

Abstract
Glioma is one of the most commonly diagnosed intracranial malignancies. The molecular mechanism underlying the development of glioma is still largely unknown. In this study, we present the first report concerning the function and mechanism of cyclin‐dependent kinase‐like 3 (CDKL3) in the development and prognosis of glioma. It is shown that CDKL3 was upregulated in glioma tissues and could independently predict poor prognosis of patients. Silencing CDKL3 in glioma cells could inhibit cell proliferation and migration and induce cell apoptosis and cell cycle arrest, whereas the overexpression of CDKL3 promoted cell proliferation. The in vivo experiments also indicated that knockdown of CDKL3 significantly suppressed tumor growth of glioma. Gene expression profiling of CDKL3 knockdown U87 cells identified RRM2 as a potential target of CDKL3, which was proved to have direct interaction with CDKL3. Given similar effects on glioma development with CDKL3, knockdown of RRM2 could rescue the effects of CDKL3 overexpression on glioma cells. Moreover, knockdown of CDKL3 or RRM2 suppressed the activity of JNK signaling, whereas CDKL3 overexpression produced the opposite effect. In conclusion, our results identified CDKL3 as a promotor for glioma, probably through the regulation of RRM2 and activation of the JNK signalling pathway, highlighting the significance of CDKL3 as a promising therapeutic target of glioma.
Keywords: CDKL3, glioma, high‐throughput sequencing, JNK signaling pathway, RRM2
Our results identified cyclin‐dependent kinase‐like 3 (CDKL3) as a tumor promotor for glioma, probably through the regulation of RRM2 and activation of the JNK signaling pathway. Our data reveal the role of CDKL3 in glioma, highlighting the significance of CDKL3 as a putative therapeutic target.
Abbreviations
- CDK
cyclin‐dependent kinase
- CDKL3
cyclin‐dependent kinase‐like 3
- co‐IP
coimmunoprecipitation
- DEG
differentially expressed gene
- EGFR
epidermal growth factor receptor
- EMT
epithelial‐mesenchymal transition
- GBM
glioblastoma
- OD
optical density
- qPCR
quantitative PCR
- qRT‐PCR
quantitative real‐time PCR
- RNA‐seq
RNA sequencing
- RR
ribonucleotide reductase
- RRM2
ribonucleotide reductase regulatory subunit M2
- shCtrl
negative control
- TCGA
The Cancer Genome Atlas
- WB
western blot
1. INTRODUCTION
Glioma is one of the most commonly diagnosed intracranial malignant tumors.1 According to global clinical statistics, the incidence rate of primary intracranial tumors is approximately 10‐21/100 000, of which glioma accounts for 40%‐50%,2, 3 and is still increasing year by year.4 Malignant gliomas (stage III or IV according to the WHO) often originate from normal brain glial cells or low‐grade astrocytomas, which commonly grow under the cerebral cortex of the supratentorial hemisphere infiltratively and invasively and often invade several lobes of the brain and deep brain tissue.5 Malignant gliomas, especially GBM, have the characteristics of high malignancy, strong proliferative activity, resistance to radiotherapy and chemotherapy, and high risk of recurrence, which result in the extremely poor prognosis and low 5‐year survival rate.6 The lack of efficient treatment for glioma could be attributed to the unclear mechanism of glioma.7 A thorough understanding of the mechanisms involved in the pathogenesis and progression of glioma can provide the theoretical basis for the development of targeted therapy and is of great significance for glioma patients.
The CDK family plays crucial roles in the regulation of the cell cycle, whose dysfunction is considered as the biological basis of aberrant cell proliferation of cancer cells.8 Cyclin‐dependent kinase‐like 3 is also a member in the CDK family.9 Recently, studies have shown that the abnormal expression of cyclin in many tumors showed the characteristics of oncogenes and is closely related to the occurrence, development, diagnosis, treatment, and prognosis of tumors.10 CDKL3 is located on human chromosome 5q31.1 and encodes a protein with a molecular weight of 52 kDa and 455 amino acid sequences. It is also known as NKIAMRE because of containing the NKIAMRE sequence, which is recognized as a universal cyclin binding domain.11 At present, the research on CDKL3 is very limited, and its specific role in tumor growth and development is still largely unclear. Until now, most studies about CDKL3 were focused on diseases in the central nervous system. For example, Dubos et al12 reported that CDKL3 could be a strong candidate for nonsyndromal autosomal dominant mental retardation. Moreover, Liu and Tao reported that CDKL3 was highly expressed in the mammalian brain during late embryonic and early postnatal stages, and the downregulation of CDKL3 decreases dendritic outgrowth and branch formation while promoting axon growth in cultured primary cortical neurons.13 It was also reported that the expression of CDKL3 was increased in anaplastic large cell lymphoma cells, suggesting that CDKL3 might be related to the occurrence and development of tumors.14 However, to the best of our knowledge, the role of CDKL3 in glioma has never been reported and remains unknown.
In this study, we comprehensively utilized RNA‐seq, data mining of public databases, and means of molecular biology to assess the relationship between CDKL3 and development as well as prognosis of glioma, and the potential underlying mechanisms.
2. MATERIALS AND METHODS
2.1. Patient recruitment
The study protocol and acquisition of tissue specimens were approved by the Ethical Review Committee of Naval Medical University (Shanghai, China). For the immunohistochemistry staining experiment, glioma specimens were consecutively recruited from patients who underwent surgical treatment at Changzheng Hospital (Shanghai, China) from January 2000 to December 2014. Normal brain tissues were taken from severe trauma patients, on whom partial resection of the brain was required to reduce increased intracranial pressure. Written informed consent was supplied by all the participants.
