

Abstract
Succinate dehydrogenase (SDH)‐deficient renal cell carcinoma (RCC) is mainly associated with a mutation in the SDHB gene and sometimes with mutations in the SDHC or SDHD genes. However, only three cases of succinate dehydrogenase A (SDHA)‐deficient RCC have been reported, and the relation between SDHA mutations and RCC has not been clarified. This study assessed the role of SDHA gene mutations in human RCC. We investigated SDHA/B/C/D gene mutations in 129 human RCCs. Targeted next‐generation sequencing and direct Sanger sequencing revealed single nucleotide variants (SNVs) of the SDHA gene with amino acid sequence variations in 11/129 tumors, while no SDHB/C/D gene mutations were found. Tumor cells with SNVs of the SDHA gene were characterized by eosinophilic cytoplasm and various patterns of proliferation. Immunohistochemistry examination found that the 11 tumors with SNVs of the SDHA gene showed significant reduction of SDHA protein and SDHB protein expression compared to the 19 tumors without SDHA or SDHB mutations (both P < .0001). Western blotting showed a greater decrease in the expression of SDHA and SDHB proteins in the 11 tumors with SNVs of the SDHA gene than in the 19 tumors without (both P < .0001). There was a positive correlation between SDHA and SDHB protein levels (P < .0001). On immunohistochemistry and Western blotting, the 11 tumors with SNVs of the SDHA gene had higher protein expression for nuclear factor E2‐related factor 2 (Nrf2) compared to the 19 tumors without the mutation (P < .01). These observations suggest that SDHA gene mutations might be associated with a subset of RCC.
Keywords: fumarate hydratase, next‐generation sequencing, nuclear factor E2‐related factor 2, renal cell carcinoma, succinate dehydrogenase
Succinate dehydrogenase (SDH) A gene mutation was identified in 11 out of 129 human renal cell carcinoma (RCC) samples by targeted next‐generation sequencing and direct Sanger sequencing, while no SDHB/C/D gene mutations were detected. Immunohistochemistry and western blotting identified a reduction of SDHA and SDHB protein expression in the tumors with SDHA gene mutations. These findings suggest that SDHA gene mutations might be linked to a subset of RCC.

Abbreviations
- 2SC
S‐(2‐succinyl)cysteine
- ACMG
American College of Medical Genetics and Genomics
- ATP
adenosine triphosphate
- ccRCC
clear cell renal cell carcinoma
- CT
computed tomography
- DMEM
Dulbecco's modified Eagle's medium
- FH
fumarate hydratase
- GAPDH
glyceraldehyde 3‐phosphate dehydrogenase
- HIF
hypoxia‐inducible factor
- HLRCC
hereditary leiomyomatosis and renal cell carcinoma
- ISUP
International Society of Urological Pathology
- Keap1
Kelch‐like ECH‐associated protein 1
- MRI
magnetic resonance imaging
- non‐ccRCC
non‐clear cell renal cell carcinoma
- Nrf2
nuclear factor E2‐related factor 2
- PBRM1
polybromo‐1
- PCR
polymerase chain reaction
- pRCC
papillary renal cell carcinoma
- pT
pathological stage
- RCC
renal cell carcinoma
- RECIST
Response Evaluation Criteria in Solid Tumors
- RET
rearranged during transfection
- ROS
reactive oxygen species
- SDH
succinate dehydrogenase
- SDHA
succinate dehydrogenase A
- SDHB
succinate dehydrogenase B
- SDHC
succinate dehydrogenase C
- SDHD
succinate dehydrogenase D
- SNV
single nucleotide variants
- TCA
tricarboxylic acid
- TNM
tumor‐lymph node‐metastasis classification of malignant tumors
- VHL
von Hippel‐Lindau
1. INTRODUCTION
The TCA cycle, which occurs in mitochondria, is a core pathway for the metabolism of sugars, lipids, and amino acids. A series of previous studies showed that altered activity and expression of TCA cycle key enzymes is associated with a subset of human cancers that show changes in cell metabolism.1, 2 Increasing evidence indicates that germline mutations of TCA cycle enzymes are associated with various hereditary and sporadic tumors.1, 2 To date, five such genes have been identified, which are fumarate hydratase (FH; 1q43 region) and the genes encoding the four subunits of succinate dehydrogenase (SDHA; 5p15 region, SDHB; 1p36.1 region, SDHC; 1q21 region, and SDHD; 11q23 region). SDH is a TCA cycle enzyme complex consisting of four protein subunits (SDHA, SDHB, SDHC, and SDHD) that is involved in the electron transport chain and is required for cellular energy metabolism. Mutations of genes encoding subunits of SDH are predominantly linked to pheochromocytoma and paraganglioma,3, 4 whereas mutations of FH are associated with leiomyoma, leiomyosarcoma, and HLRCC.5, 6, 7 These genes have been reported to act as tumor suppressors and only loss‐of‐function mutations have been detected so far.2 Intrinsic loss of the normal TCA cycle presumably promotes tumorigenesis due to metabolic alterations that arise with enforced dependence on glycolysis for energy production, mimicking the metabolic reprogramming pattern known as the Warburg effect that is associated with malignancy.8, 9, 10 The Warburg effect involves an increase of glycolysis to generate ATP for cell proliferation and enhancement of fatty acid synthesis by dephosphorylation of acetyl CoA carboxylase, an enzyme involved in the rate‐limiting step of fatty acid synthesis. Conversion of glucose metabolism from oxidation to glycolysis is one of the representative strategies cancer cells employ for generation of ATP.11
Renal cell carcinoma generally displays the Warburg effect.12 HLRCC‐associated RCCs do not possess a complete TCA cycle due to loss of FH enzyme activity and are effectively unable to perform oxidative phosphorylation, indicating that these cancers exist in a state of enforced dependence on glycolysis and are a notable example of the Warburg effect.12 Thus, FH‐deficient HLRCC might be a useful model for studying the deregulation of energy metabolism, as well as for developing new therapies to target cancers with TCA cycle enzyme deficiencies.13
SDH‐deficient RCC was accepted as a provisional entity in the 2013 ISUP Vancouver Classification, and was accepted as a unique RCC subtype by the WHO in 2016.14 Similar to FH‐deficient HLRCC‐associated RCC, SDH‐deficient RCC is characterized by impairment of oxidative phosphorylation and a metabolic shift to aerobic glycolysis.15 So far, most genomic alterations in patients with SDH‐deficient RCC have been found to affect SDHB, with the associated renal tumors being immunohistochemically negative for SDHB expression and having distinctive morphologic features,16, 17, 18, 19 while involvement of SDHC or SDHD is less common. Thus, information about SDH‐deficient RCC has largely been obtained by studying tumors with SDHB/C/D deficiency and the role of SDHA in RCC is not fully understood because only three cases of genetically confirmed SDHA‐deficient RCC have been reported.20, 21, 22 In the present study, we utilized a combination of genetic and biological techniques to investigate the role of SDHA in human RCC. Our findings could be useful for understanding the diverse roles of SDHA/B/C/D in cancer progression.
2. MATERIALS AND METHODS
2.1. Patients
We retrospectively investigated 129 patients (73 men and 56 women) with a histopathological diagnosis of RCC who underwent nephrectomy at our hospital between 2011 and 2018. Nephrectomy was performed before any other treatment. Preoperative imaging with CT and/or MRI was done for staging in all 129 patients. The postoperative follow‐up period ranged from 5 to 93 months (median: 29 months). All patients had no past or family history of paraganglioma, pheochromocytoma, or gastrointestinal stromal tumors. Of these 129 patients, 107 had clear cell RCC (ccRCC), 17 had papillary RCC type 2 (pRCC2), three had chromophobe RCC, and two had collecting duct RCC. In addition, 87 patients had a distant metastasis (M1), 103 had invasive disease (pT3/4) or lymph node involvement (pN1‐2) or both, and 81 had a poorly differentiated tumor (Fuhrman grade 3/4). On the other hand, as a control study for SDHB, but not SDHA, gene mutation, we examined a paraganglioma tissue with SDHB gene mutation with amino acid sequence abnormality (exon 3, c.274T>C, p.Ser92Pro, variant effect: missense). Furthermore, we also examined surgical samples of FH‐deficient RCC with FH, but not SDHA and SDHB, gene mutation.
