

Abstract
RNA N6‐methyladenosine (m6A) is an emerging regulatory mechanism for tumor progression in several types of cancer. However, the underlying regulation mechanisms of m6A methylation in colorectal cancer (CRC) remain unknown. Although the oncogenic function of methyl CpG binding protein 2 (MeCP2) has been reported, it is still unclear whether MeCP2 could alter RNA m6A methylation state. Here, we systematically identified MeCP2 as a prometastasis gene to regulate m6A methylation in CRC. Interestingly, MeCP2 could bind to methyltransferase‐like 14 (METTL14) to coregulate tumor suppressor Kruppel‐like factor 4 (KLF4) expression through changing m6A methylation modification. Furthermore, insulin‐like growth factor 2 mRNA‐binding protein 2 recognized the unique modified m6A methylation sites to enhance KLF4 mRNA stability. Taken together, these findings highlight the novel function of MeCP2 for regulating m6A methylation and reveal the underlying molecular mechanism for the interaction between MeCP2 and METTL14, which offers a better understanding of CRC progression and metastasis.
Keywords: colorectal cancer, m6A methylation, MeCP2, metastasis, METTL14
MeCP2 as a prometastasis gene and METTL14 as an antimetastasis gene in colorectal cancer. MeCP2 affects N6‐methyladenosine (m6A) methylation through physically interacting with METTL14. MeCP2 and METTL14 jointly regulate the target gene KLF4 through m6A methylation sites.
![]()
1. INTRODUCTION
Methyl CpG binding protein 2 (MeCP2) is a methylated DNA binding protein. MeCP2 located on the X chromosome plays key roles on neurological disorders, especially on Rett syndrome.1, 2 Recently, it has been confirmed that MeCP2 mutations could cause intellectual disability.3 Methyl CpG binding protein 2 was identified as a member of intrinsically disordered proteins (IDPs), which have rarely structured elements.4 In fact, only 40% of the protein structure of MeCP2 could form the classical tertiary structure, such as α‐helices, β‐sheets, and β‐turns.4 Overall, the high flexibility contributes to protein interaction with plentiful molecular partners.5, 6 As a critical regulation molecule in Rett syndrome and autism, MeCP2 is also associated with cancer progression. The oncogenic functions of MeCP2 have been reported in gastric cancer, colorectal cancer, hepatocellular carcinoma, and osteosarcoma.7, 8, 9, 10, 11 A tremendously influential study on an unbiased genome‐scale screen revealed that MeCP2 is a commonly amplified oncogene in human malignancies with a direct way of activating downstream pathway.12 The underlying molecular mechanisms of MeCP2 that regulate tumor progression remain unknown.
mRNA methylation modification, particularly N6‐methyladenosine (m6A), is of vital importance in mediating multiple fundamental biological processes. The m6A methylation modification is dynamically reversible by methyltransferases (methyltransferase‐like 3 [METTL3], METTL14, and WT1 associated protein [WTAP]) and demethylases (FTO and ALKBH5).13, 14, 15, 16 Moreover, RNA‐binding proteins are considered as m6A methylation “readers” and composed by diverse proteins containing YTH, KH, or hnRNP domain. N6‐methyladenosine methylation modification has diverse impacts on biological processes such as mRNA stability, alternative splicing, translation efficiency, and nuclear export.17, 18, 19, 20 Recently reports showed that m6A methylation methyltransferase complex was formed by the METTL3‐METTL14 heterodimer, and METTL14 interacted with METTL3 through their individual methyltransferase domains (MTD). In this model, METTL3 principally serves as the methyl catalytic core, while METTL14 could stabilize METTL3 to exert methyltransferase activity, which provides an RNA‐binding platform.21, 22 Furthermore, as a key regulator in m6A methylation modification, METTL14 has been confirmed to be involved in cancer progression, but the effect on tumorigenesis is controversial.23, 24, 25, 26, 27, 28, 29 However, it remains to be elucidated how m6A methylation modification and METTL14 affect cancer and what underlying molecular mechanisms or process regulations mediate these biological functional changes.
Colorectal cancer (CRC) is the third most frequently diagnosed cancer and the second most frequent cause of cancer death worldwide, accounting for approximately 10% of all cancer‐caused deaths.30 Here, we systematically identified the function of MeCP2 in facilitating metastasis of CRC. Furthermore, the interaction between MeCP2 and METTL14 regulated m6A methylation modification. Tumor suppressor Kruppel‐like factor 4 (KLF4) was found to be a coregulated downstream target by m6A methylation modification. Moreover, “reader” protein insulin‐like growth factor 2 mRNA‐binding protein 2 (IGF2BP2) recognized uniquely modified m6A methylation sites to enhance KLF4 mRNA stability. To our knowledge, this is the first study to report the novel function of MeCP2 in modifying RNA methylation in CRC carcinogenesis.
2. MATERIALS AND METHODS
Details for materials and methods are provided in Supporting information.
3. RESULTS
3.1. Expression of MeCP2 in clinical CRC samples
We first examined MeCP2 expression in paired CRC and normal tissue samples by immunoblotting, which revealed that MeCP2 was significantly upregulated in tumor tissues (Figure 1A,B). To further verify MeCP2 expression in CRC, IHC was carried out in a cohort of paired CRC and normal tissue samples. The results also showed MeCP2 had significantly higher expression in CRC tissue than normal samples (Figure 1C). Among 88 CRC samples, the IHC data showed MeCP2 protein expression was positively correlated with lymph node metastasis (P < .05) (Table S1). Moreover, we detected MeCP2 mRNA expression in another cohort of 216 paired CRC and normal tissues samples by RT‐qPCR, a total of 216 samples were classified as relatively low and high expression to normal tissue. As shown in Figure 1D, the higher MeCP2 mRNA expression was associated with a poorer prognosis. The statistical analysis of clinical pathological characteristics showed MeCP2 mRNA expression was positively correlated with TNM stages (Table S2). Overall survival data showed a poorer prognosis with higher MeCP2 expression in The Cancer Genome Atlas (TCGA) (Figure 1E). Taken together, our data showed that MeCP2 expression was increased in tumor samples and associated with a poorer prognosis; MeCP2 might act as an oncogene in CRC.
