

Abstract
Protein kinase B (AKT) hyperactivation and de novo lipogenesis are both common in tumor progression. Sterol regulatory element‐binding protein 1 (SREBP1) is the master regulator for tumor lipid metabolism, and protein arginine methyltransferase 5 (PRMT5) is an enzyme that can catalyze symmetric dimethyl arginine (SDMA) modification of the mature form of SREBP1 (mSREBP1) to induce its hyperactivation. Here, we report that SDMA‐modified mSREBP1 (mSREBP1‐SDMA) was overexpressed and correlated with Ser473‐phosphorylated AKT (AKT‐473P) expression and poor patient outcomes in human lung adenocarcinomas. Furthermore, patients with AKT‐473P and mSREBP1‐SDMA coexpression showed the worst prognosis. Mechanistic investigation revealed that AKT activation upregulated SREBP1 at both the transcriptional and post‐translational levels, whereas PRMT5 knockdown reversed AKT signaling‐mediated mSREBP1 ubiquitin‐proteasome pathway stabilization at the post‐translational level. Meanwhile, AKT activation promoted nuclear PRMT5 to the cytoplasm without changing total PRMT5 expression, and the transported cytoplasmic PRMT5 (cPRMT5) induced by AKT activation showed a strong mSREBP1‐binding ability. Immunohistochemical assay indicated that AKT‐473P and mSREBP1‐SDMA were positively correlated with cPRMT5 in lung adenocarcinomas, and high cPRMT5 levels in tumors were associated with poor patient outcomes. Additionally, PRMT5 knockdown reversed AKT activation‐induced lipid synthesis and growth advantage of lung adenocarcinoma cells both in vitro and in vivo. Finally, we defined an AKT/PRMT5/SREBP1 axis involved in de novo lipogenesis and the growth of lung cancer. Our data also support that cPRMT5 is a potential therapeutic target for hyperactive AKT‐driven lung adenocarcinoma.
Keywords: AKT, de novo lipogenesis, lung adenocarcinoma, PRMT5, SREBP1
In this research, we defined an AKT/PRMT5/SREBP1 axis involved in de novo lipogenesis and growth of lung cancer. Our data also support that PRMT5 in the cytoplasm is a potential therapeutic target for hyperactive AKT‐driven lung adenocarcinoma.

1. INTRODUCTION
Lung cancer is the most lethal malignancy worldwide, and modern targeted therapy is essential for lung cancer treatment.1, 2 As a prominent molecular feature of malignant tumors that can be induced by multiple oncogenic events (eg, EGFR or KRAS mutation), protein kinase B (AKT) hyperactivation is common in lung cancer, regulating almost all stages of tumor growth, and represents a promising antitumor target.3, 4 The compensatory resistance of tumors to AKT inhibitors, however, because of the complex AKT signaling network, compromises antitumor therapy, and only a few AKT inhibitors have been approved for antitumor treatment.5, 6 Therefore, there is an urgent need to explore novel antitumor targets for hyperactive AKT‐driven lung cancer.
De novo lipogenesis is a hallmark of tumor metabolism triggered by the overactivation of sterol regulatory element‐binding proteins (SREBPs) in tumors.7, 8 Three isoforms of SREBPs have been identified in mammals: SREBP1a and SREBP1c are produced from the sterol regulatory element‐binding transcription factor 1 (SREBF1) gene and are only different in their extreme N termini, whereas the SREBF2 gene encodes SREBP2.9 SREBP1 is the master regulator of tumor lipid metabolism and is overexpressed in certain tumors; it is the mature form of SREBP1 (mSREBP1) that directly triggers lipid enzymes (eg, ATP citrate lyase [ACLY], fatty acid synthase [FASN], and acyl‐CoA desaturase [SCD1]) gene transcription.9, 10, 11, 12 Recently, lipid hypersynthesis in lung cancer was observed, and targeting SREBP1 was shown to hamper the tumorigenesis of lung cancer and reverse gefitinib resistance in lung cancer cells.13, 14, 15 Although AKT activation has been shown to activate SREBP1 through various mechanisms to modulate lipid metabolism in several malignant tumors (eg, melanoma, glioblastoma, and breast cancer),16, 17, 18, 19, 20, 21, 22 the mechanisms underlying lipid metabolism reprogramming in hyperactive AKT‐driven lung cancer remain elusive.
Protein arginine methyltransferase 5 (PRMT5) is a newly discovered regulator of SREBP1.23, 24, 25 As a well‐characterized enzyme responsible for symmetric dimethyl arginine (SDMA) modification of histone and certain nonhistone proteins, PRMT5 can directly interact with mSREBP1 to generate SDMA‐modified mSREBP1 (mSREBP1‐SDMA), thereby inhibiting its ubiquitin‐proteasome degradation, resulting in mSREBP1 hyperactivation in cancer or immune cells, promoting tumor growth.23, 24, 25 However, it is unknown whether the PRMT5/SREBP1 pathway is regulated by AKT signaling.
In this study, for the purpose of searching for new antitumor targets for hyperactive AKT‐driven lung cancer, we investigated the regulation of AKT signaling on PRMT5/SREBP1 pathway de novo lipogenesis in lung adenocarcinoma as well as the molecular mechanisms underlying the process.
2. MATERIALS AND METHODS
2.1. Patient samples
Experiments using human tissues were approved by the Human Assurance Committee of Shanghai Chest Hospital. Informed consent was obtained from all participants, and the study was carried out in accordance with the International Ethical Guidelines for Biomedical Research Involving Human Subjects. Tissue sections were obtained by surgical resection at the Shanghai Chest Hospital between February 2008 and July 2013. Patients were staged based on the criteria of the 8th edition of the Lung Cancer Staging Manual.26
The inclusion criteria were as follows: (a) lung adenocarcinoma confirmed by pathological examination of surgical specimens; (b) tumor biopsies were paired with a matched normal tissue sample from the same patient; (c) patients were followed up until August 2016 (from 4 to 83 months) after surgery, without a history of cancer other than lung adenocarcinoma before surgery; (d) no history of antitumor treatment (eg, chemo‐ and radiotherapy) before surgery; and (e) availability of age, sex, smoking history, tumor location, pathological grade, and TNM stage.
2.2. Immunochemistry
Immunochemistry (IHC) was carried out as previously described,23, 27 and anti‐AKT‐phospho‐Ser473 (AKT‐473P) polyclonal rabbit Ab (1:500; Cell Signaling), anti‐mSREBP1‐SDMA rabbit polyclonal Ab (1:100; Hangzhou Huaan Biotechnology Co., Ltd),23 anti‐PRMT5 monoclonal rabbit Ab (1:500; Abcam), and anti‐Ki67 rabbit polyclonal Ab (1:500; Sigma‐Aldrich) were used for IHC. The signal intensity of IHC was scored blindly and independently by two researchers. The scores for immunostaining frequency (0, 0%; 1, 1%‐9%; 2, 10%‐49%; and 3, 50%‐100%) and intensity (0, negative; 1, weak; 2, moderate; and 3, strong staining) were multiplied to obtain an overall staining score (OSS). The immunostaining frequency and intensity of PRMT5 in the cytoplasm (cPRMT5) and nucleus (nPRMT5) were recorded. An OSS of 0‐3 was deemed low expression, while a score of 4‐9 was considered high expression for AKT‐473P, mSREBP1‐SDMA, cPRMT5, and nPRMT5 staining.
2.3. Cell culture and reagents
A549, NCI‐H1299, and HEK293T cells were purchased from the Type Culture Collection of the Chinese Academy of Sciences. A549, NCI‐H1299, and HEK293T cells were cultured in Ham’s F‐12K medium (Invitrogen), RPMI‐1640 medium (Gibco), and DMEM (Gibco), respectively. In general, media were supplemented with 10% FBS (Gibco), 100 mg/mL penicillin (Gibco), and 100 mg/mL streptomycin sulfate (Gibco). The cells were cultured at 37℃ in 5% CO2.
2.4. Lentivirus production
Annealing and connection of shRNAs were undertaken before insertion into the viral skeleton plasmid pLV‐U6‐EGFP‐Puro. All products were subsequently transfected into HEK293T cells using lentivirus‐packaging reagents (GenePharma), according to the manufacturer’s instructions, and previously verified shRNA‐PRMT5 and shRNA‐NC sequences were used to acquire the virus.23, 28
2.5. Knockdown of PRMT5 in lung cancer cells
Cancer cells were seeded in 6‐well plates for 24 hours prior to transduction. The cells were infected with lentivirus containing human shRNA‐PRMT5 and shRNA‐NC for 48 hours and then selected with puromycin (1 µg/mL; Sigma‐Aldrich). The noninfected cells were then killed. Cell lines with or without stable deletion of PRMT5 were subjected to subsequent experiments.
2.6. Overexpression of PRMT5 in lung cancer cells
Polymerase chain reaction‐amplified and DNA sequencing‐verified human PRMT5 from a cDNA library of HEK293T cells were cloned into HA‐tagged pcDNA4T0. The primers used in the PRMT5 clone were as follows: forward primer, 5′‐CGGGATCCAAATGGCGGCGATGGCG‐3′; and reverse primer: 5′‐CGGAATTCGGACACTTGGCACGCAGGGC‐3′. Lipofectamine 2000 reagent (Invitrogen) was used for transient transfection, following the manufacturer’s instructions.
