

Abstract
Cancer cells use autophagy for growth, survival, and cytoprotection from chemotherapy. Therefore, autophagy inhibitors appear to be good candidates for cancer treatment. Our group previously reported that macrolide antibiotics, especially azithromycin (AZM), have potent autophagy inhibitory effects, and combination treatment with tyrosine kinase inhibitors or proteasome inhibitors enhances their anti–cancer activity. In this study, we evaluated the effect of combination therapy with DNA‐damaging drugs and AZM in non–small‐cell lung cancer (NSCLC) cells. We found that the cytotoxic activities of DNA‐damaging drugs, such as doxorubicin (DOX), etoposide, and carboplatin, were enhanced in the presence of AZM in NSCLC cell lines, whereas AZM alone exhibited almost no cytotoxicity. This enhanced cell death was dependent on wild‐type‐p53 status and autophagosome‐forming ability because TP53 knockout (KO) and ATG5‐KO cells attenuated AZM‐enhanced cytotoxicity. DOX treatment upregulated lysosomal biogenesis by activating TFEB and led to lysosomal membrane damage as assessed by galectin 3 puncta assay and cytoplasmic leakage of lysosomal enzymes. In contrast, AZM treatment blocked autophagy, which resulted in the accumulation of lysosomes/autolysosomes. Thus, the effects of DOX and AZM were integrated into the marked increase in damaged lysosomes/autolysosomes, leading to prominent lysosomal membrane permeabilization (LMP) for apoptosis induction. Our data suggest that concomitant treatment with DNA‐damaging drugs and AZM is a promising strategy for NSCLC treatment via pronounced LMP induction.
Keywords: autophagy, azithromycin, lysosomal membrane permeabilization, p53, non–small‐cell lung cancer
In this paper, we showed that coadministration of DNA‐damaging drugs and azithromycin (AZM), a macrolide antibiotic, enhanced cytotoxicity due to increased lysosomal membrane permeabilization (LMP). Because DNA‐damaging drugs induce LMP, cancer cells activate autophagy to remove damaged lysosomes by lysophagy. However, AZM inhibited lysophagy, as well as autophagy, resulting in the prominent cytoplasmic accumulation of damaged lysosomes for induction of LMP‐mediated apoptosis in non–small‐cell lung cancer cells.
1. INTRODUCTION
The number of patients with lung cancer and its related mortality rate are increasing worldwide. In men, lung cancer is the most frequent cancer and the leading cause of cancer death.1 New drugs, such as tyrosine kinase inhibitors (TKI) and immune checkpoint inhibitors (ICI), have been developed for lung cancer treatment, leading to more therapeutic options.2, 3, 4 However, the indications for these drugs are limited and they not applicable in patients exhibiting interstitial pneumonia and carrying no targeting gene mutations. In addition, cancer cells exposed to TKI frequently acquire new mutations, leading to acquired resistance to TKI. Thus, DNA‐damaging drugs are still important in the treatment of such patients in the clinical setting.5, 6
Macroautophagy, hereafter referred to as autophagy, is a mechanism that degrades intracellular proteins and organelles by engulfing them with a lipid bilayer called autophagosome, these cargos are then transported and fuse with lysosomes that contain a number of hydrolases for digestion.7 Recently, the roles of autophagy in cancer cells have been clarified in many aspects.8 Genetic disruption of autophagic genes revealed that autophagy promotes tumor growth and malignancy.9 In addition, it has become evident that cancer cells use autophagy for cytoprotection against anti–cancer drugs. Autophagy is required for cancer cells to maintain their dormant state, which allows them to survive chemotherapy.10, 11 Autophagy also suppressed apoptosis by degrading pro–apoptotic proteins, such as NOXA and PUMA; therefore, inhibition of autophagy will promote the accumulation of pro–apoptotic proteins, resulting in cancer cell death by the autophagy inhibitor itself or by the combination of TNF‐related apoptosis‐inducing ligand (TRAIL) and autophagy‐related gene knockdown.12, 13 Hence, autophagy is a good target for cancer therapy, and many clinical trials for cancer chemotherapy in combination with an autophagy inhibitor, hydroxychloroquine (HCQ), are ongoing.14, 15, 16, 17, 18 Although HCQ is the only clinically available autophagy inhibitor, severe retinopathy and cardiomyopathy have been reported as adverse events of HCQ treatment.19, 20 Therefore, the development of effective and safer autophagy inhibitors is important for expanding the therapeutic options for patients with cancer.
Our group has reported that macrolide antibiotics, especially azithromycin (AZM), have a potent autophagy inhibitory effect, and that their combined use with TKI or proteasome inhibitors enhances cytotoxicity in various cancer cells, including multiple myeloma cells and pancreatic cancer cells.21, 22, 23 In this study, we examined the effect of combination therapy of DNA‐damaging drugs and a macrolide antibiotic in non–small‐cell lung cancer (NSCLC) cells.
2. MATERIALS AND METHODS
Lung cancer cell lines (A549, H226, A427, and H596) were treated with doxorubicin (DOX), etoposide (ETP), gemcitabine (GEM), or carboplatin (CBDCA) with or without azithromycin (AZM), and cell death was assessed using a live cell imaging system. Cytotoxic effects, cell death phenotypes, gene expression, and lysosomal damage induced by DNA‐damaging drugs in combination with AZM were also examined by flow cytometry, May‐Grünwald‐Giemsa staining, western blotting, immunofluorescence staining, transmission electron microscopy, real‐time quantitative PCR, and LMP assay. We also established A549/mCherry‐EGFP‐LC3, TP53‐KO A549, ATG5‐KO A549, and NOXA‐KO A549 cells using the CRISPR/Cas9 system. Details are provided in Document S1.
3. RESULTS
3.1. Azithromycin enhanced the cytotoxicity of DNA‐damaging drugs via apoptosis
The cytotoxicity of DNA‐damaging drugs DOX, ETP, CBDCA, and GEM in combination with AZM in A549, H226, and A427 lung cancer cell lines was investigated by counting the number of PI‐positive non–viable cells using a live‐cell imaging system. In A549 cells, AZM treatment alone did not induce apparent cell death, but coadministration of AZM with DOX, ETP, or CBDCA resulted in higher cell death than DOX, ETP, or CBDCA alone (Figure 1A, Figure S1A). In H226 and A427 cells, single treatment with AZM showed weak cytotoxicity, but coadministration of AZM with either DOX, ETP, or CBDCA resulted in higher cell death induction as compared to AZM or DNA‐damaging drugs alone (Figure 1B,C and Figure S1A). However, AZM did not enhance GEM‐induced cell death (Figure S1B,C). We compared the autophagy inhibitory effect between AZM and HCQ, a well‐known autophagy inhibitor. Western blotting revealed that both treatments accumulated LC3B‐II protein at the same level, indicating an equivalent autophagy inhibitory effect (Figure S2A). In addition, we compared the enhanced cytotoxicity of DOX and CBDCA by coadministration of AZM or HCQ (Figure S2B). Although HCQ enhanced the cytotoxicity of DOX and CBDCA, the cytotoxicity of HCQ itself was stronger than that of AZM, making it difficult to evaluate whether the enhanced cytotoxicity was just additive or was more than additive. These data showed that AZM itself is less toxic than HCQ. Taken together, our findings indicate that AZM enhanced cell death induced by major DNA‐damaging drugs used in the clinical setting of NSCLC cell lines. Because A549 cells showed less cytotoxicity with AZM among these cell lines, we used A549 cells in the subsequent experiments.
