

Abstract
Integrin associated protein (CD47) is an important target in immunotherapy, as it is expressed as a “don't eat me” signal on many tumor cells. Interference with its counter molecule signal regulatory protein alpha (SIRPα), expressed on myeloid cells, can be achieved with blocking Abs, but also by inhibiting the enzyme glutaminyl cyclase (QC) with small molecules. Glutaminyl cyclase inhibition reduces N‐terminal pyro‐glutamate formation of CD47 at the SIRPα binding site. Here, we investigated the impact of QC inhibition on myeloid effector cell‐mediated tumor cell killing by epidermal growth factor receptor (EGFR) Abs and the influence of Ab isotypes. SEN177 is a QC inhibitor and did not interfere with EGFR Ab‐mediated direct growth inhibition, complement‐dependent cytotoxicity, or Ab‐dependent cell‐mediated cytotoxicity (ADCC) by mononuclear cells. However, binding of a human soluble SIRPα‐Fc fusion protein to SEN177 treated cancer cells was significantly reduced in a dose‐dependent manner, suggesting that pyro‐glutamate formation of CD47 was affected. Glutaminyl cyclase inhibition in tumor cells translated into enhanced Ab‐dependent cellular phagocytosis by macrophages and enhanced ADCC by polymorphonuclear neutrophilic granulocytes. Polymorphonuclear neutrophilic granulocyte‐mediated ADCC was significantly more effective with EGFR Abs of human IgG2 or IgA2 isotypes than with IgG1 Abs, proposing that the selection of Ab isotypes could critically affect the efficacy of Ab therapy in the presence of QC inhibition. Importantly, QC inhibition also enhanced the therapeutic efficacy of EGFR Abs in vivo. Together, these results suggest a novel approach to specifically enhance myeloid effector cell‐mediated efficacy of EGFR Abs by orally applicable small molecule QC inhibitors.
Keywords: CD47, EGFR antibody, glutaminyl cyclase, immunotherapy, myeloid cell
Inhibition of CD47/signal regulatory protein alpha (SIRPα) interactions is of great interest in cancer immunotherapy. In this manuscript, we investigated the impact of glutaminyl cyclase inhibition by small molecules to interfere with CD47/SIRPa interactions in epidermal growth factor receptor Ab‐mediated tumor immunotherapy in vitro and in vivo.

Abbreviations
- Aβ
amyloid beta
- ADCC
antibody‐dependent cell‐mediated cytotoxicity
- ADCP
antibody‐dependent cellular phagocytosis
- CDC
complement‐dependent cytotoxicity
- CTX
cetuximab
- EGFR
epidermal growth factor receptor
- E:T
effector:target
- FcαR
Fc alpha receptor
- FcγR
Fc gamma receptor
- GM‐CSF
granulocyte‐macrophage colony‐stimulating factor
- GPI
glycosylphosphatidylinositol
- M‐CSF
macrophage colony‐stimulating factor
- MFI
mean fluorescence intensity
- MNC
mononuclear cell
- MTZ
matuzumab
- NHS
normal human serum
- NK
natural killer
- PANI
panitumumab
- PMN
polymorphonuclear neutrophilic granulocyte
- QC
glutaminyl cyclase
- QPCT
glutaminyl‐peptide cyclotransferase
- QPCTL
glutaminyl‐peptide cyclotransferase‐like
- RTX
rituximab
- SCC
squamous cell carcinoma
- SIRPα
signal regulatory protein alpha
- Tg
transgenic
1. INTRODUCTION
Immune checkpoint inhibition by CTLA‐4, programmed cell death protein 1, or programmed cell death protein 1 ligand 1 Abs has revolutionized cancer immunotherapy.1, 2 Innate myeloid checkpoint blockade and effective recruitment of myeloid cells could become another important therapeutic option in the future.3, 4 For example, blockade of the interaction between CD47 on lymphoma cells and SIRPα (CD172α) on myeloid effector cells by the CD47 Ab hu5F9‐G4 (magrolimab) has shown clinical efficacy in combination with rituximab in advanced lymphoma patients.5 After diligent preclinical development of this approach, 6, 7, 8 the clinical study by Advani et al5 was the first to confirm the scientific rationale for myeloid immune checkpoint blockade in tumor patients. In addition to several monoclonal CD47 Abs, CD47‐directed bispecific Abs, soluble SIRPα‐Fc molecules, and SIRPα Abs are also at different stages of development.9, 10 CD47 is a ubiquitously expressed membrane protein that interacts with SIRPα on myeloid cells to provide a “don’t eat me” signal to phagocytes.11 The intracellular signal is thereby mediated by the recruitment and activation of the tyrosine phosphatases SHP‐1 or SHP‐2, which transduce negative signals by dephosphorylation of a variety of downstream substrates, ultimately resulting in inhibition of phagocytosis and cytotoxicity.12, 13 Crystal structures of this interaction have revealed the involvement of pyro‐glutamate at the N‐terminus of CD47.14 Pyro‐glutamate formation is a common posttranslational modification of proteins, which is catalyzed by QCs such as QPCT or QPCTL.15 The contribution of QPCTL to the affinity of CD47 for SIRPα binding, and that this interaction is “druggable” by QPCT/QPCTL inhibitors, has recently been established by two independent studies.16, 17 A prototypic QPCT/QPCTL inhibitor is the small molecule SEN177, which contains a triazine ring as the QC binding motif.18
Myeloid cells kill tumor cells by ADCP or by ADCC, thereby requiring activating signals that are provided by stimulatory Fc receptors.19, 20 For human IgG Abs, these receptors are predominantly FcγRIIa (CD32a) and FcγRIIIa (CD16a), whereas human IgA Abs activate FcαRI (CD89) mainly expressed by monocytes/macrophages and PMN. Human PMNs, as the major effector cell population, are activated more effectively by human IgA than by IgG Abs,21, 22, 23, 24, 25 with human IgG2 being at least as effective as human IgG1.26, 27, 28 Thus, we investigated the impact of QC inhibition on effector mechanisms of mAbs against EGFR as a prototypic antigen on a variety of solid tumors. For ADCC and ADCP by PMN and macrophages, human Abs of IgG1, IgG2, and IgA2 isotypes were investigated to identify optimal combinations for this novel approach.
Interestingly, QC inhibition consistently improved Ab‐mediated killing of tumor cells by myeloid cells, but did not interfere with other potential effector mechanisms of these Abs. The increased killing activity by myeloid cells was observed with different Ab isotypes, suggesting a broad applicability of this novel approach. Importantly, QC inhibition also translated into increased therapeutic efficacy of EGFR Abs in vivo.
2. MATERIALS AND METHODS
Experiments with human material were approved by the Ethical Committee of the University Medical Center Schleswig‐Holstein in accordance with the Declaration of Helsinki.
2.1. Antibodies and reagents
The approved EGFR Abs PANI (human IgG2; E7.6.3, Vectibix) and CTX (chimeric human IgG1; 225, Erbitux) were obtained from Amgen and Merck, respectively. For further EGFR expression analysis and CDC experiments, EGFR Ab m425 and MTZ (humanized IgG1; Merck) were used. The CD20 Ab RTX (chimeric IgG1; c2B8, Rituxan) was purchased from Hoffmann‐La Roche. The IgA2.0 variants CTX‐IgA2 and RTX‐IgA2 were generated as described.29, 30 The QC inhibitor SEN177 was obtained from Sigma‐Aldrich and dissolved in DMSO (Thermo Fisher Scientific). Fc receptor‐specific Abs binding to CD16 (clone 3G8), CD32b (S18005H), or CD89 (A59) were purchased from BioLegend, the CD32a Ab (IV.3) was from BioCell, and the CD64 Ab (10.1) from Abs‐online. The CD47 Ab B6H12 was from eBioscience. Purified murine IgG1 (BioLegend) served as a nonspecific binding control.
