

Abstract
A user-friendly approach to sidestep the venerable Grignard addition to unactivated ketones to access tertiary alcohols by reversing the polarity of the disconnection. In this work a ketone instead acts as a nucleophile when adding to simple unactivated olefins to accomplish the same overall transformation. The scope of this coupling is broad as enabled using an electrochemical approach and the reaction is scalable, chemoselective, and requires no precaution to exclude air or water. Multiple applications demonstrate the simplifying nature of the reaction on multi-step synthesis and mechanistic studies point to an intuitive mechanism reminiscent of other chemical reductants such as SmI2 (which cannot accomplish the same reaction).
Keywords: Ketyl-Olefin, Ketyl, electrochemical Grignard, electrochemical coupling, reductive coupling
Tertiary alcohols are an abundant functional group with versatile reactivity that are found in natural products,1 pharmaceuticals,2 and a multitude of useful materials.3 Traditionally, perhaps overwhelmingly, the ketone has served as a loyal progenitor of this species (Figure 1A) for good reasons. Every undergraduate organic textbook prescribes a direct nucleophilic addition of a strong nucleophile, such as RMgX or RLi, to these electrophilic species.4 Although these incredibly robust reactions have been employed countless times, they can indirectly contribute to synthetic inefficiencies as their low chemoselectivity often necessitates the use of protecting groups.5 This dilemma is nicely illustrated (Figure 1B) by examining the patented route to steroid derivative 2.6 Although a Grignard reaction with commercially available ketone 1 is an obvious disconnection, its use introduces several protecting group additions, removals and functional group manipulations throughout the course of a seven-step sequence (only one of which forges a C–C bond).
FIGURE 1.

Tertiary alcohols from simple ketones remain a challenge for modern synthesis (A). Synthesis of 2 is emblematic of the problems with Grignard (B). Recent approaches so far do not address the problem (C). Electrochemical precedent on activated olefins (D) and a summary of this work (E).
Within the specific realm of intermolecular alkyl nucleophile additions to unactivated ketones, Grignard and related organometallic additions are fundamentally limited by their 2-electron mechanisms, which render these nucleophiles both strongly nucleophilic and often highly basic.4,5,7 Efforts to tone down their reactivity have been explored, with the most successful stemming from nucleophiles bearing activated positions (i.e. allylic, benzylic, propargylic, α-carbonyl, Figure 1C).8,9 Studies employing Zr-,10 Ti-,11 Ru-,12 and Os-13 based systems, as well as HAT chemistry,14 have also pointed to the use of olefins as precursors to species capable of adding to carbonyl groups although intermolecular additions into unactivated ketones are without precedent.15 A less intuitive approach involves an umpolung disconnection, which renders the ketone the nucleophilic group through a reductive 1-electron approach. Thus far, such approaches have relied primarily on Sm(II),7a-b Ti(III),16 or photoinduced electron transfer17 to couple activated olefins and styrenes to ketones. A general intermolecular reductive coupling of unactivated ketones and olefins is so far absent from the literature. The closest precedent for the desired transformation was disclosed by Shono and co-workers (Figure 1D).18 These reports focus predominantly on intramolecular couplings,18a-b with only a few intermolecular examples18c-d presented. To the best of our knowledge, this chemistry has not been applied in the literature, despite being available for decades, presumably due to the challenges of using a divided cell setup under an argon atmosphere and the need for at least a five-fold excess of the ketone. In this Communication, a new protocol for electrochemically driven reductive couplings of unactivated ketones and olefins is presented. This method uses a simple undivided cell tolerating exogenous air and moisture, exhibits a broad scope, and can be easily scaled (Figure 1E).