FIGURE 1.
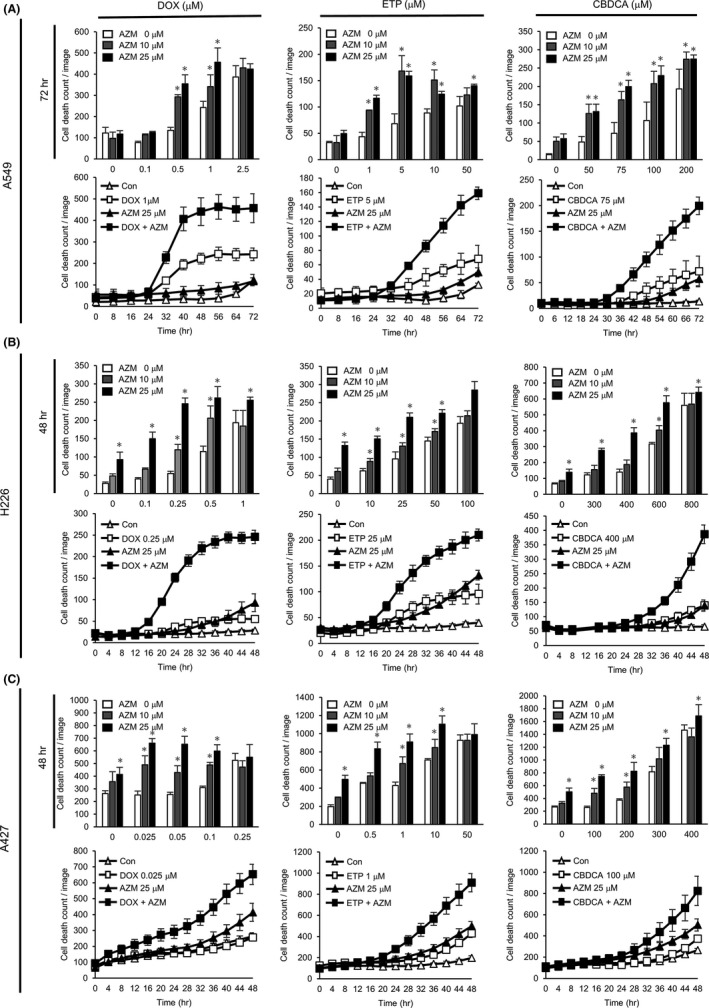
Azithromycin (AZM) enhanced doxorubicin (DOX), etoposide (ETP), and carboplatin (CBDCA)‐induced cytotoxicity in lung cancer cell lines. (A–C) A549, H226, and A427 cell death count monitored by live‐cell imaging with propidium iodide (PI) staining, following treatment with DOX, ETP, or CBDCA in combination with AZM for up to 48 or 72 h. Dose‐dependent and time‐dependent cell death numbers are shown. Dose‐dependent cell death counts at 48 or 72 h after the treatment are summarized in the top panels. Each panel shows representative data for three independent experiments. n = 4, bar = mean ± SD *P < .05 vs 0 µM AZM treatment
To determine the type of cell death induced by coadministration of AZM, we assessed the cell morphology of A549 cells using May‐Grünwald‐Giemsa staining after 72 h of drug treatment. Cells treated with DOX alone and DOX + AZM showed fragmented nuclei and chromatin condensation (Figure 2A), which are characteristic features of cells undergoing apoptosis. We also performed flow cytometry with Annexin V/PI double staining. Although AZM treatment alone did not result in Annexin V‐stained cells, combined treatment with AZM and DOX or ETP increased the number of Annexin V‐positive/PI‐negative and Annexin V‐positive/PI‐positive cells as compared to DOX or ETP alone at 48 and 72 hours (Figure 2B). In addition, the induction of cell death by the coadministration of DOX + AZM and DOX alone was significantly suppressed in the presence of Z‐VAD‐FMK, a pan‐caspase inhibitor (Figure 2C). Western blot analysis also showed that AZM enhanced the DOX‐induced cleavage of PARP and cleavage of caspase‐3 and caspase‐7 (Figure 2D). These data suggest that AZM enhanced the induction of apoptosis by DNA‐damaging drugs.
FIGURE 2.
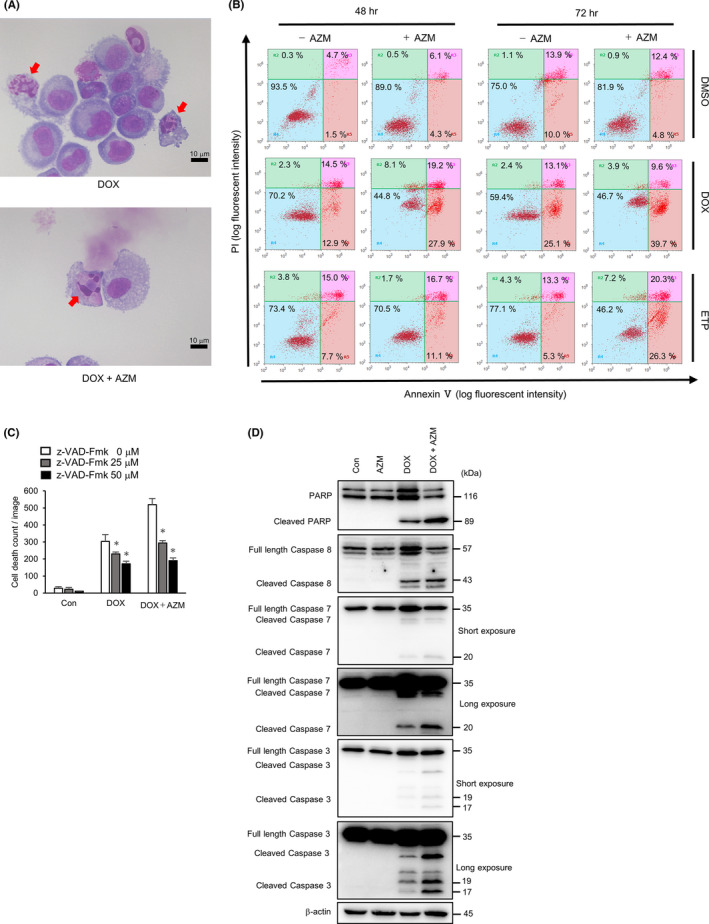
Azithromycin (AZM) enhanced apoptotic cell death in A549 cells by combination treatment with doxorubicin (DOX) or etoposide (ETP). A, Representative images of May‐Grünwald‐Giemsa stained A549 cells after DOX (1 µM) ± AZM (25 µM) treatment for 72 h. Red arrows indicate typical apoptotic dead cells. Scale bar = 10 µm. B, Flow cytometric analysis of Annexin V/PI double‐stained A549 cells treated with DOX (1 µM) or ETP (5 µM) ± AZM (25 µM) for 48 and 72 h. The vertical axis indicates the log fluorescence intensity of propidium iodide (PI), while the horizontal axis indicates the log fluorescence intensity of Annexin V. Numbers indicate the percentage of the cell numbers in each area. Representative results of three independent experiments are shown. C, A549 cells were treated with DOX (1 µM) ± AZM (25 µM), in the presence of Z‐VAD‐FMK (0, 25, 50 µM), for 48 h, and number of dead cells was assessed by IncuCyte with PI staining. n = 4, bar = mean ± SD, *P < .05. D, Cleavage of PARP and activation of caspase‐3, caspase‐7, and caspase‐8 were assessed by western blotting after A549 cells were treated with AZM (25 µM), DOX (1 µM), and the combination for 48 h. Short and long exposure images for caspase‐3 and caspase‐7 are shown. β‐actin was used as control
3.2. Enhanced cell death by combined treatment with doxorubicin and azithromycin was p53‐dependent but NOXA‐independent