2.2. Cell lines
The solid tumor cell lines A431 (SCC), MDA‐MB‐468 (breast carcinoma), Kyse‐30 (esophageal SCC), and the HEK cell line Lenti‐X 293T were obtained from the DSMZ (German Collection of Microorganisms and Cell Cultures), the colon carcinoma cell line DiFi was from ATCC. The pro B cell line Ba/F3 was a kind gift from the group of Dr. Leo Koenderman (UMC Utrecht) and was re‐authenticated by IDEXX in 2017. All cell lines were grown in media as recommended by the suppliers.
2.3. Cell growth assay
Growth of A431, Kyse‐30, and MDA‐MB‐468 cells was analyzed using the MTS proliferation assay (Promega). Cells were seeded at a density of 2 × 104 cells/well and treated with SEN177 at the indicated concentrations. To test EGFR‐directed Ab‐mediated growth inhibition in the absence or presence of SEN177, the colon cancer cell line DiFi was used. In brief, cells were seeded at a density of 2 × 104 cells/well and treated with 10 µM SEN177 or DMSO and the indicated concentrations of CTX. After incubation for 72 hours at 37℃, MTS was added and absorption at 490 nm was measured. Cell growth shown as the percentage of the control was calculated using the formula: % growth = absorption with inhibitor/Ab divided by absorption without (w/o) inhibitor/Ab ×100.
2.4. Generation of a soluble SIRPα‐IgG2σ fusion protein
A soluble human SIRPα‐IgG2σ fusion protein (referred to as SIRPα‐Fc) as well as a CD3 Ab of IgG2σ isotype (control‐IgG2σ; M. Peipp, unpublished data) were produced in‐house. The SIRPα‐Fc fusion protein consists of the N‐terminal V domain of human SIRPα (GenBank BC038510.2) fused to the Fc region (hinge‐CH2‐CH3, without CH1) of an Fc silent IgG2σ version.31 Briefly, the sequence of SIRPα‐Fc was generated by de novo synthesis (Eurofins) and subsequently cloned into the pSec‐Tag vector (Invitrogen). Protein expression was carried out by transient calcium phosphate transfection of Lenti‐X 293T cells. Both proteins were purified from culture supernatants by affinity chromatography using the CaptureSelect FcXL matrix (CaptureSelect). Afterwards, monomeric SIRPα‐Fc and control‐IgG2σ were obtained by size exclusion chromatography using a Superdex 200 26/600 column (GE Healthcare) and an ÄKTA liquid chromatography system (GE Healthcare).
2.5. Flow cytometric analyses
Epidermal growth factor receptor expression on tumor cell lines was quantified using the chimeric Ab CTX.32 CD47 expression was determined using the murine Ab B6H12. The EGFR and CD47 specific antigen binding sites per cell were determined with Abs m425 and B6H12, respectively, by indirect quantitative fluorescence flow cytometry using QIFIKIT (Dako) according to the manufacturer’s instructions. Expression of the Fc receptors FcγRIII (CD16), FcγIIa/b (CD32a/b), FcγRI (CD64), and FcαRI (CD89) on PMN and macrophages was analyzed using specific murine mAbs as described above. Binding of primary Abs was determined by FITC‐conjugated goat anti‐human or anti‐mouse Fcγ‐specific F(ab)2 fragments (Jackson ImmunoResearch Laboratories). Immunofluorescence was analyzed on a flow cytometer (Navios; Beckman Coulter). CD47 binding activity of tumor cells after treatment with SEN177 or DMSO control was analyzed using either the soluble SIRPα‐Fc fusion protein (5 µg/mL or indicated concentrations). After incubation and washing, binding of SIRPα‐Fc to CD47 was detected using phycoerythrin‐conjugated goat anti‐human Fcγ‐specific F(ab)2 fragments (Jackson ImmunoResearch Laboratories). For calculation of Ab or SIRPα binding, the following formula was used: % binding = MFI (SEN177) / MFI (DMSO) ×100.
2.6. Isolation of human effector cells and serum
Human PMN and MNC were isolated from peripheral blood of healthy volunteers using Polymorphprep (Progen) or Ficoll Paque Plus (GE Healthcare), respectively, as previously described.33, 34 Human macrophages were generated by adherence of PBMC using monocyte‐attachment medium (PromoCell). After attachment, cells were incubated for 24 hours in serum‐free X‐Vivo medium (Lonza). Subsequently, 50 ng/mL M‐CSF (PeproTech) was added for macrophage generation for at least 7 days. Human NK cells were generated from PBMC using the human NK isolation kit (Miltenyi BioTec) according to the manufacturer’s instructions. Normal human serum was prepared by using clotting activator containing S‐Monovette 9 mL Z tubes (Sarstedt).
2.7. Antibody‐dependent cell‐mediated cytotoxicity, ADCP, and CDC assays
Both ADCC and CDC were analyzed in chromium [51Cr] release assays as described previously.34 Briefly, Abs at various concentrations and medium were added to round‐bottom microtiter plates (Nunc). Assays were started by adding NHS (25% v/v) or effector cells to [51Cr] labelled target cells at indicated E:T cell ratios. In ADCC assays with PMN, 50 U/mL GM‐CSF (CellGenix) were added. After 3 hours at 37℃, [51Cr] release from triplicate samples was measured. The percentage of cytotoxicity was calculated using the formula: percentage of specific lysis = (experimental cpm − basal cpm) / (maximal cpm − basal cpm) × 100. The maximal [51Cr] release was obtained by adding 2% v/v Triton X‐100 to target cells, and basal release was measured in the absence of Abs and effector cells or NHS.
Antibody‐dependent cellular phagocytosis was analyzed by fluorescence microscopy. Briefly, macrophages were seeded at a density of 1 × 104 cells/well on µ‐slide counting plates (Ibidi). Tumor cells were labelled with 7.5 µM CSFE (CSFE Cell Division Tracker Kit; BioLegend) and added to the macrophages at an E:T cell ratio of 1:2. Antibodies (10 µg/mL) were added and cultures incubated at 37℃ for 2 hours. Antibody‐dependent cellular phagocytosis was determined as phagocytic index = number of engulfed tumor cells / number of macrophages x100, counting at least 50 macrophages.
2.8. In vivo experiments
Mice were maintained and bred in the Central Laboratory Animal Research Facility of the University of Utrecht. Experiments with IgA Abs were undertaken with male and female human FcαRI Tg mice between 11 and 34 weeks old, generated at the UMC Utrecht35 and backcrossed on a SCID background (CB17/lcr‐Prkdcscid/lcrlcoCrl; Charles River). For treatments with IgG Abs, transgene‐negative (non‐Tg) littermates were used. Mice were housed in groups in a temperature and 12:12 hour light : dark cycle controlled room, with food and water available ad libitum. All animal experiments were carried out in accordance with the international guidelines and were approved by the Animal Ethical Committee of the UMC Utrecht (AVD15002016410).
MBA‐MD‐468 cells were treated with 10 µM SEN177 or DMSO for 3 days and then labelled with 4 µM CellTrace Violet fluorescent dye (Invitrogen, Thermo Fisher Scientific) for 15 minutes at room temperature. Untreated EGFR negative murine Ba/F3 control cells were labelled with 0.125 µM of the dye. Cells were mixed at a 5:1 ratio, and a total of 6 × 106 cells in 200 µL PBS were injected intraperitoneally into each mouse. Immediately after tumor cell inoculation, a single dose of 100 µg CTX (IgG1), PANI (IgG2), or an IgA2.0 variant of CTX was given by intraperitoneal injection; PBS was used as a control. Sixteen hours later, all mice (n = 5 per group) were killed, and tumor cells recovered by peritoneal lavage using PBS containing 5 mmol/L EDTA. The ratio of CellTrace violet high MDA‐MD‐468 and control CellTrace violet low Ba/F3 cells in the peritoneum was determined by flow cytometry.
