

Abstract
Oncolytic virus therapy has emerged as a promising treatment option against cancer. To date, oncolytic viruses have been developed for malignant tumors, but the need for this new therapeutic modality also exists for benign and slow‐growing tumors. G47∆ is an oncolytic herpes simplex virus type 1 (HSV‐1) with an enhanced replication capability highly selective to tumor cells due to genetically engineered, triple mutations in the γ34.5, ICP6 and α47 genes. To create a powerful, but safe oncolytic HSV‐1 that replicates efficiently in tumors regardless of growth speed, we used a bacterial artificial chromosome system that allows a desired promoter to regulate the expression of the ICP6 gene in the G47∆ backbone. Restoration of the ICP6 function in a tumor‐specific manner using the hTERT promoter led to a highly capable oncolytic HSV‐1. T‐hTERT was more efficacious in the slow‐growing OS‐RC‐2 and DU145 tumors than the control viruses, while retaining a high efficacy in the fast‐growing U87MG tumors. The safety features are also retained, as T‐hTERT proved safe when inoculated into the brain of HSV‐1 sensitive A/J mice. This new technology should facilitate the use of oncolytic HSV‐1 for all tumors irrespective of growth speed.
Keywords: G47∆, herpes simplex virus type 1, hTERT, ICP6, oncolytic virus
We report a powerful oncolytic HSV‐1 that can replicate explosively in any tumor regardless of its growth speed using clinically used, third‐generation HSV‐1, G47∆, as the backbone. Tumor‐specific restoration of the ICP6 function led to a highly capable oncolytic HSV‐1 with intended features including safety. This new technology should facilitate the use of oncolytic HSV‐1 for all tumors whether benign or malignant.
1. INTRODUCTION
Oncolytic virus therapy using genetically engineered HSV‐1 has drastically advanced from a basic concept to clinical application.1, 2 Since the first description of the use of genetic engineering as a tool for creating an oncolytic virus,3 many oncolytic viruses have been used in patients. Talimogene laherparepvec (T‐VEC) is an oncolytic HSV‐1 with mutations in the γ34.5 and α47 genes and was approved in 2015 in the United States and Europe as a drug for patients with inoperable melanoma.4 G207 is one of the first oncolytic HSV‐1 viruses used in clinical trials,5 with deletions in both copies of the γ34.5 gene and a lacZ insertion inactivating the ICP6 gene.6 The major function of γ34.5 is the dephosphorylation of eIF2α, counteracting the host‐cell‐induced shutdown of protein synthesis mediated by protein kinase R (PKR) upon viral infection.7 This mutation permits viral replication within cancer cells but not in normal cells, because the PKR‐mediated blocking of virus replication is usually disabled in cancer cells.8 ICP6 encodes the large subunit of ribonucleotide reductase (RR) that is required for virus growth and DNA replication.9 The mammalian RR activity is upregulated only in actively dividing cells10 and appears absent in terminally differentiated cells that have stopped synthesizing DNA.11 Therefore, ICP6 inactivation permits viral replication only in dividing cells or fast‐growing tumors. Whereas these mutations cause the replication of G207 to be highly specific to cancer cells, its replication capability is significantly diminished when compared with the parental wild‐type virus. A reasonable strategy to improve the antitumor action of G207 while retaining its safety mechanisms is the tumor‐specific restoration of the functions of the γ34.5 gene and/or the ICP6 gene. G47∆ was constructed by creating a further deletion within the α47 gene of the G207 genome.12 The deletion of the overlapping US11 promoter results in placement of the late US11 gene under the control of the immediate‐early α47 promoter. Ectopic immediate‐early expression of US11 suppresses PKR‐mediated blocking of virus replication, leading to partial recovery of the deleted γ34.5 functions, and results in enhanced replication of γ34.5‐mutated viruses in tumor cells.13 Conversely, the main function of the α47 gene product is to downregulate the major histocompatibility complex (MHC) class I expression in infected host cells by binding to the transporter associated with antigen presentation (TAP).14 The α47 gene, therefore, functions for the virus to escape from immune surveillance. The loss of such function by the additional α47 deletion in G47∆ causes a further attenuation of the virus in normal cells. In fact, G47∆ has been shown to be efficacious in a variety of cancers.12, 15, 16, 17 G47Δ has shown to be safe and efficacious in a phase II study for glioblastoma, and is recently ascertained for approval in Japan as a new drug for malignant glioma. Phase I studies for prostate cancer, olfactory neuroblastoma, and malignant pleural mesothelioma have also been performed.
Whereas G47∆ successfully achieved the tumor‐specific restoration of the γ34.5 function, its ICP6 inactivation still restricts its efficient replication capability to fast‐growing tumors. In this study, we attempted to eliminate such restriction and allow the virus to replicate efficiently regardless of the dividing speed of the tumor while retaining the safety mechanisms of G47∆ by restoring the function of ICP6 in a tumor‐specific manner.