2.2. DNA extraction
Frozen tumor samples were ground to a powder in liquid nitrogen, and 30‐50 mg of the powder was used for DNA extraction with an AllPrep kit (Qiagen). DNA was quantified, and its purity was assessed with a NanoDrop ND‐1000 spectrophotometer (LabTech). Blood DNA was extracted from leukocytes according to the standard protocols.
2.3. Next‐generation sequencing
Next‐generation sequencing was performed for detection of SNVs, short insertions, and deletions (indels). We investigated mutations of SDH subunit genes (SDHA, SDHB, SDHC, and SDHD), as well as mutations of the VHL, PBRM1, RET, Akt, and FH genes, by sequencing the coding exons and intron flanking regions using both blood samples and tumor specimens, as described previously.23 The custom primers for these regions were designed using AmpliSeq Designer (Life Technologies). Library construction and sequencing were carried out with an Ion AmpliSeq Library Kit 2.0, Ion PGM IC 200 kit, and Ion PGM (Life Technologies), according to the manufacturer's instructions. Sequencing data were analyzed with Torrent Suite, and variant call was conducted with Torrent Variant Caller, Ion Reporter (v.5.1.0). Ion AmpliSeq panels cover broad research areas for germline analysis, including genes recommended by the ACMG.24 Then, the accuracy of the Ion Torrent sequencer platform in detecting SDHA gene mutations was confirmed according to a previously published method.25
2.4. Direct Sanger sequencing of SDHA, SDHB, and VHL genes
Direct Sanger sequencing of all 15 coding exons of SDHA, all eight coding exons of SDHB, and all three coding exons of VHL gene was undertaken in 11 samples to confirm the mutations identified by next‐generation sequencing. Primers were described in Table S1.
2.5. Data analysis
After each sequencing reaction, raw data were analyzed by using Torrent Suite version 4.2.1 for processing of the signals, base calling, quality score assignment, adapter trimming, mapping to GRCH37/hg19 reference, assessment of mapping quality, and variant calling. After completion of primary data analysis, a list of the sequence variants detected (SNVs and indels) was compiled in a variant call file format and presented via the web‐based user interface. The results of mapping and variant calling were visualized using Integrative Genome viewer (Broad Institute).
2.6. Immunohistochemistry
Tumor tissue specimens from 30 RCC patients (11 tumors with SNVs of SDHA gene and 19 tumors without such mutations) were subjected to immunohistochemical staining for SDHA, SDHB, and the transcription factor Nrf2. Staining of 4‐μm thick formalin‐fixed, paraffin‐embedded whole tissue sections was done with a polymer‐based detection system and a High pH K8024 Dako EnVision FLEX Mini Kit (Dako), using mouse monoclonal antibodies for SDHA (Abcam, 2E3GC12FB2AE2; 1:1000 dilution), SDHB (Abcam, 21A11AE7; 1:400 dilution), and Nrf2 (Abcam, ab‐62352; 1:1000 dilution).20 Immunostaining was interpreted as negative when tumor cells showed no cytoplasmic staining and as positive when cytoplasmic staining was present, as reported previously.20 The tumors were divided into three groups: a low expression group in which most tumor cells were negative or weakly positive for anti‐SDHA, anti‐SDHB, and anti‐Nrf2 antibodies (<30% of all tumor cells were positive), a moderate expression group in which most tumor cells showed moderate positivity for these antibodies (30%‐80% of all tumor cells were positive), and a high expression group in which many tumor cells showed strong positivity for these antibodies (>80% of all tumor cells positive).
We also performed immunostaining with anti‐SDHA, anti‐SDHB, and anti‐Nrf2 antibodies in a paraganglioma with SDHB gene mutation, but no SDHA mutation.
Furthermore, we performed immunostaining of pRCC2 with FH gene mutation derived from HLRCC family 1 (exon 2, c.247_267del) and HLRCC family 2 (exon 5, c.583T>C, p.Met195Thr) using a mouse monoclonal antibody for FH (Santa Cruz Biotechnology, J‐13; dilution 1:1000).
2.7. Western blotting
Samples of tumor tissue and normal tissue from the same 30 patients in immunohistochemistry were carefully dissected free of stromal tissue. We performed Western blotting using the same anti‐SDHA antibody, anti‐SDHB antibody, and anti‐Nrf2 antibody as immunohistochemistry, and an anti‐VHL antibody (Cell Signaling TECHNOLOGY; #68547). After protein bands were visualized by chemiluminescence, each membrane was scanned for densitometry with a precision document imaging scanner (Agfa Japan) and the data were analyzed with NIH Image software (ImageJ for Mac OS, version 1.50). Expression of SDHA, SDHB, Nrf2, and VHL was calculated relative to that of beta‐actin in the tumor tissue specimens and corresponding non‐neoplastic specimens. For quantification of protein levels, the relative amount of SDHA, SDHB, Nrf2, and VHL in tumor tissue specimens was expressed as a ratio of the optical density for the tumor specimen to that for the corresponding non‐neoplastic specimen (set at 1.0) by densitometric analysis.
2.8. DNA constructs
A full‐length human SDHA cDNA fragment was amplified from HeLa cDNA by PCR and introduced into the EcoRI and XhoI sites of the pcDNA3‐myc vector. Three new SNVs of the SDHA (p.Tyr55His, p.Gly184Arg, and p.Val657Ile) observed in the present study and a previously reported SNV (p.Arg589Trp) were investigated.22 pcDNA3‐myc‐SDHA‐Tyr55His‐SDHA‐Gly184Arg, ‐SDHA‐Val657Ile, and ‐SDHA‐Arg589Trp were constructed from pcDNA3‐myc‐SDHA with the PrimeSTAR Mutagenesis Basal Kit (Takara Bio), according to the manufacturer's protocol. The PCR products and the structures of all plasmids were confirmed by DNA sequencing.
2.9. Cell culture and transfection
HeLa cells were grown in DMEM high glucose (Wako) with 10% fetal bovine serum, 2 mM glutamine, 100 U/ml penicillin, and 0.1 mg/ml streptomycin in a 5% CO2 incubator at 37℃. Transfection of plasmid DNA into cells was performed with Lipofectamine 2000 (Thermo Fisher Scientific), according to the manufacturer's protocol, and the cells were used for experiments 24 hours post‐transfection.
2.10. Cycloheximide chase assay
HeLa cells transfected with the various plasmids were treated with cycloheximide (Sigma‐Aldrich, C1988) (20 µg/ml) for various time periods. Cells were then lysed in radioimmunoprecipitation lysis buffer containing protease inhibitor cocktail. Cell lysates were subjected to SDS‐PAGE followed by Western blotting with anti‐myc (MBL, M171‐3, 1:4000 dilution) and anti‐GAPDH antibodies (MBL, 562, 1:1000 dilution). Immunoreactive bands were detected with the ECL Prime Western Blotting Detection Reagent (GE Healthcare) or Clarity Western ECL substrate (Bio‐Rad) and captured with an Amersham Imager 600 (Amersham). Band intensities were measured with NIH Image software (ImageJ for Mac OS, version 1.50).
2.11. Statistical analysis
Associations among SDHA gene mutation and SDHA, SDHB or Nrf2 protein expression on immunohistochemistry were analyzed by Pearson's chi‐square test for contingency tables using commercially available software. Western blotting data were analyzed by the Mann‐Whitney U test for comparisons between SDHA gene mutation and SDHA, SDHB or Nrf2 protein expression, and Spearman's rank correlation coefficient analysis was employed to determine the relations between SDHA, SDHB, and Nrf2. P < .05 was considered to indicate statistical significance.
