

Abstract

SARS-CoV-2 entry into host cells relies on the spike (S) protein binding to the human ACE2 receptor. In this study, we investigated the structural dynamics of the viral S protein at the fusion peptide (FP) domain and small molecule binding for therapeutics development. Following comparative modeling analysis and docking studies of our previously identified fusion inhibitor chlorcyclizine, we performed a pharmacophore-based virtual screen and identified two novel chemotypes of entry inhibitors targeting the FP. The compounds were evaluated in the pseudoparticle viral entry assay and SARS-CoV-2 cytopathic effect assay and showed single-digital micromole inhibition against SARS-CoV-2 as well as SARS-CoV-1 and MERS. The characterization of the FP binding site of SARS-CoV-2 S protein provides a promising target for the structure-based development of small molecule entry inhibitors as drug candidates for the treatment of COVID-19.
Keywords: SARS-CoV-2, high-throughput screening, virtual screening, COVID-19, spike protein, fusion peptide
Novel coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has become a serious threat to global public health.1 In response to the pandemic, significant efforts have been made to investigate the viral infection for vaccine development, and millions of drug and chemical compounds have been screened to search for effective drug candidates.2−4 To date, while many candidates are currently undergoing clinical trials, no effective therapeutics are available for the treatment of COVID-19. The clinical utility of remdesivir, the only drug approved by FDA for COVID-19, is still limited and does not reduce mortality in severe patients.5 The emergence and rapid spread of SARS-CoV-2 variants currently pose greater concerns and urgent needs to develop effective antivirals.6,7
SARS-CoV-2 entry into host cells occurs in several steps, beginning with spike (S) protein binding to the human ACE2 receptor.8 Upon the cleavage of S protein by host proteases, the membrane fusion and reconstruction processes are initiated at the fusion peptide (FP) and heptad repeat (HR) domain, followed by internalization and the pH-dependent endocytic pathway. Therefore, the S protein is a vital target for blocking viral entry into host cells.9 A number of HR mimetics of peptides have been developed as entry inhibitors of coronaviruses including SARS-CoV-2 which mainly target the HR domain to inhibit the membrane fusion process.10,11 Several small molecules have also been identified as effective entry inhibitors of SARS-CoV-2 through drug repurposing screening and virtual screening.12,13 However, the molecular mechanism of these small molecule entry inhibitors against coronavirus remains unclear.
We have previously reported chlorcyclizine (CCZ) as a potent hepatitis C virus (HCV) entry inhibitor.14,15 Through site-directed mutagenesis studies and computational modeling, we have elucidated the mechanism of action of CCZ on inhibition of HCV entry directly targeting the HCV E1 fusion loop and blocking the viral fusion process.16 CCZ also showed inhibitory activities against SARS-CoV-2, SARS-CoV-1, and MERS in the pseudoparticle (PP) entry assay, indicating that CCZ is a broad antiviral entry inhibitor.12,17
To investigate whether CCZ utilizes the same mechanism of inhibition against coronavirus, we performed a comparative structural analysis of the FP in SARS-CoV-2 S protein. The FP fragment (815–852) is located downstream of the S2′ cleavage site, characterized with a short helix at the beginning and a dynamic loop region. The loop is found to be highly flexible and typically disordered in the prefusion structure of the trimer complex.18 A packed conformation of FP is also observed in the soluble S structure in the RBD-down conformation.19 Analysis of these available structures revealed a well-defined hydrophobic pocket at the FP helix region and the adjacent HR1 domain (Figure 1). The hydrophobic pocket is partially formed by the extended FP loop in a closed conformation in the packed structure, while it is rather open and extended to a hydrophobic groove along the adjacent HR1 helix in the disordered structure.
Figure 1.

Comparison of the structure of SARS-CoV-2 S protein in the RBD-down conformation (PDB 6XR8) and RBD-up conformation (PDB 6VSB). The FP is colored in magenta, and the binding pocket is rendered in hydrophobic surface. The chemical structure of CCZ and the predicted binding model in the FP pocket are shown on the right panel.