FIGURE 1.
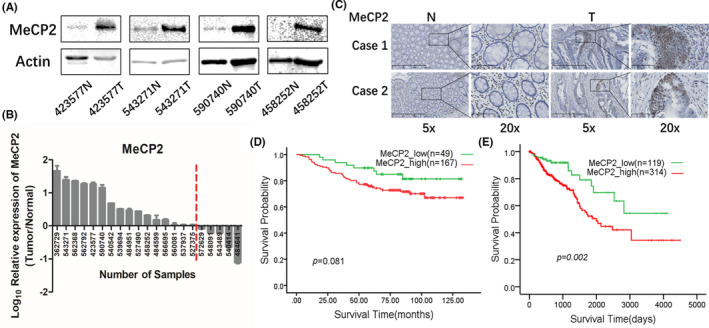
Expression of methyl CpG binding protein 2 (MeCP2) in clinical colorectal cancer (CRC) samples. A, Expression of MeCP2 in paired CRC and normal tissue samples. B, Quantification analysis of (A) and the quantitative value greater than zero means higher expression of MeCP2 in CRC tissues compared with paired normal tissues. C, Representative immunohistochemistry images of MeCP2 in paired normal tissues (N) and CRC tissues (T). D, Kaplan‐Meier plots for overall survival of MeCP2 in clinical CRC samples. E, Kaplan‐Meier plots for overall survival of MeCP2 in The Cancer Genome Atlas database
3.2. Methyl CpG binding protein 2 promotes metastasis in CRC cells
Recently, some studies have revealed MeCP2 as an oncogene in CRC.9, 31 To further determine the biological roles of MeCP2 in CRC, we detected the protein and mRNA expression of MeCP2 in six CRC cell lines (Figure S1). Then shRNA was used to knock down MeCP2 in HT29, HCT8, and SW620 cells (Figures 2A,B and S2G). Knockdown of MeCP2 significantly decreased the potential of migration and invasion (Figures 2C,D and S2H). To avoid off‐target effects of shRNA, we used siRNA to silence MeCP2 (Figure S2A,B), which showed a similar biological phenotype in that MeCP2 knockdown could significantly repress migration and invasion in HT29 and HCT8 cells (Figure S2C,D). Furthermore, overexpression of MeCP2 was constructed in HCT116, RKO, and DLD1 cells (Figures 2E,F and S2I). Overexpression of MeCP2 could significantly promote migration and invasion (Figure 2G,H). Collectively, these data suggested that MeCP2 could promote migration and invasion of CRC cells.
FIGURE 2.

Methyl CpG binding protein 2 (MeCP2) knockdown inhibited migration and invasion of colorectal cancer (CRC) cells. MeCP2 knockdown enhanced N6‐methyladenosine (m6A) methylation in CRC cells. A, Left, western blots of MeCP2 protein expression in HT29 cells; right, RT‐qPCR of MeCP2 mRNA expression in HT29 cells. B, Left, western blots of MeCP2 protein expression in HCT8 cells; right, RT‐qPCR of MeCP2 mRNA expression in HCT8 cells. C, Transwell assay for investigating migration and invasive properties in HT29 cells. D, Transwell assay for investigating migration and invasive properties in HCT8 cells. E, Left, western blots of MeCP2 protein expression in HCT116 cells; right, RT‐qPCR of MeCP2 mRNA expression in HCT116 cells. F, Left, western blots of MeCP2 protein expression in RKO cells; right, RT‐qPCR of MeCP2 mRNA expression in RKO cells. G, Transwell assay for investigating migration and invasive properties in HCT116 cells. H, Transwell assay for investigating migration and invasive properties in RKO cells. I, Left, dot blots of m6A methylation level in HT29 cells; right, immunohistochemistry (IHC) of m6A methylation level in HT29 cells. J, Left, dot blots of m6A methylation level in HCT8 cells; right, IHC of m6A methylation level in HCT8 cells. K, Left, dot blots of m6A methylation level in HCT116 cells; right, IHC of m6A methylation level in HCT116 cells. L, Left, dot blots of m6A methylation level in RKO cells; right, IHC of m6A methylation level in RKO cells. EV, empty vector; PARP, poly(ADP‐ribose) polymerase
3.3. Methyl CpG binding protein 2 reduces m6A methylation level in CRC cells
Previous studies reported the roles of MeCP2 in DNA methylation, however, whether MeCP2 could regulate RNA methylation has not been clarified. First, we used dot blots to detect the m6A methylation level. The results showed that knockdown of MeCP2 upregulated the m6A methylation level in HT29 and HCT8 cells by shRNA (Figure 2I,J) and siRNA (Figure S2E,F). Consistently, overexpression of MeCP2 downregulated the m6A methylation level in HCT116 and RKO cells (Figure 2K,L). To confirm the results above, we also used IHC to detect m6A methylation levels. Immunohistochemistry using anti‐m6A showed an increased m6A methylation level in MeCP2‐silenced HT29 and HCT8 cells, whereas forced expression of MeCP2 in HCT116 and RKO cells displayed a significantly decreased m6A methylation level (Figures 2I‐L and S2E,F). All these results indicated that MeCP2 could inhibit RNA m6A methylation in CRC cells.
