

Abstract
Enhancement of vascular permeability is indispensable for cancer metastasis. Weakened endothelial barrier function enhances vascular permeability. Circulating tumor cells moving in the microvasculature tend to invade into stromal tissue at the location where vascular permeability is enhanced. Many basic studies have identified permeability factors by using gene‐modified animals and cells. These factors directly/indirectly interact with endothelial cells. Here, we review vascular permeability factors and their molecular mechanisms. Interactions between tumor cells and endothelial cells are also discussed in the process of extravasation, one of the most critical steps in tumor metastasis. In some cases, primary tumors can manipulate permeability in a remote organ by secreting permeability factors. In addition, the importance of glycocalyx, which covers the endothelial cell surface, in controlling vascular permeability and tumor metastasis is also described. Furthermore, analysis of the hyperpermeable region found in a mouse model study is introduced. It clearly showed that tumor‐bearing mouse lungs had a hyperpermeable region due to the influence of a remote primary tumor, and fibrinogen deposition was observed in that region. Given that fibrinogen was reported to be a permeability factor and a key regulator of inflammation, eliminating fibrinogen deposition may prevent future metastasis.
Keywords: endothelial cell, fibrinogen, metastasis, permeability, premetastatic soil
Interactions between tumor cells and endothelial cells are discussed to specify the roles of primary tumor and tumor cells in the process of metastasis. The importance of glycocalyx, which covers the endothelial cell surface, in controlling vascular permeability and tumor metastasis is also described. Furthermore, analysis of the hyperpermeable region found in a mouse model study is introduced.
1. INTRODUCTION
Continuous efforts have been made to explore anticancer treatments for primary tumors, which have contributed to increasing the survival rate of cancer patients.1 Nevertheless, metastatic tumors remain evasive, and new treatments for tumor metastasis need to be introduced. Tumor metastasis is considered to account for the majority of cancer‐related deaths.2 At present, we cannot predict the precise locations and timing of metastasis. It has been reported that tumor metastasis starts at an early stage of primary tumor growth,3 which has been supported by some experimental models.4, 5 Tumor metastasis is classified into two phases, premetastatic and postmetastatic phases, based on the engraftment of tumor cells in the metastatic region. Model animal studies of the premetastatic phase revealed that a chain of reactions occurs to facilitate metastasis before the appearance of tumor cells in a remote organ.6, 7, 8, 9 Thus, to prevent metastasis, there is a need to reveal the detailed mechanisms of the premetastatic phase at the molecular level. The entire process of the premetastatic phase is composed of multiple steps including invasion into surrounding tissue in the primary site, intravasation, circulation in the bloodstream, and extravasation of tumor cells in a remote organ. Vascular permeability is highly correlated with the extravasation of tumor cells because tumor cells must break through the endothelial cell barrier. Some invaded tumor cells in the secondary site may form a metastatic colony, supported by the permeability‐related inflammatory microenvironment. Here, we focus on a molecular perspective of vascular permeability and related progress in our recent research.
2. VASCULAR PERMEABILITY FACTORS (VPFs)
Microvessels are maintained by endothelial cells.10 They are supported by the basement membrane and pericytes. The function of endothelial cells as blood vessel components requires a solid cell‐cell contact, mainly driven by tight junctions and adherens junctions. Molecular studies found that the former is mediated by occludin and claudin, among others, and the latter is mediated by vascular endothelial (VE)‐cadherin. Vascular permeability is controlled by vesicle transport across the endothelial cell layer (transendothelial permeability) or the tuning of endothelial cell‐cell junctions (paracellular permeability). Molecular ligands or mechanistic stimulation (shear stress) give impetus to increase vascular permeability. Many molecules have been reported to be permeability factors (Table 1, which includes mechanism‐based categories shown in Figure 1), which weaken cell‐cell junctions of the microvessel endothelial cells. The following briefly describes all the listed molecules. Proline‐glycine‐proline tripeptide is produced from collagen and further acetylated under physiological conditions.11 This tripeptide promotes VE‐cadherin phosphorylation to enhance vascular permeability in a CXC chemokine receptor 2 (CXCR2) signaling system–dependent manner. ADAM10, 17, and MMP3 degrade junction proteins.12, 13, 14 Angiomodulin produced in human bladder carcinoma cells and fibroblasts binds integrin ανβ3 to induce actin stress fibers, resulting in loosening of the VE‐cadherin–mediated intercellular junctions.15 Epithelial tumor cells produce high levels of angptl4, which binds integrin α5β1, claudin‐5, and VE‐cadherin to disrupt endothelial cell‐cell interactions. The angptl4–integrin α5β1 binding activates Rac1/PAK signaling to weaken cell‐cell contact.16 BMP6 is highly expressed in malignant prostate