Similar to CCZ binding to the FP of HCV E1, the FP hydrophobic pocket of SARS-COV-2 S protein provided a potential binding site for small molecule intervention. To examine the dynamics and conformational changes of FP upon inhibitor binding, we first modeled the flexible loop in the trimer complex of the S protein structure. Different conformations of the loop in the closed, open, and intermediate states can be seen (Figure S1). Although these ensembles of loop conformations were modeled in an aqueous system which may behave differently in the membrane surface, the intrinsic dynamics of the FP implied its vital functional role in the membrane fusion process. Molecular dynamics (MD) simulations showed that, while the unstructured FP loop is highly fluctuated, the hydrophobic pocket at the FP hinge region remained rather stable in the prefusion structure of S protein.
We performed ensemble docking of CCZ to the modeled FP conformations to identify the plausible binding interaction with SARS-CoV-2. As shown in Figure 1, the small molecule fits well in the hydrophobic pocket with the FP loop preferably in a closed conformation. Remarkably, an H-bond is formed between the piperazine N atom with Asp867 in the pocket, the same as what was observed in the HCV E1 binding model.16 Another interesting binding interaction can be seen with Phe833 from the FP loop, which points into the hydrophobic pocket and forms a π–π stacking interaction with the phenyl group.
We further explored the CCZ binding at the FP in SARS-CoV-1 and MERS. While the FP loop in these two viruses is also highly flexible and typically disordered in the structural complex, a similar hydrophobic pocket is found at the FP region (Figure 2). Docking of CCZ revealed the same binding mode as observed in SARS-CoV-2, that the molecule is accommodated well in the FP hydrophobic pocket. Moreover, the key interacting residues Asp867 and Phe833 in SARS-CoV-2 are highly conserved in SARS-CoV-1 and MERS.
Figure 2.

Comparison of the FP binding pocket of the spike protein of SARS-CoV-2 (PDB 6XR8), SARS-CoV-1 (PDB 5WRG), and MERS (PDB 6NB3). The spike protein is rendered in ribbons with the FP colored in magenta and the HR1 domain in blue. The FP in the structure of SARS-CoV-1 is disordered. The entry inhibitor CCZ bound in the FP pocket and key interacting residues Asp867/Asp849/Glu944 are shown.
Inspired by the encouraging results, we performed virtual screening to search for novel inhibitors targeting the FP binding pocket. A pharmacophore model was first generated based on the predicted binding interaction of CCZ at the FP binding pocket of SARS-CoV-2 (Figure S2). The NCATS in-house library consisting of nearly 200,000 drug-like compounds was screened, and a total of 10,000 hits were extracted from the pharmacophore-based and 3D-shape-based searching, followed by a stepwise docking of the hits to the FP binding site of SARS-CoV-2. The top-scored 2000 compounds from docking were selected and clustered. All compounds from each cluster and singletons were visually inspected. Finally, 120 compounds were prioritized for further evaluation.
The selected compounds were tested in the SARS-CoV-2 PP entry assay and the live virus cytopathic effect (CPE) assay.12,20 The PP entry assay was developed using pseudotyped murine leukemia virus (MLV) harboring SARS-CoV-2 S protein and was performed in a HEK293-ACE2 cell line. A counter assay with vesicular stomatis virus G protein (VSV-G) PP was used to eliminate false positives that inhibit luciferase reporter expression and compounds that do not act on the spike-mediated entry. A compound cytotoxicity counter assay was carried out in parallel with both PP and CPE assays to detect nonspecific compound cytotoxicity in the respective cells. Of 120 compounds tested, a few hits exhibited specific activity against SARS-CoV-2 PPs with at least 5-fold selectivity over that against the VSV-G PPs, while also showing activity in the CPE assay. These active hits varied in potency, efficacy, and cytotoxicity, and two chemotypes of compounds had robust activities in both CPE and PP assays (Figure 3).
Figure 3.

Identified inhibitors from VS and dose–response activities in the SARS-CoV-2 PP entry assay and the CPE assay.
Clobenztropine (C1) belongs to the same class of H1 antihistamines as CCZ. It showed activities in the SARS-CoV-2 PP assay with an IC50 of 12.6 μM and in the CPE assay with an IC50 of 8.9 μM. The quinazoline-based compound D3-βArr C2 is a positive allosteric modulator for thyrotropin receptor (TSHR),21 which is a structurally appealing chemotype with no previously reported antiviral activity. It showed an IC50 of 15.8 μM in the SARS-CoV-2 PP assay and 10.0 μM in the CPE assay, respectively. The two inhibitors are structurally distinct but share a similar 3D triangular molecule shape as CCZ. Figure 4 shows the predicted binding models of these inhibitors bound at the FP site. Both compounds fit into the pocket by forming extensive hydrophobic and aromatic stacking interactions. Similar to CCZ binding, an H-bond is formed between Asp867 and the N atom of the polar headgroup. Another key interaction is also seen with Phe833, which forms a π–π stacking interaction with the chlorophenyl and quinazoline groups of C1 and C2. While the S enantiomer of C2 was initially evaluated, we also tested the R enantiomer of C2, which showed similar activities in the CPE and PP assays. The results were consistent with the predicted binding interactions of these two enantiomers at the FP binding pocket (Figure S3).