2.2. Cell lines and cell culture
Human glioma U87, U251, U373, and SHG44 and human glial HBE cell lines were purchased from BeNa Technology. Cells were cultured in DMEM (Corning) with 10% FBS (Invitrogen). The incubator was maintained at 37°C with 95% CO2 and humidity 70%‐80%. Cell medium was replaced every 72 hours.
2.3. High‐throughput sequencing
Total RNA from tissues or cells (3 v 3) was extracted using TRIzol (Thermo Fisher Scientific). Then RNA quantity and quality were assessed with a Thermo Nanodrop 2000 (Thermo Fisher Scientific) and an Agilent 2100 Bioanalyzer (Agilent). Affymetrix PrimeView Human Gene Expression Arrays (Thermo Fisher Scientific) were used for microarray analysis to obtain gene expression profiles according to the manufacturer’s instructions. Significantly differentially expressed genes were selected based on this condition: P < .05 and absolute fold change greater than 2. The KEGG pathway enrichment analysis was carried out for all significantly differentially expressed genes.
2.4. Target shRNA sequence design and cell infection
For overexpressing CDKL3, the CDKL3 construct was generated by subcloning PCR amplified full‐length human CDKL3 cDNA into vector. For silencing of CDKL3 (or other genes), the shRNA sequence was packaged into the linearized vector BR‐V‐115 (Shanghai Biosciences) according to the Fermentas T4 DNA Ligase instructions. The plasmids were confirmed and sequenced by PCR and the EndoFree Maxi Plasmid Kit (Qiagen) was used to extract positive plasmid containing the target RNA interference. The targeting sequences and shRNA sequences used in construction are listed in Table S1. The U87 and U251 cell lines were cultured in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin at 37°C with 5% CO2. U87 and U251 cells were then cultured in 6‐well plates and transfected with shCDKL3 or shRRM2 or shCtrl or CDKL3 construct. The transfected cells were cultured for another 5 days. The target gene knockdown or overexpression efficiency was evaluated in both cell lines by qRT‐PCR and WB analysis.
2.5. Immunohistochemistry array
The construction of the microarray and the following immunohistochemistry were carried out as described previously by Shanghai Biochip Company.39 Glioma and normal tissue sections from glioma patients were deparaffinized, repaired with citrate antigen, blocked, and treated with 5% goat serum for 15 minutes at room temperature. The tissue sections were stained by diluted primary anti‐CDKL3 (1:200; Bioss) or anti‐RRM2 (1:400; Abcam) at 4°C overnight. Tissue sections were then stained with DAB, counterstained with hematoxylin, and dehydrated with ethanol. Images were captured using an ImageScope v11 (Leica). All sections were scored by two independent pathologists in a blinded fashion. In particular, the intensity of positive‐staining cytoplasm or nucleus of CDKL3 was scored on a scale of 0‐3 (0, negative; 1, light brown; 2, medium brown; and 3, dark brown), which was multiplied by the corresponding value of positive percentage (1, less than 25%; 2, 25%‐50%; 3, 50%‐75%; and 4, more than 75%). The two scores of cellular cytoplasm and nucleus were added to estimate the expression level of CDKL3, which was divided into the high or low cut by the median total score 4.
2.6. Quantitative real‐time PCR
Total RNA was extracted using TRIzol reagent according to the manufacturer’s protocol. The purity and integrity of the RNA was assessed by Nanodrop 2000/2000C spectrophotometry. We used HiScript qRT SuperMix for qPCR (+gDNA wiper; Vazyme) for generating cDNA according to the manufacturer’s protocol. The qRT‐PCR was carried out using SYBR Premix Ex Taq (Takara) and the Mx3000P qPCR System (Agilent). GADPH was used as an endogenous reference. The primers used in qRT‐PCR were as follows: CDKL3‐F, 5′‐ TCAAAGGAGGAAGAGGAGA‐3′; CDKL3‐R, 5′‐ AGTTAGATTGATGGGTGGC‐3′; RRM2‐F, 5′‐ AAGAAACGAGGACTGATGC‐3′; RRM2‐R, 5′‐ CTGTCTGCCACAAACTCAA‐3′; GAPDH‐F, 5′‐TGACTTCAACAGCGACACCCA‐3′; and GAPDH‐R: 5′‐CACCCTGTTGCTGTAGCCAAA‐3′.
2.7. Western blot analysis and co‐IP assay
Cells were lysed with ice‐cold RIPA Buffer, then total proteins were extracted. Proteins were added to the gel for electrophoresis, and then transferred to a PVDF membrane. After blocking with 5% nonfat milk at room temperature for 1 hour, primary Abs were bound overnight at 4°C. The next day, the PVDF membrane was incubated with corresponding secondary Abs at room temperature for 2 hours. Each membrane was visualized with the ECL‐plus WB system (General Electric), and were detected by an X‐ray imaging analyzer (Eastern Kodak). The Abs used in WB analysis are listed in Table S2.
Before co‐IP experiments, U87 cells were transfected with lentivirus expressing Flag‐tagged CDKL3. Cells were lysed with RIPA buffer containing protease inhibitors. Immunoprecipitation using anti‐Flag or anti‐RRM2 Abs and the resulting immunocomplexes were then subjected to SDS‐PAGE and analyzed by WB analysis using the anti‐Flag and anti‐RRM2 Abs.
2.8. Cell proliferation
Cell viability were assessed using an MTT assay and Celigo cell counting assay.
For MTT, after the trypsinization of the cells in each experimental group in the logarithmic growth phase, cells (2000 cells/well) were seeded into a 96‐well plate overnight. Four hours before culture termination, 20 μL 5 mg/mL MTT (Genview) was added to each well. Absorbance values at 490 nm were determined for each well using an enzyme‐connected immunodetector (Tecan) and reference wavelength was 570 nm. The absorbance is associated with the percentage of vital cells. The cell viability ratio was calculated by the following formula: cell viability (%) = OD (treated) / OD (control) × 100%.