3.4. Methyl CpG binding protein 2 interacted with m6A methylation methyltransferase METTL14
To explore the intrinsic relationship between MeCP2 and RNA m6A methylation, we undertook coimmunoprecipitation (co‐IP) assays in MeCP2‐overexpressing cells to investigate whether MeCP2 could interact with the methyltransferases (METTL3, METTL14, and WTAP) and demethylases (ALKBH5 and FTO). There was a significant interaction between MeCP2 and METTL14, but not with the others (Figures 3A,B and S3A). Next, the endogenous interaction between MeCP2 and METTL14 was also confirmed by co‐IP (Figures 3C and S3B). As shown in Figure 3D, both Flag‐MeCP2 with HA‐METTL14 and HA‐MeCP2 with Flag‐METTL14 had reciprocal interaction with each other in 293T cells. Some previous reports have suggested that METTL14 and METTL3 proteins could form a stable heterodimer core complex.21, 22 Interestingly, the presence of excess MeCP2 will occupy reciprocity between METTL3 and METTL14 (Figures 3E and S3C), which indicated that there was a competitive relationship between MeCP2 and METTL3 with METTL14. METTL14 executes its function on RNA methylation by its methyl binding domain, which also plays a major role in interacting with METTL3, so we constructed truncated proteins to determine whether it is the same domain interacting with MeCP2. We generated several truncated constructs of METTL14 and coexpressed them with MeCP2 in 293T cells (Figure 3F). Interestingly, truncated protein containing aa 1‐395 and 165‐456, which included the whole MTD domain in METTL14, could interact with MeCP2. As expected, the MTD of METTL14 was also crucial to interact with METTL3. Thus, we defined the MTD as a competitive structure of METTL14 for MeCP2 and METTL3 to interact with METTL14. Recently, MeCP2 was reported as an IDP.4 As an important characteristic of IDP is their inability to acquire a stable secondary structure, we thought that the structural flexibility enabled MeCP2 to interact with METTL14. We then undertook the immunofluorescence assay, which showed MeCP2 and METTL14 colocating in the nucleus (Figure 3G). The overexpression of MeCP2 affected the METTL3 expression at the protein level in HCT116, HT29, and SW620 cells (Figures 3H and S3D), but not at the transcriptional level (Figure 3I). Otherwise, the protein expression of METTL3 was not changed in other cell lines. The discrepant results need to be studied further. In addition, the mRNA expression of other RNA methylation participants was no different. Collectively, these findings strongly clarified that the influence on RNA methylation by MeCP2 was dependent on the interaction between MeCP2 and METTL14.
FIGURE 3.

Interaction of methyl CpG binding protein 2 (MeCP2) with methyltransferase‐like 14 (METTL14). A, Western blots of the interaction between exogenous MeCP2 and endogenous N6‐methyladenosine (m6A) methylation methyltransferases and demethylases. B, Western blots of the interaction of endogenous METTL14 with exogenous MeCP2. C, Western blots of the interaction of endogenous MeCP2 with endogenous METTL14. D, Western blots of the interaction of exogenous MeCP2 with exogenous METTL14. E, Western blots of the interaction of MeCP2 with METTL14 and METTL3 with METTL14. F, Upper, schematic diagram of the functional domains of METTL14 and truncated protein of METTL14. Lower, western blots of the interaction of truncated protein of METTL14 with full‐length MeCP2 and full‐length METTL3. HM, α‐helical motif; MTD, methyltransferase domain. G, Immunofluorescence staining for METTL14 and Flag‐MeCP2 in HCT116, RKO, and DLD1 cells. Nuclei were stained with DAPI. H, Western blots of METTL3 and METTL14 protein expression in HCT116, RKO, HT29, and HCT8 cells with MeCP2 overexpression vector or shRNA. I, RT‐qPCR of m6A methylation methyltransferases and demethylases mRNA expression. EV, empty vector; IP, immunoprecipitant; PARP, poly(ADP‐ribose) polymerase
3.5. Methyltransferase‐like 14 inhibits metastasis in CRC cells
Previous studies showed METTL14 inhibited tumor metastasis in colorectal cancer.32, 33, 34 Next, we focused on the function of METTL14 in CRC progression and tumorigenesis. Overall survival data showed a poorer prognosis with lower METTL14 expression in TCGA (Figure 4A). Two contradictory prognosis trends between METTL14 and MeCP2 for CRC progression and tumorigenesis reminded us that they functioned with opposing biological roles in CRC. Detection of protein expression by western blots and mRNA expression by RT‐qPCR were then carried out in several CRC cell lines (Figure S4A,B). We applied shRNA to stably knock down the expression of METTL14 in HCT116 and RKO cells (Figure 4B,C). Although the expression of MeCP2 was almost unchanged by METTL14 knockdown in both protein (Figure 4B,C) and mRNA levels (Figure 4D), METTL14 silencing promoted CRC cell migration and invasion, indicating a tumor suppressor role for METTL14 in CRC (Figure 4E,F). As a classical RNA methyltransferase, METTL14 silencing significantly reduced m6A methylation levels both by dot blots and IHC (Figure 4G,H). Recently, a study showed that MeCP2 increased the level of pAKT and activated the PI3K pathway.12 In another study, downregulated METTL14 correlated with increasing levels of pAKT and activation of the PI3K pathway.24 Therefore, we investigated the pAKT change after treatment with 100 ng epidermal growth factor for 4 hours in HCT116 cells (Figure S4C), and then detected pAKT in MeCP2‐overexpressing and METTL14‐knockdown HCT116 cells (Figure S4D). The results implied that MeCP2 overexpression and METTL14 knockdown have a similar effect on activation of pAKT, which shares an identical regulation model. Overall, we concluded that METTL14 functioned as a tumor suppressor to inhibit CRC progression and tumorigenesis.
FIGURE 4.