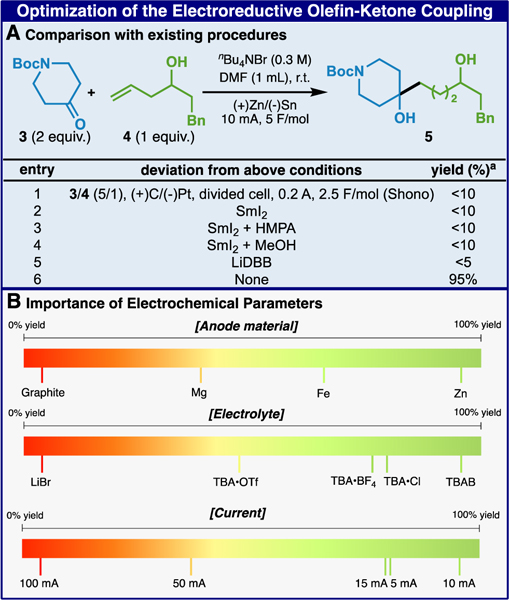
Explorations began by studying Shono’s original conditions18c on a medicinally-relevant model substrate pair: homoallylic alcohol 4 and piperidone 3 (Table 1A). In principle, the use of Grignard chemistry to carry out this assembly would necessitate the use of a protecting group on 4 and perhaps other precautions due to the enolizability of 3, hence, more gentle methodologies were sought. Revisiting the electrochemical method developed by Shono for less ornate substrates,18c only resulted in low yields (Table 1A, entry 1). This method was pursued with some rigor (see SI for a full listing); however, the yield could not be improved beyond 17%. Chemical reductants such as SmI2 and LiDBB were examined next, and while these methods have been shown to have success in similar intramolecular scenarios, they were found to be unsatisfactory for this purpose (entries 2–5, Table 1a). Developing this chemistry following the guiding principles from our own forays19,20 into electrochemistry, specifically deeply reductive electrochemistry,20 allowed us to hone in on the sacrificial anode, electrolyte, current density and concentration needed to facilitate a high yielding olefin-ketone coupling (Table 1A, see the SI for a full listing). As graphically illustrated in Table 1B, these three variables were crucial to the success of this transformation which, after optimization, let to 95% isolated yield of adduct 5 (Table 1A, entry 6). The use of an inexpensive sacrificial anode (Zn) was ideal and, in contrast to prior work, a lower current ensured broad functional group tolerance (10 mA vs. 200 mA). Notably, unlike prior precedent, only 2 equivalents of the ketone are required, inexpensive electrodes are employed, and an operationally simple undivided cell is used. No precautions are taken to exclude air or moisture and in fact the reaction can be run open to the air (cap removed). Finally, the linear versus branched selectivity is remarkable (>15:1 in most of the cases).
TABLE 1.
Optimization of the reductive ketone olefin coupling. Comparison to known chemical methods (A) and a graphical optimization overview of the newly developed electrochemical protocol (B).
|
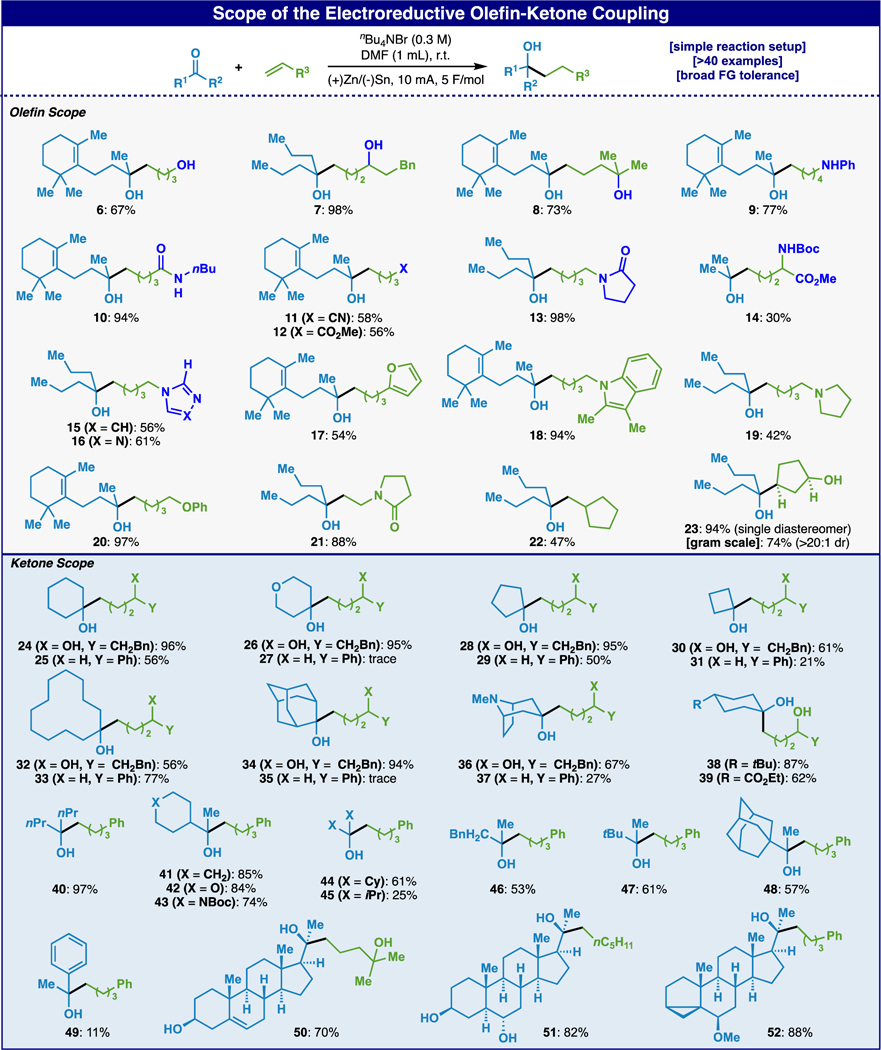
With these results in hand, the scope of the ketone-olefin coupling was investigated (Table 2). Several functionalities on the olefin were tolerated; free alcohols (1°, 2°, and 3°; 6 to 8), aniline (9), amides (10, 13, 21), nitrile (11), ester (12), protected amino acid (14), and heterocycles (15–19) (moderate to high yields). Most of these functional groups would be challenging to employ using canonical 2e- tactics such as Grignard. The reaction tolerated mono-substituted olefins but performed less successfully with poly-substituted olefins with compound 22 and 23 being the only ones affording good yields. A plausible reason for this lack of reactivity with more substituted olefins could be due to a slower rate of addition (for steric reasons) compared to the lifetime of the ketyl radical.21 In the case of cyclopentene-3-ol, an interesting finding was that the reaction took place in high yield with perfect syn diastereoselectivity. The analogous TBS-protected olefin did not react, nor did cyclopentene itself. The directing effect of homo-allylic alcohols in this chemistry is notable and perhaps relevant to the mechanism of the reaction (vide infra).
