

Abstract
The small GTPases RalA and RalB are members of the Ras family and activated downstream of Ras. Ral proteins are found in GTP‐bound active and GDP‐bound inactive forms. The activation process is executed by guanine nucleotide exchange factors, while inactivation is mediated by GTPase‐activating proteins (GAPs). RalGAPs are complexes that consist of a catalytic α1 or α2 subunit together with a common β subunit. Several reports implicate the importance of Ral in pancreatic ductal adenocarcinoma (PDAC). However, there are few reports on the relationship between levels of RalGAP expression and malignancy in PDAC. We generated RalGAPβ‐deficient PDAC cells by CRISPR‐Cas9 genome editing to investigate how increased Ral activity affects malignant phenotypes of PDAC cells. RalGAPβ‐deficient PDAC cells exhibited several‐fold higher Ral activity relative to control cells. They had a high migratory and invasive capacity. The RalGAPβ‐deficient cells grew more rapidly than control cells when injected subcutaneously into nude mice. When injected into the spleen, the RalGAPβ‐deficient cells formed larger splenic tumors with more liver metastases, and unlike controls, they disseminated into the abdominal cavity. These results indicate that RalGAPβ deficiency in PDAC cells contributes to high activities of RalA and RalB, leading to enhanced cell migration and invasion in vitro, and tumor growth and metastasis in vivo.
Keywords: pancreatic ductal adenocarcinoma, Ral, RalGAP, Ras, small GTPase
Ral is a Ras family small GTPase that have recently been noted as a novel signaling factor that regulates progression of pancreatic ductal adenocarcinoma (PDAC). In this study, by characterizing newly generated RalGAP‐deficient PDAC cells, we first demonstrated that Ral GTPase–activating proteins played a critical role in not only cell migration and invasion in vitro but also tumor growth and metastasis in vivo in PDAC.

Abbreviations
- Cdc42
cell division control protein 42 homolog
- Co‐IP
co‐immunoprecipitation
- GAP
GTPase‐activating protein
- GAPDH
glyceraldehyde‐3‐phosphate dehydrogenase
- GDP
guanosine diphosphate
- GEF
guanine nucleotide exchange factor
- GTP
guanosine triphosphate
- KO
knockout
- PDAC
pancreatic ductal adenocarcinoma
- Rac1
Ras‐related C3 botulinum toxin substrate 1
- Ral
Ras‐like
- RalBP1
Ral‐binding protein 1
- RalGDS
Ral guanine nucleotide dissociation stimulator
- RalGPS
RalGEF with pleckstrin homology domain and Src‐homologous 3 binding motif
- Ras
rat sarcoma
- RGL
RalGDS‐like
- Rho
Ras homologous
- SEM
standard error of the mean
1. INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is one of the most aggressive cancers, with a poor prognosis. It is known that one of the triggers for its development is an activating mutation of KRAS, followed by mutation of the tumor suppressor p53, which causes progression of the disease.1 Other factors affecting development and progression of PDAC have been extensively investigated, including the involvement of RalA and RalB, which belong to the Ras family of small GTPases.2, 3
Ral proteins are found in GTP‐bound active and GDP‐bound inactive forms. The activation process is executed by a guanine nucleotide exchange reaction mediated by guanine nucleotide exchange factors (GEFs), while inactivation is via the GTP hydrolysis reaction mediated by GTPase‐activating proteins (GAPs).4, 5 Thus far, six RalGEFs have been identified, four of them being direct effector molecules of Ras, which means Ras activation stimulates Ral activity.5 RalGAPs are complexes that consist of a catalytic RalGAPα1 or α2 subunit together with a common β subunit.6 Although the RalGAPβ subunit has no catalytic domain, it is indispensable for stability of RalGAPα subunits and the function of the complex.6 It has been demonstrated that Ral regulates multiple cellular functions such as endocytosis, exocytosis, cell growth, and cytoskeletal reorganization via its effectors, including the exocyst complex, Ral‐binding protein 1 (RalBP1), and others.5, 6, 7, 8, 9, 10
Ral plays an important role in cancer development and progression.3, 10, 11 The Ral pathway is indispensable for Ras‐induced transformation of human cells.10, 12 In an experimental chemically induced skin cancer model, it has been shown that carcinogenesis is essentially absent in mice deficient in RalGDS (one of the RalGEFs).13 We have shown that downregulation of RalGAP causes bladder cancer progression and enhances invasion and metastasis.11
It has been reported that Ral regulates PDAC progression.2, 3 RalA function is critical for PDAC initiation, whereas RalB function is more important for its metastasis.2 Rgl2, one of the RalGEFs highly expressed in PDAC tumor tissue and cell lines, is essential for anchorage‐independent growth and invasion of PDAC cells in vitro.3 The Ral GTPase pathway was also involved in pancreatic tumor chemo‐ and radio‐resistance.14 However, there are few reports on the relationship between the level of expression of RalGAPs and malignancy in PDAC. Therefore, we investigated the effects of RalGAPβ suppression using RalGAPβ‐deficient PDAC cells generated using CRISPR‐Cas9 technology.
2. MATERIALS AND METHODS
The materials and methods used in this study are described in the “Supplementary Methods.”
Data are shown as means ± standard errors (SEM). A two‐tailed unpaired Welch's t‐test was used for comparisons between two groups (Figures 7A,B and 8C,E). One‐way ANOVA followed by Tukey's multiple comparison test or Dunnett's multiple comparison test was performed for comparisons between three or more groups (Figures 2C,E,G, 3C,E,G, 4C,E and 5A,D). P‐value <0.05 was considered statistically significant.