For the Celigo cell counting assay, cells were plated in 96‐well plates at a density of 2 × 103 cells per well at 72 hours after transfection. The GFP‐positive cells were counted using a Celigo Adherent Cell Cytometer (Nexcelom).
2.9. Colony formation assay
U87 and U251 cells in the logarithmic growth phase were trypsinized and diluted to single‐cell suspensions. Following this, 400‐1000 cells/mL were seeded into 6‐well plates and cultured for 15 days until the number of most colonies was greater than 50 cells. During the cell culture, the cell status and change were observed every 3 days. The colonies were fixed with methanol for 30 minutes at room temperature and stained with Giemsa for 20 minutes. The number of colonies (more than 50 cells/colony) were counted and photographs were taken with a digital camera.
2.10. Apoptosis assay
U87 and U251 cells (at least 5 × 105) were suspended and seeded in 6 cm dishes and cultured until cells covered approximately 70% of the plate. The cells were digested with trypsin and resuspended, then stained with the addition of 10 μL annexin V‐APC for 15 minutes in the dark. The percentages of cell phases were measured using FACScan (Millipore) to assess the apoptotic rate, and results were analyzed.
2.11. Cell cycle
Cells were harvested in the exponential growth phase, and single‐cell suspensions containing 1 × 106 cells were fixed with prechilled 70% alcohol. Cell cycle was monitored using propidium iodide staining and measured with a Guava easyCyte HT flow cytometer (Millipore). The results were analyzed with ModFit 3.0 software (Verity Software House).
2.12. Wound healing assay
Cells were spread at the bottom of 96‐well plates. A line wound was made by scraping 100 μL tips across the confluent cell layer. After sucking cell culture and washing the cells three times to remove detached cells and debris, serum‐free medium was added into the 96‐well plates and incubated under the usual culture conditions for 48 hours. Wound closure photographs were captured using a light microscope (DFC500; Leica) at indicated time points and the outcomes were analyzed.
2.13. Transwell assay
Cell migration assays was also undertaken using Transwell chambers (Corning) in a 24‐well plate. For the migration assay, 5 × 104 cells in serum‐free medium were placed in the upper chamber of the insert; DMEM supplemented with 10% FBS (800 μL) was used as the chemoattractant in the lower chamber. After 24 hours, the medium was removed, and the cells remaining on the upper surface of the membrane were removed. Cells were stained with Giemsa (Sigma Diagnostics) and then captured using microscopic inspection (Olympus, MicroPublisher 3.3 RTV) according to the manufacturer’s instructions.
2.14. Tumorigenesis in vivo
Animal studies were approved by the Ethical Committee of Naval Medical University and were carried out in accordance with guidelines for animal care and protection and protocols. BALB/c nude mice (6 weeks old) were purchased from Shanghai Jiesijie Experimental Animals Co., Ltd. The U87 cells (5 × 106 per mouse) transfected by shCDKL3 or the negative control were subcutaneously or intracranially injected into BALB/c nude mice. Tumor growth was monitored and measured in volume (calculated by the equation π/6 × L × W2, where L represents the longest dimension and W is the dimension perpendicular to length) at the indicated days (7, 10, 13, 16, 19, 22, 25, 28 days) postinoculation, by a caliper.
2.15. Statistical analysis
The data are expressed as mean ± SD (n ≥ 3) and analyzed using GraphPad Prism 6 software (GraphPad Software). The significance of the differences between groups with continuous data was evaluated by the Student’s t test. Wilcoxon’s rank‐sum test was used to compare the difference of counting data between two groups. Spearman’s analysis was used to examine the relationship between CDKL3 expression and glioma grade. Overall survival curves were plotted by the Kaplan‐Meier method and compared by log‐rank test. The identification of relevant prognostic factors was carried out by univariate analysis, multivariate analysis, and stepwise backward Cox regression model. Variables with a value of P < .2 on univariate analysis were added into the multivariate analysis. Overall survival was calculated in months from the initial date of diagnosis to the time of death, regardless of cause. P values less than .05 were considered statistically significant.
3. RESULTS
3.1. Identification of CDKL3 as potential promoter in glioma
In order to explore the molecular mechanism of glioma, global gene expression profiling of clinical glioma tissues and corresponding paracarcinoma tissues was obtained by RNA‐seq. In general, by using the threshold of fold change greater than or equal to 2 or less than or equal to 0.5, and P value <.05, 3469 DEGs were identified, including 1652 upregulated genes and 1817 downregulated genes (Figure 1A). Subsequently, 10 of the significantly upregulated genes were chosen for further high‐content screening by delivering corresponding shRNAs into U87 cells followed by detecting inhibitory rates of cell proliferation. As shown in Figure S1, shCDKL3 induced the most remarkable inhibition of cell proliferation, which was, therefore, focused.
FIGURE 1.