Figure 4.

Predicted binding model of C1 (clobenztropine) and C2 (D3-βArr) in the FP binding pocket.
To further investigate the two potential fusion inhibitors, we evaluated a number of derivatives using the same PP and CPE assays. The analogue compounds were retrieved from the structural clusters in the pharmacophore-based virtual screening. Three clobenztropine-related compounds—benztropine, difluorobenztropine, and clemastine—were tested (Figure 5). All of these small molecules are H1 antihistamine agents, which also showed robust anti-HCV activity in our previous drug repurposing screen.14 Compared to clobenztropine, benztropine showed decreased activities in both CPE and PP assays due to lack of halogen bonding interaction between chlorine and Asp830, while difluorobenztropine had an increase in potencies and efficacies in both assays owning to the dual fluorine binding. Clemastine adopted the same binding mode as the analogues. The compound has two stereocenters in the R configuration, which showed the most favorable binding conformation in the FP pocket (Figure S4).
Figure 5.

Activities of clobenztropine analogues in the CPE and PP assays. na = not active.
A diversity of C2 analogue compounds was tested in the CPE and PP assays (Table 1). The predicted binding interactions of these inhibitors at the FP binding pocket also provided a structural basis and SAR of this interesting chemotype against SARS-CoV-2 (Figure S5). Most analogues showed comparable activities to C2 with IC50 ranging from 10 to 20 μM in both assays and were selective to SARS-CoV-2. Modifications of the quinazoline core displayed better activities, likely because of better binding interactions with the flexible FP loop, but showed cytotoxicity at high concentration (C3 and C7). Substitutions on the phenyl group (C5 and C6) are tolerated, but the para-position might be preferable because it points out to the solvent-exposed region. Consistent with the predicted binding models of these analogues at the FP pocket, replacement of the piperazine headgroup (C9 and C10) resulted in a significant decrease of activities in both assays.
Table 1. Activities of C2 Derivatives in the PP and CPE Assays.
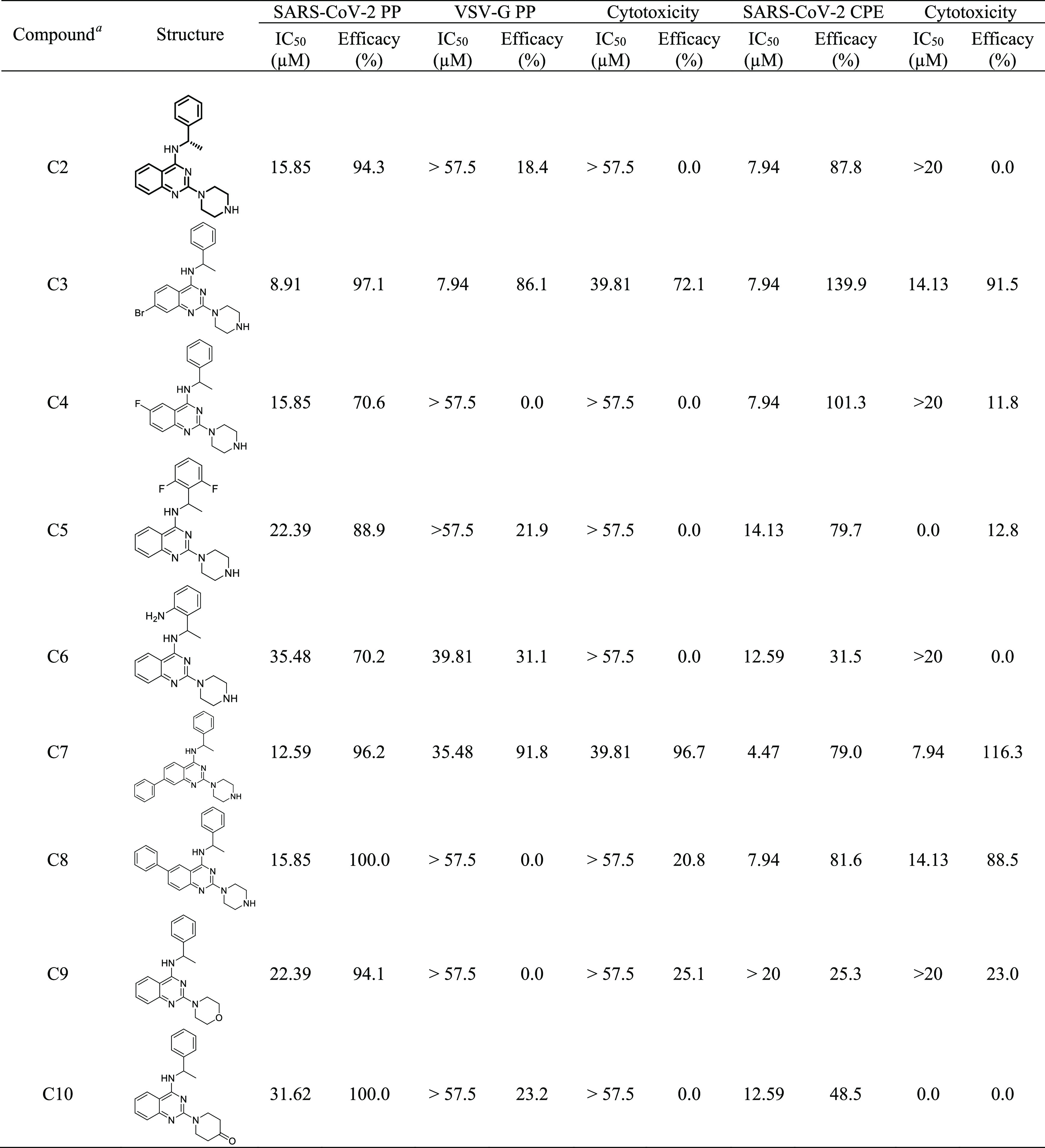
All compounds tested except C2 (S) are racemic mixtures.