FIGURE 7.
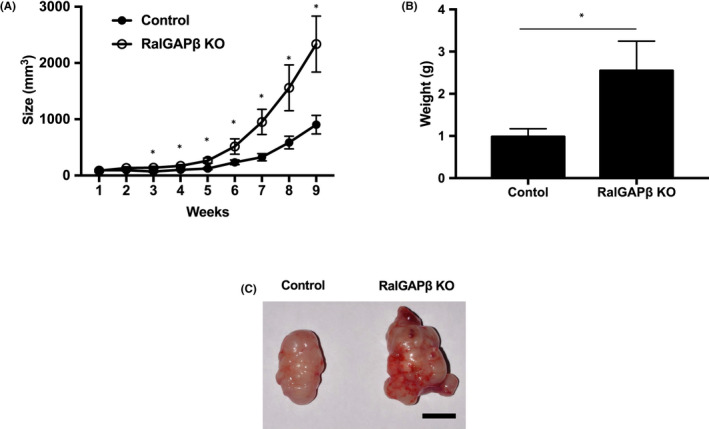
RalGAPβ deficiency increases tumorigenesis in vivo. RalGAPβ‐deficient (KO) and control MIA PaCa‐2 cells were used in a subcutaneous injection model. Cells were injected into the bilateral thorax of 12 nude mice, euthanized 9 weeks later. A, Subcutaneous tumor volume (mm3) versus time (wk). B, Weight (g) of subcutaneous tumor. C, Typical macroscopic image of subcutaneous tumor. Scale bars represent 10 mm. *P < 0.05
FIGURE 8.
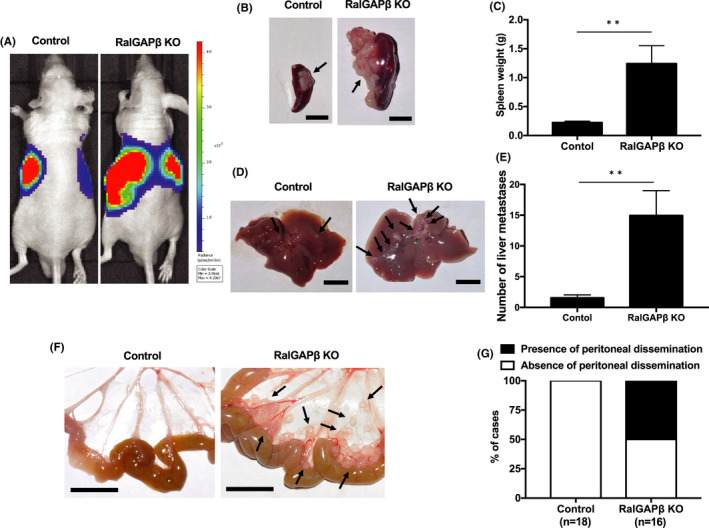
RalGAPβ deficiency increases tumorigenesis and metastasis in vivo. RalGAPβ‐deficient (KO) and control MIA PaCa‐2 cells stably expressing firefly luciferase were injected into the spleens of 34 nude mice (control, n = 18; RalGAPβ KO, n = 16), euthanized 6 weeks later. A, Typical luminescence image in vivo. B, Typical macroscopic image of spleen. C, Weight (g) of spleen. D, Typical macroscopic image of the liver surface. E, Number of metastatic foci on the liver surface. F, Typical macroscopic image of peritoneal dissemination. G, Presence of peritoneal dissemination. Scale bars represent 10 mm. **P < 0.01
FIGURE 2.
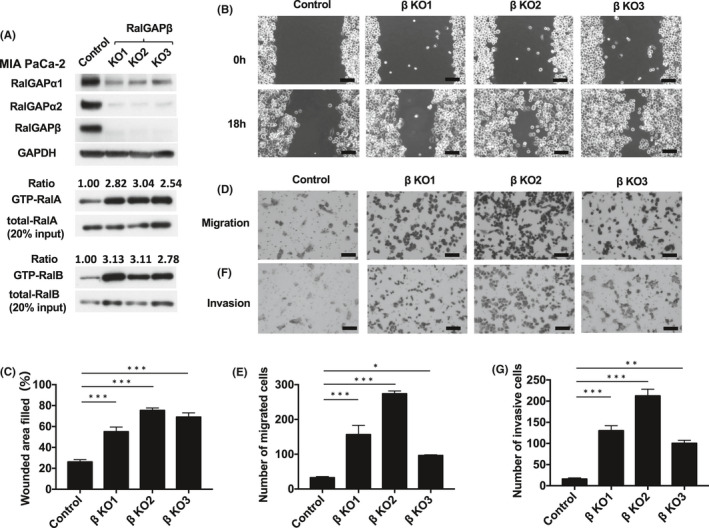
RalGAPβ deficiency increases RalA and RalB activity and enhances migration and invasion of MIA PaCa‐2. A, GTP‐bound RalA and RalB of RalGAPβ‐deficient (KO, knockout) and control MIA PaCa‐2 cells assessed by the pull‐down assay. Total RalA and RalB, RalGAPα1, RalGAPα2, RalGAPβ, and GAPDH evaluated by Western blotting. GTP‐bound RalA and RalB was evaluated by the pull‐down assay. The ratios of GTP‐bound RalA/B to total RalA/B of control cells are set at unity. B‐C, Motility of RalGAPβ‐deficient and control MIA PaCa‐2 cells evaluated using the wound healing assay. Representative images (B) and quantification (C) of the wounded area. D‐E, Migration of RalGAPβ‐deficient and control MIA PaCa‐2 cells evaluated using transwell assays. Representative images (D) and quantification (E) in the form of cell numbers. F‐G, Invasion by RalGAPβ‐deficient and control MIA PaCa‐2 cells evaluated using transwell assays. Representative images (F) and quantification (G) in the form of cell numbers. Scale bars represent 100 µm. Data are mean ± SEM of three independent experiments. *P < 0.05. **P < 0.01. ***P < 0.001
FIGURE 3.