2.7. Xenograft study
NCI‐H1299 cells with or without PRMT5 knockdown (5 × 106 cells in 100 µL RPMI‐1640) were injected subcutaneously into the left armpit region of BALB/c nude mice (24 [Sh‐NC‐NCI‐H1299 × 12; Sh‐PRMT5‐NCI‐H1299 × 12] male, 5‐week‐old mice, obtained from Shanghai Laboratory Animal Center). SC79 (100 mg/kg, in corn oil to 200 µL) or corn oil (200 µL, as the control) were intraperitoneally injected 30 minutes before and every other day after the cell injection (SC79 [−]/Sh‐NC‐NCI‐H1299 × 6; SC79 [+]/Sh‐NC‐NCI‐H1299 × 6; SC79 [−]/Sh‐PRMT5‐NCI‐H1299 × 6; SC79 [+]/Sh‐PRMT5‐NCI‐H1299 × 6). Tumors appeared on the 10th day after injection, and the tumor size was measured every 3 days. Mice were killed on the 28th day after injection, and the subcutaneous tumors were removed and weighed. The mice were handled in accordance with the guidelines published by the Animal Ethics Committee of the Shanghai Chest Hospital.
2.8. Data analyses
Data were analyzed using GraphPad Prism 6 (GraphPad Software) or SPSS 19.0 (SPSS) software. Paired or unpaired two‐tailed t tests were used to assess the differences between the two groups. A χ2 test was used to determine the correlation between tumor protein expression and patient features. Kaplan‐Meier analysis was used for the survival assay. *P‐value <.05 and **P‐value <.005 indicate statistical significance.
3. RESULTS
3.1. Mature SREBP1‐SDMA is overexpressed and positively correlated with AKT‐473P in lung adenocarcinomas
To determine whether AKT signaling regulates the PRMT5/SREBP1 pathway in lung adenocarcinoma, we analyzed the expression of AKT‐473P29 and PRMT5/SREBP1 pathway‐mediated mSREBP1‐SDMA in 102 pairs of human lung adenocarcinoma tissues and adjacent nontumor tissues by IHC assay. A validated Ab specifically recognizing PRMT5‐catalyzed SDMA‐modified SR (the Arg321 site methylated in mSREBP1a corresponds to Arg297 in mSREBP1c) GEKRTAHNC motif was used for IHC to detect PRMT5‐catalyzed SDMA‐modified mSREBP1.23 As a result, AKT‐473P and mSREBP1‐SDMA were both higher in tumor tissues than in adjacent nontumor tissues (Figures 1A‐D and S1) and were found to be positively correlated in tumors (r = .721, P < .001, Table S1). Moreover, high AKT‐473P and high mSREBP1‐SDMA were both correlated with a relatively late T stage and poor patient outcomes (high mortality rate and short survival time) (P = .017 to <.001; Table 1 and Figure 1E,F). High AKT‐473P was also related to a relatively late N stage (P = .027) and late TNM stage (P < .001) (Table 1). However, the associations of AKT‐473P and mSREBP1‐SDMA with other patient features (eg, age and sex) were not significant (P > .05; Table 1). Furthermore, among patients with different AKT‐473P and mSREBP1‐SDMA coexpression status, those with AKT‐473P and mSREBP1‐SDMA coexpression status had the poorest prognosis (P = .030 to <.001; Figure 1G).
FIGURE 1.
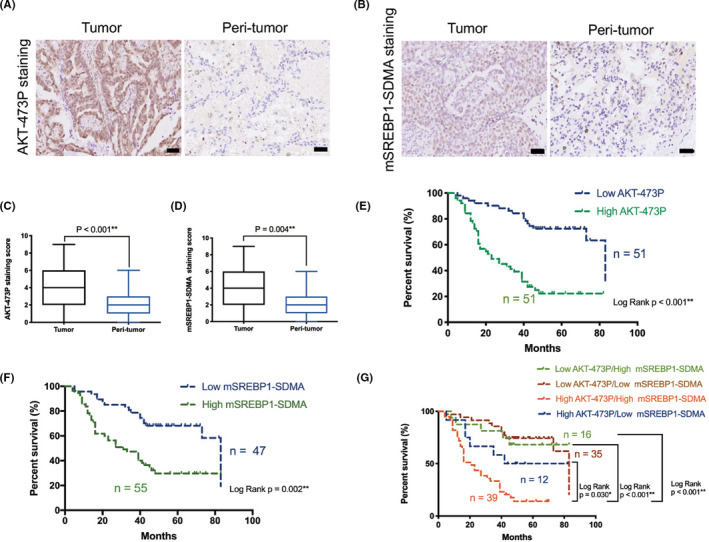
Mature sterol regulatory element‐binding protein 1 (mSREBP1) and symmetric dimethyl arginine (SDMA) overexpressed and positively correlated with Ser473‐phosphorylated protein kinase B (AKT‐473P) in human lung adenocarcinoma tissues. A, B, Representative histopathologic sections of human adenocarcinoma and adjacent non‐tumor lung tissues stained with (A) AKT‐473P and (B) mSREBP1‐SDMA Abs. Scale bar = 50 μm. C, D, Semiquantitative analyses of (C) AKT‐473P and (D) mSREBP1‐SDMA staining scores in human adenocarcinoma and adjacent nontumor lung tissues. E, Kaplan‐Meier analysis of overall survival depending on the AKT‐473P level in lung adenocarcinoma tissues, and the mortality rate and median survival of low and high AKT‐473P expression groups were 33.3% (17/51) and 76.5% (39/51), and 83 and 23 mo, respectively. F, Kaplan‐Meier analysis of overall survival depending on the mSREBP1‐SDMA level in lung adenocarcinoma tissues, and the mortality rate and median survival of low and high mSREBP1‐SDMA expression groups were 38.3% (18/47) and 69.1% (38/55), and 83 and 31 mo, respectively. G, Kaplan‐Meier analysis of overall survival depending on the AKT‐473P and mSREBP1‐SDMA coexpression status. Mortality rate and median survival of the four groups were: 31.2% (5/16) and 66 mo of low AKT‐473P and high mSREBP1‐SDMA expression group; 34.3% (12/35) and 83 mo of low AKT‐473P and low mSREBP1‐SDMA expression group; 50.0% (6/12) and 62 mo of high AKT‐473P and low mSREBP1‐SDMA expression group; and 84.6% (33/39) and 21 mo of AKT‐473P and mSREBP1‐SDMA co‐high expression group, respectively
TABLE 1.
Correlation between mSREBP1‐symmetric dimethyl arginine (SDMA) and Ser473‐phosphorylated protein kinase B (AKT‐473P) levels with other clinicopathological characteristics in 102 lung adenocarcinomas
| Variable | Number | mSREBP1‐SDMA | AKT‐473P | ||||
|---|---|---|---|---|---|---|---|
| Low expression | High expression | P value | Low expression | High expression | P value | ||
| Age (y) | |||||||
| ≤60 | 52 | 20 | 32 | 26 | 26 | ||
| >60 | 50 | 27 | 23 | .116 | 25 | 25 | 1.000 |
| Gender | |||||||
| Female | 46 | 25 | 21 | 27 | 19 | ||
| Male | 56 | 22 | 34 | .129 | 24 | 32 | .111 |
| Smoking history | |||||||
| Yes | 47 | 25 | 22 | 27 | 20 | ||
| No | 55 | 21 | 34 | .104 | 24 | 31 | .164 |
| Tumor location | |||||||
| Left | 31 | 17 | 14 | 20 | 11 | ||
| Right | 71 | 30 | 41 | .241 | 31 | 40 | .053 |
| Pathological grade | |||||||
| I‐II | 59 | 28 | 31 | 26 | 33 | ||
| III | 43 | 19 | 24 | .743 | 25 | 18 | .160 |
| T stage | |||||||
| T1 | 50 | 28 | 22 | 31 | 19 | ||
| T2‐4 | 52 | 19 | 33 | .049* | 20 | 32 | .017* |
| N stage | |||||||
| N0 | 59 | 31 | 28 | 35 | 24 | ||
| N1‐3 | 43 | 16 | 27 | .125 | 16 | 27 | .027* |
| M stage | |||||||
| M0 | 99 | 47 | 52 | 51 | 48 | ||
| M1 | 3 | 0 | 3 | .247 | 0 | 3 | .243 |
| TNM stage | |||||||
| I‐II | 71 | 37 | 34 | 45 | 26 | ||
| III‐IV | 31 | 10 | 21 | .061 | 6 | 25 | <.001** |
| Mortality | |||||||
| Survived | 46 | 29 | 17 | 34 | 12 | ||
| Died | 56 | 18 | 38 | <.001** | 17 | 39 | <.001** |
P <. 05; **P < .005.
The above data revealed a close correlation between AKT signaling and the PRMT5/SREBP1 pathway, and the importance of the PRMT5/SREBP1 pathway in hyperactive AKT‐driven lung adenocarcinoma progression.