cancer. It binds the ALK2/BMPRII receptor complex to promote c‐Src–dependent VE‐cadherin phosphorylation.17 CCL2 is produced by many different cells, including cancer cells, fibroblasts, and endothelial cells. It binds CCR2 to activate actin‐myosin contraction.18 CXCL12 (SDF‐1) is expressed in fibroblasts, and it binds CXCR4 to reduce ZO‐1, occludin, and VE‐cadherin expression.19 CXCL5 is expressed in immune cells and fibroblasts. It binds CXCR2 to activate p38 MAPK signaling.20 Ephrin‐A1 is cleaved by ADAM12 to become a soluble form, which is capable of binding EphA1/A2 receptor to disrupt preexisting Ephrin‐Eph interaction between endothelial cells.21, 22 Fibrinogen binds ICAM‐1 and integrin α5β1 to induce F‐actin formation via ERK phosphorylation. Its Bβ chain 15‐42 also binds the extracellular domain of VE‐cadherin to disrupt cell‐cell interactions.23, 24 Hepatocyte growth factor (HGF) is expressed in cancer cells. It binds the c‐met receptor to increase the phosphorylation of ZO‐1 and to decrease the ZO‐1 protein level.25 Histamine is mainly produced in mast cells, and it binds the H1R receptor to activate actomyosin via RhoA‐ROCK signaling.26 HMGB1 is produced in macrophages and monocytes, and it binds RAGE to activate Src signaling to increase VE‐cadherin phosphorylation.27 IFNγ is produced in natural killer (NK) cells and T cells. It binds the Interferon‐gamma receptor complex to increase caldesmon phosphorylation and to induce caldesmon relocalization to alter the interaction of caldesmon with actin and myosin. At the same time, it induces the reorganization and downregulation of VE‐cadherin.28 IL1β is produced in macrophages, and it binds IL1R to activate RhoA signaling, leading to VE‐cadherin phosphorylation. Integrin ανβ5 blocking was shown to suppress the IL1β‐induced permeability. It was considered that, in this case, the stress fiber formation was integrin ανβ5‐dependent.29 IL6, produced in macrophages, binds the IL6R‐gp130 heterodimer to activate STAT3. STAT3 does not directly regulate the transcription of junction proteins, ZO‐1, and occludin, but upregulates vascular endothelial growth factor (VEGF) expression. Thus, IL6 makes an indirect contribution to permeability.30 IL8 is produced in cells involved in innate immune responses. It binds CXCR1 and CXCR2 to phosphorylate the VEGFR2 receptor in a VEGF‐independent manner. This process is mediated by Src, and VEGFR2 activation leads to RhoA activation to facilitate junction protein reorganization.31 Laminin 2 is a component of the basal membrane and is overexpressed in invasive carcinomas. N‐terminal proteolytic fragment of the γ2 chain has higher permeability than the full‐length protein. It may bind syndecan‐1 to induce actin stress fiber formation via the RhoA‐ROCK pathway.32 A high concentration of LPSs (100 μg/ml) was shown to downregulate claudin‐5 and VE‐cadherin levels, but a low concentration (0.01 μg/ml) increased these protein levels to decrease the permeability. The low‐dose effect was canceled by PI3K/Akt inhibition.33 Mannose‐associated serine protease 1 (MASP‐1) and thrombin interact with protease‐activated receptor 1 (PAR1) to mobilize intracellular Ca2+ and promote myosin light chain phosphorylation, leading to actin rearrangement via ROCK activation to retract cell‐cell junction proteins.34, 35 Platelet‐activating factor (PAF) activates NO synthase to generate NO. NO is used to modify a cysteine residue of vasodilator‐stimulated phosphoprotein (VASP), an actin regulatory protein. In endothelial cells, VASP associates with actin stress fibers, adherens junctions, tight junctions, and focal adhesions. VASP nitrosylation affects the architecture of the cell.36 Pancreatic adenocarcinoma–upregulated factor (PAUF) is expressed in human pancreatic ductal adenocarcinoma. It binds the CXCR4/TLR2 complex to upregulate eNOS and activate Src to increase VE‐cadherin phosphorylation.37 Resistin is produced in PBMCs and macrophages. It binds TLR4 to activate p38, NADPH oxidase, and cAMP‐response element binding (CREB) to reduce ZO‐1 and occludin production.38 S100A4 released from inflammatory cells was found to reduce the expression of occludin.39 S100A8/A9 is also released from inflammatory cells. These proteins share similar structures but require different receptor proteins. S100A8 binds TLR4, whereas S100A9 binds RAGE. These signals induce actin stress fiber formation in a p38– and ERK‐phosphorylation–dependent manner.40 Stem cell factor (SCF) is expressed in many types of cell. It binds cKit receptor to phosphorylate cKit. At the same time, NOS is activated to produce NO, and NO induces β‐catenin and p120‐catenin S‐nitrosylation, leading to the dissociation of these proteins from VE‐cadherin.41 Semaphorin 3A, produced in glioma stem‐like cells, binds the neuropilin 1 (NPR1)–plexin A1 (plxA1) complex to activate Src to internalize VE‐cadherin.42 Secreted protein acidic and rich in cysteine (SPARC) is one of the tumor‐derived permeability factors promoting tumor cell extravasation in the metastatic organ. SPARC binds vascular cell adhesion molecule 1 (VCAM1) on endothelial cells to activate the p38 MAPK cascade.43 TNFα is produced in monocytes and macrophages. TNFα stimulation was found to activate p38 and ERK, and ROCK inhibition prevented an increase of permeability.44 VEGF, produced in cancer cells, binds VEGFR2 to activate Src signaling, leading to VE‐cadherin phosphorylation and VE‐cadherin internalization.45
TABLE 1.