A549, H226, and A427 cells carry wild‐type TP53.24 DOX and ETP induce DNA damage, and it was known that both drugs activate p53, resulting in the upregulation of downstream pro–apoptotic genes. To verify whether this is true, we performed western blot analysis. The results showed that DOX and ETP treatment increased p53 and phospho‐p53, as well as MDM2 and p21, which are transcriptionally regulated by p53 in A549 cells, but not in H596 cells carrying TP53 mutation (G245C) (Figure S3A). Thus, p53 in H596 was non–functional, and the enhancing effect of AZM on DOX‐induced or ETP‐induced cell death was much lower than that in A549 cells (Figure S3B). The same results were obtained by flow cytometry with Annexin V/PI staining after the combination treatment, showing no difference in the number of live cells in the DOX‐treated or ETP‐treated fraction, in the presence or absence of AZM (Figure S3C). To further confirm this observation, we established TP53‐KO A549 cells using the CRISPR‐Cas9 system (Figure 3A) and evaluated cell death after the coadministration of AZM and DOX (Figure 3B). In wild‐type (WT) A549 cells, AZM enhanced DOX‐induced cell death at 48 and 72 hours, whereas in TP53‐KO A549 cells, no significant enhancement in DOX cytotoxicity occurred with the coadministration of AZM (Figure 3B,C). This was supported by flow cytometry results, showing that AZM did not enhance DOX‐induced cell death in TP53‐KO A549 cells (Figure 3D). Taken together, these results demonstrated that cell death enhancement by AZM is p53‐dependent. TP53‐KO cells started to die after 32 hours of exposure to DOX, which indicated that long exposure was required to obtain the same cytotoxicity effect observed in WT A549 cells (Figure 3C,D).
FIGURE 3.
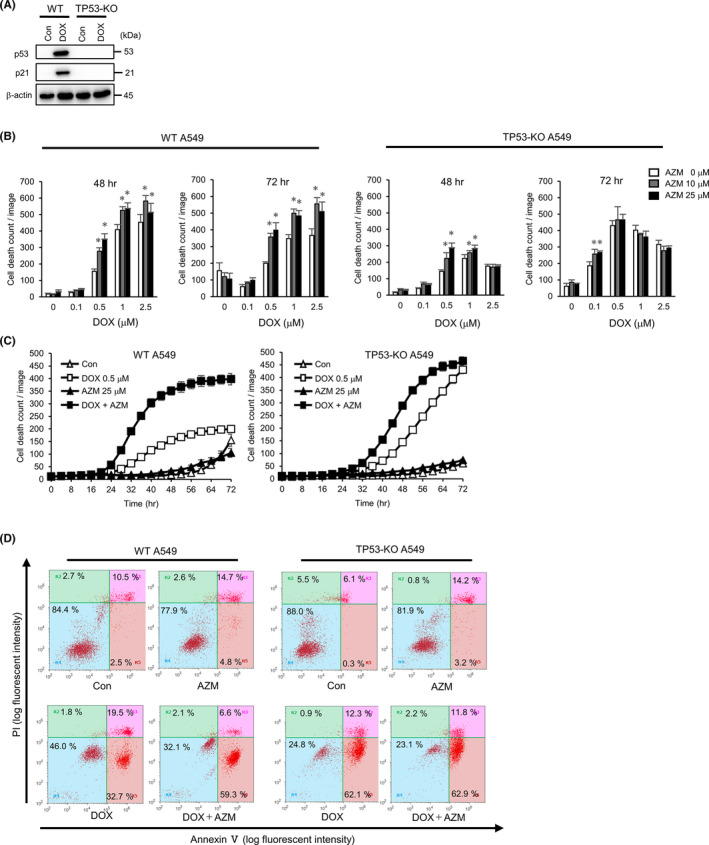
Enhanced cytotoxicity by azithromycin (AZM) was dependent on p53 status. A, TP53‐KO in A549 cells was confirmed by western blotting. Expression of p53, p21 was assessed after doxorubicin (DOX) (1 µM) treatment for 24 h. β‐actin was used as control. B, Wild‐type (WT) and TP53‐KO A549 cells were treated with DOX in combination with AZM for 72 h. Dead cell number was assessed by IncuCyte with propidium iodide (PI) staining. Representative data for three independent experiments are shown. n = 4, bar = mean ± SD, *P < .05. vs 0 µM AZM treatment. C, Summary of time dependency of cell death is shown in (B). n = 4, bar = mean ± SD D, Flow cytometric analysis of WT and TP53‐KO A549 cells after Annexin V/PI double staining, following DOX (1 µM) ± AZM (25 µM) treatment for 72 h. Numbers indicate the percentage of the cell numbers in each area. Representative data for three independent experiments are shown
The pro–apoptotic proteins PUMA and NOXA are transcriptionally regulated by p53.25 AZM has an effect on autophagy inhibition and NOXA and PUMA have been reported to be degraded via autophagy as well as by proteasomes;12, 13, 26 therefore, these pro–apoptotic proteins may be involved in AZM‐enhanced cell death. To confirm this, we performed western blotting and observed that the expression of NOXA, but not PUMA, was increased by AZM and was further enhanced by coadministration of AZM with DOX or ETP (Figure S4A). To examine the contribution of NOXA to pronounced cell death induction, NOXA‐KO A549 cells were established (Figure S4B), and induction of cell death was assessed using a live cell imaging system (Figure S4C,D). Contrary to our expectations, NOXA‐KO cells exhibited significantly enhanced cell death, similar to WT A549 cells (Figure S4C,D). NOXA protein expression was, indeed, upregulated by AZM with DOX, or ETP, even in TP53‐KO A549 cells (Figure S4E), although the induction of NOXA mRNA was strongly suppressed compared to that in WT A549 cells. These results indicated that NOXA did not contribute to the pronounced cell death induced by AZM.