2.9. Data processing and statistical analyses
The ADCC, CDC, and flow cytometric data were generated from at least three independent experiments. Both ADCC and CDC were carried out in triplicate with effector cells or NHS from different healthy donors. Graphical and statistical analyses were undertaken using GraphPad Prism 6.0 (GraphPad Software). Group data are reported as mean values ± SEM. Significance was determined by two‐way ANOVA repeated measures test with Bonferroni's post hoc correction or unpaired t test. The EC50 values were calculated from dose‐response curves and reported as mean values ± SEM.
3. RESULTS
3.1. Glutaminyl cyclase inhibition specifically affects CD47‐SIRPα interactions
Glutaminyl cyclase‐mediated pyro‐glutamate formation at the N‐terminus of CD47 has been described to be involved in SIRPα binding (Figure 1A).14 We initially studied the effect of the prototypic QC inhibitor SEN177 on the survival of the tumor cell lines A431 (SCC), Kyse‐30 (esophageal SCC), and MDA‐MB‐468 (breast cancer). All three cell lines are EGFR/CD47 double positive (Figure S1). Using increasing SEN177 concentrations of up to 50 µM over 72 hours, the inhibitor had no direct toxic effects (Figure 1B). For finding the optimal use of SEN177, the dose and time dependent inhibition of soluble SIRPα‐Fc binding on MDA‐MB‐468 cells was investigated. A concentration of 10 µM SEN177 is required to effectively interrupt CD47‐SIRPα interactions (Figure 1C), the maximum inhibition was achieved after 72 hours (Figure 1D). A 50% or higher inhibition of SIRPα‐Fc binding persisted for up to 48 hours after withdrawal of SEN177, while after 96 hours SIRPα‐Fc binding was reconstituted by more than 75% (Figure 1E). Importantly, saturating concentrations of SIRPα‐Fc were not able to overcome SEN177‐induced binding reduction (Figure 1F). At evaluated treatment conditions (10 µM SEN177 for 72 hours), SIRPα‐Fc binding on A431 cells was reduced by 36.2 ± 10.0% and by 56.9 ± 7.5% and 56.9 ± 8.3% for Kyse‐30 and MDA‐MB‐468 cells, respectively (Figure 1G). Neither the expression of the target antigen EGFR nor CD47 were affected by QC inhibition as measured by staining with the mAbs CTX and B6H12, respectively (Figure S1C). Together, these data indicate effective and consistent inhibition of N‐terminal pyro‐glutamate formation of CD47 by QC inhibitor treatment.
FIGURE 1.
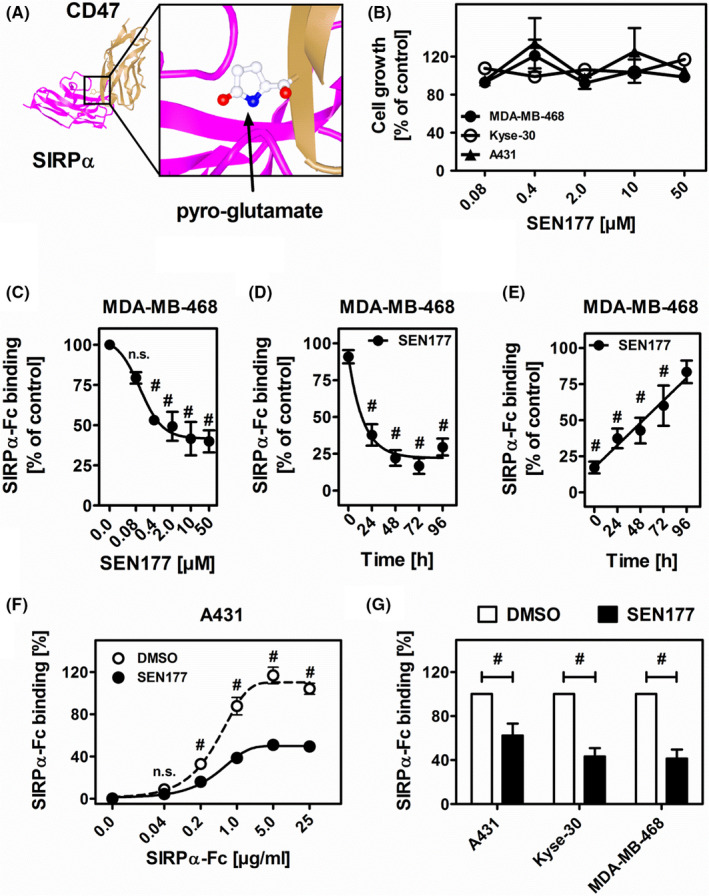
Glutaminyl cyclase inhibition by SEN177 reduces signal regulatory protein alpha (SIRPα) binding to CD47‐positive tumor cells. A, Spatial interactions between the N‐terminal pyro‐glutamate modification at CD47 (brown) and its binding site for SIRPα (purple) (adapted from NCBI Structure PDB ID 2JJT). B, Treatment of MDA‐MB‐468, Kyse‐30, and A431 cancer cells with SEN177 (0.08‐50 µM) for 3 d does not affect cell growth as measured by MTS assays. Mean values are shown as percentage of DMSO control ± SEM of three independent experiments. C, Treatment of MDA‐MB‐468 cells with SEN177 (0.08‐50 µM, 72 h) results in dose‐dependent reduction of SIRPα‐Fc (5 µg/mL) binding compared to DMSO control, as analyzed by indirect immunofluorescence analyses. D, Time‐dependent (0‐96 h) inhibition of SIRPα‐Fc (5 µg/mL) binding to SEN177 (10 µM) treated MDA‐MB‐468 cells. E, Reconstitution of SIRPα‐Fc (5 µg/mL) binding after SEN177 (10 µM, 72 h) treatment of MDA‐MB‐468 cells when cell culture medium was refreshed and SIRPα‐Fc binding to target cells was analyzed at the indicated time points. F, Dose‐dependent binding of soluble SIRPα‐Fc (0.04‐25 µg/mL) on SEN177 (10 µM, 72 h) treated A431 cells is significantly reduced compared to DMSO treated cells. G, Reduction of SIRPα‐Fc (5 µg/mL) binding on different SEN177 (10 µM, 72 h) treated tumor cell lines. Mean values as a percentage of the DMSO control ± SEM of at least three independent experiments are displayed. #P ≤ .05 (two‐way ANOVA). n.s., nonsignificant difference
3.2. Glutaminyl cyclase inhibition does not interfere with direct growth inhibition, CDC, and NK cell‐mediated ADCC triggered by EGFR Abs
Next, we analyzed the effect of QC inhibition on potential effector mechanisms of EGFR‐directed Abs. As previously described, CTX is capable of killing tumor cells by multiple effector mechanisms including direct growth inhibition, CDC (in combination with another EGFR Ab), or ADCC.36 SEN177 did not alter the ability of CTX to induce concentration‐dependent growth inhibition on DiFi colon cancer cells (Figure 2A). Control experiments confirmed reduced SIRPα‐Fc binding (60.9 ± 1.8%) after SEN177 treatment (10 µM for 3 days) also on DiFi cells (data not shown). Complement‐dependent cytotoxicity against A431 cells triggered by the combination of CTX and MTZ (both of human IgG1 isotype) was also not affected by SEN177 treatment (Figure 2B). In addition, no effects of SEN177 on ADCC against A431 was observed using either whole PBMC (including NK cells) or with enriched purified NK cells (Figure 2C).
FIGURE 2.