Methyltransferase‐like 14 (METTL14) knockdown promoted migration and invasion in colorectal cancer cells. A, Kaplan‐Meier plots for overall survival of METTL14 in The Cancer Genome Atlas database. B, Left, western blots of METTL14 protein expression in HCT116 cells; right, RT‐qPCR of METTL14 mRNA expression in HCT116 cells. C, Left, western blots of METTL14 protein expression in RKO cells; right, RT‐qPCR of METTL14 mRNA expression in RKO cells. D, RT‐qPCR of MeCP2 mRNA expression in HCT116 or RKO cells. E, Transwell assay for investigating migration and invasive properties in HCT116 cells. F, Transwell assay for investigating migration and invasive properties in RKO cells. G, Left, dot blots of m6A methylation level in HCT116 cells; right, immunohistochemistry (IHC) of N6‐methyladenosine (m6A) methylation level in HCT116 cells. H, Left, dot blots of m6A methylation level in RKO cells; right, IHC of m6A methylation level in RKO cells. PARP, poly(ADP‐ribose) polymerase
3.6. Expression of KLF4 is regulated in an m6A methylation‐involved manner
To explore the potential targets coregulated by METTL14 and MeCP2, RNA sequencing (RNA‐seq) was undertaken in METTL14‐knockdown HCT116 cells and MeCP2‐knockdown HT29 cells to screen their common regulated target genes (Figure 5A). The results revealed 600 upregulated or downregulated genes in METTL14‐knockdown HCT116 cells and 2297 upregulated or downregulated genes in MeCP2‐knockdown HT29 cells (Figure 5B). In our hypothetical model, METTL14‐knockdown cells are functionally equal to MeCP2‐overexpressing cells, so the target genes’ expression should present the opposite trend in these two RNA‐seq datasets. A total of 32 genes were downregulated in METTL14‐knockdown and upregulated in MeCP2‐knockdown cells, and 44 genes were upregulated in METTL14‐knockdown and downregulated in MeCP2‐knockdown cells. Enrichment analysis for the 76 genes revealed related signaling pathway (Figure 5C), including some important cancer metastasis‐related genes, for example TGFB2, KLF4, IGFBP6, and BCL6 (Figure 5D). Reverse transcription‐PCR was used to verify these genes expression changes in HCT116 and HT29 cells (Figure 5E). Next, we sought m6A methylation binding sites from a published m6A sequencing database.35 RRACH (R = G, A and H = A, C or U) motif has been proved as the most common m6A methylation modification site, so we checked the specific A in this motif from all target candidates. It is well established that KLF4 has a significantly antineoplastic effect in carcinoma, especially in CRC, so we selected KLF4 as a common target for further study. First, protein and mRNA levels of KLF4 regulated by MeCP2 and METTL14 were determined (Figure 5F,G). We found several potential m6A methylation sites in KLF4 and designed a series of primers to investigate m6A methylation. Methylated RNA immunoprecipitation combined with RT‐qPCR was carried out using Ab against m6A methylation. As shown in Figure 5H, both METTL14 knockdown and MeCP2 overexpression significantly decreased the enrichment of KLF4 m6A methylation levels. The m6A methylation modification site was located at a coding sequence near a terminator, which agreed with a previous study.13, 36 Moreover, KLF4 has been reported as a prognostic predictor to function as a tumor suppressor in CRC.37, 38, 39 Here, we stably silenced KLF4 in HCT116 cells and overexpressed KLF4 in HT29 cells (Figure 5I,J). Cell migration and invasion assays revealed that KLF4 could inhibit the ability of invasive properties in CRC cells (Figure 5K,L), which was consistent with previous studies.40, 41 From these collective results, we concluded that MeCP2 and METTL14 regulated the expression of KLF4 in an m6A methylation‐involved manner, further contributing to CRC progression and tumorigenesis.
FIGURE 5.

Kruppel‐like factor 4 (KLF4) inhibited migration and invasion in colorectal cancer cells as a coregulated target of methyltransferase‐like 14 (METTL14) and methyl CpG binding protein 2 (MeCP2) in an N6‐methyladenosine (m6A)‐dependent manner. A, Schematic diagram of experimental approach of screening regulated target genes. B, RNA‐sequencing (RNA‐seq) identified upregulated and downregulated genes in HCT116 or HT29 cells. C, Top regulated biological process terms both regulated by METTL14 and MeCP2 identified by gene ontology enrichment analysis of the differentially expressed genes based on DAVID online tools (P < .01). D, Rank of multipathway genes of the regulated genes. For each gene, the total number of pathways regulated by METTL14 and MeCP2 is indicated. E, RT‐qPCR to determine the relative mRNA expression of oncogenes coregulated by METTL14 and MeCP2, including KLF4, ID2, IGFBP6, ARHGAP18, IRS2, and BCL6. F, RT‐qPCR of KLF4 mRNA expression in HCT116 or HT29 cells. G, Western blots of KLF4 protein expression in HCT116 or HT29 cells. H, MeRIP‐qPCR analysis of m6A methylation enrichment of KLF4 in HCT116 or HT29 cells. I, Left, western blots of KLF4 protein expression in HCT116 cells; right, RT‐qPCR of KLF4 mRNA expression in HCT116 cells. J, Left, western blots of KLF4 protein expression in HT29 cells; right, RT‐qPCR of KLF4 mRNA expression in HT29 cells. K, Transwell assay for investigating migration and invasive properties in HCT116 cells. L, Transwell assay for investigating migration and invasive properties in HT29 cells. EV, empty vector; PARP, poly(ADP‐ribose) polymerase
3.7. Methyl CpG binding protein 2 promotes metastasis through METTL14‐mediated antimetastasis in vivo and in vitro
We next investigated the function of MeCP2 and METTL14 in vivo and in vitro. Stably transfected HCT116 cells were constructed and categorized as the luciferase‐labeled control group (Empty vehicle, Scramble, and METTL14‐sh‐Empty vehicle) or experimental group (MeCP2‐Overexpression, METTL14‐sh, METTL14‐sh‐MeCP2‐Overexpression) (Figures 6A and S5A). In vitro, we observed a significantly increased migration and invasion with either MeCP2 overexpression or METTL14 knockdown. Surprisingly, METTL14 knockdown abolished the effect that MeCP2 overexpression promoted CRC cell migration and invasion (Figure 6B). In vivo, the group of HCT116 cells labeled with luciferase were surgically injected into the spleen of nude mice. After 2 months, the nude mice were subjected to D‐luciferin injection and the luciferase signals were monitored and quantified. The MeCP2‐overexpressing and METTL14‐knockdown cells showed more prometastatic signals than the control group (Figure 6C). After surgical dissection, more metastatic foci were observed in their livers (Figure 6D,E). By H&E staining, more and larger metastatic foci were present in the liver of mice in the experimental group compared to the control group (Figure 6F). Furthermore, we used IHC to investigate the expression of MeCP2, METTL14, and KLF4 in serial sections from the same samples with H&E staining (Figure 6F). Further verification was carried out by IHC of MeCP2, METTL14, and KLF4 in paired CRC and normal tissue samples in patients (Figure 6G). The samples with high expression of MeCP2 synchronously carried low expression of METTL14 or KLF4. These data in vivo suggested that MeCP2 might be considered as a prometastatic gene and METTL14 as an antimetastatic gene, which needs to be verified in more animal assays.