Vascular permeability factors
| Permeability factor | Mechanism | Category (Figure 1A) | Origin | References |
|---|---|---|---|---|
| Ac‐ProGlyPro | VE‐cadherin phosphorylation | 5 | Extracellular matrix | 11 |
| Angiomodulin | Junction protein reorganization | 4 | Human bladder carcinoma cells and fibroblast | 12 |
| Angptl4 | VE‐cadherin, claudin‐5 binding | 1 | Epithelial tumor cells | 13 |
| BMP6 | VE‐cadherin phosphorylation | 5 | Malignant prostate cancer | 14 |
| CCL2 | Junction protein reorganization | 4, 5 | Cancer cells, fibroblast, and endothelial cells | 15 |
| CXCL12 | Reduction of gene expressions | 3 | Fibroblast | 16 |
| CXCL5 | Junction protein reorganization | 4 | Immune cells and fibroblasts | 17 |
| Ephrin‐A1 | EphA1/A2‐EphrinA1 disruption, VE‐cadherin downregulation | 1, 3 | Cancer cells | 18, 19 |
| Fibrinogen | Junction protein reorganization, VE‐cadherin binding | 1, 4 | Liver cells | 20, 21 |
| HGF | ZO‐1 reduction, ZO‐1 phosphorylation | 3, 5 | Cancer cells | 22 |
| Histamine | Junction protein reorganization | 4 | Mast cells | 23 |
| HMGB1 | VE‐cadherin phosphorylation | 5 | Macrophages and monocytes | 24 |
| IFNγ | Junction protein reorganization | 4 | NK cells and T cells | 25 |
| IL‐1β | Junction protein reorganization | 4, 5 | Macrophages | 26 |
| IL‐6 | VEGF upregulation | 5 | Macrophages | 27 |
| IL‐8 | Junction protein reorganization | 4, 5 | Cells involved in innate immune responses | 28 |
| Laminin2 | Junction protein reorganization | 4 | Basal membrane | 29 |
| LPS | VE‐cadherin, claudin‐5 downregulation | 3 | 30 | |
| MASP‐1 | Junction protein reorganization | 4 | Liver cells | 31 |
| PAF | VASP nitrosylation | 4 | Macrophages | 33 |
| PAUF | VE‐cadherin phosphorylation | 5 | Human pancreatic ductal adenocarcinoma | 34 |
| Resistin | ZO‐1, occludin reduction | 3 | PBMCs and macrophages | 35 |
| S100A4 | ZO‐1, occludin reduction | 3 | Inflammatory cells | 36 |
| S100A8 | Junction protein reorganization | 4, 5 | Inflammatory cells | 37 |
| S100A9 | Junction protein reorganization | 4, 5 | Inflammatory cells | 37 |
| SCF | Junction protein reorganization | 4 | Many types of cell | 38 |
| Sema3A | VE‐cadherin phosphorylation | 5 | Glioma stem–like cells | 39 |
| SPARC | Junction protein reorganization | 4 | Cancer cells | 40 |
| Thrombin | Junction protein reorganization | 4 | Liver cells | 32, 46 |
| TNFα | Junction protein reorganization | 4 | Macrophages and monocytes | 41 |
| VEGF | VE‐cadherin phosphorylation | 4, 5 | Cancer cells | 42, 47 |
Abbreviations: MASP‐1, mannose‐associated serine protease 1; NK, natural killer; PAF, platelet‐activating factor; PAUF, atic adenocarcinoma–upregulated factor; SCF, stem cell factor; SPARC, secreted protein acidic and rich in cysteine; TNFα, tumor necrosis factor‐alpha; VASP, vasodilator‐stimulated phosphoprotein; VE, vascular endothelial; VEGF, vascular endothelial growth factor.
FIGURE 1.

Mechanisms of vascular permeability and tumor metastasis. A, Five types of paracellular permeability are illustrated. B, Interactions between tumor cells and endothelial cells are depicted. In B‐2, tumor cell–secreted factors are derived from the primary tumor and circulating tumor cells. C, Glycocalyx protects endothelial cells from tumor cell–secreted factors and tumor cells
3. MECHANISMS OF ACTION OF PERMEABILITY FACTORS
In this review, the focus is on paracellular permeability because tumor cells pass intercellular gaps in the case of endothelial extravasation. There are several types of molecular mechanism that account for enhanced vascular permeability upon stimulation by a permeability factor. These mechanisms are not mutually exclusive, so in some cases one permeability factor can elicit two different effects to increase permeability. Figure 1 shows an outline of the mechanisms of permeability, the details of which are described below.