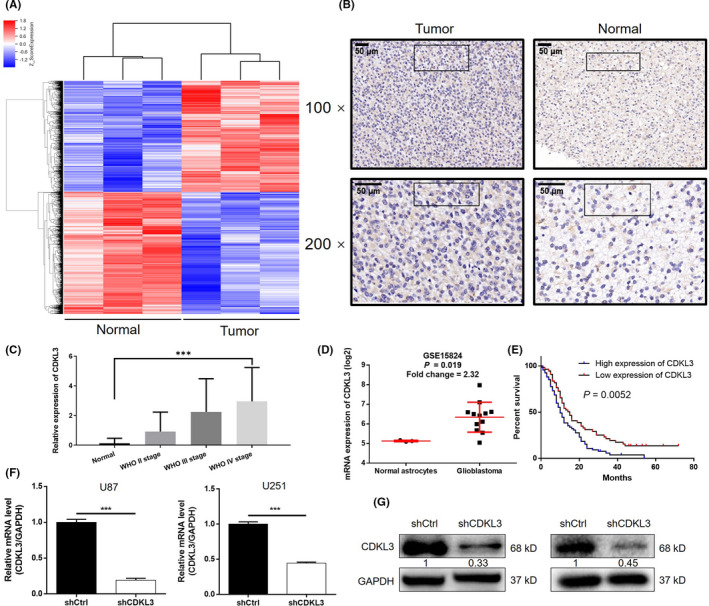
Cyclin‐dependent kinase‐like 3 (CDKL3) is upregulated in glioma and correlates with prognosis. A, RNA sequencing heatmap of glioma and normal tissues (3 v 3) that identified 3469 differentially expressed genes, including 1652 upregulated genes and 1817 downregulated genes. B, Expression of CDKL3 in tumor and normal tissues was detected by immunohistochemistry analysis. C, Immunohistochemistry staining score of CDKL3 expression in 128 glioblastoma cases is significantly higher than that in 16 normal brain tissues (P < .001). CDKL3 expression is positively related to pathological grade of glioma (P < .001). D, Data collected from the GSE15824 dataset shows the upregulated expression of CDKL3 in glioma cells compared with normal astrocytes. E, CDKL3 expression correlates with survival time of glioblastoma patients. Kaplan‐Meier survival analysis in 128 GBM cases stratified by CDKL3 expression indicates that high level of CDKL3 expression was associated with shorter overall survival (10 vs 15 months, P = .0052). F, G, Knockdown efficiency of CDKL3 in U87 and U251 cells was evaluated by (F) quantitative PCR and (G) western blot analysis. Data are presented as mean ± SD. ***P < .001. shCtrl, normal control
Subsequently, a total of 259 glioma tissues (including 79 grade II, 36 grade III, and 128 grade IV glioma cases) and 16 normal brain tissues were recruited for further detection. As determined by immunohistochemistry analysis, CDKL3 expression was found to be significantly elevated in glioblastoma (2.9609 ± 2.274 vs 0.125 ± 0.3415, P < .001) and positively correlated with pathological grade (P < .001; Figure 1B,C, Table 1). Notably, the upregulation of CDKL3 in glioma was also confirmed in the GSE15824 dataset (P = .019; Figure 1D). The Cox proportional hazards regression model revealed high expression of CDKL3 (hazard ratio = 1.731, P = .009) as an independent risk factor for glioblastoma patients (Table 2). Furthermore, Kaplan‐Meier survival analysis indicated that high CDKL3 expression was associated with poor prognosis of glioblastoma patients (10 vs 15 months, P = .0052; Figure 1E).
TABLE 1.
Clinicopathological characteristics of 128 glioblastoma patients
| Characteristic | Patients (N = 128)a | CDKL3 expression | |
|---|---|---|---|
| Low | High | ||
| Age (y) | |||
| <60 | 97 (75.8) | 50 | 47 |
| ≥60 | 31 (24.2) | 10 | 21 |
| Gender | |||
| Male | 87 (68.0) | 42 | 45 |
| Female | 41 (32.0) | 18 | 23 |
| Resection degree | |||
| Total | 90 (70.3) | 40 | 50 |
| Partial | 38 (29.7) | 20 | 18 |
| Chemotherapy | |||
| Yes | 93 (72.7) | 43 | 50 |
| No | 35 (27.3) | 17 | 18 |
| Radiotherapy | |||
| Yes | 93 (72.7) | 45 | 48 |
| No | 33 (25.8) | 13 | 20 |
| Unknown | 2 (1.5) | 1 | 1 |
| IDH1 | |||
| Mutant | 3 (2.4) | 1 | 2 |
| Wild‐type | 124 (97.6) | 59 | 65 |
| Survival status | |||
| Live | 12 (9.4) | 9 | 3 |
| Dead | 110 (85.9) | 46 | 64 |
| Unknown | 6 (4.7) | 5 | 1 |
Abbreviation: CDKL3, cyclin‐dependent kinase‐like 3.
Data are shown as n (%).
TABLE 2.
Multivariate Cox regression analysis of hazard ratio (HR) in patients with glioma
| Characteristic | HR value | 95% CI | P valuea |
|---|---|---|---|
| CDKL3 | 1.731 | 1.15‐2.606 | .009 |
| Resection | 1.541 | 1.009‐2.354 | .045 |
| Chemotherapy | 0.681 | 0.439‐1.056 | .086 |
| Age | 1.726 | 1.097‐2.717 | .018 |
| Radiotherapy | 0.642 | 0.418‐0.986 | .043 |
Abbreviations: CDKL3, cyclin‐dependent kinase‐like 3; CI, confidence interval.
Log‐rank test.