FIGURE 6.

Methyl CpG binding protein 2 (MeCP2) promotes colorectal cancer (CRC) metastasis and methyltransferase‐like 14 (METTL14) inhibits CRC metastasis both in vitro and in vivo. A, Left, western blots of MeCP2 and METTL14 protein expression in HCT116 cells; right, RT‐qPCR of MeCP2 and METTL14 mRNA expression in HCT116 cells. B, Transwell assay for investigating migration and invasive properties in HCT116 cells. C, Bioluminescence images of the spleen‐liver distant metastasis model, which were established by splenic injection of HCT116 cells in nude mice. D, Images of the spleen‐liver distant metastasis model by dissecting nude mice from (C). Arrows indicate liver and spleen metastatic foci. E, Bioluminescence images of the spleen‐liver distant metastasis model. F, Representative H&E staining and immunohistochemistry (IHC) images of MeCP2, METTL14, or Kruppel‐like factor 4 (KLF4) of liver tissue from spleen‐liver metastasis model of nude mice from (C). G, Representative images of IHC images of MeCP2, METTL14, or KLF4 in CRC samples. EV, empty vector; N, normal tissue; PARP, poly(ADP‐ribose) polymerase; T, tumor tissue
3.8. Methyl CpG binding protein 2 and METTL14 enhances KLF4 mRNA stability in an m6A methylation and IGF2BP2‐dependent way
To further clarify the relationship between MeCP2, METTL14, and KLF4, we measured the stability of KLF4 mRNA by blocking nascent RNA synthesis using actinomycin D. The results suggested that METTL14 knockdown and MeCP2 overexpression strikingly reduced the stability of KLF4 mRNA; however, MeCP2 knockdown maintained the stability (Figure 7A). Generally, the “reader” proteins of m6A methylation modification could determine the destiny of modified mRNA, which alter mRNA stability or protein translation efficiency. The IGF2BPs (IGF2BP1/2/3) constitute an RNA‐binding protein family mainly acting on inhibition of mRNA decay.42, 43, 44 The IGF2BPs were reported to recognize the consensus GGAC sequence of targeted mRNAs in an m6A‐dependent manner.45 But whether IGF2BPs could regulate the stability of m6A methylation modification in KLF4 mRNA is not known. We changed the potential m6A methylation binding sites from A to C for a mutagenesis. After analyzing the KLF4 mRNA coding sequence near the terminator in which the m6A methylation modification was confirmed to locate, we found four candidate motifs with RRACH. Then we constructed two luciferase reporters with the mutant motifs including MUT‐1 and MUT‐2 (Figure 7B). Compared with forced expression of the WT, MUT‐1 presented a significantly reduced luciferase activity. Substantial METTL14 knockdown and MeCP2 overexpression reduced luciferase activity of the reporter, whereas MeCP2 knockdown augmented luciferase activity of the reporter. MUT‐2 with only m6A methylation modification sites presented similar luciferase activity as WT (Figure 7C). Furthermore, the percentage of MUT‐1 to WT represented that compared to control groups, the influence on regulation of IGF2BPs in treated groups (MeCP2‐overexpressing and METTL14‐knockdown) of MUT‐1 were significantly shrunk (Figure 7C). The results above suggested that the m6A methylation binding sites A were exactly regulated by METTL14 and MeCP2. Two specific motifs, RRACH and GGAC, were crucial to regulate the stability of KLF4 mRNA, so MeCP2 and METTL14 regulated the mRNA stability of KLF4 in an m6A‐dependent manner. In order to determine which member of the IGF2BP family regulated m6A methylation, we designed specific primers spanning partly HA‐tagged sequences and used RNA immunoprecipitation to verify the protein‐RNA binding (Figure 7D). We detected the expression of IGF2BPs in HCT116 and HT29 cells, which showed extremely low expression of IGF2BP1 (Figure 7E). In the IGF2BP2‐ and IGF2BP3‐overexpressing 293T cells, both IGF2BP2 and IGF2BP3 could bind to KLF4 mRNA, but only IGF2BP2 could specifically recognize m6A methylation modification in KLF4 mRNA (Figure 7F).
FIGURE 7.