2. MATERIALS AND METHODS
2.1. Cells and viruses
Vero (African green monkey kidney), U87MG and OS‐RC‐2 cells were obtained from the ATCC (USA) and maintained in DMEM supplemented with 10% FCS. DU145 cells were grown in MEM supplemented with 10% FBS, 1% non‐essential amino acids and 1% sodium pyruvate. Viruses were grown in Vero cells and virus titers were determined as described previously.12
2.2. Plasmids
The ICP6 gene fragment was amplified by PCR (primers: 5′‐GTCGACGCCGCGTCTGTTGAAAT‐3′ and 5′‐AGGCCTCACAGCGCGCAGCTCA‐3′) to obtain a 3.4‐kb fragment. The sequence of ICP6 was confirmed on an ABI 377 DNA auto‐sequencer (Applied Biosystems, USA). The shuttle vector SV‐pro was constructed to contain a 45‐bp FRT adaptor, a 50 bp loxP adaptor, the lacZ gene from pcDNA6E/Uni‐lacZ (TaKaRa Bio Inc Shiga, Japan), the 3.4‐kb ICP6 gene, a 3989‐bp fragment of lambda HindIII DNA, and a multiple cloning site with recognition sequences for the following restriction enzymes: ScaI, SpeI, SbfI, AflII, Acc65I, NheI, SwaI, and SalI. The hTERT core promoter fragment (−367 to +56) was amplified by PCR using the primers, 5′‐TCGGGTTACCCCACAGCCTA‐3′ and 5′‐AGGGCTTCCCACGTGCGCA‐3′ from pGL3‐378 (kindly provided by Dr. Satoru Kyo, Kanazawa University, Kanazawa, Japan).18 The sequence of the hTERT promoter was confirmed and inserted into the SpeI site of SV‐pro to generate SV‐hTERT.
2.3. Construction of recombinant HSV‐1 with a desired promoter
Mutagenesis of the T‐BAC plasmid was performed by a two‐step replacement procedure. A mixture of T‐BAC plasmid and SV‐hTERT was incubated with Cre recombinase (NEB, M0298, USA) at 37℃ for 30 min and electroporated into E. coli DH10B. Bacteria were streaked onto LB plates containing chloramphenicol (Cm; 15 µg/mL) and kanamycin (Kan; 10 µg/mL) and incubated at 37℃ overnight. The DNA integrity of the recombinant T‐BAC/hTERT plasmids was confirmed by gel analyses following endonuclease digestions (Figure S1). Transfections were performed on Vero cells using T‐BAC/hTERT DNA and pOG44 (ThermoFisher Scientific, V600520) with Lipofectamine (ThermoFisher Scientific, 11 688 027, USA) in accordance with the manufacturer’s instructions. Transfected cells were incubated and the progeny viruses were confirmed for GFP negativity using an inverted fluorescence microscope, and lacZ positivity by X‐gal staining. Two rounds of limiting dilution were performed to pick out a single clone. Recombinant viruses were harvested and the structure of the viral DNA was confirmed by HindIII digestion. The structure of the recombinant viruses was also confirmed by Southern blot analyses (Figure 2C). Following HindIII digestion, DNA fragments were separated by electrophoresis on 0.6% (w/v) agarose gels in 1× Tris‐borate‐EDTA buffer for 18 h at 30 V. DNA probes of pcDNA6E/Uni‐lacZ digested with EcoRI (corresponding to lacZ sequence), or a fragment of the hTERT PCR products were labeled using an AlkPhos Direct kit with CDP‐Star detection (GE Healthcare, RPN3690, USA).
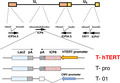
FIGURE 2.
The features of T‐hTERT. A, Southern blotting analyses of viruses probed with lacZ: 12.5 kb for G47∆ (lane 1), 16 kb for T‐pro (lane 2), 16.6 kb for Nos. 1‐4 of T‐hTERT candidates (lanes 3‐6), negative for T‐BAC (lane 7) and 20.1 kb for T‐BAC product (lane 8). No. 3 of T‐hTERT (lane 5) had a weak band. B, Southern blotting analyses of viruses probed with hTERT DNA fragments: negative for G47∆ (lane 1), negative for T‐pro (lane 2), 16.6 kb for Nos. 1‐4 of T‐hTERT candidates (lanes 3‐6), negative for T‐BAC (lane 7) and 20.1 kb for T‐BAC product (lanes 8, 9). C, DNA fragments from EcoRI‐digested pcDNA6E/Uni‐lacZ corresponding to the lacZ sequence was used as the hybridization probe for T‐pro candidates. All bands seen are as expected: 12.5 kb for G47∆ (lane 2), 16 kb for Nos. 1‐5 of T‐pro candidates (lane 3‐7). Lane 1 is a kb‐marker. D, Western blot analyses. Three cells lines were infected with viruses; G47∆ (lane 1), R47∆ (lane 2), T‐hTERT (lane 3), T‐pro (lane 4) and T‐01 (lane 5). T‐hTERT‐infected cells and R47∆‐infected cells showed similar levels of the 140‐kDa band corresponding to RR
2.4. Western blotting
Approximately 5 × 105 OS‐RC‐2 and DU145 cells were infected with G47∆, R47∆, T‐hTERT, T‐pro or T‐01 at an multiplicity of infection (MOI) of 0.02. After 48 h, cells were washed and treated with a RIPA buffer (150 mmol/L NaCl, 1% NP‐40, 0.5% deoxycholate (DOC), 50 mmol/L Tris‐HCl pH 8.0) on ice for 10 min and centrifuged. The cell lysates (8 μg) were electrophoresed on a 10% SDS‐polyacrylamide gel and transferred onto an Immobilon‐P transfer membrane (Merck, INCP00010, Germany) using a semi‐dry transfer blot system (ATTO, Tokyo, Japan). After blocking with TBS containing 0.1% (v/v) Tween20 and 5% (w/v) skim milk for 1 h, the membranes were incubated with a primary antibody to Q3299 (1:250, corresponding to the viral RR) for 1 h, washed, and incubated with the ECL peroxidase labeled anti‐rabbit antibody (1:10 000, Promega, NA934VS, USA) and HRP‐avidin. Specific proteins were detected using an enhanced chemiluminescence system (Fujifilm, Tokyo, Japan).