3.2. Knockdown of CDKL3 inhibits glioma development in vitro
Given the upregulation of CDKL3 in glioma tissues, its endogenous expression was detected in HEB human glial cells and glioma cell lines including U87, U251, U373 and SHG44. Consistently, higher expression of CDKL3 in glioma cells than in normal cells was observed (P < .05; Figure S2A). Subsequently, lentivirus vectors were used to deliver shCDKL3 (for silencing CDKL3) or shCtrl (as negative control) to U87 and U251 cells. The transfection efficiency was detected through fluorescence imaging (>80%; Figure S2B). The knockdown efficiencies of CDKL3 in U87 and U251 cells were estimated through qPCR and WB analysis (Figure 1F,G), respectively. The MTT assay showed significantly inhibited cell growth in the shCDKL3 group compared with the shCtrl group (P < .001; Figure 2A). Similar results were also obtained from the detection of colony formation, which clearly showed fewer colonies in the shCDKL3 group (P < .01; Figure 2B). Moreover, cells with CDKL3 depletion showed a significantly increased apoptosis rate (3‐fold for U87, 2‐fold for U251 cells, P < .001; Figure 2C). The application of a human apoptosis Abs array revealed that knockdown of CDKL3 could induce cell apoptosis through regulating Bax, Bad, heat shock protein 27, and caspase 8 (Figures 2D and Figure S3). Moreover, CDKL3 knockdown significantly induced arrest of G2 phase of the cell cycle (Figure 2E). Subsequently, wound healing and Transwell assays were both carried out to evaluate the impact of CDKL3 on cell migration. As shown in Figure 3A,B, knockdown of CDKL3 could significantly inhibit cell motility (P < .01). The inhibition of cell migration was also rationalized by the downregulation of EMT‐related biomarkers including N‐cadherin, vimentin, and Snail in the shCDKL3 group (Figure 3C). Moreover, the inhibition of glioma by CDKL3 knockdown was further confirmed in SHG44 and U373 cells and similar results were obtained (Figures S4‐S6). The inhibited stemness and promoted inflammatory response of glioma cells after transfection with shCDKL3 were also displayed by the downregulation of stemness biomarkers (CD133, MELK, and Sox2) and upregulation of proinflammatory cytokines such as Toll‐like receptor 4, nuclear factor‐κB, and c‐Myc (Figures 3D and S7). All these results indicated that CDKL3 may play an essential role in the development and progression of glioma.
FIGURE 2.
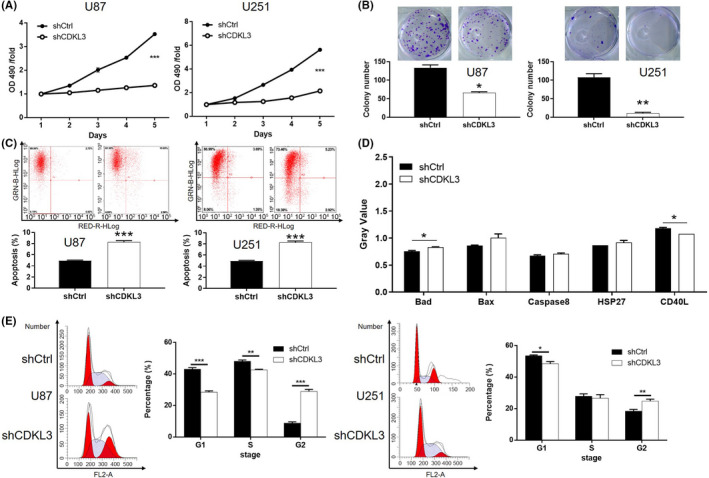
Knockdown of cyclin‐dependent kinase‐like 3 (CDKL3) inhibits cell proliferation and induces apoptosis of glioma cells. A, Effect of CDKL3 knockdown on cell proliferation of U87 and U251 cells was evaluated by MTT assay. B, Effect of CDKL3 knockdown on colony formation of U87 and U251 cells was detected by Giemsa staining. C, Cell apoptosis of U87 and U251 cells transfected with shCDKL3 or normal control (shCtrl) was examined by flow cytometry. D, Human apoptosis Ab array analysis was undertaken in U87 cells with or without CDKL3 knockdown. E, Cell cycle distribution of U87 and U251 cells transfected with shCDKL3 or shCtrl were examined by flow cytometry. Representative images were selected from at least three independent experiments. Data are presented as mean ± SD. *P < .05, **P < .01, ***P < .001
FIGURE 3.
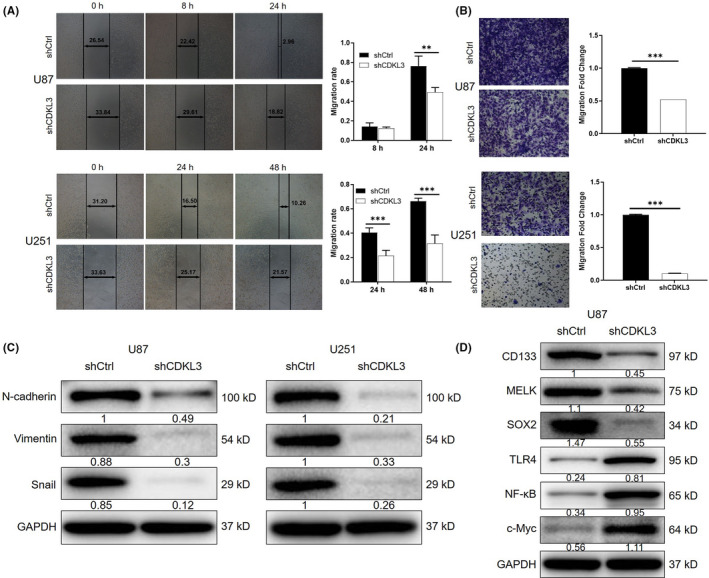
Knockdown of cyclin‐dependent kinase‐like 3 (CDKL3) inhibits cell migration. A, B, Inhibition effect of CDKL3 on glioma cell migration was confirmed by (A) wound healing assay and (B) Transwell assay. C, Western blot (WB) analysis was used to detect the expression of epithelial‐mesenchymal transition‐related proteins including E‐cadherin, vimentin, and Snail in U87 and U251 cells. D, WB analysis was utilized to detect the expression of stemness‐ and inflammation‐related proteins including CD133, MELK, SOX2, Toll‐like receptor 4 (TLR4), nuclear factor‐κB (NF‐κB), and c‐Myc in U87 cells. Representative images were selected from at least three independent experiments. Data are presented as mean ± SD. **P < .01, ***P < .001. shCtrl, normal control
3.3. Overexpression of CDKL3 promotes cell proliferation and colony formation of glioma cells
To further explore the role of CDKL3 in the development of glioma, PCR amplified full‐length human CDKL3 cDNA was subcloned into vector for the construction of CDKL3 overexpressed cell models based on U87 and U251 cells. The overexpression efficiencies of CDKL3 in both cell lines was confirmed to be larger than 2‐fold by qPCR (P < .001), which was also verified by WB analysis (Figure S8A). We found that CDKL3 overexpression significantly promoted cell proliferation (Figure S8B). Moreover, cells with entopic expression of CDKL3 showed significantly enhanced colony formation ability (Figure S8C). Collectively, these results provided extra evidence of the promotion effects of CDKL3 on glioma.