3.3. Enhanced cytotoxicity by azithromycin was attenuated by disruption of autophagy
Previous reports have shown that autophagy acts as a cytoprotective factor when cancer cells are exposed to DNA‐damaging drugs.27, 28 It has also been reported that autophagy was induced after ETP‐treatment via p53 activation.29 Thus, we determined that inhibition of autophagy by AZM contributed to the pronounced cytotoxicity. Hence, we established A549 cells stably expressing mCherry‐EGFP‐LC3 for autophagy evaluation; mCherry‐EGFP‐LC3 probes localized on the autophagosomal membrane and showed yellow puncta signals after autophagosome formation. When autophagosomes fuse with lysosomes, EGFP is quenched due to the lysosomal acidic condition, thus showing the red puncta signal under confocal microscopy.30 GEM, DOX, and ETP treatment increased the number of mCherry+ puncta, and in the DOX‐treated cells, the color of puncta changed to red (Figure 4A,B), indicating an increased number of autophagosomes/autolysosomes in response to GEM, DOX, and ETP treatment. Western blot analysis showed that these drugs decreased the expression of p62, which is a substrate of autophagy. Because AZM inhibited autophagy, AZM treatment accumulated LC3B‐II and p62. Coadministration with AZM resulted in increased expression of LC3B‐II compared to AZM alone, indicating that autophagy was inducted in response to these DNA‐damaging drugs (Figure 4C).
FIGURE 4.
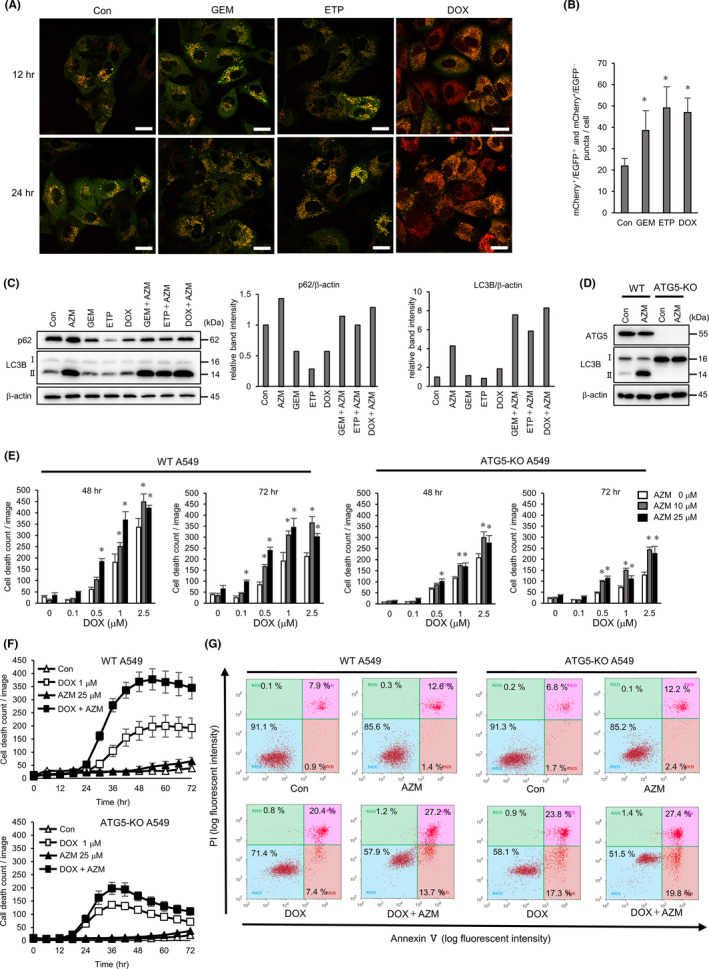
Azithromycin (AZM) enhanced cell toxicity by suppressing DNA‐damaging drug‐induced autophagy. A, Confocal microscopy of A549 cells expressing mCherry‐EGFP‐LC3 after treatment with gemcitabine (GEM) (1 µM), etoposide (ETP) (5 µM), or doxorubicin (DOX) (1 µM) for 12 or 24 h. Representative images of each treatment are shown. Scale bar = 20 µm. B, Number of red puncta (mCherry+ /EGFP‐ and mCherry+ /EGFP+) in each treated cells at 24 h is summarized. n = 5 (fields), bar = mean ± SD, *P < .05. vs control treatment. C, Western blotting of p62 and LC3B protein expression in A549 cells treated with indicated drugs to evaluate autophagy flux. Band intensity of p62 and LC3B‐II was measured and standardized by β‐actin expression and summarized in right columns. D, Western blotting analysis of ATG5‐KO in A549 cells. Expression of ATG5‐ATG12 and LC3B were analyzed after the control or 25 µM AZM treatment for 24 h. E, Wild‐type (WT) and ATG5‐KO A549 cells were treated with DOX ± AZM for up to 72 h. Number of dead cells was assessed by IncuCyte with propidium iodide (PI) staining. Representative data of three independent experiments are shown. n = 4, bar = mean ± SD, *P < .05 vs 0 µM AZM treatment. F, Time dependency of cell death shown in (E) is summarized. G, Flow cytometry analysis with the Annexin V/PI double‐stained WT or ATG5‐KO A549 cells after DOX (1 µM) ± AZM (25 µM) treatment for 72 h. Numbers indicate the percentage of the cell numbers in each area. Representative data of three independent experiments are shown
To address the role of autophagy in the enhanced cytotoxicity of the drug combination, we assessed the effect of AZM and DOX in ATG5‐KO A549 cells lacking the autophagosome‐forming ability (Figure 4D). Compared to WT A549 cells, the attenuation of the enhancement by the drug combination was apparent in ATG5‐KO A549 cells (Figure 4E,F). However, ATG5‐KO A549 cells started to die earlier than WT cells in response to DOX treatment (Figure 4F). Flow cytometry also showed no enhancement by coadministration but increased the number of Annexin V‐positive cells, indicating that ATG5‐KO A549 cells were more sensitive to DOX than WT A549 cells (Figure 4G). Thus, our results suggest that AZM may have promoted the cytotoxicity of DNA‐damaging drugs by blocking cytoprotective autophagy.
3.4. Lysosomal membrane permeabilization is enhanced by combined treatment with doxorubicin and azithromycin
It has been reported that LMP leads to the release of lysosomal hydrolases into the cytosol following the induction of various types of cell death, including apoptosis.29, 31 To assess whether blocking autophagy using AZM contributes to pronounced cytotoxicity by inhibiting the clearance of damaged lysosomes with LMP, we used the galectin‐3 (Gal3) puncta assay.31, 32 Because Gal3 can translocate to damaged lysosomes, we can observe punctate signals of Gal3 by immunofluorescence staining when the lysosomes are damaged. Because LAMP2 is a lysosomal membrane protein, the colocalization of Gal3 and LAMP2 signals indicates the damaged lysosomal membrane.32 The positive control, which was treated with lysosomotropic reagent LLOMe, exhibited Gal3 and LAMP2 colocalization (Figure 5A). AZM treatment increased enlarged lysosomes/autolysosomes but did not cause Gal3 and LAMP2 colocalization. DOX treatment resulted in the punctation of colocalized LAMP2 and Gal3, indicating LMP induction (Figure 5A,B). It is noteworthy that combined treatment with AZM and DOX increased the number of puncta, indicating pronounced LMP induction (Figure 5A,B). The same enhancement in LMP was observed in coadministration of CBDCA with AZM (Figure S5). Transmission electron microscopy (TEM) showed that AZM treatment resulted in autolysosome accumulation, which was probably due to blocking autophagy flux; in contrast, DOX treatment resulted in an increased number of lysosomes. Of note, after combination treatment with AZM and DOX, many enlarged autolysosomes containing undigested cytoplasmic remnants, including lysosomes, were observed (Figure 5C), suggesting that the lysosomes damaged by DOX, which should have been eliminated by lysophagy, had accumulated since AZM blocked lysophagy. Supporting this, western blot analysis showed that coadministration of AZM with DOX further increased LAMP1 protein expression compared to AZM or DOX alone (Figure 5D).
