

Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) mutations can impact infectivity, viral load, and overall morbidity/mortality during infection. In this analysis, we look at the mutational landscape of the SARS-CoV-2 receptor-binding domain, a structure that is antigenic and allows for viral binding to the host. We develop a bioinformatics platform and analyze 104 193 Global Initiative on Sharing All Influenza Data sequences acquired on 15 October 2020, with a majority of sequences (96%) containing point mutations. We report high frequency mutations with improved binding affinity to ACE2 including S477N, N439K, V367F, and N501Y and address the potential impact of RBD mutations on antibody binding. The high frequency S477N mutation is present in 6.7% of all SARS-CoV-2 sequences, co-occurs with D614G, and is currently present in 14 countries. To address RBD-antibody interactions, we take a subset of human-derived antibodies and define their interacting residues using PDBsum. Our analysis shows that RBD mutations were found in approximately 9% of our dataset, with some mutations improving RBD-ACE2 interactions. We also show that antibody-mediated immunity against SARS-CoV-2 enlists broad coverage of the RBD, with multiple antibodies targeting a variety of RBD regions. These data suggest that it is unlikely for neutralization/RBD antibody binding to be significantly impacted, as a whole, in the presence of RBD point mutations that conserve the RBD structure.
Keywords: COVID-19, SARS-CoV-2, bioinformatics, receptor-binding domain, mutations, antigenic evolution
Statement of Significance
SARS-CoV2 is responsible for the current COVID-19 pandemic. In this work, we developed a MATLAB program to analyze SARS-CoV-2 RBD mutations and conducted a thorough analysis of all SARS-CoV-2 RBD mutations using the GISAID database. We found four high frequency variants with improved binding to ACE2—S477N, N439K, V367F, and N501Y and cross-referenced antibody interaction data with RBD mutations.
INTRODUCTION
Coronaviruses encompass a large family of viruses that can infect both humans and animals. To date, there are seven coronaviruses that are known to infect humans, causing relatively mild to severe respiratory infections. Human coronaviruses 229E, NL63, OC43, and HKU1 typically replicate within the upper respiratory tract, resulting in infections that resemble the common cold [1–5]. Severe acute respiratory syndrome coronavirus (SARS-CoV), Middle East respiratory syndrome coronavirus, and the most recent SARS-CoV-2 can replicate within the lower respiratory tract, causing a pneumonia that can be fatal [1, 6].
SARS-CoV-2 is responsible for the current COVID-19 pandemic that has spread to 218 countries, infecting more than 140 million people and causing more than 3 million deaths (WHO). This virus can be transmitted through respiratory droplets and is most commonly characterized by fever and cough but is also associated with pulmonary embolisms, kidney injury, and gastrointestinal symptoms [7–11]. Importantly, SARS-CoV-2 has a high transmission efficiency and can lead to mortality, especially when infecting the elderly or individuals with underlying medical conditions [12, 13].
Upon entry into the respiratory tract, coronaviruses use a homotrimeric spike glycoprotein (S protein) on the surface of the virion to mediate an interaction with the host receptor angiotensin-converting enzyme 2 (ACE2) and the protease TMPRSS2 [14, 15]. These interactions facilitate viral envelope fusion to the cell membrane via the endosomal pathway [16, 17]. The release of (+) sense RNA into the host cytoplasm allows for viral RNA translation and replication. Viral proteins and genomic RNA are subsequently assembled into virions and released from cells via vesicle transport. This infectious cycle causes host cells to undergo inflammatory pyroptotic cell death resulting in an aggressive inflammatory response and damage to the airways [1].
The S glycoprotein is essential for the initial interaction and internalization of the SARS-CoV-2 virus by the host making it an important structure on the virion [18–21]. The S protein is composed of the S1 and S2 domains with S1 containing the receptor-binding domain (RBD) that directly interacts with the peptidase domain (PD) of the ACE2 protein (Fig. 1a and b). The S2 domain contains a fusion peptide, two heptad repeats, a transmembrane region, and an intracellular region (Fig. 1a). RBD-mediated receptor binding causes dissociation of the S1 domain and allows for the S2 domain to reach a post-fusion state enabling viral membrane fusion [16, 17].
Figure 1.
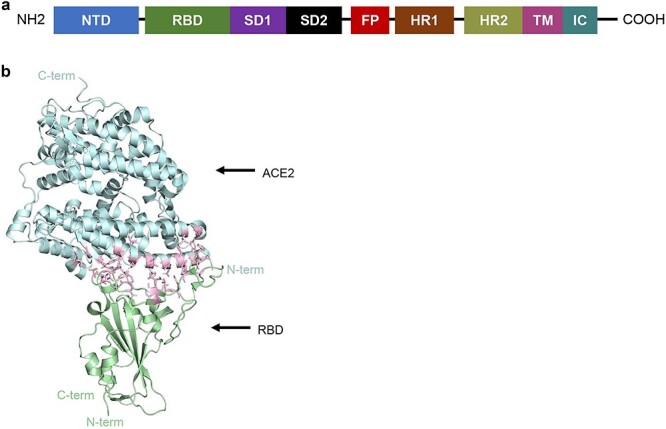
Structure of SARS-CoV-2 RBD bound to ACE2 receptor. (a) Annotated spike monomer. NTD, N-terminal domain; SD1 and SD2, subdomain 1 and subdomain 2; FP, fusion peptide; HR1 and 2, heptad repeat 1 and heptad repeat 2; TM, transmembrane region; IC, intracellular domain. The RBD spans amino acids 330–531. (b) SARS-CoV-2 RBD interaction with ACE2 [22]. ACE2 in cyan. RBD in green. Interacting residues are shown inpink.