3.2. Activation of AKT increases PRMT5‐mediated protein stabilization of mSREBP1
Protein arginine methyltransferase 5‐mediated mSREBP1 SDMA modification is essential for its ubiquitin‐proteasome pathway stabilization.23 The positive correlation between AKT‐473P and mSREBP1‐SDMA in tumors indicates that AKT signaling could influence SDMA modification and the subsequent ubiquitin‐proteasome degradation of mSREBP1. We then used SC79,30 a small molecule that can irreversibly induce AKT phosphorylation and activation by binding to its pleckstrin homology (p‐h) domain, to activate AKT in NCI‐H1299 or A549 cells, and detected the SDMA and ubiquitin modifications, and the expression of protein mSREBP1. As a result, SC79 promoted SDMA modification and expression of mSREBP1 while blocking its ubiquitin modification (Figure 2A,B). However, the precursor of SREBP1 (preSREBP1) was only slightly changed by SC79 (Figure 2B). We then used cycloheximide to inhibit mSREBP1 synthesis in NCI‐H1299 cells with or without PRMT5 deletion, and to detect the degradation rates of mSREBP1. As a result, SC79 hampered mSREBP1 degradation in cells without PRMT5 deletion, whereas in cells with PRMT5 deletion, the degradation of mSREBP1 was only slightly changed and was faster than that in cells without PRMT5 deletion and with SC79 stimulation (Figure 2C,D). We also used MG132 to inhibit ubiquitin‐proteasome degradation of mSREBP1 in NCI‐H1299 cells with or without PRMT5 deletion, with or without SC79 stimulation, and to detect the expression of mSREBP1. As a result, MG132 weakened the upregulation effect of SC79‐induced AKT activation on mSREBP1 and hampered the restraining effect of PRMT5 knockdown on AKT activation‐induced mSREBP1 expression (Figure 2E).
FIGURE 2.
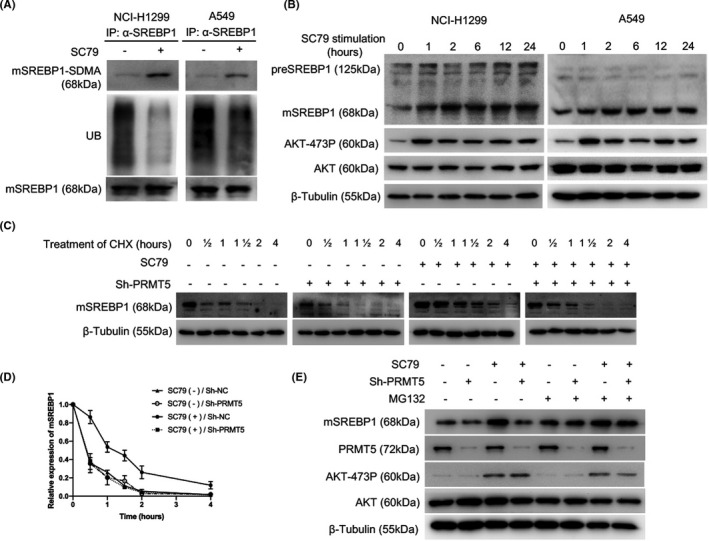
Activation of protein kinase B (AKT) increases the protein arginine methyltransferase 5 (PRMT5)‐mediated protein stabilization of mature sterol regulatory element‐binding protein 1 (mSREBP1). A, After cells were cultured in medium without FBS for 24 h, SC79 (4 µg/mL; Selleck) was added (DMSO as control), and the cells were harvested at 24 h. Lysates of NCI‐H1299 or A549 cells were immunoprecipitated (IP) with anti‐SREBP1 Ab, and mSREBP1 expression levels were then normalized. Indicated Abs were used to analyze post‐translational modification of mSREBP1. UB, ubiquitin modification. B, After cells were cultured in medium without FBS for 24 h, SC79 (4 µg/mL; Selleck) was added (DMSO as control), and the cells were harvested at 0, 1, 2, 6, 12, and 24 h. Lysates of NCI‐H1299 or A549 cells were immunoblotted with Abs against SREBP1, Ser473‐phosphorylated AKT (AKT‐473P), or AKT. β‐Tubulin was used as a loading control. C, NCI‐H1299 cells with or without PRMT5 stable deletion were cultured in medium without FBS for 24 h. SC79 (4 µg/mL) was then added (DMSO as control) for 24 h. Subsequently, cycloheximide (CHX, 100 µg/mL; Sigma‐Aldrich) was added, and the cells were harvested at 0, 0.5, 1, 1.5, 2, and 4 h. Cell lysates were immunoblotted with an anti‐SREBP1 Ab. β‐Tubulin was used as a loading control. D, NCI‐H1299 cells with or without PRMT5 stable deletion were cultured in medium without FBS for 24 hours. SC79 (4 μg/ml) was then added (DMSO as control) for 24 h. Subsequently, CHX (100 μg/ml) was added, and the cells were harvested at 0, ½, 1, 1½, 2, and 4 h. The mSREBP1 degradation rate in the four NCI‐H1299 cell groups was then determined. The mSREBP1 expression level was normalized to the β‐Tubulin level before being normalized to the time = 0 control, and the degradation rates are presented as a percentage. This experiment was repeated for three times, and the mean and SD of degradation rates were shown. E, NCI‐H1299 cells with or without PRMT5 stable deletion were cultured in medium without FBS for 24 h. SC79 (4 µg/mL) was then added (DMSO as control) for 24 h. MG132 (100 µmol/L; Sigma‐Aldrich) was added, and cells were harvested at 2 h. Cell lysates were immunoblotted with Abs against SREBP1, PRMT5, AKT‐473P, or AKT. β‐Tubulin was used as a loading control
Here, we showed that AKT activation increases the SDMA modification of mSREBP1, hampering its ubiquitin‐proteasome pathway degradation by PRMT5.
3.3. Protein arginine methyltransferase 5 participates in AKT activation‐induced mSREBP1 activation at post‐translational level
To determine whether PRMT5 participates in AKT signaling‐mediated mSREBP1 activation in lung cancer cells, we used GSK591, a specific inhibitor of PRMT5, to inhibit PRMT5 enzyme activity in NCI‐H1299 cells with or without SC79 stimulation, and then detected the expression of SREBP1 and its downstream lipogenic enzymes. As expected, AKT activation increased the protein expression of mSREBP1 and its downstream lipogenic enzymes ACLY, FASN, and SCD1, and this process could be reversed by GSK591 (Figure 3A). Consistently, AKT activation‐induced mSREBP1 and lipogenic enzyme expression could also be reversed by silencing PRMT5 in cancer cells (Figure 3B). Both preSREBP1 and PRMT5 were changed little by SC79, and SC79‐induced AKT‐473P was changed little by PRMT5 inhibitor or PRMT5 knockdown in NCI‐H1299 cells (Figure 3A,B). Moreover, the luciferase reporter assay confirmed that SC79 increased the transcriptional activity of SREBP1, whereas PRMT5 knockdown restrained SC79‐induced SREBP1 activation (Figure 3C). At the gene level, SREBF1, ACLY, FASN, and SCD1 mRNA levels were upregulated by SC79 to different degrees, and the SC79‐induced increase in lipogenic gene expression was higher than that of SREBF1. Knockdown of PRMT5 reversed the SC79‐induced increase in ACLY, FASN, and SCD1 mRNA expression, whereas SC79‐induced SREBF1 expression was slightly altered (Figure 3D‐G). PRMT5 expression was also slightly changed by SC79 (Figure 3H).
FIGURE 3.
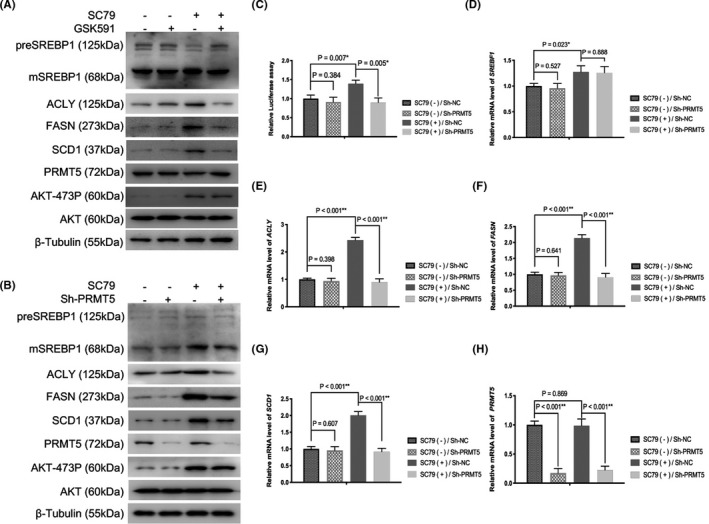
Inhibition of protein arginine methyltransferase 5 (PRMT5) reverses protein kinase B (AKT) activation‐induced mature sterol regulatory element‐binding protein 1 (mSREBP1) activation at the post‐translational level. A, After NCI‐H1299 cells were cultured in medium without FBS for 24 h, SC79 (4 µg/mL) was added for 16 h (DMSO as control), then the cells were treated with or without GSK591 (1 µmol/L; Selleck) for 8 h. Cell lysates were immunoblotted with Abs against SREBP1, ATP citrate lyase (ACLY), fatty acid synthase (FASN), acyl‐CoA desaturase (SCD1), PRMT5, Ser473‐phosphorylated AKT (AKT‐473P), or AKT. β‐Tubulin was used as a loading control. B, NCI‐H1299 cells with or without PRMT5 stable deletion were cultured in medium without FBS for 24 h. SC79 (4 µg/mL) was then added (DMSO as control), and cell lysates were immunoblotted with Abs against SREBP1, ACLY, FASN, SCD1, PRMT5, AKT‐473P, or AKT at 24 h. β‐Tubulin was used as a loading control. C, NCI‐H1299 cells with or without PRMT5 stable deletion were cultured in medium without FBS for 24 h. SC79 (4 µg/mL) was then added (DMSO as control; Sh‐NC) for 24 h. Luciferase activities were then analyzed. D‐H, NCI‐H1299 cells with or without PRMT5 stable deletion were cultured in medium without FBS for 24 h. SC79 (4 µg/mL) was then added (DMSO as control; Sh‐NC) for 24 h. mRNA levels of indicated genes were then analyzed. D, SREBF1. E, ACLY. F, FASN. G, SCD1. H, PRMT5
The AKT inhibitor MK2206‐2HCL was then used in NCI‐H1299 cells with or without exogenous PRMT5 expression to further investigate the regulatory effect of AKT signaling on the PRMT5/SREBP1 pathway. At the protein level, MK2206‐2HCL decreased the expression of mSREBP1, ACLY, FASN, and SCD1, while almost unchanging preSREBP1 and PRMT5 levels in cells with or without exogenous PRMT5 expression. Exogenous PRMT5 increased the expression of mSREBP1, ACLY, FASN, and SCD1; however, preSREBP1 in cells without MK2206‐2HCL treatment were almost unchanged. In cells treated with MK2206‐2HCL, the effect of exogenous PRMT5 on mSREBP1 and lipogenic enzymes was blocked (Figure S2A). The luciferase reporter assay showed that MK2206‐2HCL decreased the transcriptional activity of SREBP1 in cells with or without exogenous PRMT5 expression; exogenous PRMT5 increased the transcriptional activity of SREBP1 in cells without MK2206‐2HCL treatment, whereas in cells treated with MK2206‐2HCL, the effect of exogenous PRMT5 on SREBP1 activation was blocked (Figure S2B). At the gene level, MK2206‐2HCL decreased the expression of SREBF1, ACLY, FASN, and SCD1 to different degrees in cells with or without exogenous PRMT5 expression, whereas PRMT5 expression was unchanged. The effect of MK2206‐2HCL on SREBF1 was weaker than on lipogenic genes; exogenous PRMT5 increased the expression of ACLY, FASN, and SCD1, whereas SREBF1 expression was unaffected in cells without MK2206‐2HCL treatment. In cells treated with MK2206‐2HCL, the effect of exogenous PRMT5 on lipogenic enzymes was blocked (Figure S2C‐G).