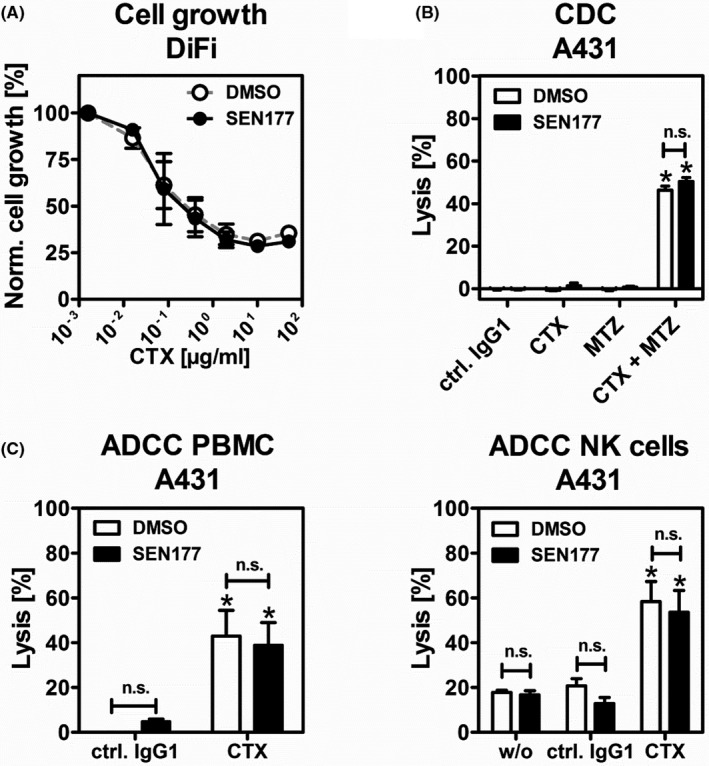
Glutaminyl cyclase inhibition does not affect epidermal growth factor receptor (EGFR) Ab‐triggered growth inhibition, complement‐dependent cytotoxicity (CDC), or Ab dependent cell‐mediated cytotoxicity (ADCC). A, Colon cancer cell line DiFi was treated with 10 µM SEN177 or DMSO as a control in combination with increasing concentrations of cetuximab (CTX) for 3 d. Cell growth was measured in a colorimetric, MTS‐based assay. Mean values are shown as percentage of control without Ab ± SEM of three independent experiments. B, SEN177 (10 µM, 72 h) or DMSO pretreated A431 cells served as target cells in CDC experiments. Normal human serum (25% v/v) was used as a source of complement. The EGFR Abs CTX and matuzumab (MTZ) or their combination were applied. Rituximab was used as a human isotype control Ab (ctrl. IgG1). C, In ADCC assays, A431, PBMC, or purified natural killer (NK) cells were used as effector cells in classical chromium release experiments (effector : target cell ratios of 40:1 and 10:1, respectively). In B and C, all Abs were used at a final concentration of 10 µg/mL. Mean values ± SEM of at least three independent experiments are shown. Data were analyzed by two‐way ANOVA. *P ≤ .05, control vs specific Abs. n.s., nonsignificant difference between DMSO control and SEN177 treated cells
3.3. Glutaminyl cyclase inhibition improves ADCP by macrophages
CD47‐SIRPα interactions regulate macrophage activation by tumor‐directed Abs.37 Thus, the effects of QC inhibition on ADCP mediated by EGFR Abs of different isotypes, CTX (IgG1), PANI (IgG2), and CTX‐IgA2 were analyzed. Macrophages were generated from peripheral blood monocytes by adhesion and subsequently differentiated to uncommitted M0 macrophages by M‐CSF. These macrophages displayed high expression of FcγRIIa (CD32a) and FcγRI (CD64), intermediate expression of FcγRIIb (CD32b, expressed by 44.7 ± 8.7% of macrophages) and FcαRI (CD89), and low levels of FcγRIII (CD16) as determined by indirect immunofluorescence (Figure 3A). The cancer cell lines MDA‐MB‐468 and Kyse‐30 were treated for 3 days with 10 µM SEN177 or DMSO and subsequently used as target cells in ADCP assays. A431 cells were excluded from these experiments as they were not phagocytosed by macrophages, which could be explained by their larger size compared to MDA‐MB‐48 and Kyse‐30 cells, or by other tumor cell characteristics.38 Glutaminyl cyclase inhibition significantly increased the ability of all three EGFR Abs to induce ADCP (Figure 3B). Compared to DMSO treated cells, the ADCP capacity of CTX was improved 2.2‐fold for MDA‐MB‐468 and 1.9‐fold for Kyse‐30. With PANI the improvement was 2.2‐fold and 2.4‐fold, and 2.5‐fold and 1.7‐fold with CTX‐IgA2.
FIGURE 3.
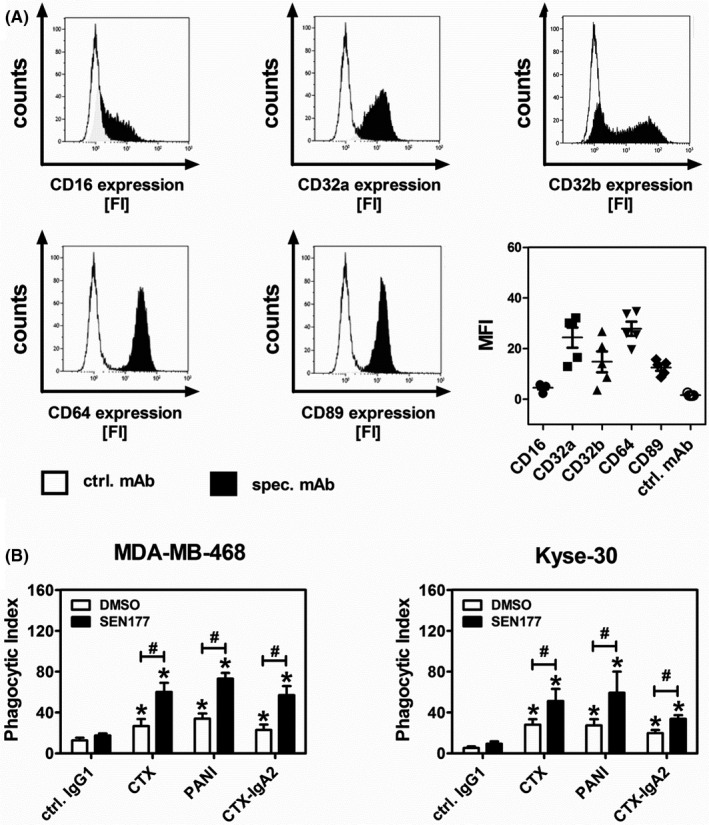
Glutaminyl cyclase inhibition enhances tumor cell phagocytosis by macrophages using epidermal growth factor receptor (EGFR) Abs of different isotypes. A, Fc receptor profiling of macrophage colony‐stimulating factor (M‐CSF) differentiated macrophages was undertaken by indirect flow cytometry using Abs (10 µg/mL) against the indicated Fc receptors. Results shown are histograms from one representative experiment with an isotype control Ab (ctrl. mAb, white area) and the specific Ab (spec. mAb, black area). FI, fluorescence intensity. A summary of the mean fluorescence intensities (MFI) ± SEM obtained from experiments with five different donors is provided. B, Tumor cell lines MDA‐MB‐468 and Kyse‐30 were treated with SEN177 (10 µM) or DMSO for 3 d before serving as target cells in Ab‐dependent cellular phagocytosis experiments with M‐CSF differentiated macrophages at an effector : target cell ratio of 1:2. EGFR Abs of human IgG1 (cetuximab [CTX]), IgG2 (panitumumab [PANI]), and IgA2 (CTX‐IgA2) as well as a nonbinding control Ab (ctrl. IgG1) were used at a concentration of 50 µg/mL. Mean values ± SEM of at least three independent experiments are shown. Data were analyzed by two‐way ANOVA. *P ≤ .05, control vs specific Abs; #P ≤ .05, DMSO vs SEN177 treated cells
3.4. Glutaminyl cyclase inhibition improves PMN‐mediated ADCC by different Ab isotypes
CD47‐SIRPα interactions also regulate the cytotoxic activity of PMN as effector cells in ADCC.27, 39 In contrast to M0 macrophages, PMN freshly isolated from human peripheral blood showed high expression of FcγRIII (CD16), followed by FcαRI (CD89) and FcγRIIa (CD32a), whereas FcγIIb (CD32b) and FcγRI (CD64) were hardly expressed (Figure 4A), confirming previous findings.25, 40 Cetuximab, PANI, and CTX‐IgA2 were investigated in ADCC assays with GM‐CSF stimulated PMN against tumor cell lines, which were pretreated with SEN177 for 3 days. As shown in Figure 4B, PMN‐mediated ADCC against Kyse‐30 cells was significantly improved by QC inhibition, in particular when the IgG2 and IgA2 Abs were used. The effect on cell lysis with the IgG1 Ab CTX was much less pronounced. The PMN‐mediated lysis of A431 and MDA‐MB‐468 cells was consistently increased following QC inhibition with PANI (Figure 4C). The IgA2 isotype yielded the highest cell lysis rates among all Abs, and the ADCC was significantly improved by QC inhibition against MDA‐MB‐468 cells (Figure 4C). Antibody‐dependent cell‐mediated cytotoxicity with CTX yielded the lowest tumor cell lysis rates and was not significantly affected by SEN177. This is in line with the finding that the GPI‐linked FcγRIIIb (CD16b) receptor expressed by PMN does not trigger ADCC.40
FIGURE 4.