2.5. Virus yield studies
For viral yield studies, OS‐RC‐2 or DU145 cells were seeded onto 6‐well plates at 5 × 105 cells/well. Cells were infected with either T‐hTERT, T‐01, or T‐pro in duplicate wells at an MOI of 0.01. After 48 h of infection, the cells were scraped and lysed by 3 cycles of freezing and thawing. The progeny virus was titrated on Vero cells by plaque assay as described previously.12 Results represent the average of duplicates.
For cell growth curve studies, OS‐RC‐2, DU145 or U87MG cells were seeded onto 4 6‐well plates at 2 × 105 cells/well. Cells were grown in medium containing 10% (v/v) FCS. The number of surviving cells was counted daily with a Coulter Counter (Beckman Coulter, USA).
2.6. Animal studies
All animal experiment protocols were approved by the committee for Ethics of Animal Experimentation and were in accordance with the Guideline for Animal Experiments in the University of Tokyo. Six‐wk‐old female A/J mice and female athymic mice (BALB/c nu/nu) were purchased from CLEA Japan (Tokyo, Japan). All animals were caged in groups of 5 or fewer. Subcutaneous tumor therapy was performed as described elsewhere.19 Subcutaneous tumors were generated by injecting 1 × 107 cells (OS‐RC‐2 and DU145) or 2 × 106 cells (U87MG) subcutaneously into the left flanks of athymic mice. When established subcutaneous tumors reached approximately 5‐6 mm in diameter, 2 × 105 pfu of T‐hTERT, T‐pro, T‐01 or mock was inoculated into the tumor on days 0 and 3 in 3 tumor models including OS‐RC‐2 (n = 6), DU145 (n = 6), and U87MG (n = 6). The body weight of each mouse was measured every week. When subcutaneous OS‐RC‐2 tumors reached 8‐9 mm in diameter, 2 × 105 pfu of each virus was inoculated twice on days 0 and 3 in the OS‐RC‐2 large tumor model (n = 10). The tumor volume (length ×width × height) was measured twice a week. Mice were sacrificed when the maximum diameter of the tumor reached 24 mm.
2.7. Intracerebral inoculation safety studies
Mock, strain F (1 × 103 pfu), T‐01 (1 × 106 pfu) or T‐hTERT (1 × 106 pfu) in a volume of 5 μL was injected over a 5‐min period into the right hemispheres of the brains of 6‐wk‐old female A/J mice (n = 5) with a Kopf stereotactic frame (BioResearchCenter, Aichi, Japan). Cages were then blinded and mice were monitored daily for clinical manifestations for 3 wk.
2.8. Statistical analysis
Significance of data comparisons between treatment groups was calculated using Student t test (two‐tailed) or by two‐way ANOVA with Sidak multiple comparisons test. A value of P < .05 was considered statistically significant. All statistical analyses were performed using JMP Pro v.11.0.0 (SAS Institute, USA).
3. RESULTS
3.1. Construction of T‐hTERT
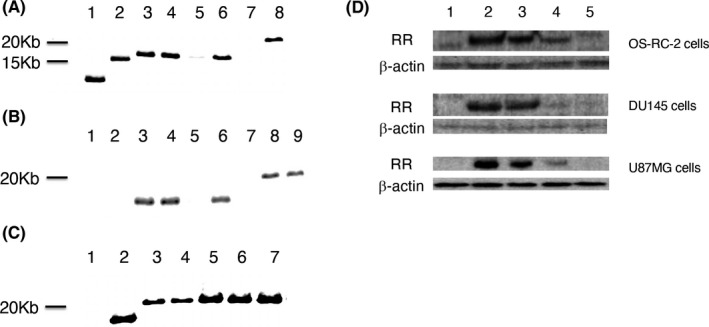
We newly constructed an oncolytic HSV‐1, termed T‐hTERT, derived from G47∆ by inserting the ICP6 gene controlled by the human telomerase reverse transcriptase (hTERT) promoter (Figure 1A). Expression of hTERT is observed at high levels in tumor cells but not in normal cells,20 and telomerase activity presents an optimal tumor‐specific trait for the regulation of oncolytic viruses.21 We previously utilized a bacterial artificial chromosome (BAC) and 2 recombinase systems (Cre/loxP and Flp/FRT) to construct “armed” oncolytic HSV‐1 viruses to express desired transgenes using G47∆ as the backbone.19 The system has been since reconstructed with modifications (T‐BAC system) to produce G47∆‐derived armed oncolytic HSV‐1 with improved replication capabilities. T‐hTERT has the lacZ gene and the hTERT promoter‐driven ICP6 gene, placed in opposite directions, inserted into the deleted ICP6 locus, in addition to deletions in both copies of the γ34.5 gene and a deletion in the α47 gene. T‐01 is a control virus created by this system that contains an empty expression cassette instead of a cassette with a transgene (Figure 1A).