Collectively, AKT activation activates SREBP1 at both the transcriptional and post‐transcriptional levels, and PRMT5 plays a crucial role in AKT activation‐induced mSREBP1 activation at the post‐transcriptional level.
3.4. Activation of AKT promotes interaction between PRMT5 and mSREBP1 by changing PRMT5 subcellular localization
As described above, PRMT5 participates in AKT activation‐induced mSREBP1 activation at the post‐transcriptional level. The interaction of PRMT5 and mSREBP1 is the first step in the PRMT5/SREBP1 pathway of mSREBP1 activation. We investigated whether AKT signaling influences the interaction between PRMT5 and mSREBP1. The immunoprecipitation (IP) assay confirmed that SC79 upregulated the interaction between both endogenous and exogenous PRMT5 with endogenous mSREBP1 in NCI‐H1299 cells (Figures 4A and S3A). The proximity ligation assay also revealed the SC79‐induced increase in the interaction between endogenous and exogenous PRMT5 with endogenous mSREBP1 (Figures 4B and S3B). It has been reported that PRMT5 in the cytoplasm, but not in the nucleus, is essential for lung cancer malignancy,31, 32 which led us to examine the changes in the subcellular localization of PRMT5. Interestingly, SC79 increased both endogenous and exogenous cPRMT5 while decreasing endogenous and exogenous nPRMT5 in NCI‐H1299 cells; meanwhile, cytoplasmic and nuclear mSREBP1 levels were both upregulated (Figures 4C and S3C). Moreover, the IP assay revealed that, compared with cPRMT5 in cells without SC79 stimulation or nPRMT5 in cells with or without SC79 stimulation, cPRMT5 in cells with SC79 stimulation showed the strongest mSREBP1‐binding ability (Figures 4D and S3D).
FIGURE 4.
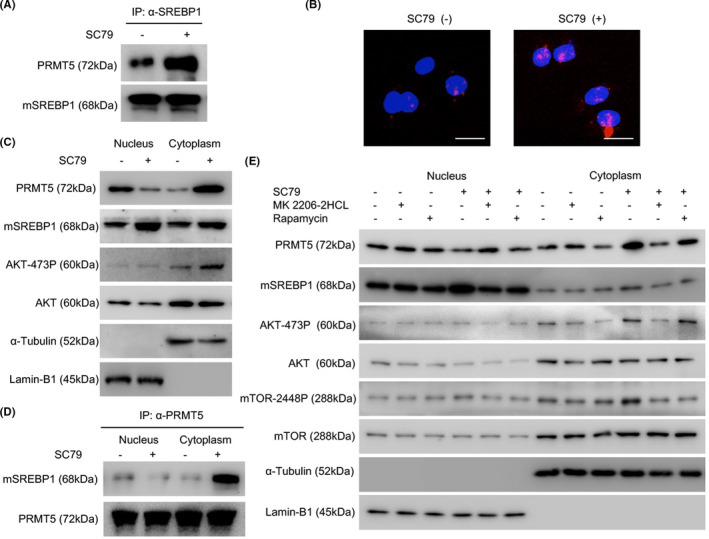
Activation of protein kinase B (AKT) promotes the interaction between endogenous protein arginine methyltransferase 5 (PRMT5) and mature sterol regulatory element‐binding protein 1 (mSREBP1) by changing PRMT5 subcellular localization. A, After culture of NCI‐H1299 cells in medium without FBS for 24 h, SC79 (4 µg/mL) was added (DMSO as control), and the cells were harvested at 24 h. Cell lysates were immunoprecipitated with anti‐SREBP1 Ab, and the mSREBP1 expression level was then normalized. An anti‐PRMT5 Ab was used to analyze the interaction between mSREBP1 and PRMT5. B, After the culture of NCI‐H1299 cells in medium without FBS for 24 h, SC79 (4 µg/mL) was added (DMSO as control), and a proximity ligation assay was carried out at 24 h. Scale bar = 25 µm. C, After culture of NCI‐H1299 cells in medium without FBS for 24 h, SC79 (4 µg/mL) was added (DMSO as control) for 24 h. The cytoplasmic and nuclear fractions were then separated and immunoblotted with Abs against SREBP1, PRMT5, Ser473‐phosphorylated AKT (AKT‐473P), or AKT. α‐Tubulin was used as a cytoplasmic fraction loading control while Lamin‐B1 served as the nuclear fraction loading control. D, After culture of NCI‐H1299 cells in medium without FBS for 24 h, SC79 (4 µg/mL) was added (DMSO as control) for 24 h. The cytoplasmic and nuclear fractions were then separated and immunoprecipitated with anti‐PRMT5 Ab, and the PRMT5 expression levels were then normalized. The anti‐SREBP1 Ab was used to analyze the mSREBP1 and PRMT5 interaction in the cytoplasm and nucleus. E, After culture of NCI‐H1299 cells in medium without FBS for 24 h, SC79 (4 µg/mL) was added (DMSO as control) for 12 h, MK2206‐2HCL (100 nmol/L; Selleck) or rapamycin (1 µmol/L; Selleck) was then added (DMSO as control) for 24 h. The cytoplasmic and nuclear fractions were then separated and immunoblotted with Abs against SREBP1, PRMT5, mTOR, mTOR‐2448P, AKT‐473P, or AKT. α‐Tubulin was used as a cytoplasmic fraction loading control. Lamin‐B1 served as the nuclear fraction loading control
The AKT inhibitor MK2206‐2HCL or mTOR inhibitor rapamycin was added to inhibit AKT or its downstream mTOR activation (detected by an Ab against mTOR‐phospho‐Ser2448 [mTOR‐2448P]),33 respectively, in NCI‐H1299 cells, and to investigate whether SC79‐induced AKT activation was the mechanism underlying SC79‐triggered PRMT5 subcellular relocalization in lung cancer cells. As a result, MK2206‐2HCL and rapamycin both restrained SC79‐induced subcellular relocalization of PRMT5 (Figure 4E), indicating that it is SC79‐induced AKT/mTOR pathway activation, rather than other pathways, that regulate PRMT5 subcellular localization.
Taken together, we showed that AKT activation promotes the transport of nPRMT5 to the cytoplasm. The transported cPRMT5 induced by AKT signaling shows a strong mSREBP1‐binding ability, promoting the interaction between PRMT5 and mSREBP1 in lung adenocarcinoma cells.
3.5. Cytoplasmic PRMT5 is positively correlated with AKT‐473P and mSREBP1‐SDMA in lung adenocarcinomas
The upregulation of AKT activation on PRMT5 cytoplasmic localization led us to examine the correlations between AKT‐473P and mSREBP1‐SDMA with cPRMT5 and nPRMT5 (Figure 5A) in human lung adenocarcinomas by IHC assay. There was a positive correlation between AKT‐473P and cPRMT5 levels in lung adenocarcinomas (r = .353, P = .001), whereas the correlation between AKT‐473P and nPRMT5 was not significant (P = .472) (Tables S2 and S3). Similarly, mSREBP1‐SDMA was positively correlated with cPRMT5 (r = .313, P < .001) rather than with nPRMT5 (P = .454) in lung adenocarcinomas (Tables S4 and S5). Moreover, high cPRMT5 expression was related to a relatively poor pathological grade, late N stage, and late TNM stage of tumors (P = .042 to <.001), whereas high nPRMT5 expression was related to a relatively better pathological grade of lung adenocarcinoma (P = .035) (Table 2). However, the associations between cPRMT5 or nPRMT5 and other patient features (eg, age and sex) were not significant (P > .05; Table 2). Furthermore, high expression of cPRMT5 (P < .001) rather than nPRMT5 (P > .05) was correlated with poor outcomes (high mortality rate and short survival time) in patients with lung adenocarcinoma (Figure S5B,C and Table 2).
