

Summary
Deep learning has emerged as the technique of choice for identifying hidden patterns in cell imaging data, but is often criticized as ‘black-box’. Here, we employ a generative neural network in combination with supervised machine learning to classify patient-derived melanoma xenografts as ‘efficient’ or ‘inefficient’ metastatic, validate predictions regarding melanoma cell lines with unknown metastatic efficiency in mouse xenografts, and use the network to generate in silico cell images that amplify the critical predictive cell properties. These exaggerated images unveiled pseudopodial extensions and increased light scattering as hallmark properties of metastatic cells. We validated this interpretation using live cells spontaneously transitioning between states indicative of low and high metastatic efficiency. This study illustrates how the application of Artificial Intelligence can support the identification of cellular properties that are predictive of complex phenotypes and integrated cell functions, but are too subtle to be identified in the raw imagery by a human expert.
A record of this paper’s Transparent Peer Review process is included in the Supplemental Information.
eTOC Blurb
We “reverse engineered” a convolutional neural network (CNN) autoencoder to identify cellular properties that distinguish aggressive from less aggressive metastatic melanoma using label free movies of living cells. This was achieved by amplifying in synthetic cell images the cellular features that define metastatic efficiency but are too subtle to be identified in the raw imagery. The CNN and classifier were validated by comparing predicted and experimental spreading of new melanoma cell lines xenografted into mice.
Graphical Abstract
Introduction
Recent machine learning studies have impressively demonstrated that label-free images contain information on the molecular organization within the cell (Cheng et al., 2021; Christiansen et al., 2018; Guo et al., 2019; LaChance and Cohen, 2020; Ounkomol et al., 2018; Sullivan and Lundberg, 2018; Yuan et al., 2018). These studies relied on generative models that transform label-free to fluorescent images, which can indicate the organization and, in some situations, even the relative densities of molecular structures. Models were trained by using pairs of label-free and fluorescence images subject to minimizing the error between the fluorescence ground-truth image and the model-generated image. Other studies used similar concepts to enhance imaging resolution by learning a mapping from low-to-high resolution (Belthangady and Royer, 2019; Fang et al., 2019; Nehme et al., 2018; Ouyang et al., 2018; Wang et al., 2019; Weigert et al., 2018). Common to all these studies is the concept that the architecture of a deep convolutional neural network can extract from the label-free or low-resolution cell images unstructured hidden information - also referred to as latent information - that is predictive of the molecular organization of a cell or its high-resolution image, yet escapes the human eye.
We wondered whether this paradigm could be applied also to the prediction of complex cell states that result from the convergence of numerous structural and molecular factors. We combined unsupervised generative deep neural networks and supervised machine learning to train a classifier that can predict the metastatic efficiency of human melanoma cells. The power of cell appearance for determining cell states that correlate with function has been the basis of decades of histopathology (Chan, 2014a; López, 2013a; Travis et al., 2013). Cell appearance has been established as an explicit predictor of signaling states that are directly implicated in the regulation of cell morphogenesis (Bakal et al., 2007; Goodman and Carpenter, 2016; Gordonov et al., 2015; Pascual-Vargas et al., 2017; Scheeder et al., 2018; Sero and Bakal, 2017; Yin et al., 2013). Whether cell appearance is also informative of a broader spectrum of cell signaling programs, such as those driving processes in metastasis, is less clear, although very recent work, using conventional shape-based machine learning of fluorescently labeled cell lines, suggests this may be the case (Wu et al., 2020).
The paradigm of extracting latent information via deep convolutional neural networks from label-free and time-resolved image sequences holds particularly strong promise for a task of this complexity. The design of cell appearance metrics that encode the state of, e.g., a cellular signal that promotes cell survival or proliferation, exceeds human intuition. The flip side of learning information that classifies well but is non-intuitive is the discomfort of relying on a ‘black box’. Especially in a clinical setting, the lack of a straightforward meaning of key drivers of a classifier is a widely perceived weakness of deep learning systems. Here, we demonstrate a mechanism to overcome this problem: By generating “in silico” cell images that were never observed experimentally we “reverse engineered” the physical properties of the latent image information that discriminates melanoma cells with low versus high metastatic efficiency. These results demonstrate that the internal encoding of latent variables in a deep convolutional neural network can be mapped to physical entities predictive of complex cell states. More broadly, they highlight the potential of “interpreted artificial intelligence” to augment investigator-driven analysis of cell behavior with an entirely novel set of hypotheses.
Results
Label-free imaging of living patient-derived xenograft (PDX) melanoma cells and cell lines
To test whether the latent information extracted from label-free live cell movies can predict the metastatic propensity of melanoma, we relied on a previously established patient-derived xenotransplantation (PDX) assay, in which tumor samples from stage III melanoma patients were taken and repeatedly transplanted between immuno-compromised mice (Quintana et al., 2012). All tumors grew and eventually seeded metastases in the xenograft model. Whereas some tumors seeded widespread metastases in various distant organs, referred to as a PDX with high metastatic efficiency, other tumors mainly seeded only lung metastases, referred to as a PDX with low metastatic efficiency. Low efficiency PDXs originated from patients that were cured after surgery and chemotherapeutic treatment. High efficiency PDXs originated from patients with fatal outcome (Quintana et al., 2012).
For this study, we had access to a panel of nine PDXs, seven of which had known metastatic efficiency and matching patient outcome. For the remaining two PDXs, the metastatic efficiency, including patient outcome, was unknown (Table S1). To define the genomic states of the PDXs with known metastatic efficiency, we sequenced a panel of ~1400 clinically actionable genes and found that the PDXs span the genomic landscape of melanoma mutations, including mutations in BRAF (5/6), CKIT (2/6), NRAS (1/6), TP53 (2/6), and copy number variation (CNV) in CDKN2A (6/6) and PTEN (3/6) (Hayward et al., 2017; Hodis et al., 2012) (Table S2). For one PDX (m528), we were unable to generate sufficient genomic material for sequencing, although the cell culture was sufficiently robust for single cell imaging.
In order to prevent morphological homogenization and to better mimic the collagenous ECM of the dermal stroma, we imaged cells on top of a thick slab of collagen. The cells were plated sparsely to focus on cell-autonomous behaviors with minimal interference from interactions with other cells (Methods). For each plate, we recorded with a 20X/0.8NA lens phase contrast movies of at least 2 hours duration, sampled at 1 minute intervals (Fig. 1A, Video S1–2). Each recording sampled 10–20 randomly distributed fields of view from 1–4 plates of different cell types, each containing 8–20 individual cells.
Figure 1. Unsupervised learning of a latent vector that encodes characteristic features of individual melanoma cells.

(A) Top: Snapshot of a representative field of view of m481 PDX cells. Scale bar = 50 μm. Bottom: Time-lapse sequence of a single cell undergoing dynamic blebbing. Scale bar = 50 μm. (B) Representative time-lapse images of single cells from PDX tumors exhibiting low (m498) and high (m634) metastatic efficiency. Sequential images were each acquired 1 minute apart. (C) Design of the adversarial autoencoder, comprising an encoder (dark red) to extract from single cell images a 56-dimensional latent vector, so that a decoder can reconstruct from the vector a similar image. The “adversarial” component (top) penalizes randomly generated latent cell descriptors q(z) that the network fails to distinguish from latent cell descriptors drawn from the distribution of observed cells p(z). (D) Examples of cell reconstructions. Raw cell images (top): beginning of epoch #110K (trained on 10,000 images), around midway training of epoch #11M (after 1,000,000 images), at the end of epoch #3, epoch #6, and epoch #46. (E) Convergence of autoencoder loss (binary cross-entropy between raw and reconstructed image). Epoch is a full data set training cycle that consists of ~1.7 million images. Mini-batch is the number of images processed on the GPU at a time. Each mini-batch includes 50 cell images randomly selected for each network parameter learning update. For every epoch, the images order is scrambled and then partitioned into ordered sets of 50 for each mini-batch.
We complemented the PDX data set with equivalently acquired time-lapse sequences of two untransformed melanocyte cell lines and six melanoma cell lines. The former served as a control to test whether the latent information allows at minimum the distinction of untransformed and metastatic cells. The latter served as a control to test whether the latent information allows the distinction of different cell populations, which, by the long-term selection of passaging in the lab, likely have drifted to a spectrum of molecular and regulatory states that differs from the PDX.
In total, our combined data set comprises time-lapse image sequences of more than 12,000 single melanoma cells, resulting in approximately 1,700,000 raw images. The cells were typically not migratory but displayed variable morphology and local dynamics (Video S3). Many of the cells were characterized by an overall round cell shape and dynamic surface blebbing (Fig. S1A, Video S1–2), regardless of whether they belonged to the melanoma group with high or low metastatic efficiency (Fig. 1B, Fig. S1B), which is consistent with reports of primary melanoma behavior in vivo (Pinner and Sahai, 2008; Sadok et al., 2015; Sahai and Marshall, 2003) and on soft substrates in vitro (Cantelli et al., 2015; Welf et al., 2016). Thus, we speculated that cell shape or motion might not be informative of the metastatic state of a melanoma cell.
Nonetheless, we still noted textural variation and dynamics between individual cell images. Thus, we wondered whether these images contain visually unstructured signal that could predict the metastatic propensity of a cell.
Design of adversarial autoencoders for unsupervised feature extraction
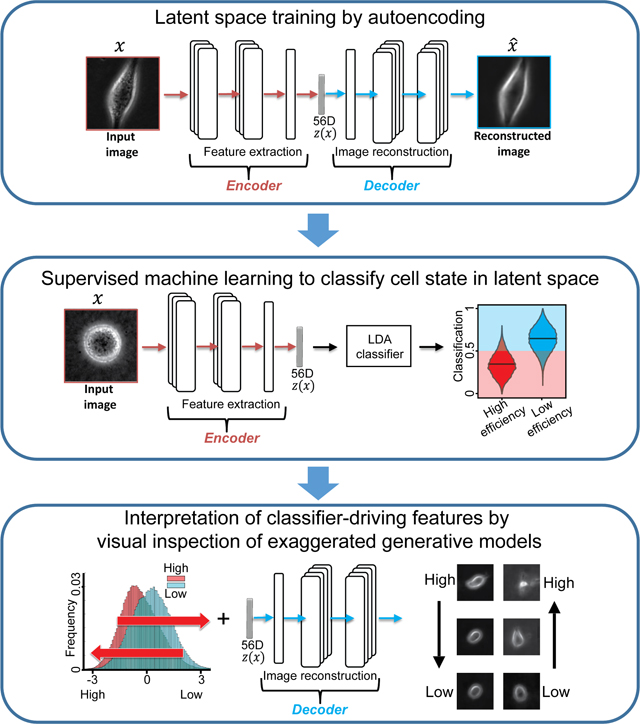
After detection and tracking of single cells over time (Methods), we used the cropped single cell images as atomic units to train an adversarial autoencoder (Makhzani et al., 2015) (Fig. 1C, Methods). The autoencoder comprises a deep convolutional neural network to “encode” the image data of a single cell in a vector of latent information, from which a structurally symmetric deep convolutional neural network “decodes” synthetic images (Fig. 1C). The networks are trained to minimize the discrepancy between input and reconstructed images. The adversarial component penalizes randomly generated latent cell descriptors q(z) that the network fails to distinguish from latent cell descriptors drawn from the distribution of observed cells p(z), thus ensuring regularization of the latent information space. Our network architecture employed the part of the network previously used to reconstruct landmarks of the cell nucleus and cytoplasm (Johnson et al., 2017) in fluorescence microscopy images. We supplied the network with phase-contrast images instead of fluorescence images and found that the adversarial autoencoder displayed fast convergence in reconstructing phase-contrast cell images (Fig. 1D–E, Video S4, Fig. S1C). Furthermore, the trained network’s latent space defined a faithful metric for discriminating images of cells that appear morphologically different (Methods, Fig. S2). The network training was agnostic to the subsequent classification task. The goal of this step was to determine for each melanoma cell an unsupervised latent cell descriptor that holds a compressed representation of a cell image for further classification of cell states.
The latent cell descriptor can discriminate between different cell categories
In our label-free imaging assay, the latent space cell descriptors seemed to be distorted by batch effects related to inconsistencies in different imaging sessions such as operator, microscope, and gel preparation (Methods, Fig. S3). These systematic but meaningless variations in the data are a major hurdle in classification tasks (Boutros et al., 2015; Caicedo et al., 2017; Chandrasekaran et al., 2020). To address this issue, we transformed the auto-encoder latent space into a classifier space that was robust to inter-day confounding factors, but discriminated between different cell categories. A cell category was defined as a set of multiple cell types with a common property. For example, the category “cell line” comprises six different cell types: A375, MV3, WM3670, WM1361, WM1366, and SKMEL2. The discrimination was accomplished by training supervised machine learning models on the normalized latent cell descriptor using Linear Discriminant Analysis (LDA) at the single cell level. Our intuition was that the diversity of the training data, in terms of cell categories and range of batch effects, makes the LDA classifier space robust. We validated the models in multiple rounds of training and testing, each round with the imaging data of one cell type (i.e., a specific cell line or PDX) designated as the test-set, while the rest of the data was used as the training set (Fig. 2A). Hence, the discriminative model was trained with information fully independent of the cell type it was tested on (Jones, 2019).