We further evaluated the antiviral activity of these compounds in VSV-based PP entry assays including SARS-CoV-1, SARS-CoV-2, and MERS. Different from the previously used MLV-based PP assays in a HEK293-ACE2 cell line, the VSV PPs harboring the spike protein were generated and tested in human hepatoma cells (Huh7), which are susceptible to SARS-CoV-2 infection.22 As shown in Figure 6, the two series of inhibitors and analogues were active against three viruses with IC50 in single-digital micromole. While the higher efficacy and potency against SARS-CoV-2 in the VSV-based PP assay were likely due to differences in pseudoparticle and host cell lines, the results were generally consistent in both MLV- and VSV-based PP assays, and activities of these inhibitors against the three viruses in the VSV-based PP assay were comparable to those determined in the MLV-based PP assays. C2 showed the best activity against SARS-CoV-1 with an IC50 of 0.96 μM, while C4 was the most potent analogue against SARS-CoV-2 with an IC50 of 1.70 μM. Notably, most of these inhibitors were slightly more potent and selective to SARS-CoV-1, which may be associated with a more flexible FP loop in SARS-COV-1. The activities and structure–activity relationship of C2 analogues against SARS-CoV-1 and MERS were also consistent, as observed in the MLV-based assay against SARS-CoV-2 (Figure S6).
Figure 6.

Activities of (A) clobenztropine and analogues and (B) D3-βArr and analogues in the PP entry assay against SARS2, SARS1, and MERS.
It is worth noting that all inhibitors of the two series of compounds showed a similar molecule shape and binding interactions in the FP binding pocket, reiterating our hypothesis that these CCZ-mimicking small molecules share the same mechanism of action targeting the conserved FP binding pocket. Xiao et al. reported five drug compounds from drug-repurposing screening which inhibited SARS-CoV-2 S protein-mediated cell fusion.23 Remarkably, these fusion inhibitors such as fendiline and vortioxetine are also structurally similar to CCZ with a triangle molecule shape. Docking studies showed that these two drugs fit well in the FP binding pocket (Figure S7). The unique FP hydrophobic pocket together with the HR1 domain appears to be the key driving force for such CCZ-like small molecule binding. It is postulated that inhibitor binding at the FP domain blocks the conformational changes of the S protein, thus preventing the folding/unfolding and membrane fusion process. The induced conformational changes and dual solubility of FP at the water–membrane interface play an important role in the viral postfusion process.24 Since the ordered and closed conformation of FP is directly associated with the RBD-down conformation, an alternative mechanism of action is that small molecule binding stabilizes the FP loop in the closed conformation, thereby locking the S protein in the RBD-down inactive form and blocking receptor–RBD binding.