Permeability factors directly interact with an extracellular domain of adhesion molecules (Figure 1A‐1). Fibrinogen, cAngptl4, and Ephrin‐A1 have such direct binding effect on endothelial cells to disrupt endothelial cell‐cell contacts to increase gaps between endothelial cells.16, 21, 24
Permeability factors digest junction proteins (Figure 1A‐2). Metalloproteinases, such as MMP3, ADAM10, and ADAM17, are expressed in endothelial cells. These enzymes are produced as proenzymes and later processed by other proteases to make active forms. Active enzymes can degrade junction proteins to reduce cell‐cell contact and increase vascular permeability.12, 13, 14
Permeability factors downregulate expression of genes encoding junction proteins (Figure 1A‐3). Many permeability factors intervene in the gene regulatory system of endothelial cells to downregulate junction proteins. The detailed mechanisms have generally not been intensively investigated. In the case of resistin, it is assumed that resistin‐TLR4 signaling finally phosphorylates the CREB protein to reduce ZO‐1 transcription.38
Permeability factors trigger junction protein reorganization (Figures 1, 2, 3A‐4). Many permeability factors can activate intracellular signaling to affect cell morphology. For instance, thrombin can interact with PARs, belonging to a family of seven‐transmembrane receptors expressed on the surface of endothelial cells. PAR activation results in an increase of actin stress fiber formation and cell contraction. This cellular morphological change enlarges intercellular gaps and, as a result, increases endothelial permeability.46
In some cases, junction protein reorganization is accompanied by the phosphorylation of junction proteins (Figures 1, 2, 3A‐5). In particular, the relationship between the phosphorylation of VE‐cadherin and permeability has been extensively studied. VEGF, originally designated as VPF, is the most powerful permeability stimulator.47 VEGF is known to interact with an endothelial surface receptor, VEGFR2, to increase the phosphorylation of VE‐cadherin. Phosphorylated VE‐cadherin is then internalized in the cell, so that the cell‐cell contact is lost.
FIGURE 2.

Hyperpermeability regions in the premetastatic phase. Lungs isolated from tumor‐bearing mouse after Evans blue dye was injected via the tail vein. Hyperpermeable regions were stained deeply. Note that these regions were not observed in non–tumor‐bearing mouse lungs. In the case of CCL2 knockout mice, such hyperpermeable regions were not observed because tumor‐dependent vascular permeability was mediated by the CCL2‐CCR2 signaling pathway. Immunostaining revealed that CCL2 expression was upregulated in the hyperpermeable region. In this region, fibrinogen deposition was also observed (upper panel). The CCL2 and CCR2 system regulates permeability accompanied by cytokines and inflammatory factors via the toll‐like 4/MD2 complex. Endothelial contractility is regulated by focal adhesion kinase (FAK) (lower panel)
FIGURE 3.

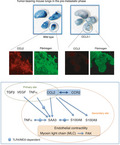
Hepato‐entrained natural killer (NK) cells. System‐tracing NK cells in liver to lungs. The liver of mice expressing the photoconvertible protein Kikume Green‐Red (KikGR) was exposed to violet light. Some of the liver NK cells were educated by primary tumor–derived factors exhibiting FX expression and relocated to the lungs (upper panel). In the lungs, educated NK cells were observed in the hyperpermeable region where fibrinogen deposits accumulated (middle panel). The fibrinogen was eliminated by FXa in the NK cells, resulting in the suppression of metastasis (lower panel)
4. METASTASIS AND VASCULAR PERMEABILITY
Next, interactions between tumor cells and endothelial cells are discussed. Tumor cells press to increase permeability in a remote organ where metastasis tends to occur. The interactions are classified into three types. The first is direct, but the other two are indirect interactions.
1. Tumor‐cell–mediated permeability (Figure 1B‐1)
Tumor cells directly attaching to an endothelial cell can modulate its barrier function. Amyloid precursor protein expressed on the surface of tumor cells was shown to bind death receptor 6 (DR6), also known as tumor necrosis factor receptor superfamily member 21 (TNFRSF21), to promote endothelial cell death. This type of cell death is receptor‐interacting serine/threonine‐protein kinase 3 (RIPK3)‐dependent programmed cell death, which is referred to as necroptosis. Tumor cells, capable of inducing endothelial necroptosis, enhance vascular permeability and their extravasation in the metastatic organ.48
2. Permeability mediated by primary tumor–secreted factors (Figure 1B‐2)
Primary tumor and tumor‐associated cells secrete many molecules, some of which reach distant organs where they act as permeability factors (Table 1). The molecular species may vary depending on the type and stage of the primary tumor. A mouse model study using lung endothelial cell–specific blockade of focal adhesion kinase (FAK) found that tumor‐derived hyperpermeability was not observed in this mouse lung. Moreover, labeled tumor cell homing assay revealed that tumor cell recruitment in these gene‐modified mice was lower than in wild‐type mice.49
3. Resident cell–mediated permeability (Figure 1B‐3)
As stated above, tumor cells secrete inflammatory cytokines, which influence resident cells. Some of them directly bind to endothelial cells, but others may modulate other tissue‐resident cells. In mouse lungs, alveolar macrophages and lung endothelial cells release a tissue‐specific molecule, serum amyloid A3 (SAA3), upon stimulation of tumor‐secreted factors. SAA3 further acts as a TLR4 ligand in alveolar epithelial type II cells to generate TNFα, and locally produced TNFα increases vascular permeability.50 To confirm the importance of resident cells, we performed an experiment in which bone marrow–derived cells (BMDCs), isolated from tumor‐bearing transgenic mice that expressed green fluorescent protein (GFP), were injected into the tail vein of non–tumor‐bearing and tumor‐bearing wild‐type mice to determine the distribution in the lungs. BMDCs in tumor‐bearing mice had to be primed by the primary tumor so that their behavior was controlled by tumor‐derived factors, but their pattern of spread in the non–tumor‐bearing mouse lungs was very low with a diffuse pattern compared with that in tumor‐bearing lungs. This indicated that resident endothelial cell–triggered permeability was induced by a remote primary tumor (unpublished data).