Figure 2. Discrimination of different melanoma cell categories:

melanoma cell line versus melanocytes (B-D), cell lines versus clonal expanded cell lines (E-G), and cell lines versus PDXs (H-J). (A) Blinding the cell type. A cell type was defined as a specific cell line or PDX. Categories encompass multiple cell types. Multiple rounds of training and testing were performed. In each round, data from one cell type was used as the test dataset, defining a single observation that was composed of many single cell classifications. The training set contained the rest of the data relevant for the task (e.g., all melanoma cell lines and all PDXs when discriminating these two categories). The trained model was completely blind to the cell type used in each test set. The trained model classified each single cell in the test set. (B) Receiver-Operator Characteristic (ROC) curve for the distinction of the category ‘cell lines’ from the category ‘melanocytes’. AUC = 0.635. (C) Accuracy in predicting for a cell type its association with the category ‘cell lines’ versus the category ‘melanocytes’. Each data point indicates the outcome of testing a particular cell type by the fraction of individual cells classified as ‘cell line’. N = 8 cell types: 6 melanoma cell lines, 2 melanocyte lines. 7/8 successful predictions. Wilcoxon rank-sum and Binomial statistical tests on the null hypothesis that the classifier scores of a cell line and of melanocytes are drawn from the same distribution, p = 0.071 (Wilcoxon), p = 0.035 (Binomial), see Methods for justification of the statistical tests. (D) Bootstrap distribution of the prediction of a cell type as a member of the ‘cell lines’ category. For each cell type, we generated 1000 observations by repeatedly selecting 20 random cells and recorded the fraction of these cells that were classified as ‘cell lines’. Horizontal line – median. Wilcoxon rank-sum test p < 0.0001 rejecting the null hypothesis that the classifiers scores of observations from the two categories stem from the same distribution. This analysis demonstrated the ability to discriminate cell lines versus melanocytes from random samples of 20 cells in a cell type. (E) ROC curve for the distinction of the category ‘cell lines’ from the category ‘clonal’ (expansion line). (F) Accuracy in predicting for a cell type its association with the category ‘cell lines’ versus the category ‘clonal’. Each data point indicates the outcome of testing a particular cell type by the fraction of individual cells classified as ‘cell line’. N = 10 cell types: 6 melanoma cell lines, 4 clonal expansion lines. 10/10 successful predictions. Wilcoxon rank-sum and Binomial statistical test on the null hypothesis that the classifier scores of a cell line and of a clonal expansion line are drawn from the same distribution, p = 0.010 (Wilcoxon), p < 0.001 (Binomial). (G) Bootstrap distribution of the prediction of a cell type as a member of the ‘cell lines’ category. See panel D. Horizontal line - median. Wilcoxon rank-sum test p < 0.0001 rejecting the null hypothesis that the classifiers scores of observations from the two categories stem from the same distribution. (H) ROC curve for the distinction of the category ‘cell lines’ from the category ‘PDXs’. AUC = 0.714. (I) Accuracy in predicting for a cell type its association with the category ‘cell lines’ versus the category ‘PDXs’. Each data point indicates the outcome of testing a particular cell type by the fraction of individual cells classified as ‘cell line’. N = 15 cell types: 6 cells lines, 9 PDXs. 14/15 successful predictions. Wilcoxon rank-sum and Binomial statistical test on the null hypothesis that the classifier scores of cell lines and of PDX are drawn from the same distribution, p < 0.0004 (Wilcoxon), p < 0.0005 (Binomial). (J) Bootstrap distribution of the prediction of a cell type as a member of the ‘cell lines’ category. See panel D. Horizontal line – median. Wilcoxon rank-sum test p < 0.0001 rejecting the null hypothesis that the classifiers scores of observations from the two categories stem from the same distribution. For all panels we used the time-averaged latent space vector over the entire movie as a cell’s descriptor.
The number of cells from each category was balanced during training to eliminate sampling bias. To overcome the limited statistical power due to the small number of cell types (two melanocytes, four clonal expansions, six cell lines and nine PDXs), we also considered test datasets defined by all cells from one cell type imaged in one day. In this case, the training dataset included the remainder of all imaging data, except cells of any type imaged on the same day or cells of the same type on any other day (Fig. S4A). These approaches were successful in discriminating transformed melanoma cell lines from non-transformed melanocyte cell lines (Fig. 2B–D, Fig. S4B–C), melanoma cell lines from clonal expansions of these cell lines (Fig. 2E–G, Fig. S4D–E, Methods), and melanoma cell lines from patient-derived xenografts (PDX) (Fig. 2H–J, Fig. S4F–G). We also found that in pairwise comparisons most cell types could be discriminated from one another (Fig. S4H). Our latent space descriptor surpassed simple shape-based descriptors attained by phase contrast single cell segmentation (Winter et al., 2016), and it did not benefit from either explicit incorporation of temporal information or mean square displacement analysis of trajectories (Methods, Figs. S5). Based on these findings we used the time-averaged latent space cell descriptors as the basic feature set for cell classification throughout the remainder of our study.
Although the classification performance was moderate at the single cell level (e.g., AUC of cell lines versus PDXs was 0.71, Fig. 2H), each imaging session included enough cells to accurate categorize cells at the population level (e.g., 14/15 successful cell lines versus PDXs predictions at the population level, Fig. 2I). Altogether, these results established that the latent cell descriptor captures information on the functional cell state that is distinct for different cell categories and types.
Classification of melanoma metastatic efficiency
Equipped with the latent space cell descriptors and LDA classifiers, we tested our ability to predict the metastatic efficiency of single cells from melanoma stage III PDXs (Fig. 3A). Our approach was able to perfectly discriminate between the categories melanomas with high versus low metastatic efficiency (Fig. 3B–D). It was also successful at distinguishing single cells from PDXs with low versus high metastatic efficiency that were imaged on a single day (small n), by classifiers that were blind to the PDX and to the day of imaging (Fig. S4A, Fig. 3E–G). Cell shape information (Fig. S6A) and mean square displacement analysis of trajectories (Fig. S6B–C) could not stratify PDXs along these two categories. Classifiers trained with the latent space cell descriptor were robust to artificial blurring (Fig. 3H), and illumination changes (Fig. 3I). These results established the potential of the proposed imaging and analytical pipeline as a diagnostic, live cytometry approach.
Figure 3. Discrimination of PDXs with low versus high metastatic efficiency as defined by the correlation between outcomes in mouse and man.

(A) (Quintana et al., 2012). Classifiers were trained to predict metastatic efficiency at the single cell level (panels B, E). The association of a particular PDX with either the category ‘Low’ [metastatic efficiency] or the category ‘High’ [metastatic efficiency] was determined at the population level – either considering the fraction of all cells of a PDX predicted as ‘Low’ (C, F) or a bootstrap sample of 20 cells (D, G). (B) Receiver Operating Characteristic (ROC) curve for single cell classification. AUC = 0.71. (C) Accuracy in predicting for a single PDX (cell type) its association with the category ‘Low’ versus the category ‘High’. Each data point indicates the outcome of testing a particular cell type by the fraction of individual cells classified as ‘Low’. N = 7 PDXs: 4 low efficiency, 3 high efficiency metastasizers. 7/7 predictions are correct. Wilcoxon rank-sum and Binomial statistical test on the null hypothesis that the classifier scores of PDX with low versus high metastatic efficiency are drawn from the same distribution, p = 0.0571 (Wilcoxon), p ≤ 0.00782 (Binomial), see Methods for justification of the statistical tests. (D) Bootstrap distribution of the prediction of a PDX as a member of the ‘Low’ category. For each PDX we generated 1000 observations by repeatedly selecting 20 random cells and recorded the fraction of these cells that were classified as ‘Low’. Horizontal line - median. Wilcoxon rank-sum test p < 0.0001 rejecting the null hypothesis that the classifiers scores of observations from the two categories stem from the same distribution. This analysis demonstrated the ability to predict metastatic efficiency from samples of 20 random cells. (E-G) Discrimination results using classifiers that were blind to the cell type and day of imaging (Fig. S4A, more observations, smaller n - number of cells for each observation). (E) Receiver Operating Characteristic (ROC) curve; AUC = 0.723. (F) Accuracy in predicting for one PDX on a particular day (cell type) its association with the category ‘Low’ versus the category ‘High’. Each data point indicates the outcome of testing one PDX on a particular day by the fraction of individual cells classified as ‘Low’. N = 49 cell types and days: 25 low metastatic efficiency, 24 high metastatic efficiency. 32/49 predictions were correct. Wilcoxon rank-sum and Binomial statistical test on the null hypothesis that the classifier scores of PDX with low versus high metastatic efficiency are drawn from the same distribution p = 0.0042 (Wilcoxon), p ≤ 0.0222 (Binomial). (G) Bootstrap distribution of the prediction of a PDX imaged in one day as member of the ‘Low’ category. See panel D. Horizontal line - median. Wilcoxon rank-sum test p < 0.0001 rejecting the null hypothesis that the classifiers scores of observations from the two categories stem from the same distribution. (H) Robustness of classifier against image blur. Blur was simulated by filtering the raw images with Gaussian kernels of increased size. The PDX m528 was used to compute AUC changes as a function of blur. Representative blurred image (middle) and its reconstruction (bottom). (I) Robustness of classifier to illumination changes. AUC as a function of altered illumination (top). Representative image of m528 cell after simulated illumination alteration (middle), and its reconstruction (bottom).
Identification of classification-driving features in autoencoder latent space
Our results thus far established the predictive power of the latent cell descriptor for the diagnosis of metastatic potential. However, the power of these deep networks to recognize statistically meaningful image patterns that escape the attention of a human observer is also its biggest weakness (Belthangady and Royer, 2019; Caicedo et al., 2017; Chandrasekaran et al., 2020): What is the information extracted in the latent space that drives the accurate classification of low versus high metastatic PDXs? When we plotted a series of cell snapshots from one PDX in rank order of the LDA-based classifier score of metastatic efficiency, there was no pattern that could intuitively explain the score shift (Fig. 4A). This outcome was not too surprising given that much of the cell appearance is likely unrelated to metastasis-enabling functions, including the image signals associated with batch effects (Boyd et al., 2020) (Fig. S3).
Figure 4.

Metastatic efficiency is encoded by a single component of the latent space cell descriptor. (A) Gallery of snapshots of cells from a PDX (m610) ordered by their corresponding classifier score. (B) Approach: Each feature in the latent space cell descriptor is correlated with the score of the classifier trained to distinguish PDXs with high versus low metastatic efficiency. (C) Correlation between all 56 features (y-axis) and classifier scores for 7 PDXs (x-axis). (D) Value of feature #56 and classifier scores for individual cells color-grouped by PDX. (E) Distribution of the correlations from panel B; feature #56 (red arrow) is an obvious outlier. Left: distribution. Right: plot of log frequency for better visualization of feature #56. (F) Normalized correlation values (Z-scores) all 56 features (y-axis) and classifier scores (x-axis). Z-scores are calculated using the mean value and standard deviation of the distribution of correlation values in panel D. (G) Distribution of feature #56 values for cells grouped by association with a PDX. (H) Distribution of feature #56 values for cells grouped by association with low and high metastatic efficiency. (I) Gallery of snapshots of cells from PDX m610 in ascending order of the normalized value of feature #56. Note, high metastatic efficiency relates to negative, low metastatic efficiency to positive values of feature #56.
To probe which features encapsulated in the latent cell descriptor are most discriminative of the metastatic state we first correlated each of the 56 features to the classifier score (Fig. 4B–C). The correlations were calculated independently for each PDX using a classifier blind to the PDX (see Fig. 2A). For all 7 PDXs the last feature #56 stood out as highly negatively correlated to the classifier scores (Fig. 4C–D). The correlation values fell outside the range of correlations observed for any other feature (Fig. 4E–F). The distributions of values of feature #56 for individual cells clearly separated tumors with high versus low metastatic efficiency (Fig. 4G & H). However, as with the classifier score (Fig. 4A), a series of random cell snapshots from one PDX in rank order of feature #56 values did not reveal a cell image pattern that could intuitively explain the meaning of this feature (Fig. 4I). This suggests that feature #56 encoded a multifaceted image property reflecting the metastatic potential of melanoma PDXs that cannot readily be grasped by visual inspection.
Interpretation of classification-driving latent feature using generative models and spontaneous cell plasticity
Neither a series of cell images rank-ordered by classification scores of high vs low metastatic efficiency nor a series rank-ordered by feature #56 offered a visual clue as to which image properties may determine a cell’s metastatic efficiency. We concluded that the natural variation of feature #56 values in our data was too low to give such clues and/or that the natural variation of features unrelated to metastatic efficiency largely masked image shifts related to the variation of feature #56 between PDXs with low and high metastatic efficiency. To glean some of the image properties that are controlled by feature #56 we exploited the network decoder to generate a series of “in silico” cell images in which, given a particular location of a cell in the latent space, feature #56 was gradually altered while fixing all other features (Fig. 5A). As expected, the changes in feature #56 negatively correlated with the changes they caused in the classifier score, regardless of the metastatic efficiency of the cells from which the images were derived (Fig. 5B). The generative modeling brought two advantages over our previous attempts of visually interpreting feature #56: First, it allowed us to observe ‘pure’ image changes along a principal axis of metastatic efficiency change. Second, it allowed us to shift the value of feature #56 outside the value range of the natural distribution and thus to analyze the exaggerated cell images for emergent properties in cell appearance. Upon morphing a PDX cell classified as low metastatic efficiency within a normalized z-score range for feature #56 of [−3.5, 3.5], we observed two properties emerging with the high metastatic efficiency domain. The formation of pseudopodial extensions and changes in the level of cellular light scattering as observed by brighter image intensities at the cell periphery and interior (Fig. 5C). The pseudopodial activity was visually best appreciated when compiling the morphing sequences into videos that shift a cell classified as low metastatic towards the high metastatic efficiency domain (Video S5) and, vice versa, a cell classified as highly metastatic towards the low metastatic efficiency domain (Video S6).