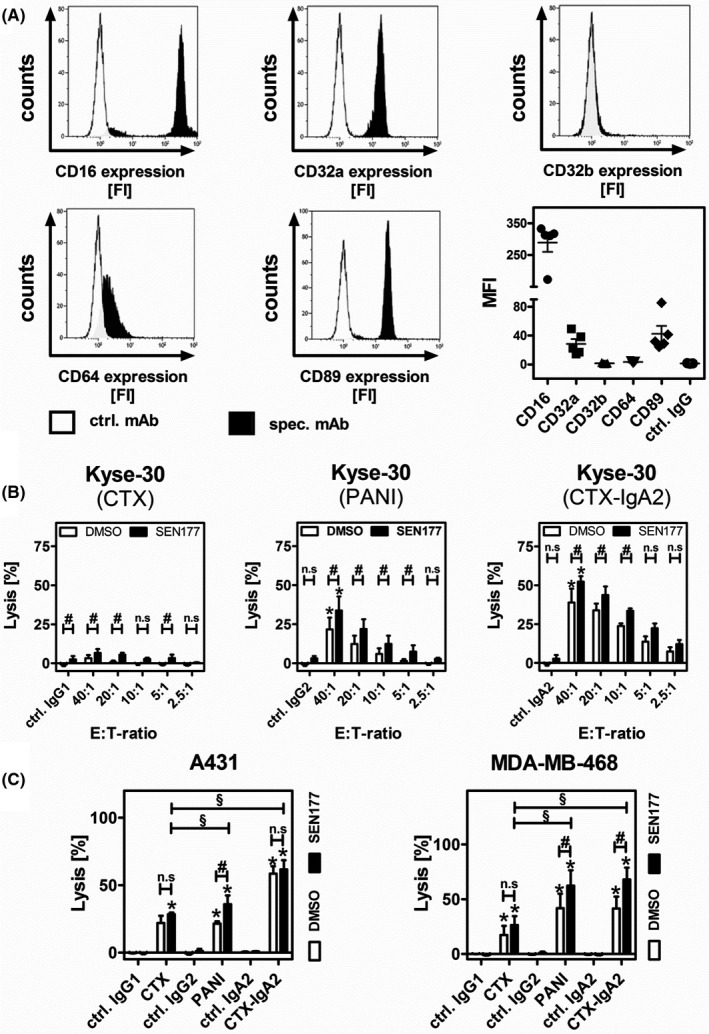
Glutaminyl cyclase inhibition improves polymorphonuclear neutrophilic granulocyte (PMN)‐mediated Ab‐dependent cell‐mediated cytotoxicity (ADCC) by epidermal growth factor receptor Abs depending on their isotype. A, Expression of Fc receptors on PMN was analyzed by flow cytometry. Results are presented as histograms from one representative experiment showing the fluorescence intensity (FI) and as a summary of the mean fluorescence intensities (MFI) ± SEM obtained from five experiments each with a different donor. B, Kyse‐30 cells were treated with SEN177 (10 µM) or an equivalent amount of DMSO as a control for 3 d and subsequently served as target cells in PMN‐mediated ADCC experiments using varying effector : target (E:T) cell ratios (fixed E:T ratio of 40:1 for isotype control). C, Tumor cell lines A431 and MDA‐MB‐468 were treated with SEN177 (10 µM) or DMSO for 3 d and subsequently used as target cells in PMN‐mediated ADCC experiments. E:T cell ratios of 40:1 (A431) and 80:1 (MDA‐MB‐468) were applied. In ADCC experiments, all Abs and isotype controls (ctrl.) were used at a concentration of 1 µg/mL. Bars represent mean values ± SEM of at least three independent experiments. Data were analyzed by two‐way ANOVA. *P ≤ .05, control vs specific Abs; #P ≤ .05, DMSO vs SEN177 treated cells; §P ≤ .05 between isotypes (SEN177 treated cells). CTX, cetuximab; PANI, panitumumab
3.5. Glutaminyl cyclase inhibition in tumor cells translated into enhanced EGFR Ab efficacy in vivo
The effect of QC inhibition on Ab therapy against tumors in vivo was investigated in xenografted mice treated with human EGFR Abs of different isotypes. Experiments with IgA Abs were carried out in human FcαRI transgenic (CD89 Tg) SCID mice; for all other Abs, non‐Tg littermates were used. As the prototypic QC inhibitor SEN177 has significant off‐target toxicity in mice (hazard code GHS06), MDA‐MD‐468 tumor cells were pretreated with SEN177 (10 µM) or DMSO in vitro for 3 days, and then mixed with untreated murine Ba/F3 cells (ratio 5:1) as a recovery control. Tumor cells were labeled with a fluorescent dye and injected into the peritoneal cavity of SCID mice, immediately followed by treatment with either PBS or EGFR Abs of IgG1 (CTX), IgG2 (PANI), or IgA2 (CTX‐IgA2) isotype. After 16 hours, mice were killed and tumor load in the peritoneal cavity was analyzed by measuring the ratio of MDA‐MB‐468 and Ba/F3 cells (Figure 5A). At this time point, CD47‐SIRPα interaction was significantly inhibited (>50%) as shown by reduced SIRPα‐Fc binding after SEN177 withdrawal (Figure 1E). Importantly, all three EGFR Abs—IgG1, IgG2, and IgA2—significantly reduced the MDA‐MB‐468 tumor load compared to PBS treated control mice (Figure 5B,C). The tumor load was decreased by QC inhibition compared to DMSO controls in all Ab‐treated groups, but these differences were not statistically significant, probably due to the low number of mice in each group (n = 5). However, when all mice who received EGFR Abs were analyzed together, the tumor cell recovery of SEN177 treated cells was significantly (P < .05) reduced compared to DMSO treated cells. No significant difference was observed in the group of mice treated with PBS (Figure 5D). Together, these results indicate that QC inhibition enhances the therapeutic efficacy of EGFR Abs in vivo.
FIGURE 5.