3. RESULTS
3.1. Next‐generation sequencing
Targeted next‐generation sequencing of coding exons revealed numerous SNVs, de novo mutations, and somatic/blood mutations in the resected primary tumor tissue samples. The blood/tumor sequence traces for the SDHA variants to allow assessment for loss of heterozygosity are shown in Table S2. SNVs of the SDHA gene causing amino acid sequence variants (missense mutations) were detected in 11 (six pRCC2 and five ccRCC) out of 129 patients, while there were no mutations of the SDHB, SDHC, SDHD, or FH genes. We found three SNVs of the SDHA gene (exon 3, c.163T>C, p.Tyr55His, variant effect: missense; exon 5, c.550G>A, p.Gly184Arg, variant effect: missense; and exon 15, c.1969G>A, p.Val657Ile, variant effect: missense) (Table 1). These three SNVs of SDHA gene have not been identified in previous studies of SDHA‐deficient RCC.20, 21, 22 Interestingly, the missense mutation of SNVs of the SDHA gene was identical between somatic and blood DNA in all 11 patients. The SNV was also consistent within two families. In brief, the blood and somatic SNVs of the SDHA gene were identical in a father (case 4‐1) and his son (case 4‐2), while family members from another pedigree had the same SNV of the SDHA gene from blood and the two RCC patients in this family (daughter and father: cases 5‐1 and 5‐2, respectively) showed identical somatic and blood SNV of the SDHA gene.
TABLE 1.
Patients and tumour characteristics
| Case | Age/gender | Tumor size (cm) | Fuhrman grade | TNM stagea | Histologyb | Outcomec | SDHA | VHL | Others | RCC Family histoly | Uterine leiomyomatosis | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Tumor/blood | Immunohistochemistry | Tumor | Tumor | |||||||||
| 1 | 48/F | 9 | 3 | pT3aN1M1 (PUL, OSS) | Eosinophilic variant of ccRCC | DOD, 7 mo | c.163T>C p.Tyr55His | Lower | None | RET: c.3275A>G, p.Asn1092Ser | None | None |
| 2 | 63/M | 11 | 4 | pT3bN0M1 (PUL) | pRCC2 | DOD, 13 mo | c.1969G>A p.Val657Ile | Moderate | None | PBRM1: c.3215_3216insT, p.Ala1073fs | None | |
| 3 | 67/M | 11 | 3 | pT3aN0M1 (PUL) | ccRCC | AWD, 95 mo | c.163T>C p.Tyr55His | Moderate | c.449delA, p.Asn150fs | None | ||
| 4‐1 | 66/M | 6 | 3 | pT1bN0M0 | ccRCC | NED, 97 mo | c.1969G>A p.Val657Ile | Lower | c.292T>A, p.Tyr98Asn | RCC in son (case 4‐2) and paternal cousin | ||
| 4‐2 | 46/M | 9 | 3 | pT3aN0M1 (PUL) | ccRCC | AWD, 38 mo | c.1969G>A p.Val657Ile | Lower | c.563T>C, p.Leu188Pro | RCC in father (case 4‐1) | ||
| 5‐1 | 34/F | 8 | 2 | pT3aN0M0 | pRCC2 | NED, 90 mo | c.550G>A p.Gly184Arg | Moderate | None | RET: c.2166G>A, p.Val706Met | RCC in father (case 5‐2) | None |
| 5‐2 | 66/M | 13 | 3 | pT3aN2M1 (PUL, OSS) | pRCC2 | AWD, 19 mo | c.550G>A p.Gly184Arg | Moderate | None | Akt1: c.726delG, p.Glu242fs RET: c.2166G>A, p.Val706Met | RCC in daughter (case 5‐1) | |
| 6 | 32/M | 3 | 2 | pT1aN0M0 | pRCC2 | NED, 27 mo | c.1969G>A p.Val657Ile | Lower | None | None | ||
| 7 | 69/M | 9 | 3 | pT2aN0M0 | pRCC2 | NED, 27 mo | c.1969G>A p.Val657Ile | Moderate | None | PBRM1: c.4337_4338insG, p.Gly1447fs | RCC in father | |
| 8 | 49/F | 12 | 3 | pT3aN0M1 (PUL, OSS) | Eosinophilic variant of ccRCC | AWD, 7 mo | c.550G>A p.Gly184Arg | Moderate | None | PBRM1: c.2567G>C, p.Arg856Pro; c.4337_4338insG, pGly1447fs RET: c.1465G>A, p.Asp489Asn | None | None |
| 9 | 71/F | 2 | 3 | pT1aN0M0 | pRCC2 | NED, 7 mo | c.163T>C p.Tyr55His | Lower | None | None | Hysterectomy | |
Metastatic (M) lesions: PUL, lung; OSS, bone.
Histology: ccRCC, clear cell renal cell carcinoma; pRCC2, papillary type 2 renal cell carcinoma.
Outcome: DOD, dead of disease; AWD, alive with disease; NED, no evidence of disease.
3.2. Direct Sanger sequencing
The SNVs of the SDHA and VHL genes acquired by targeted next‐generation sequencing were identified to those by direct Sanger sequencing in 11 samples (Figure S1). The mutation of the SDHB gene was not confirmed by direct Sanger sequencing.
3.3. Clinicopathological characteristics
The clinicopathological characteristics of the patients with tumors showing SNVs of the SDHA gene are summarized in Table 1.
Renal cell carcinoma with SNVs of the SDHA gene displayed diverse histological features, including tumor cells with pale eosinophilic cytoplasm, vascular stroma, and various patterns of proliferation from formation of tubular structures to solid alveolar nests. Both clear cells and strongly eosinophilic cells were found at different sites (Figure 1).
FIGURE 1.
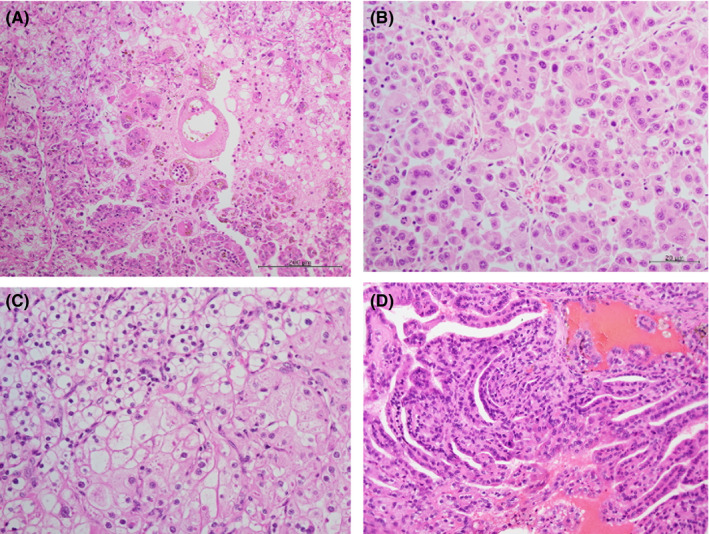
Diverse histological patterns. Tumor cells have diverse histological patterns; small alveolar structure of cells with eosinophilic granular cytoplasm in case 1 (A), acinar architecture composed of eosinophilic granular cells with large nuclei exhibiting prominent nucleoli in case 4‐2 (B), sheet arrangement with mixed clear and eosinophilic cytoplasm in case 8 (C), and a single layer of cuboidal cells cover a fibrovascular stalks and form papillary structures in case 9 (D)
These tumors were infiltrative and were composed of polygonal cells that contained large pleomorphic nuclei with prominent nucleoli. RCCs with SNVs of the SDHA gene featured tumor cells with large, heavily stained (eosinophilic) nucleoli surrounded by a noticeable unstained space or halo within a normally stained nucleus (Figure 2). These features were similar to that of HLRCC‐associated RCCs with FH gene mutations (Figure S2A,C).
FIGURE 2.