Viral entry is a complex process with a number of protein targets in virus and host cells involved in the membrane fusion mechanism and endocytic pathway.25,26 While we identified small molecule entry inhibitors from the structure-based virtual screening targeting the FP binding site and confirmed their inhibitory activities in the PP entry assays and CPE assay, further experimental studies are required to elucidate the mechanism of action of these potential fusion inhibitors. As we have previously validated the CCZ binding in the FP pocket of HCV,16 site-directed mutagenesis studies of the key residues in the FP pocket such as Asp867 and Phe833 would provide insight on the molecular basis of inhibition in the viral fusion process.
In summary, through comparative structural modeling analysis, we have revealed a fusion-peptide hydrophobic pocket of S protein in SARS-CoV-2 that also exists in SARS-CoV-1 and MERS. The FP binding site and interactions with small molecule were explored by the fusion inhibitor CCZ. Further structure-based virtual screening led to the identification of two chemotypes of inhibitors, which may serve as a starting point for further development of potent inhibitors against SARS-CoV-2. The characterization of the coronavirus S protein FP binding pocket provides a novel and promising target for the structure-based design of small molecule entry inhibitors as drug candidates for the treatment of COVID-19.
Acknowledgments
This work was supported by the Intramural Research Programs of the National Center for Advancing Translational Sciences and NIDDK, National Institutes of Health.
Glossary
Abbreviations
- SARS-CoV-2
severe acute respiratory syndrome coronavirus 2
- FP
fusion peptide
- HR
heptad repeat
- CCZ
chlorcyclizine
- PP
pseudoparticle
- VS
virtual screening
- SAR
structure–activity relationship
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00263.
Dynamics of the fusion peptide loop; predicted binding models of C1 and C2 analogues; activities of C2 analogues in the PP entry against SARS-CoV-2, SARS-CoV-1, and MERS; physicochemical properties and source of identified inhibitors; and experimental methods (PDF)
The authors declare no competing financial interest.
This article is made available via the ACS COVID-19 subset for unrestricted RESEARCH re-use and analyses in any form or by any means with acknowledgement of the original source. These permissions are granted for the duration of the World Health Organization (WHO) declaration of COVID-19 as a global pandemic.
Supplementary Material
References
- Lai C. C.; Shih T. P.; Ko W. C.; Tang H. J.; Hsueh P. R. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): The epidemic and the challenges. Int. J. Antimicrob. Agents 2020, 55 (3), 105924. 10.1016/j.ijantimicag.2020.105924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AlNaamani K.; AlSinani S.; Barkun A. N. Medical research during the COVID-19 pandemic. World J. Clin Cases 2020, 8 (15), 3156–3163. 10.12998/wjcc.v8.i15.3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A. K.; Brindisi M.; Shahabi D.; Chapman M. E.; Mesecar A. D. Drug Development and Medicinal Chemistry Efforts toward SARS-Coronavirus and Covid-19 Therapeutics. ChemMedChem 2020, 15 (11), 907–932. 10.1002/cmdc.202000223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupferschmidt K.; Cohen J. Race to find COVID-19 treatments accelerates. Science 2020, 367 (6485), 1412–1413. 10.1126/science.367.6485.1412. [DOI] [PubMed] [Google Scholar]
