centromere α satellite transcripts were as previously described (Liu et al., 2015; Xue et al., 2013).
Gene expression, aneuploidy correlation, patient survival and drug sensitivity analyses
For analyses of Suv39 and Suv420 isoform expression in cancer, RNA-seqv2 Illumina Hiseq RSEM-normalized data were acquired through FireBrowse (firebrowse.org) and aneuploidy scores obtained from (Taylor et al., 2018). Both datasets were imported into R statistical programming environment version 3.3.1 and run through a custom analysis pipeline to reformat and combine the datasets based on tumor sample. The Pearson correlation coefficient between aneuploidy score and gene expression was determined as described in Taylor et al. (2018), and linear regression analysis performed to determine statistical significance where P<0.0025 reflects a significant correlation. RPKM-normalized expression values of Suv39 and Suv420 isoforms in normal and cancer contexts were obtained from FireBrowse and filtered to select and graph only cancer subtypes where paired normal tissue samples were available. The Kaplan–Meier Plotter platform (kmplot.com/analysis; Nagy et al., 2018; Györffy et al., 2010) was used to query survival data relative to Suv39 and Suv420 isoform expression to compare samples for top and bottom quartile expression for their respective genes. Hochberg's step-up method corrected P-values were determined and significance defined as P≤0.0053. Gene expression data were downloaded from the Broad Cancer Cell Line Encyclopedia (CCLE) 2 January 2019 run containing expression data of 84,434 genes from 1457 different cancer cell lines. Drug screening half-maximal inhibitory concentration (IC50) area under the fitted dose–response curve (AUC) data were downloaded from the Genomics of Drug Sensitivity in Cancer database GDSC1 run containing 518 different drugs across 988 different cell lines. Both sets of data were then imported into R statistical programming environment version 3.3.1 and run through an analysis pipeline to reformat and combine the datasets based on cancer cell lines present in both. The datasets were sorted to independently identify the top and bottom quartile in terms of Suv39 or Suv420 expression. For both the high and low expression groups for each gene, the drug IC50 of each individual cell line and the mean±s.e.m. of all cell lines in each group were graphed for each drug of interest. Significance was determined using an unpaired two-tailed Student's t-test corrected for multiple comparisons using the Bonferroni correction method, where P<0.00625 is significant.
Supplementary Material
Acknowledgements
We thank members of the Manning laboratory for critical reading and feedback on the manuscript.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: A.L.M.; Methodology: C.P.H., S.H., N.M.H., E.A.C., A.L.M.; Validation: C.P.H.; Formal analysis: C.P.H., S.H., E.A.C.; Investigation: C.P.H., N.M.H.; Data curation: C.P.H.; Writing - original draft: C.P.H., A.L.M.; Writing - review & editing: C.P.H., S.H., N.M.H., E.A.C., A.L.M.; Visualization: C.P.H., N.M.H.; Supervision: A.L.M.; Project administration: A.L.M.; Funding acquisition: A.L.M.
Funding
This work was supported by the Richard and Susan Smith Family Foundation [Award for Excellence in Biomedical Research] and the Division of Cancer Prevention, National Cancer Institute [R00CA182731] to A.L.M. Deposited in PMC for release after 12 months.