Structural studies have resolved in detail the interactions between SARS-CoV-2 RBD and human ACE2, pinpointing essential contact residues (Table 1) [22–26]. Mutations in either the RBD or ACE2 can have an effect on the affinity of this interaction and may therefore impact infectivity, viral load, and overall morbidity/mortality during infection [27, 28]. In this work, we sought to provide insight into RBD mutations using the Global Initiative on Sharing All Influenza Data (GISAID) database [29, 30]. Multiple works have characterized the mutational landscape of the full SARS-CoV-2 genome or the S coding sequence using the GISAID database downloaded at various timepoints [27, 31–34]. SARS-CoV-2 regions with high mutation frequency have been shown to include ORF1a, ORF1b, S, ORF3a, and N coding sequences [32]. Mutational analyses led to the discovery of a D614G mutation found on the S protein that was later shown to be responsible for higher infectivity in a pseudotyped viral infection assay, showed higher Ct values in patients, and was shown to enhance viral load in the upper respiratory tract of patients [33–39].
Table 1.
High frequency RBD mutations that improve ACE2 binding
| ACE2 residue | SARS-CoV-2 RBD residue |
|---|---|
| S19 | A475, G476 |
| Q24 | A475, G476, N487 |
| T27 | F456, Y473, A475, Y489 |
| F28 | Y489 |
| D30 | K417, L455, F456 |
| K31 | L455, F456, Y480, E484, F490, Q493 |
| H34 | Y453, L455, Q493 |
| E35 | Q493 |
| E37 | Y505 |
| D38 | Y449, G496, Q498 |
| Y41 | T500, N501, Q498 |
| Q42 | G446, Y449, Q498 |
| L45 | Q498, T500 |
| L79 | F486 |
| M82 | F486 |
| Y83 | F486, N487, Y489 |
| N330 | T500 |
| K353 | G496, N501, G502, Y505 |
| G354 | Y502, Y505 |
| D355 | T500, G502 |
| R357 | T500 |
| R393 | Y505 |
Noting the significance of these findings, it is essential that we continue to assess SARS-CoV-2 mutations as the GISAID database expands. Here we report an analysis of 104 596 GISAID RBD sequences acquired on 15 October 2020. We use a combination of bioinformatics and published affinity data to determine the mutational landscape and affinities of SARS-CoV-2 RBD mutants to wild-type ACE2 proteins [27]. Finally, it has been reported that the antibody neutralization sensitivity of some RBD mutants including A475V, F490L, and V483A (among others) is reduced, suggesting that SARS-CoV-2 is mutating to evade neutralization/RBD binding [33, 40]. Here we analyze RBD amino acid mutations found at antibody contact sites and assess the breadth of antibody-mediated immunity across the RBD structure [41]. Specifically, our work focuses on mutations found at interacting residues between the RBD and the structurally characterized human antibodies BD23, COVA2-39, CV07-250, B38, CV30, CB6, P2B2F6, CV07-270, BD-368-2, and S309 [41–52].
MATERIALS AND METHODS
Database development
A MATLAB program was developed (https://github.com/LMSE/COVID_19_Mutations) to analyze viral sequences reported on the (GISAID) database [29, 30]. The GISAID database contains thousands of SARS-CoV-2 sequences isolated from patients around the world. All complete and high coverage sequences were downloaded from 1 September 2019 to 15 October 2020, totaling 110 761 sequences. The data were curated by removing duplicates, sequences with gaps, sequences found in animals, and sequences with a similarity score lower than 97%, yielding a 106 941-sequence dataset. The developed code generates all three reading frames for input nucleotide sequences and enables analysis of amino acid and nucleotide mutations to a reference strain. Furthermore, the code analyzes each sequence’s metadata to extract and store the collection date and the isolation location of samples reported in the database. We hope that the bioinformatic workflow and code presented here will be a valuable community tool for analyzing the spread of mutations in SARS-CoV2 sequence.
Analyses overview
Our program was used to assess the similarity of patient sequences to the reference genome of SARS-CoV-2 (NC_045512.2). Two analyses were conducted in this work. First, RBD fragment nucleotide sequences from patient samples were locally aligned to the reference genome RBD fragment, nucleotides 22544–23146. For the extended RBD analysis, nucleotides 22544–23407 were used. The RBD fragment was subsequently translated to its amino acid sequence comprising 201 amino acid residues. These RBD sequences were locally aligned to the reference SARS-CoV-2 RBD amino acid sequence. The second dataset expanded the alignment and analyzed amino acid residues 22544–23407 in order to include the D614 amino acid residue. The methodology for the second dataset was the same as the first. For affinity analysis, all RBD mutants with a frequency higher than 0.0067% or mutations at the RBD-ACE2 binding interface were analyzed (Supplementary Table 1).