- Eastman R. T.; Roth J. S.; Brimacombe K. R.; Simeonov A.; Shen M.; Patnaik S.; Hall M. D. Remdesivir: A Review of Its Discovery and Development Leading to Emergency Use Authorization for Treatment of COVID-19. ACS Cent. Sci. 2020, 6 (5), 672–683. 10.1021/acscentsci.0c00489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Beltran W. F.; Lam E. C.; St Denis K.; Nitido A. D.; Garcia Z. H.; Hauser B. M.; Feldman J.; Pavlovic M. N.; Gregory D. J.; Poznansky M. C.; Sigal A.; Schmidt A. G.; Iafrate A. J.; Naranbhai V.; Balazs A. B. Multiple SARS-CoV-2 variants escape neutralization by vaccine-induced humoral immunity. Cell 2021, 184, 2523. 10.1016/j.cell.2021.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G. L.; Wang Z. Y.; Duan L. J.; Meng Q. C.; Jiang M. D.; Cao J.; Yao L.; Zhu K. L.; Cao W. C.; Ma M. J. Susceptibility of Circulating SARS-CoV-2 Variants to Neutralization. N. Engl. J. Med. 2021, 384, 2354. 10.1056/NEJMc2103022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Yang P.; Liu K.; Guo F.; Zhang Y.; Zhang G.; Jiang C. SARS coronavirus entry into host cells through a novel clathrin- and caveolae-independent endocytic pathway. Cell Res. 2008, 18 (2), 290–301. 10.1038/cr.2008.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang T.; Bidon M.; Jaimes J. A.; Whittaker G. R.; Daniel S. Coronavirus membrane fusion mechanism offers a potential target for antiviral development. Antiviral Res. 2020, 178, 104792. 10.1016/j.antiviral.2020.104792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia S.; Zhu Y.; Liu M.; Lan Q.; Xu W.; Wu Y.; Ying T.; Liu S.; Shi Z.; Jiang S.; Lu L. Fusion mechanism of 2019-nCoV and fusion inhibitors targeting HR1 domain in spike protein. Cell. Mol. Immunol. 2020, 17 (7), 765–767. 10.1038/s41423-020-0374-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia S.; Liu M.; Wang C.; Xu W.; Lan Q.; Feng S.; Qi F.; Bao L.; Du L.; Liu S.; Qin C.; Sun F.; Shi Z.; Zhu Y.; Jiang S.; Lu L. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020, 30 (4), 343–355. 10.1038/s41422-020-0305-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C. Z.; Xu M.; Pradhan M.; Gorshkov K.; Petersen J. D.; Straus M. R.; Zhu W.; Shinn P.; Guo H.; Shen M.; Klumpp-Thomas C.; Michael S. G.; Zimmerberg J.; Zheng W.; Whittaker G. R. Identifying SARS-CoV-2 Entry Inhibitors through Drug Repurposing Screens of SARS-S and MERS-S Pseudotyped Particles. ACS Pharmacol Transl Sci. 2020, 3 (6), 1165–1175. 10.1021/acsptsci.0c00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H.; Wang Y.; Chen C. Z.; Xu M.; Guo H.; Itkin M.; Zheng W.; Shen M. Identification of SARS-CoV-2 viral entry inhibitors using machine learning and cell-based pseudotyped particle assay. Bioorg. Med. Chem. 2021, 38, 116119. 10.1016/j.bmc.2021.116119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S.; Lin B.; Chu V.; Hu Z.; Hu X.; Xiao J.; Wang A. Q.; Schweitzer C. J.; Li Q.; Imamura M.; Hiraga N.; Southall N.; Ferrer M.; Zheng W.; Chayama K.; Marugan J. J.; Liang T. J. Repurposing of the antihistamine chlorcyclizine and related compounds for treatment of hepatitis C virus infection. Sci. Transl. Med. 2015, 7 (282), 282ra49. 10.1126/scitranslmed.3010286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S.; Xiao J.; Dulcey A. E.; Lin B.; Rolt A.; Hu Z.; Hu X.; Wang A. Q.; Xu X.; Southall N.; Ferrer M.; Zheng W.; Liang T. J.; Marugan J. J. Discovery, Optimization, and Characterization of Novel Chlorcyclizine Derivatives for the Treatment of Hepatitis C Virus Infection. J. Med. Chem. 2016, 59 (3), 841–53. 10.1021/acs.jmedchem.5b00752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z.; Rolt A.; Hu X.; Ma C. D.; Le D. J.; Park S. B.; Houghton M.; Southall N.; Anderson D. E.; Talley D. C.; Lloyd J. R.; Marugan J. C.; Liang T. J. Chlorcyclizine Inhibits Viral Fusion of Hepatitis C Virus Entry by Directly Targeting HCV Envelope Glycoprotein 1. Cell Chem. Biol. 2020, 27 (7), 780–792. 10.1016/j.chembiol.2020.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brimacombe K. R.; Zhao T.; Eastman R. T.; Hu X.; Wang K.; Backus M.; Baljinnyam B.; Chen C. Z.; Chen L.; Eicher T.; Ferrer M.; Fu Y.; Gorshkov K.; Guo H.; Hanson Q. M.; Itkin Z.; Kales S. C.; Klumpp-Thomas C.; Lee E. M.; Michael S.; Mierzwa T.; Patt A.; Pradhan M.; Renn A.; Shinn P.; Shrimp J. H.; Viraktamath A.; Wilson K. M.; Xu M.; Zakharov A. V.; Zhu W.; Zheng W.; Simeonov A.; Mathe E. A.; Lo D. C.; Hall M. D.; Shen M.. An OpenData portal to share COVID-19 drug repurposing data in real time. 2020, bioRxiv 2020.06.04.135046. bioRxiv e-Print archive. https://www.biorxiv.org/content/10.1101/2020.06.04.135046v1.