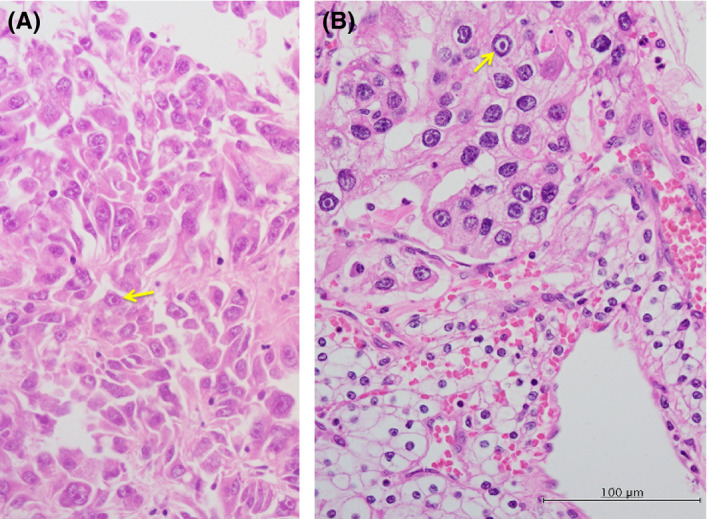
Prominent pale eosinophilic cytoplasmic inclusions within the tumor cells. Some neoplastic cells contained cytoplasmic eosinophilic inclusions. The tumor cells are pleomorphic appearance with cytoplasmic eosinophilic inclusions (yellow arrows) in case 2 (A) or enlarged, heavily stained (eosinophilic) nucleoli surrounded by a noticeable unstained space or halo within normally stained nuclei (yellow arrows) in case 8 (B)
3.4. Immunohistochemistry
In non‐tumor tissues, epithelial cells of renal tubular showed a strong reaction for anti‐SDHA and anti‐SDHB antibodies, while the glomerulus showed a weak reaction (Figure S3). On the other hand, in a paraganglioma with SDHB, but not SDHA, gene mutation, which was examined as a control study, homogeneous deletion of SDHB protein was noted, while SDHA protein expression was normal (Figure 3A).
FIGURE 3.
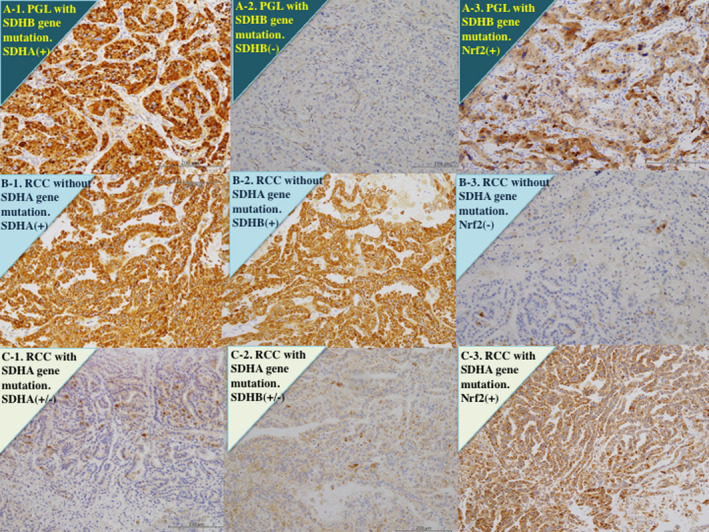
Immunohistochemistry for succinate dehydrogenase (SDH) A, SDHB, and nuclear factor E2‐related factor 2 (Nrf2). A, Paraganglioma (PGL) with only SDHB gene mutation, but not SDHA. Tumor cells show diffusely strong staining for an anti‐SDHA antibody (A‐1) and an anti‐Nrf2 antibody (A‐3), but very weak reaction for an anti‐SDHB antibody (A‐2). B, Clear cell renal cell carcinoma (RCC) without SDHA gene mutation. Tumor cells show diffusely strong staining for an anti‐SDHA antibody (B‐1) and an anti‐SDHB antibody (B‐2), but very weak staining for an anti‐Nrf2 antibody (B‐3). C, Clear cell RCC with SDHA gene mutation (case 1). Tumor cells show heterogeneous reaction with weak to moderate staining for an anti‐SDHA antibody (C‐1) and an anti‐SDHB antibody (C‐2), while those show strong staining for an anti‐Nrf2 antibody (C‐3)
Renal cell carcinomas without SDHA gene mutation showed strong immunostaining by anti‐SDHA and anti‐SDHB antibodies and weak immunostaining by anti‐Nrf2 antibody (Figure 3B, Figure S3). In contrast, tumors with SNVs of the SDHA gene of missense mutations showed a significant decrease in the expression of SDHA protein (P < .0001) and SDHB protein (P < .0001), and a significant increase in the expression for Nrf2 protein (P = .0067, Figures 3C and Figure S3, and Table 2). There was a significant positive correlation between SDHA and SDHB protein expression (P < .0001, Table 2). Even when tumors had the same SNVs of missense mutation, some showed weak positivity for SDHA and SDHB, while others were moderately positive (Table 1). On the other hand, we found an inverse relationship of immunostaining between Nrf2 and SDHA (P = .0023, Table 2) and SDHB (P = .0025, Table 2).
TABLE 2.
Relationship between succinate dehydrogenase (SDH) A gene mutation and proteins (n = 30)
| Immunohistochemistry | |||||
|---|---|---|---|---|---|
| SDHA protein | |||||
| Strong (diffused positive) (n = 17) | Moderate (n = 7) | Lower (n = 6) | P value | ||
| SDHA gene | Mutation (−) (n = 19) | 17 | 1 | 1 | <.0001 |
| Mutation (+) (n = 11) | 0 | 6 | 5 | ||
| SDHB protein | |||||
| Strong (diffused positive) (n = 17) | Moderate (n = 5) | Lower (n = 8) | P value | ||
| SDHA gene | Mutation (−) (n = 19) | 17 | 1 | 1 | <.0001 |
| Mutation (+) (n = 11) | 0 | 4 | 7 | ||
| SDHB protein | |||||
| Strong (diffused positive) (n = 17) | Moderate (n = 5) | Lower (n = 8) | P value | ||
| SDHA protein | Strong (diffused positive) (n = 17) | 17 | 0 | 0 | <.0001 |
| Moderate (n = 7) | 0 | 5 | 2 | ||
| Lower (n = 6) | 0 | 0 | 6 | ||
| Nrf2 protein | |||||
| High (n = 17) | Low (n = 13) | P value | |||
| SDHA gene | Mutation (−) (n = 19) | 7 | 12 | .0067 | |
| Mutation (+) (n = 11) | 10 | 1 | |||
| SDHA protein | |||||
| Strong (diffused positive) (n = 17) | Moderate (n = 7) | Lower (n = 6) | P value | ||
| Nrf2 protein | High (n = 17) | 5 | 6 | 6 | .0023 |
| Low (n = 13) | 12 | 1 | 0 | ||
| SDHB protein | |||||
| Strong (diffused positive) (n = 17) | Moderate (n = 5) | Lower (n = 8) | P value | ||
| Nrf2 protein | High (n = 17) | 5 | 5 | 7 | .0024 |
| Low (n = 13) | 12 | 0 | 1 |
In HLRCC family 1 with FH gene deletion (c.247_267del), tumor cells displayed FH deficiency (Figure S2B), while the tumor cells of HLRCC family 2 with SNVs of FH gene missense mutation (c.583T>C) demonstrated a weak reaction to anti‐FH antibody (Figure S2D). The HLRCC‐associated RCCs without SDHA gene mutation showed strong staining not only for anti‐SDHA and anti‐SDHB antibodies but also for anti‐Nrf2 antibody (Figure S4).
3.5. Western blotting
In ccRCC without SDHA and VHL gene mutations, SDHA and SDHB were similarly expressed in cancer and non‐tumor tissues (Figure 4A). In ccRCC with SNVs of the SDHA gene, expressions of SDHA and SDHB were lower in cancer tissues than in non‐tumor tissues, regardless of a VHL gene mutation (Figure 4A). In pRCC2 without SDHA gene mutation, SDHA and SDHB were similarly expressed in cancer and non‐tumor tissues (Figure 4A). In pRCC2 with SNVs of the SDHA gene, SDHA and SDHB were decreased in cancer tissues than non‐tumor tissues (Figure 4A). The 11 tumors with SNVs of the SDHA gene showed greater decreased expressions for SDHA and SDHB than the 19 tumors without (Figure 4B,C, both P < .0001). In contrast, the tumors with SNVs of the SDHA gene showed higher expression for Nrf2 than those without the mutation (Figure 4D, P < .0001). There was a significant positive correlation between SDHA and SDHB protein expression (Figure 4E, P < .0001) but an inverse relationship between Nrf2 and SDHA (Figure 4F, P = .0009) and SDHB (Figure 4G, P = .0013) protein expression.