Peer review history
The peer review history is available online at https://journals.biologists.com/jcs/article-lookup/doi/10.1242/jcs.249763
References
- Abe, Y., Sako, K., Takagaki, K., Hirayama, Y., Uchida, K. S. K., Herman, J. A., Deluca, J. G. and Hirota, T. (2016). HP1-assisted Aurora B kinase activity prevents chromosome segregation errors. Dev. Cell 36, 487-497. 10.1016/j.devcel.2016.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker, D. J., Jin, F., Jeganathan, K. B. and Van Deursen, J. M. (2009). Whole chromosome instability caused by Bub1 insufficiency drives tumorigenesis through tumor suppressor gene loss of heterozygosity. Cancer Cell 16, 475-486. 10.1016/j.ccr.2009.10.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnhart, M. C., Kuich, P. H. J. L., Stellfox, M. E., Ward, J. A., Bassett, E. A., Black, B. E. and Foltz, D. R. (2011). HJURP is a CENP-A chromatin assembly factor sufficient to form a functional de novo kinetochore. J. Cell Biol. 194, 229-243. 10.1083/jcb.201012017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard, P., Maure, J. F., Partridge, J. F., Genier, S., Javerzat, J. P. and Allshire, R. C. (2001). Requirement of heterochromatin for cohesion at centromeres. Science 294, 2539-2542. 10.1126/science.1064027 [DOI] [PubMed] [Google Scholar]
- Berry, W. L. and Janknecht, R. (2013). KDM4/JMJD2 histone demethylases: epigenetic regulators in cancer cells. Cancer Res. 73, 2936-2942. 10.1158/0008-5472.CAN-12-4300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black, J. C., Van Rechem, C. and Whetstine, J. R. (2012). Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol. Cell 48, 491-507. 10.1016/j.molcel.2012.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blower, M. D. (2016). Centromeric transcription regulates Aurora-B localization and activation. Cell Rep. 15, 1624-1633. 10.1016/j.celrep.2016.04.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broad, A. J., Deluca, K. F. and Deluca, J. G. (2020). Aurora B kinase is recruited to multiple discrete kinetochore and centromere regions in human cells. J. Cell Biol. 219, e201905144. 10.1083/jcb.201905144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg, K. D., Mitchell, T. R. H., Upadhyay, A. K., Jakob, C. G., Jhala, M. A., Comess, K. M., Lasko, L. M., Li, C., Tuzon, C. T., Dai, Y.et al. (2017). The SUV4-20 inhibitor A-196 verifies a role for epigenetics in genomic integrity. Nat. Chem. Biol. 13, 317-324. 10.1038/nchembio.2282 [DOI] [PubMed] [Google Scholar]
- Cahill, D. P., Lengauer, C., Yu, J., Riggins, G. J., Willson, J. K., Markowitz, S. D., Kinzler, K. W. and Vogelstein, B. (1998). Mutations of mitotic checkpoint genes in human cancers. Nature 392, 300-303. 10.1038/32688 [DOI] [PubMed] [Google Scholar]
- Cahill, D. P., Da Costa, L. T., Carson-Walter, E. B., Kinzler, K. W., Vogelstein, B. and Lengauer, C. (1999). Characterization of MAD2B and other mitotic spindle checkpoint genes. Genomics 58, 181-187. 10.1006/geno.1999.5831 [DOI] [PubMed] [Google Scholar]
- Carretero, M., Ruiz-Torres, M., Rodríguez-Corsino, M., Barthelemy, I. and Losada, A. (2013). Pds5B is required for cohesion establishment and Aurora B accumulation at centromeres. EMBO J. 32, 2938-2949. 10.1038/emboj.2013.230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll, C. W. and Straight, A. F. (2006). Centromere formation: from epigenetics to self-assembly. Trends Cell Biol. 16, 70-78. 10.1016/j.tcb.2005.12.008 [DOI] [PubMed] [Google Scholar]
- Cheeseman, I. M., Chappie, J. S., Wilson-Kubalek, E. M. and Desai, A. (2006). The conserved KMN network constitutes the core microtubule-binding site of the kinetochore. Cell 127, 983-997. 10.1016/j.cell.2006.09.039 [DOI] [PubMed] [Google Scholar]
- Choi, C.-M., Seo, K. W., Jang, S. J., Oh, Y.-M., Shim, T.-S., Kim, W. S., Lee, D.-S. and Lee, S.-D. (2009). Chromosomal instability is a risk factor for poor prognosis of adenocarcinoma of the lung: fluorescence in situ hybridization analysis of paraffin-embedded tissue from Korean patients. Lung Cancer 64, 66-70. 10.1016/j.lungcan.2008.07.016 [DOI] [PubMed] [Google Scholar]