FIGURE 5.
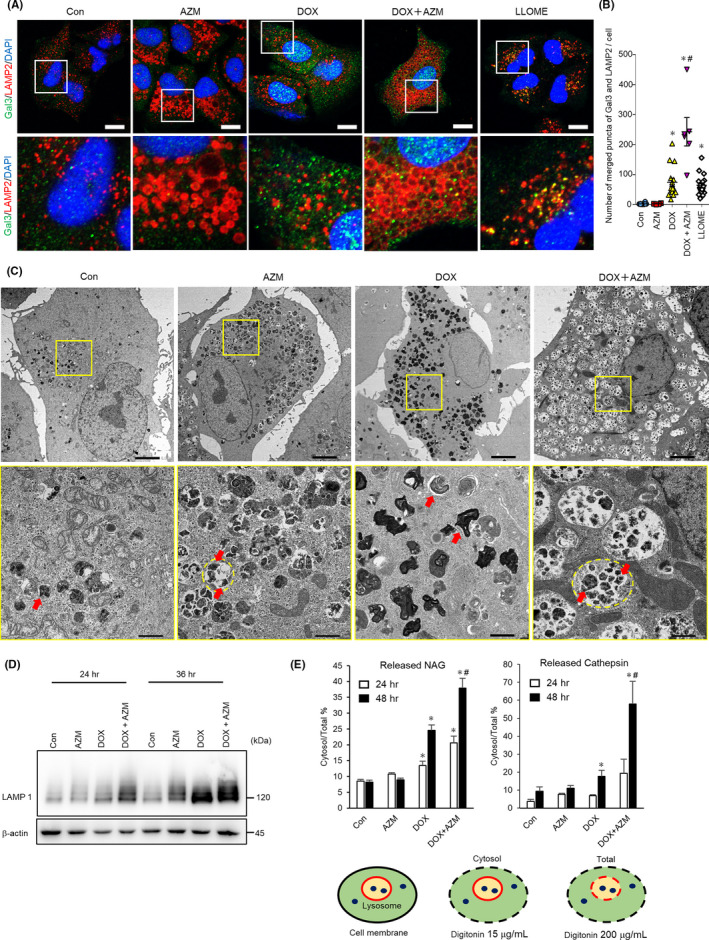
Coadministration of azithromycin (AZM) enhanced doxorubicin (DOX)‐induced lysosomal membrane permeabilization in A549 cells. A, Colocalization of Gal3 and LAMP2 was assessed by immunofluorescent staining and confocal microscopic observation. A549 cells were treated with DOX (1 µM) ± AZM (25 µM) for 48 h and then stained for Gal3 (Green), LAMP2 (Red), and nuclei (blue) with DAPI. A549 cells treated with LLOMe (1 mM) for 4 h were used as positive control. Scale bar = 10 µm. The boxed area was enlarged in the bottom panels. B, Colocalized signals of LAMP2 and Gal3 were calculated and summarized. n for Con, AZM, DOX, and DOX + AZM are 14, 4, 15, and 6, respectively, bar = mean ± SE, *P < .05 vs control, #P < .05 vs DOX. C, A549 cells treated with DOX (1 µM) ± AZM (25 µM) for 48 h were observed with transmission electron microscopy. Scale bar = 5 µm for top panels. The enlarged images of the boxed area are shown below. Red arrows indicate lysosomes and a yellow dashed line indicates autolysosome. Scale bar = 1 µm for bottom panels. D, LAMP1 expression level assessed by western blotting. A549 cells were treated with indicated reagents for 24 or 36 h. E, Lysosomal membrane permeabilization (LMP) was measured by assessing the ratio of cytosolic β‐N‐acetyl‐glucosaminidase (NAG) or cathepsin activity per total cellular activity. A549 cells were treated with DOX (1 µM) ± AZM (25 µM) for 24 or 48 h, and then LMP was assessed. Representative data of three independent experiments are shown. n = 4, bar = mean ± SD, *P < .05 vs control, #P < .05 vs DOX. At the bottom, a schema of this experiment is shown. Plasma membrane and lysosomal membrane are shown in black and red, respectively. Lysosomal enzymes are shown in blue dots. Different concentrations of digitonin permeabilized different membranes are shown with a dashed line
To confirm the leakage of lysosomal contents via LMP, we measured cytosolic cathepsin and N‐acetyl‐glucosaminidase (NAG) activity released from lysosomes. Lysosomal leakage was detectable by DOX treatment but not by AZM. AZM and DOX combination treatment further enhanced lysosomal leakage compared to treatment with DOX alone (Figure 5E). These results suggested that the increased cell death by AZM was due to the enhanced LMP via inhibition of lysosomal clearance (lysophagy), leading to the accumulation of damaged lysosomes.
3.5. Enhanced lysosomal membrane permeabilization by doxorubicin and azithromycin coadministration was mediated through p53‐dependent TFEB activation
Because our data suggested that AZM enhanced DOX‐induced cell death by increasing LMP, we clarified how DOX induced LMP and how AZM enhanced this effect by comparing LMP between WT and TP53‐KO A549 cells. We found that LMP was significantly suppressed in TP53‐KO cells (Figure 6A). This suggests that LMP was induced, at least in part, in a p53‐dependent manner. Brady et al (2018) reported that TFEB, which is a master transcription factor for lysosome and autophagosome biogenesis, is activated by the p53 pathway, and contributes to LMP.29 Immunofluorescent staining showed that DOX, but not AZM, induced nuclear translocation of TFEB, whereas this translocation was attenuated in TP53‐KO cells (Figure 6B). In the case of gene expression, the autophagy‐related and lysosome‐related genes (LAMP1, LAMP2, ATG2A, ATG2B, CTSD, and MAP1LC3B), which are all transcriptionally regulated by TFEB, were upregulated after 24 hours of treatment with DOX in WT cells (Figure 6C). We also observed increased lysosomes and LAMP1 protein expression following DOX treatment (Figure 5C,D). In TP53‐KO A549 cells, the expression of these TFEB‐related genes was not increased at 24 hours, consistent with the nuclear translocation of TFEB, but was upregulated at 48 hours of DOX and DOX plus AZM treatment. This suggested that TFEB might also activate the p53‐independent pathway after longer exposure to DOX (Figure 6C). In addition, the delayed cell death and suppressed LMP in DOX‐treated TP53‐KO A549 cells (Figure 3C, Figure 6A) might be due to the retarded transcriptional activation of TFEB in response to these drugs. Therefore, lysosomal biogenesis by TFEB appeared to contribute to LMP induction by DOX plus AZM treatment.