TABLE 2.
Scope of the electroreductive olefin-ketone coupling.
|
In a similar fashion, ketones bearing several different substituents were tolerated (moderate to high yields); ethers (26), protected amines (36–37), esters (39), carbamates (43), alcohols (50–51), cyclopropanes (52). When 4-substituted cyclohexanones were used, single diastereomers were isolated with the selectivity reminiscent of SmI2 promoted reactions (anti, 38–39).22 Even cyclic ketones of varying ring sizes (24–39) worked well, which are often challenging for other methods; reduction products are often observed when sterically hindered ketones react with Grignards. For acyclic ketones, the sterics of the substituents showed a minor impact on the reaction yields (40–48), although only 25% yield of the desired product was isolated when very hindered diisopropylketone (45) was used. Notably, unprotected steroidal substrates 50–52 delivered a single diastereomeric product in high yield (see SI for structure confirmation).
This reductive coupling could also be applied to simplify real-world challenges in medicinal chemistry (Scheme 1A). Thus, the synthesis of a simple vitamin D analog sidechain 55 was reported through a seven-step route wherein only one of those steps formed a C–C bond (Scheme 1A-1).23 In contrast, commercially available oxazolidinone 53 could be allylated and reduced to yield (S)-2 methyl-4-penten-1-ol 54. Coupling of 54 with acetone under the developed electrochemical conditions then smoothly furnished sidechain 55. Of the three steps required to access 55, two forged key C–C bonds. Next, the synthesis of DNA-binding metabolite 58 required a five-step sequence with two protecting groups and air-sensitive SmI2 to forge a key C–C bond (73% ee, Scheme 1A-2).24 Using the electrochemical strategy outlined above, commercially available aldehyde 56 could be converted to the same product in only 3 steps via simple Brown allylation, followed by electrochemical addition of acetone/TBAF work-up and a final oxidative lactonization (72% yield, 93% ee). Finally, the steroidal example6 mentioned in Figure 1 could be addressed in a similar way from the same starting material (Scheme 1A-3). Thus, electrochemical addition of 1 to Teoc-protected amine 59 delivered a single diastereomeric tertiary alcohol that, after CsF-induced deprotection delivered 2 in only 2 steps. Clearly, the success of the above applications benefits from the chemoselective (FG tolerant) nature of the electrochemical ketone-olefin coupling. The reaction can be conducted on 100 gram-scale (>1 mol) using a flow system affording a comparable yield to the batch reaction(Scheme 1B, see SI for details).
SCHEME 1.

(A) Electrochemical ketone-olefin coupling facilitates rapid access to medicinally relevant structures such as a vitamin D sidechain (1), a DNA-binding metabolite (2), and a hedgehog signaling modulator (3). (B) Batch and flow scale-up. aIsolated yield
The mechanism of this useful reaction (Scheme 2) was next interrogated through the observation of certain side-products (Scheme 2A), deuterium labeling (Scheme 2B), kinetics, and voltammetric studies (Scheme 2D & 2E). A notable limitation of this chemistry was that ketones bearing alpha-substituents (such as 60) were not tolerated and elimination of the alpha-substituent was observed (62), suggestive of a ketyl radical intermediate. Using allyl alcohol (64), the bis addition adduct 65 was observed, perhaps pointing to a carbanion intermediate wherein ZnBr2 generated from anodic oxidation could assist in the departure of the primary alcohol and regeneration of another olefin. Deuterium labeling using acetone-d6 led to 80% incorporation at the highlighted position (Scheme 2B) further supporting a carbanion intermediate. When regular acetone was used in the same experiment but with deuterated DMF, no deuterated product was observed. Kinetic studies revealed zero-order dependence on all components except current indicating that reduction is purely electrochemical.
SCHEME 2.

Mechanistic insights from byproducts (A), deuterium labeling (B), proposed reaction mechanism (C), and voltammetry studies (D & E). See SI for details. affording a comparable yield to the batch reaction(Scheme 1B, see SI for details).