FIGURE 5.
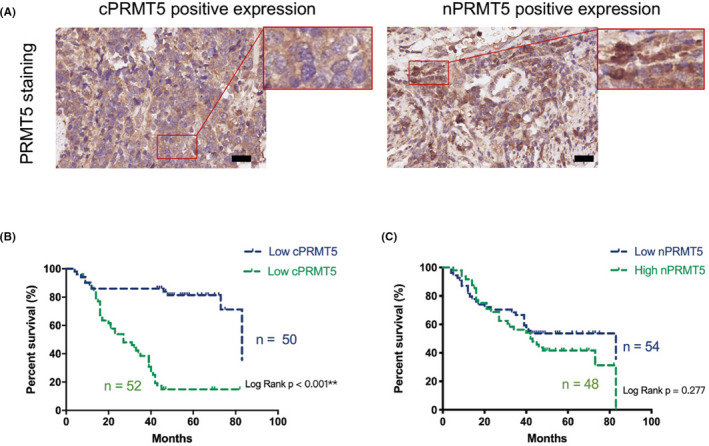
High expression of cytoplasmic protein arginine methyltransferase 5 (cPRMT5) in lung adenocarcinoma is associated with poor patient prognosis. A, Representative histopathologic sections of human adenocarcinoma with high expression of (left) cPRMT5 and (right) nuclear PRMT5 (nPRMT5). Scale bar = 50 μm. B, Kaplan‐Meier analysis of overall survival depending on the cPRMT5 level in lung adenocarcinoma tissues, and the mortality rate and median survival of low and high cPRMT5 expression groups were 24.0% (12/50) and 84.6% (44/52), and 83 and 27 mo, respectively. C, Kaplan‐Meier analysis of overall survival depending on the nPRMT5 level in lung adenocarcinoma tissues, and the mortality rate and median survival of low and high nPRMT5 expression groups were 48.1% (26/54) and 62.5% (30/48), and 83 and 42.5 mo, respectively
TABLE 2.
Correlation between levels of protein arginine methyltransferase 5 in the cytoplasm (cPRMT5) and nucleus (nPRMT5) with other clinicopathological characteristics in 102 lung adenocarcinomas
| Variable | Number | cPRMT5 | nPRMT5 | ||||
|---|---|---|---|---|---|---|---|
| Low expression | High expression | P‐value | Low expression | High expression | P‐value | ||
| Age (y) | |||||||
| ≤60 | 52 | 22 | 30 | 27 | 25 | ||
| >60 | 50 | 28 | 22 | .167 | 27 | 23 | .843 |
| Gender | |||||||
| Female | 46 | 27 | 19 | 27 | 19 | ||
| Male | 56 | 23 | 33 | .076 | 27 | 29 | .291 |
| Smoking history | |||||||
| Yes | 47 | 27 | 20 | 29 | 18 | ||
| No | 55 | 23 | 32 | .116 | 25 | 30 | .101 |
| Tumor location | |||||||
| Left | 31 | 18 | 13 | 16 | 15 | ||
| Right | 71 | 32 | 39 | .227 | 38 | 33 | .859 |
| Pathological grade | |||||||
| I‐II | 59 | 34 | 25 | 26 | 33 | ||
| III | 43 | 16 | 27 | .042* | 28 | 15 | .035* |
| T stage | |||||||
| T1 | 50 | 24 | 26 | 25 | 25 | ||
| T2‐4 | 52 | 26 | 26 | .840 | 29 | 23 | .560 |
| N stage | |||||||
| N0 | 59 | 37 | 22 | 35 | 24 | ||
| N1‐3 | 43 | 13 | 30 | .001** | 19 | 24 | .130 |
| M stage | |||||||
| M0 | 99 | 50 | 49 | 52 | 47 | ||
| M1 | 3 | 0 | 3 | .243 | 2 | 1 | 1.000 |
| TNM stage | |||||||
| I‐II | 71 | 45 | 26 | 38 | 33 | ||
| III‐IV | 31 | 5 | 26 | <.001** | 16 | 15 | .859 |
| Mortality | |||||||
| Survived | 46 | 38 | 8 | 28 | 18 | ||
| Died | 56 | 12 | 44 | <.001** | 26 | 30 | .146 |
P <. 05; **P <.005.
The above data indicate a close correlation between AKT activation‐induced PRMT5 cytoplasmic localization with mSREBP1 SDMA modification and the malignant progression of lung adenocarcinoma.
3.6. Activation of AKT induces lipid production in lung adenocarcinoma cells through PRMT5
The adjusting role of AKT signaling in the PRMT5/SREBP1 pathway led us to examine whether AKT signaling regulates lipid production through PRMT5. We found that SC79 elevated the levels of multiple fatty acid types (eg, C16:0, C16:1n9, and C16:1n7) in NCI‐H1299 cells, which could be reversed by PRMT5 knockdown (Figure 6A). SC79 also increased the triglyceride (TG) level in NCI‐H1299 cells, and this increase could also be reversed by PRMT5 knockdown (Figure 6B). Consistently, oil red O staining showed that SC79 increased lipid accumulation in NCI‐H1299 cells, and this process could be restrained by PRMT5 knockdown (Figure 6C). In addition, the AKT inhibitor MK2206‐2HCL inhibited fatty acid (eg, C16:0, C16:1n9, and C16:1n7) synthesis, TG levels, and lipid accumulation in NCI‐H1299 cells with or without exogenous PRMT5 expression. Exogenous PRMT5 increased fatty acid synthesis, TG levels, and lipid accumulation in NCI‐H1299 cells without MK2206‐2HCL treatment, whereas in NCI‐H1299 cells treated with MK2206‐2HCL, the upregulation effect of exogenous PRMT5 on lipid synthesis was blocked (Figure S4).
FIGURE 6.
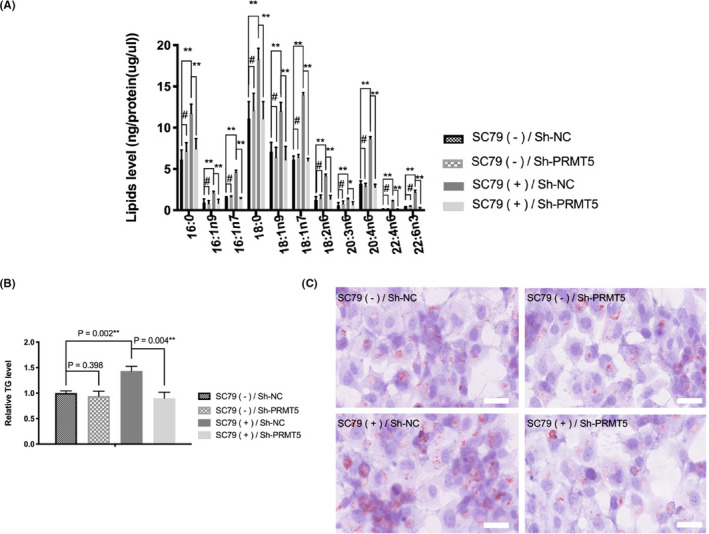
Protein arginine methyltransferase 5 (PRMT5) knockdown reverses protein kinase B (AKT) activation‐induced lipid production in lung adenocarcinoma cells. NCI‐H1299 cells with or without PRMT5 stable deletion were cultured in medium without FBS for 24 h. SC79 (4 µg/mL) was added (DMSO as control [Sh‐NC]) for 36 h, and intracellular lipids in the NCI‐H1299 cells were analyzed. A, Lipid metabolites were analyzed by gas chromatography‐mass spectrometry, and results were normalized by total protein levels. # P > .05; *P < .05; **P < .005. B, Triglyceride (TG) levels were quantitated and normalized by total protein levels and levels in control cells. C, Representative oil red O staining images. Scale bar = 50 µm
The above data proved that AKT activation induces lipid production through PRMT5 in lung adenocarcinoma cells.
3.7. Activation of AKT induces lung adenocarcinoma cells growth through PRMT5
A growth assay was also undertaken to verify the dependency of AKT activation‐induced cancer cell growth on PRMT5. In vitro, SC79 enhanced the proliferation, tumorigenicity, and motility of NCI‐H1299 cells in vitro, whereas PRMT5 knockdown reversed the SC79‐induced growth advantage of cancer cells (Figures 7A‐C and S5). Moreover, the AKT inhibitor MK2206‐2HCL inhibited the proliferation, tumorigenicity, and motility of NCI‐H1299 cells with or without exogenous PRMT5 expression. Exogenous PRMT5 promoted the proliferation, tumorigenicity, and motility of cancer cells without MK2206‐2HCL treatment, whereas in NCI‐H1299 cells treated with MK2206‐2HCL, the effect of exogenous PRMT5 on cell growth was blocked (Figure S6A‐C). In vivo, tumors with SC79‐stimulated AKT hyperactivation grew faster and larger in volume and were heavier than tumors without SC79 stimulation, whereas SC79 stimulation resulted in comparable tumor volumes and weights when PRMT5 was silenced in cancer cells (Figures 7D‐F and S7). We also confirmed that tumors with SC79 stimulation were more proliferative than tumors without SC79 stimulation, whereas PRMT5 knockdown almost completely blocked the growth advantage rendered by SC79 (Figure 7G), based on Ki‐67 IHC.34 Oil red O staining of tumors was also carried out, and SC79 increased lipid accumulation in tumors, whereas PRMT5 knockdown inhibited SC79‐induced intracellular lipid accumulation (Figure 7G). Taken together, we proved that AKT activation induces the growth of lung adenocarcinoma cells through PRMT5 in vivo and in vitro.