3.4. Knockdown of CDKL3 inhibits glioma tumor growth in vivo
To further investigate the effects of CDKL3 on glioma growth in vivo, a mouse xenograft model was constructed through subcutaneous injection of U87 cells with or without CDKL3 knockdown. It was observed that the tumors formed by U87 cells with knockdown of CDKL3 showed slower growth rate compared with tumors in the shCtrl group (Figure 4A). The inhibition of tumor growth by CDKL3 knockdown was also verified by the smaller weight of tumors in the shCDKL3 group (Figure 4B,C). Furthermore, low expression of Ki‐67 and CDKL3 was observed in shCDKL3 tumors (Figure 4D). More importantly, the mouse model constructed through intracranial injection also revealed suppressed tumor growth on brain tissue (Figure 4E,F). All of these results indicated that knockdown of CDKL3 could inhibit the development of glioma in vivo.
FIGURE 4.
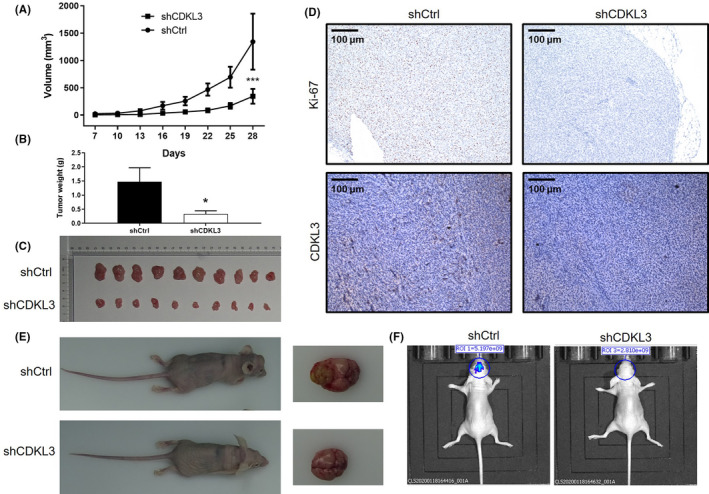
Knockdown of cyclin‐dependent kinase‐like 3 (CDKL3) inhibits tumor growth in vivo. A, Volume of tumors in the normal control (shCtrl) group and shCDKL3 group was measured at 7, 10, 13, 16, 19, 22, 25, 28 days postinjection. B, Average weight of tumors removed from shCtrl and shCDKL3 groups. C, Tumor images taken from mice in shCtrl and shCDKL3 groups. D, Immunohistochemistry was used to detect the expression of Ki‐67 and CDKL3 in tumor tissues removed from mice in shCtrl and shCDKL3 groups. E, Intracranial growth of tumor on brain tissue of mice in shCtrl and shCDKL3 groups. F, Before they were killed, mice were subjected to in vivo imaging to evaluate the growth of intracranial growth of glioma. Data are presented as mean ± SD. *P < .05, ***P < .001
3.5. Cyclin‐dependent kinase‐like 3 could promote development of glioma by regulating RRM2 and the JNK signaling pathway
Global gene expression profiling of U87 cells with or without CDKL3 knockdown was undertaken by RNA‐seq. Six hundred and thirty‐three DEGs, including 153 upregulated genes and 480 downregulated genes, were identified (Figure S9). Moreover, the analysis showed the enrichment of DEGs in the MAPK signaling pathway, which is a frequently reported pathway in glioma (Figure S10). After the verification through WB analysis, we speculated that CDKL3 might promote the development of glioma through upregulating RRM2, EGFR, and PPM1A and activating the JNK signaling pathway (Figure 5A,B). Consistently, the obvious activation of the JNK signaling pathway was manifested in CDKL3 overexpressed glioma cells (Figure 5B). Additionally, we found that JNK activator anisomycin could reactivate the activity of the JNK pathway that had been inhibited by CDKL3 knockdown (Figure 5C). The results of MTT assay also indicated that treating cells with anisomycin could impair the inhibition effects of CDKL3 knockdown on proliferation of U87 and U251 cells (P < .01, Figure 5D), elucidating the involvement of the JNK pathway in the glioma regulation mechanism of CDKL3.
FIGURE 5.