Figure 5. Generative modeling of cell images to interpret the meaning of feature #56.

(A) Approach: alter feature #56 while fixing all other features in the latent space cell descriptor to identify interpretable cell image properties encoded by feature #56. (B) Shifts in feature #56 (y-axis, measured in z-score) negatively correlated with variation in the classifier scores. (C) In silico cells generated by decoding the latent cell descriptor of a representative m498 PDX cell under gradual shifts in feature #56 (“Recon.”). Visualization of the intensity differences between consecutive virtual cells (Izscore - Izscore+0.5), only positive difference values are shown (“Diff+”). Changes in feature #56 are indicated in units of the z-score. The corresponding classifier’s score and value of feature #56 are shown. (D) Approach: correlating temporal fluctuations of each feature to fluctuations in the classifiers’ score. (E) Summary of correlations. Y-axis - different classifiers for each PDX. X-axis - features. Bin (x,y) records the Pearson correlation coefficients between temporal fluctuations in feature #x and the score of classifier #y over all cells of the PDX. (F) Normalization of correlation coefficients as a Z-score. Mean value and standard deviation are derived from the correlation values in panel E. (G) Following a m610 PDX cell spontaneously switching from the low to the high metastatic efficiency domain (as predicted by the classifier). Live imaging for 10 minutes. Left (top-to-bottom): raw cell image, diff+ images, classifier’s score, feature #56 values. Right: visualization of the classifier score as a function of time, switching from “low” to “high” in less than 10 minutes.
Repeating the morphing for many PDX cells (Fig. S7, Video S7) underscores pseudopod formation and enhanced light scattering as the systematic factors that distinguish cells with low feature #56 values/high metastatic efficiency from those with high feature #56 values/low metastatic efficiency. Moreover, by variation of all other latent space features one-by-one we visually confirmed this combination of morphological properties was specifically controlled by feature #56 (Fig. S8).
To corroborate our conclusion from synthetic images we tested whether “plastic” cells, which change their classifier score during the time course of acquisition from low to high efficiency or vice versa, displayed visually identifiable image transitions. First, we verified that temporal fluctuations in feature #56 negatively correlated with the temporal fluctuations in the classifier scores (Fig. 5D–F). Second, we confirmed that PDX cells spontaneously transitioning from a predicted low to a predicted high metastatic efficiency displayed increased light scattering (Fig. 5G, Video S8). We were not able to conclusively validate the enhanced protrusive activity in the time courses of experimental data. The subtlety and perhaps also the subcellular localization of this phenotype requires visualization outside the natural variation of the latent feature space.
Generalizing the interpretation to high dimensions
When we applied the same feature-to-score correlation analysis to classifiers trained for discrimination of cell lines from PDXs, we found the three features #26, #27, and #36 as classification-driving (Fig. S9A–B). This result underscores two key properties of our interpretation of the latent space: First, distinct classification tasks are driven by different feature subsets in the latent space cell descriptor, which capture distinguishing cell properties. In all generality, the classification task is driven not by a single but by multiple latent space cell descriptors. To enable interpretation of such multi-feature drivers, we generalized the traversal of the latent space by computing a trajectory that follows in every location the gradient of the classifier score. Since LDA is a linear classifier the gradient follows throughout the entire latent space the directions determined by the classifier coefficients (Fig. S9C–D). Thus, we traversed the latent space up and down in steps that are weighted by the LDA coefficients (Methods). For the classifier distinguishing PDXs from cell lines, the latent space traversal to positions beyond the natural variation in the data suggests that PDX cells exhibit a wider range of non-round morphologies than cell lines (Fig. S9E). However, for one cell the simulated PDX image outside the natural data range displays an artefactual break-up of the cell volume, indicating an example of occasional failure of the described extrapolation strategy.
As a second test case, we trained another (unsupervised) adversarial autoencoder (Fig. 1C) to capture an alternative latent space representation of cell appearance. The network training was performed on the same dataset of PDXs, cell lines, clones and melanocyte images as the first network, and was followed by training LDA classifiers to discriminate between high and low metastatic efficient PDXs, each blind to the PDX in test. Because of the stochasticity in selecting mini-batches, the training converged to a different latent space cell image representation. In this representation, several features, and not only feature #56, correlated with the classifier score (Fig. S10A), as also reflected by multiple LDA coefficients with high magnitudes (Fig. S10B–C). Tracing PDX cells along the LDA coefficients to latent space locations outside the natural variation of the data confirmed light scattering and pseudopodial extensions as the determinants between cells with high versus low metastatic efficiency by shifting feature #56 in the latent representation determined by the original autoencoder network (compare Fig. S10D). These results establish the generalization of in silico latent features amplification to higher-dimensional discriminant feature sets.
PDX-trained classifier can predict the metastatic potential of melanoma cell lines in mouse xenografts
We were interested in the capacity of PDX-trained classifiers to predict the spontaneous metastasis of tumor-forming melanoma cell line xenografts. We hypothesized that, despite the distinct morphologies of PDX and cell lines indicated by the classifier in Fig. 2H–J, the core differentiating properties between low and high efficiency metastatic PDXs would be conserved for melanoma cell lines. Using the PDX-trained classifiers, A375, a BRAFV600E-mutated and NRAS wild-type melanoma cell line, originally excised from a primary malignant tumor (Davies et al., 2002; Ghandi et al., 2019; Giard et al., 1973; Kozlowski et al., 1984; Rozenberg et al., 2010; Tanami et al., 2004), was predicted as the most aggressive metastasizer (Fig. 6A). MV3, a BRAF wild-type and NRAS-mutated melanoma cell line, originally excised from a metastatic lymph node and described as highly metastatic (Quax et al., 1991; Schrama et al., 2008; van Muijen et al., 1991), was predicted by the PDX-trained classifiers as the least aggressive (Fig. 6A). Consistent with our previous analyses of the influence of the latent space features on classification, feature #56 was lower for A375 than for MV3 (Fig. 6B). We subcutaneously injected luciferase-labeled versions of A375 and MV3 cells into the flanks of NSG mice (Methods). Both cell models formed robust primary tumors at the site of injection (Fig. 6C–D) as well as metastases in the lungs and in multiple other remote organs (Fig. 6E–F). Bioluminescence imaging of individual excised organs showed a higher spreading to organs other than the lungs in mice injected with A375 cells compared to those injected with MV3 cells (Fig. 6E–F). It was previously determined that the most robust measure of metastatic efficiency in this model was visually identifiable macrometastases in organs other than the lungs (Quintana et al., 2012). As confirmation that the A375 cells metastasized more efficiently in this model, we found macrometastases in other organs in 5/5 mice xenografted with A375 cells versus in 1/5 mice xenografted with MV3 cells (Fig. 6G). Intriguingly, primary tumors in MV3-injected mice grew much faster than in A375-injected mice (Fig. 6H), in contrast to being less aggressive in spreading to remote organs, suggesting that primary tumor growth is uncoupled from the ability to produce remote metastases (Ganesh et al., 2020; Quintana et al., 2012; Viceconte et al., 2017). Under the assumption that overall tumor burden would be limiting for metastatic dissemination instead of time after injection, we conclude, in agreement with the prediction of our classifier, that A375 cells are more metastatically efficient than MV3 cells in this model. Broadly, these data confirm that properties captured by the latent space cell descriptor define a specific gauge of the metastatic potential of melanoma that is independent of the tumorigenic potential.
Figure 6. PDX-trained classifiers predict the potential for spontaneous metastasis of mouse xenografts from melanoma cell lines.

(A) All 7 PDX-trained classifiers consistently predicted that among the 6 analyzed cell lines A375 has the highest and MV3 the lowest metastatic efficiency. (B) The distribution of single cell values of feature #56 is lower for A375 than the distribution of values for MV3 cells. (C, E) Bioluminescence (BLI) of NSG mouse sacrificed 24–35 days after subcutaneous transplantation of 100 Luciferase-GFP+ cells from the A375 melanoma cell line (C) versus from the MV3 cell line (E). (D, F) Bioluminescence of organs dissected from the A375 xenografted mouse (D) and from the MV3-xenografted mouse (F). 1, Gastrointestinal Tract (GI); 2, Lungs and Heart; 3, Pancreas and Spleen; 4, Liver; 5, Kidneys and Adrenal glands. In the MV3, mouse metastases were mostly found in the lungs. Black shades are mats on which the organs and mice are imaged (Methods). (G) Summary of metastatic efficiency for A375 and MV3 melanoma cell lines in 5 mice. “BLI Lungs”: Detection of BLI in the lungs. “BLI other organs”: BLI in multiple organs beyond the lungs. “Remote macro mets”: Macrometastases in remote organs (excluding lungs), identification of “visceral metastasis”, macrometastases visually identifiable without BLI, the measure used to define metastatic efficiency to the PDXs in (Quintana et al., 2012). (H) Primary tumors in MV3 xenografts grow faster than in A375 xenografts. Mice were sacrificed 24 days after injection with MV3, 35 days after injection with A375 cells. N = 5 mice for A375 and MV3 cell line. Statistics for tumor size after 24 days p-value = 0.0079 (Wilcoxon rank-sum test), fold = 1.6241.
Image-based classifiers are more predictive of metastatic potential than the mutational profile
Following initial diagnosis, it is standard practice for a melanoma biopsy to undergo mutational sequencing analysis to determine the best course of therapy. But, to our knowledge it has not been determined if there is a general mutational profile associated with more aggressively metastatic disease. While metastatic melanoma are expected to harbor a ‘standard’ set of primary mutations, such as those in BRAF or NRAS (Jakob et al., 2012) – and indeed all our PDX models and metastatic cell lines do contain an activating mutation in either one of these genes (Table S2) – we were curious as to whether secondary mutations in the genomic profiles of these cell models would encode information on the metastatic efficiency. To address this question we examined the distributions of genomic distances among the PDX cell models and two cell lines vis-à-vis the distance distributions in the latent feature space. The conclusion from these experiments was that the states of oncogenic/likely-oncogenic mutations in the 20 most mutated genes in melanoma (Hodis et al., 2012) were insufficient for a prediction of the metastatic efficiency (Fig. S11). In fact, the oncogenic/likely-oncogenic mutations in the genes were not more predictive than non-oncogenic mutations or an unbiased analysis of a full panel of 1400 genes for metastatic states. Thus, image-based classifiers can identify more metastatically aggressive cancers, which is not currently possible for clinical diagnostics based on genomics.
Discussion
Visually unstructured properties of cell image appearance enable robust cell type classification
Morphology has long been a cue for cell biologists and pathologists to recognize cell category and abnormalities related to disease (Bakal et al., 2007; Chan, 2014b; Eddy et al., 2018; Gordonov et al., 2015; Gurcan et al., 2009; López, 2013b; Pavillon et al., 2018; Wu et al., 2020; Yin et al., 2013). In this study, we rely on the exquisite sensitivity of deep learned artificial neural networks in recognizing subtle but systematic image patterns to classify different cell categories and cell states. To assess this potential we chose phase contrast light microscopy, an imaging modality that uses simple transmission of white or monochromatic light through an unlabeled cell specimen and thus minimizes experimental interference with the sensitive patient samples that we used in our study. A further advantage of phase contrast microscopy is that the imaging modality captures visually unstructured properties, which relate to a variety of cellular properties, including surface topography, organelle organization, cytoskeleton density and architecture, and interaction with fibrous extracellular matrix.
Our cell type classification rests on the combination of an unsupervised deep learned autoencoder for extraction of meaningful but visually hidden features followed by conventional supervised classifier that discriminates between distinct cell categories. The choice of this two-step implementation allowed us to construct several different cell classifiers for different tasks using a one-time learned, common feature space. Thus, the task of distinguishing, for example, melanoma cell lines from normal melanocytes could benefit from the information extracted from PDXs, while PDXs could be divided into groups with high versus low metastatic propensity with the support of information extracted from melanoma cell lines and untransformed melanocytes. Accordingly, sensitive classifiers could be trained on relatively small data subsets – much smaller than would be required to train an ab initio deep-learned classifier for the same task. The approach is not only data-economical, but it greatly reduces computational costs as the deep learning procedure is performed only once on the full dataset. Indeed, in our study we learned a single latent feature space using time lapse sequences from over 12,000 cells (~1.7 million snapshots); and then trained classifiers on data subsets that included labeled categories smaller than 1,000 cells. As an additional benefit of the orthogonalization of unsupervised feature extraction and supervised classifier training, we were able to evaluate the performance of our classifiers by repeated leave-one-out validation, verifying that the discriminative model training is completely independent of the cell type at test. A similar evaluation strategy, requiring the repeated re-training of a deep learned classifier, would likely become computationally prohibitive.