FIGURE 7.
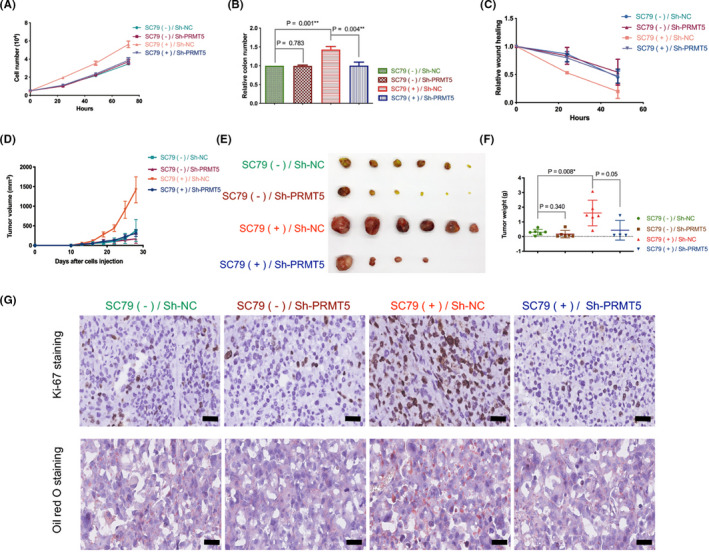
Protein arginine methyltransferase 5 (PRMT5) knockdown reverses protein kinase B (AKT) activation‐induced growth of lung adenocarcinoma cells. A, NCI‐H1299 cells with or without PRMT5 stable deletion were cultured in medium without FBS for 24 h. SC79 (4 µg/mL) was then added (DMSO as control; Sh‐NC), and the number of cells was determined at 0, 24, 48, and 72 h. B, NCI‐H1299 cells with or without PRMT5 stable deletion were cultured in medium without FBS for 24 h. SC79 (4 µg/mL) was then added (DMSO as control), and cell colony formation assays were carried out at 14 days. The relative number of colonies was calculated, and the number of the control group was designated as 1. C, NCI‐H1299 cells with or without PRMT5 stable deletion were cultured in medium without FBS for 24 h. SC79 (4 µg/mL) was then added (DMSO as control), and cell wound‐healing assays were carried out at 0, 24, 48 h. Relative wound healing was normalized by the 0 h point of each group, which was designated 1. D‐G, Nude mice were injected with NCI‐H1299 cells with or without PRMT5 stable deletion and intraperitoneally injected with or without SC79 (100 mg/kg). D, Growth curve of in vivo tumor volume. E, Image of tumors isolated from nude mice. F, Weight of tumors when mice were killed. G, Representative images of Ki‐67 and oil red O staining of tumor samples. Scale bar = 50 μm
4. DISCUSSION
Hyperactivation of AKT is a crucial molecular characteristic of lung cancer, existing in approximately half of lung cancer patients, and is associated with aggressive tumor behavior and poor patient outcomes.35, 36 This was also observed in this study. Protein kinase B can be activated by multiple oncogenic events through a PI3K‐associated phosphorylation cascade to promote cancer cell proliferation and survival through oncogenic signaling as well as metabolic reprogramming (eg, aerobic glycolysis and de novo lipogenesis).37, 38 In this study, we sought to explore the mechanisms underlying de novo lipogenesis in hyperactive AKT‐driven lung cancer and to identify new therapeutic targets for hyperactive AKT‐driven lung cancer. Finally, we demonstrated that the AKT/PRMT5/SREBP1 axis is involved in de novo lipogenesis and growth of lung adenocarcinoma, and proposed that cPRMT5 is a promising therapeutic target for hyperactive AKT‐driven lung cancer.
The PRMT5/SREBP1 pathway is a newly discovered oncogenic signaling pathway, and mSREBP1‐SDMA is a newly identified prognostic biomarker for malignant tumor.23, 25 Thus far, it is unclear whether the PRMT5/SREBP1 pathway is regulated by AKT signaling in tumors. In this study, we found that mSREBP1‐SDMA was overexpressed and associated with large tumor size and poor patient outcomes in lung adenocarcinoma. More importantly, mSREBP1‐SDMA was positively correlated with AKT‐473P expression in lung adenocarcinoma. Furthermore, patients with AKT‐473P and mSREBP1‐SDMA coexpression showed the worst prognosis. Together with our in vitro data showing that AKT activation elevated SDMA modification of mSREBP1 in lung cancer cells, it is reasonable to believe that AKT signaling regulates the PRMT5/SREBP1 pathway in lung adenocarcinoma, and that PRMT5‐mediated SDMA modification of mSREBP1 is essential for the progression of hyperactive AKT‐driven lung cancer.
As the major adjustor for de novo lipogenesis, SREBP1 is translated as a precursor protein after gene transcription, and is then cleaved into the mature form mSREBP1 in the endoplasmic reticulum (ER) to trigger downstream lipid enzyme gene transcription, finally degrading through the ubiquitin‐proteasome pathway.39 Sterol regulatory element‐binding protein 1 can be regulated at transcriptional, translational, and post‐translational cleavage and degradation levels, and can be controlled by multiple signaling pathways (eg, glucose, insulin, sirtuin 1, insig‐1, and polyunsaturated fatty acids), including AKT signaling.16, 17, 22, 40, 41, 42, 43, 44 However, although there is evidence that AKT signaling regulates lipid synthesis through SREBP1 in several malignancies, the mechanisms by which AKT signaling regulates SREBP1 activity seem to vary among tumors. For example, AKT activation hampers the post‐translational degradation of mSREBP1 in melanoma,16 whereas in breast cancer, AKT activation upregulates SREBF1 at the transcriptional level.17 Activation of AKT also promotes post‐translational cleavage of preSREBP1 in glioblastoma.45 Recently, AKT activation was reported to induce SREBP1 activation in lung adenocarcinoma cells, but the mechanisms underlying this process remain elusive.15 Consistently, our data confirmed that AKT activation promotes mSREBP1 expression and activation in lung adenocarcinoma cells. Moreover, in addition to previously reported findings, our data indicated that, in lung adenocarcinoma, AKT hyperactivation not only upregulates SREBF1 gene expression at the transcriptional level, but also hampers the ubiquitin‐proteasome pathway degradation of mSREBP1 at the post‐translational level through PRMT5, inducing mSREBP1 overactivation in cancer cells, resulting in lipid synthesis and uncontrolled growth of cancer cells.
As a well‐proven oncogenic enzyme, PRMT5 is universally present in the nucleus and cytoplasm of cancer cells, contributing to tumorigenesis by directly catalyzing the SDMA modification of oncogenic signaling. We previously reported that PRMT5 is also an SREBP1 regulator, and therefore is involved in de novo lipogenesis of cancer cells,23 which was also observed in this study. However, recent evidence indicates that cPRMT5 rather than nPRMT5 associated with poor differentiation and poor outcomes in non‐small‐cell lung cancer (NSCLC).31, 32 To date, the mechanisms affecting PRMT5 subcellular localization in lung cancer remain unknown. Significantly, in this study, we found that in lung cancer cells, although AKT activation had little effect on the gene and protein expression of PRMT5, it promoted the transport of nPRMT5 to the cytoplasm. Furthermore, AKT activation‐induced cPRMT5 showed strong mSREBP1‐binding and SDMA modification abilities. We also confirmed that AKT inhibition blocked PRMT5‐induced mSREBP1 activation, lipid synthesis, and growth of lung adenocarcinoma cells, but did not change total expression of PRMT5. Moreover, the IHC assay revealed positive correlations between AKT‐473P and mSREBP1‐SDMA with cPRMT5 in human lung adenocarcinomas. Notably, similar to previous reports that cPRMT5 was associated with poor differentiation and patient outcomes in NSCLC,31, 32 we found that high cPRMT5 expression was associated with a relatively poor pathological grade, late N stage, and late TNM stage of lung adenocarcinoma, and a higher mortality rate and shorter survival time of lung adenocarcinoma patients. We also observed a correlation between high nPRMT5 expression and a relatively better pathological grade of lung adenocarcinoma, which is similar to the previously reported correlation between high nPRMT5 expression and low‐grade lung cancer subtypes.31, 32 Together, AKT activation could activate PRMT5 by inducing its transfer from the nucleus to the cytoplasm at the post‐transcriptional level, enhancing the interaction between PRMT5 and mSREBP1 in the cytoplasm, resulting in mSREBP1 overactivation in lung adenocarcinoma cells. Our data also indicate that cPRMT5 could play a crucial role in lung adenocarcinoma development.