- Chung, N. G., Kim, M. S., Yoo, N. J. and Lee, S. H. (2012). Somatic mutation of STAG2, an aneuploidy-related gene, is rare in acute leukemias. Leukemia Lymphoma 53, 1234-1235. 10.3109/10428194.2011.645819 [DOI] [PubMed] [Google Scholar]
- Cimini, D., Howell, B., Maddox, P., Khodjakov, A., Degrassi, F. and Salmon, E. D. (2001). Merotelic kinetochore orientation is a major mechanism of aneuploidy in mitotic mammalian tissue cells. J. Cell Biol. 153, 517-527. 10.1083/jcb.153.3.517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimini, D., Moree, B., Canman, J. C. and Salmon, E. D. (2003). Merotelic kinetochore orientation occurs frequently during early mitosis in mammalian tissue cells and error correction is achieved by two different mechanisms. J. Cell Sci. 116, 4213-4225. 10.1242/jcs.00716 [DOI] [PubMed] [Google Scholar]
- Cimini, D., Wan, X., Hirel, C. B. and Salmon, E. D. (2006). Aurora kinase promotes turnover of kinetochore microtubules to reduce chromosome segregation errors. Curr. Biol. 16, 1711-1718. 10.1016/j.cub.2006.07.022 [DOI] [PubMed] [Google Scholar]
- Cucco, F. and Musio, A. (2016). Genome stability: what we have learned from cohesinopathies. Am. J. Med. Genet. C Semin. Med. Genet. 172, 171-178. 10.1002/ajmg.c.31492 [DOI] [PubMed] [Google Scholar]
- Cuellar, T. L., Herzner, A.-M., Zhang, X., Goyal, Y., Watanabe, C., Friedman, B. A., Janakiraman, V., Durinck, S., Stinson, J., Arnott, D.et al. (2017). Silencing of retrotransposons by SETDB1 inhibits the interferon response in acute myeloid leukemia. J. Cell Biol. 216, 3535-3549. 10.1083/jcb.201612160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai, J., Sullivan, B. A. and Higgins, J. M. (2006). Regulation of mitotic chromosome cohesion by Haspin and Aurora B. Dev. Cell 11, 741-750. 10.1016/j.devcel.2006.09.018 [DOI] [PubMed] [Google Scholar]
- Davoli, T., Xu, A. W., Mengwasser, K. E., Sack, L. M., Yoon, J. C., Park, P. J. and Elledge, S. J. (2013). Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell 155, 948-962. 10.1016/j.cell.2013.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rop, V., Padeganeh, A. and Maddox, P. S. (2012). CENP-A: the key player behind centromere identity, propagation, and kinetochore assembly. Chromosoma 121, 527-538. 10.1007/s00412-012-0386-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deluca, J. G. (2017). Aurora A kinase function at kinetochores. Cold Spring Harb. Symp. Quant. Biol. 82, 91-99. 10.1101/sqb.2017.82.034991 [DOI] [PubMed] [Google Scholar]
- Deluca, J. G., Gall, W. E., Ciferri, C., Cimini, D., Musacchio, A. and Salmon, E. D. (2006). Kinetochore microtubule dynamics and attachment stability are regulated by Hec1. Cell 127, 969-982. 10.1016/j.cell.2006.09.047 [DOI] [PubMed] [Google Scholar]
- Deluca, K. F., Lens, S. M. and Deluca, J. G. (2011). Temporal changes in Hec1 phosphorylation control kinetochore-microtubule attachment stability during mitosis. J. Cell Sci. 124, 622-634. 10.1242/jcs.072629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ditchfield, C., Johnson, V. L., Tighe, A., Ellston, R., Haworth, C., Johnson, T., Mortlock, A., Keen, N. and Taylor, S. S. (2003). Aurora B couples chromosome alignment with anaphase by targeting BubR1, Mad2, and Cenp-E to kinetochores. J. Cell Biol. 161, 267-280. 10.1083/jcb.200208091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert, C. A., Gravdahl, D. J. and Megee, P. C. (2007). The enhancement of pericentromeric cohesin association by conserved kinetochore components promotes high-fidelity chromosome segregation and is sensitive to microtubule-based tension. Genes Dev. 21, 278-291. 10.1101/gad.1498707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenech, M., Kirsch-Volders, M., Natarajan, A. T., Surralles, J., Crott, J. W., Parry, J., Norppa, H., Eastmond, D. A., Tucker, J. D. and Thomas P. (2011). Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis 26, 125-132. 10.1093/mutage/geq052 [DOI] [PubMed] [Google Scholar]