FIGURE 4.
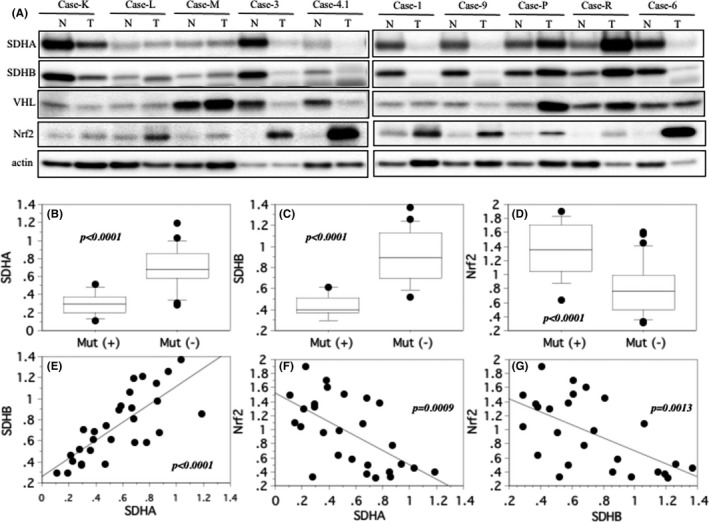
Western blotting for succinate dehydrogenase (SDH) A, SDHB, and nuclear factor E2‐related factor 2 (Nrf2). A, Case‐K, ‐L, ‐M: clear cell renal cell carcinoma (ccRCC) without mutations of SDHA, SDHB, and von Hippel‐Lindau (VHL) genes. SDHA and SDHB are expressed to the same extent in non‐tumor (N) and tumor (T) tissues, or more prevalent in non‐tumor (N) tissues. Nrf2 are expressed to the same extent in non‐tumor (N) and tumor (T) tissues, or more prevalent in tumor (T) tissues. Case‐3, −4.1: ccRCC with SDHA and VHL gene mutation. SDHA, SDHB, and VHL are expressed lower in tumor (T) tissues, while Nrf2 are higher expressed in tumor (T) tissues. Case‐1, −9: ccRCC with SDHA gene mutation. SDHA and SDHB are expressed lower in tumor (T) tissues, and Nrf2 are abundant in tumor (T) tissues. Case‐P, ‐R: papillary renal cell carcinoma type 2 (pRCC2) without mutations of SDHA, SDHB and VHL genes. SDHA and SDHB are expressed to the same extent in non‐tumor (N) and tumor (T) tissues, or less in non‐tumor (N) tissues, and Nrf2 are increased in tumor (T) tissues. Case‐6: pRCC2 with SDHA gene mutation. The tumor (T) tissues show less reaction for anti‐SDHA and ‐SDHB antibodies, but strong reaction for anti‐Nrf2 antibody. B, C, D, The central line indicates the median value, the box shows the interquartile range, the bars display the full range, and the points are outliers. Ratio of the optical density of the tumor specimen to that of the corresponding non‐neoplastic specimen (set at 1.0) in western blotting for SDHA, SDHB, and Nrf2. E, F, G, Spearman's rank correlation coefficient analysis. There was a positive correlation between expressions of SDHA and SDHB, and there was an inverse correlation between Nrf2 and SDHA or SDHB in the tumor tissues
3.6. Cycloheximide chase assay
Some SDHB mutants identified in pheochromocytoma and paraganglioma may undergo faster degradation than wild‐type SDHB.26 The same may be true for the SDHA mutants identified here, in which case the expression level of the SDHA mutants in tumors would be decreased. To test this possibility, we compared the protein stability of wild‐type SDHA and SDHA mutants by transfecting pcDNA‐myc‐SDHA or ‐SDHA mutant (p.Gly184Arg, p.Val657Ile, and p.Tyr55His) into HeLa cells and then treating the cells with cycloheximide chase assay to inhibit protein synthesis 24 hours post‐transfection. The expression levels of SDHA were measured at various times by Western blot analysis. The expression levels of p.Gly184Arg and p.Val657Ile SDHA but not of p.Tyr55His SDHA decreased faster than the expression level of wild‐type SDHA (Figure S5). A similar result was obtained by using another SDHA mutant, p.Arg589Trp SDHA (Figure S5), the expression level of which was reported to be decreased in tumors.22
4. DISCUSSION
As represented by the malignant phenotype of FH‐deficient HLRCC‐associated RCC, germline mutations of TCA cycle enzyme genes have been suggested to have a role in several aspects of carcinogenesis and tumor progression,27 but there has been limited investigation of the genetic and genomic profile of SDH‐deficient RCC, particularly SDHA‐associated RCC. The following findings were obtained in the present study. First, 11 out of 129 RCCs had SNVs of the SDHA gene of missense mutations causing amino acid changes with a decrease in SDHA and SDHB protein expression, without mutations of SDHB/C/D or FH. Second, the tumor cells with SNVs of the SDHA gene generally had eosinophilic cytoplasm with a clear space (halo) around the nucleus. The mechanism underlying the association of SDH gene mutations with RCC has not been elucidated. However, FH is the enzyme that follows SDH in the mitochondrial TCA cycle, so loss or reduction of SDH function and consequent TCA cycle impairment may result in a similar metabolic shift to aerobic glycolysis in FH‐deficient HLRCC‐associated RCC and SDH‐deficient RCC, suggesting that these cancers could share common features and be similarly aggressive.12, 15, 27
The kidney is one of the most common sites for oncocytic tumors characterized by abundant mitochondria.1 Most of the SDHA‐deficient RCCs had eosinophilic cytoplasm and seemed to be oncocytic tumors.20, 21, 22 In the present study, some of the tumors with SDHA mutation demonstrated histopathological features associated with HLRCC, ie, tubulo‐papillary architecture and cells with a large nucleus containing an eosinophilic nucleolus surrounded by a clear halo,28, 29 although the number of such cells was small and there were variations among the tumors. RCC associated with HLRCC is an aggressive form of inherited renal cancer with type 2 papillary or collecting duct histology that usually has an unfavorable prognosis.5, 6, 7 Three patients with SDHA‐deficient RCCs with nuclear grade 3/4 who underwent surgical resection developed systemic metastases within a few years, and the metastatic foci had a poorer response for systemic therapy, indicating that SDHA‐deficient RCCs had biological aggressive features.20, 21, 22 On the other hand, in the present cases with 11 RCCs, six pRCC2, and five ccRCC, with SNVs of the SDHA gene, the clinicopathological features, such as histological grade, stage, a reduction in SDHA expression and outcome, were diverse and showed a variety of features for each case, while the prognosis of these tumors might not be worse than that of HLRCC‐associated RCC.5, 6, 7 It is unclear whether these differences are due to a complete deletion of SDHA, a reduction of SDHA, or SNVs of the SDHA gene. Thus, clinicopathological features of RCCs with SNVs of SDHA gene cannot be unequivocally concluded at this time. Although the number of patients was too small for drawing definite conclusions, the similarities and differences between these types of RCC are considered here.