Unique N6‐methyladenosine (m6A) methylation modification elevates Kruppel‐like factor 4 (KLF4) mRNA stability through an insulin‐like growth factor 2 mRNA‐binding protein 2 (IGF2BP2)‐dependent mechanism. A, KLF4 mRNA stability assay by RT‐qPCR of KLF4 mRNA expression at the indicated time points after treatment with actinomycin D in HCT116 or HT29 cells. B, Schematic diagram of the luciferase reporter constructs used in the mRNA stability assay containing KLF4 mRNA of the WT or MUT (A‐to‐C mutation). C, Relative luciferase activity of WT or MUT KLF4 luciferase reporter in HCT116 or HT29 cells. Firefly luciferase activity was measured and normalized to Renilla luciferase activity. D, Schematic diagram of the overexpression vector constructs carried out in RNA immunoprecipitation (RIP) containing KLF4 mRNA of the WT or MUT (A‐to‐C mutation). E, RT‐qPCR of IGF2BP mRNA expression in HCT116 and HT29 cells. F, RIP with RT‐qPCR analysis of the interaction between empty vector (EV), IGF2BP2, or IGF2BP3 with Flag‐tag and KLF4‐WT or KLF4‐MUT with HA‐tag in 293T cells. Enrichment of KLF4‐WT or KLF4‐MUT with HA‐tag was normalized to input. G, Schematic model of the mechanism of MeCP2 and METTL14 in regulation of KLF4, which participated through IGF2BP2 in an m6A methylation modification‐dependent manner. CRC, colorectal cancer
4. DISCUSSION
In this study, we identified MeCP2 as an oncogene in CRC progression by interacting with METTL14 and reducing m6A methylation modification. KLF4 as an m6A methylation modification downstream target gene that inhibits CRC metastasis. Additionally, RNA‐binding protein IGF2BP2 could recognize specific motif and trigger protection procedures to delay degradation of KLF4 mRNA. The reduced m6A methylation modification attenuated mRNA stability of KLF4, resulting in the facilitation of CRC migration and invasion (Figure 7G).
Although recent experimental evidence has shown that MeCP2 contributes to cancer proliferation and metastasis,31, 46, 47 the biological mechanism of MeCP2 on carcinogenesis is still unclear. Here, we found a novel mechanism through indirect influence on m6A methylation modification. Surprisingly, it is the interaction between MeCP2 and METTL14 that impacts m6A methylation levels. Methyl CpG binding protein 2 is an IDP, which acts as scaffolding for the recruitment of other proteins and works as an interaction hub.48 Both MeCP2 and METTL3 could competitively interact with MTD of METTL14. However, we observed that METTL3 expression was increased in MeCP2‐knockdown HT29 cells and reduced in MeCP2‐overexpressing HCT116 cells, but not in HCT8 or RKO cells. The reason for the discrepant results might be the genetic background of different cell lines. In addition, whether other functions of MeCP2 exist in carcinogenesis remains to be elucidated.
An increasing number of studies have suggested that m6A methylation modification is closely related to diverse biological processes, especially in various types of cancers. Although some genes have been reported as the new m6A methylation regulation molecules, their function and mechanism have not been clearly illuminated. Therefore, we investigated the interaction between MeCP2 and the canonical methyltransferases (METTL3, METTL14, and WTAP) and demethylases (ALKBH5 and FTO) in the present study, which showed that only METTL14 could bind to MeCP2. Compared with METTL3, METTL14 might present higher methyltransferase activity due to the roles of methyltransferase activity itself and the effect on METTL3.16, 49 Although more studies have focused on the m6A methylation regulation function of METTL3, METTL14 might have more effect on downstream events than METTL3. Previous studies showed that METTL3 was upregulated while METTL14 was downregulated in CRC tissues,50, 51 which was consistent with our biological experimental data that METTL14 has a suppressive effect on CRC.
KLF4 is a notable tumor suppressor gene and regulates intestinal epithelial homeostasis, which is especially enriched in the gut.52, 53 The expression of KLF4 is significantly reduced in diverse cancer including CRC and gastric cancer.39, 54 KLF4 is strongly negatively correlated with survival and prognosis in CRC.37 In our study, we combined m6A methylation modification with KLF4 by the regulation of mRNA stability. The m6A methylation nucleotides are located on a unique consensus motif, RRACH.13, 55 The specific m6A methylation sites of KLF4 were found to locate in a CDS region near stop codons. The mRNA stability assays and dual‐luciferase reporter assays were carried out to illustrate that m6A methylation modification enhanced KLF4 mRNA stability.56 The IGF2BPs (IGF2BP1/2/3) were confirmed as m6A methylation “reader” proteins, which could recognize the unique binding motif GGAC.42, 43, 44 Our data first clarified that only IGF2BP2, but not IGF2BP1 or 3, directly bound to the specific modified m6A methylation sites in KLF4 coding sequence regions in an m6A‐dependent manner. Our data determined that MUT‐1 with RRACH and GGAC was the specific site for IGF2BP2 to bind to KLF4 mRNA. However, to a certain extent, IGF2BP2 showed a limited effect on the binding of KLF4 and further exploration on underlying m6A methylation molecular mechanisms deserves extensive study.
In summary, the expression of MeCP2 was upregulated in CRC cells and MeCP2 could promote the metastasis of CRC cells. High expression of MeCP2 indicated poor prognosis of patients. The expression of METTL14 was downregulated in CRC cells and METTL14 could inhibit the metastasis of CRC cells. Low expression of METTL14 indicated poor prognosis of patients. Methyl CpG binding protein 2 and METTL14 presented a protein‐protein interaction, and the level of m6A methylation in CRC cells was ultimately affected through this interaction. Methyl CpG binding protein 2 and METTL14 could jointly regulate the two specific m6A methylation sites on KLF4. The m6A methylation reader protein IGF2BP2 could specifically recognize the two unique m6A methylation modification sites on KLF4 mRNA, change the stability of KLF4 mRNA, as well as KLF4 mRNA and protein levels, and ultimately regulate the metastasis ability of CRC cells. With further research, the MeCP2‐METTL14/IGF2BP2/KLF4 axis could become a novel cancer therapeutic target.
DISCLOSURE
The authors have no conflict of interest.
Supporting information
FIGURE S1
FIGURE S2
FIGURE S3
FIGURE S4
FIGURE S5
Supplementary Material
ACKNOWLEDGMENTS
This work is supported by grants from Basic Public Welfare Research Project of Zhejiang Province (LGF20H160023), the National Natural Science Foundation of China (91859204) and the 111 Project (B13026).
Wang S, Gan M, Chen C, et al. Methyl CpG binding protein 2 promotes colorectal cancer metastasis by regulating N6‐methyladenosine methylation through methyltransferase‐like 14. Cancer Sci. 2021;112:3243–3254. 10.1111/cas.15011
Shuo Wang and Meifu Gan contributed equally to this work.