It has been shown that PRMT5 promotes AKT activation by directly interacting with PRMT5 in lung cancer cells, and the colocalization of PRMT5 with AKT‐473P in the nucleus has also been observed.46 In this study, we used SC79,30 an AKT activator that directly binds to its p‐h domain to trigger irreversible AKT phosphorylation, to induce AKT overactivation in A549 and NCI‐H1299 cells. Unlike the basal AKT‐473P of lung cancer cells, which might be restrained by PRMT5 knockdown or inhibition,46 the SC79‐induced AKT‐473P was stable and almost unchanged by PRMT5 knockdown or inhibition. As a result, we showed the regulatory effect of AKT activation on PRMT5 cytoplasmic localization and activation in lung adenocarcinoma cells. Combining our data with data from Zhang et al,46 there may be a positive AKT/PRMT5 feedback loop in lung adenocarcinoma, similar to that reported in lymphoma.47 However, the mechanisms underlying the regulation of AKT activation on PRMT5 in this feedback loop in lymphoma and lung adenocarcinoma are different: in lymphoma,47 AKT activation directly promotes PRMT5 gene expression at the transcriptional level, whereas in lung adenocarcinoma, AKT activation promotes the localization of PRMT5 in the cytoplasm to enhance its enzyme activity at the post‐translational level. Collectively, there could be a positive AKT/PRMT5 feedback loop in cancer development, and the mechanisms underlying this feedback loop in different cancer types might also be different. However, it is still unknown whether PRMT5 interacts with AKT‐473P in the cytoplasm; the interaction of PRMT5 and AKT‐473P in the cytoplasm, the biological effects of cPRMT5 and nPRMT5, and the mechanisms underlying Akt signaling‐mediated PRMT5 subcellular relocalization in lung adenocarcinoma cells merit further research.
Tumor growth involves uncontrolled cell proliferation and metabolic reprogramming, including de novo lipogenesis.48, 49 In contrast to the recycled lipids in normal cells, newly synthesized lipids in cancer cells can promote tumor growth by providing biological membranous blocks, metabolism substrates, and protein modification donors.50 Recently, an increase in lipid synthesis in lung adenocarcinoma has been observed,13 but the underlying mechanisms remain elusive. In this study, we found that AKT signaling contributes to de novo lipogenesis in lung adenocarcinoma cells through PRMT5/SREBP1 pathway mSREBP1 activation, not only strengthening the importance of AKT hyperactivation for metabolic reprogramming in lung adenocarcinoma, but also promoting new mechanisms underlying de novo lipogenesis in hyperactive AKT‐driven lung adenocarcinoma.
As a well‐known oncogenic tumor driver, AKT hyperactivation has been shown to be a promising therapeutic target for cancer treatment. However, clinical trials of AKT inhibitors have recently been limited because of the compensatory resistance of tumors to AKT inhibitors.5 Meanwhile, PRMT5 has recently received significant attention as a safe and effective antitumor target in cancer research, and certain PRMT5 inhibitors (GSK3326595 and JNJ‐64619178) have already entered clinical trials.51, 52 In this study, we showed that PRMT5 knockdown could reverse AKT activation‐induced lipid synthesis and growth advantage of lung adenocarcinoma cells both in vitro and in vivo, suggesting that PRMT5 is a promising therapeutic target for hyperactive AKT‐driven lung adenocarcinoma, supporting the development of clinical trials investigating the therapeutic effect of PRMT5 inhibitors on hyperactive AKT‐driven lung cancer. Our data also indicate that the cytoplasmic localization of PRMT5 is crucial for lung cancer development; therefore, the exploration of new drugs targeting cPRMT5 or hampering PRMT5 cytoplasmic localization is also promising for the treatment of hyperactive AKT‐driven lung cancer.
Overall, we defined an AKT/PRMT5/SREBP1 axis involved in lung adenocarcinoma de novo lipogenesis and progression (Figure 8). The effects of AKT signaling on SREBP1 begin at the gene level and promote translation and subsequent cleavage of the preSREBP1 in the ER, leading to the formation of mSREBP1, while AKT activation promotes nuclear PRMT5 transport to the cytoplasm. Cytoplasmic PRMT5 induced by AKT signaling shows strong mSREBP1‐binding and SDMA modification ability, inhibiting ubiquitin‐proteasome pathway degradation of mSREBP1; as a result, mSREBP1 is stabilized and shows increased activity in cancer cells, triggering de novo lipogenesis and malignant growth of lung adenocarcinoma cells. Our data also support that cPRMT5 is a potential therapeutic target for hyperactive AKT‐driven lung adenocarcinoma.
FIGURE 8.
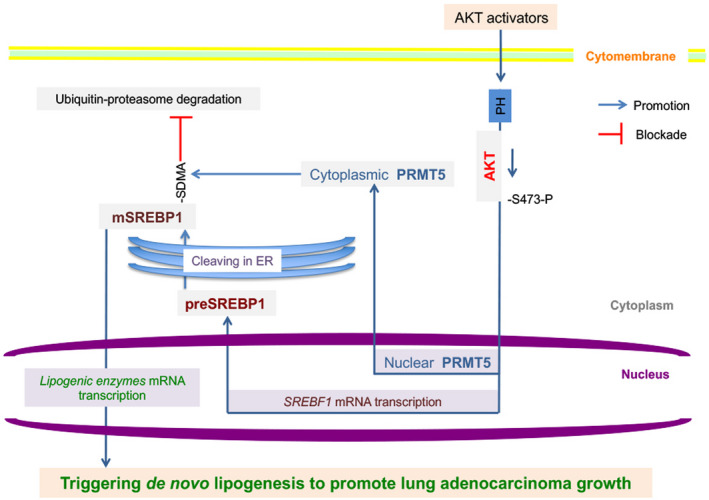
Involvement of the protein kinase B (AKT)/protein arginine methyltransferase 5 (PRMT5)/sterol regulatory element‐binding protein 1 (SREBP1) axis in lung cancer de novo lipogenesis and progression. ER, endoplasmic reticulum; PH, p‐h domain of AKT; SDMA, symmetric dimethyl arginine
DISCLOSURE
The authors declare that they have no competing interest.
Supporting information
Fig S1‐S7
Table S1‐S7
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Youth Medical Talents‐Medical Imaging Practitioners Program (Grant No. SHWRS(2020)_087), the National Natural Science Foundation of China (Grant Nos. 81602415, 81830052, and 81903065), the Natural Science Foundation of Shanghai (Grant No. 18ZR1435200), the Nurture projects for basic research of Shanghai Chest Hospital (Grant Nos. 2020YNJCM07, 2020YNJCQ06, and 2020YNJCQ11), the Shanghai Sailing Program (Grant No. 20YF1444500), and the special project of integrated traditional Chinese and Western medicine in general hospital of Shanghai Health Committee (Grant No. ZHYY‐ZXYJHZX‐202023). We also thank Li Zhao, Ruixue Zhang, and Mengqin Shen for technical assistance.
Liu L, Yan H, Ruan M, et al. An AKT/PRMT5/SREBP1 axis in lung adenocarcinoma regulates de novo lipogenesis and tumor growth. Cancer Sci. 2021;112:3083–3098. 10.1111/cas.14988
Liu Liu and Hui Yan contributed equally to this work.
Contributor Information
Gang Huang, Email: huanggang0311@163.com.
Wenhui Xie, Email: shxknuclear@126.com.