FIGURE 1.
Construction of T‐hTERT. A, The structure of T‐hTERT. G47∆‐derived HSV‐1 contains deletions in both copies of the γ34.5 gene, a deletion in the α47 gene and a deletion in the ICP6 gene. The lacZ gene and the hTERT promoter‐driven ICP6 gene, placed in opposite directions, are inserted into the deleted ICP6 locus. T‐pro is a control virus with no promoter to drive the inserted ICP6 gene, and T‐01 is also a control virus with an empty expression cassette without a functioning ICP6. Thicker arrows indicate transcribed regions. B, A diagram describing the modified T‐BAC system for constructing G47∆‐derived oncolytic HSV‐1 with the ICP6 gene regulated by a desired promoter. C, Representative gel electrophoreses digested with HindIII. Instead of the band at 14 986 bp in T‐BAC (lane 1), bands are observed at 19 521 bp in T‐BAC/pro (lane 2) and 20 024 bp in T‐BAC/hTERT (lane 3)
In this study, the shuttle vector of the T‐BAC system was further modified to allow a rapid, reliable and simultaneous construction of multiple G47∆‐derived oncolytic HSV‐1 in which the ICP6 expression was regulated by a desired promoter (Figure 1B). The first step of this system was to insert the entire sequence of the shuttle vector into the loxP site of T‐BAC by Cre‐mediated recombination. The second step was to co‐transfect the integrant with a plasmid expressing FLP into Vero cells to excise the BAC sequence flanked by the FRT sites. Four clones of the constructed HSV‐1 were isolated by 2 rounds of limiting dilution, and the construct was confirmed by digestion with HindIII (Figures 1C and S1), and by Southern blot analyses (Figure 2), and No. 2 of T‐hTERT candidates was selected. T‐pro is a control virus with no promoter that regulates the inserted ICP6 sequence, created in the same manner (Figures 1, 2 and S1). Western blot analyses of virus‐infected tumor cell lines revealed that T‐hTERT‐infected cells, but not G47∆‐, T‐01‐ or T‐pro‐infected cells, produced high levels of RR corresponding to the 140‐kDa band similarly to those infected with R47∆, a double‐mutated HSV‐1 with deletions in γ34.5 and α47 but with an intact ICP6 (Figure 2D). The hTERT promoter, therefore, functions to express ICP6 sufficiently in tumor cells. A faint band of 140 kDa by T‐pro in each tumor cell line suggested that a leaky expression of RR may occur without any promoter.
3.2. Enhanced replication capability of T‐hTERT in vitro
The doubling time of cell proliferation of U87MG in vitro was approximately 1 d, whereas it was approximately 2 d for OS‐RC‐2 and DU145 by regression line analysis (data not shown). OS‐RC‐2 and DU145 cells grow significantly more slowly in vitro than U87MG cells (P < .05). So, we determined the yield of progeny virus in human tumor cell lines, OS‐RC‐2 (renal cell carcinoma) and DU145 (prostate cancer), both slow‐growing tumor cells, and U87MG (malignant glioma), fast‐growing tumor cells, 48 h after infection at an MOI of 0.01 (Figure 3). T‐hTERT produced higher yields than T‐01, resulting in 38‐fold and 227‐fold increases in titer in DU145 and OS‐RC‐2, respectively. T‐hTERT produced higher yields than T‐pro, resulting in 1.8‐fold and 3.6‐fold increases in titer in DU145 and OS‐RC‐2, respectively. T‐hTERT showed the highest virus yield among those tested. The unexpected yields of T‐pro may be because T‐pro contains the full sequence of ICP6, despite the absence of a regulating promoter. RR expression in T‐pro‐infected DU145 cells was very limited, but an expression level of RR and a virus replication capability may not show a linear correlation, and the expression of RR required for virus replication is not presumed to be very high. In U87MG, there was no significant difference among these 3 viruses.
FIGURE 3.