Funding information
Basic Public Welfare Research Project of Zhejiang Province, Grant/Award Number: LGF20H160023; National Natural Science Foundation of China, Grant/Award Number: 91859204; 111 Project, Grant/Award Number: B13026.
Contributor Information
Honghe Zhang, Email: honghezhang@zju.edu.cn.
Maode Lai, Email: lmp@zju.edu.cn.
REFERENCES
- 1.Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X‐linked MECP2, encoding methyl‐CpG‐binding protein 2. Nat Genet. 1999;23:185‐188. [DOI] [PubMed] [Google Scholar]
- 2.Ballestar E, Wolffe AP. Methyl‐CpG‐binding proteins. Targeting specific gene repression. Eur J Biochem. 2001;268:1‐6. [DOI] [PubMed] [Google Scholar]
- 3.Ramocki MB, Zoghbi HY. Failure of neuronal homeostasis results in common neuropsychiatric phenotypes. Nature. 2008;455:912‐918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adams VH, McBryant SJ, Wade PA, Woodcock CL, Hansen JC. Intrinsic disorder and autonomous domain function in the multifunctional nuclear protein, MeCP2. J Biol Chem. 2007;282:15057‐15064. [DOI] [PubMed] [Google Scholar]
- 5.Das S, Mukhopadhyay D. Intrinsically unstructured proteins and neurodegenerative diseases: conformational promiscuity at its best. IUBMB Life. 2011;63:478‐488. [DOI] [PubMed] [Google Scholar]
- 6.Gsponer J, Futschik ME, Teichmann SA, Babu MM. Tight regulation of unstructured proteins: from transcript synthesis to protein degradation. Science. 2008;322:1365‐1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao L, Liu Y, Tong D, et al. MeCP2 promotes gastric cancer progression through regulating FOXF1/Wnt5a/beta‐catenin and MYOD1/Caspase‐3 signaling pathways. EBioMedicine. 2017;16:87‐100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao LY, Tong DD, Xue M, et al. MeCP2, a target of miR‐638, facilitates gastric cancer cell proliferation through activation of the MEK1/2‐ERK1/2 signaling pathway by upregulating GIT1. Oncogenesis. 2017;6:e368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Song N, Li K, Wang Y, Chen Z, Shi L. Lentivirus mediated knockdown of MeCP2 inhibits the growth of colorectal cancer cells in vitro. Mol Med Rep. 2016;13:860‐866. [DOI] [PubMed] [Google Scholar]
- 10.Zhao LY, Zhang J, Guo B, et al. MECP2 promotes cell proliferation by activating ERK1/2 and inhibiting p38 activity in human hepatocellular carcinoma HEPG2 cells. Cell Mol Biol (Noisy‐le‐grand). 2013;Suppl 59:OL1876‐1881. [PubMed] [Google Scholar]
- 11.Meng G, Lv Y, Dai H, Zhang X, Guo QN. Epigenetic silencing of methyl‐CpG‐binding protein 2 gene affects proliferation, invasion, migration, and apoptosis of human osteosarcoma cells. Tumour Biol. 2014;35:11819‐11827. [DOI] [PubMed] [Google Scholar]
- 12.Neupane M, Clark AP, Landini S, et al. MECP2 is a frequently amplified oncogene with a novel epigenetic mechanism that mimics the role of activated RAS in malignancy. Cancer Discov. 2016;6:45‐58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dominissini D, Moshitch‐Moshkovitz S, Schwartz S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A‐seq. Nature. 2012;485:201‐206. [DOI] [PubMed] [Google Scholar]
- 14.Jia G, Fu YE, Zhao XU, et al. N6‐methyladenosine in nuclear RNA is a major substrate of the obesity‐associated FTO. Nat Chem Biol. 2011;7:885‐887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu J, Yue Y, Han D, et al. A METTL3‐METTL14 complex mediates mammalian nuclear RNA N6‐adenosine methylation. Nat Chem Biol. 2014;10:93‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Y, Li Y, Toth JI, Petroski MD, Zhang Z, Zhao JC. N6‐methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol. 2014;16:191‐198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang X, Lu Z, Gomez A, et al. N6‐methyladenosine‐dependent regulation of messenger RNA stability. Nature. 2014;505:117‐120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang X, Zhao B, Roundtree I, et al. N(6)‐methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161:1388‐1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xiao W, Adhikari S, Dahal U, et al. Nuclear m(6)A reader YTHDC1 regulates mRNA splicing. Mol Cell. 2016;61:507‐519. [DOI] [PubMed] [Google Scholar]
- 20.Zhao XU, Yang Y, Sun B‐F, et al. FTO‐dependent demethylation of N6‐methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. 2014;24:1403‐1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang P, Doxtader KA, Nam Y. Structural basis for cooperative function of Mettl3 and Mettl14 methyltransferases. Mol Cell. 2016;63:306‐317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang X, Feng J, Xue Y, et al. Structural basis of N(6)‐adenosine methylation by the METTL3‐METTL14 complex. Nature. 2016;534:575‐578. [DOI] [PubMed] [Google Scholar]
- 23.Weng H, Huang H, Wu H, et al. METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m(6)A modification. Cell Stem Cell. 2018;22(2):191‐205.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu J, Eckert MA, Harada BT, et al. m(6)A mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nat Cell Biol. 2018;20:1074‐1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang C, Zhang M, Ge S, et al. Reduced m6A modification predicts malignant phenotypes and augmented Wnt/PI3K‐Akt signaling in gastric cancer. Cancer Med. 2019;8:4766‐4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gong D, Zhang J, Chen Y, et al. The m(6)A‐suppressed P2RX6 activation promotes renal cancer cells migration and invasion through ATP‐induced Ca(2+) influx modulating ERK1/2 phosphorylation and MMP9 signaling pathway. J Exp Clin Cancer Res. 2019;38:233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu L, Wu D, Ning J, Liu W, Zhang D. Changes of N6‐methyladenosine modulators promote breast cancer progression. BMC Cancer. 2019;19:326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma JZ, Yang F, Zhou CC, et al. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N(6) ‐methyladenosine‐dependent primary MicroRNA processing. Hepatology. 2017;65:529‐543. [DOI] [PubMed] [Google Scholar]