- Fioriniello, S., Marano, D., Fiorillo, F., D'esposito, M. and Della Ragione, F. (2020). Epigenetic factors that control pericentric heterochromatin organization in mammals. Genes 11, 595. 10.3390/genes11060595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischle, W., Tseng, B. S., Dormann, H. L., Ueberheide, B. M., Garcia, B. A., Shabanowitz, J., Hunt, D. F., Funabiki, H. and Allis, C. D. (2005). Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature 438, 1116-1122. 10.1038/nature04219 [DOI] [PubMed] [Google Scholar]
- Frescas, D., Guardavaccaro, D., Kuchay, S. M., Kato, H., Poleshko, A., Basrur, V., Elenitoba-Johnson, K. S., Katz, R. A. and Pagano, M. (2008). KDM2A represses transcription of centromeric satellite repeats and maintains the heterochromatic state. Cell Cycle 7, 3539-3547. 10.4161/cc.7.22.7062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem, N. J., Godinho, S. A. and Pellman, D. (2009). A mechanism linking extra centrosomes to chromosomal instability. Nature 460, 278-282. 10.1038/nature08136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, C., Furge, K., Koeman, J., Dykema, K., Su, Y., Cutler, M. L., Werts, A., Haak, P. and Vande Woude, G. F. (2007). Chromosome instability, chromosome transcriptome, and clonal evolution of tumor cell populations. Proc. Natl. Acad. Sci. USA 104, 8995-9000. 10.1073/pnas.0700631104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghandi, M., Huang, F. W., Jané-Valbuena, J., Kryukov, G. V., Lo, C. C., Mcdonald, E. R., Barretina, J., Gelfand, E. T., Bielski, C. M., Li, H.et al. (2019). Next-generation characterization of the cancer cell line encyclopedia. Nature 569, 503-508. 10.1038/s41586-019-1186-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glynn, E. F., Megee, P. C., Yu, H.-G., Mistrot, C., Unal, E., Koshland, D. E., Derisi, J. L. and Gerton, J. L. (2004). Genome-wide mapping of the cohesin complex in the yeast Saccharomyces cerevisiae. PLoS Biol. 2, e259. 10.1371/journal.pbio.0020259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregan, J., Polakova, S., Zhang, L., Tolić-Nørrelykke, I. M. and Cimini, D. (2011). Merotelic kinetochore attachment: causes and effects. Trends Cell Biol. 21, 374-381. 10.1016/j.tcb.2011.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guler, G. D., Tindell, C. A., Pitti, R., Wilson, C., Nichols, K., Kaiwai Cheung, T., Kim, H.-J., Wongchenko, M., Yan, Y., Haley, B.et al. (2017). Repression of stress-induced LINE-1 expression protects cancer cell subpopulations from lethal drug exposure. Cancer Cell 32, 221-237.e13. 10.1016/j.ccell.2017.07.002 [DOI] [PubMed] [Google Scholar]
- Guse, A., Carroll, C. W., Moree, B., Fuller, C. J. and Straight, A. F. (2011). In vitro centromere and kinetochore assembly on defined chromatin templates. Nature 477, 354-358. 10.1038/nature10379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Györffy, B., Lanczky, A., Eklund, A. C., Denkert, C., Budczies, J., Li, Q. and Szallasi, Z. (2010). An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res. Treat. 123, 725-731. 10.1007/s10549-009-0674-9 [DOI] [PubMed] [Google Scholar]
- Hadders, M. A., Hindriksen, S., Truong, M. A., Mhaskar, A. N., Wopken, J. P., Vromans, M. J. M. and Lens, S. M. A. (2020). Untangling the contribution of Haspin and Bub1 to Aurora B function during mitosis. J. Cell Biol. 219, e201907087. 10.1083/jcb.201907087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagstrom, K. A. and Meyer, B. J. (2003). Condensin and cohesin: more than chromosome compactor and glue. Nat. Rev. Genet. 4, 520-534. 10.1038/nrg1110 [DOI] [PubMed] [Google Scholar]