Application of cell type classification to the prediction of metastatic efficiency
Among the cell classification tasks, we were able to distinguish the metastatic efficiency of stage III melanoma harvested from a xenotransplantation assay that had previously been shown to maintain the patient outcome (Quintana et al., 2012). While the distinction was perfect at the level of PDXs, at the single cell level the classifier accuracy dropped to 70%. This is not necessarily a weakness of the classifier but speaks to the fact that tumor cells grown from a single cell clone are not homogeneous in function and/or appearance. Our estimates of classifier accuracy relies on leave-one-out strategies where the training set and the test set were completely non-overlapping, both with regards to the classified cell category and to the days the classified category was imaged. Thus, it can be assumed that the reported accuracies can be reproduced on new, independent PDXs.
Besides numerical testing, we validated the accuracy of our classifiers high versus low metastatic efficiency in a fully orthogonal experiment. We applied the PDX-trained classifiers to predict the metastatic efficiency of well-established melanoma cell lines and validated their predictions in mouse xenografts. We emphasize that the PDX-trained classifier has never encountered a cell line and that despite the significant differences between cell lines and PDXs (Fig. 2H–J), the classifier correctly predicted high metastatic potential for the cell line A375 and low potential for MV3 (Fig. 6). Moreover, a recent paper that demonstrated the use of in vivo barcoding as a readout for metastatic potential of cancer cell lines engrafted in mice showed that A375 is more aggressive than SKMEL2 (Jin et al., 2020), in agreement with our classifier’s prediction (Fig. 6A). Intriguingly, the aggressiveness in primary tumor growth was reversed between A375 and MV3, supporting the notion that tumorigenesis and metastasis are unrelated phenomena (Ganesh et al., 2020; Jin et al., 2020; Quintana et al., 2012; Viceconte et al., 2017) (Fig. 6H). This shows that the latent feature space encodes cell properties that specifically contribute to cell functions required for metastatic spreading and that these features are orthogonal to features that distinguish cell lines from PDX models.
Interpretation of latent features discriminating high and low metastatic cell propensity
Deep Learning Artificial Neural Networks have revolutionized machine learning and computer vision as powerful tools for complex pattern recognition, but there is increasing mistrust in results produced by ‘black-box’ neural networks(Belthangady and Royer, 2019). Aside from increasing the confidence, the interpretation of the properties – also referred to as ‘mechanisms’ – of the pattern recognition process can potentially generate insight of a biological/physical phenomenon that escapes the analysis driven by human intuition.
In medical imaging the quest for interpretability has been responded by identifying image sub-regions of special importance for trained deep neural networks (Ash et al., 2018; Courtiol et al., 2019; Cruz-Roa et al., 2013; Fu et al., 2019; Pan et al., 2019; Shamai et al., 2019). A similar idea was implemented in fluorescent microscopy images, in the context of classification of protein subcellular localization, to visualize the supervised network activation patterns (Kraus et al., 2017). Localization of sub-regions that were particularly important for the classifier result permitted a visual assessment and pathological interpretation of distinctive image properties. Such approaches are only suitable when the classification-driving information is localized in one image region over another, and when highlighting the region is sufficient to establish a biological hypothesis. For cellular phenotyping, this is not the case. Because of the orthogonalization of feature space construction and classifier training we could elegantly extract visual cues for the inspection of classifier-relevant cell appearances. By exploiting the single cell variation of the latent feature space occupancy and the associated variation in the scoring of a classifier discriminating high from low metastatic melanoma, we identified feature #56 as predominant in prescribing metastatic propensity. Of note, the feature-to-classifier correlation analysis is not restricted to determining a single discriminatory feature (Fig. S9, S10) and is directly applicable to non-linear classifiers.
Visual inspection of cell images ranked by the classifier score or feature #56 did not reveal any salient cell image appearance that would distinguish efficiently from inefficiently metastasizing cells (Fig. 4A,I). These particular image properties were masked by cell appearances that are unrelated to the metastatic function. Moreover, the function-driving feature #56 represents a nonlinear combination of multiple image properties that are not readily discernible. To test whether feature #56 encodes image properties that are human-interpretable but buried in the intrinsic heterogeneity of cell image appearances, we exploited the generative power of our autoencoder. We ‘shifted’ cells along the latent space axis of feature #56 while leaving the other 55 feature values fixed. The approach also allowed us to examine how cell appearances would change with feature #56 values outside the natural range of our experimental data. Hence, the combination of purity and exaggeration allowed us to generate human discernible changes in image appearance that correspond to a shift in metastatic efficiency.
The outcome of a single feature, i.e., feature #56, driving the classification between two cell categories is by chance. As we show for the classification of PDXs versus cell lines, multiple features may strongly correlate with the classifier score. In this case, interpretation by visual inspection of exaggerated images has to be achieved by traversing the latent space in trajectories that follow in every location the gradient of the classifier score. In the particular case of the LDA classifier, the gradient is spatially invariant and follows the combination of the LDA coefficients. Thus, the proposed mechanism of visual latent space interpretation does not hinge on the identification of a single driver feature.
Once exaggerated in silico images offered a glimpse of key image properties distinguishing efficient from inefficient metastasizers, we could validate the predicted appearance shifts in experimental data. This was especially important to exclude the possibility that our extrapolation of feature values introduced image artifacts. We screened our data set for cells whose classification score and feature #56 values drifted from a low to high metastatic state or vice versa. We supposed that during such spontaneous dynamic events the variation in cell image appearances would be dominated, for a brief time window, by the variation in feature #56 and only marginally influenced by other features. Therefore, time-resolved data may present transitions in cell image appearance comparable to those induced by selective manipulation of latent space values along the direction of feature #56. It is highly unlikely to find a similarly pure transition between a pair of cells, explaining why we were unable to discern differences between cells with low and high metastatic efficiency in feature #56 ordered cell image series (Fig 4A).
Analyses of appearance shifts in both exaggerated in silico images and selected experimental images unveiled cellular properties of highly metastatic melanoma. First, these cells seemed to form pseudopodial extensions (Fig. 5C, Fig. S7, Video S5, Video S6). Because of its subtlety, this phenotype was more difficult to discern visually during spontaneous transitions of cell states (Fig. 5G). Second, images of cells in a highly metastatic state displayed brighter cell peripheral and interior signals, indicative of alteration in cellular light scattering. Because light scattering affects the image signal globally, this phenotype was clearly apparent in simulations (Fig. 5C, Fig. S7, Video S5, Video S6) and in experimental time lapse sequences of transitions between cells states (Fig. 5G, Video S8). Neither one of the two cell phenotypes follows a mathematically intuitive formalism that could be implemented as an ab initio feature detector. This highlights the power of deep learned networks in extracting complex cell function-driving image appearances.
Pseudopodial extensions play critical roles in cell invasion and migration. However, at least in a simplified migration assay in tissue culture dishes, the highly metastatic cell population did not exhibit enhanced migration (Fig. S6). Recent work has suggested mechanistic links between enhanced branched actin formation in lamellipodial and enhanced cell cycle progression (Mohan et al., 2019; Molinie et al., 2019), especially in micro-metastases. Therefore, we offer as a hypothesis that the connection between pseudopod formation and metastatic efficiency predicted by our analysis relates to the lamellipodia-driven upregulation of proliferation and survival signals (Nikolaou and Machesky, 2020; Swaminathan et al., 2020).
The observation that light scattering can indicate metastatic efficiency suggests that the cellular organelles and processes captured by light scattering are relevant to the metastatic process (Schürmann et al., 2015). Indeed, differences in light scattering upon acetic acid treatment are often used to detect cancerous cells in patients (Marina et al., 2012). Although the mechanisms underlying light scattering of cells are unclear, intracellular organelles such as phase separated droplets (Falke et al., 2019) or lysosomes will be detected by changes to light scattering (Choi et al., 2007). With the establishment of our machine-learning based classifier, we are set to systematically probe the intersection of hypothetical metastasis-driving molecular processes, actual metastatic efficiency, and cell image appearance in follow-up studies.
STAR Methods
RESOURCE AVAILABILITY
Lead contact:
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Assaf Zaritsky (assafza@bgu.ac.il) or Gaudenz Danuser (gaudenz.Danuser@utsouthwestern.edu).
Materials Availability:
This study did not generate new materials.
Data and Code Availability:
Raw image data, raw single cell images, corresponding metadata, the trained neural network, and the feature representation of all cells source data have been deposited at the Image Data Resource (Williams et al., 2017), https://idr.openmicroscopy.org, and are publicly available under the accession numbers: idr0109.
Original source code and test data is publicly available at https://github.com/DanuserLab/openLCH (doi: https://doi.org/10.5281/zenodo.4619858)
Scripts used to generate the figures presented in this paper are not provided in this paper but are available from the Lead Contact on request.
Any additional information required to reproduce this work is available from the Lead Contact.
METHOD DETAILS
Patient-derived xenograft (PDX) melanoma cells
Populations of primary melanoma cells were created from tumors grown in murine xenograft models as described previously (Quintana et al., 2010). Briefly, cells were suspended in Leibovitz’s L-15 Medium (ThermoFisher) containing mg/ml bovine serum albumin, 1% penicillin/streptomycin, 10 mM HEPES and 25% high protein Matrigel (product 354248; BD Biosciences). Subcutaneous injections of human melanoma cells were performed in the flank of NOD.CB17-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice (Jackson Laboratory). These experiments were performed according to protocols approved by the animal use committees at the University of Texas Southwestern Medical Center (protocol 2011–0118). After surgical removal, tumors were mechanically dissociated and subjected to enzymatic digestion for 20 min with 200 U ml−1 collagenase IV (Worthington), 5 mM CaCl2, and 50 U ml−1 DNase at 37oC. Cells were filtered through a 40 μm cell strainer to break up cell clumps and washed through the strainer to remove cells from large tissue pieces.
Cell culture and origin
Cell cultures were grown on polystyrene tissue culture dishes to confluence at 37°C and 5% CO2. Melanoma cells derived from murine PDX models were gifts from Sean Morrison (UT Southwestern Medical Center, Dallas, TX) and cultured in medium containing the Melanocyte Growth Kit and Dermal Cell Basal Medium from ATCC. Primary melanocytes were obtained from ATCC (PCS-200–013) and grown in medium containing the Melanocyte Growth Kit and Dermal Cell Basal Medium from ATCC. The m116 melanocytes, a gift from J. Shay (UT Southwestern Medical Center, Dallas), were derived from fetal foreskin and were cultured in medium 254 (Fisher). A375 cells were obtained from ATCC (CRL-1619). SK-Mel2 cells were obtained from ATCC (HTB-68). MV3 cells were a gift from Peter Friedl (MD Anderson Cancer Center, Houston, TX). MV3 and A375 cells were cultured in DMEM with 10% FBS. WM3670, WM1361, and WM1366 were obtained directly from the Wistar Institute and cultured in the recommended medium (80% MCDB1653, 20%, 2% FBS, CaCl2 and bovine insulin).
PDX-derived cell culture
We found that melanoma cell cultures derived from PDX tumors exhibited variable responses to traditional cell culture practices. Although some of the cell cultures retained high viability and proliferated readily, others exhibited extensive cell death and failed to proliferate. We determined that frequent media changes (<24 hrs) and subculturing only at high (>50%) confluence dramatically increased the viability and proliferation of PDX-derived cell cultures. Although we observed no correlation between metastatic efficiency and robustness in cell culture, we followed these general cell culture practices for all PDX-derived cultures.
Clonal cell line experiments
To create cell populations “cloned” from a single cell, cells were released from the culture dish via trypsinization and passed through a cell strainer (Fischer; 07–201-430) to ensure single-cell solution, counted and then seeded on a 10 cm polystyrene tissue culture dish at low density of 350,000 cells/10 ml of phenol-red free DMEM. Single cells were identified via phase-contrast microscopy. The single cells were isolated using cloning rings (Sigma; C1059) and expanded within the ring. For clonal medium changes, the medium was aspirated within the cloning rings. Subsequently, conditioned medium from a culture dish with corresponding confluent cells were passed through a filter (Fischer; 568–0020), which removed any cells and cell debris and then added to each cloning ring. Once confluent within the cloning ring, the clonal populations were released via trypsinization inside the cloning ring, transferred to individual cell culture dishes, and allowed to expand until confluence.
Bioluminescence imaging of NSG mice with melanoma cell lines
Injection of melanoma cells, monitoring of mice, dissection of mice, and imaging were all done as described in Quintana & Piskounova et al. (Quintana et al., 2012). Briefly, 100 Luciferase-GFP+ cells were injected into the right flank. Mice were monitored until the tumor at the site of injection reached 2 cm in diameter. Mice injected with MV3 were sacrificed 24 days after injection and A375 sacrificed 35 days after injection. The stomach, gut, rectum, and esophagus were labeled as the gastrointestinal tract. The black shades are mats that were used to image the mice’s organs. Some mouse/organ images have mats with (Fig. 6D) and without (Fig. 6F) gridlines.