4. Glycocalyx
Glycocalyx is another important element regarding vascular permeability (Figure 1C). The luminal surface of endothelial cells is covered by glycocalyx, which is composed of proteoglycans, heparan sulfate, chondroitin sulfate, and hyaluronic acid. In healthy cells, the glycocalyx lining completely covers the cell surface to function as a barrier for endothelial cells.51 These extracellular matrixes capture exotic molecules, so that permeability factors cannot easily access cell surface receptors. In inflammation, glycocalyx components are degraded by heparanase, MMP, and thrombin, and endothelial cell surface receptors are exposed in the blood flow to allow permeability factors to access endothelial cells. Cleaved fragments of glycocalyx components may function as permeability factors. In a recent study, it was demonstrated that thrombin‐cleaved syndecan‐3/4 ectodomain fragments increased permeability in a Rho‐kinase–dependent manner.52 The endothelial barrier function is also effective for protecting endothelial cells from circulating tumor cells. Cell adhesion molecules expressed on endothelial cells are buried in the glycocalyx layer in a normal state. Circulating tumor cells capable of binding to intracellular adhesion molecule 1 (ICAM‐1) or P‐selectin cannot access the endothelial cells until the thickness of the glycocalyx layer is reduced.53
5. ANALYZING THE HYPERPERMEABLE REGION IN A MOUSE MODEL SYSTEM
The premetastatic soil has been studied in several organs, and it is widely believed that almost all organs might be influenced by primary tumors.54, 55 Molecular analysis has proven that proteins, exosomes, DNA, RNA, as well as further degradation of proteins may play important roles in premetastatic soil formation.9, 56 Cytokines were first recognized as signature molecules responsible for the formation of premetastatic soil, and permeability factors including VEGF, transforming growth factor‐β (TGF‐β), tumor necrosis factor‐alpha (TNFα), and SDF‐1 were also revealed to be involved in its formation.7, 8, 57, 58 To characterize the premetastatic region, Evans blue dye injection in mouse tail has been utilized for visualization. The assay results showed a blue gradation image of the lungs, indicating dye leakage in the stromal tissue (Figure 2). The dark blue area has higher permeability than the light blue area because the accumulation of Evans blue dye stains the area dark blue. Lung metastasis assay, with the injection of labeled tumor cells via the tail vein, performed after the Evans blue injection, revealed that metastasis was prone to occur in the hyperpermeable area. Comparisons of dye leakage patterns between non–tumor‐bearing and tumor‐bearing mouse lungs showed that the former lungs had a much smaller hyperpermeable area than the latter lungs, suggesting that the occurrence of the area relies on a tumor‐bearing state. Comparisons of hyperpermeable and less permeable areas revealed that the expression level of chemokine CC motif ligand 2 (CCL2) was upregulated in the hyperpermeable area.59 As indicated in Table 1, a CCL2–CC motif chemokine receptor 2 (CCR2) system triggers junction protein reorganization to increase vascular permeability. Our study confirmed the phosphorylation of FAK, which is an upstream reaction of VE‐cadherin phosphorylation, in the hyperpermeable area. In addition, an endothelial‐specific CCR2‐deficient model clearly demonstrated that endothelial CCR2 expression is required for tumor cell extravasation and pulmonary metastasis via permeability.18
6. FIBRINOGEN DEPOSITION IN THE HYPERPERMEABLE AREA
We also focused on fibrinogen deposition in the hyperpermeable area in lungs taken from tumor‐bearing mice. Fibrinogen was also reported as a vascular permeable factor (Table 1). We recently found that a subgroup of NK cells with fibrinogen‐binding molecules was enriched in the hyperpermeable and fibrinogen deposition–rich area (Figure 3).59 These cells were first primed in the tumor‐bearing mouse liver to gain coagulation factor 10 (FX) with the ability to eliminate fibrinogen, and then efficiently relocated from the liver to the lungs. The educated NK cells are a specific population possessing the ability to eliminate fibrinogen because it has been reported that tumor cells with fibrinogen protected the tumoricidal activity of NK cells.60, 61 The problem to be solved in this context is that this relocation into premetastatic lungs decreases as the primary tumor progresses.62 This indicates that the character of these NK cells is affected by the primary tumor to some extent, which can potentially be altered by introducing external factors.
7. PERSPECTIVES
Distinct premetastatic hyperpermeable soil was detected by the deposition of fibrinogen in a mouse model. This soil contains various different immune cells as a result of the primary tumor–derived stimulation; however, unfortunately, many cells are considered to be “protumor” or “immunosuppressive,” but only a fraction of “antitumor” cells are present in the region. Ideally, antimetastatic immune cells should have strong tumoricidal activity and mobility toward the pre‐/postmetastatic soil to eliminate the premetastatic soil. An imminent solution should be adaptive transfer of the subgroup of educated NK. It is also worth trying to improve NK cells after expansion by tissue culture techniques before transplantation. Although very limited data are available for human samples, a fibrinogen‐positive area and fibrinogen‐binding molecule–expressing immune cells were found in cancer patients. These findings are very similar to those in mouse model studies. This type of cell in human specimens has not been identified, but it is highly possible that single‐cell transcriptomic analysis could reveal a subgroup of NK cells with the ability to eliminate fibrinogen deposition. In addition, because the cancer‐related fibrinogen may be modified, the sweeper mechanism would be effectively developed in NK cells.