FIGURE 6.
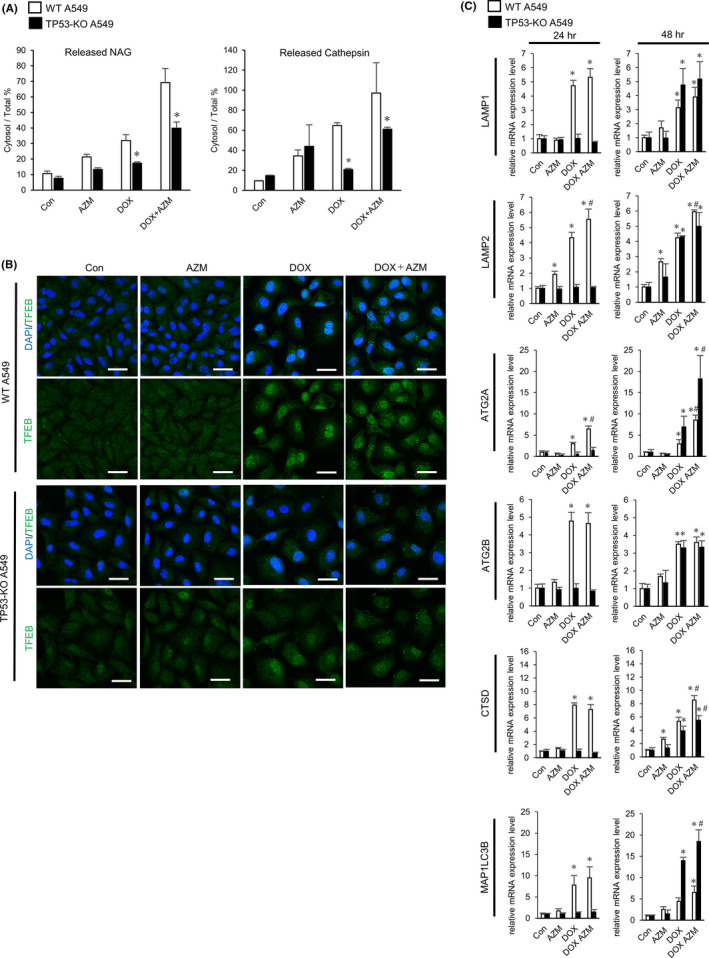
Enhanced lysosomal membrane permeabilization (LMP) by doxorubicin (DOX) and azithromycin (AZM) coadministration was mediated by p53‐dependent TFEB activation. A, LMP in wild‐type (WT) A549 cells and TP53‐KO A549 cells were measured by assessing released cytosolic NAG or cathepsin activity. Cells were treated with DOX (1 µM) ± AZM (25 µM) for 48 h. Representative data of three independent experiments are shown. n = 3, bar = mean ± SD, *P < .05 vs WT. B, Confocal microscopy of WT or TP53‐KO A549 cells treated with 1 µM DOX, with or without 25 µM for 24 h, and immunostained for TFEB (green). Nuclei were counterstained with DAPI (blue). Scale bar = 20 µm. C, Relative mRNA expression level of TFEB target genes in WT or TP53‐KO A549 cells, assessed by real‐time PCR. Each cell was treated with DOX (1 µM) ± AZM (25 µM) for 24 h or 48 h. n = 4, bar = mean ± SD, *P < .05. vs Control treatment, #P < .05 vs DOX treatment
4. DISCUSSION
In the present study, we demonstrated that combined treatment with DNA‐damaging drugs and AZM resulted in prominent induction of apoptosis in NSCLC cell lines (Figure 1). We also showed that DNA‐damaging drugs caused lysosomal membrane damage in a p53‐dependent manner, resulting in the promotion of lysosomal biogenesis via TFEB activation (Figures 5, 6). In contrast, AZM treatment increased the number of autolysosomes by inhibiting autophagic flux, which also suppressed the clearance of damaged lysosomes via lysophagy (Figure 5A,C). Therefore, coadministration of AZM with DNA‐damaging drugs enhanced lysosomal membrane damage, as shown in the galectin 3 puncta assay, as well as the cytoplasmic release of lysosomal enzymes (Figure 5B,E). Thus, the simultaneous treatment with DNA‐damaging drugs and AZM led to the accumulation of a large number of damaged lysosomes/autolysosomes, resulting in the pronounced LMP that enhanced apoptosis induction (Figure 7).
FIGURE 7.
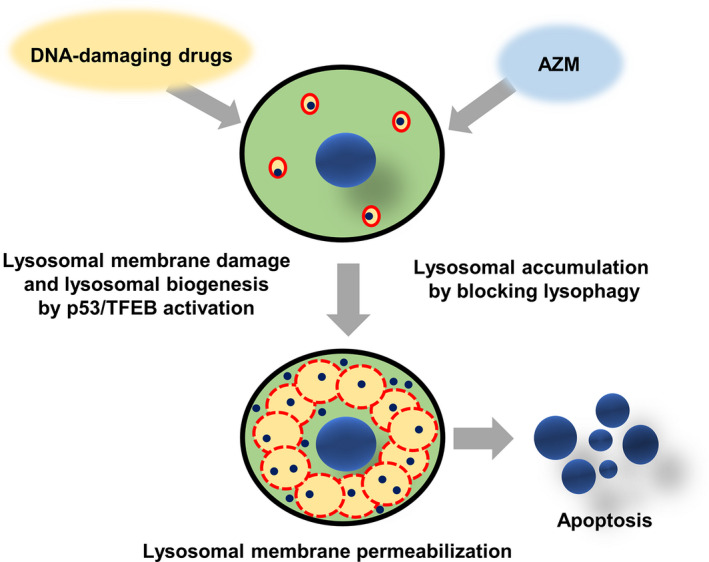
Schematic diagram of azithromycin (AZM)‐enhanced lysosomal membrane permeabilization (LMP) and cell death induction of DNA‐damaging drugs. DNA‐damaging drugs induce lysosomal membrane damage and lysosomal biogenesis via p53/TFEB activation. AZM blocks autophagy, as well as lysophagy, leading to the accumulation of lysosomes/autolysosomes. Combination treatment with DNA‐damaging drugs and AZM results in the accumulation of damaged lysosomes having LMP, leading to the leakage of lysosomal enzymes, followed by pronounced induction of apoptosis. Red circles: lysosomes/autolysosomes. Dashed red line circle: lysosomes/autolysosomes with LMP. Blue dots: lysosomal enzymes
According to Wang et al (2018), LMP causes the release of lysosomal contents, such as cathepsins to the cytoplasm, which induces various types of cell death, including apoptosis.31 In terms of induction of apoptosis, de Castro et al (2016) reported that cathepsins released from lysosomes into the cytoplasm induce the proteolytic activation of substrates, such as Bid and Bax, leading to activation of caspases via the mitochondria.33 According to Johansson et al (2010), the released cathepsins can directly activate caspases independent of the mitochondrial pathway.34 Although the leakage of lysosomal cathepsin B, D, and L was reported to induce apoptosis,34 CA‐074Me, a specific cathepsin B inhibitor, did not suppress cell death induction in the present study (data not shown). Therefore, other cathepsin(s) or lysosomal enzymes may be involved in apoptosis induction. Further studies are required to clarify the molecular mechanisms that connect LMP and apoptosis. Recently, we also showed that AZM enhanced the anti–cancer effect of lansoprazole (LPZ), a proton pump inhibitor, by upregulating LMP, although AZM or LPZ alone did not induce LMP in A549 cells.35 Taken together, these results suggest that AZM induces lysosomal membrane fragility in cancer cells. Furthermore, it has been reported that malignant transformation alters lysosomal structure and functions and makes cancer cells more sensitive to lysosome‐targeting reagents,36, 37 suggesting that AZM could be a good candidate for cancer therapy.