Sequence alignment
The program locally aligns the nucleotide sequence of patient samples and their three reading frames to a reference sequence. The algorithm executes local alignment in parallel using the MATLAB bioinformatics toolbox [53]. The alignment algorithm uses a pam250 scoring matrix to obtain the similarity scores of patient samples [54]. Similarity scores are then normalized to percentage to yield more meaningful datasets. For each sample, the reading frame with the maximum similarity score to the standard amino acid sequence is selected as a baseline for identifying mutations.
Antibody interacting residues
The PDBsum server was used to obtain interacting residues on the protein-protein interface between antibodies and the RBD of SARS-CoV-2 [55]. Specifically, antibodies B38 (PDB: 7BZ5), CV30 (PDB: 6XE1), CB6 (PDB: 7C01), BD23 (PDB: 7BYR), COVA2-39 (PDB: 7JMP), CV07-250 (PDB: 6XKQ), P2B-2F6 (PDB: 7BWJ), CV07-270 (PDB: 6XKP), BD-368-2 (PDB: 7CHE), and S309 (PDB: 6WPS). Detailed interactions can be found in Supplementary Table 2.
RESULTS
SARS-CoV-2 nucleotide mutations
A total of 9 275 RBD mutants were found across 104 193 sequences (8.7%) when aligned against the Wuhan reference strain (NC_045512.2). Of the 9 275 RBD mutants, there were 8 871-point mutations, 179 double mutants, and 14 mutants with three to six mutations each for a total of 9 064 mutant SARS-CoV-2 strains. These data are consistent with previous work showing a relatively high number of mutations at the S protein for SARS-CoV-2 [32]. Analysis of mutational frequency showed that approximately 75.2% of all nucleotide mutations were G1430A, 6.8% were C1317A, 0.9% were G1144T, and 42 additional mutants ranged between 0.1 and 0.9% frequency (Fig. 2a; Supplementary Table 3). Whole genome mutational analysis of SARS-CoV-2 previously showed a high number of C->U transitions and suggested that host-driven antiviral editing mechanisms may be driving this preference [56, 57]. In the context of RBD analysis alone, the most common mutation was a G->A transition (7.4% of mutants), followed by a C->A transversion 0.67% of the time, a G->T transversion 0.51% of the time and a C->U transition 0.42% of the time (Fig. 2b; Supplementary Table 3). Although seemingly contrasting, these data are complementary in suggesting a high C->U transition frequency when excluding the high number of G1430A mutations in this dataset, as it represents a potentially selected mutation. Collectively, our analysis of RBD sequences shows a large number of mutations occurring at the RBD with one mutant predominating the SARS-CoV-2 population.
Figure 2.
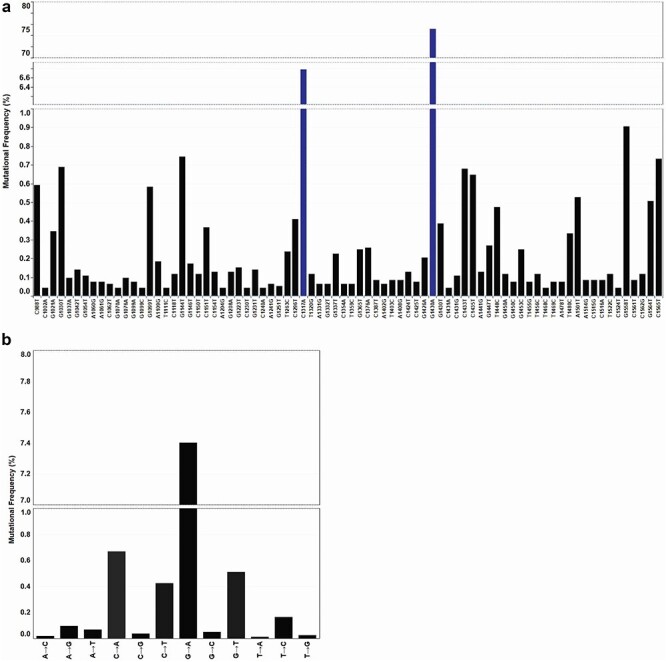
SARS-CoV-2 RBD nucleotide mutations. (a) RBD nucleotide point mutations. Top 70 high frequency mutations across all 243 nucleotide mutations located within the RBD. Frequency reported as a function of 9 064 mutant sequences. Line breaks/blue bars represent large changes in mutation frequency. (b) Nucleotide transversion and transitions within the RBD. Frequency of transitions and transversions as a function of a total 104 193 sequences.