Finally, a series of voltammetric studies were performed (Scheme 2D & 2E) to understand how traditionally nucleophilic ketyl radical can serve as competent coupling partners with unactivated olefins, as well as to provide evidence for the overall electrochemical mechanistic sequence as proposed in Scheme 2C. We hypothesized that the change in its electronic property and reactivity can be facilitated by a strong adsorption of the ketyl radical to the Sn electrode. Cyclic voltammetry studies were performed using Sn and glassy carbon (GC) as working electrodes with acetophenone25 as the source of ketyl radical. Pre-peaks on the CV were observed using Sn as the working electrode but not observed using GC as the working electrode. These pre-peaks are distinct characteristics of an electron transfer where the product (ketyl radical) is strongly adsorbed into the working electrode.26 Furthermore, the current response observed in the pre-peak in Sn was found to be dependent on the concentration of ketone (see SI).27 This result also rationalizes the effectiveness of using Sn-cathode over other electrode materials (see SI). Square-wave voltammetry (SWV) studies were performed and the results are summarized in Scheme 2E. The addition of alkene 61 to acetophenone showed an anodic shift in the cathodic peak potential denoting a chemical reaction with the ketyl radical after one-electron reduction. However, even at high frequencies (100 Hz), the expected second reduction peak was not observed. We hypothesized that one crucial role of the sacrificial Zn-anode is to provide Zn2+ as a thermodynamic sink for the second electron reduction. SWV analysis in the presence of catalytic amounts of ZnBr2 showed three distinct reduction peaks where the third peak can be the reduction of the ZnBr2-coordinated ketone (see SI). Taken together, these results suggest an ECEC-type electrochemical mechanism where the ketyl radical formation (E) takes place at the Sn-cathode with strong adsorption characteristic followed by radical addition (C) into the olefin. A second one-electron reduction (second E) of the radical anion to the dianion followed by protonation (second C) and then workup delivers the final product. The enhanced reactivity of homoallylic alcohols may be due to improved binding of the olefin substrate to the cathode surface.
In summary, a chemoselective, scalable method to combine unactivated olefins and ketones has been developed that subverts the issues encountered using Grignard reagents in conventional retrosynthetic analysis. The scope of this reaction is broad and it is operationally simple to perform. A number of applications demonstrate that the utility extends beyond that of a simple tactical change as when strategically employed, it can dramatically reduce overall step count. Mechanistic studies point to an intuitive electrochemically driven reductive pathway that initiates upon the formation of a ketyl radical, addition to the olefin, and further reduction to a stabilized carbanion prior to workup. This work is thus another example of how strongly reducing chemistry can be uniquely facilitated and enabled in complex settings under electrochemical control when classical chemical reagents fail.
Supplementary Material
ACKNOWLEDGMENT
Financial support for this work was provided by NIH (GM-118176), NSF (CCI Phase 1 grant 1740656 and Phase II grant 2002158), George E. Hewitt Foundation (P.H), and Swedish Research Council (B.K.P). Authors are grateful to Dr. Dee-Hua Huang and Dr. Laura Pasternack (Scripps Research) for assistance with nuclear magnetic resonance (NMR) spectroscopy, to Dr. Jason Chen, Brittany Sanchez and Emily Sturgell (Scripps Automated Synthesis Facility) for assistance with HPLC, HRMS and LCMS, to Dr. James R. Gage, Dr. Yi Hsiao and Dr. Enxuan zhang (Asymchem Inc.) for assistance with scale-up reaction.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website. The Supporting Information contains all experimental procedures, analysis, and compound characterization data.
REFERENCES