REFERENCES
- 1.Miller KD, Nogueira L, Mariotto AB, et al. Cancer treatment and survivorship statistics, 2019. CA Cancer J Clin. 2019;69:363‐385. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7‐30. [DOI] [PubMed] [Google Scholar]
- 3.Fumarola C, Bonelli MA, Petronini PG, Alfieri RR. Targeting PI3K/AKT/mTOR pathway in non small cell lung cancer. Biochem Pharmacol. 2014;90:197‐207. [DOI] [PubMed] [Google Scholar]
- 4.Song M, Bode AM, Dong Z, Lee MH. AKT as a therapeutic target for cancer. Cancer Res. 2019;79:1019‐1031. [DOI] [PubMed] [Google Scholar]
- 5.Shariati M, Meric‐Bernstam F. Targeting AKT for cancer therapy. Expert Opin Investig Drugs. 2019;28:977‐988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang J, Xu‐Monette ZY, Jabbar KJ, et al. AKT hyperactivation and the potential of AKT‐targeted therapy in diffuse large B‐cell lymphoma. Am J Pathol. 2017;187:1700‐1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stoiber K, Naglo O, Pernpeintner C, et al. Targeting de novo lipogenesis as a novel approach in anti‐cancer therapy. Br J Cancer. 2018;118:43‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DeBose‐Boyd RA, Ye J. SREBPs in lipid metabolism, insulin signaling, and beyond. Trends Biochem Sci. 2018;43:358‐368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng C, Geng F, Cheng X, Guo D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun (Lond). 2018;38:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perone Y, Farrugia AJ, Rodriguez‐Meira A, et al. SREBP1 drives Keratin‐80‐dependent cytoskeletal changes and invasive behavior in endocrine‐resistant ERalpha breast cancer. Nat Commun. 2019;10:2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhai D, Cui C, Xie L, Cai L, Yu J. Sterol regulatory element‐binding protein 1 cooperates with c‐Myc to promote epithelial‐mesenchymal transition in colorectal cancer. Oncol Lett. 2018;15:5959‐5965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Min X, Wen J, Zhao L, et al. Role of hepatoma‐derived growth factor in promoting de novo lipogenesis and tumorigenesis in hepatocellular carcinoma. Mol Oncol. 2018;12:1480‐1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vriens K, Christen S, Parik S, et al. Evidence for an alternative fatty acid desaturation pathway increasing cancer plasticity. Nature. 2019;566:403‐406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li J, Yan H, Zhao L, et al. Inhibition of SREBP increases gefitinib sensitivity in non‐small cell lung cancer cells. Oncotarget. 2016;7:52392‐52403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang B, Wu J, Guo P, et al. Down‐regulation of SREBP via PI3K/AKT/mTOR pathway inhibits the proliferation and invasion of non‐small‐cell lung cancer cells. Onco Targets Ther. 2020;13:8951‐8961. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 16.Yamauchi Y, Furukawa K, Hamamura K, Furukawa K. Positive feedback loop between PI3K‐Akt‐mTORC1 signaling and the lipogenic pathway boosts Akt signaling: induction of the lipogenic pathway by a melanoma antigen. Cancer Res. 2011;71:4989‐4997. [DOI] [PubMed] [Google Scholar]
- 17.Furuta E, Pai SK, Zhan R, et al. Fatty acid synthase gene is up‐regulated by hypoxia via activation of Akt and sterol regulatory element binding protein‐1. Cancer Res. 2008;68:1003‐1011. [DOI] [PubMed] [Google Scholar]
- 18.Guo D, Reinitz F, Youssef M, et al. An LXR agonist promotes glioblastoma cell death through inhibition of an EGFR/AKT/SREBP‐1/LDLR‐dependent pathway. Cancer Discov. 2011;1:442‐456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang LH, Chung HY, Su HM. Docosahexaenoic acid reduces sterol regulatory element binding protein‐1 and fatty acid synthase expression and inhibits cell proliferation by inhibiting pAkt signaling in a human breast cancer MCF‐7 cell line. BMC Cancer. 2017;17:890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bai PS, Xia N, Sun H, Kong Y. Pleiotrophin, a target of miR‐384, promotes proliferation, metastasis and lipogenesis in HBV‐related hepatocellular carcinoma. J Cell Mol Med. 2017;21:3023‐3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ru P, Hu P, Geng F, et al. Feedback loop regulation of SCAP/SREBP‐1 by miR‐29 modulates egfr signaling‐driven glioblastoma growth. Cell Rep. 2016;16:1527‐1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng C, Ru P, Geng F, et al. Glucose‐mediated N‐glycosylation of SCAP is essential for SREBP‐1 activation and tumor growth. Cancer Cell. 2015;28:569‐581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu L, Zhao X, Zhao L, et al. Arginine methylation of SREBP1a via PRMT5 promotes de novo lipogenesis and tumor growth. Cancer Res. 2016;76:1260‐1272. [DOI] [PubMed] [Google Scholar]
- 24.Webb LM, Sengupta S, Edell C, et al. Protein arginine methyltransferase 5 promotes cholesterol biosynthesis‐mediated Th17 responses and autoimmunity. J Clin Invest. 2020;130:1683‐1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang X, Wu J, Wu C, et al. The LINC01138 interacts with PRMT5 to promote SREBP1‐mediated lipid desaturation and cell growth in clear cell renal cell carcinoma. Biochem Biophys Res Commun. 2018;507:337‐342. [DOI] [PubMed] [Google Scholar]
- 26.Goldstraw P, Chansky K, Crowley J, et al. The IASLC Lung Cancer Staging Project: proposals for revision of the TNM stage groupings in the forthcoming (eighth) edition of the TNM classification for lung cancer. J Thorac Oncol. 2016;11:39‐51. [DOI] [PubMed] [Google Scholar]
- 27.Zhu Z, Zhao X, Zhao L, et al. p54(nrb)/NONO regulates lipid metabolism and breast cancer growth through SREBP‐1A. Oncogene. 2016;35:1399‐1410. [DOI] [PubMed] [Google Scholar]
- 28.Yang H, Zhao X, Zhao L, et al. PRMT5 competitively binds to CDK4 to promote G1‐S transition upon glucose induction in hepatocellular carcinoma. Oncotarget. 2016;7:72131‐72147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor‐mTOR complex. Science. 2005;307:1098‐1101. [DOI] [PubMed] [Google Scholar]
- 30.Jo H, Mondal S, Tan D, et al. Small molecule‐induced cytosolic activation of protein kinase Akt rescues ischemia‐elicited neuronal death. Proc Natl Acad Sci USA. 2012;109:10581‐10586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shilo K, Wu X, Sharma S, et al. Cellular localization of protein arginine methyltransferase‐5 correlates with grade of lung tumors. Diagn Pathol. 2013;8:201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ibrahim R, Matsubara D, Osman W, et al. Expression of PRMT5 in lung adenocarcinoma and its significance in epithelial‐mesenchymal transition. Hum Pathol. 2014;45:1397‐1405. [DOI] [PubMed] [Google Scholar]
- 33.Alzahrani AS. PI3K/Akt/mTOR inhibitors in cancer: at the bench and bedside. Semin Cancer Biol. 2019;59:125‐132. [DOI] [PubMed] [Google Scholar]
- 34.Paulus W. GFAP, Ki67 and IDH1: perhaps the golden triad of glioma immunohistochemistry. Acta Neuropathol. 2009;118:603‐604. [DOI] [PubMed] [Google Scholar]
- 35.Tsurutani J, Fukuoka J, Tsurutani H, et al. Evaluation of two phosphorylation sites improves the prognostic significance of Akt activation in non‐small‐cell lung cancer tumors. J Clin Oncol. 2006;24:306‐314. [DOI] [PubMed] [Google Scholar]
- 36.David O, Jett J, LeBeau H, et al. Phospho‐Akt overexpression in non‐small cell lung cancer confers significant stage‐independent survival disadvantage. Clin Cancer Res. 2004;10:6865‐6871. [DOI] [PubMed] [Google Scholar]
- 37.Hoxhaj G, Manning BD. The PI3K‐AKT network at the interface of oncogenic signalling and cancer metabolism. Nat Rev Cancer. 2020;20:74‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Revathidevi S, Munirajan AK. Akt in cancer: mediator and more. Semin Cancer Biol. 2019;59:80‐91. [DOI] [PubMed] [Google Scholar]
- 39.Eberle D, Hegarty B, Bossard P, Ferre P, Foufelle F. SREBP transcription factors: master regulators of lipid homeostasis. Biochimie. 2004;86:839‐848. [DOI] [PubMed] [Google Scholar]
- 40.Yellaturu CR, Deng X, Cagen LM, et al. Insulin enhances post‐translational processing of nascent SREBP‐1c by promoting its phosphorylation and association with COPII vesicles. J Biol Chem. 2009;284:7518‐7532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guo S, Ma B, Jiang X, Li X, Jia Y. Astragalus polysaccharides inhibits tumorigenesis and lipid metabolism through miR‐138‐5p/SIRT1/SREBP1 pathway in prostate cancer. Front Pharmacol. 2020;11:598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ouyang S, Mo Z, Sun S, Yin K, Lv Y. Emerging role of Insig‐1 in lipid metabolism and lipid disorders. Clin Chim Acta. 2020;508:206‐212. [DOI] [PubMed] [Google Scholar]
- 43.Tang M, Floyd S, Cai H, Zhang M, Yang R, Dang R. The status of omega‐3 PUFAs influence chronic unpredicted mild stress‐induced metabolic side effects in rats through INSIG/SREBP pathway. Food Funct. 2019;10:4649‐4660. [DOI] [PubMed] [Google Scholar]
- 44.Krycer JR, Sharpe LJ, Luu W, Brown AJ. The Akt‐SREBP nexus: cell signaling meets lipid metabolism. Trends Endocrinol Metab. 2010;21:268‐276. [DOI] [PubMed] [Google Scholar]
- 45.Guo D, Prins RM, Dang J, et al. EGFR signaling through an Akt‐SREBP‐1‐dependent, rapamycin‐resistant pathway sensitizes glioblastomas to antilipogenic therapy. Sci Signal. 2009;2:ra82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang S, Ma Y, Hu X, Zheng Y, Chen X. Targeting PRMT5/Akt signalling axis prevents human lung cancer cell growth. J Cell Mol Med. 2019;23:1333‐1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhu F, Guo H, Bates PD, et al. PRMT5 is upregulated by B‐cell receptor signaling and forms a positive‐feedback loop with PI3K/AKT in lymphoma cells. Leukemia. 2019;33:2898‐2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Faubert B, Solmonson A, DeBerardinis RJ. Metabolic reprogramming and cancer progression. Science. 2020;368:eaaw5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Warburg O. On the origin of cancer cells. Science. 1956;123:309‐314. [DOI] [PubMed] [Google Scholar]
- 50.Buckley D, Duke G, Heuer TS, et al. Fatty acid synthase – modern tumor cell biology insights into a classical oncology target. Pharmacol Ther. 2017;177:23‐31. [DOI] [PubMed] [Google Scholar]
- 51.Richters A. Targeting protein arginine methyltransferase 5 in disease. Future Med Chem. 2017;9:2081‐2098. [DOI] [PubMed] [Google Scholar]
- 52.Li X, Wang C, Jiang H, Luo C. A patent review of arginine methyltransferase inhibitors (2010–2018). Expert Opin Ther Pat. 2019;29:97‐114. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S7
Table S1‐S7
Supplementary Material