SARS-CoV-2 amino acid mutations
To determine the effects of these nucleotide mutations, we looked at the amino acid output. Of the 9 275 nucleotide mutations, approximately 1.7% of them were synonymous, whereas the remaining 98.3% modified the amino acid sequence of the RBD. The G1430A mutation led to the S477N mutant which represented 76.5% of all 9 121 amino acid mutations. C1317A at 5% and G1558T at 0.9% lead to N439K and A520S mutations, respectively (Fig. 3; Supplementary Table 3). Noting the high frequency of these residue modifications, we wanted to assess their relevance in the context of ACE2 binding affinity. Published data on deep mutational scanning of the RBD had already reported RBD-ACE2 affinity data for all possible RBD mutants. Importantly, at the time, mutations at residues S477N and N439K represented 0.09 and 0.4% of the total GISAID sequences (31 570 total sequences) [27]. Moreover, these data showed that the S477N and the N439K mutant RBDs had a higher affinity for the ACE2 receptor than their WT counterparts, with only S477N showing improved expression in yeast cells. Here we report that these mutations now represent 6.7% (S477N) and 0.6% (N439K) of the total GISAID population (Fig. 3; Supplementary Table 3). Although no significant frequency change has been observed for the N439K mutant, there has been a more than 70-fold increase in the presence of the S477N mutation, suggesting that selection may be playing a role in the propagation of this RBD mutant. Collectively, 13 mutations found in our GISAID analysis coded for an RBD that improved binding affinity to the ACE2 receptor, these included S477N, N439K, V367F, and N510Y among others (Table 2; Supplementary Table 1). The potential negative impact of improved ACE2 binding during SARS-CoV-2 infection led us to analyze whether the high frequency S477N mutant co-occurred with the D614G spike mutation (a high frequency mutation producing more infectious SARS-CoV-2 particles). As expected, all S proteins carrying the S477N mutation also carried the D614G mutation providing evidence for this co-occurrence (Supplementary Table 4). We elaborate on this in the next section.
Figure 3.
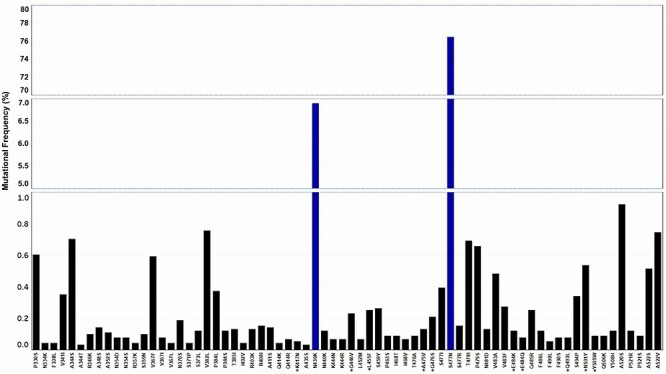
SARS-CoV-2 RBD amino acid mutations. Top 70 high frequency RBD amino acid mutations of a total 205 amino acid mutants. Line breaks/blue bars represent large changes in mutation frequency. Residues with a star represent ACE2 interacting amino acids. Frequency reported as a function of 9 121 amino acid mutations.
Table 2.
| RBD-ACE2 binding residue | Mutation | Count | Frequency of mutation/104 193 sequences (%) | Deep mutational scanning binding affinity21 |
|---|---|---|---|---|
| * | Y453F | 3 | 0.002879 | 0.25 |
| * | N501Y | 49 | 0.047028 | 0.24 |
| * | Y505W | 8 | 0.007678 | 0.13 |
| V367F | 54 | 0.051827 | 0.07 | |
| N440K | 11 | 0.010557 | 0.07 | |
| Y508H | 11 | 0.010557 | 0.07 | |
| S477N | 6 974 | 6.693348 | 0.06 | |
| * | E484K | 11 | 0.010557 | 0.06 |
| * | Q493L | 7 | 0.006718 | 0.05 |
| N439K | 629 | 0.603687 | 0.04 | |
| S359N | 9 | 0.008638 | 0.04 | |
| N354S | 7 | 0.006718 | 0.04 | |
| * | E484Q | 7 | 0.006718 | 0.03 |
Global distribution of RBD mutations
We wanted to analyze the global distribution of individual RBD mutants as a function of country (Fig. 4a; Supplementary Table 3 and Supplementary Fig. 1). Approximately 69% of all SARS-CoV-2 sequences in Australia contained the S477N mutation in the RBD (Fig. 4b). England, Scotland, and Switzerland showed an incidence of 4.7, 0.9, and 0.8% for this mutation, respectively. The highest incidence of N439K mutants was found in England (4.1%) followed by Scotland, Ireland, and Wales (Fig. 4c). There was no incidence of a sequenced N439K/S477N double mutant. Overall, the S477N mutation was present in 14 countries with N439K present in 10 (Supplementary Table 4). To look at the association of D614G with S477N or N439K, we reanalyzed the dataset by increasing the length of our RBD nucleotide sequence to include the D614G codon (see Analyses overview). In both cases, all SARS-CoV-2 sequences containing either the S477N mutation or N439K mutation also contained the D614G mutation. As expected, no triple mutant was found. The S477N/D614G double mutant was found in 14 countries with a high prevalence in Australia (Supplementary Table 4). The N439K/D614G double mutant was found in 10 countries with a high prevalence in England (Supplementary Table 5).
Figure 4.