- (1).a) For selected reviews, see: Nicolaou KC; Montagnon T. Molecules That Changed the World. Wiley-VCH; (2008); [Google Scholar]; b) Arimoto H; Uemura D. In: Quaternary Stereocenters, Challenges and Solutions for Organic Synthesis; (Eds.: Christoffers J, Baro A), Wiley-VCH, Weinheim, 2005, chap.1, pp 1–24; [Google Scholar]; c) de Vries JG in: Quaternary Stereocenters, Challenges and Solutions for Organic Synthesis; (Eds.: Christoffers J, Baro A), Wiley-V CH, Weinheim, 2005, chap. 2, pp 25–50. [Google Scholar]
- (2).a) For selected reviews, see: Motwani HV; De Rosa M; Odell LR; Hallberg A; Larhed M. Aspartic protease inhibitors containing tertiary alcohol transition-state mimics. Eur. J. Med. Chem 2015, 90, 462–490; [DOI] [PubMed] [Google Scholar]; b) Talele TT; Natural-Products-Inspired Use of the gem-Dimethyl Group in Medicinal Chemistry. J. Med. Chem 2018, 61, 2166–2210; [DOI] [PubMed] [Google Scholar]; c) Cramer J; Sager CP; Ernst B. Hydroxyl Groups in Synthetic and Natural-Product-Derived Therapeutics: A Perspective on a Common Functional Group. J. Med. Chem 2019, 62, 8915–8930. [DOI] [PubMed] [Google Scholar]
- (3).(a) For selected reviews, see: Chen L; Yin X-P; Wang C-H; Zhou J. Catalytic functionalization of tertiary alcohols to fully substituted carbon centres. Org. Biomol. Chem 2014, 12, 6033–6048.; [DOI] [PubMed] [Google Scholar]; (b) Naredla RR; Klumpp DA Contemporary Carbocation Chemistry: Applications in Organic Synthesis. Chem. Rev 2013, 113, 6905–6948. [DOI] [PubMed] [Google Scholar]
- (4).For a selected example, see: Vollhardt K; Schore N. Organic Chemistry: Structure and Function; Freeman WH: New York, 2014. [Google Scholar]
- (5).(a) For selected reviews, see: Rappoport Z, Marek I, Eds. The Chemistry of Organomagnesium Compounds; Wiley-VCH: Weinheim, Germany, 2008; [Google Scholar]; (b) Seyferth D. The Grignard Reagents. Organometallics 2009, 28, 1598–1605; [Google Scholar]; (c) Silverman GS; Rakita PE Handbook of Grignard Reagents; CRC Press: New York, 1996; [Google Scholar]; (d) Knochel P; Dohle W; Gommermann N; Kneisel FF; Kopp F; Korn T; Sapountzis I; Vu VA Highly Functionalized Organomagnesium Reagents Prepared through Halogen-Metal Exchange. Angew. Chem., Int. Ed 2003, 42, 4302–4320; [DOI] [PubMed] [Google Scholar]; (e) Rappoport Z; Marek I, Eds. The Chemistry of Organolithium Compounds, Wiley-VCH, 2004; [Google Scholar]; (f) Luisi R; Capriati V. Eds. Lithium Compounds in Organic Synthesis – From Fundamentals to Applications, Wiley-VCH, 2014. [Google Scholar]
- (6).Xiao W; Epperson M; Farouz F; Stappenbeck F; Thorsett E. Oxysterol compounds. WO2012/024584A2.
- (7).(a) For reviews of nucleophiles other than R-MgX or R-Li, see: Szostak M; Fazakerley NJ; Parmar D; Procter DJ Cross-Coupling Reactions Using Samarium(II) Iodide. Chem. Rev 2014, 114, 5959–6039; [DOI] [PubMed] [Google Scholar]; (b) Nicolaou KC; Ellery SP; Chen JS Samarium Diiodide Mediated Reactions in Total Synthesis. Angew. Chem. Int. Ed 2009, 48, 7140–7165; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Reetz MT; Westermann J; Steinbach R; Wenderoth B; Peter R; Ostarek R; Maus S. Chemoselective Addition of Organotitaniurn Reagents to Carbonyl Compounds. Chem. Ber 1985, 118, 1421–1440; [Google Scholar]; (d) Knochel P; Jones P. Organozinc Reagents; Oxford University Press: Oxford, 1999; [Google Scholar]; (e) Weidmann B; Seebach D. Organometallic Compounds of Titanium and Zirconium as Selective Nucleophilic Reagents in Organic Synthesis. Angew. Chem. Int. Ed 1983, 22, 31–45; [Google Scholar]; (f) Marek I. Titanium and Zirconium in Organic Synthesis. Wiley-VCH, 2002; [Google Scholar]; (g) Liu H-S; Shia K-S; Shang X; Zhu B-Y; Shang X; Zhu B-Y Organocerium Compounds in Synthesis. Tetrahedron 1999, 55, 3803–3830. [Google Scholar]
- (8).(a) For selected reviews, see: Holmes A; Schwartz LA; Krische MJ Intermolecular Metal-Catalyzed Reductive Coupling of Dienes, Allenes, and Enynes with Carbonyl Compounds and Imines. Chem. Rev 2018, 118, 6026–6052; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yus M; Gonzalez-Go ´mez JC.; Foubelo F. Catalytic Enantioselective Allylation of Carbonyl Compounds and Imines. Chem. Rev 2011, 111, 7774–7854; [DOI] [PubMed] [Google Scholar]; (c) Shibashaki M; Kanai M. Asymmetric Synthesis of Tertiary Alcohols and -Tertiary Amines via Cu-Catalyzed C-C Bond Formation to Ketones and Ketimines. Chem. Rev 2008, 108, 2853–2873; [DOI] [PubMed] [Google