CONFLICT OF INTEREST
The authors have no conflict of interest.
Tomita T, Kato M, Hiratsuka S. Regulation of vascular permeability in cancer metastasis. Cancer Sci. 2021;112:2966–2974. 10.1111/cas.14942
REFERENCES
- 1.Arnold M, Rutherford MJ, Bardot A, et al. Progress in cancer survival, mortality, and incidence in seven high‐income countries 1995–2014 (ICBP SURVMARK‐2): a population‐based study. Lancet Oncol. 2019;20(11):1493‐1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dillekås H, Rogers MS, Straume O. Are 90% of deaths from cancer caused by metastases? Cancer Med. 2019;8(12):5574‐5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Friberg S, Nyström A. Cancer metastases: early dissemination and late recurrences. Cancer Growth Metastasis. 2015;8:43‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weng D, Penzner JH, Song B, Koido S, Calderwood SK, Gong J. Metastasis is an early event in mouse mammary carcinomas and is associated with cells bearing stem cell markers. Breast Cancer Res. 2012;14(1):R18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hüsemann Y, Geigl JB, Schubert F, et al. Systemic spread is an early step in breast cancer. Cancer Cell. 2008;13(1):58‐68. [DOI] [PubMed] [Google Scholar]
- 6.Liu Y, Cao X. Characteristics and significance of the pre‐metastatic niche. Cancer Cell. 2016;30(5):668‐681. [DOI] [PubMed] [Google Scholar]
- 7.Hiratsuka S, Nakamura K, Iwai S, et al. MMP9 induction by vascular endothelial growth factor receptor‐1 is involved in lung‐specific metastasis. Cancer Cell. 2002;2(4):289‐300. [DOI] [PubMed] [Google Scholar]
- 8.Kaplan RN, Riba RD, Zacharoulis S, et al. VEGFR1‐positive haematopoietic bone marrow progenitors initiate the pre‐metastatic niche. Nature. 2005;438(7069):820‐827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peinado H, Zhang H, Matei IR, et al. Pre‐metastatic niches: organ‐specific homes for metastases. Nat Rev Cancer. 2017;17(5):302‐317. [DOI] [PubMed] [Google Scholar]
- 10.Wallez Y, Huber P. Endothelial adherens and tight junctions in vascular homeostasis, inflammation and angiogenesis. Biochim Biophys Acta. 2008;1778(3):794‐809. [DOI] [PubMed] [Google Scholar]
- 11.Hahn CS, Scott DW, Xu X, et al. The matrikine N‐α‐PGP couples extracellular matrix fragmentation to endothelial permeability. Sci Adv. 2015;1(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schulz B, Pruessmeyer J, Maretzky T, et al. ADAM10 regulates endothelial permeability and T‐Cell transmigration by proteolysis of vascular endothelial cadherin. Circ Res. 2008;102(10):1192‐1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koenen RR, Pruessmeyer J, Soehnlein O, et al. Regulated release and functional modulation of junctional adhesion molecule A by disintegrin metalloproteinases. Blood. 2009;113(19):4799‐4809. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Q, Zheng M, Betancourt CE, et al. Increase in blood‐brain barrier (BBB) permeability is regulated by MMP3 via the ERK signaling pathway. Oxid Med Cell Longev. 2021;2021:6655122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Komiya E, Sato H, Watanabe N, et al. Angiomodulin, a marker of cancer vasculature, is upregulated by vascular endothelial growth factor and increases vascular permeability as a ligand of integrin αvβ3. Cancer Med. 2014;3(3):537‐549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang R‐L, Teo Z, Chong HC, et al. ANGPTL4 modulates vascular junction integrity by integrin signaling and disruption of intercellular VE‐cadherin and claudin‐5 clusters. Blood. 2011;118(14):3990‐4002. [DOI] [PubMed] [Google Scholar]
- 17.Benn A, Bredow C, Casanova I, Vukičević S, Knaus P. VE‐cadherin facilitates BMP‐induced endothelial cell permeability and signaling. J Cell Sci. 2016;129(1):206‐218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roblek M, Protsyuk D, Becker PF, et al. CCL2 is a vascular permeability factor inducing CCR2‐dependent endothelial retraction during lung metastasis. Mol Cancer Res. 2019;17(3):783‐793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ahirwar DK, Nasser MW, Ouseph MM, et al. Fibroblast‐derived CXCL12 promotes breast cancer metastasis by facilitating tumor cell intravasation. Oncogene. 2018;37(32):4428‐4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu M, Ma X, Jiang D, Wang L, Zhan Q, Zhao J. CXC chemokine ligand 5 (CXCL5) disrupted the permeability of human brain microvascular endothelial cells via regulating p38 signal. Microbiol Immunol. 2021;65(1):40‐47. [DOI] [PubMed] [Google Scholar]
- 21.Ieguchi K, Tomita T, Omori T, et al. ADAM12‐cleaved ephrin‐A1 contributes to lung metastasis. Oncogene. 2014;33(17):2179‐2190. [DOI] [PubMed] [Google Scholar]