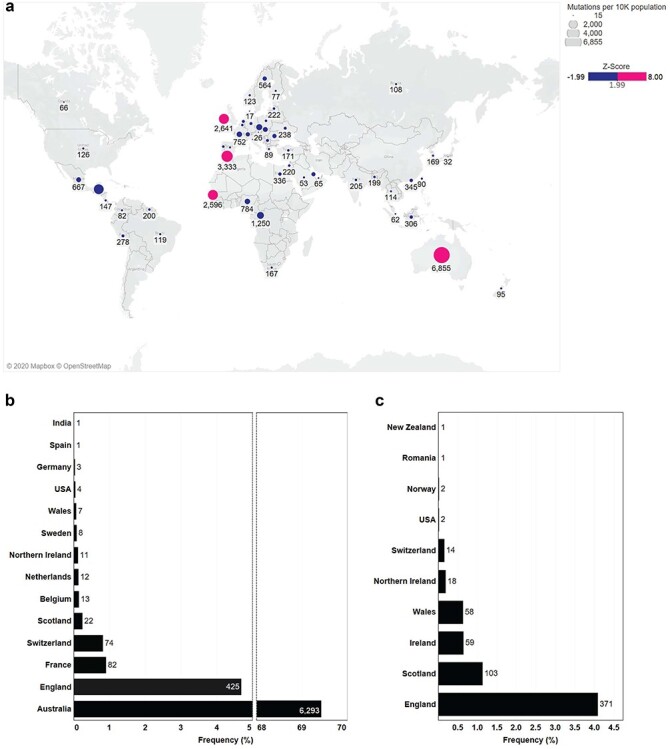
Global distribution of RBD mutations. (a) Frequency of all RBD amino acid mutations by country. Precise Z-score values and frequencies can be found in Supplementary Figure 1, raw data located in Supplementary Table 1. (b) Frequency of S477N mutations by country as a function of a total 6 974 S477N amino acid mutants. (c) Frequency of N439K mutations by country as a function of a total 629 N439K amino acid mutants.
Mutations in antibody interacting residues found on the RBD
Structural data of human antibodies interacting with the RBD of SARS-CoV-2 were coupled with our mutational analysis. RBD mutations were found at 75% of all antibody interaction sites (Supplementary Table 2). High frequency RBD mutations included sites S477 (6.7%), V483 (0.069%), A344 (0.065%), T478 (0.065%), and N501 (0.050%) (Fig. 5). S477 interacts with antibodies CV07-250, CV30, CB6, and BD-368-2; V483 interacts with COVA2-39 and P2B-2F6; A344 interacts with S309; T478 interacts with CV07-250; and N501 interacts with CV07-250, B38, and BD-368-2 (Fig. 5). A number of high frequency mutants such as N439 and A520 did not show any direct interactions to antibodies in our analysis but may impact binding via minor structural changes [33, 40] (Supplementary Table 2). Furthermore, no single point mutation on the RBD was found to have a direct interaction with every antibody in our analysis.
Figure 5.
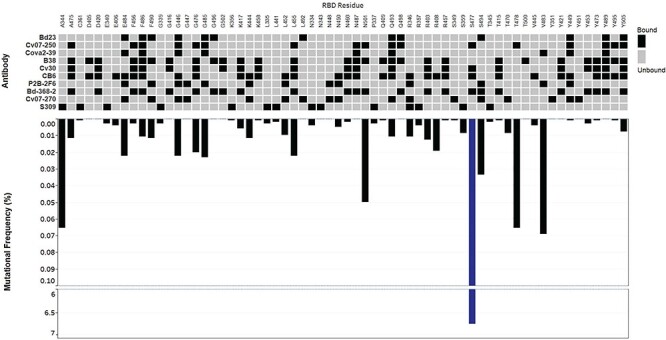
RBD mutations at antibody contact sites. Inverted bar graphs show frequency of RBD amino acid mutations as a function of 104 193 sequences. Line breaks represent large changes in mutational frequency. Heat map presents antibody interactions with amino acid contact residues of theRBD.
DISCUSSION
Analyzing the GISAID database for SARS-CoV-2 mutations is essential as the pandemic continues. In this analysis, we provide a snapshot of all publicly available GISAID SARS-CoV-2 RBD sequences with a focus on emerging mutations that may have a negative health impact to the host. Coupling our data with previous mutational scanning work has led to multiple RBD mutants with improved affinities for their ACE2 binding target [27] (Supplementary Table 1). Some of the highest frequency mutations with improved binding to the ACE2 receptor included S477N, N439K, V367F, and N501Y. The S477N mutation represented the highest frequency of all with N501Y displaying the highest binding affinity [27] (Supplementary Table 1). For future analysis, it may be important to look at the potential frequency changes that occur at RBD mutations which improve ACE2 binding affinity (Table 2). The GISAID database contained no mutations at residues N487, T500, Y489, G502, and G496. This was expected as mutations at any of these key sites reduce RBD affinity for the ACE2 receptor [27]. These data strongly suggest that selection is playing a role in improving SARS-CoV-2 infectivity, with some key exceptions.
There were no SARS-CoV-2 mutations found at the Q498 residue of the RBD. This is perplexing as the Q498H, Q498Y, and Q498F all improve RBD expression and ACE2 binding; moreover, the Q498H mutation boasts the highest affinity for ACE2 of any RBD mutant [27]. Furthermore, there were a number of mutations that could significantly improve RBD-ACE2 binding but were either completely absent in our data or of extremely low frequency, including N501F, Y453F, T385R, Q493M, and Q414A, among others [27] (Supplementary Table 3). These contradictions may be explained by the transmission-mortality trade-off theory [58]. This theory suggests that fitness is not defined by a high mortality rate due to symptoms, but rather a high transmission rate or R0. It is possible that mutations at Q498 would negatively impact viral fitness by increasing mortality and reducing SARS-CoV-2 transmission. If correct, this theory suggests that the mortality rate of SARS-CoV-2 should decrease over time while transmission rate should improve. This may be the case with the recently increased frequency of a new spike deletion variant ΔH69/ΔV70 (unpublished work). We would like to note here that following the completion of this work a number of analyses have been conducted leading to the discovery of new SARS-CoV2 variants.