Quantification of metastatic efficiency in NSG mice
We used three measures to assess metastatic efficiency (Quintana et al., 2012). First, detection of BLI in the lungs. Second, detection of BLI in multiple organs beyond the lungs. Third, identification of “visceral metastasis”, macrometastases visually identifiable without BLI, see details in (Quintana et al., 2012). We refrained from a more quantitative analysis of the BLI intensity for two reasons: 1) cells from some tumors lose expression of luciferase and 2) differences in melanin expression in melanoma cells and in tissue absorption can affect luminescence independent of cell density.
Targeted sequencing cancer-related genes and copy number variation analysis
Targeted sequencing of exons of 1385 cancer-related genes was performed by the Genomics and Molecular Pathology Core at UT Southwestern Medical Center as previously described (Zhang et al., 2020). Sequencing was performed on 6 out of 7 PDXs and the two cell lines A375 and MV3. Due to the difficulty in expanding the cells of PDX m528 in culture, we were not able to sequence this PDX. From the raw variant calling files, high confidence variants were determined by filtering variants found to have (a) strand bias, (b) depth of coverage < 20 reads and alt allele frequency < 20%. Common variants were filtered if they were in > 1% allele frequency in any population (Karczewski et al., 2020). Oncogenic potential was assess using oncokb-annotator (https://github.com/oncokb/oncokb-annotator). Summary tables of high-confidence variants of melanoma PDXs and cell lines were assembled in Table S2.
Live cell imaging
Live cell phase contrast imaging was performed on a Nikon Ti microscope equipped with an environmental chamber held at 37oC and 5% CO2 in 20x magnification (pixel size of 0.325μm). In order to prevent morphological homogenization and to better mimic the collagenous ECM of the dermal stroma, we imaged cells on top of a thick slab of collagen. Collagen slabs were made from rat tail collagen Type 1 (Corning; 354249) at a final concentration of 3 mg/mL, created by mixing with the appropriate volume of 10x PBS and water and neutralized with 1N NaOH. A total of 200 μL of collagen solution was added to the glass bottom portion of a Gamma Irradiated 35MM Glass Bottom Culture Dish (MatTek P35G-0–20-C). The dish was then placed in an incubator at 37°C for 15 minutes to allow for polymerization.
Cells were seeded on top of the collagen slab at a final cell count of 5000 cells in 400 uL of medium per dish. This solution was carefully laid on top of the collagen slab, making sure not to disturb the collagen or spill any medium off of the collagen and onto the plastic of the MatTek dish. The dish was then placed in a 37°C incubator for 4 hours. Following incubation, one mL of medium was gently added to the dish. The medium was gently stirred to suspend debris and unattached cells. The medium was then drawn off and gently replaced with two mL of fresh medium.
Single cell detection and tracking
We took advantage of the observation that image regions associated with “cellular foreground” had lower temporal correlation than the background regions associated with the collagen slab because of their textured and dynamic nature. This allowed us to develop an image analysis pipeline that detected and tracked cells without segmenting the cell outline. This approach allowed us to deal with the vast variability in the appearance of the different cell models and batch imaging artifacts in the phase-contrast images. The detection was performed in super-pixels with a size equivalent to a 10 × 10 μm patch. For each patch in every image, we recorded two measurements, one temporal- and the other intensity-dependent (see details later), generating two corresponding downsampled images reflecting the local probability of a cell being present. We used these as input to a particle tracking software, which detected and tracked local maxima of particularly high probability (Aguet et al., 2013). The first measurement captures the patch’s maximal spatial cross-correlation from frame t to frame t+1 within a search radius that can capture cell motion up to 60 μm/hour. The second measurement used the mean patch intensity in the raw image to capture the slightly brighter intensity of cells in relation to the background in phase-contrast imaging. Notably, our reduced resolution in the segmentation-free detection and tracking approach would break for imaging in higher cell densities. A bounding box of 70 × 70 μm around each cell was defined and used for single cell segmentation and feature extraction (details will follow). We excluded cells within 70μm from the image boundaries to avoid analyzing cells entering or leaving the field of view and to avoid the characteristic uneven illumination in these regions. Tracking of single cells over 8 hours was performed manually using the default settings in CellTracker v1.1 (Piccinini et al., 2016).
Unsupervised feature extraction with Adversarial Autoencoders
We have developed an unsupervised, generative representation for capturing cell image features using Adversarial Autoencoders (AAE) (Goodfellow et al., 2014; Makhzani et al., 2015). The autoencoder learns a compressed representation of cell images by encoding the images using a series of convolution and pooling layers leading ultimately to a lower dimensional embedding, or latent space. Points in the embedding space can then be decoded by a symmetric series of layers flowing in the opposite direction to reconstruct an image that, once trained, ideally appears nearly identical to the original input (Hinton et al., 2006). The training/optimization of the AAE is regularized (by using a second network during training) such that points close together in the embedding space will generate images sharing close visual resemblance/features (Makhzani et al., 2015). This convenient property can also generate synthetic/imaginary cell images to interpolate the appearance of cells from different regions of the space. We used the architecture from Johnson et al. (Johnson et al., 2017), that was based on the network presented in (Makhzani et al., 2015). Johnson’s network includes an AAE that learns to reconstruct landmarks of the cell nucleus and cytoplasm. The adversarial component teaches the network to discriminate between features derived from real cells and those drawn randomly from the latent space. We trained the regularized AAE with bounding boxes of phase-contrast single cell images (of size 70μm x 70 μm, or 217 × 217 pixels) that were rescaled to 256×256 pixels. The network was trained to extract a 56-dimensional image encoding representation of cell appearance. This representation and its variation over time were used as descriptors for cell appearance and action. We adapted Torch code from https://github.com/AllenCellModeling/torch_integrated_cell (Arulkumaran, 2017; Johnson et al., 2017) for unsupervised AAEs, and adjusted it to execute on our high-performance computing cluster. Torch (Collobert et al., 2011) is a Lua script-based scientific computing framework oriented towards machine learning algorithms with an underlying C/CUDA implementation.
The adversarial autoencoder latent vector preserves a visual similarity measure
We verified that the 56-dimensional latent vector preserves a visual similarity measure for cell appearance, i.e., increasing distances between two data points in the latent space correspond to increasing differences between the input images. We first validated that variations in the latent vector cause variations in cell appearances (Fig. S2A). To accomplish this we numerically perturbed the latent vector after encoding a cell image with varying amounts of noise and calculated the mean squared error between the raw and reconstructed images. As expected, the mean squared error between reconstructed and raw images monotonically increased with increasing amount of noise added in the latent space (Fig. S2B). Hence, the trained encoder generates a locally differentiable latent space. Second, we interpolated a linear trajectory in the latent space between two experimentally observed cells, as well as between two random points, and confirmed, visually and quantitatively, that the decoded images gradually transform from one image to the other (Fig. S2C–D, Video S9). Hence, the trained encoder generates a latent space without discontinuities. Third, we calculated the latent space distances between a cell at time t and the same cell at t+100 minutes and between a cell at time t and a neighboring cell in the same sample at time t. The distances between time-shifted latent space vectors for the same cell were shorter than those between neighboring cells (Fig. S2E). Hence, the combined effects of time variation in global imaging parameters and of morphological changes on displacements in the latent space tend to be smaller than the difference between cells.
Determining batch effects (inter-day variability)
In the case of the presented label-free imaging assay, batch effects may arise from uncontrolled experimental variables such as variations in the properties of the collagen gel, illumination artifacts, or inconsistencies in the phase ring alignment between sessions. Autoencoders are known to be very effective in capturing subtle image patterns. Therefore, they may pick up batch effects that mask image appearances related to the functional state of a cell. Under the assumption that intra-patient/cell line variability in image appearance is less than inter-patient/cell line appearance, we expect the latent cell descriptors of the same cell category on different days to be more similar than the descriptors of different cell categories imaged on the same day.
To test how strong batch effects may be in our data, we simultaneously imaged four different PDXs in an imaging session that we replicated on different days. Every cell was represented by the time-averaged latent space vector over the entire movie. We then computed the Euclidean distance as a measure of dissimilarity between descriptors from the same PDX imaged on different days to the distribution of Euclidean distances between different PDXs imaged on the same day (Fig. S3A). For three of the four tested PDXs we could not find a clear difference between the intra-PDX/inter-day similarity and the intra-day/inter-PDX similarity (Fig. S3B). Only PDX m610 displayed greater intra-PDX/inter-day similarity than intra-day/inter-PDX similarity. Consistent with this assessment, visualization of all time-averaged cell descriptors over all PDXs and days using PCA (Jolliffe, 2011) or tSNE (Maaten and Hinton, 2008) projections neither showed cell clusters associated with different PDXs nor with different imaging days, except for m610 (Fig. S3C–D). These results suggest that the latent space cell descriptors are impacted by both experimental batch effects and putative differences in the functional states between PDXs.
Single cell segmentation in phase-contrast imaging and shape feature extraction
To compare the performance of the deep-learned cell descriptors to conventional, shape-based descriptors of cell states (Bakal et al., 2007; Goodman and Carpenter, 2016; Gordonov et al., 2015; Pascual-Vargas et al., 2017; Scheeder et al., 2018; Sero and Bakal, 2017; Yin et al., 2013) we segmented phase contrast cell images of multiple cell types with diverse appearances.
Label-free cell segmentation is a challenging task, especially in the diverse landscape of shapes and appearance of the different melanoma cell systems we used. We used the LEVER (Winter et al., 2016) (downloaded from https://git-bioimage.coe.drexel.edu/opensource/lever), a designated phase-contrast cell segmentation algorithm to segment single cells within the bounding boxes identified by the previously described segmentation-free cell tracking. Briefly, the LEVER segmentation is based on minimum cross entropy thresholding and additional post-processing. While the segmentation was not perfect, it generally performed robustly to cells from different origins and varied imaging conditions (Fig. S5A–B). We used MATLAB’s function regionprops to extract 13 standard shape features from the segmentation masks produced by LEVER. These included: Area, MajorAxisLength, MinorAxisLength, Eccentricity, Orientation, ConvexArea, FilledArea, EulerNumber, EquivDiameter, Solidity, Extent, Perimeter, PerimeterOld.
Encoding temporal information
We compared three different approaches to incorporating temporal information when using either the autoencoder-based representation or the shape-based representation of cell appearance (Fig. S5C). First, static snapshot images ignoring the temporal information. Second, averaging the cell static descriptors along a cell’s trajectory, canceling noise for cells that do not undergo dramatic changes. Notably, the resulting cell descriptor matches the static descriptor in size and features. Accordingly, classifiers that were trained on average temporal descriptors could be applied to static snapshot descriptors (see Figs. 4–5). In the third encoding we relied on the ‘bag of words’ (BOW) approach (Sivic and Zisserman, 2009), in which each trajectory is represented by the distribution of discrete cell states, termed ‘code words’. A ‘dictionary’ of 100 code words was predetermined by k-means clustering (MacQueen, 1967) on the full dataset of cell descriptors.
We found that purely shape-based descriptors could not distinguish cell lines from PDXs (Fig. S5D). This indicates that the autoencoder latent space captures information from the phase-contrast images that is missed by the shape features. Incorporation of temporal information, especially the time-averaging, slightly (but significantly) boosted the classification performance of LDA models derived from latent space cell descriptors (Fig. S5E). This outcome is consistent with computer vision studies concluding that explicit modeling of time may lead to only marginal gains in classification performance (Karpathy et al., 2014).
Dimensionality reduction
We used tSNE (Fig. S3C) and PCA (Fig. S3D) for dimensionality reduction. Each cell was represented by its time-averaged descriptors in the latent space. For tSNE we used a GPU-accelerated implementation, https://github.com/CannyLab/tsne-cuda (Chan et al., 2018).
Discrimination analysis
We used Matlab’s vanilla implementation of Linear Discriminant Analysis (LDA) for the discrimination tasks (Figs. 2–3) and to identify the cellular phenotypes that correlate with low or high metastatic efficiency (Figs. 4–5). The feature vector for each cell was given by the normalized latent cell descriptor extracted by the autoencoder. Normalization of each latent cell descriptor component to a z-score feature was accomplished as follows. The mean (μ) and standard deviation (σ) of a latent cell descriptor component were calculated across the full data set of cropped cell images and used to calculate the corresponding z-score measure: xnorm = (x −μ)/σ, i.e., the variation from the mean values in units of standard deviation that can later be compared across different features.
For each classification task, the training data was kept completely separate from the testing data. Training and testing sets were assigned according to two methodologies. First, hold out all data from one cell type and train the classifier using all other cell types (Fig. 2A). Second, hold out all data from one cell type imaged in one day as the test set (“cell type - day”, e.g., Fig. 3F) and train the classifier on all other cell types excluding the data imaged on the same day as the test set (Fig. S4A). This second approach trained models that had never seen the cell type or data imaged on the same day of testing. In both classification settings we balanced the instances from each category for training by randomly selecting an equal number of observations from each class. This scheme was used for classification tasks involving categories containing more than one cell type: cell lines versus melanocytes, cell lines versus clonally expanded cell lines, cell lines versus PDXs, low versus high metastatic efficiency in PDXs (Figs. 2–3). For statistical analysis, all the cells in a single test set are considered as a single independent observation. Hence, “cell type - day” testing sets provide more independent observations (N) at the cost of fewer cells imaged in each day compared to testing set of the form of “cell type”.