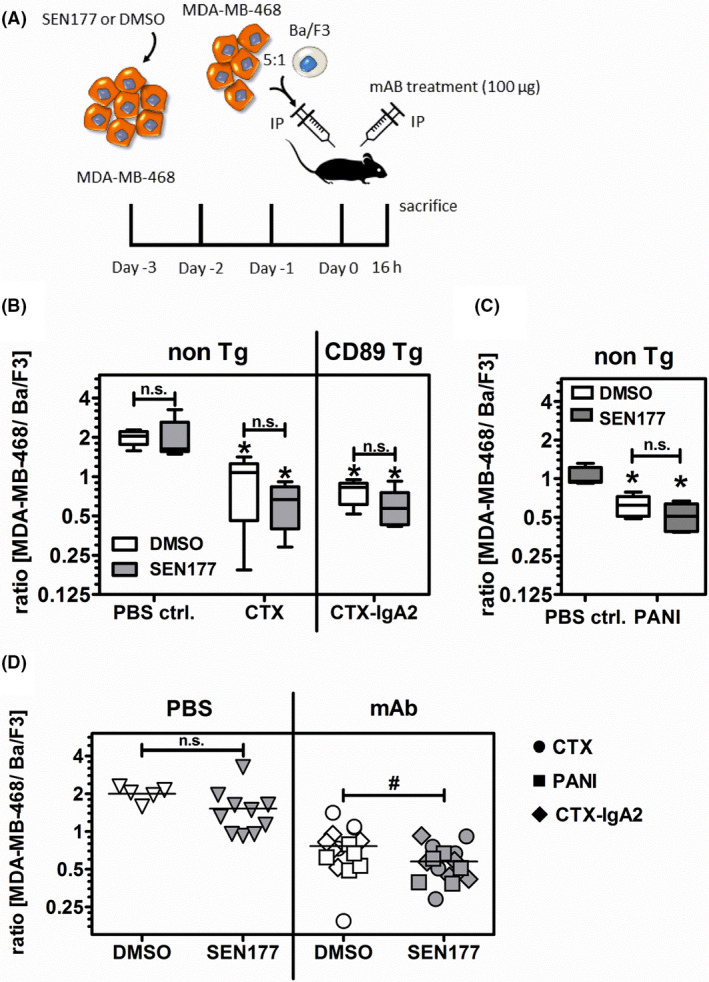
Glutaminyl cyclase inhibition in tumor cells improves epidermal growth factor receptor (EGFR) Ab therapeutic efficacy in a xenogeneic tumor model. A, MBA‐MD‐468 tumor cells were incubated with 10 µM SEN177 or DMSO for 3 d prior to intraperitoneal (IP) injection into FcαRI transgenic (CD89 Tg) or nontransgenic (non‐Tg) SCID mice. Untreated EGFR‐negative murine Ba/F3 cells served as a recovery control. Immediately after cell inoculation, mice were treated with 100 µg EGFR Abs or PBS as a control and killed 16 h later. B, C, Numbers of tumor cells in mice were evaluated by flow cytometry of cells from the peritoneal fluid 16 h after treatment with Abs or PBS as indicated. Results are presented as the median ratio of MBA‐MD‐468 to Ba/F3 cells (n = 5 per group) with an initial ratio of 5:1. D, Tumor cell recovery from mice treated with EGFR Abs irrespective of their isotype was significantly (P ≤ .05) reduced in mice who received SEN177 pretreated vs DMSO treated tumor cells (right panel), while a nonsignificant difference was observed in mice treated with PBS (left panel). Data shown are the individual and mean values of the experiments shown in (B) and (C) with 15 mice per group (Ab treatment panel) and five mice (DMSO) and 10 mice (SEN177) in the PBS treated panel. Data were analyzed by two‐tailed, unpaired t test. *P ≤ .05, PBS control vs specific Abs; #P ≤ .05, DMSO vs SEN177 treatment. n.s., not significant. CTX, cetuximab; PANI, panitumumab
4. DISCUSSION
Here, we investigated the impact of different EGFR Ab isotypes, human IgG1, IgG2, and IgA2, on tumor cell killing by myeloid effector cells during interference with CD47/SIRPα interactions by QC inhibition. After QC inhibition by SEN177, tumor cell killing by PMN was significantly enhanced with human IgA2 and IgG2 Abs, but only marginally with human IgG1, the most commonly used Ab isotype in the clinic. These differences between isotypes were less pronounced for macrophages, which were similarly effective with all three isotypes and enhanced in their efficacy by QC inhibition (benefits of at least 2‐fold with QC inhibition compared to control cells). Similarly, Chao et al reported that the combination of RTX and a CD47 blocking Ab improved the phagocytic index of B‐lymphoma cells by approximately 2‐fold.8 Interestingly, this combination resulted in clinical responses, with nearly 50% of patients achieving an objective response and 36% a complete response.5 Moreover, the increase in PMN‐mediated tumor cell lysis in combination with QC inhibition was similar to what could be achieved in the presence of CD47 blocking Abs, as shown by Treffers et al.41 Tumor cell killing by different effector cell populations is known to depend on the isotype of the tumor‐directed Abs, which bind to different Fc receptors on effector cells.42 For example, human NK cells are optimally activated by human IgG1 Ab binding to FcγRIIIa. However, human unstimulated NK cells typically do not express SIRPα and, therefore, do not benefit from CD47/SIRPα inhibition.11 Human IgG1 is suboptimal in the activation of human PMN, probably because the highly expressed FcγRIIIb receptor on PMN functions as a decoy receptor for this isotype.34, 40 Antibody‐dependent cell‐mediated cytotoxicity by human PMN is more effectively activated by human IgG2, predominantly by binding to FcγRIIa (CD32).26, 27 Optimal PMN activation occurs with human IgA Abs, which bind to FcαRI (CD89) and more effectively trigger ERK activation than Fcγ receptors do.25 Interestingly, the presented results indicate that the previously reported Ab isotype hierarchy in triggering ADCC by PMN (IgA2 > IgG2 > IgG1)25, 27, 30 is not altered by QC inhibition (Figure 4B,C).
Human Ab isotypes have been investigated in mice,43 but results need to be interpreted with caution as Fc receptor isoforms and expression profiles on effector cells differ between humans and mice.42 To better address these issues, mice with “humanized” Fc receptor expression profiles have been generated.44, 45
Myeloid checkpoint blockade offers novel opportunities to improve cancer immunotherapy.3, 4 Interference with CD47/SIRPα interactions by the CD47 Ab hu5F9‐G4 (magrolimab) is currently the most clinically advanced approach. Crystallographic studies have shown that an N‐terminal pyro‐glutamate in CD47 is located at the binding site for its ligand SIRPα (CD172α).14 The formation of this pyro‐glutamate is catalyzed by the enzyme QC,46 which can be inhibited by small molecules such as SEN177.18 A genetic screen has demonstrated the impact of QPCTL for the binding affinity of CD47 to SIRPα and its functional relevance for tumor cell killing.16, 17 Interestingly, inhibition of QC enzymes affects posttranslational pyro‐glutamate formation in many proteins, such as several chemokines, hormones, and other secreted proteins or peptides such as Aβ.46 The prototypic QC inhibitor SEN177 used in this manuscript is not suitable for in vivo studies due to off‐target toxicity, but more specific and less toxic molecules have been developed.47 The clinically most advanced QC inhibitor PQ912 is developed for the treatment of Alzheimer’s disease. Although numerous molecules could be affected by QC inhibition, which could result in potential side‐effects, the application of the QC inhibitor PQ912 in humans shows that it is well tolerated with only mild anemia commonly observed.48 In mouse models of Alzheimer’s disease transgenic for both human amyloid precursor protein and human glutaminyl cyclase, treatment with the QC inhibitor PQ912 resulted in dose‐related nearly complete QC occupancy in the brain with a reduction of pyroglutamyl‐modified Aβ peptides and neurological improvement after 4 months of treatment.49 Additional studies with QC inhibitors in clinical development need to determine whether a combination of QC inhibitors with tumor‐directed Abs will hold promise in tumor treatment. For specific disruption of CD47‐SIRPα interactions, mAbs against CD47 or SIRPα and soluble SIRPα‐Fc fusion proteins are most commonly applied today, with some of them being in clinical trials.50 Additionally, Wang et al recently described a CD47 targeted peptide (RS17), which is based on the sequence of thrombomodulin, another ligand of CD47.51 Alternatively, molecules targeting SIRPα, such as soluble high affinity CD47 variants, are being developed.52
In addition to the selection of the most appropriate Ab isotype, other criteria are critical to make the combination of CD47 inhibition and tumor‐directed Abs therapeutically successful. For example, tumors should contain a significant myeloid cell infiltrate,53, 54 or sufficient myeloid cells need to be recruited by specific therapeutic approaches.41, 55, 56 Myeloid cell infiltrates can vary widely with respect to their macrophage and PMN content, even within a well‐defined tumor entity such as triple negative breast cancer.57 Several genetic alterations in tumor cells have been reported to increase myeloid cell infiltration,58 which is often correlated with a worse prognosis for patients.53 Furthermore, the selection of appropriate target antigens59 or even particular epitopes on given antigens60, 61 could determine the extent of cytotoxicity that can be obtained.