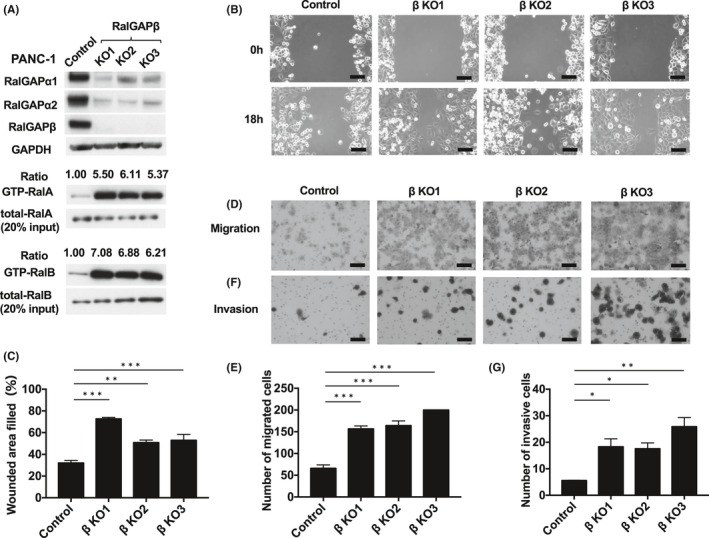
RalGAPβ deficiency increases RalA and RalB activity and enhances migration and invasion of PANC‐1 cells. A, GTP‐bound RalA and RalB of RalGAPβ‐deficient (KO) and control PANC‐1 cells measured by the pull‐down assay. Total RalA and RalB, RalGAPα1, RalGAPα2, RalGAPβ, and GAPDH in these cells and control cells was evaluated by Western blotting. GTP‐bound RalA and RalB was evaluated by the pull‐down assay. The ratios of GTP‐bound RalA/B to total RalA/B of control cells were set at unity. B‐C, Motility of RalGAPβ‐deficient and control PANC‐1 cells evaluated using the wound healing assay. Representative images (B) and quantification (C) of the wounded area. D‐E, Migration by RalGAPβ‐deficient and control PANC‐1 cells evaluated using the transwell migration assay. Representative images (D) and quantification (E) in the form of cell numbers. F‐G, Invasion by RalGAPβ‐deficient and control PANC‐1 cells evaluated using the transwell invasion assay. Representative images (F) and quantification (G) in the form of cell numbers. Scale bars represent 100 µm. Data are means ± SEM of three independent experiments. *P <0 .05. **P < 0.01. ***P < 0.001
FIGURE 4.
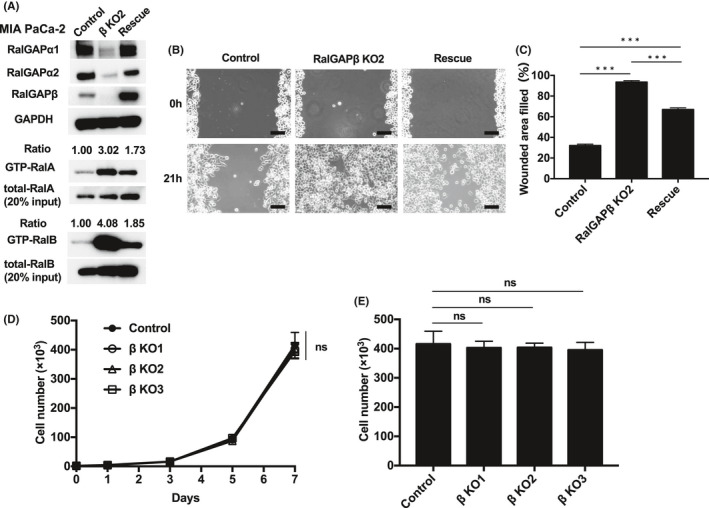
Rescue of RalGAPβ expression restores RalGAPα1 and RalGAPα2 expression and reduces migration (MIA PaCa‐2). A, Full‐length RalGAPβ was expressed in RalGAPβ‐deficient MIA PaCa‐2 cells using a lentiviral system (Rescue). Western blotting was performed to analyze the indicated proteins. B‐C, Migration of the control, RalGAPβ‐deficient (KO), and rescued MIA PaCa‐2 cells analyzed in the wound healing assay. Representative images (B) and quantification (C) of the wounded area. Scale bars represent 100 µm. D‐E, Cells were counted to evaluate the proliferation rate of RalGAPβ‐deficient and control MIA PaCa‐2 cells on days 1, 3, 5, and 7 after seeding. Cell numbers on each day (D) and day 7 (E). Data are mean ± SEM of three independent experiments. ns, not significant. ***P < 0.001
FIGURE 5.