- Hahn, M., Dambacher, S., Dulev, S., Kuznetsova, A. Y., Eck, S., Worz, S., Sadic, D., Schulte, M., Mallm, J.-P., Maiser, A.et al. (2013). Suv4-20h2 mediates chromatin compaction and is important for cohesin recruitment to heterochromatin. Genes Dev. 27, 859-872. 10.1101/gad.210377.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauf, S., Cole, R. W., Laterra, S., Zimmer, C., Schnapp, G., Walter, R., Heckel, A., Van Meel, J., Rieder, C. L. and Peters, J. M. (2003). The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J. Cell Biol. 161, 281-294. 10.1083/jcb.200208092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilig, C. E., Löffler, H., Mahlknecht, U., Janssen, J. W. G., Ho, A. D., Jauch, A. and Krämer, A. (2010). Chromosomal instability correlates with poor outcome in patients with myelodysplastic syndromes irrespectively of the cytogenetic risk group. J. Cell Mol. Med. 14, 895-902. 10.1111/j.1582-4934.2009.00905.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindriksen, S., Lens, S. M. A. and Hadders, M. A. (2017). The ins and outs of Aurora B inner centromere localization. Front. Cell Dev. Biol. 5, 112. 10.3389/fcell.2017.00112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinshaw, S. M. and Harrison, S. C. (2018). Kinetochore function from the bottom up. Trends Cell Biol. 28, 22-33. 10.1016/j.tcb.2017.09.002 [DOI] [PubMed] [Google Scholar]
- Hirota, T., Lipp, J. J., Toh, B.-H. and Peters, J.-M. (2005). Histone H3 serine 10 phosphorylation by Aurora B causes HP1 dissociation from heterochromatin. Nature 438, 1176-1180. 10.1038/nature04254 [DOI] [PubMed] [Google Scholar]
- Huang, H., Lampson, M., Efimov, A. and Yen, T. J. (2018). Chromosome instability in tumor cells due to defects in Aurora B mediated error correction at kinetochores. Cell Cycle 17, 2622-2636. 10.1080/15384101.2018.1553340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ideue, T., Cho, Y., Nishimura, K. and Tani, T. (2014). Involvement of satellite I noncoding RNA in regulation of chromosome segregation. Genes Cells 19, 528-538. 10.1111/gtc.12149 [DOI] [PubMed] [Google Scholar]
- Jambhekar, A., Emerman, A. B., Schweidenback, C. T. and Blower, M. D. (2014). RNA stimulates Aurora B kinase activity during mitosis. PLoS ONE 9, e100748. 10.1371/journal.pone.0100748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen, A., Colmenares, S. U. and Karpen, G. H. (2018). Heterochromatin: guardian of the genome. Annu. Rev. Cell Dev. Biol. 34, 265-288. 10.1146/annurev-cellbio-100617-062653 [DOI] [PubMed] [Google Scholar]
- Kallio, M. J., Mccleland, M. L., Stukenberg, P. T. and Gorbsky, G. J. (2002). Inhibition of Aurora B kinase blocks chromosome segregation, overrides the spindle checkpoint, and perturbs microtubule dynamics in mitosis. Curr. Biol. 12, 900-905. 10.1016/S0960-9822(02)00887-4 [DOI] [PubMed] [Google Scholar]
- Kang, J., Chaudhary, J., Dong, H., Kim, S., Brautigam, C. A. and Yu, H. (2011). Mitotic centromeric targeting of HP1 and its binding to Sgo1 are dispensable for sister-chromatid cohesion in human cells. Mol. Biol. Cell 22, 1181-1190. 10.1091/mbc.e11-01-0009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor, T. M., Mayer, T. U., Coughlin, M. L. and Mitchison, T. J. (2000). Probing spindle assembly mechanisms with monastrol, a small molecule inhibitor of the mitotic kinesin, Eg5. J. Cell Biol. 150, 975-988. 10.1083/jcb.150.5.975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly, A. E., Ghenoiu, C., Xue, J. Z., Zierhut, C., Kimura, H. and Funabiki, H. (2010). Survivin reads phosphorylated histone H3 threonine 3 to activate the mitotic kinase Aurora B. Science 330, 235-239. 10.1126/science.1189505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, M. S., Kim, S. S., Je, E. M., Yoo, N. J. and Lee, S. H. (2012). Mutational and expressional analyses of STAG2 gene in solid cancers. Neoplasma 59, 524-529. 10.4149/neo_2012_067 [DOI] [PubMed] [Google Scholar]
- Kleyman, M., Kabeche, L. and Compton, D. A. (2014). STAG2 promotes error correction in mitosis by regulating kinetochore-microtubule attachments. J. Cell Sci. 127, 4225-4233. 10.1242/jcs.151613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krenn, V. and Musacchio, A. (2015). The Aurora B Kinase in chromosome bi-orientation and spindle checkpoint signaling. Front. Oncol. 5, 225. 10.3389/fonc.2015.00225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupershmit, I., Khoury-Haddad, H., Awwad, S. W., Guttmann-Raviv, N. and Ayoub, N. (2014). KDM4C (GASC1) lysine demethylase is associated with mitotic chromatin and regulates chromosome segregation during mitosis. Nucleic Acids Res. 42, 6168-6182. 10.1093/nar/gku253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuukasjarvi, T., Karhu, R., Tanner, M., Kahkonen, M., Schaffer, A., Nupponen, N., Pennanen, S., Kallioniemi, A., Kallioniemi, O. P. and Isola, J. (1997). Genetic heterogeneity and clonal evolution underlying development of asynchronous metastasis in human breast cancer. Cancer Res. 57, 1597-1604. [PubMed] [Google Scholar]