- 29.Cui QI, Shi H, Ye P, et al. m(6)A RNA methylation regulates the self‐renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. 2017;18:2622‐2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394‐424. [DOI] [PubMed] [Google Scholar]
- 31.Luo D, Ge W. MeCP2 promotes colorectal cancer metastasis by modulating ZEB1 transcription. Cancers. 2020;12(3):758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen X, Xu MU, Xu X, et al. METTL14‐mediated N6‐methyladenosine modification of SOX4 mRNA inhibits tumor metastasis in colorectal cancer. Mol Cancer. 2020;19:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen X, Xu MU, Xu X, et al. METTL14 suppresses CRC progression via regulating N6‐methyladenosine‐dependent primary miR‐375 processing. Mol Ther. 2020;28:599‐612. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34.Yang X, Zhang S, He C, et al. METTL14 suppresses proliferation and metastasis of colorectal cancer by down‐regulating oncogenic long non‐coding RNA XIST. Mol Cancer. 2020;19:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xuan J‐J, Sun W‐J, Lin P‐H, et al. RMBase v2.0: deciphering the map of RNA modifications from epitranscriptome sequencing data. Nucleic Acids Res. 2018;46:D327‐D334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3' UTRs and near stop codons. Cell. 2012;149:1635‐1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Patel NV, Ghaleb AM, Nandan MO, Yang VW. Expression of the tumor suppressor Kruppel‐like factor 4 as a prognostic predictor for colon cancer. Cancer Epidemiol Biomarkers Prev. 2010;19:2631‐2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ton‐That H, Kaestner KH, Shields JM, Mahatanankoon CS, Yang VW. Expression of the gut‐enriched Kruppel‐like factor gene during development and intestinal tumorigenesis. FEBS Lett. 1997;419:239‐243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wei D, Gong W, Kanai M, et al. Drastic down‐regulation of Kruppel‐like factor 4 expression is critical in human gastric cancer development and progression. Cancer Res. 2005;65:2746‐2754. [DOI] [PubMed] [Google Scholar]
- 40.Shao H, Dong D, Shao F. Long non‐coding RNA TUG1‐mediated down‐regulation of KLF4 contributes to metastasis and the epithelial‐to‐mesenchymal transition of colorectal cancer by miR‐153‐1. Cancer Manag Res. 2019;11:8699‐8710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhai F, Cao C, Zhang L, Zhang J. miR‐543 promotes colorectal cancer proliferation and metastasis by targeting KLF4. Oncotarget. 2017;8:59246‐59256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Degrauwe N, Suva ML, Janiszewska M, Riggi N, Stamenkovic I. IMPs: an RNA‐binding protein family that provides a link between stem cell maintenance in normal development and cancer. Genes Dev. 2016;30:2459‐2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chatterji P, Rustgi AK. RNA binding proteins in intestinal epithelial biology and colorectal cancer. Trends Mol Med. 2018;24:490‐506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cao J, Mu Q, Huang H. The roles of insulin‐like growth factor 2 mRNA‐binding protein 2 in cancer and cancer stem cells. Stem Cells Int. 2018;2018:4217259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang H, Weng H, Sun W, et al. Recognition of RNA N(6)‐methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20:285‐295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang H, Li J, He J, et al. Methyl‐CpG‐binding protein 2 drives the Furin/TGF‐beta1/Smad axis to promote epithelial‐mesenchymal transition in pancreatic cancer cells. Oncogenesis. 2020;9:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tong D, Zhang J, Wang X, et al. MeCP2 facilitates breast cancer growth via promoting ubiquitination‐mediated P53 degradation by inhibiting RPL5/RPL11 transcription. Oncogenesis. 2020;9:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bah A, Forman‐Kay JD. Modulation of intrinsically disordered protein function by post‐translational modifications. J Biol Chem. 2016;291:6696‐6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. N(6)‐methyladenosine‐dependent RNA structural switches regulate RNA‐protein interactions. Nature. 2015;518:560‐564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li Y, Xiao J, Bai J, et al. Molecular characterization and clinical relevance of m(6)A regulators across 33 cancer types. Mol Cancer. 2019;18:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu X, Liu L, Dong Z, et al. Expression patterns and prognostic value of m(6)A‐related genes in colorectal cancer. Am J Transl Res. 2019;11:3972‐3991. [PMC free article] [PubMed] [Google Scholar]
- 52.Ghaleb AM, McConnell BB, Kaestner KH, Yang VW. Altered intestinal epithelial homeostasis in mice with intestine‐specific deletion of the Kruppel‐like factor 4 gene. Dev Biol. 2011;349:310‐320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shields JM, Christy RJ, Yang VW. Identification and characterization of a gene encoding a gut‐enriched Kruppel‐like factor expressed during growth arrest. J Biol Chem. 1996;271:20009‐20017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhao W, Hisamuddin IM, Nandan MO, Babbin BA, Lamb NE, Yang VW. Identification of Kruppel‐like factor 4 as a potential tumor suppressor gene in colorectal cancer. Oncogene. 2004;23:395‐402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Harper JE, Miceli SM, Roberts RJ, Manley JL. Sequence specificity of the human mRNA N6‐adenosine methylase in vitro. Nucleic Acids Res. 1990;18:5735‐5741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Han Y, Feng J, Xia L, et al. CVm6A: a visualization and exploration database for m(6)As in cell lines. Cells. 2019;8(2):168. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1
FIGURE S2
FIGURE S3
FIGURE S4
FIGURE S5
Supplementary Material