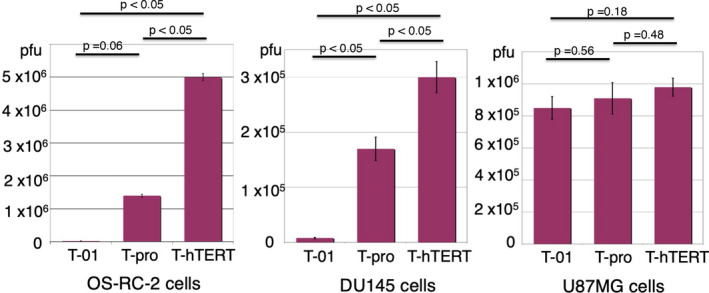
Virus yield studies of T‐hTERT. OS‐RC‐2, DU145 or U87MG cells were seeded at 5 × 105 cells/well and infected with T‐hTERT, T‐pro, or T‐01 at an MOI of 0.01. After 48 h of infection, the progeny virus was titrated. T‐hTERT produced higher yields than T‐pro and T‐01 in both OS‐RC‐2 cells (2.2 × 104, 1.4 × 106 and 5.0 × 106 pfu for T‐01, T‐pro and T‐hTERT, respectively) and DU145 cells (7.8 × 103, 1.7 × 105 and 3.0 × 105 pfu for T‐01, T‐pro and T‐hTERT, respectively). Results represent the average of duplicates. P‐value was calculated using a two‐sided ttest
3.3. In vivo efficacy of T‐hTERT in subcutaneous tumor models
Three human tumor cells lines were used in athymic mice; OS‐RC‐2 and DU145, again, representing slow‐growing tumors in vivo and U87MG, representing a fast‐growing tumor. When established subcutaneous tumors reached approximately 5‐6 mm in diameter, T‐hTERT, T‐pro, T‐01 (2 × 105 pfu) or mock were inoculated into the tumor twice on days 0 and 3. In the OS‐RC‐2 model, all viruses caused a significant inhibition of tumor growth compared with mock. T‐hTERT was significantly more efficacious than T‐pro and T‐01 (P < .05 vs T‐pro on days 16‐51, and vs T‐01 on days 10‐58; Figure 4A). Also, in the DU145 model, all viruses caused a significant inhibition of tumor growth compared with mock. T‐hTERT was significantly more efficacious than T‐01 (P < .05 on days 59‐87; Figure 4B), although not statistically more than T‐pro (P = .07 on days 77‐87). In the U87MG model, in which tumors grew more rapidly than in the DU145 and OS‐RC‐2 models, all viruses caused a significant inhibition of tumor growth compared with mock (Figure 4C). However, as expected, there was no difference in efficacy between T‐hTERT and other viruses, T‐pro and T‐01, indicating that T‐01 and T‐pro, even without ICP6 expression, can readily replicate in U87MG tumors, presumably due to sufficient upregulation of RR of the host cells. There was no difference in body weight between the T‐hTERT group and other treatment groups in all 3 tumor models (data not shown).
FIGURE 4.
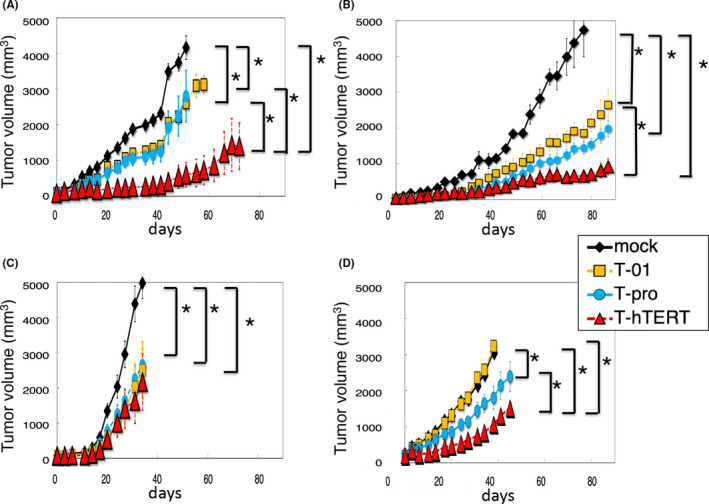
The in vivo efficacy of T‐hTERT in 3 subcutaneous tumor models. OS‐RC‐2 and DU145 represent slow‐growing tumors and U87MG represents a fast‐growing tumor. When established subcutaneous tumors reached approximately 5‐6 mm (A‐C), or 8‐9 mm (D) in diameter, T‐hTERT, T‐pro, T‐01 (2 × 105 pfu) or mock was inoculated into the tumor twice on days 0 and 3. A, OS‐RC‐2 model. T‐hTERT was significantly more efficacious than T‐pro or T‐01 (n = 6/group). B, DU145 model. T‐hTERT was significantly more efficacious than T‐01, although not statistically more than T‐pro (n = 6/group). C, U87MG model. T‐hTERT was as efficacious as T‐pro and T‐01 (n = 6/group). D, OS‐RC‐2 large tumor model. T‐hTERT was significantly more efficacious than T‐pro and T‐01 (n = 10/group). Mock (black rhombuses), T‐01 (yellow squares), T‐pro (blue circles), or T‐hTERT (red triangles) was used. The results represent the means. The bars represent the standard error of the mean (SEM). *P < .05
To investigate the efficacy of T‐hTERT in large tumors of a slow‐growing type, when subcutaneous OS‐RC‐2 tumors reached approximately 8‐9 mm in diameter, 2 × 105 pfu of each virus was inoculated twice on days 0 and 3. While T‐01 did not exhibit a significant efficacy compared with mock at this dose, T‐hTERT and T‐pro caused a significant inhibition of tumor growth compared with mock. The T‐hTERT treatment was significantly more efficacious than T‐pro and T‐01 (P < .05 vs. T‐pro on days 6‐41, and vs T‐01 on days 6‐38; Figure 4D). This suggests that T‐hTERT can exert its efficacy even if administered after slow‐growing tumors have grown to a relatively large size.