- 22.Ieguchi K, Tomita T, Takao T, et al. Analysis of ADAM12‐mediated Ephrin‐A1 cleavage and its biological functions. Int J Mol Sci. 2021;22(5):2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tyagi N, Roberts AM, Dean WL, Tyagi SC, Lominadze D. Fibrinogen induces endothelial cell permeability. Mol Cell Biochem. 2008;307(1–2):13‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sahni A, Arévalo MT, Sahni SK, Simpson‐Haidaris PJ. The VE‐cadherin binding domain of fibrinogen induces endothelial barrier permeability and enhances transendothelial migration of malignant breast epithelial cells. Int J Cancer. 2009;125(3):577‐584. [DOI] [PubMed] [Google Scholar]
- 25.Martin TA, Mansel RE, Jiang WG. Antagonistic effect of NK4 on HGF/SF induced changes in the transendothelial resistance (TER) and paracellular permeability of human vascular endothelial cells. J Cell Physiol. 2002;192(3):268‐275. [DOI] [PubMed] [Google Scholar]
- 26.Ehringer WD, Edwards MJ, Miller FN. Mechanisms of alpha‐thrombin, histamine, and bradykinin induced endothelial permeability. J Cell Physiol. 1996;167(3):562‐569. [DOI] [PubMed] [Google Scholar]
- 27.Huang W, Liu Y, Li L, et al. HMGB1 increases permeability of the endothelial cell monolayer via RAGE and Src family tyrosine kinase pathways. Inflammation. 2012;35(1):350‐362. [DOI] [PubMed] [Google Scholar]
- 28.Ng CT, Fong LY, Yong YK, Hakim MN, Ahmad Z. Interferon‐γ induces biphasic changes in caldesmon localization as well as adherens junction organization and expression in HUVECs. Cytokine. 2018;111:541‐550. [DOI] [PubMed] [Google Scholar]
- 29.Ganter MT, Roux J, Miyazawa B, et al. Interleukin‐1beta causes acute lung injury via alphavbeta5 and alphavbeta6 integrin‐dependent mechanisms. Circ Res. 2008;102(7):804‐812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yun J‐H, Park SW, Kim K‐J, et al. Endothelial STAT3 activation increases vascular leakage through downregulating tight junction proteins: implications for diabetic retinopathy. J Cell Physiol. 2017;232(5):1123‐1134. [DOI] [PubMed] [Google Scholar]
- 31.Petreaca ML, Yao M, Liu Y, Defea K, Martins‐Green M. Transactivation of vascular endothelial growth factor receptor‐2 by interleukin‐8 (IL‐8/CXCL8) is required for IL‐8/CXCL8‐induced endothelial permeability. Mol Biol Cell. 2007;18(12):5014‐5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sato H, Oyanagi J, Komiya E, Ogawa T, Higashi S, Miyazaki K. Amino‐terminal fragments of laminin γ2 chain retract vascular endothelial cells and increase vascular permeability. Cancer Sci. 2014;105(2):168‐175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zheng X, Zhang W, Hu X. Different concentrations of lipopolysaccharide regulate barrier function through the PI3K/Akt signalling pathway in human pulmonary microvascular endothelial cells. Sci Rep. 2018;8(1):9963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Debreczeni ML, Németh Z, Kajdácsi E, et al. MASP‐1 increases endothelial permeability. Front Immunol. 2019;10:991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Nieuw Amerongen GP, Draijer R, Vermeer MA, van Hinsbergh VW. Transient and prolonged increase in endothelial permeability induced by histamine and thrombin: role of protein kinases, calcium, and RhoA. Circ Res. 1998;83(11):1115‐1123. [DOI] [PubMed] [Google Scholar]
- 36.Zamorano P, Marín N, Córdova F, et al. nitrosylation of VASP at cysteine 64 mediates the inflammation‐stimulated increase in microvascular permeability. Am J Physiol Heart Circ Physiol. 2017;313(1):H66‐H71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim SJ, Lee Y, Kim NY, et al. atic adenocarcinoma upregulated factor, a novel endothelial activator, promotes angiogenesis and vascular permeability. Oncogene. 2013;32(31):3638‐3647. [DOI] [PubMed] [Google Scholar]
- 38.Jamaluddin MS, Yan S, Lü J, Liang Z, Yao Q, Chen C. Resistin increases monolayer permeability of human coronary artery endothelial cells. PLoS One. 2013;8(12):e84576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nishioku T, Furusho K, Tomita A, et al. Potential role for S100A4 in the disruption of the blood‐brain barrier in collagen‐induced arthritic mice, an animal model of rheumatoid arthritis. Neuroscience. 2011;189:286‐292. [DOI] [PubMed] [Google Scholar]
- 40.Wang L, Luo H, Chen X, Jiang Y, Huang Q. Functional characterization of S100A8 and S100A9 in altering monolayer permeability of human umbilical endothelial cells. PLoS One. 2014;9(3):e90472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim JY, Choi J‐S, Song S‐H, et al. Stem cell factor is a potent endothelial permeability factor. Arterioscler Thromb Vasc Biol. 2014;34(7):1459‐1467. [DOI] [PubMed] [Google Scholar]