- Lam, A. L., Boivin, C. D., Bonney, C. F., Rudd, M. K. and Sullivan, B. A. (2006). Human centromeric chromatin is a dynamic chromosomal domain that can spread over noncentromeric DNA. Proc. Natl. Acad. Sci. USA 103, 4186-4191. 10.1073/pnas.0507947103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampson, M. A., Renduchitala, K., Khodjakov, A. and Kapoor, T. M. (2004). Correcting improper chromosome-spindle attachments during cell division. Nat. Cell Biol. 6, 232-237. 10.1038/ncb1102 [DOI] [PubMed] [Google Scholar]
- Liang, C., Zhang, Z., Chen, Q., Yan, H., Zhang, M., Zhou, L., Xu, J., Lu, W. and Wang, F. (2020). Centromere-localized Aurora B kinase is required for the fidelity of chromosome segregation. J. Cell Biol. 219, e201907092. 10.1083/jcb.201907092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, H., Qu, Q., Warrington, R., Rice, A., Cheng, N. and Yu, H. (2015). Mitotic transcription installs Sgo1 at centromeres to coordinate chromosome segregation. Mol. Cell 59, 426-436. 10.1016/j.molcel.2015.06.018 [DOI] [PubMed] [Google Scholar]
- Liu, Y., Chen, C., Xu, Z., Scuoppo, C., Rillahan, C. D., Gao, J., Spitzer, B., Bosbach, B., Kastenhuber, E. R., Baslan, T.et al. (2016). Deletions linked to TP53 loss drive cancer through p53-independent mechanisms. Nature 531, 471-475. 10.1038/nature17157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning, A. L., Yazinski, S. A., Nicolay, B., Bryll, A., Zou, L. and Dyson, N. J. (2014). Suppression of genome instability in pRB-deficient cells by enhancement of chromosome cohesion. Mol. Cell 53, 993-1004. 10.1016/j.molcel.2014.01.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins, N. M. C., Bergmann, J. H., Shono, N., Kimura, H., Larionov, V., Masumoto, H. and Earnshaw, W. C. (2016). Epigenetic engineering shows that a human centromere resists silencing mediated by H3K27me3/K9me3. Mol. Biol. Cell 27, 177-196. 10.1091/mbc.E15-08-0605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins, N. M. C., Cisneros-Soberanis, F., Pesenti, E., Kochanova, N. Y., Shang, W.-H., Hori, T., Nagase, T., Kimura, H., Larionov, V., Masumoto, H.et al. (2020). H3K9me3 maintenance on a human artificial chromosome is required for segregation but not centromere epigenetic memory. J. Cell Sci. 133, jcs242610. 10.1242/jcs.242610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcclelland, S. E., Burrell, R. A. and Swanton, C. (2009). Chromosomal instability: a composite phenotype that influences sensitivity to chemotherapy. Cell Cycle 8, 3262-3266. 10.4161/cc.8.20.9690 [DOI] [PubMed] [Google Scholar]
- Meppelink, A., Kabeche, L., Vromans, M. J., Compton, D. A. and Lens, S. M. (2015). Shugoshin-1 balances Aurora B kinase activity via PP2A to promote chromosome bi-orientation. Cell Rep. 11, 508-515. 10.1016/j.celrep.2015.03.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercadante, D. L., Crowley, E. A. and Manning, A. L. (2019). Live cell imaging to assess the dynamics of metaphase timing and cell fate following mitotic spindle perturbations. J. Vis. Exp. 10.3791/60255 [DOI] [PubMed] [Google Scholar]
- Muro, Y., Masumoto, H., Yoda, K., Nozaki, N., Ohashi, M. and Okazaki, T. (1992). Centromere protein B assembles human centromeric alpha-satellite DNA at the 17-bp sequence, CENP-B box. J. Cell Biol. 116, 585-596. 10.1083/jcb.116.3.585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy, Á., Lánczky, A., Menyhárt, O. and Győrffy, B. (2018). Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci. Rep. 8, 9227. 10.1038/s41598-018-27521-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negrini, S., Gorgoulis, V. G. and Halazonetis, T. D. (2010). Genomic instability — an evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 11, 220-228. 10.1038/nrm2858 [DOI] [PubMed] [Google Scholar]