There have only been three previous reports about SDHA‐deficient RCC, and immunohistochemistry suggested complete deletion of SDHA in all cases.20, 21, 22 The SDHA gene is located at p15.33 and comprises 15 exons at the coordinates chr5:218356 to 256815. The previously reported SDHA gene deletions were located at chr5:218437 through chr5:235454, at splice site 622‐2_622‐2delA, and at c.91C>T (p.Arg31*) with a somatic missense variant c.1765C > T (p.Arg589Trp).20, 21, 22
In the present study, we found three new SNVs of SDHA in human RCC by next‐generation sequencing and direct Sanger sequencing, but we did not detect any somatic or blood mutations of SDHB, SDHC, or SDHD. We performed next‐generation sequencing that targeted the coding exons and detected numerous SNVs, de novo mutations, and germline/somatic mutations in the coding regions. A nonsynonymous substitution is a nucleotide change that alters the amino acid sequence of a protein. Missense mutations are nonsynonymous substitutions that arise from point mutations, which are mutations of an SNV that result in substitution of a different amino acid in the protein encoded by the gene. We confirmed such SNVs by direct Sanger sequencing. We also found that the blood (germline) mutations of SDHA detected were identical to those seen in the sporadic somatic in the tumors in all 11 patients. However, it is not clear if the mutations were monoallelic or biallelic in the tumor, and if those in the blood were germline mutations. If the tumors have biallelic inactivation (germline on one allele and sporadic on the other), negative immunohistochemistry staining for SDHA would be expected. SDHA appears to act as a tumor suppressor gene in relation to paraganglioma and pheochromocytoma.30 SDHA‐deficient RCC is characterized by inactivation of the SDHA gene, which is usually due to germline mutation with the addition of a somatic second hit (double hit with different mutations) and leads to dysfunction of the SDH complex and SDHA is deleted.20, 21, 22 Therefore, SDHA gene deletion may be linked with carcinogenesis in SDHA‐deficient RCC. In contrast, none of our 11 cases had somatic SNVs that were different from germline SNVs, and none showed complete loss of SDHA protein. Therefore, the presented cases were not categorized as SDHA‐deficient RCC.
We observed a reduction in SDHA expression only with immunohistochemistry and Western blotting, arguing against a double hit. When mutations are mono‐allelic, it is difficult to discriminate between germline and sporadic mutation. Because the previously reported SDHA‐deficient RCC had two variants of germline SNV of p.Arg31* in conjunction with somatic p.Arg589Trp,22 in the cycloheximide chase assay we transfected p.Arg589Trp as a control. In this assay, degradation of SDHA protein was enhanced for p.Gly184Arg, p.Val657Ile, and p.Arg589Trp compared with wild‐type, but not for p.Tyr55His. Furthermore, p.Gly184Arg and p.Val657Ile reduced SDHA proteins by almost the same amount as p.Arg589Trp did. These findings suggest that p.Gly184Arg and p.Val657Ile might be involved in reducing protein content, whereas p.Tyr55His might be involved in a different process. However, the detailed mechanism of protein reduction by p.Gly184Arg and p.Val657Ile, for example destabilization by conformational change, ubiquitination, binding molecule change or localization change, has yet to be elucidated. Moreover, the possibility that the amount of protein decreased because of a reduction in mRNA could not be ruled out. Thus, it is currently unclear which gene regulates SDHA enzyme activity, but it is likely that SDHA mutations associated with amino acid abnormalities as in our patients do not necessarily cause complete SDHA enzyme deficiency. On the other hand, HLRCC is an aggressive RCC characterized by biallelic inactivation of FH, and FH acts as a tumor suppressor and its activity is very low to absent in tumors from patients with HLRCC. However, various FH gene alterations causing loss of FH activity have been reported, including missense, frameshift, and nonsense mutations, as well as whole‐gene deletion, and not all tumors had complete loss of FH expression in HLRCC cases.5, 6, 7 In fact, as shown in Figure S2 in this study, tumor cells with FH gene deletion showed no FH expression in HLRCC family 1, while tumor cells with FH gene missense mutation displayed weak to moderate FH positivity in HLRCC family 2. We found that missense mutations of SNVs of SDHA gene did not necessarily lead to complete suppression of SDHA protein expression, and even tumors with the same missense mutation showed variable (weak to moderate) immunohistochemical staining for SDHA. Even without complete deletion, reduction of SDHA activity due to gene mutation might play a role in tumorigenesis of RCC, and identifying loss‐ or gain‐of‐function mutations that affect other key enzymes may help to shed light on metabolic changes related to SDHA‐associated tumorigenesis. The present study showed that SDHA gene mutation was identical between somatic and blood DNA in all 11 patients. Somatic and blood mutations of SDHA were identical in cases 4‐1 and 4‐2. Similarly, cases 5‐1 and 5‐2 displayed identical somatic and blood SNVs. The high mutation rate in HLRCC families has led to screening of at‐risk individuals for early detection of RCC, allowing initiation of therapy while the tumor is still small.5, 6, 7 SDHA gene mutation may be autosomal dominant like the mutations causing HLRCC, suggesting that family members with blood SDHA mutation should be kept under active surveillance for early detection of RCC.
In addition to reduction of SDHA protein, loss of SDHB protein has been observed in SDHA‐deficient RCC by immunohistochemistry.20, 21, 22 In the present study, no SDHB gene mutation associated with an amino acid abnormality was recognized, but the tumors with SNVs of SDHA gene also showed a weaker reaction for SDHB protein by immunohistochemistry and western blotting. It has been reported that the entire mitochondrial complex 2 becomes unstable if biallelic inactivation of any of its components leads to degradation of the SDHB subunit, which means that biallelic inactivation of SDHA results in loss of both SDHA and SDHB.3, 4, 30 In contrast, tumors with SDHB, SDHC, or SDHD mutations have been reported to exhibit loss of SDHB staining but retain SDHA staining, although it is not clear why SDHA protein expression remains stable in the presence of SDHB, SDHC, or SDHD mutations. In the current study, SDHB gene mutation associated with an amino acid abnormality was recognized in a paraganglioma, but not SDHA mutation, and SDHB protein was completely deleted while SDHA protein expression was normal. Taken together, these findings suggest that SDHA gene deletion may lead to combined deficiency of SDHA and SDHB proteins and that missense mutations of SDHA may suppress SDHA and SDHB protein expression, indicating the existence of a pathway by which the SDHA gene mediates or modifies expression of both SDHA and SDHB protein in cells with SDHA mutation. Furthermore, even if SDHA and SDHB protein are not complete deleted, some of the reduction of these two proteins may be involved in the development of RCCs with SDHA gene mutation. We did not examine the expression for SDHB proteins by cycloheximide chase assay using transfection of SDHA variants such as p.Arg589Trp, so this topic needs to be investigated to confirm this hypothesis in the future.