3.4. The safety of T‐hTERT
HSV‐1 sensitive A/J mice were inoculated intracerebrally with mock, strain F (1 × 103 pfu), T‐01 (1 × 106 pfu) or T‐hTERT (1 × 106 pfu) (n = 5/group). Strain F is a parental wild‐type HSV‐1. Treated groups were blinded, and each mouse was monitored daily for clinical manifestations for 3 wk. All strain F‐inoculated mice deteriorated rapidly and became moribund within 7 d of inoculation. All T‐hTERT‐inoculated mice as well as all T‐01‐ and mock‐inoculated mice survived without any abnormal manifestations. These results indicated that T‐hTERT is as safe as T‐01 when inoculated into the brain of A/J mice at this dose (Figure 5).
FIGURE 5.
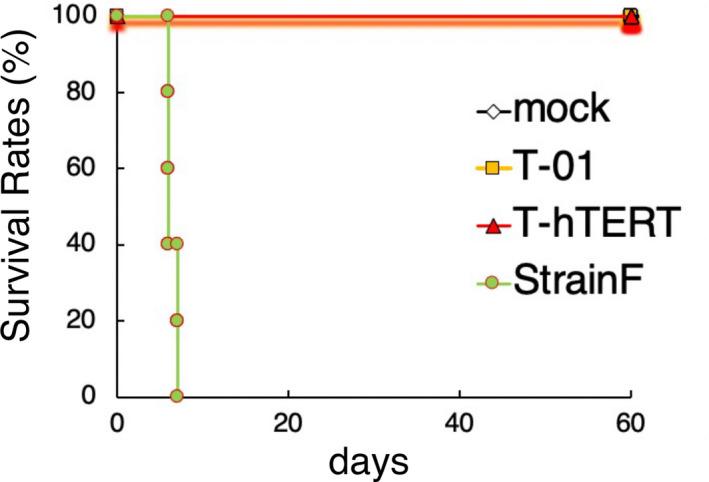
Safety of T‐hTERT with intracerebral inoculation. Mock, strain F (1 × 103 pfu), T‐01 (1 × 106 pfu) or T‐hTERT (1 × 106 pfu) was injected into the brains of A/J mice. Strain F is a parental wild‐type HSV‐1 virus. All mice inoculated with T‐01 or T‐hTERT survived and stayed healthy for the 3‐wk observation period, whereas all strain F‐inoculated mice deteriorated rapidly and became moribund within 7 d of inoculation
4. DISCUSSION
Needless to say, the key for developing useful oncolytic HSV‐1 is to acquire high antitumor efficacy without compromising safety and to obtain a therapeutic window as wide as possible, the difference between the dose needed to obtain the efficacy and that to exert toxicity. A natural strategy is to enable the virus to replicate explosively in tumors and still be completely incapacitated in normal cells. G47∆ succeeded in drastically widening the therapeutic window by adding a third genetically engineered mutation to the double‐mutated G207, therefore conferring further attenuation in normal cells, but improving its replication capability in tumor cells using the second‐site suppressor for γ34.5 mutation.8 In the majority of tumor cell lines tested, G47∆ replicates better than G207, resulting in higher virus yields and greater cytopathic effects, and G47∆ is efficacious in vivo for a variety of solid tumors.12, 15, 16, 17 G47Δ has been shown to act synergistically when combined with antineoplastic agents such as paclitaxel, temozolomide, and trichostatin.22, 23 G47∆ is also suitable as the backbone for arming various transgenes, the expression of which can potentially increase the toxicity.19, 24, 25
A theoretical downside to G47∆ is that, because of ICP6 inactivation, the replication capability of G47∆ may depend on the RR activity of the host tumor cell. We found that R47∆, in which the only difference between G47∆ was the intact ICP6 gene, showed higher virus yields than G47∆ in certain tumor cells.12 Others showed an induced elevation of RR enhanced the replication of G207.26 G47∆, therefore, may replicate well in fast‐growing tumors, but not so well in slow‐growing or benign tumors. G47Δ has been shown to kill cancer stem cells efficiently,27, 28 but may not replicate in cancer stem cells so well, as they are usually slow dividing. Development of oncolytic virus therapy has been aimed mainly at intractable cancer, but the need for this new therapeutic modality also exists for benign tumors such as skull base tumors in which tumors repeatedly appear despite resection, and slow‐growing tumors such as prostate cancer in which patients often avoid surgery because of age or complication risks. An attractive antitumor mechanism of oncolytic viruses, especially G47∆, is the induction of systemic and specific antitumor immunity in the course of oncolytic activity, so an oncolytic virus suited for benign tumors would be most useful for hereditary benign tumors such as neurofibromatosis type 1 and familial adenomatous polyposis.