- Nowell, P. C. (1976). The clonal evolution of tumor cell populations. Science 194, 23-28. 10.1126/science.959840 [DOI] [PubMed] [Google Scholar]
- O'carroll, D., Scherthan, H., Peters, A. H. F. M., Opravil, S., Haynes, A. R., Laible, G., Rea, S., Schmid, M., Lebersorger, A., Jerratsch, M.et al. (2000). Isolation and characterization of Suv39h2, a second histone H3 methyltransferase gene that displays testis-specific expression. Mol. Cell. Biol. 20, 9423-9433. 10.1128/MCB.20.24.9423-9433.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohzeki, J.-i., Bergmann, J. H., Kouprina, N., Noskov, V. N., Nakano, M., Kimura, H., Earnshaw, W. C., Larionov, V. and Masumoto, H. (2012). Breaking the HAC Barrier: histone H3K9 acetyl/methyl balance regulates CENP-A assembly. EMBO J. 31, 2391-2402. 10.1038/emboj.2012.82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohzeki, J., Larionov, V., Earnshaw, W. C. and Masumoto, H. (2019). De novo formation and epigenetic maintenance of centromere chromatin. Curr. Opin. Cell Biol. 58, 15-25. 10.1016/j.ceb.2018.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea-Resa, C., Bury, L., Cheeseman, I. M. and Blower, M. D. (2020). Cohesin removal reprograms gene expression upon mitotic entry. Mol. Cell 78, 127-140.e7. 10.1016/j.molcel.2020.01.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfau, S. J. and Amon, A. (2012). Chromosomal instability and aneuploidy in cancer: from yeast to man. EMBO Rep. 13, 515-527. 10.1038/embor.2012.65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluta, A. F., Saitoh, N., Goldberg, I. and Earnshaw, W. C. (1992). Identification of a subdomain of CENP-B that is necessary and sufficient for localization to the human centromere. J. Cell Biol. 116, 1081-1093. 10.1083/jcb.116.5.1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopalan, H. and Lengauer, C. (2004). Aneuploidy and cancer. Nature 432, 338-341. 10.1038/nature03099 [DOI] [PubMed] [Google Scholar]
- Schotta, G., Lachner, M., Sarma, K., Ebert, A., Sengupta, R., Reuter, G., Reinberg, D. and Jenuwein, T. (2004). A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev. 18, 1251-1262. 10.1101/gad.300704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma, A. B., Dimitrov, S., Hamiche, A. and Van Dyck, E. (2019). Centromeric and ectopic assembly of CENP-A chromatin in health and cancer: old marks and new tracks. Nucleic Acids Res. 47, 1051-1069. 10.1093/nar/gky1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotillo, R., Hernando, E., Díaz-Rodríguez, E., Teruya-Feldstein, J., Cordón-Cardo, C., Lowe, S. W. and Benezra, R. (2007). Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell 11, 9-23. 10.1016/j.ccr.2006.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotillo, R., Schvartzman, J.-M., Socci, N. D. and Benezra, R. (2010). Mad2-induced chromosome instability leads to lung tumour relapse after oncogene withdrawal. Nature 464, 436-440. 10.1038/nature08803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan, B. A. and Karpen, G. H. (2004). Centromeric chromatin exhibits a histone modification pattern that is distinct from both euchromatin and heterochromatin. Nat. Struct. Mol. Biol. 11, 1076-1083. 10.1038/nsmb845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanton, C., Nicke, B., Schuett, M., Eklund, A. C., Ng, C., Li, Q., Hardcastle, T., Lee, A., Roy, R., East, P.et al. (2009). Chromosomal instability determines taxane response. Proc. Natl. Acad. Sci. USA 106, 8671-8676. 10.1073/pnas.0811835106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, A., Gao, K., Chu, L., Zhang, R., Yang, J. and Zheng, J. (2017). Aurora kinases: novel therapy targets in cancers. Oncotarget 8, 23937-23954. 10.18632/oncotarget.14893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, A. M., Shih, J., Ha, G., Gao, G. F., Zhang, X., Berger, A. C., Schumacher, S. E., Wang, C., Hu, H., Liu, J.et al. (2018). Genomic and functional approaches to understanding cancer aneuploidy. Cancer Cell 33, 676-689.e3. 10.1016/j.ccell.2018.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, G. E., Renjith, M. R. and Manna, T. K. (2017). Kinetochore-microtubule interactions in chromosome segregation: lessons from yeast and mammalian cells. Biochem. J. 474, 3559-3577. 10.1042/BCJ20170518 [DOI] [PubMed] [Google Scholar]