We used bootstrapping to statistically test the ability to predict metastatic efficiency from samples of 20 random cells. This was performed for “cell type” (Fig. 3D) or “cell type - day” (Fig. 3G) test sets. For each test set, we generated 1000 observations by repeatedly selecting 20 random cells (with repetitions), recorded the fraction of these cells that were classified as low efficiency and the 95% confidence interval of the median. Statistical significance in all settings was inferred using two statistical tests using each test set classifier’s mean score: (1) The nonparametric Wilcoxon signed-rank test, considering the null hypothesis that the classifiers scores of observations from the two categories stem from the same distribution; (2) The Binomial test, considering the null hypothesis that the classifier prediction is random in respect to the ground truth labels. For inference of phenotypes that correlate with metastatic efficiency (Fig. 5) we used the classifier that was trained on the mean latent cell description along its trajectory (which proved to be superior to training with single snapshots) on latent cell descriptors derived from single snapshots, which hold the same, just noisier features.
The area under the Receiver Operating Characteristic (ROC) curve was recorded to assess and compare the discriminative accuracy of different tasks (Figs. 2–3). The true-positive rate (TPR) or sensitivity is the percentage of “low” metastatic cells classified correctly. The false-positive rate (FPR) or (1-specificity) is the percent of “high” metastatic cells incorrectly classified as “low”. Area under the ROC curve (AUC) was used as a measure of discrimination power. Note that the scores of all cells from all relevant cell types were pooled together for this analysis. Different classifiers can produce different scores, which means that our analysis provides a lower bound (pessimistic estimation). ROC analysis could not be applied for individual (held-out) test sets because they consist of only a single ground truth label.
We used the web-application PlotsOfData (Postma and Goedhart, 2019) to generate all boxplots.
In silico traversal weighted according to LDA coefficients
To generalize the in silico cell image amplification to multiple features, we traversed the high dimensional latent space according to the corresponding LDA coefficients. More specifically, we moved up/down the classifier’s score gradient by adding/subtracting multiples of one standard deviation of the unit vector weighted according to the LDA classifier coefficients.
Correlating classifier scores to genomic mutation markers
We calculated a distance matrix to assess the similarity between all pairs of PDXs and the cell lines A375 and MV3. The distances were calculated in terms of the classifier score and of genomic mutation panels. m528 was excluded from the analysis due missing sequencing data (see above). For the distance matrix of the classifier score, we calculated the Jensen-Shannon (JS) divergence (Lin, 1991) between the distributions of single cell classifier scores using the corresponding PDX-based classifiers (see discrimination analysis section in the Methods). For the cell lines, a new classifier was trained using all cells from all seven PDXs. This classifier was used to determine the classifier score for A375 and MV3. For each cell type, the distribution was approximated with a 25 bin histogram. JS divergence was calculated on pairs of cell type classifier score distributions.
To calculate distance matrices based genomic mutations we considered three panels of established melanoma genomic mutation markers. Two genomic mutation panels were derived from variation of exomes associated with 1385 cancer-related genes (see above). Mutations in commonly mutated genes in melanoma (Hodis et al., 2012) were annotated using OncoKB (Chakravarty et al., 2017) and divided into (i) oncogenic or likely oncogenic (Table S3, Fig. S11B) and (ii) benign or unannotated (“non-oncogenic”) (Table S4, Fig. S11C). Mutational based genetic distances were derived by converting mutation scores to a binary state (1=presence, 0=absence) and computing the Jaccard index (Jaccard, 1912) between cell types. In Fig. S11D we calculated distances using MASH (Ondov et al., 2016), which compared the K-mer profiles between samples, thus giving a distance of the raw sequence data, without biases introduced in the alignment and variant calling analysis.
The distance matrices derived from classifier scores and mutational states were correlated (Pearson correlation) to assess whether the genomic mutation state and image-derived classifier scores for low and high metastatic efficacies were linked.
QUANTIFICATION AND STATISTICAL ANALYSIS
For each classification task, the training data was kept completely separate from the testing data. For statistical analysis, all the cells in a single test set were considered as a single independent observation. We used bootstrapping to statistically test the ability to predict a category from samples of 20 random cells (Fig. 2D, Fig. 2G, Fig. 2J, Fig. 3D, Fig. 3G). Statistical significance in category classification of “cell type” (Fig. 2C, Fig. 2F, Fig. 2I, Fig. 3C) or “cell type - day” (Fig. 3F, Fig. S4C, Fig. S4E, Fig. S4G) was inferred using two statistical tests. (1) The nonparametric Wilcoxon signed-rank test, considering the null hypothesis that the classifiers scores of observations from the two categories stem from the same distribution; (2) The Binomial test, considering the null hypothesis that the classifier prediction is random in respect to the ground truth labels. The purpose of testing two different null hypotheses was to increase thoroughness, especially given the small sample sizes (number of cell types). Statistical significance of discrimination using cell shape and temporal information (Fig. S5D–F, Fig. S6A) was inferred using the Wilcoxon signed-rank test. Full details on the statistical issues can be found in sub-section entitled Discrimination analysis in the Methods. Statistical details of all experiments can be found in the figure legends including the statistical tests used, exact value of n, and clear descriptions of what n represents.
Supplementary Material
Table S1. Panel of melanoma cell types used for this study: cell lines, melanocytes and patient-derived xenograft (PDX), Related to the STAR Methods section. Cells with high metastatic efficiency were derived from patients that exhibited metastases within 22 months, whereas cells with low metastatic efficiency were derived from patients that developed distant metastases within 22 to 50 months (Quintana et al., 2012).
Table S2. Genomics Variants in PDX and two cell lines. PDX-SNVs-Indel and CellLines-SNVs-Indels: Single nucleotide variants (SNVs) and Insertions/Deletions (Indel)s of high-quality variants identified were filtered for common variants in > 1% of any population in GnomAD, Related to the STAR Methods section. The information provided by the columns is as follows: Cell Type, corresponding sample labels in Table S1; Hugo Symbol, the HUGO Gene Nomenclature Committee approved gene name (symbol); Chromosome, the affected chromosome; Start, the mutation start coordinate; Variant_Classification translational effect of variant allele; Depth, the read depth across this locus in tumor BAM; RefCt, the Read depth supporting the reference allele in tumor BAM; AltCt, read depth supporting the variant allele in tumor BAM; Tumor_MAF, mutational allele frequency (AltCt/Depth) and GT, the genotype encoded as alleles values separated by either of ”/” or “|”, e.g. The allele values are 0 for the reference allele (what is in the reference sequence), 1 for the ALT. PDX-CNVs and CellLines-CNVs: Copy number variants (CNVs) identified were filtered for genes mutated in > 5% of patients and > 10 patients in TCGA (Hoadley et al., 2018): The information provided by the columns is as follows: Cell Type, corresponding sample labels in Table S1; Hugo_Symbol, the HUGO Gene Nomenclature Committee approved gene name (symbol); Chromosome, the affected chromosome; Start, the mutation start coordinate; End, the mutational end position; Abberation is the type of copy number alteration as gain or loss; CN, the copy number of the gene; Score is calculated by CNV Kit as the sum of the weights of the bins supporting the CNV; Cytoband, the position on the chromosomal cytogentic band.
Table S3. Oncogenic Genomics Variants in PDX and two cell lines in commonly mutated genes (Hodis et al., 2012), Related to the STAR Methods section. The information provided by the columns is as follows: Annotation, the oncogenic effect, Hugo_Symbol, the HUGO Gene Nomenclature Committee approved gene name (symbol); Variant Type, CNV=Copy Number Variation, SNV=Single Nucleotide Variant and Indel= Insertion or Deletion; Variant, for SNVs and InDels, this is the HSGS protein change and is the aberration type (gain or loss) which has the predicted oncogenic effect; each following column is the Cell Type corresponding to Table S1 and the genotype or copy of the Cell Type.
Table S4. Non-oncogenic Genomics Variants in PDX and two cell lines in commonly mutated genes (Hodis et al., 2012), Related to the STAR Methods section. The information provided by the columns is as follows: Hugo_Symbol, the HUGO Gene Nomenclature Committee approved gene name (symbol); Variant Type, CNV=Copy Number Variation, SNV=Single Nucleotide Variant and Indel= Insertion or Deletion; Variant, for SNVs and InDels, this is the HSGS protein change and is the aberration type (gain or loss) which has the predicted oncogenic effect; each following column is the Cell Type corresponding to Table S1 and the genotype or copy of the Cell Type.
Video S1. Time lapse sequence of a representative field of view of high metastatic m481 PDX cells, Related to Figure 1.
Video S2. Time lapse sequence of a representative field of view of low metastatic m610 PDX cells, Related to Figure 1.
Video S3. Heterogeneous morphology and local dynamics of patient derived melanoma cells. Shown are time lapse of single cells cropped from the field of view and used as the input for the autoencoder, Related to Figure 1.
Video S4. Reconstruction evolution and convergence during autoencoder training, Related to Figure 1. Raw (top) and reconstructed (bottom) images during the autoencoder training minimizing the binary cross-entropy error between the two.
Video S5. Morphing m498 PDX cell in silico from low to high metastatic efficiency by decoding the latent cell descriptor under gradual shifts in feature #56, Related to Figure 5. The corresponding classifier’s score (“ClassifierScore”) and value of feature #56 in units of the z-score (“f56”) are shown.
Video S6. Morphing m405 PDX cell in silico from high to low metastatic efficiency by decoding the latent cell descriptor under gradual shifts in feature #56, Related to Figure 5. The corresponding classifier’s score (“ClassifierScore”) and value of feature #56 in units of the z-score (“f56”) are shown.
Video S7. Morphing 100 m498 cells by gradually decreasing feature #56 (increasing classifier score), Related to Figure 5.
Video S8. Time lapse of a m610 PDX cell spontaneously switching from the low to the high metastatic efficiency domain (as predicted by the classifier), Related to the Figure 5. Live imaging for 10 minutes.
Video S9. Cell morphing in silico, Related to the STAR Methods section. Following the decoded cell image gradually morphing along an interpolated linear trajectory in the latent space between two cells.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Leibovitz’s L-15 Medium | Gibco | 21083027 |
| Matrigel | BD Biosciences | 354248 |
| Collagenase IV | Gibco | 17104019 |
| DNase | Fisher | 89836 |
| penicillin streptomycin | Gibco | 15140148 |
| fetal bovine serum | Gibco | 16000044 |
| DMEM | Gibco | 12430054 |
| puromycin | Gibco | A1113802 |
| G418 | ThermoFisher | 10131027 |
| trypsin/EDTA | Gibco | 15400054 |
| medium 254 | Fisher | M254500 |
| Melanocyte Growth Kit | ATCC | PCS-200-041 |
| Dermal Cell Basal Medium | ATCC | PCS-200-030 |
| MCDB 153 | Sigma | M7403 |
| Bovine Insulin | Sigma | I-5500 |
| rat tail collagen | Corning | 354249 |
| Experimental Models: Cell Lines | ||
| Primary melanocytes | ATCC | PCS-200-013 |
| m116 melanocytes | Laboratory or Jerry Shay, UT Southwestern | N/A |
| Human Melanoma MV3 | Laboratory of Peter Friedl, MD Anderson Houston TX | N/A |
| A375 | ATCC | CRL-1619 |
| SK-Mel2 | ATCC | HTB-68 |
| WM3670 | Wistar Institute | WC00119 |
| WM1361 | Wistar Institute | WC00075 |
| WM1366 | Wistar Institute | WC00078 |
| Experimental Models: Organisms/Strains | ||
| NSG Mice | Jackson Laboratories | 005557 |
| Recombinant DNA | ||
| FUW lentiviral expression vector containing dsRed2 and luciferase | Laboratory of Sean Morrison, UT Southwestern | N/A |
| Software and Algorithms | ||
| Source code and test data | This work | Zenodo (Github) 4619858 |
| Other | ||
| Raw and processed data | This work | Image Data Resource (IDR) idr0109 |
Highlights.