Together, QPCTL inhibition by orally applicable and well‐tolerated small molecules could provide exciting opportunities to increase the therapeutic efficacy of tumor‐directed Abs by interfering with CD47/SIRPα interactions. For instance, advantages of orally applicable small molecules over CD47 blocking Abs could be less common infusion reactions, higher patient compliance, and better access to tumor cells due to improved tumor penetration, especially in the solid tumor microenvironment. The relative benefit on neutrophil recruitment has been shown to depend on the selected isotype of the tumor‐targeting Ab. These approaches could be particularly promising for tumor entities with a predominant myeloid cell infiltrate.53
DISCLOSURE
The authors declare no potential conflicts of interest.
Supporting information
Fig S1
ACKNOWLEDGMENT
These studies were supported by the German Research Foundation (DFG) (grant number: VA 124/9‐1). Deutsche Forschungsgemeinschaft, Grant/Award Number: (VA 124/9‐1).
Baumann N, Rösner T, Jansen JHM, et al. Enhancement of epidermal growth factor receptor antibody tumor immunotherapy by glutaminyl cyclase inhibition to interfere with CD47/signal regulatory protein alpha interactions. Cancer Sci. 2021;112:3029–3040. 10.1111/cas.14999
Niklas Baumann and Thies Rösner contributed equally to this work.
Funding information
German Research Foundation, Grant/Award Number: VA 124/9‐1.
REFERENCES
- 1.Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8:1069‐1086. [DOI] [PubMed] [Google Scholar]
- 2.Chen DS, Mellman I. Elements of cancer immunity and the cancer‐immune set point. Nature. 2017;541:321‐330. [DOI] [PubMed] [Google Scholar]
- 3.Mantovani A, Longo DL. Macrophage checkpoint blockade in cancer ‐ Back to the future. N Engl J Med. 2018;379:1777‐1779. [DOI] [PubMed] [Google Scholar]
- 4.van den Berg TK , Valerius T. Myeloid immune‐checkpoint inhibition enters the clinical stage. Nat Rev Clin Oncol. 2019;16:275‐276. [DOI] [PubMed] [Google Scholar]
- 5.Advani R, Flinn I, Popplewell L, et al. CD47 blockade by Hu5F9‐G4 and rituximab in non‐Hodgkin's lymphoma. N Engl J Med. 2018;379:1711‐1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu J, Wang L, Zhao F, et al. Pre‐clinical development of a humanized anti‐CD47 antibody with anti‐cancer therapeutic potential. PLoS One. 2015;10:e0137345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chao MP, Alizadeh AA, Tang C, et al. Therapeutic antibody targeting of CD47 eliminates human acute lymphoblastic leukemia. Cancer Res. 2011;71:1374‐1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chao MP, Alizadeh AA, Tang C, et al. Anti‐CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non‐Hodgkin lymphoma. Cell. 2010;142:699‐713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matlung HL, Szilagyi K, Barclay NA, van den Berg TK . The CD47‐SIRPα signaling axis as an innate immune checkpoint in cancer. Immunol Rev. 2017;276:145‐164. [DOI] [PubMed] [Google Scholar]
- 10.Weiskopf K. Cancer immunotherapy targeting the CD47/SIRPα axis. Eur J Cancer. 2017;76:100‐109. [DOI] [PubMed] [Google Scholar]
- 11.Barclay AN, Van den Berg TK. The interaction between signal regulatory protein alpha (SIRPα) and CD47: structure, function, and therapeutic target. Annu Rev Immunol. 2014;32:25‐50. [DOI] [PubMed] [Google Scholar]
- 12.Okazawa H, Motegi S, Ohyama N, et al. Negative regulation of phagocytosis in macrophages by the CD47‐SHPS‐1 system. J Immunol. 2005;174:2004‐2011. [DOI] [PubMed] [Google Scholar]
- 13.Matozaki T, Murata Y, Okazawa H, Ohnishi H. Functions and molecular mechanisms of the CD47‐SIRPα signalling pathway. Trends Cell Biol. 2009;19:72‐80. [DOI] [PubMed] [Google Scholar]
- 14.Hatherley D, Graham SC, Turner J, Harlos K, Stuart DI, Barclay AN. Paired receptor specificity explained by structures of signal regulatory proteins alone and complexed with CD47. Mol Cell. 2008;31:266‐277. [DOI] [PubMed] [Google Scholar]
- 15.Schilling S, Manhart S, Hoffmann T, Ludwig HH, Wasternack C, Demuth HU. Substrate specificity of glutaminyl cyclases from plants and animals. Biol Chem. 2003;384:1583‐1592. [DOI] [PubMed] [Google Scholar]
- 16.Logtenberg MEW, Jansen JHM, Raaben M, et al. Glutaminyl cyclase is an enzymatic modifier of the CD47‐ SIRPα axis and a target for cancer immunotherapy. Nat Med. 2019;25:612‐619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu Z, Weng L, Zhang T, et al. Identification of glutaminyl cyclase isoenzyme isoQC as a regulator of SIRPα‐CD47 axis. Cell Res. 2019;29:502‐505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pozzi C, Di Pisa F, Benvenuti M, Mangani S. The structure of the human glutaminyl cyclase‐SEN177 complex indicates routes for developing new potent inhibitors as possible agents for the treatment of neurological disorders. J Biol Inorg Chem. 2018;23:1219‐1226. [DOI] [PubMed] [Google Scholar]
- 19.Weiskopf K, Weissman IL. Macrophages are critical effectors of antibody therapies for cancer. MAbs. 2015;7:303‐310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Treffers LW, Hiemstra IH, Kuijpers TW, van den Berg TK , Matlung HL. Neutrophils in cancer. Immunol Rev. 2016;273:312‐328. [DOI] [PubMed] [Google Scholar]
- 21.Huls G, Heijnen IA, Cuomo E, et al. Antitumor immune effector mechanisms recruited by phage display‐derived fully human IgG1 and IgA1 monoclonal antibodies. Cancer Res. 1999;59:5778‐5784. [PubMed] [Google Scholar]
- 22.Dechant M, Beyer T, Schneider‐Merck T, et al. Effector mechanisms of recombinant IgA antibodies against epidermal growth factor receptor. J Immunol. 2007;179:2936‐2943. [DOI] [PubMed] [Google Scholar]
- 23.Boross P, Lohse S, Nederend M, et al. IgA EGFR antibodies mediate tumour killing in vivo. EMBO Mol Med. 2013;5:1213‐1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lohse S, Loew S, Kretschmer A, et al. Effector mechanisms of IgA antibodies against CD20 include recruitment of myeloid cells for antibody‐dependent cell‐mediated cytotoxicity and complement‐dependent cytotoxicity. Br J Haematol. 2018;181:413‐417. [DOI] [PubMed] [Google Scholar]
- 25.Brandsma AM, Bondza S, Evers M, et al. Potent Fc receptor signaling by IgA leads to superior killing of cancer cells by neutrophils compared to IgG. Front Immunol. 2019;10:704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schneider‐Merck T, Lammerts van Bueren JJ, Berger S, et al. Human IgG2 antibodies against epidermal growth factor receptor effectively trigger antibody‐dependent cellular cytotoxicity but, in contrast to IgG1, only by cells of myeloid lineage. J Immunol. 2010;184:512‐520. [DOI] [PubMed] [Google Scholar]
- 27.Rosner T, Kahle S, Montenegro F, et al. Immune effector functions of human IgG2 antibodies against EGFR. Mol Cancer Ther. 2018;18:77‐88. [DOI] [PubMed] [Google Scholar]