As shown in Figure 3 and Figure S3, the enhanced cytotoxicity of the two‐drug combination was attenuated in TP53‐KO A549 cells or H596 cells that carry TP53 mutations, suggesting that the pronounced cytotoxicity was TP53‐dependent. Previous literature demonstrated that nuclear translocation of TFEB in response to ETP treatment was attenuated in TP53‐KO murine embryonic fibroblast (MEF) as compared with WT MEF.29 It was also reported that DNA damage activates sestrin, a downstream gene of p53, causing a reduction in mTORC1 activity, followed by TFEB activation.38 Thus, TFEB appears to be regulated by the p53‐sestrin‐mTORC1 axis. Meanwhile, our data showed that TFEB activation in response to DOX was delayed compared to WT A549 cells, but still occurred even in TP53‐KO A549 cells, indicating that TFEB was activated p53‐independently at a late stage (Figure 6). Similar to this delay, TP53‐KO cells required longer exposure time with DOX to exhibit sufficient cytotoxic effect (Figure 3C) and showed lower LMP by DOX at 48 hours than WT cells (Figure 6A). However, after 72 hours of treatment, TP53‐KO A549 cells exhibited high sensitivity to DOX alone, without showing enhancement by AZM. Therefore, other types of cell death mechanisms, such as mitotic catastrophe,39 may be involved after longer exposure due to the disruption of p53 and DOX‐induced DNA damage. These data suggest that DNA‐damaging drugs cause two different types of cell death: p53‐dependent LMP‐induced cell death, which is enhanced by AZM, and p53‐independent cell death. Although we showed that DNA‐damaging drugs induced LMP and TP53‐KO suppressed it, it is not clear whether p53 activation is sufficient to induce LMP or if other p53‐independent pathways are also needed. Further study is needed to clarify this.
It has been reported that autophagy in macrophages digests Mycobacterium tuberculosis and prevents infection in the lung.40 Renna et al (2011) reported that long‐term administration of AZM to patients with cystic fibrosis makes them more susceptible to mycobacterium infection. These authors revealed that AZM blocked autophagy of alveolar macrophages, which leads to inhibition of the digestion of mycobacterium.41 From a different point of view, this adverse event of AZM suggested the possibility of using AZM as an autophagy inhibitor. Our group has reported that macrolide antibiotics have a potent autophagy inhibitory effect.22, 42 In addition, our report revealed that AZM exhibited the most potent autophagy inhibition as compared to other macrolide antibiotics, such as clarithromycin and EM900.22 AZM did not inhibit the fusion of autophagosomes and lysosomes, but inhibited autolysosomal digestion by raising the lysosomal pH (Takano et al, unpublished). This effect appears to be consistent with the findings of TEM, which showed that many autolysosomes contained undigested cytoplasmic remnants, including lysosomes (Figure 5C). Although HCQ used in clinical trials as an autophagy inhibitor has adverse events, including retinopathy and cardiomyopathy,19, 20 AZM has been well used in the clinical setting for a long time, with almost no severe adverse events. As a drug repositioning agent, AZM appears to be a promising autophagy inhibitor.
It has become evident that cancer cells in dormancy or in a diapause‐like state can tolerate chemotherapy, and autophagy plays a critical role in maintaining this state.10, 11, 43 Thus, aside from lysosomes, autophagy is also a good target for cancer therapy. This is the first study to demonstrate that cancer cells induced apoptosis via LMP using DNA‐damaging drugs in combination with AZM. This effect is based on: (a) initiation of lysophagy by lysosomal membrane damage; (b) concomitantly blocking lysophagy and accumulating lysosomes at the late stage; and (c) upregulation of lysosome biogenesis via TFEB activation, all of which are integrated into the prominent LMP induction. Therefore, this drug combination has potential to be a new treatment strategy for intractable tumors, including lung cancer, and further in vivo studies and clinical trials are warranted.
DISCLOSURE
Authors declare no conflicts of interest for this article.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by the MEXT program for the Strategic Research Foundation at Private Universities (S1411011, 2014–2018) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (to KM); by JSPS KAKENHI Grant Numbers 17K15031 and 20K07298 (to NT); and by a Grant‐in‐Aid from Tokyo Medical University Cancer Research (to NT). This research was also supported by AMED under Grant Number JP18lm0203004 (to NT).