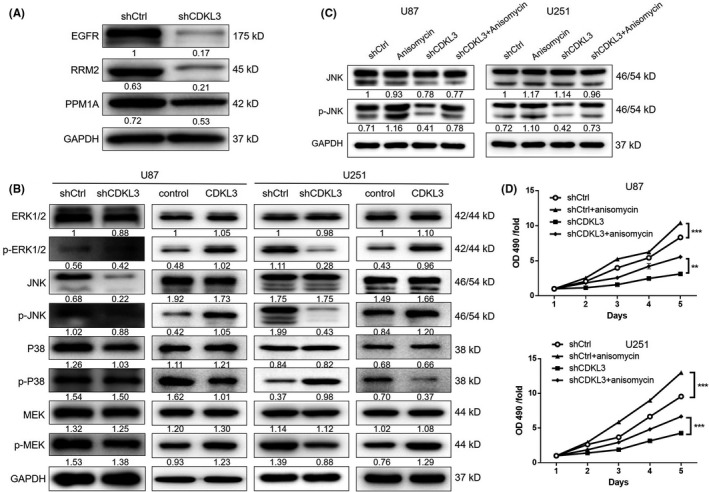
Exploration of the downstream pathway underlying the effect of cyclin‐dependent kinase‐like 3 (CDKL3) on glioma. A, Western blotting (WB) was used to verify the expression of epidermal growth factor receptor (EGFR), ribonucleotide reductase regulatory subunit M2 (RRM2), and PPM1A, which were selected from the differentially expressed genes in U87 cells. B, WB was used to detect the expression and phosphorylation of MAPK signaling pathway‐related proteins including ERK1/2, p‐ERK1/2, JNK, p‐JNK, P38, p‐P38, MEK, and p‐MEK in U87 and U251 cells. C, D, JNK pathway activator (anisomycin) was used to treat glioma cells transfected with normal control (shCtrl) or shCDKL3. Activation of the JNK signaling pathway by anisomycin was confirmed by WB (C) and the cells were subsequently subjected to detection of cell proliferation (D). Data are presented as mean ± SD. **P < .01, ***P < .001
3.6. Knockdown of RRM2 inhibits proliferation, apoptosis, cell cycle, and migration of glioma cells
Combining the association analysis of CDKL3‐related genes and the data mining of the TCGA‐GBM database, RRM2 was identified as a promising target of CDKL3 (Figure 6A,B). Importantly, the direct interaction between CDKL3 and RRM2 was proven by co‐IP assay (Figure 6C). Therefore, three shRNAs targeting RRM2 were designed and, as shown in Figure S11, shRRM2‐1 had the highest RRM2 knockdown efficiency of 97.1% and was used in all follow‐up experiments. The knockdown of RRM2 in U87 (97.1%) and U251 (67.7%) cells was further verified by both qPCR and WB analyses (Figure 6D, P < .01). As expected, the inhibition of glioma cell proliferation by shRRM2 was proven by MTT assay (P < .01, Figure 6E). The increased apoptotic cell ratio in U87 (2‐fold) and U251 (5.2‐fold) cells of the shRRM2 group was observed (P < .01; Figure 7A). Similar to CDKL3, cells with RRM2 knockdown displayed decreased cell percentage in G1 phase and increased cell percentage in G2 phase (P < .01; Figure 7B). Wound healing assay indicated the suppression of cell migration ability by RRM2 knockdown (P < .01; Figure 7C). Immunohistochemistry analysis of RRM2 expression also showed the upregulation of RRM2 in glioma tissues (Figure 7D and Table S3). Moreover, we found that knockdown of RRM2 could suppress the activity of the JNK pathway, which was similar with CDKL3 (Figure 7E). Therefore, these results indicated that RRM2 could be a potential downstream of CDKL3 in the regulation of glioma.
FIGURE 6.
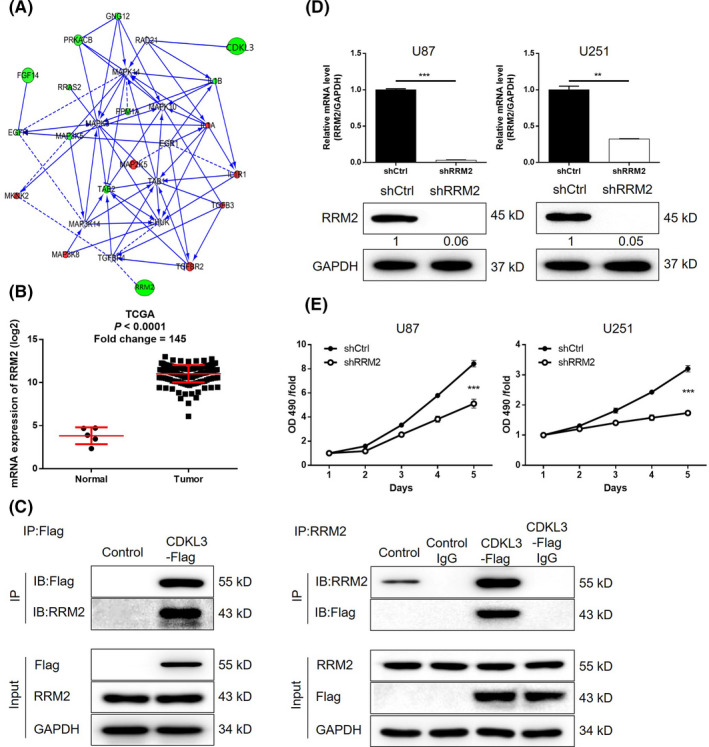
Knockdown of ribonucleotide reductase regulatory subunit M2 (RRM2) inhibits cell proliferation and induces cell apoptosis of glioma cells. A, Prediction of cyclin‐dependent kinase‐like 3 (CDKL3)‐related interaction network was carried out based on high‐throughput sequencing. B, Upregulation of RRM2 was verified by data mining of The Cancer Genome Atlas (TCGA) database. C, Interaction between CDKL3 and RRM2 was confirmed by coimmunoprecipitation in U87 cells. CDKL3 was tagged with a Flag. Immunoprecipitation was carried out using anti‐Flag or anti‐RRM2. Immunoprecipitants (IP) were probed with anti‐Flag and anti‐RRM2. D, Knockdown efficiencies of RRM2 in U87 and U251 cells were evaluated by quantitative PCR and western blot analysis. E, MTT assay was used to detect the effect of RRM2 knockdown on cell proliferation of U87 and U251cells. Representative images were selected from at least three independent experiments. Data are presented as mean ± SD. **P < .01, ***P < .001. OD, optical density
FIGURE 7.