One possibility is that not only the gene mutation itself but also an unknown interaction between SDHA and other signaling pathways leads to loss or substantial reduction of SDHA protein function. In the present study, we found a difference in the ratio of SNVs of the SDHA gene in five ccRCC (0.9%) and in six pRCC2 (3.5%). So far, no SDH‐deficient RCCs have been shown to harbor mutations in the VHL gene.20, 21, 22 Furthermore, FH‐deficient RCCs have been well documented to be associated with pRCC2 or collecting duct carcinoma,5, 6, 7 no FH‐deficient RCCs of the clear cell subtype (ccRCC) have been reported. FH catalyzes sequential steps in the TCA cycle (oxidation of succinate to fumarate by SDH, followed by hydration of fumarate to malate by FH), and it is tempting to speculate that their roles in the TCA cycle are relevant to their tumor suppressor activity.1, 2 Although the association of SDH mutation with RCC has not yet been elucidated, two mechanisms have been suggested to explain how mutations of mitochondrial tumor suppressor genes could contribute to tumorigenesis, which are oxidative stress resulting from increased mitochondrial production of ROS, and metabolic signaling via TCA cycle metabolites as intracellular messengers.1, 2 Some SDH mutations lead to generation of ROS.1, 2 There is growing evidence that Nrf2 is the major regulator of antioxidant and detoxification pathways for ROS, and has a pivotal role in tumor proliferation, invasion, and chemoresistance, with elevated tumor expression of Nrf2 protein being linked to a poor prognosis.31, 32 It was reported that activation of Nrf2‐dependent antioxidant pathways is a key step in the development of FH‐deficient pRCC2. In HLRCC, FH deficiency leads to succination of Kelch‐like ECH‐associated protein 1 (Keap1), stabilization of Nrf2, and induction of stress‐response genes to promote survival of FH‐deficient cells, indicating that FH‐deficient HLRCC‐associated RCC has an antioxidant phenotype.12, 33, 34, 35 Several studies have shown that immunohistochemical staining for 2SC and FH can aid in the detection of FH gene aberrations in RCC.29 2SC is a chemical modification of proteins formed by a Michael addition reaction between the TCA cycle intermediate, fumarate, and thiol groups in proteins, a process known as protein succination, and a succinated protein causes irreversible inactivation of the protein.36 In the present study, the tumor cells with SDHA gene mutation and HLRCC‐associated pRCC2 with FH gene mutation showed higher expression of Nrf2 than the tumors without SDHA or FH gene mutations, indicating that a sustained activation of Nrf2 has a role in RCC with SDH or FH gene mutations. Thus, future studies should examine the expression of 2SC in RCC with SDHA gene mutation to assess the roles of the Keap1/Nrf2 pathway. Furthermore, changes of metabolic signaling with induction of pseudo hypoxia were recently proposed as an alternative mechanism of carcinogenesis, and pseudo hypoxia may be related to the development and progression of HLRCC.12, 27 Succinate production is increased in SDH‐deficient tumors and succinate is known to inhibit prolyl hydroxylase, which degrades HIF under normoxic conditions, leading to induction of HIF.1, 2 Generation of ROS by SDH mutations also leads to inhibition of prolyl hydroxylase, indicating that increased ROS production can mediate pseudo hypoxia in tumors with SDH mutation as well as in HLRCC, in which FH‐associated changes might develop in a HIF‐dependent manner.12, 27 Accordingly, we should also study the HIF pathway in RCCs with SDHA mutation in the future. In addition, a cardinal feature of ccRCC is a very high frequency of VHL inactivation caused by gene deletion, mutation, and/or silencing via promoter methylation, leading to HIF accumulation, and the VHL gene is thought to be driver gene as a tumor suppressor.37 Among five ccRCCs with SDHA gene mutation, three tumors had VHL gene mutation in the current study. On the other hand, to date no SDHA‐deficient RCCs have been shown to harbor VHL mutations.20, 21, 22 This contrasts with our finding where three tumors had VHL mutations. In the present study, gene mutations in SDHA, SDHB, and VHL were investigated using both next‐generation sequencing and direct Sanger sequencing, and both sequencing methods identified the mutations. Western blotting showed that ccRCC without SDHA gene mutation had almost similar expression of SDHA in cancer tissues and non‐tumor tissue. In contrast, the tumors with SDHA gene mutation had lower SDHA expression in cancer tissues than in non‐tumor tissues, regardless of the presence or not of a VHL mutation. This was also observed in a pRCC2 without VHL mutation. Biallelic gene mutation for SDHA is directly responsible for SDHA‐deficient RCC. However, even if complete deletion of SDHA by a double hit is not confirmed, it appears likely that an SDHA gene mutation might be linked to a subset of SDHA‐mutated RCCs, regardless of a VHL mutation. Furthermore, a common feature of SDH and VHL mutations is their capacity to mediate a pseudo‐hypoxic response, and inactivation of SDH also leads to HIF stabilization, through the inhibition of their hydroxylation by prolyl‐4‐hydroxylases, necessary for their recognition by VHL protein.38 Therefore, it is necessary to study the signal crosstalk between the VHL‐HIF pathway and SDHA.
The present study had several limitations, including its retrospective design, a relatively small number of subjects, and a follow‐up period that was too short to allow definite conclusions regarding the possible influence of SDHA mutation on the prognosis of RCC. SDH‐deficient RCC is very rare and among the various types of SDH‐deficient RCC, SDHA‐deficient RCC is the rarest. In the present study, we found three SNVs of the SDHA gene, and the frequency of SDHA gene mutation (11 of 129 RCC, 8.5%) was uncommonly high and may indicate sequencing artifacts; however, we identified the SNVs of SDHA gene mutation in the tumors by next‐generation sequencing, for which an artifact rate of 0.5%‐1.7% was reported,39, 40 and all SNVs of the SDHA gene identified by next‐generation sequencing were also confirmed by direct Sanger sequencing. Furthermore, the unsolved problem was the criteria for assigning pathogenicity to the SDHA missense substitutions. In the present study, the variants were assessed according to ACMG criteria.24, 25 Ion AmpliSeq panels covered broad research areas for germline analysis, including genes recommended by the ACMG, and the accuracy in detecting SDHA gene mutations was confirmed according to a previously published method.24, 25 As shown in Table S3, at present the ClinVar and OMIM databases suggest a link between SDHA gene mutations and benign diseases, while the COSMIC database shows a link between SDHA gene mutations and leukemia, malignant lymphoma, adenocarcinoma, and hemangioblastoma. Thus, so far, the association of SDHA gene mutation with malignancy has not been fully elucidated. We showed that HLRCC‐associated and SDHA mutation‐associated RCCs might have morphological similarities, suggesting the functions and mutations of both FH and SDH should be evaluated in more detail to support development of new treatment options for these mitochondria‐associated hereditary RCCs. Our findings need to be confirmed by further investigations, preferably large‐scale prospective controlled trials.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
ETHICAL APPROVAL AND CONSENT TO PARTICIPATE
This study was conducted in accordance with the Declaration of Helsinki and was approved by the Institutional Ethics Review Board of Dokkyo Medical University Hospital (approval no. 24017.R‐6‐5.). Before surgery, written consent regarding the use of surgical samples and blood as well as data publication was requested, and protection of patient privacy in future studies was guaranteed through a signed consent form approved by our institutional Committee on Human Rights in Research. Surviving patients provided written consent for analysis using the surgical samples and peripheral blood through a signed form approved by our institutional Committee on Human Rights in Research. In cases of patient death, consent for publication was sought orally from next of kin of the participant. All clinical samples were anonymized before analysis to guarantee protection of patient privacy. Consequently, each patient in this cohort signed the consent form approved by our institutional Committee on Human Rights in Research (approval no. 24017.R‐6‐5.).
CONSENT FOR PUBLICATION
Each patient signed an informed consent and subsequent publication form that was approved by our institutional Committee on Human Rights in Research. All clinical samples were anonymized before analysis to guarantee protection of patient privacy.
Supporting information
Figure S1
Figure S2
Figure S3
Figure S4
Figure S5
Table S1
Table S2
Table S3
ACKNOWLEDGMENTS
The authors are especially grateful to Chiaki Matsuyama, Kin‐ichi Matsuyama, Kie Kageyama, Yoshiko Omata, Mayumi Kirihara, and Junka Hamano for their excellent assistance in this study.
Kamai T, Higashi S, Murakami S, et al. Single nucleotide variants of succinate dehydrogenase A gene in renal cell carcinoma. Cancer Sci. 2021;112:3375–3387. 10.1111/cas.14977
Funding information
This work was supported in part by Japanese Science Progress Society KAKENHI Grants (17K11156, 20K09546) to T. Kamai.
DATA AVAILABILITY STATEMENT
The data that support the findings of the current study are available from the corresponding author upon reasonable request.