Toriyama K, Takano N, Kokuba H, et al. Azithromycin enhances the cytotoxicity of DNA‐damaging drugs via lysosomal membrane permeabilization in lung cancer cells. Cancer Sci. 2021;112:3324–3337. 10.1111/cas.14992
REFERENCES
- 1.Bade BC, Dela Cruz CS. Lung cancer 2020: epidemiology, etiology, and prevention. Clin Chest Med. 2020;41:1‐24. [DOI] [PubMed] [Google Scholar]
- 2.Ruiz‐Cordero R, Devine WP. Targeted therapy and checkpoint immunotherapy in lung cancer. Surg Pathol Clin. 2020;13:17‐33. [DOI] [PubMed] [Google Scholar]
- 3.Sgambato A, Casaluce F, Maione P, Gridelli C. Targeted therapies in non‐small cell lung cancer: a focus on ALK/ROS1 tyrosine kinase inhibitors. Expert Rev Anticancer Ther. 2018;18:71‐80. [DOI] [PubMed] [Google Scholar]
- 4.Yoneda K, Imanishi N, Ichiki Y, Tanaka F. Treatment of non‐small cell lung cancer with EGFR‐mutations. J UOEH. 2019;41:153‐163. [DOI] [PubMed] [Google Scholar]
- 5.Shah RR. Tyrosine kinase inhibitor‐induced interstitial lung disease: clinical features, diagnostic challenges, and therapeutic dilemmas. Drug Saf. 2016;39:1073‐1091. [DOI] [PubMed] [Google Scholar]
- 6.Otsubo K, Okamoto I, Hamada N, Nakanishi Y. Anticancer drug treatment for advanced lung cancer with interstitial lung disease. Respir Investig. 2018;56:307‐311. [DOI] [PubMed] [Google Scholar]
- 7.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728‐741. [DOI] [PubMed] [Google Scholar]
- 8.Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017;17:528‐542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amaravadi R, Kimmelman AC, White E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016;30:1913‐1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rehman SK, Haynes J, Collignon E, et al. Colorectal cancer cells enter a diapause‐like DTP state to survive chemotherapy. Cell. 2021;184:226‐242.e221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vera‐Ramirez L, Vodnala SK, Nini R, Hunter KW, Green JE. Autophagy promotes the survival of dormant breast cancer cells and metastatic tumour recurrence. Nat Commun. 2018;9:1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang J, Cui D, Gu S, et al. Autophagy regulates apoptosis by targeting NOXA for degradation. Biochim Biophys Acta. 2018;1865:1105‐1113. [DOI] [PubMed] [Google Scholar]
- 13.Thorburn J, Andrysik Z, Staskiewicz L, et al. Autophagy controls the kinetics and extent of mitochondrial apoptosis by regulating PUMA levels. Cell Rep. 2014;7:45‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu R, Ji Z, Xu C, Zhu J. The clinical value of using chloroquine or hydroxychloroquine as autophagy inhibitors in the treatment of cancers: A systematic review and meta‐analysis. Medicine. 2018;97:e12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Poklepovic A, Gewirtz DA. Outcome of early clinical trials of the combination of hydroxychloroquine with chemotherapy in cancer. Autophagy. 2014;10:1478‐1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vogl DT, Stadtmauer EA, Tan KS, et al. Combined autophagy and proteasome inhibition: a phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy. 2014;10:1380‐1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haas NB, Appleman LJ, Stein M, et al. Autophagy inhibition to augment mTOR inhibition: a phase I/II trial of everolimus and hydroxychloroquine in patients with previously treated renal cell carcinoma. Clin Cancer Res. 2019;25:2080‐2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang P, Burikhanov R, Jayswal R, et al. Neoadjuvant administration of hydroxychloroquine in a phase 1 clinical trial induced plasma Par‐4 levels and apoptosis in diverse tumors. Genes Cancer. 2018;9:190‐197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yusuf IH, Sharma S, Luqmani R, Downes SM. Hydroxychloroquine retinopathy. Eye (London, England). 2017;31:828‐845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yogasundaram H, Putko BN, Tien J, et al. Hydroxychloroquine‐induced cardiomyopathy: case report, pathophysiology, diagnosis, and treatment. Can J Cardiol. 2014;30:1706‐1715. [DOI] [PubMed] [Google Scholar]
- 21.Moriya S, Komatsu S, Yamasaki K, et al. Targeting the integrated networks of aggresome formation, proteasome, and autophagy potentiates ER stress‐mediated cell death in multiple myeloma cells. Int J Oncol. 2015;46:474‐486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mukai S, Moriya S, Hiramoto M, et al. Macrolides sensitize EGFR‐TKI‐induced non‐apoptotic cell death via blocking autophagy flux in pancreatic cancer cell lines. Int J Oncol. 2016;48:45‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tanaka H, Hino H, Moriya S, et al. Comparison of autophagy inducibility in various tyrosine kinase inhibitors and their enhanced cytotoxicity via inhibition of autophagy in cancer cells in combined treatment with azithromycin. Biochem Biophys Rep. 2020;22:100750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barretina J, Caponigro G, Stransky N, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603‐607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shibue T, Suzuki S, Okamoto H, et al. Differential contribution of Puma and Noxa in dual regulation of p53‐mediated apoptotic pathways. EMBO J. 2006;25:4952‐4962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ploner C, Kofler R, Villunger A. Noxa: at the tip of the balance between life and death. Oncogene. 2008;27(Suppl 1):S84‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu HM, Shao LJ, Jiang ZF, Liu RY. Gemcitabine‐induced autophagy protects human lung cancer cells from apoptotic death. Lung. 2016;194:959‐966. [DOI] [PubMed] [Google Scholar]
- 28.Nagata M, Arakawa S, Yamaguchi H, et al. Dram1 regulates DNA damage‐induced alternative autophagy. Cell Stress. 2018;2:55‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brady OA, Jeong E, Martina JA, Pirooznia M, Tunc I, Puertollano R. The transcription factors TFE3 and TFEB amplify p53 dependent transcriptional programs in response to DNA damage. eLife. 2018;7:e40856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hansen TE, Johansen T. Following autophagy step by step. BMC Biol. 2011;9:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang F, Gomez‐Sintes R, Boya P. Lysosomal membrane permeabilization and cell death. Traffic (Copenhagen, Denmark). 2018;19:918‐931. [DOI] [PubMed] [Google Scholar]
- 32.Aits S, Kricker J, Liu B, et al. Sensitive detection of lysosomal membrane permeabilization by lysosomal galectin puncta assay. Autophagy. 2015;11:1408‐1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Castro MA , Bunt G, Wouters FS. Cathepsin B launches an apoptotic exit effort upon cell death‐associated disruption of lysosomes. Cell Death Discov. 2016;2:16012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johansson AC, Appelqvist H, Nilsson C, Kågedal K, Roberg K, Öllinger K. Regulation of apoptosis‐associated lysosomal membrane permeabilization. Apoptosis. 2010;15:527‐540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takeda A, Takano N, Kokuba H, et al. Macrolide antibiotics enhance the antitumor effect of lansoprazole resulting in lysosomal membrane permeabilization‐associated cell death. Int J Oncol. 2020;57:1280‐1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kallunki T, Olsen OD, Jäättelä M. Cancer‐associated lysosomal changes: friends or foes? Oncogene. 2013;32:1995‐2004. [DOI] [PubMed] [Google Scholar]
- 37.Domagala A, Fidyt K, Bobrowicz M, Stachura J, Szczygiel K, Firczuk M. Typical and atypical inducers of lysosomal cell death: a promising anticancer strategy. Int J Mol Sci. 2018;19(8):2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008;134:451‐460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vitale I, Galluzzi L, Castedo M, Kroemer G. Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat Rev Mol Cell Biol. 2011;12:385‐392. [DOI] [PubMed] [Google Scholar]
- 40.Vergne I, Singh S, Roberts E, et al. Autophagy in immune defense against Mycobacterium tuberculosis. Autophagy. 2006;2:175‐178. [DOI] [PubMed] [Google Scholar]
- 41.Renna M, Schaffner C, Brown K, et al. Azithromycin blocks autophagy and may predispose cystic fibrosis patients to mycobacterial infection. J Clin Invest. 2011;121:3554‐3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moriya S, Che XF, Komatsu S, et al. Macrolide antibiotics block autophagy flux and sensitize to bortezomib via endoplasmic reticulum stress‐mediated CHOP induction in myeloma cells. Int J Oncol. 2013;42:1541‐1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Recasens A, Munoz L. Targeting cancer cell dormancy. Trends Pharmacol Sci. 2019;40:128‐141. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material