Scholar]; (d) Denmark SE; Fu J. Catalytic Enantioselective Addition of Allylic Organometallic Reagents to Aldehydes and Ketones. Chem. Rev 2003, 103, 2763–2793; [DOI] [PubMed] [Google Scholar]; (e) Pu L; Yu H-B Catalytic Asymmetric Organozinc Additions to Carbonyl Compounds. Chem. Rev 2001, 101, 757–824; [DOI] [PubMed] [Google Scholar]; (f) Diner C; Szabó KJ Recent Advances in the Preparation and Application of Allylboron Species in Organic Synthesis. J. Am. Chem. Soc 2017, 139, 2–14; [DOI] [PubMed] [Google Scholar]; (g) Leonori D; Aggarwal VK Lithiation−Borylation Methodology and Its Application in Synthesis. Acc. Chem. Res 2014, 47, 3174–3183; [DOI] [PubMed] [Google Scholar]; (h) Riant O; Hannedouche J. Asymmetric catalysis for the construction of quaternary carbon centres: nucleophilic addition on ketones and ketimines. Org. Biomol. Chem 2007, 5, 873–888; [DOI] [PubMed] [Google Scholar]; (i) Liu Y-L; Lin X-T Recent Advances in Catalytic Asymmetric Synthesis of Tertiary Alcohols via Nucleophilic Addition to Ketones. Adv. Syn. Catal 2019, 361, 876–918. [Google Scholar]
- (9).(a) For recent examples, see: Miller JJ; Sigman MS Design and Synthesis of Modular Oxazoline Ligands for the Enantioselective Chromium-Catalyzed Addition of Allyl Bromide to Ketones. J. Am. Chem. Soc 2007, 129, 2752–2753; [DOI] [PubMed] [Google Scholar]; (b) Barnett DS; Moquist PN; Schaus SE Angew.Chem. Int. Ed 2009, 48, 8679–8682; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Shi S-L; Xu L-W; Oisaki K; Kanai M; Shibasaki MJ Am. Chem. Soc 2010, 132, 6638–6639; [DOI] [PubMed] [Google Scholar]; (c) Saxena A; Choi B; Lam HW Enantioselective Copper Catalyzed Reductive Coupling of Alkenylazaarenes with Ketones. J. Am. Chem. Soc 2012, 134, 8428–8431; [DOI] [PubMed] [Google Scholar]; (d) Meng F; Jang H; Jung B; Hoveyda AH Cu-Catalyzed Chemoselective Preparation of 2-(Pinacolato)boron-Substituted Allylcopper Complexes and their In Situ Site-, Diastereo-, and Enantioselective Additions to Aldehydes and Ketones. Angew. Chem. Int. Ed 2013, 52, 5046–5051; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Zhang Y; Li N; Qu B; Ma S; Lee H; Gonnella NC; Gao J; Li W; Tan Z; Reeves JT; Wang J; Lorenz JC; Li G; Reeves DC; Premasiri A; Grinberg N; Haddad N; Lu BZ; Song JJ; Senanayake CH Org. Lett 2013, 15, 1710–1713. [DOI] [PubMed] [Google Scholar]; (f) Yang Y; Perry IB; Lu G; Liu P; Buchwald SL Copper-Catalyzed Asymmetric Addition of Olefin Derived Nucleophiles To Ketones. Science 2016, 353, 144–150; [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Robbins DW; Lee KA; Silverio DL; Volkov A; Torker S; Hoveyda AH Practical and Broadly Applicable Catalytic Enantioselective Additions of Allyl‐B(pin) Compounds to Ketones and α-Ketoesters. Angew. Chem. Int. Ed 2016, 55, 9610–9614; [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Tsai EY; Liu RY; Yang Y; Buchwald SL A Regio- and Enantioselective CuH-Catalyzed Ketone Allylation with Terminal Allenes. J. Am. Chem. Soc 2018, 140, 2007–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Li K; Shao X; Tseng L; Malcolmson SJ 2-Azadienes as Reagents for Preparing Chiral Amines: Synthesis of 1,2-Amino Tertiary Alcohols by Cu-Catalyzed Enantioselective Reductive Couplings with Ketones. J. Am. Chem. Soc 2018, 140, 598–601; [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Li C; Liu RY; Jesikiewicz LT; Yang Y; Liu P; Buchwald SL J. Am. Chem. Soc 2019, 141, 5062–5070; [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Brito GA; Jung W-O, Yoo M; Krische MJ Enantioselective Iridium-Catalyzed Allylation of Acetylenic Ketonesvia 2-Propanol-Mediated ReductiveCoupling of Allyl Acetate: C14-C23 of Pladienolide D. Angew. Chem. Int. Ed 2019, 58, 18803–18807; [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Schewarz JL; Kleinmans R; Paulisch TO; Glorius F. 1,2-Amino Alcohols via Cr/Photoredox Dual-Catalyzed Addition of α‑Amino Carbanion Equivalents to Carbonyls. J. Am. Chem. Soc 2020, 142, 2168–2174. [DOI] [PubMed] [Google Scholar]
- (10).(a) Negishi E; Takahashi T. Organozirconium Compounds in Organic Synthesis. Synthesis 1988, 1–19;; (b) Hirao Y; Katayama Y; Mitsunuma H; Kanai M. Chromium-Catalyzed Linear-Selective Alkylation of Aldehydes with Alkenes. Org. Lett 2020, 22, 8584–8588. [DOI] [PubMed] [Google Scholar]