- Wrapp D.; Wang N.; Corbett K. S.; Goldsmith J. A.; Hsieh C. L.; Abiona O.; Graham B. S.; McLellan J. S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367 (6483), 1260–1263. 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y.; Zhang J.; Xiao T.; Peng H.; Sterling S. M.; Walsh R. M. Jr.; Rawson S.; Rits-Volloch S.; Chen B. Distinct conformational states of SARS-CoV-2 spike protein. Science 2020, 369 (6511), 1586–1592. 10.1126/science.abd4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C. Z.; Shinn P.; Itkin Z.; Eastman R. T.; Bostwick R.; Rasmussen L.; Huang R.; Shen M.; Hu X.; Wilson K. M.; Brooks B. M.; Guo H.; Zhao T.; Klump-Thomas C.; Simeonov A.; Michael S. G.; Lo D. C.; Hall M. D.; Zheng W. Drug Repurposing Screen for Compounds Inhibiting the Cytopathic Effect of SARS-CoV-2. Front. Pharmacol. 2021, 11, 592737. 10.3389/fphar.2020.592737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann S.; Eliseeva E.; Boutin A.; Barnaeva E.; Ferrer M.; Southall N.; Kim D.; Hu X.; Morgan S. J.; Marugan J. J.; Gershengorn M. C. Discovery of a Positive Allosteric Modulator of the Thyrotropin Receptor: Potentiation of Thyrotropin-Mediated Preosteoblast Differentiation In Vitro. J. Pharmacol. Exp. Ther. 2018, 364 (1), 38–45. 10.1124/jpet.117.244095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case J. B.; Rothlauf P. W.; Chen R. E.; Liu Z.; Zhao H.; Kim A. S.; Bloyet L. M.; Zeng Q.; Tahan S.; Droit L.; Ilagan M. X. G.; Tartell M. A.; Amarasinghe G.; Henderson J. P.; Miersch S.; Ustav M.; Sidhu S.; Virgin H. W.; Wang D.; Ding S.; Corti D.; Theel E. S.; Fremont D. H.; Diamond M. S.; Whelan S. P. J. Neutralizing Antibody and Soluble ACE2 Inhibition of a Replication-Competent VSV-SARS-CoV-2 and a Clinical Isolate of SARS-CoV-2. Cell Host Microbe 2020, 28 (3), 475–485. 10.1016/j.chom.2020.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X.; Wang C.; Chang; Wang Y.; Dong X.; Jiao T.; Zhao Z.; Ren L.; Dela Cruz C. S.; Sharma L.; Lei X.; Wang J. Identification of Potent and Safe Antiviral Therapeutic Candidates Against SARS-CoV-2. Front. Immunol. 2020, 11, 586572. 10.3389/fimmu.2020.586572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton D. J.; Wrobel A. G.; Xu P.; Roustan C.; Martin S. R.; Rosenthal P. B.; Skehel J. J.; Gamblin S. J. Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature 2020, 588 (7837), 327–330. 10.1038/s41586-020-2772-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil C.; Ginex T.; Maestro I.; Nozal V.; Barrado-Gil L.; Cuesta-Geijo M. A.; Urquiza J.; Ramirez D.; Alonso C.; Campillo N. E.; Martinez A. COVID-19: Drug Targets and Potential Treatments. J. Med. Chem. 2020, 63 (21), 12359–12386. 10.1021/acs.jmedchem.0c00606. [DOI] [PubMed] [Google Scholar]
- Tharappel A. M.; Samrat S. K.; Li Z.; Li H. Targeting Crucial Host Factors of SARS-CoV-2. ACS Infect. Dis. 2020, 6 (11), 2844–2865. 10.1021/acsinfecdis.0c00456. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.