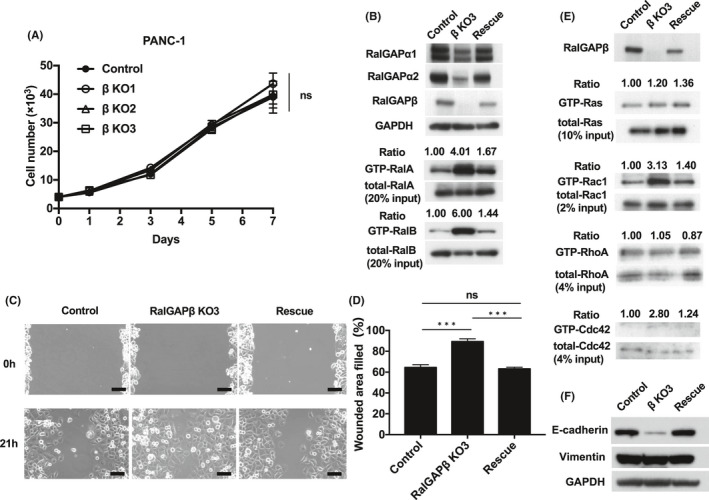
Rescue of RalGAPβ expression restores RalGAPα1 and RalGAPα2 expression and reduces migration of the rescued cells. RalGAPβ‐deficient cells exhibit Rac1 and Cdc42 activity (PANC‐1). A, Cells counted to evaluate the proliferation rate of RalGAPβ‐deficient and control PANC‐1 cells on days 1, 3, 5, and 7 after seeding. B, Full‐length RalGAPβ was expressed in RalGAPβ‐deficient PANC‐1 cells using a lentiviral system (Rescue). Western blotting was performed to analyze the indicated proteins. The ratios of GTP‐bound RalA/B to total RalA/B of control cells were set at unity (C‐D) Migration of the control, RalGAPβ‐deficient (KO), and rescued PANC‐1 cells analyzed in the wound healing assay. Representative images (C) and quantification (D) of the wounded area. E, GTP‐bound active Ras, Rac1, RhoA, and Cdc42 measured by the pull‐down assay in control, RalGAPβ‐deficient (KO), and rescued PANC‐1 cells. The ratios of GTP‐bound small GTPase to total small GTPase of control cells were set at unity. F, E‐cadherin, Vimentin, and GAPDH in the control, RalGAPβ‐deficient (KO), and rescued PANC‐1 cells was evaluated by Western blotting. Scale bars represent 100 µm. Data are means ± SEM of three independent experiments. ns, not significant. ***P < 0.001
All the genetic recombination experiments and the animal experiments were conducted with the approval by the relevant committees of Tohoku University.
3. RESULTS
3.1. Ral activity is negatively regulated by RalGAPs
We examined the level of expression and activity of Ral in six PDAC cell lines. Total RalA protein levels were similar in all six lines, whereas RalB levels were higher in AsPC‐1, BxPC‐3, SUIT‐2, and SW‐1990 cells and very low in PANC‐1 and MIA PaCa‐2 cells shown by Western blotting (Figure 1A). The activity of RalA and RalB paralleled in each cell line although the activity levels were various (Figure 1A). RalA activity (GTP‐RalA) was relatively strong in PANC‐1 and MIA PaCa‐2, intermediate in AsPC‐1 and SW‐1990, and low in BxPC‐3 and SUIT‐2 cells (Figure 1A). RalB activities (GTP‐RalB) were difficult to compare among the cell lines due to their weak signals in the Western blot (Figure 1A).
FIGURE 1.
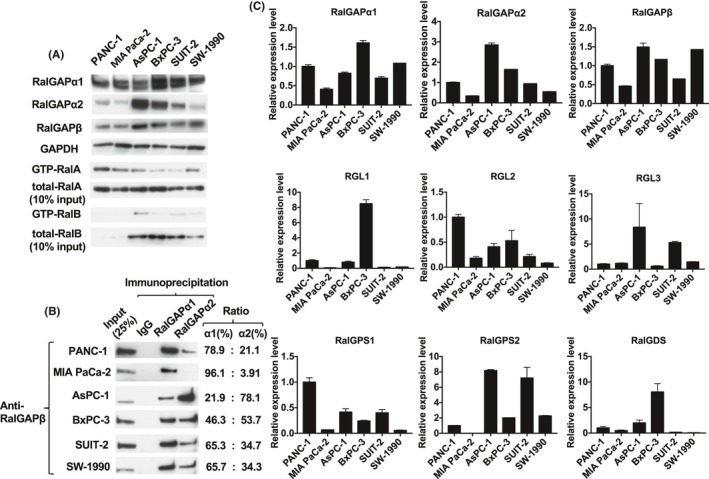
Ral activity is associated with downregulated expression of RalGAPs. A, GTP‐bound active RalA and RalB analyzed by the pull‐down assay. GTP‐bound RalA and RalB, total RalA and RalB, RalGAPα1, RalGAPα2, RalGAPβ, and GAPDH in six pancreatic ductal adenocarcinoma (PDAC) cell lines was evaluated by Western blotting. B, Relative levels of RalGAPα1 and RalGAPα2 in the six PDAC cell lines, showing RalGAPβ complexed with RalGAPα1 and RalGAPα2 after immunoprecipitation with anti‐RalGAPα1, anti‐RalGAPα2 antibody, or control immunoglobulin. C, RalGAPα1, RalGAPα2, RalGAPβ, and RalGEFs including RalGDS, RalGPS1, RalGPS2, RGL1, RGL2, and RGL3 mRNA from the PDAC cell lines was quantified using GAPDH as the reference. Means of the mRNA levels in PANC‐1 cells are set at unity. Data are representative of three independent experiments with similar results
KRAS mutation is a well‐known driver of pancreatic cancer development.15 We therefore examined KRAS, HRAS, and NRAS sequences in the six PDAC cell lines because Ral is activated downstream of Ras.14 As shown in Table 1, activating mutations in KRAS (G12C or G12D) were found in all lines, with the exception of BxPC‐3 that had wild‐type KRAS. We found no mutations in HRAS or NRAS in all PDAC cells lines analyzed.