- (11).(a) Kablaoui NM; Buchwald SL Reductive Cyclization of Enones by a Titanium Catalyst. J. Am. Chem. Soc 1995, 117, 6785–6786; [Google Scholar]; (b) Kablaoui NM; Buchwald SL Development of a Method for the Reductive Cyclization of Enones by a Titanium Catalyst. J. Am. Chem. Soc 1996, 118, 3182–3291; [Google Scholar]; c) Crowe WE; Rachita MJ Titanium-Catalyzed Reductive Cyclization of δ,ε-Unsaturated Ketones and Aldehydes. J. Am. Chem. Soc 1995, 117, 6787–6788. [Google Scholar]
- (12).Yamaguchi E; Mowat J; Luong T; Krische MJ Regio- and Diastereoselective C–C coupling of α-Olefins and Styrenes to 3-Hydroxy-2-oxindoles by Ru-Catalyzed Hydrohydroxyalkylation. Angew. Chem. Int. Ed. Engl 2013, 52, 8428–8431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Park BY; Luong T; Sato H; Krische MJ Osmium(0) Catalyzed C–C Coupling of Ethylene and α-olefins with Diols, Ketols or Hydroxy Esters via Transfer Hydrogenation. J. Org. Chem 2016, 81, 8585–8594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Saladrigas M; Bosch C; Saborit GV; Bonjoch J; Bradshaw B. Radical Cyclization of Alkene-Tethered Ketones Initiated by Hydrogen-Atom Transfer. Angew. Chem. Int. Ed 2018, 57, 182–186. [DOI] [PubMed] [Google Scholar]
- (15).Nguyen KD; Park BY; Luong T; Sato H; Garza VJ; Krische MJ Metal-Catalyzed Reductive Coupling of Olefin-Derived Nucleophiles: Reinventing Carbonyl Addition. Science 2016, 354, aah5133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).a) For selected reviews, see: McCallum T; Wu X; Lin S. Recent Advances in Titanium Radical Redox Catalysis. J. Org. Chem 2019, 84, 14369–14380; [DOI] [PubMed] [Google Scholar]; b) Castro Rodríguez M; Rodríguez Garcia I; Rodríguez Maecker RN; Pozo Morales L; Oltra JE; Rosales Martínez A. Cp2TiCl: An Ideal Reagent for Green Chemistry? Org. Process Res. Dev 2017, 21, 911–923.; [Google Scholar]; c) Morcillo SP; Miguel D; Campaña AG; Álvarez de Cienfuegos L; Justicia J; Cuerva JM Recent applications of Cp2TiCl in natural product synthesis. Org. Chem. Front 2014, 1, 15–33. [Google Scholar]
- (17).(a) Belotti D; Cossy J; Pete J-P; Portella C. Photoreductive Cyclization of δ,ε-Unsaturated Ketones. Tetrahedron Lett. 1985, 26, 4591–4594; [Google Scholar]; (b) Belotti D; Cossy J; Pete J-P; Portella C. Synthesis of Bicyclic Cylopetanols by Photoredutive Cyclization of δ,ε-Unsaturated Ketones. J. Org. Chem 1986, 51, 4196–4200; [Google Scholar]; (c) Cossy J; Belotti D; Pete J-P Photoreductive cyclization of N,N-Dialkyl β-Oxoamides. Tetrahedron Lett. 1987, 28, 4545–4546; [Google Scholar]; (d) Cossy J; Belotti D. Photoreductive Cyclization: Application to the Total Synthesis of (±)-Actinidine. Tetrahedron Lett. 1988, 29, 6113–6114; [Google Scholar]; (e) Cossy J. A Short Access to Iridoid Precursors. Tetrahedron Lett. 1989, 30, 4113–4116; [Google Scholar]; (f) Cossy J; Aclinou P; Bellosta V; Furet N; Baranne-Lafont J; Sparfel D; Souchaud C. Radical Anoin Ring Opening Reactions via Photochemically Induced Electron Transfer. Tetrahedron Lett. 1991, 32, 1315–1316; [Google Scholar]; (g) Cossy J; Leblanc C. First Efficeint Synthesis of Iso-oxy-skytanthine. Tetrahedron Lett. 1991, 32, 3051–3052; [Google Scholar]; (h) Cossy J; Madaci A; Pete J-P Tetrahedron Lett. 1994, 35, 1541–1544; [Google Scholar]; (i) Pandey G; Hajra S; Ghorai MK J. Org. Chem 1997, 62, 5966–5973; [Google Scholar]; (j) Seo H; Jamison TF Catalytic Generation and Use of Ketyl Radical from Unactivated Aliphatic Carbonyl Compounds. Org. Lett 2019, 21, 10159–10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).(a) Shono T; Mitani M. Electroorganic Chemistry. VIII. Intramolecular Cycloaddition of Nonconjugated Olefinic Ketones to Form Cyclic Tertiary Alcohols. J. Am. Chem. Soc 1971, 93, 5284–5286; [Google Scholar]; (b) Shono T; Nishiguchi I; Ohmizu H; Mitani M. Electroorganic Chemistry. 31. Reductive Cyclization of Nonconjugated Olefinic Ketones to Cyclic Tertiary Alcohols. J. Am. Chem. Soc 1978, 100, 545–550; [Google Scholar]; (c) Shono T; Kashimura S; Mori Y; Hayashi T; Soejima T; Yamaguchi Y. Electroreductive Intermolecular Coupling of Ketones with Olefins. J. Org. Chem 1989, 54, 6001–6003; [Google Scholar]; (d) Shono T; Morishima Y; Moriyoshi N; Ishifune M. Electroreductively Promoted Diastereoselective Coupling of Ketones with Allylic Alcohols. Synthesis of Optically Active 1,4-Diols. J. Org. Chem 1994, 59, 273–275. [Google Scholar]