- 28.Arce Vargas F, Furness AJS, Litchfield K, et al. Fc effector function contributes to the activity of human anti‐CTLA‐4 antibodies. Cancer Cell. 2018;33:649‐63 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meyer S, Nederend M, Jansen JH, et al. Improved in vivo anti‐tumor effects of IgA‐Her2 antibodies through half‐life extension and serum exposure enhancement by FcRn targeting. MAbs. 2016;8:87‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lohse S, Meyer S, Meulenbroek LA, et al. An anti‐EGFR IgA that displays improved pharmacokinetics and myeloid effector cell engagement in vivo. Cancer Res. 2016;76:403‐417. [DOI] [PubMed] [Google Scholar]
- 31.Vafa O, Gilliland GL, Brezski RJ, et al. An engineered Fc variant of an IgG eliminates all immune effector functions via structural perturbations. Methods. 2014;65:114‐126. [DOI] [PubMed] [Google Scholar]
- 32.Schlaeth M, Berger S, Derer S, et al. Fc‐engineered EGF‐R antibodies mediate improved antibody‐dependent cellular cytotoxicity (ADCC) against KRAS‐mutated tumor cells. Cancer Sci. 2010;101:1080‐1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oberg HH, Kellner C, Gonnermann D, et al. Tribody [(HER2)2xCD16] is more effective than trastuzumab in enhancing γδ T cell and natural killer cell cytotoxicity against HER2‐expressing cancer cells. Front Immunol. 2018;9:814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Derer S, Glorius P, Schlaeth M, et al. Increasing FcγRIIa affinity of an FcγRIII‐optimized anti‐EGFR antibody restores neutrophil‐mediated cytotoxicity. MAbs. 2014;6:409‐421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Egmond M , van Vuuren AJ , Morton HC, et al. Human immunoglobulin A receptor (FcαRI, CD89) function in transgenic mice requires both FcR γ chain and CR3 (CD11b/CD18). Blood. 1999;93:4387‐4394. [PubMed] [Google Scholar]
- 36.Peipp M, Dechant M, Valerius T. Effector mechanisms of therapeutic antibodies against ErbB receptors. Curr Opin Immunol. 2008;20:436‐443. [DOI] [PubMed] [Google Scholar]
- 37.Takimoto CH, Chao MP, Gibbs C, et al. The macrophage 'do not eat me' signal, CD47, is a clinically validated cancer immunotherapy target. Ann Oncol. 2019;30:486‐489. [DOI] [PubMed] [Google Scholar]
- 38.Sosale NG, Rouhiparkouhi T, Bradshaw AM, Dimova R, Lipowsky R, Discher DE. Cell rigidity and shape override CD47's "self"‐signaling in phagocytosis by hyperactivating myosin‐II. Blood. 2015;125:542‐552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao XW, van Beek EM , Schornagel K, et al. CD47‐signal regulatory protein‐alpha (SIRPα) interactions form a barrier for antibody‐mediated tumor cell destruction. Proc Natl Acad Sci U S A. 2011;108:18342‐18347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Treffers LW, van Houdt M , Bruggeman CW, et al. FcγRIIIb restricts antibody‐dependent destruction of cancer cells by human neutrophils. Front Immunol. 2018;9:3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Treffers LW, Ten Broeke T, Rosner T, et al. IgA‐mediated killing of tumor cells by neutrophils is enhanced by CD47‐SIRPα checkpoint inhibition. Cancer Immunol Res. 2019;8:120‐130. [DOI] [PubMed] [Google Scholar]
- 42.Bruhns P. Properties of mouse and human IgG receptors and their contribution to disease models. Blood. 2012;119:5640‐5649. [DOI] [PubMed] [Google Scholar]
- 43.Overdijk MB, Verploegen S, Ortiz Buijsse A, et al. Crosstalk between human IgG isotypes and murine effector cells. J Immunol. 2012;189:3430‐3438. [DOI] [PubMed] [Google Scholar]
- 44.Smith P, DiLillo DJ, Bournazos S, Li F, Ravetch JV. Mouse model recapitulating human Fcγ receptor structural and functional diversity. Proc Natl Acad Sci U S A. 2012;109:6181‐6186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee CH, Kang TH, Godon O, et al. An engineered human Fc domain that behaves like a pH‐toggle switch for ultra‐long circulation persistence. Nat Commun. 2019;10:5031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schilling S, Wasternack C, Demuth HU. Glutaminyl cyclases from animals and plants: a case of functionally convergent protein evolution. Biol Chem. 2008;389:983‐991. [DOI] [PubMed] [Google Scholar]
- 47.Hoang VH, Tran PT, Cui M, et al. Discovery of potent human glutaminyl cyclase inhibitors as anti‐Alzheimer's agents based on rational design. J Med Chem. 2017;60:2573‐2590. [DOI] [PubMed] [Google Scholar]
- 48.Scheltens P, Hallikainen M, Grimmer T, et al. Safety, tolerability and efficacy of the glutaminyl cyclase inhibitor PQ912 in Alzheimer's disease: results of a randomized, double‐blind, placebo‐controlled phase 2a study. Alzheimers Res Ther. 2018;10:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hoffmann T, Meyer A, Heiser U, et al. Glutaminyl cyclase inhibitor PQ912 improves cognition in mouse models of Alzheimer's disease‐studies on relation to effective target occupancy. J Pharmacol Exp Ther. 2017;362:119‐130. [DOI] [PubMed] [Google Scholar]
- 50.Zhang W, Huang Q, Xiao W, et al. Advances in anti‐tumor treatments targeting the CD47/SIRPα axis. Front Immunol. 2020;11:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang X, Wang Y, Hu J, Xu H. An antitumor peptide RS17‐targeted CD47, design, synthesis, and antitumor activity. Cancer Med. 2021;10:2125‐2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ho CC, Guo N, Sockolosky JT, et al. "Velcro" engineering of high affinity CD47 ectodomain as signal regulatory protein alpha (SIRPα) antagonists that enhance antibody‐dependent cellular phagocytosis. J Biol Chem. 2015;290:12650‐12663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gentles AJ, Newman AM, Liu CL, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. 2015;21:938‐945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Singhal S, Bhojnagarwala PS, O'Brien S, et al. Origin and role of a subset of tumor‐associated neutrophils with antigen‐presenting cell features in early‐stage human lung cancer. Cancer Cell. 2016;30:120‐135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Turaj AH, Hussain K, Cox KL, et al. Antibody tumor targeting is enhanced by CD27 agonists through myeloid recruitment. Cancer Cell. 2017;32(6)777‐791.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dahal LN, Dou L, Hussain K, et al. STING activation reverses lymphoma‐mediated resistance to antibody immunotherapy. Cancer Res. 2017;77:3619‐3631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim IS, Gao Y, Welte T, et al. Immuno‐subtyping of breast cancer reveals distinct myeloid cell profiles and immunotherapy resistance mechanisms. Nat Cell Biol. 2019;21:1113‐1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wellenstein MD, de Visser KE . Cancer‐cell‐intrinsic mechanisms shaping the tumor immune landscape. Immunity. 2018;48:399‐416. [DOI] [PubMed] [Google Scholar]
- 59.Carter PJ, Lazar GA. Next generation antibody drugs: pursuit of the 'high‐hanging fruit'. Nat Rev Drug Discov. 2018;17:197‐223. [DOI] [PubMed] [Google Scholar]
- 60.Cleary KLS, Chan HTC, James S, Glennie MJ, Cragg MS. Antibody distance from the cell membrane regulates antibody effector mechanisms. J Immunol. 2017;198:3999‐4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li J, Stagg NJ, Johnston J, et al. Membrane‐proximal epitope facilitates efficient T cell synapse formation by anti‐FcRH5/CD3 and is a requirement for myeloma cell killing. Cancer Cell. 2017;31:383‐395. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1