Generative deep network encoded latent representation of live imaged melanoma cells
Supervised ML classified metastatic efficiency using latent cell representations
Validated classifier prediction on melanoma cell lines in mouse xenografts
Interpreted metastasis driving features in amplified generative cell image models
Acknowledgements
This work was supported by the Cancer Prevention and Research Institute of Texas (CPRIT R160622 to GD), the National Institutes of Health (R35GM126428 to GD; K25CA204526 to ESW), and the Israeli Council for Higher Education (CHE) via Data Science Research Center, Ben-Gurion University of the Negev, Israel (to AZ). We thank Sean Morrison for PDX-derived cell models. We thank Andrew R. Cohen for LEVER.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Inclusion and diversity
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper self-identifies as living with a disability. One or more of the authors of this paper received support from a program designed to increase minority representation in science.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aguet F, Antonescu CN, Mettlen M, Schmid SL, and Danuser G. (2013). Advances in analysis of low signal-to-noise images link dynamin and AP2 to the functions of an endocytic checkpoint. Developmental cell 26, 279–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arulkumaran K. (2017). Autoencoders. [Google Scholar]
- Ash JT, Darnell G, Munro D, and Engelhardt BE (2018). Joint analysis of gene expression levels and histological images identifies genes associated with tissue morphology. bioRxiv, 458711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakal C, Aach J, Church G, and Perrimon N. (2007). Quantitative morphological signatures define local signaling networks regulating cell morphology. Science 316, 1753–1756. [DOI] [PubMed] [Google Scholar]
- Belthangady C, and Royer LA (2019). Applications, promises, and pitfalls of deep learning for fluorescence image reconstruction. Nature methods, 1–11. [DOI] [PubMed] [Google Scholar]
- Boutros M, Heigwer F, and Laufer C. (2015). Microscopy-based high-content screening. Cell 163, 1314–1325. [DOI] [PubMed] [Google Scholar]
- Boyd JC, Pinheiro A, Del Nery E, Reyal F, and Walter T. (2020). Domain-invariant features for mechanism of action prediction in a multi-cell-line drug screen. Bioinformatics 36, 1607–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caicedo JC, Cooper S, Heigwer F, Warchal S, Qiu P, Molnar C, Vasilevich AS, Barry JD, Bansal HS, and Kraus O. (2017). Data-analysis strategies for image-based cell profiling. Nature methods 14, 849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantelli G, Orgaz JL, Rodriguez-Hernandez I, Karagiannis P, Maiques O, Matias-Guiu X, Nestle FO, Marti RM, Karagiannis SN, and Sanz-Moreno V. (2015). TGF-β-induced transcription sustains amoeboid melanoma migration and dissemination. Current biology 25, 2899–2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarty D, Gao J, Phillips S, Kundra R, Zhang H, Wang J, Rudolph JE, Yaeger R, Soumerai T, and Nissan MH (2017). OncoKB: a precision oncology knowledge base. JCO precision oncology 1, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DM, Rao R, Huang F, and Canny JF (2018). t-SNE-CUDA: GPU-Accelerated t-SNE and its Applications to Modern Data. Paper presented at: 2018 30th International Symposium on Computer Architecture and High Performance Computing (SBAC-PAD) (IEEE). [Google Scholar]
- Chan JK (2014a). The wonderful colors of the hematoxylin-eosin stain in diagnostic surgical pathology. Int J Surg Pathol 22, 12–32. [DOI] [PubMed] [Google Scholar]
- Chan JK (2014b). The wonderful colors of the hematoxylin–eosin stain in diagnostic surgical pathology. International journal of surgical pathology 22, 12–32. [DOI] [PubMed] [Google Scholar]
- Chandrasekaran SN, Ceulemans H, Boyd JD, and Carpenter AE (2020). Image-based profiling for drug discovery: due for a machine-learning upgrade? Nat Rev Drug Discov. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S, Fu S, Kim YM, Song W, Li Y, Xue Y, Yi J, and Tian L. (2021). Single-cell cytometry via multiplexed fluorescence prediction by label-free reflectance microscopy. Science Advances 7, eabe0431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi W, Fang-Yen C, Badizadegan K, Oh S, Lue N, Dasari RR, and Feld MS (2007). Tomographic phase microscopy. Nature methods 4, 717–719. [DOI] [PubMed] [Google Scholar]
- Christiansen EM, Yang SJ, Ando DM, Javaherian A, Skibinski G, Lipnick S, Mount E, O’Neil A, Shah K, and Lee AK (2018). In silico labeling: Predicting fluorescent labels in unlabeled images. Cell 173, 792–803. e719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collobert R, Kavukcuoglu K, and Farabet C. (2011). Torch7: A matlab-like environment for machine learning. Paper presented at: BigLearn, NIPS workshop. [Google Scholar]
- Courtiol P, Maussion C, Moarii M, Pronier E, Pilcer S, Sefta M, Manceron P, Toldo S, Zaslavskiy M, and Le Stang N. (2019). Deep learning-based classification of mesothelioma improves prediction of patient outcome. Nature medicine, 1–7. [DOI] [PubMed] [Google Scholar]
- Cruz-Roa AA, Arevalo Ovalle JE, Madabhushi A, and González Osorio FA (2013). A deep learning architecture for image representation, visual interpretability and automated basal-cell carcinoma cancer detection. Med Image Comput Comput Assist Interv 16, 403–410. [DOI] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, and Bottomley W. (2002). Mutations of the BRAF gene in human cancer. Nature 417, 949. [DOI] [PubMed] [Google Scholar]
- Eddy CZ, Wang X, Li F, and Sun B. (2018). The morphodynamics of 3D migrating cancer cells. arXiv preprint arXiv:180710822. [Google Scholar]
- Falke S, Brognaro H, Martirosyan A, Dierks K, and Betzel C. (2019). A multi-channel in situ light scattering instrument utilized for monitoring protein aggregation and liquid dense cluster formation. Heliyon 5, e03016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang L, Monroe F, Novak SW, Kirk L, Schiavon C, Seungyoon BY, Zhang T, Wu M, Kastner K, and Kubota Y. (2019). Deep Learning-Based Point-Scanning Super-Resolution Imaging. bioRxiv, 740548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Jung AW, Torne RV, Gonzalez S, Vohringer H, Jimenez-Linan M, Moore L, and Gerstung M. (2019). Pan-cancer computational histopathology reveals mutations, tumor composition and prognosis. bioRxiv, 813543. [DOI] [PubMed] [Google Scholar]
- Ganesh K, Basnet H, Kaygusuz Y, Laughney AM, He L, Sharma R, O’Rourke KP, Reuter VP, Huang Y-H, and Turkekul M. (2020). L1CAM defines the regenerative origin of metastasis-initiating cells in colorectal cancer. Nature Cancer 1, 28–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghandi M, Huang FW, Jané-Valbuena J, Kryukov GV, Lo CC, McDonald ER, Barretina J, Gelfand ET, Bielski CM, and Li H. (2019). Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giard DJ, Aaronson SA, Todaro GJ, Arnstein P, Kersey JH, Dosik H, and Parks WP (1973). In vitro cultivation of human tumors: establishment of cell lines derived from a series of solid tumors. Journal of the National Cancer Institute 51, 1417–1423. [DOI] [PubMed] [Google Scholar]
- Goodfellow I, Pouget-Abadie J, Mirza M, Xu B, Warde-Farley D, Ozair S, Courville A, and Bengio Y. (2014). Generative adversarial nets. Paper presented at: Advances in neural information processing systems. [Google Scholar]
- Goodman A, and Carpenter AE (2016). High-Throughput, Automated Image Processing for Large-Scale Fluorescence Microscopy Experiments. Microscopy and Microanalysis 22, 538–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordonov S, Hwang MK, Wells A, Gertler FB, Lauffenburger DA, and Bathe M. (2015). Time series modeling of live-cell shape dynamics for image-based phenotypic profiling. Integrative Biology 8, 73–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S-M, Krishnan AP, Folkesson J, Ivanov I, Chhun B, Cho N, Leonetti M, and Mehta SB (2019). Revealing architectural order with polarized light imaging and deep neural networks. bioRxiv, 631101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurcan MN, Boucheron LE, Can A, Madabhushi A, Rajpoot NM, and Yener B. (2009). Histopathological image analysis: A review. IEEE reviews in biomedical engineering 2, 147–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward NK, Wilmott JS, Waddell N, Johansson PA, Field MA, Nones K, Patch A-M, Kakavand H, Alexandrov LB, and Burke H. (2017). Whole-genome landscapes of major melanoma subtypes. Nature 545, 175. [DOI] [PubMed] [Google Scholar]
- Hinton GE, Osindero S, and Teh Y-W (2006). A fast learning algorithm for deep belief nets. Neural computation 18, 1527–1554. [DOI] [PubMed] [Google Scholar]
- Hoadley KA, Yau C, Hinoue T, Wolf DM, Lazar AJ, Drill E, Shen R, Taylor AM, Cherniack AD, and Thorsson V. (2018). Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell 173, 291–304. e296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat J-P, Nickerson E, Auclair D, Li L, and Place C. (2012). A landscape of driver mutations in melanoma. Cell 150, 251–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaccard P. (1912). The distribution of the flora in the alpine zone. 1. New phytologist 11, 37–50. [Google Scholar]
- Jakob JA, Bassett RL Jr, Ng CS, Curry JL, Joseph RW, Alvarado GC, Rohlfs ML, Richard J, Gershenwald JE, and Kim KB (2012). NRAS mutation status is an independent prognostic factor in metastatic melanoma. Cancer 118, 4014–4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Demere Z, Nair K, Ali A, Ferraro GB, Natoli T, Deik A, Petronio L, Tang AA, Zhu C, et al. (2020). A metastasis map of human cancer cell lines. Nature 588, 331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson GR, Donovan-Maiye RM, and Maleckar MM (2017). Generative Modeling with Conditional Autoencoders: Building an Integrated Cell. arXiv preprint arXiv:170500092. [Google Scholar]
- Jolliffe I. (2011). Principal component analysis (Springer; ). [Google Scholar]
- Jones DT (2019). Setting the standards for machine learning in biology. Nature Reviews Molecular Cell Biology, 1–2. [DOI] [PubMed] [Google Scholar]
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, and Birnbaum DP (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. bioRxiv, 531210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpathy A, Toderici G, Shetty S, Leung T, Sukthankar R, and Fei-Fei L. (2014). Large-scale video classification with convolutional neural networks. Paper presented at: Proceedings of the IEEE conference on Computer Vision and Pattern Recognition. [Google Scholar]
- Kozlowski JM, Fidler IJ, Campbell D, Xu Z. l., Kaighn ME, and Hart IR (1984). Metastatic behavior of human tumor cell lines grown in the nude mouse. Cancer research 44, 3522–3529. [PubMed] [Google Scholar]
- Kraus OZ, Grys BT, Ba J, Chong Y, Frey BJ, Boone C, and Andrews BJ (2017). Automated analysis of high-content microscopy data with deep learning. Mol Syst Biol 13, 924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaChance J, and Cohen DJ (2020). Practical Fluorescence Reconstruction Microscopy for High-Content Imaging. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J. (1991). Divergence measures based on the Shannon entropy. IEEE Transactions on Information theory 37, 145–151. [Google Scholar]
- López JI (2013a). Renal tumors with clear cells. A review. Pathol Res Pract 209, 137–146. [DOI] [PubMed] [Google Scholar]
- López JI (2013b). Renal tumors with clear cells. A review. Pathology-Research and Practice 209, 137–146. [DOI] [PubMed] [Google Scholar]
- Maaten L.v.d., and Hinton G. (2008). Visualizing data using t-SNE. Journal of machine learning research 9, 2579–2605. [Google Scholar]
- MacQueen J. (1967). Some methods for classification and analysis of multivariate observations. Paper presented at: Proceedings of the fifth Berkeley symposium on mathematical statistics and probability; (Oakland, CA, USA.). [Google Scholar]
- Makhzani A, Shlens J, Jaitly N, Goodfellow I, and Frey B. (2015). Adversarial autoencoders. arXiv preprint arXiv:151105644. [Google Scholar]
- Marina OC, Sanders CK, and Mourant JR (2012). Effects of acetic acid on light scattering from cells. Journal of biomedical optics 17, 085002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan AS, Dean KM, Isogai T, Kasitinon SY, Murali VS, Roudot P, Groisman A, Reed DK, Welf ES, and Han SJ (2019). Enhanced Dendritic Actin Network Formation in Extended Lamellipodia Drives Proliferation in Growth-Challenged Rac1P29S Melanoma Cells. Developmental cell 49, 444–460. e449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinie N, Rubtsova SN, Fokin A, Visweshwaran SP, Rocques N, Polesskaya A, Schnitzler A, Vacher S, Denisov EV, and Tashireva LA (2019). Cortical branched actin determines cell cycle progression. Cell research 29, 432–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nehme E, Weiss LE, Michaeli T, and Shechtman Y. (2018). Deep-STORM: super-resolution single-molecule microscopy by deep learning. Optica 5, 458–464. [Google Scholar]
- Nikolaou S, and Machesky LM (2020). The stressful tumour environment drives plasticity of cell migration programmes, contributing to metastasis. J Pathol 250, 612–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ondov BD, Treangen TJ, Melsted P, Mallonee AB, Bergman NH, Koren S, and Phillippy AM (2016). Mash: fast genome and metagenome distance estimation using MinHash. Genome biology 17, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ounkomol C, Seshamani S, Maleckar MM, Collman F, and Johnson GR (2018). Label-free prediction of three-dimensional fluorescence images from transmitted-light microscopy. Nature methods 15, 917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang W, Aristov A, Lelek M, Hao X, and Zimmer C. (2018). Deep learning massively accelerates super-resolution localization microscopy. Nature biotechnology 36, 460–468. [DOI] [PubMed] [Google Scholar]