TABLE 1.
RAS mutation status in six PDAC cell lines. Amino acid mutations in hot spots, including G12, G13, A59, Q61, K117, A146 in HRAS, NRAS, and KRAS were evaluated. Activating mutations in KRas (G12C or G12D) were found in all lines except BxPC‐3
| Cell lines | SNP | AA mutation | SNP | AA mutation | SNP | AA mutation |
|---|---|---|---|---|---|---|
| HRAS | HRAS | NRAS | NRAS | KRAS | KRAS | |
| PANC‐1 | H27H homo, CAT→CAC | wild | wild | wild | G12D hetero, GGT→GAT | G12D hetero |
| MIA PaCa‐2 | H27H hetero, CAT→CAC | wild | wild | wild | G12C homo, GGT→TGT | G12C homo |
| AsPC‐1 | wild | wild | wild | wild | G12D homo, GGT→GAT | G12D homo |
| BxPC‐3 | H27H homo, CAT→CAC | wild | wild | wild | wild | wild |
| SUIT‐2 | H27H homo, CAT→CAC | wild | wild | wild | G12D hetero, GGT→GAT | G12D hetero |
| SW‐1990 | wild | wild | wild | wild | G12D homo, GGT→GAT | G12D homo |
Abbreviations: AA, amino acid; SNP, single‐nucleotide polymorphism.
The activity of small GTPase is regulated by GEFs and GAPs. Therefore, we analyzed expression levels of these molecules. Western blotting showed that PANC‐1, MIA PaCa‐2, and SW‐1990 cells possessed low levels of each subunit of RalGAP and tended to exhibit high RalA activity. The mRNA levels of RalGAP subunits matched well with their protein levels (Figure 1A,C).
RalGAPs are heterodimers consisting of either an α1 or an α2 catalytic subunit and a common β subunit.6 To analyze which catalytic subunit is dominant in PDAC cells, we performed co‐immunoprecipitation (IP) with anti‐RalGAPα1 or anti‐RalGAPα2 antibody and evaluated the amounts of associated common RalGAPβ subunits. As shown in Figure 1B, in PANC‐1, MIA PaCa‐2, SUIT‐2, and SW‐1990 cells, the levels of RalGAPα1 were higher than those of RalGAPα2. On the other hand, RalGAPα1 was present at lower levels than RalGAPα2 in AsPC‐1 and BxPC‐3 cells. Thus, the dominant catalytic subunit of RalGAP was various in the different PDAC cell lines. No correlation was seen between the mRNA levels of each RalGEF (RalGDS, RalGPS1, RalGPS2, RGL1, RGL2, or RGL3) and RalA/B activity in the six PDAC cell lines examined (compare Figure 1A with Figure 1C).
3.2. RalGAPβ deficiency decreases expression of RalGAPα1 and RalGAPα2 and increases RalA and RalB activity
We generated RalGAPβ‐deficient PANC‐1 and MIA PaCa‐2 clonal cell lines using the CRISPR/Cas9 system (Figure S1A). Genetic deletion of RalGAPβ was confirmed by genomic sequencing (Figure S1B) and Western blotting (Figures 2A and 3A).
The expression levels of both RalGAPα1 and RalGAPα2 were also dramatically decreased in RalGAPβ‐deficient MIA PaCa‐2 (Figure 2A) and PANC‐1 cells (Figure 3A). Reconstituting the expression of RalGAPβ in these RalGAPβ‐deficient cells restored RalGAPα1 and RalGAPα2 protein levels (Figures 4A and 5B) because RalGAPβ is necessary for protein stability of RalGAPα1 and RalGAPα2 as shown previously.6 Accordingly, the activity of RalA and RalB was markedly increased in RalGAPβ‐deficient cells relative to controls (Figures 2A and 3A). Ral activity of BxPC‐3 cells without KRAS mutation were also increased by RalGAPβ knockdown (Figure S2A‐C).
3.3. RalGAPβ deficiency enhances cell migration and invasion, possibly due to Rac1 and Cdc42 activity
We next examined the effect of RalGAPβ deficiency on the migration and invasion capacity of PDAC cells. In the wound healing assay, all three RalGAPβ‐deficient MIA PaCa‐2 cells exhibited enhanced cell migration relative to control cells in vitro (Figure 2B‐C). Likewise, these cells also exhibited enhanced cell migration in the transwell migration assay (Figure 2D‐E) and higher invasive activity in the Matrigel invasion assay (Figure 2F‐g). When we reconstituted RalGAPβ expression in RalGAPβ‐deficient MIA PaCa‐2 cells to a similar level to the control cells (Figure 4A), both Ral activity and cell migration capacity levels returned nearly to the levels observed in control cells (Figure 4A‐C). Proliferation rates of the RalGAPβ‐deficient MIA PaCa‐2 cells in the culture dishes were not altered compared with the controls (Figure 4D‐E). These findings, namely enhanced cell migration and invasion and no change in cell proliferation in vitro, were observed in RalGAPβ‐deficient PANC‐1 cells as well (Figures 3B‐G and 5A‐D).
As epithelial‐mesenchymal transition (EMT), in addition to cell motility, is also involved in the acquisition of invasive phenotypes of cancer cells, we analyzed the expression levels of EMT markers, E‐cadherin and vimentin, in RalGAP‐deficient cells. The expression of E‐cadherin was markedly decreased in RalGAPβ‐deficient PANC‐1 cells compared with control or rescued cells (Figure 5F), while there was no difference in the expression level of vimentin among these cells (Figure 5F).