Finally, we expected that in some cases RBD mutations may impact the affinity of antibodies to the RBD [33, 40]. To that extent, we combined our RBD mutation analysis with previously published work on RBD-antibody binding to address the potential for immune deficiencies [41]. Specifically, we analyzed 10 human-derived antibodies and assessed the frequency of RBD mutations at antibody contact sites. In our analysis, the pool of human-derived antibodies showed that adaptive immunity against SARS-CoV-2 enlists broad coverage of the RBD. Moreover, no single RBD point mutation was found to have a direct interaction with every antibody in our analysis. This suggests that immunization with wild type and potentially any RBD point mutant that conserves structure will elicit the development of RBD antibodies that can bind the RBD/neutralize SARS-CoV-2, providing prophylaxis in immunocompetent individuals and significantly reducing morbidity or mortality should re-infection occur [33, 40]. It should be noted that antibody neutralization may vary based on certain mutations as shown with the E484K mutation, which in our dataset was found in 11 SARS-CoV-2 sequences [59]. The extent to which some of these mutations modify transmission efficiency and infectivity remains to be seen, but in the context of natural infection or vaccine immunization, both wild type and point mutant RBD antigens represent excellent targets for the development of neutralizing antibodies/antibodies that bind to theRBD.
CONCLUSION
In conclusion, we found a number of high frequency SARS-CoV-2 RBD mutations with improved binding affinities to the ACE2 receptor including S477N, N439K, V367F, and N501Y. We show that S477N and N439K mutations represent high frequency mutations in Australia and England, respectively. And, we analyzed RBD-antibody contact sites, showing that human antibodies produced against the RBD interact with different RBD regions. Collectively, our data suggest that antibodies produced post SARS-CoV-2 infection or vaccination should provide protection against RBD point mutants via neutralization/RBD binding.
SUPPLEMENTARY DATA
Supplementary data are available at ABT Online.
DATA AVAILABILITY
The authors confirm that the data supporting the findings of this study are available within the article and/or its supplementary materials.
CONFLICT OF INTEREST STATEMENT
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
ACKNOWLEDGMENTS
Map data copyrighted OpenStreetMap contributors and available from https://www.openstreetmap.org.
Contributor Information
Marios Mejdani, Department of Chemical Engineering and Applied Chemistry, University of Toronto, Toronto, ON M5S 3E5, Canada.
Kiandokht Haddadi, Department of Chemical Engineering and Applied Chemistry, University of Toronto, Toronto, ON M5S 3E5, Canada.
Chester Pham, Department of Chemical Engineering and Applied Chemistry, University of Toronto, Toronto, ON M5S 3E5, Canada.
Radhakrishnan Mahadevan, Department of Chemical Engineering and Applied Chemistry, University of Toronto, Toronto, ON M5S 3E5, Canada; Institute of Biomedical Engineering, University of Toronto, Toronto, ON M5S 3G9, Canada.
FUNDING
This work was supported by the Precision Medicine Initiative (PRiME) at the University of Toronto internal fellowship number PRMF2020-006. We gratefully acknowledge the authors from the originating laboratories responsible for obtaining the specimens and the submitting laboratories where genetic sequence data were generated and shared via the GISAID Initiative, on which this research is based.
References
- 1.Tay, MZ, Poh, CM, Renia, L et al. The trinity of COVID-19: immunity, inflammation and intervention. Nat Rev Immunol 2020; 20: 363–74. 10.1038/s41577-020-0311-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang, G, Deering, C, Macke, M et al. Human coronavirus 229E infects polarized airway epithelia from the apical surface. J Virol 2000; 74: 9234–9. 10.1128/jvi.74.19.9234-9239.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abdul-Rasool, S, Fielding, BC. Understanding Human Coronavirus HCoV-NL63. Open Virol J 2010; 4: 76–84. 10.2174/1874357901004010076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van der Hoek, L, Pyrc, K, Jebbink, MF et al. Identification of a new human coronavirus. Nat Med 2004; 10: 368–73. 10.1038/nm1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vabret, A, Dina, J, Gouarin, S et al. Detection of the new human coronavirus HKU1: a report of 6 cases. Clin Infect Dis 2006; 42: 634–9. 10.1086/500136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Standl, F, Jockel, KH, Brune, B et al. Comparing SARS-CoV-2 with SARS-CoV and influenza pandemics. Lancet Infect Dis 2021; 21: e77. 10.1016/S1473-3099(20)30648-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tan, ND, Qiu, Y, Xing, XB et al. Associations between angiotensin-converting enzyme inhibitors and angiotensin II receptor blocker use, gastrointestinal symptoms, and mortality among patients with COVID-19. Gastroenterology 2020; 159: 1170–1172 e1171. 10.1053/j.gastro.2020.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Léonard-Lorant, I, Delabranche, X, Séverac, F et al. Acute pulmonary embolism in patients with COVID-19 at CT angiography and relationship to d-dimer levels. Radiology 2020; 296: E189–91. 10.1148/radiol.2020201561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ng, JJ, Luo, Y, Phua, K et al. Acute kidney injury in hospitalized patients with coronavirus disease 2019 (COVID-19): A meta-analysis. J Infect 2020; 81: 647–79. 