The aim of this study was to obtain a powerful oncolytic HSV‐1 that can replicate explosively in any tumor regardless of its growth speed, using G47∆ as the backbone to retain the safety features. T‐hTERT is such a G47∆‐derived virus, in which the hTERT promoter regulates the ICP6 gene such that RR is self‐provided in a tumor‐specific manner whether benign or malignant. T‐hTERT was more efficacious in the slow‐growing OS‐RC‐2 and DU145 tumors than both T‐pro, a control virus with an intact ICP sequence without a promoter, and T‐01, a control virus without the ICP6 gene, whereas the efficacy of T‐hTERT was comparable with these control viruses in the fast‐growing U87MG tumors. In slow‐growing tumor cells in culture, T‐hTERT also showed significantly higher virus yields than both control viruses, whereas T‐pro showed somewhat higher virus yields than T‐01. However, in vivo, there was no significant difference between T‐01 and T‐pro in both slow‐growing tumors. As RR has been shown to reflect the growth rate of host cells,29 it stands to reason that T‐hTERT showed increased efficacy in slow‐growing tumors by self‐providing the RR in tumor cells with low host RR activities. Regulating a virus gene using a tumor‐specific promoter has been reported: A nestin promoter was used to regulate 1 copy of the γ34.5 gene in double‐mutated HSV‐1, resulting in glioma‐selective improvement of the oncolytic activity,30 and a midkine promoter has been used in a similar fashion.31
The tumor‐specific restoration of the ICP6 function compensates for the shortcomings of G47∆: T‐hTERT may be a more potent oncolytic virus than G47∆ that can be used even for benign tumors. The safety features of G47∆ seem retained, as T‐hTERT was as safe as the ICP6‐inactivated control viruses when inoculated into the brain of HSV‐1 sensitive A/J mice. It is yet to be investigated whether the hTERT promoter, among other tumor‐specific promoters, is best suited for regulating the ICP6 gene with regard to tumor specificity, strength, and timing. We believe that this new technology would facilitate the use of oncolytic HSV‐1 in tumors of all types, including slow‐growing and benign tumors, and therefore promote the new therapeutic modality to become a regular choice for all tumor patients.
CONFLICT OF INTEREST
Tomoki Todo owns the patent right for G47∆ in multiple countries including Japan.
Supporting information
Fig S1
ACKNOWLEDGMENTS
This work was supported in part by research grants from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan (grant number 09028017 for TT), the Japan Agency of Medical Research and Development (AMED) (grant numbers JP15cm0106015h0005 and JP20cm0106252h0002 for TT) and Grant‐in‐Aid for Scientific Research from the Japan Society for the Promotion of Science (JSPS Kakenhi) (grant numbers 08049157 and 09013291 for HF). We thank Dr. Yasushi Ino for his helpful advice and Sayaka Kanaami for her technical assistance.
Fukuhara H, Takeshima Y, Todo T. Triple‐mutated oncolytic herpes virus for treating both fast‐ and slow‐growing tumors. Cancer Sci. 2021;112:3293–3301. 10.1111/cas.14981
REFERENCES
- 1.Kirn D, Martuza RL, Zwiebel J. Replication‐selective virotherapy for cancer: Biological principles, risk management and future directions. Nat Med. 2001;7:781‐787. [DOI] [PubMed] [Google Scholar]
- 2.Fukuhara H, Ino Y, Todo T. Oncolytic virus therapy: A new era of cancer treatment at dawn. Cancer Sci. 2016;107:1373‐1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martuza RL, Malick A, Markert JM, Ruffner KL, Coen DM. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science. 1991;252:854‐856. [DOI] [PubMed] [Google Scholar]
- 4.Coffin R. Interview with Robert Coffin, inventor of T‐VEC: the first oncolytic immunotherapy approved for the treatment of cancer. Immunotherapy. 2016;8:103‐106. [DOI] [PubMed] [Google Scholar]
- 5.Markert JM, Medlock MD, Rabkin SD, et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther. 2000;7:867‐874. [DOI] [PubMed] [Google Scholar]
- 6.Mineta T, Rabkin SD, Yazaki T, Hunter WD, Martuza RL. Attenuated multi‐mutated herpes simplex virus‐1 for the treatment of malignant gliomas. Nat Med. 1995;1:938‐943. [DOI] [PubMed] [Google Scholar]
- 7.Cassady KA, Gross M, Roizman B. The second‐site mutation in the herpes simplex virus recombinants lacking the gamma34.5 genes precludes shutoff of protein synthesis by blocking the phosphorylation of eIF‐2alpha. J Virol. 1998;72:7005‐7011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mohr I, Sternberg D, Ward S, Leib D, Mulvey M, Gluzman Y. A herpes simplex virus type 1 gamma34.5 second‐site suppressor mutant that exhibits enhanced growth in cultured glioblastoma cells is severely attenuated in animals. J Virol. 2001;75:5189‐5196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goldstein DJ, Weller SK. Herpes simplex virus type 1‐induced ribonucleotide reductase activity is dispensable for virus growth and DNA synthesis: isolation and characterization of an ICP6 lacZ insertion mutant. J Virol. 1988;62:196‐205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thelander L, Reichard P. Reduction of ribonucleotides. Annu Rev Biochem. 1979;48:133‐158. [DOI] [PubMed] [Google Scholar]