We next examined cellular morphology by rhodamine‐phalloidin staining of actin fibers. We found that RalGAPβ‐deficient cells exhibited spindle‐like shape with filopodia and lamellipodia‐like structures in contrast to the round shape of both control cells and RalGAPβ‐rescued cells (Figure 6A).
FIGURE 6.
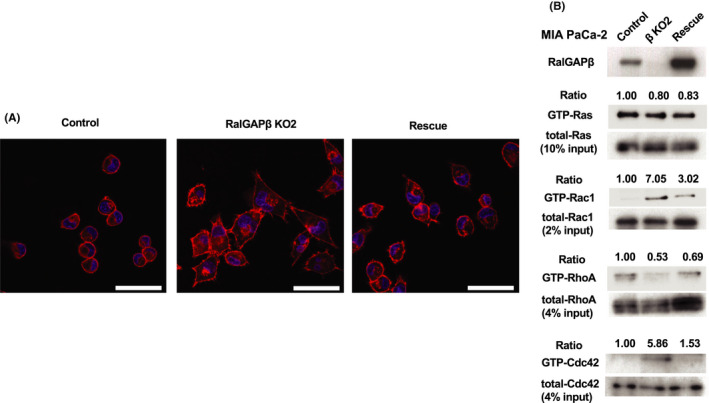
Morphology of RalGAPβ‐deficient cells and RalGAPβ rescued cells, and activity of Rac1 and Cdc42. A, Rhodamine‐phalloidin staining of actin fibers in control, RalGAPβ‐deficient (KO), and rescued MIA PaCa‐2 cells. Scale bars represent 50 µm. B, GTP‐bound active Ras, Rac1, RhoA, and Cdc42 measured by the pull‐down assay in control, RalGAPβ‐deficient (KO), and rescued MIA PaCa‐2 cells. Ratios of GTP‐bound small GTPase to total small GTPase of control cells are set at unity. Data are representative of three independent experiments with similar results
It is well known that cell migration and cell shape are regulated by the Rho family small GTPases.17 We next analyzed activities of various small GTPases in the cells. We found that the activity of Rac1 and Cdc42, but not RhoA or Ras, was increased in the RalGAPβ‐deficient cells, as shown in Figures 5E and 6B.
3.4. RalGAPβ deficiency increases tumorigenesis and metastasis in vivo
In the final set of experiments in this study, we investigated the effect of RalGAPβ deficiency on the growth, invasion, and metastasis of MIA PaCa‐2 cells in vivo. RalGAPβ‐deficient and control cells were injected subcutaneously into nude mice. The sizes and weights of tumors derived from RalGAPβ‐deficient cells were significantly increased relative to tumors formed by control cells (Figure 7A‐C).
Next, RalGAPβ‐deficient and control cells stably expressing firefly luciferase were injected into the spleens of nude mice and analyzed after 6 weeks. In vivo imaging revealed that the luminescence signals reflecting the presence of the tumor were stronger not only in the spleen but also in the liver of mice injected with the RalGAPβ‐deficient cells compared with those injected with control cells (Figure 8A). At 6 weeks after injection, mice were sacrificed, and the tumors in the abdominal cavity were evaluated. The spleens of mice injected with RalGAPβ‐deficient cells were significantly heavier than those injected with control cells (1.24 ± 0.31 g with the RalGAPβ‐deficient cells vs 0.22 ± 0.02 g with control cells; P < .01, Figure 8B‐C). We then examined the livers and found far more metastatic foci on their surface of mice injected with RalGAPβ‐deficient cells than those with control cells (P < .01, Figure 8D‐E). Furthermore, eight of 16 mice injected with RalGAPβ‐deficient cells exhibited peritoneal dissemination foci while none with control cells did (Figure 8F‐g). Thus, RalGAPβ‐deficient PDAC cells exogenously implanted in spleen exhibited faster local growth with more liver metastases and peritoneal dissemination than control cells.
4. DISCUSSION
In this study, we utilized RalGAPβ‐deficient cells to study the role of Ral and RalGAPs in the invasion and metastasis of PDAC cells. When the RalGAPβ subunit, the common subunit of the RalGAP complexes, was disrupted, the expression of both RalGAPα1 and RalGAPα2 was markedly reduced (Figure 2A and 3A). On the other hand, when the α2 subunit was knocked out, the expression of RalGAPβ was reduced as shown previously.11 Thus, both the α and β subunits are necessary for stable expression of each other. Ral activity was increased by several times in both RalGAPβ‐deficient PANC‐1 and MIA PaCa‐2 cells, compared with their control cells. Because active mutations of RAL genes are rare in cancer, Ral activity may be controlled to a high degree by varying levels of its regulatory proteins, such as RalGEFs and RalGAPs.16
It is well known that most PDACs possess activating mutations in KRAS genes 1 and that Ral is activated downstream of Ras. We analyzed KRAS gene sequence in six PDAC cell lines (Table 1) and detected KRAS activating mutations in five of the cell lines, except for BxPC‐3. The low activity of Ral in BxPC‐3 cells could be due to lack of KRAS mutation in this cell line.
However, Ral activity in cells with KRAS mutations were various (Figure 1A), suggesting that KRAS mutation is not only a factor that determines the activity of Ral. Despite harboring KRAS mutation, SUIT‐2 cells showed low Ral activity comparable to that of BxPC‐3 cells. This suggests that the activating mutation of KRAS may have less impact on Ral activity than expected.