10.1016/j.jinf.2020.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grant, MC, Geoghegan, L, Arbyn, M et al. The prevalence of symptoms in 24,410 adults infected by the novel coronavirus (SARS-CoV-2; COVID-19): a systematic review and meta-analysis of 148 studies from 9 countries. PLoS One 2020; 15: e0234765. 10.1371/journal.pone.0234765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chowdhury, MA, Hossain, N, Kashem, MA et al. Immune response in COVID-19: a review. J Infect Public Health 2020; 13: 1619–29. 10.1016/j.jiph.2020.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mueller, AL, McNamara, MS, Sinclair, DA. Why does COVID-19 disproportionately affect older people. Aging (Albany NY) 2020; 12: 9959–81. 10.18632/aging.103344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harrison, AG, Lin, T, Wang, P. Mechanisms of SARS-CoV-2 transmission and pathogenesis. Trends Immunol 2020; 41: 1100–1115. 10.1016/j.it.2020.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benton, DJ, Wrobel, AG, Xu, P et al. Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature 2020; 588: 327–30. 10.1038/s41586-020-2772-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Letko, M, Marzi, A, Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat Microbiol 2020; 5: 562–9. 10.1038/s41564-020-0688-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wrapp, D, Wang, N, Corbett, KS et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020; 367: 1260–3. 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fan, X, Cao, D, Kong, L et al. Cryo-EM analysis of the post-fusion structure of the SARS-CoV spike glycoprotein. Nat Commun 2020; 11: 3618. 10.1038/s41467-020-17371-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Antonopoulos, A, Broome, S, Sharov, V et al. Site-specific characterisation of SARS-CoV-2 spike glycoprotein receptor binding domain. Glycobiology 2020; 31: 181–7. 10.1093/glycob/cwaa085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walls, AC, Park, YJ, Tortorici, MA et al. Structure, function, and antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020; 181: 281–292 e286. 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Watanabe, Y, Allen, JD, Wrapp, D et al. Site-specific glycan analysis of the SARS-CoV-2 spike. Science 2020; 369: 330–3. 10.1126/science.abb9983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Turoňová, B, Sikora, M, Schürmann, C et al. In situ structural analysis of SARS-CoV-2 spike reveals flexibility mediated by three hinges. Science 2020; 370: 203–8. 10.1126/science.abd5223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lan, J, Ge, J, Yu, J et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020; 581: 215–20. 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- 23.Othman, H, Bouslama, Z, Brandenburg, JT et al. Interaction of the spike protein RBD from SARS-CoV-2 with ACE2: similarity with SARS-CoV, hot-spot analysis and effect of the receptor polymorphism. Biochem Biophys Res Commun 2020; 527: 702–8. 10.1016/j.bbrc.2020.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shang, J, Ye, G, Shi, K et al. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020; 581: 221–4. 10.1038/s41586-020-2179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang, Q, Zhang, Y, Wu, L et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 2020; 181: 894–904 e899. 10.1016/j.cell.2020.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yan, R, Zhang, Y, Li, Y et al. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020; 367: 1444–8. 10.1126/science.abb2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Starr, TN, Greaney, AJ, Hilton, SK et al. Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell 2020; 182: 1295–1310 e1220. 10.1016/j.cell.2020.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ali, F, Elserafy, M, Alkordi, MH et al. ACE2 coding variants in different populations and their potential impact on SARS-CoV-2 binding affinity. Biochem Biophys Rep 2020; 24: 100798. 10.1016/j.bbrep.2020.100798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shu, Y, McCauley, J. GISAID: Global Initiative on Sharing All Influenza Data - from vision to reality. Euro Surveill 2017; 22: 1–3. 10.2807/1560-7917.ES.2017.22.13.30494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Elbe, S, Buckland-Merrett, G. Data, disease and diplomacy: GISAID's innovative contribution to global health. Glob Chall 2017; 1: 33–46. 10.1002/gch2.1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alouane, T, Laamarti, M, Essabbar, A et al. Genomic diversity and hotspot mutations in 30,983 SARS-CoV-2 genomes: moving toward a universal vaccine for the “Confined Virus”? Pathogens 2020; 9: 829. 10.3390/pathogens9100829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim, JS, Jang, JH, Kim, JM et al. Genome-wide identification and characterization of point mutations in the SARS-CoV-2 Genome. Osong Public Health Res Perspect 2020; 11: 101–11. 10.24171/j.phrp.2020.11.3.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li, Q, Wu, J, Nie, J et al. The impact of mutations in SARS-CoV-2 spike on viral infectivity and antigenicity. Cell 2020; 182: 1284–1294 e1289. 10.1016/j.cell.2020.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Korber, B, Fischer, WM, Gnanakaran, S et al. Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell 2020; 182: 812–827 e819. 10.1016/j.cell.2020.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eaaswarkhanth, M, Al Madhoun, A, Al-Mulla, F. Could the D614G substitution in the SARS-CoV-2 spike (S) protein be associated with higher COVID-19 mortality? Int J Infect Dis 2020; 96: 459–60. 