- 11.Engstrom Y, Rozell B, Hansson HA, Stemme S, Thelander L. Localization of ribonucleotide reductase in mammalian cells. Embo J. 1984;3:863‐867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Todo T, Martuza RL, Rabkin SD, Johnson PA. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc Natl Acad Sci U S A. 2001;98:6396‐6401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He B, Chou J, Brandimarti R, Mohr I, Gluzman Y, Roizman B. Suppression of the phenotype of gamma (1)34.5‐ herpes simplex virus 1: failure of activated RNA‐dependent protein kinase to shut off protein synthesis is associated with a deletion in the domain of the alpha47 gene. J Virol. 1997;71:6049‐6054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.York IA, Roop C, Andrews DW, Riddell SR, Graham FL, Johnson DC. A cytosolic herpes simplex virus protein inhibits antigen presentation to CD8+ T lymphocytes. Cell. 1994;77:525‐535. [DOI] [PubMed] [Google Scholar]
- 15.Sugawara K, Iwai M, Yajima S, et al. Efficacy of a Third‐Generation Oncolytic Herpes Virus G47Δ in Advanced Stage Models of Human Gastric Cancer. Mol Ther Oncolytics. 2020;17:205‐215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamada T, Tateishi R, Iwai M, Koike K, Todo T. Neoadjuvant Use of Oncolytic Herpes Virus G47Δ Enhances the Antitumor Efficacy of Radiofrequency Ablation. Mol Ther Oncolytics. 2020;21:612‐623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fukuhara H, Martuza RL, Rabkin SD, Ito Y, Todo T. Oncolytic herpes simplex virus vector G47delta in combination with androgen ablation for the treatment of human prostate adenocarcinoma. Clin Cancer Res. 2005;11:7886‐7890. [DOI] [PubMed] [Google Scholar]
- 18.Takakura M, Kyo S, Kanaya T, et al. Cloning of human telomerase catalytic subunit (hTERT) gene promoter and identification of proximal core promoter sequences essential for transcriptional activation in immortalized and cancer cells. Cancer Res. 1999;59:551‐557. [PubMed] [Google Scholar]
- 19.Fukuhara H, Ino Y, Kuroda T, Martuza RL, Todo T. Triple gene‐deleted oncolytic herpes simplex virus vector double‐armed with interleukin 18 and soluble B7–1 constructed by bacterial artificial chromosome‐mediated system. Cancer Res. 2005;65:10663‐10668. [DOI] [PubMed] [Google Scholar]
- 20.Kyo S, Takakura M, Fujiwara T, Inoue M. Understanding and exploiting hTERT promoter regulation for diagnosis and treatment of human cancers. Cancer Sci. 2008;99:1528‐1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doloff JC, Waxman DJ, Jounaidi Y. Human telomerase reverse transcriptase promoter‐driven oncolytic adenovirus with E1B–19 kDa and E1B–55 kDa gene deletions. Hum Gene Ther. 2008;19:1383‐1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zeng WG, Li JJ, Hu P, Lei L, Wang JN, Liu RB. An oncolytic herpes simplex virus vector, G47Δ, synergizes with paclitaxel in the treatment of breast cancer. Oncol Rep. 2013;29:2355‐2361. [DOI] [PubMed] [Google Scholar]
- 23.Liu TC, Castelo‐Branco P, Rabkin SD, Martuza RL. Trichostatin A and oncolytic HSV combination therapy shows enhanced antitumoral and antiangiogenic effects. Mol Ther. 2008;16:1041‐1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu TC, Zhang T, Fukuhara H, et al. Oncolytic HSV armed with platelet factor 4, an antiangiogenic agent, shows enhanced efficacy. Mol Ther. 2006;14:789‐797. [DOI] [PubMed] [Google Scholar]
- 25.Liu TC, Zhang T, Fukuhara H, et al. Dominant‐negative fibroblast growth factor receptor expression enhances antitumoral potency of oncolytic herpes simplex virus in neural tumors. Clin Cancer Res. 2006;12:6791‐6799. [DOI] [PubMed] [Google Scholar]
- 26.Petrowsky H, Roberts GD, Kooby DA, et al. Functional interaction between fluorodeoxyuridine‐induced cellular alterations and replication of a ribonucleotide reductase‐negative herpes simplex virus. J Virol. 2001;75:7050‐7058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheema TA, Wakimoto H, Fecci PE, et al. Multifaceted oncolytic virus therapy for glioblastoma in an immunocompetent cancer stem cell model. Proc Natl Acad Sci USA. 2013;110:12006‐12011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zeng W, Hu P, Wu J, et al. The oncolytic herpes simplex virus vector G47Δ effectively targets breast cancer stem cells. Oncol Rep. 2013;29:1108‐1114. [DOI] [PubMed] [Google Scholar]
- 29.Elford HL, Freese M, Passamani E, Moriis HP. Ribonucleotide reductase and cell proliferation: Variations of ribonucleotide reductase activity with tumor growth rate in a series of rat hepatomas. J Biol Chem. 1970;245:5228‐5233. [PubMed] [Google Scholar]
- 30.Kambara H, Okano H, Chiokka EA, Saeki Y. An oncolytic HSV‐1 mutant expressing ICP34.5 under control of a nestin promoter increases survival of animals even when symptomatic from a brain tumor. Cancer Res. 2005;65:2832‐2839. [DOI] [PubMed] [Google Scholar]
- 31.Maldonado AR, Klanke C, Jegga AG, et al. Molecular engineering and validation of an oncolytic herpes simplex virus type 1 transcriptionally targeted to midkine‐positive tumors. J Gene Med. 2010;12:612‐623. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1