It has been reported that the high expression of Rgl2, a RalGEF, induces strong Ral activity in pancreatic cells.3 However, we did not detect any specific RalGEFs including Rgl2 in the mRNA levels, which were correlated with Ral activity in the six PDAC cell lines examined. (Figure 1A,C).
We performed the wound healing assay of six PDAC cell lines and found that the migration rates were highly various among cell lines (Figure S3A‐B). SUIT‐2 cells with low RalA activity exhibited high migration activity, and MIA PaCa‐2 cells with high RalA activity showed low migration activity. Thus, the migration activity did not seem simply associated with Ral activities among various cell lines. This could be due to many factors involved in the regulation of migration capacity. Nevertheless, in a specific cell line such as PANC‐1 cells or MIA PaCa‐2 cells, the migration activity is clearly associated with Ral activity, shown by experiments with RalGAPβ cells (Figures 1A and S3A‐B). Thus, we consider that Ral activity plays an important role in the regulation of migration of PDAC cells. Although PANC‐1 cells and MIA PaCa‐2 cells, with which we examined the cell motility as described above, possess KRAS mutation, KRAS of BxPC‐3 cells was wild‐type. Ral activity and migration capacity of BxPC‐3 cells were also increased by RalGAPβ knockdown (Figure S2A‐C), suggesting that these phenotypes are not specific to PDAC cells with KRAS mutation.
In the RalGAPβ‐deficient PDAC cells, activity of Rac1 and Cdc42, both implicated in cell motility,17 was increased (Figure 5E and 6B). This could stimulate the cell migration and invasion capacity in RalGAPβ‐deficient PDAC cells. RalBP1, a Ral effector, has been reported to regulate the activity of Rac and Cdc42,18, 19, 20 and therefore the increased activity of Rac and Cdc42 activities in RalGAPβ‐deficient cells might be mediated by RalBP1. In addition, Ral also regulates membrane traffic through another effector, the exocyst complex.21 This Ral‐activated membrane traffic may also contribute to enhanced cell motility and invasion capacity by providing membrane into the leading edge of the migrating cells. Further investigation is required to elucidate the mechanism.
In invasion and metastasis of cancer cells, it is well understood that one of the critical processes is EMT. We examined the expression levels of representative EMT markers, E‐cadherin and vimentin, in RalGAP‐deficient cells. The expression levels of E‐cadherin were markedly decreased in RalGAPβ‐deficient PANC‐1 cells compared with control or rescued cells although the levels of vimentin were not altered (Figure 5F) suggesting that RalGAPβ‐deficiency may lead cells toward EMT. This may contribute to the higher invasion and metastasis capacity in RalGAPβ‐deficient cells.
The anchorage‐dependent cell growth in culture dishes has not been affected by Ral activity, demonstrated by Lim et al22 and us (Figures 4D‐E and 5A). In contrast, RalGAPβ‐deficient PDAC cells exhibited much faster local growth than control cells when implanted subcutaneously as well as when introduced into the spleen (Figure 7A‐C and 8B‐C). Previously, we had shown that the sizes of chemically induced bladder tumors in RalGAPα2‐deficient mice are much larger than in wild‐type mice.11 The precise mechanism is unclear, although it may be due to Ral‐enhanced tumor angiogenesis because it has been reported that mice deficient in RalBP1 (one of the Ral effectors) exhibit impaired tumor angiogenesis.23, 24
RalGAPβ‐deficient PDAC cells formed significantly more liver metastases and peritoneal dissemination when injected into the spleen (Figure 8D‐G). Although the association between low RalGAP expression levels and liver metastasis remains unclear, the lower mRNA expression of RalGAPα1 in the pancreas cancer tissues is correlated with worse prognosis of pancreatic cancer patients in The Cancer Genome Atlas (TCGA) (https://www.cancer.gov/about‐nci/organization/ccg/research/structural‐genomics/tcga) (Figure S4A). The low expression of RalGAPα2 and β also tends to be associated with poor prognosis (Figure S4B,C). These data suggest that lower expression of RalGAPs could be involved in the poor prognosis of pancreas cancer patients.
In conclusion, RalGAPβ deficiency in PDAC cells enhances RalA and RalB activity, cell migration, and invasion in vitro, and tumor growth and metastasis in vivo. Ral inhibitors have been developed for use as antitumor drugs in PDAC.25 For the further development of such drugs, our RalGAPβ‐deficient PDAC cell implantation models could be useful for evaluating how such drugs affect Ral‐dependent tumor growth and metastasis.
DISCLOSURE
All the authors have no conflict of interest related to the study.
Supporting information
Supplementary Material
Appendix S1
ACKNOWLEDGEMENTS
We also thank Bryan Welm and Feng Zhang for providing the plasmids.
Yoshimachi S, Shirakawa R, Cao M, et al. Ral GTPase–activating protein regulates the malignancy of pancreatic ductal adenocarcinoma. Cancer Sci. 2021;112:3064–3073. 10.1111/cas.14970
Funding information
This study was supported by the Japan Society for the Promotion of Science (JSPS) Grant‐in‐Aid for Scientific Research JP16H05148 (RS) and JP16K08574 (HH). This work was also supported by Anzai Memorial Grant for Diabetes Research from Gonryo Foundation for the Promotion of Medical Science (RS).