- 42.Le Guelte A, Galan‐Moya E‐M, Dwyer J, et al. Semaphorin 3A elevates endothelial cell permeability through PP2A inactivation. J Cell Sci. 2012;125(Pt 17):4137‐4146. [DOI] [PubMed] [Google Scholar]
- 43.Tichet M, Prod’Homme V, Fenouille N, et al. Tumour‐derived SPARC drives vascular permeability and extravasation through endothelial VCAM1 signalling to promote metastasis. Nat Commun. 2015;6:6993. [DOI] [PubMed] [Google Scholar]
- 44.Nwariaku FE, Rothenbach P, Liu Z, Zhu X, Turnage RH, Terada LS. Rho inhibition decreases TNF‐induced endothelial MAPK activation and monolayer permeability. J Appl Physiol. 2003;95(5):1889‐1895. [DOI] [PubMed] [Google Scholar]
- 45.Gavard J, Gutkind JS. VEGF controls endothelial‐cell permeability by promoting the beta‐arrestin‐dependent endocytosis of VE‐cadherin. Nat Cell Biol. 2006;8(11):1223‐1234. [DOI] [PubMed] [Google Scholar]
- 46.Amado‐Azevedo J, de Menezes RX, van Nieuw Amerongen GP, van Hinsbergh VWM, Hordijk PL. A functional siRNA screen identifies RhoGTPase‐associated genes involved in thrombin‐induced endothelial permeability. PLoS One. 2018;13(7):e0201231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Senger DR, Galli SJ, Dvorak AM, Perruzzi CA, Harvey VS, Dvorak HF. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science. 1983;219(4587):983‐985. [DOI] [PubMed] [Google Scholar]
- 48.Strilic B, Yang L, Albarrán‐Juárez J, et al. Tumour‐cell‐induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature. 2016;536(7615):215‐218. [DOI] [PubMed] [Google Scholar]
- 49.Hiratsuka S, Goel S, Kamoun WS, et al. Endothelial focal adhesion kinase mediates cancer cell homing to discrete regions of the lungs via E‐selectin up‐regulation. Proc Natl Acad Sci USA. 2011;108(9):3725‐3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tomita T, Sakurai Y, Ishibashi S, Maru Y. Imbalance of Clara cell‐mediated homeostatic inflammation is involved in lung metastasis. Oncogene. 2011;30(31):3429‐3439. [DOI] [PubMed] [Google Scholar]
- 51.Chelazzi C, Villa G, Mancinelli P, De Gaudio AR, Adembri C. Glycocalyx and sepsis‐induced alterations in vascular permeability. Crit Care. 2015;19:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jannaway M, Yang X, Meegan JE, Coleman DC, Yuan SY. Thrombin‐cleaved syndecan‐3/‐4 ectodomain fragments mediate endothelial barrier dysfunction. PLoS One. 2019;14(5):e0214737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mensah SA, Nersesyan AA, Harding IC, et al. Flow‐regulated endothelial glycocalyx determines metastatic cancer cell activity. FASEB J. 2020;34(5):6166‐6184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Costa‐Silva B, Aiello NM, Ocean AJ, et al. atic cancer exosomes initiate pre‐metastatic niche formation in the liver. Nat Cell Biol. 2015;17(6):816‐826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Eveno C, Hainaud P, Rampanou A, et al. Proof of prometastatic niche induction by hepatic stellate cells. J Surg Res. 2015;194(2):496‐504. [DOI] [PubMed] [Google Scholar]
- 56.McAllister SS, Weinberg RA. The tumour‐induced systemic environment as a critical regulator of cancer progression and metastasis. Nat Cell Biol. 2014;16(8):717‐727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hiratsuka S, Watanabe A, Aburatani H, Maru Y. Tumour‐mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat Cell Biol. 2006;8(12):1369‐1375. [DOI] [PubMed] [Google Scholar]
- 58.Seubert B, Grünwald B, Kobuch J, et al. Tissue inhibitor of metalloproteinases (TIMP)‐1 creates a premetastatic niche in the liver through SDF‐1/CXCR4‐dependent neutrophil recruitment in mice. Hepatology. 2015;61(1):238‐248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hiratsuka S, Ishibashi S, Tomita T, et al. Primary tumours modulate innate immune signalling to create pre‐metastatic vascular hyperpermeability foci. Nat Commun. 2013;4:1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Palumbo JS, Talmage KE, Massari JV, et al. Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell‐mediated elimination of tumor cells. Blood. 2005;105(1):178‐185. [DOI] [PubMed] [Google Scholar]
- 61.Zheng S, Shen J, Jiao Y, et al. Platelets and fibrinogen facilitate each other in protecting tumor cells from natural killer cytotoxicity. Cancer Sci. 2009;100(5):859‐865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hiratsuka S, Tomita T, Mishima T, et al. Hepato‐entrained B220+CD11c+NK1.1+ cells regulate pre‐metastatic niche formation in the lung. EMBO Mol Med. 2018;10(7):e8643. [DOI] [PMC free article] [PubMed] [Google Scholar]