- Thompson, S. L. and Compton, D. A. (2008). Examining the link between chromosomal instability and aneuploidy in human cells. J. Cell Biol. 180, 665-672. 10.1083/jcb.200712029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang, L. W. K., Hu, N. and Underhill, D. A. (2010). Comparative analyses of SUV420H1 isoforms and SUV420H2 reveal differences in their cellular localization and effects on myogenic differentiation. PLoS ONE 5, e14447. 10.1371/journal.pone.0014447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukahara, T., Tanno, Y. and Watanabe, Y. (2010). Phosphorylation of the CPC by Cdk1 promotes chromosome bi-orientation. Nature 467, 719-723. 10.1038/nature09390 [DOI] [PubMed] [Google Scholar]
- Wang, Z., Cummins, J. M., Shen, D., Cahill, D. P., Jallepalli, P. V., Wang, T.-L., Parsons, D. W., Traverso, G., Awad, M., Silliman, N.et al. (2004). Three classes of genes mutated in colorectal cancers with chromosomal instability. Cancer Res. 64, 2998-3001. 10.1158/0008-5472.CAN-04-0587 [DOI] [PubMed] [Google Scholar]
- Wang, F., Dai, J., Daum, J. R., Niedzialkowska, E., Banerjee, B., Stukenberg, P. T., Gorbsky, G. J. and Higgins, J. M. (2010). Histone H3 Thr-3 phosphorylation by Haspin positions Aurora B at centromeres in mitosis. Science 330, 231-235. 10.1126/science.1189435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, E., Ballister, E. R. and Lampson, M. A. (2011). Aurora B dynamics at centromeres create a diffusion-based phosphorylation gradient. J. Cell Biol. 194, 539-549. 10.1083/jcb.201103044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver, B. A. A., Silk, A. D., Montagna, C., Verdier-Pinard, P. and Cleveland, D. W. (2007). Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell 11, 25-36. 10.1016/j.ccr.2006.12.003 [DOI] [PubMed] [Google Scholar]
- Weber, S. A., Gerton, J. L., Polancic, J. E., Derisi, J. L., Koshland, D. and Megee, P. C. (2004). The kinetochore is an enhancer of pericentric cohesin binding. PLoS Biol. 2, e260. 10.1371/journal.pbio.0020260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welburn, J. P. I., Vleugel, M., Liu, D., Yates, J. R., Lampson, M. A., Fukagawa, T. and Cheeseman, I. M. (2010). Aurora B phosphorylates spatially distinct targets to differentially regulate the kinetochore-microtubule interface. Mol. Cell 38, 383-392. 10.1016/j.molcel.2010.02.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue, J., Wijeratne, S. S. K. and Zempleni, J. (2013). Holocarboxylase synthetase synergizes with methyl CpG binding protein 2 and DNA methyltransferase 1 in the transcriptional repression of long-terminal repeats. Epigenetics 8, 504-511. 10.4161/epi.24449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagishi, Y., Honda, T., Tanno, Y. and Watanabe, Y. (2010). Two histone marks establish the inner centromere and chromosome bi-orientation. Science 330, 239-243. 10.1126/science.1194498 [DOI] [PubMed] [Google Scholar]
- Yang, W., Soares, J., Greninger, P., Edelman, E. J., Lightfoot, H., Forbes, S., Bindal, N., Beare, D., Smith, J. A., Thompson, I. R.et al. (2013). Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 41, D955-D961. 10.1093/nar/gks1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama, Y., Hieda, M., Nishioka, Y., Matsumoto, A., Higashi, S., Kimura, H., Yamamoto, H., Mori, M., Matsuura, S. and Matsuura, N. (2013). Cancer-associated upregulation of histone H3 lysine 9 trimethylation promotes cell motility in vitro and drives tumor formation in vivo. Cancer Sci. 104, 889-895. 10.1111/cas.12166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, M., Li, Y., Lin, S., Chen, Y., Qian, Y., Zhao, Z. and Fan, H. (2019). H3K9me3, H3K36me3, and H4K20me3 expression correlates with patient outcome in esophageal squamous cell carcinoma as epigenetic markers. Dig. Dis. Sci. 64, 2147-2157. 10.1007/s10620-019-05529-2 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.