- (19).(a) For reviews, see: Horn EJ; Rosen BR; Baran PS Synthetic Organic Electrochemistry: An Enabling and Innately Sustainable Method. ACS Cent. Sci 2016, 2, 302–308; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yan M; Kawamata Y; Baran PS Synthetic Organic Electrochemical Methods Since 2000: On the Verge of a Renaissance. Chem. Rev 2017, 117, 13230–13319; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kingston C; Palkowitz MD; Takahira Y; Vantourout JC; Peters BK; Kawamata Y; Baran PS A Survival Guide for the “Electro-curious”. Acc. Chem. Res 2020, 53, 72–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Peters BK; Rodriguez KX; Reisberg SH; Beil SB; Hickey DP; Kawamata Y; Collins M; Starr J; Chen L; Udyavara S; Klunder K; Gorey TJ; Ander SL; Neurock M; Minteer SD; Baran PS Scalable and Safe Synthetic Organic Electroreduction Inspired by Li-ion Battery Chemistry. Science 2019, 363, 838–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Leifert D.; Studer A. The Persistent Radical Effect in Organic Synthesis. Angew. Chem. Int. Ed 2020, 59, 74–108. [DOI] [PubMed] [Google Scholar]
- (22).(a) Fukuzawa S; Nakanishi A; Fujinami T; Sakai S. Samarium(II) Di-iodide Induced Reductive Coupling of α,β-Unsaturated Esters with Carbonyl Compounds Leading to a Facile Synthesis of γ-Lactone. J. Chem. Soc. Perkin Trans I. 1988, 1669–1675; [Google Scholar]; (b) Sono M; Shoji T; Tamaki T; Kishi S; Tori M. The Stereochemistry of Electrolysis and Samarium Diiodide-Induced Cyclization Between Carbonyl and Enone System in Inter- and Intramolecular Coupling. Heterocycles. 2007, 72, 517–528. [Google Scholar]
- (23).(a) Plonska-Ocypa K; Grzywacz P; Sicinski RR; Plum LA; DeLuca HF Synthesis and biological evaluation of a des-C,D-analog of 2-methylene-19-nor-1α,25-(OH)2D3. J. Steroid Biochem. Mol. Biol 2007, 103, 298–304; [DOI] [PubMed] [Google Scholar]; (b) Plonska-Ocypa K; Sicinski RR; Plum LA; Grzywacz P; Frelek J; Clagett-Dame M; DeLuca HF 13-Methyl-substituted des-C,D Analogs of (20S)-1a,25-Dihydroxy-2-methylene-19-norvitamin D3(2MD): Synthesis and Biological Evaluation. Bioorg. Med. Chem 2009, 17, 1747–1763. [DOI] [PubMed] [Google Scholar]
- (24).(a) For isolation, see: Maul C; Sattler I; Zerlin M; Hinze C; Koch C; Maier A; Grabley S; Thiericke R. Biomolecular-chemical Screening: A Novel Screening Approach for the Discovery of Biologically Active Secondary Metabolites. III. New DNA-binding Metabolites. J. Antibiot 1999, 52, 1124–1134; For previous syntheses, see: [DOI] [PubMed] [Google Scholar]; (b) Kerrigan NJ; Hutchison PC; Heightman TD; Procter DJ Application of an Ephedrine Chiral Linker in a Solid-phase, ‘Asymmetric Catch-release’ Approach to γ-Butyrolactones. Chem. Commun 2003, 1402–1403; [DOI] [PubMed]; (c) Kerrigan NJ; Hutchison PC; Heightman TD; Procter DJ Development of a Solid-phase, ‘Asymmetric Resin-capture-release’ Process: Application of an Ephedrine Chiral Resin in an Approach to γ-Butyrolactones. Org. Biomol. Chem 2004, 2, 2476–2482. [DOI] [PubMed] [Google Scholar]
- (25).Acetophenone was used despite the low yields when using this ketone because various dialkyl ketones failed to give a clear reduction peak under CV studies.For a list of dialkyl ketones attempted, see SI Fig. S9. For low electrochemical activity of dialkyl ketone on CV scale, see:Bondue CJ; Koper MTM Electrochemical reduction of the carbonyl functional group: The importance of adsorption geometry, molecular structure, and electrode surface structure. J. Am. Chem. Soc 2019, 141, 12071–12078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Wopschall RH; Shain I. Effects of adsorption of electroactive species in stationary electrode polarography. Anal. Chem 1967, 39, 1514–1527. [Google Scholar]
- (27). [The observed adsorption phenomena of the ketyl radical unto Sn electrode was also supported by CV analysis using various scan rates and chronoamperometric studies (for results and discussion, see SI Figs S7 & S8).]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.