- Pan C, Schoppe O, Parra-Damas A, Cai R, Todorov MI, Gondi G, von Neubeck B, Böğürcü-Seidel N, Seidel S, and Sleiman K. (2019). Deep Learning Reveals Cancer Metastasis and Therapeutic Antibody Targeting in the Entire Body. Cell 179, 1661–1676. e1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual-Vargas P, Cooper S, Sero J, Bousgouni V, Arias-Garcia M, and Bakal C. (2017). RNAi screens for Rho GTPase regulators of cell shape and YAP/TAZ localisation in triple negative breast cancer. Scientific Data 4, 170018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavillon N, Hobro AJ, Akira S, and Smith NI (2018). Noninvasive detection of macrophage activation with single-cell resolution through machine learning. Proceedings of the National Academy of Sciences 115, E2676–E2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccinini F, Kiss A, and Horvath P. (2016). CellTracker (not only) for dummies. Bioinformatics 32, 955–957. [DOI] [PubMed] [Google Scholar]
- Pinner S, and Sahai E. (2008). Imaging amoeboid cancer cell motility in vivo. Journal of microscopy 231, 441–445. [DOI] [PubMed] [Google Scholar]
- Postma M, and Goedhart J. (2019). PlotsOfData—A web app for visualizing data together with their summaries. PLoS biology 17, e3000202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quax P, Van Muijen G, Weening-Verhoeff E, Lund L, Danø K, Ruiter D, and Verheijen J. (1991). Metastatic behavior of human melanoma cell lines in nude mice correlates with urokinase-type plasminogen activator, its type-1 inhibitor, and urokinase-mediated matrix degradation. The Journal of cell biology 115, 191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana E, Piskounova E, Shackleton M, Weinberg D, Eskiocak U, Fullen DR, Johnson TM, and Morrison SJ (2012). Human melanoma metastasis in NSG mice correlates with clinical outcome in patients. Science translational medicine 4, 159ra149–159ra149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana E, Shackleton M, Foster HR, Fullen DR, Sabel MS, Johnson TM, and Morrison SJ (2010). Phenotypic heterogeneity among tumorigenic melanoma cells from patients that is reversible and not hierarchically organized. Cancer cell 18, 510–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozenberg GI, Monahan KB, Torrice C, Bear JE, and Sharpless NE (2010). Metastasis in an orthotopic murine model of melanoma is independent of RAS/RAF mutation. Melanoma research 20, 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadok A, McCarthy A, Caldwell J, Collins I, Garrett MD, Yeo M, Hooper S, Sahai E, Kuemper S, and Mardakheh FK (2015). Rho kinase inhibitors block melanoma cell migration and inhibit metastasis. Cancer research 75, 2272–2284. [DOI] [PubMed] [Google Scholar]
- Sahai E, and Marshall CJ (2003). Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nature cell biology 5, 711–719. [DOI] [PubMed] [Google Scholar]
- Scheeder C, Heigwer F, and Boutros M. (2018). Machine learning and image-based profiling in drug discovery. Current opinion in systems biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrama D, Keller G, Houben R, Ziegler CG, Vetter-Kauczok CS, Ugurel S, and Becker JC (2008). BRAFV600E mutations in malignant melanoma are associated with increased expressions of BAALC. Journal of carcinogenesis 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schürmann M, Scholze J, Müller P, Chan CJ, Ekpenyong AE, Chalut KJ, and Guck J. (2015). Refractive index measurements of single, spherical cells using digital holographic microscopy. In Methods in cell biology (Elsevier), pp. 143–159. [DOI] [PubMed] [Google Scholar]
- Sero JE, and Bakal C. (2017). Multiparametric analysis of cell shape demonstrates that β-PIX directly couples YAP activation to extracellular matrix adhesion. Cell systems 4, 84–96. e86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamai G, Binenbaum Y, Slossberg R, Duek I, Gil Z, and Kimmel R. (2019). Artificial intelligence algorithms to assess hormonal status from tissue microarrays in patients with breast cancer. JAMA network open 2, e197700-e197700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivic J, and Zisserman A. (2009). Efficient visual search of videos cast as text retrieval. IEEE transactions on pattern analysis and machine intelligence 31, 591–606. [DOI] [PubMed] [Google Scholar]
- Sullivan DP, and Lundberg E. (2018). Seeing More: A Future of Augmented Microscopy. Cell 173, 546–548. [DOI] [PubMed] [Google Scholar]
- Swaminathan K, Campbell A, Papalazarou V, Jaber-Hijazi F, Nixon C, McGhee E, Strathdee D, Sansom OJ, and Machesky LM (2020). The RAC1 Target NCKAP1 Plays a Crucial Role in the Progression of Braf;Pten-Driven Melanoma in Mice. J Invest Dermatol. [DOI] [PubMed] [Google Scholar]
- Tanami H, Imoto I, Hirasawa A, Yuki Y, Sonoda I, Inoue J, Yasui K, Misawa-Furihata A, Kawakami Y, and Inazawa J. (2004). Involvement of overexpressed wild-type BRAF in the growth of malignant melanoma cell lines. Oncogene 23, 8796. [DOI] [PubMed] [Google Scholar]
- Travis WD, Brambilla E, Noguchi M, Nicholson AG, Geisinger K, Yatabe Y, Ishikawa Y, Wistuba I, Flieder DB, Franklin W, et al. (2013). Diagnosis of lung adenocarcinoma in resected specimens: implications of the 2011 International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society classification. Arch Pathol Lab Med 137, 685–705. [DOI] [PubMed] [Google Scholar]
- van Muijen GN, Jansen KF, Cornelissen IM, Smeets DF, Beck JL, and Ruiter DJ (1991). Establishment and characterization of a human melanoma cell line (MV3) which is highly metastatic in nude mice. International journal of cancer 48, 85–91. [DOI] [PubMed] [Google Scholar]
- Viceconte N, Dheur M-S, Majerova E, Pierreux CE, Baurain J-F, van Baren N, and Decottignies A. (2017). Highly aggressive metastatic melanoma cells unable to maintain telomere length. Cell reports 19, 2529–2543. [DOI] [PubMed] [Google Scholar]
- Wang H, Rivenson Y, Jin Y, Wei Z, Gao R, Gunaydin H, Bentolila LA, Kural C, and Ozcan A. (2019). Deep learning enables cross-modality super-resolution in fluorescence microscopy. Nat Methods 16, 103–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigert M, Schmidt U, Boothe T, Müller A, Dibrov A, Jain A, Wilhelm B, Schmidt D, Broaddus C, Culley S, et al. (2018). Content-aware image restoration: pushing the limits of fluorescence microscopy. Nature methods 15, 1090. [DOI] [PubMed] [Google Scholar]
- Welf ES, Driscoll MK, Dean KM, Schäfer C, Chu J, Davidson MW, Lin MZ, Danuser G, and Fiolka R. (2016). Quantitative multiscale cell imaging in controlled 3D microenvironments. Developmental cell 36, 462–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams E, Moore J, Li SW, Rustici G, Tarkowska A, Chessel A, Leo S, Antal B, Ferguson RK, Sarkans U, et al. (2017). The Image Data Resource: A Bioimage Data Integration and Publication Platform. Nat Methods 14, 775–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter M, Mankowski W, Wait E, Temple S, and Cohen AR (2016). LEVER: software tools for segmentation, tracking and lineaging of proliferating cells. Bioinformatics 32, 3530–3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P-H, Gilkes DM, Phillip JM, Narkar A, Cheng TW-T, Marchand J, Lee M-H, Li R, and Wirtz D. (2020). Single-cell morphology encodes metastatic potential. Science Advances 6, eaaw6938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Z, Sadok A, Sailem H, McCarthy A, Xia X, Li F, Garcia MA, Evans L, Barr AR, and Perrimon N. (2013). A screen for morphological complexity identifies regulators of switch-like transitions between discrete cell shapes. Nature cell biology 15, 860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H, Cai L, Wang Z, Hu X, Zhang S, and Ji S. (2018). Computational modeling of cellular structures using conditional deep generative networks. Bioinformatics 35, 2141–2149. [DOI] [PubMed] [Google Scholar]
- Zhang W, Williams TA, Bhagwath AS, Hiermann JS, Peacock CD, Watkins DN, Ding P, Park JY, Montgomery EA, and Forastiere AA (2020). GEAMP, a novel gastroesophageal junction carcinoma cell line derived from a malignant pleural effusion. Laboratory Investigation 100, 16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Panel of melanoma cell types used for this study: cell lines, melanocytes and patient-derived xenograft (PDX), Related to the STAR Methods section. Cells with high metastatic efficiency were derived from patients that exhibited metastases within 22 months, whereas cells with low metastatic efficiency were derived from patients that developed distant metastases within 22 to 50 months (Quintana et al., 2012).
Table S2. Genomics Variants in PDX and two cell lines. PDX-SNVs-Indel and CellLines-SNVs-Indels: Single nucleotide variants (SNVs) and Insertions/Deletions (Indel)s of high-quality variants identified were filtered for common variants in > 1% of any population in GnomAD, Related to the STAR Methods section. The information provided by the columns is as follows: Cell Type, corresponding sample labels in Table S1; Hugo Symbol, the HUGO Gene Nomenclature Committee approved gene name (symbol); Chromosome, the affected chromosome; Start, the mutation start coordinate; Variant_Classification translational effect of variant allele; Depth, the read depth across this locus in tumor BAM; RefCt, the Read depth supporting the reference allele in tumor BAM; AltCt, read depth supporting the variant allele in tumor BAM; Tumor_MAF, mutational allele frequency (AltCt/Depth) and GT, the genotype encoded as alleles values separated by either of ”/” or “|”, e.g. The allele values are 0 for the reference allele (what is in the reference sequence), 1 for the ALT. PDX-CNVs and CellLines-CNVs: Copy number variants (CNVs) identified were filtered for genes mutated in > 5% of patients and > 10 patients in TCGA (Hoadley et al., 2018): The information provided by the columns is as follows: Cell Type, corresponding sample labels in Table S1; Hugo_Symbol, the HUGO Gene Nomenclature Committee approved gene name (symbol); Chromosome, the affected chromosome; Start, the mutation start coordinate; End, the mutational end position; Abberation is the type of copy number alteration as gain or loss; CN, the copy number of the gene; Score is calculated by CNV Kit as the sum of the weights of the bins supporting the CNV; Cytoband, the position on the chromosomal cytogentic band.
Table S3. Oncogenic Genomics Variants in PDX and two cell lines in commonly mutated genes (Hodis et al., 2012), Related to the STAR Methods section. The information provided by the columns is as follows: Annotation, the oncogenic effect, Hugo_Symbol, the HUGO Gene Nomenclature Committee approved gene name (symbol); Variant Type, CNV=Copy Number Variation, SNV=Single Nucleotide Variant and Indel= Insertion or Deletion; Variant, for SNVs and InDels, this is the HSGS protein change and is the aberration type (gain or loss) which has the predicted oncogenic effect; each following column is the Cell Type corresponding to Table S1 and the genotype or copy of the Cell Type.
Table S4. Non-oncogenic Genomics Variants in PDX and two cell lines in commonly mutated genes (Hodis et al., 2012), Related to the STAR Methods section. The information provided by the columns is as follows: Hugo_Symbol, the HUGO Gene Nomenclature Committee approved gene name (symbol); Variant Type, CNV=Copy Number Variation, SNV=Single Nucleotide Variant and Indel= Insertion or Deletion; Variant, for SNVs and InDels, this is the HSGS protein change and is the aberration type (gain or loss) which has the predicted oncogenic effect; each following column is the Cell Type corresponding to Table S1 and the genotype or copy of the Cell Type.
Video S1. Time lapse sequence of a representative field of view of high metastatic m481 PDX cells, Related to Figure 1.
Video S2. Time lapse sequence of a representative field of view of low metastatic m610 PDX cells, Related to Figure 1.
Video S3. Heterogeneous morphology and local dynamics of patient derived melanoma cells. Shown are time lapse of single cells cropped from the field of view and used as the input for the autoencoder, Related to Figure 1.
Video S4. Reconstruction evolution and convergence during autoencoder training, Related to Figure 1. Raw (top) and reconstructed (bottom) images during the autoencoder training minimizing the binary cross-entropy error between the two.
Video S5. Morphing m498 PDX cell in silico from low to high metastatic efficiency by decoding the latent cell descriptor under gradual shifts in feature #56, Related to Figure 5. The corresponding classifier’s score (“ClassifierScore”) and value of feature #56 in units of the z-score (“f56”) are shown.
Video S6. Morphing m405 PDX cell in silico from high to low metastatic efficiency by decoding the latent cell descriptor under gradual shifts in feature #56, Related to Figure 5. The corresponding classifier’s score (“ClassifierScore”) and value of feature #56 in units of the z-score (“f56”) are shown.
Video S7. Morphing 100 m498 cells by gradually decreasing feature #56 (increasing classifier score), Related to Figure 5.
Video S8. Time lapse of a m610 PDX cell spontaneously switching from the low to the high metastatic efficiency domain (as predicted by the classifier), Related to the Figure 5. Live imaging for 10 minutes.
Video S9. Cell morphing in silico, Related to the STAR Methods section. Following the decoded cell image gradually morphing along an interpolated linear trajectory in the latent space between two cells.
Data Availability Statement
Raw image data, raw single cell images, corresponding metadata, the trained neural network, and the feature representation of all cells source data have been deposited at the Image Data Resource (Williams et al., 2017), https://idr.openmicroscopy.org, and are publicly available under the accession numbers: idr0109.
Original source code and test data is publicly available at https://github.com/DanuserLab/openLCH (doi: https://doi.org/10.5281/zenodo.4619858)
Scripts used to generate the figures presented in this paper are not provided in this paper but are available from the Lead Contact on request.
Any additional information required to reproduce this work is available from the Lead Contact.