REFERENCES
- 1.Lu L, Zeng J. Evaluation of Kras and p53 expression in pancreatic adenocarcinoma using the cancer genome atlas. PLoS ONE. 2017;12(7):e0181532. 10.1371/journal.pone.0181532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lim KH, O’Hayer K, Adam SJ, et al. Divergent roles for RalA and RalB in malignant growth of human pancreatic carcinoma cells. Curr Biol. 2006;16:2385‐2394. [DOI] [PubMed] [Google Scholar]
- 3.Vigil D, Martin TD, Williams F, et al. Aberrant overexpression of the Rgl2 Ral small GTPase‐specific guanine nucleotide exchange factor promotes pancreatic cancer growth through Ral‐dependent and Ral‐independent mechanisms. J Biol Chem. 2010;285:34729‐34740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bos JL, Rehmann H, Wittinghofer A. GEFs and GAPs: critical elements in the control of small G proteins. Cell. 2007;129:865‐877. [DOI] [PubMed] [Google Scholar]
- 5.Shirakawa R, Horiuchi H. Ral GTPases: crucial mediators of exocytosis and tumourigenesis. J Biochem. 2015;157:285‐299. [DOI] [PubMed] [Google Scholar]
- 6.Shirakawa R, Fukai S, Kawato M, et al. Tuberous sclerosis tumor suppressor complex‐like complexes act as GTPase‐activating proteins for Ral GTPases. J Biol Chem. 2009;284:21580‐21588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Synek L, Sekeres J, Zarsky V. The exocyst at the interface between cytoskeleton and membranes in eukaryotic cells. Front Plant Sci. 2014;4:543. 10.3389/fpls.2013.00543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Balasubramanian N, Meier JA, Scott DW, et al. RalA‐exocyst complex regulates integrin‐dependent membrane raft exocytosis and growth signaling. Curr Biol. 2010;20:75‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yan C, Theodorescu D. RAL GTPases: biology and potential as therapeutic targets in cancer. Pharmacol Rev. 2018;70:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guin S, Theodorescu D. The RAS‐RAL axis in cancer: evidence for mutation‐specific selectivity in non‐small cell lung cancer. Acta Pharmacol Sin. 2015;36:291‐297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saito R, Shirakawa R, Nishiyama H, et al. Downregulation of Ral GTPase‐activating protein promotes tumor invasion and metastasis of bladder cancer. Oncogene. 2013;32:894‐902. [DOI] [PubMed] [Google Scholar]
- 12.Rangarajan A, Hong SJ, Gifford A, Weinberg RA. Species‐ and cell type‐specific requirements for cellular transformation. Cancer Cell. 2004;6:171‐183. [DOI] [PubMed] [Google Scholar]
- 13.Gonzalez‐Garcia A, Pritchard CA, Paterson HF, et al. RalGDS is required for tumor formation in a model of skin carcinogenesis. Cancer Cell. 2005;7:219‐226. [DOI] [PubMed] [Google Scholar]
- 14.Jonckheere N, Vasseur R, Van Seuningen I. The cornerstone K‐RAS mutation in pancreatic adenocarcinoma: from cell signaling network, target genes, biological processes to therapeutic targeting. Crit Rev Oncol Hematol. 2017;111:7‐19. [DOI] [PubMed] [Google Scholar]
- 15.Zeitouni D, Pylayeva‐Gupta Y, Der CJ, Bryant KL. KRAS mutant pancreatic cancer: No lone path to an Eeective treatment. Cancers (Basel). 2016;8(4):45. 10.3390/cancers8040045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao P, Liu S, Yoshida R, et al. Ral GTPase activation by downregulation of RalGAP enhances oral squamous cell carcinoma progression. J Dent Res. 2019;98:1011‐1019. [DOI] [PubMed] [Google Scholar]
- 17.Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509‐514. [DOI] [PubMed] [Google Scholar]
- 18.Cantor SB, Urano T, Feig LA. Identification and characterization of Ral‐binding protein 1, a potential downstream target of Ral GTPases. Mol Cell Biol. 1995;15:4578‐4584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jullien‐Flores V, Dorseuil O, Romero F, et al. Bridging Ral GTPase to Rho pathways. RLIP76, a Ral effector with CDC42/Rac GTPase‐activating protein activity. J Biol Chem. 1995;270:22473‐22477. [DOI] [PubMed] [Google Scholar]
- 20.Matsubara K, Hinoi T, Koyama S, Kikuchi A. The post‐translational modifications of Ral and Rac1 are important for the action of Ral‐binding protein 1, a putative effector protein of Ral. FEBS Lett. 1997;410:169‐174. [DOI] [PubMed] [Google Scholar]
- 21.Zuo X, Zhang J, Zhang Y, et al. Exo70 interacts with the Arp2/3 complex and regulates cell migration. Nat Cell Biol. 2006;8:1383‐1388. [DOI] [PubMed] [Google Scholar]
- 22.Lim KH, Baines AT, Fiordalisi JJ, et al. Activation of RalA is critical for Ras‐induced tumorigenesis of human cells. Cancer Cell. 2005;7:533‐545. [DOI] [PubMed] [Google Scholar]
- 23.Martin TD, Chen XW, Kaplan RE, et al. Ral and Rheb GTPase activating proteins integrate mTOR and GTPase signaling in aging, autophagy, and tumor cell invasion. Mol Cell. 2014;53:209‐220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee S, Wurtzel JG, Singhal SS, et al. RALBP1/RLIP76 depletion in mice suppresses tumor growth by inhibiting tumor neovascularization. Cancer Res. 2012;72:5165‐5173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yan C, Liu D, Li L, et al. Discovery and characterization of small molecules that target the GTPase Ral. Nature. 2014;515:443‐447. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Appendix S1