REFERENCES
- 1.Eng C, Kiuru M, Fernandez MJ, Aaltonen LA. A role for mitochondrial enzymes in inherited neoplasia and beyond. Nat Rev Cancer. 2003;3:193‐202. [DOI] [PubMed] [Google Scholar]
- 2.Gottlieb E, Tomlinson IP. Mitochondrial tumor suppressors: a genetic and biochemical update. Nat Rev Cancer. 2005;5:857‐866. [DOI] [PubMed] [Google Scholar]
- 3.Barletta JA, Hornick JL. Succinate dehydrogenase‐deficient tumors: diagnostic advances and clinical implications. Adv Anat Pathol. 2012;19:193‐203. [DOI] [PubMed] [Google Scholar]
- 4.Gill AJ. Succinate dehydrogenase (SDH) and mitochondrial driven neoplasia. Pathology. 2012;44:285‐292. [DOI] [PubMed] [Google Scholar]
- 5.Launonen V, Vierimaa O, Kiuru M, et al. Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc Natl Acad Sci USA. 2001;98:3387‐3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tomlinson IP, Alam NA, Rowan AJ, et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet. 2002;30:406‐410. [DOI] [PubMed] [Google Scholar]
- 7.Alam NA, Rowan AJ, Wortham NC, et al. Genetic and functional analyses of FH mutations in multiple cutaneous and uterine leiomyomatosis, hereditary leiomyomatosis and renal cancer, and fumarate hydratase deficiency. Hum Mol Genet. 2003;12:1241‐1252. [DOI] [PubMed] [Google Scholar]
- 8.Briere JJ, Favier J, Gimenez‐Roqueplom AP, Rustin P. Tricarboxylic acid cycle dysfunction as a cause of human diseases and tumor formation. Am J Physiol Cell Physiol. 2006;291:C1114‐C1120. [DOI] [PubMed] [Google Scholar]
- 9.Levine AJ, Puzio‐Kuter AM. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science. 2010;330:1340‐1344. [DOI] [PubMed] [Google Scholar]
- 10.Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11:85‐95. [DOI] [PubMed] [Google Scholar]
- 11.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;334:1029‐1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Linehan WM, Rouault TA. Molecular pathways: fumarate hydratase‐deficient kidney cancer‐targeting the Warburg effect in cancer. Clin Cancer Res. 2013;19:3345‐3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang Y, Valera V, Sourbier C, et al. A novel fumarate hydratase‐deficient HLRCC kidney cancer cell line, UOK268: a model of the Warburg effect in cancer. Cancer Genet. 2012;205:377‐390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Srigley JR, Delahunt B, Eble JN, et al. ISUP Renal Tumor Panel. The International Society of Urological Pathology (ISUP) Vancouver classification of renal neoplasia. Am J Surg Pathol. 2013;37:1469‐1489. [DOI] [PubMed] [Google Scholar]
- 15.Ricketts CJ, Shuch B, Vocke CD, et al. Succinate dehydrogenase kidney cancer: an aggressive example of the Warburg effect in cancer. J Urol. 2012;188:2063‐2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ricketts C, Woodward ER, Killick P, et al. Germline SDHB mutations and familial renal cell carcinoma. J Natl Cancer Inst. 2008;100:1260‐1262. [DOI] [PubMed] [Google Scholar]
- 17.Gill AJ, Hes O, Papathomas T, et al. Succinate dehydrogenase (SDH)‐deficient renal carcinoma: a morphologically distinct entity. A clinicopathologic series of 36 tumors from 27 patients. Am J Surg Pathol. 2014;38:1588‐1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Williamson SR, Eble JN, Amin MB, et al. Succinate dehydrogenase‐deficient renal cell carcinoma: detailed characterization of 11 tumors defining a unique subtype of renal cell carcinoma. Mod Pathol. 2015;28:80‐94. [DOI] [PubMed] [Google Scholar]
- 19.Saxena N, Maio N, Crooks DR, et al. SDHB‐deficient cancers: the role of mutations that impair iron sulfur cluster delivery. J Natl Cancer Inst. 2016;108(1):djv287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yakirevich E, Ali SM, Mega A, et al. A novel SDHA‐deficient renal cell carcinoma revealed by comprehensive genomic profiling. Am J Surg Pathol. 2015;39:858‐863. [DOI] [PubMed] [Google Scholar]
- 21.Ozluk Y, Taheri D, Matoso A, et al. Renal carcinoma associated with a novel succinate dehydrogenase A mutation: a case report and review of literature of a rare subtype of renal carcinoma. Hum Pathol. 2015;46:1951‐1955. [DOI] [PubMed] [Google Scholar]
- 22.McEvoy SR, Koe L, Choong DY, et al. SDH‐deficient renal cell carcinoma associated with biallelic mutation in succinate dehydrogenase A: comprehensive genetic profiling and its relation to therapy response. NPJ Precis Oncol. 2018;2:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kamai T, Abe H, Arai K, et al. Radical nephrectomy and regional lymph node dissection for locally advanced type 2 papillary renal cell carcinoma in an at‐risk individual from a family with hereditary leiomyomatosis and renal cell cancer. a case report. BMC Cancer. 2016;16(1):232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Casey RT, Ten Hoopen R, Ochoa E, et al. SDHC epi‐mutation testing in gastrointestinal stromal tumours and related tumours in clinical practice. Sci Rep. 2019;9:10244. https://www.thermofisher.com/tw/en/home/life‐science/sequencing/next‐generation‐sequencing/ion‐torrent‐next‐generation‐sequencing‐workflow/ion‐torrent‐next‐generation‐sequencing‐select‐targets/ampliseq‐target‐selection/ion‐ampliseq‐on‐demand‐panels‐targeted‐sequencing.html [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elliott AM, Radecki J, Moghis B, Li X, Kammesheidt A. Rapid detection of the ACMG/ACOG‐recommended 23 CFTR disease‐causing mutations using ion torrent semiconductor sequencing. J Biomol Tech. 2012;23:24‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang C, Matro JC, Huntoon KM, et al. Missense mutations in the human SDHB gene increase protein degradation without altering intrinsic enzymatic function. FASEB J. 2012;26:4506‐4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Srinivaan R, Ricketts CJ, Sourbier C, Linehan WM. New strategies in renal cell carcinoma: targeting the genetic and metabolic basis of disease. Clin Cancer Res. 2015;21:10‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Merino MJ, Torres‐Cabala C, Pinto P, Linehan WM. The morphologic spectrum of kidney tumors in hereditary leiomyomatosis and renal cell carcinoma (HLRCC) syndrome. Am J Sur Pathol. 2007;31:1578‐1585. [DOI] [PubMed] [Google Scholar]
- 29.Chen YB, Brannon AR, Toubaji A, et al. Hereditary leiomyomatosis and renal cell carcinoma syndrome‐associated renal cancer: recognition of the syndrome by pathologic features and the utility of detecting aberrant succination by immunohistochemistry. Am J Surg Pathol. 2014;38:627‐637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burnichon N, Briere JJ, Libe R, et al. SDHA is a tumor suppressor gene causing paraganglioma. Hum Mol Genet. 2010;19:3011‐3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sporn MB, Liby KT. NRF2 and cancer: the good, the bad and the importance of context. Nat Rev Cancer. 2012;12:564‐571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DeNicola GM, Karreth FA, Humpton TJ, et al. Oncogene‐induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475:106‐109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ooi A, Wong JC, Petillo D, et al. An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell. 2011;20:511‐523. [DOI] [PubMed] [Google Scholar]
- 34.Adam J, Hatipoglu E, O'Flaherty L, et al. Renal cyst formation in Fh1‐deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell. 2011;20:524‐537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang O, Maxwell PH, Pollard PJ. Renal cell carcinoma: translational aspects of metabolism and therapeutic consequences. Kidney Int. 2013;2013:667‐681. [DOI] [PubMed] [Google Scholar]
- 36.Alderson NL, Wang Y, Blatnik M, et al. S‐(2‐Succinyl)cysteine: A novel chemical modification of tissue proteins by a Krebs cycle intermediate. Arch Biochem Biophys. 2006;450:1‐8. [DOI] [PubMed] [Google Scholar]
- 37.Cancer Genome Atlas Research Network . Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liao L, Testa JR, Yang H. The roles of chromatin‐remodelers and epigenetic modifiers in kidney cancer. Cancer Genet. 2015;208:206‐214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Quail MA, Smith M, Coupland P, et al. A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genom. 2012;13:341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Song L, Huang W, Kang J, Huang Y, RenH DK. Comparison of error correction algorithms for Ion Torrent PGM data: application to hepatitis B virus. Sci Rep. 2017;7(1):8106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Figure S3
Figure S4
Figure S5
Table S1
Table S2
Table S3
Data Availability Statement
The data that support the findings of the current study are available from the corresponding author upon reasonable request.