10.1016/j.ijid.2020.05.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Omotuyi, IO, Nash, O, Ajiboye, OB et al. Atomistic simulation reveals structural mechanisms underlying D614G spike glycoprotein-enhanced fitness in SARS-COV-2. J Comput Chem 2020; 41: 2158–61. 10.1002/jcc.26383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Plante, JA, Liu, Y, Liu, J et al. Spike mutation D614G alters SARS-CoV-2 fitness. Nature 2020; 592: 116–21. 10.1038/s41586-020-2895-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yurkovetskiy, L, Wang, X, Pascal, KE et al. Structural and functional analysis of the D614G SARS-CoV-2 spike protein variant. Cell 2020; 183: 739–751 e738. 10.1016/j.cell.2020.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang, L, Jackson, CB, Mou, H et al. SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat Commun 2020; 11: 6013. 10.1038/s41467-020-19808-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weisblum, Y, Schmidt, F, Zhang, F et al. Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. Elife 2020; 9: 1–31. 10.7554/eLife.61312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yuan, M, Liu, H, Wu, NC et al. Recognition of the SARS-CoV-2 receptor binding domain by neutralizing antibodies. Biochem Biophys Res Commun 2020; 538: 192–203. 10.1016/j.bbrc.2020.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cao, Y, Su, B, Guo, X et al. Potent neutralizing antibodies against SARS-CoV-2 identified by high-throughput single-cell sequencing of convalescent patients’ B cells. Cell 2020; 182: 73–84 e16. 10.1016/j.cell.2020.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu, Y, Wang, F, Shen, C et al. A noncompeting pair of human neutralizing antibodies block COVID-19 virus binding to its receptor ACE2. Science 2020; 368: 1274–8. 10.1126/science.abc2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yuan, M, Liu, H, Wu, NC et al. Structural basis of a shared antibody response to SARS-CoV-2. Science 2020; 369: 1119–23. 10.1126/science.abd2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hurlburt, NK, Seydoux, E, Wan, YH et al. Structural basis for potent neutralization of SARS-CoV-2 and role of antibody affinity maturation. Nat Commun 2020; 11: 5413. 10.1038/s41467-020-19231-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shi, R, Shan, C, Duan, X et al. A human neutralizing antibody targets the receptor-binding site of SARS-CoV-2. Nature 2020; 584: 120–4. 10.1038/s41586-020-2381-y. [DOI] [PubMed] [Google Scholar]
- 47.Ju, B, Zhang, Q, Ge, J et al. Human neutralizing antibodies elicited by SARS-CoV-2 infection. Nature 2020; 584: 115–9. 10.1038/s41586-020-2380-z. [DOI] [PubMed] [Google Scholar]
- 48.Pinto, D, Park, YJ, Beltramello, M et al. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature 2020; 583: 290–5. 10.1038/s41586-020-2349-y. [DOI] [PubMed] [Google Scholar]
- 49.Wu, NC et al. An alternative binding mode of IGHV3-53 antibodies to the SARS-CoV-2 receptor binding domain. bioRxiv 2020; 33: 1–8. 10.1101/2020.07.26.222232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brouwer, PJM, Caniels, TG, van der Straten, K et al. Potent neutralizing antibodies from COVID-19 patients define multiple targets of vulnerability. Science 2020; 369: 643–50. 10.1126/science.abc5902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kreye, J, Reincke, SM, Kornau, HC et al. A therapeutic non-self-reactive SARS-CoV-2 antibody protects from lung pathology in a COVID-19 hamster model. Cell 2020; 183: 1058–1069.e1069. 10.1016/j.cell.2020.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.du, S, Cao, Y, Zhu, Q et al. Structurally resolved SARS-CoV-2 antibody shows high efficacy in severely infected hamsters and provides a potent cocktail pairing strategy. Cell 2020; 183: 1013–1023 e1013. 10.1016/j.cell.2020.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cai, JJ, Smith, DK, Xia, X et al. MBEToolbox: a MATLAB toolbox for sequence data analysis in molecular biology and evolution. BMC Bioinformatics 2005; 6: 64. 10.1186/1471-2105-6-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pearson, WR. Selecting the right similarity-scoring matrix. Curr Protoc Bioinformatics 2013; 43: 3.5.1–9. 10.1002/0471250953.bi0305s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Laskowski, RA. PDBsum: summaries and analyses of PDB structures. Nucleic Acids Res 2001; 29: 221–2. 10.1093/nar/29.1.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Simmonds, P. Rampant C-->U hypermutation in the genomes of SARS-CoV-2 and other coronaviruses: causes and consequences for their short- and long-term evolutionary trajectories. mSphere 2020; 5: e00408–20. 10.1128/mSphere.00408-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Matyasek, R, Kovarik, A. Mutation patterns of human SARS-CoV-2 and Bat RaTG13 coronavirus genomes are strongly biased towards C>U transitions, indicating rapid evolution in their hosts. Genes (Basel) 2020; 11: 1–13. 10.3390/genes11070761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bull, JJ, Lauring, AS. Theory and empiricism in virulence evolution. PLoS Pathog 2014; 10: e1004387. 10.1371/journal.ppat.1004387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Greaney, AJ, Starr, TN, Gilchuk, P et al. Complete mapping of mutations to the SARS-CoV-2 spike receptor-binding domain that escape antibody recognition. Cell Host Microbe 2021; 29: 44–57 e49. 10.1016/j.chom.2020.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and/or its supplementary materials.