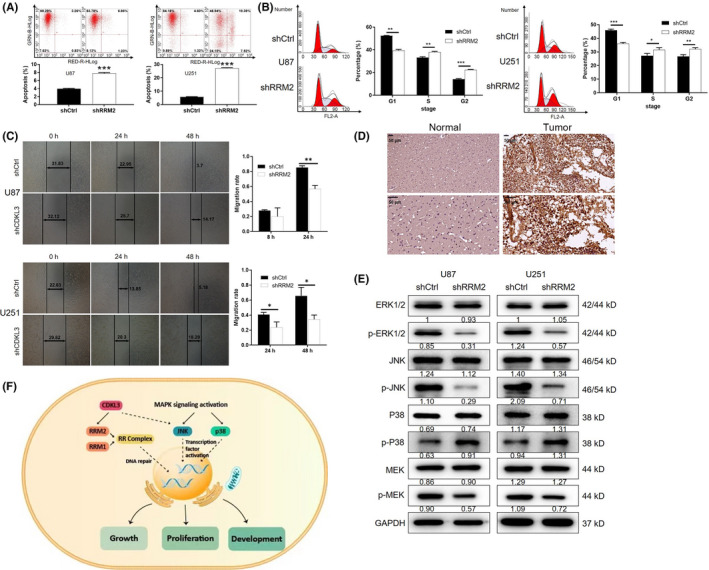
Knockdown of ribonucleotide reductase regulatory subunit M2 (RRM2) inhibits cell migration and rescues the promotion effect of cyclin‐dependent kinase‐like 3 (CDKL3) overexpression. A, B, Flow cytometry was used to examine (A) cell apoptosis and (B) cell cycle of U87 and U251 cells in both normal control (shCtrl) and shCDKL3 groups. C, Wound healing assay was used to detect the cell migration ability of U87 and U251 cells transfected with shCtrl or shCDKL3. D, Immunohistochemistry was used to detect the expression of RRM2 in glioma tumor tissues. E, Western blot analysis was used to detect the expression and phosphorylation of MAPK signaling pathway related proteins including ERK1/2, p‐ERK1/2, JNK, p‐JNK, P38, p‐P38, MEK, and p‐MEK in U87 and U251 cells with or without RRM2 knockdown. F, Schematic diagram of the CDKL3‐induced regulation of glioma. Representative images were selected from at least three independent experiments. Data are presented as mean ± SD. *P < .05, **P < .01, ***P < .001
3.7. Knockdown of RRM2 impairs the promotion of glioma by CDKL3 overexpression
In order to provide fundamental evidence for the regulation of glioma by the CDKL3/RRM2 axis, U87 and U251 cells with simultaneous overexpression of CDKL3 and knockdown of RRM2 were constructed, in which CDKL3 was slightly upregulated and RRM2 was downregulated (Figure S12). The MTT assay was used to detect proliferation of cells in control, shCtrl, CDKL3, and CDKL3 + shRRM2 groups. As shown in Figure S13, the results indicated that knockdown of RRM2 could partially reverse the promoted cell growth by CDKL3 overexpression. Notably, the elevation of JNK pathway activity in the CDKL3 + shRRM2 group was consistent with the accelerated cell growth (Figure S12).
4. DISCUSSION
Glioma is one of the most common malignancies in the central nervous system.5 It is characterized by high invasiveness and the tendency of low‐grade to high‐grade transformation, which has been considered as a major threat to human health.15 The lack of specific radiotherapy or chemotherapy results in a high risk of recurrence and poor overall treatment efficiency and prognosis.16 Although the molecular mechanism is complex, accumulating evidence showed that the regulation of abnormally expressed or mutated genes could inhibit or even reverse the malignant development of glioma in vitro or in vivo. For example, Azambuja et al17 reported that CD73 downregulation decreased glioma cell migration and invasion by reducing expression of metalloproteinase‐2 and vimentin and reduced cell proliferation. Moreover, CD73 inhibition also decreased in vivo progression of rat glioblastoma. Talamillo et al18 reported that ODZ1 allowed glioblastoma to sustain invasiveness through a Myc‐dependent transcriptional upregulation of RhoA. Although some biomarkers for glioma have been recognized, such as O6‐methylguananine‐DNA methyltransferase (MGMT) and EGFR, the overall 5‐year survival rate is still far from satisfactory.19, 20 In this study, high‐throughput sequencing of glioma tissues and normal tissues was carried out for screening DEGs and CDKL3 was identified by further high‐content screening.
In this study, our results indicated that knockdown of CDKL3 could significantly inhibit cell proliferation and cell migration ability and induce cell apoptosis and cell cycle arrest of glioma cells. In addition, CDKL3 knockdown inhibited the expression of EMT‐ and stemness‐related proteins and promoted the expression of inflammation‐related cytokines. The promotion effect of CDKL3 on glioma was also proved by the facilitated cell proliferation and colony formation by CDKL3 overexpression. The suppressed tumor growth in vivo was also elucidated in mouse models constructed by either intravenous or intracranial injection of glioma cells. Cyclin‐dependent kinase‐like 3, which is a protein serine kinase with homology to MAPKs and CDKs, could be involved in the regulation of cell growth and/or differentiation.21 In 2003, Yee et al22 found that the expression of CDKL3 mRNA could be detected in mouse embryo, and the expression reached the highest level at birth, followed by a gradual decrease. Moreover, Dubos et al12 reported the distinctly higher transcriptional level of CDKL3 in lymphoid stem cells of female patients with nonspecific mental retardation compared with healthy persons, indicating the functions of CDKL3 in diseases of the central nervous system. It has also been reported that the SKP1‐CDKL3 fusion, which was predicted by sequencing, has a potential role in cancer.23 Another work indicated that CDKL3 was relevant to enhanced cell growth of HeLa cells.24
Combining the detection of the global gene expression profile of U87 cells transfected with shCtrl or shCDKL3 and data mining of TCGA, RRM2 was proposed as the downstream of the regulation effect of CDKL3 on glioma. In this study, we confirmed the direct interplay between CDKL3 and RRM2 by co‐IP. The detection of cellular functions revealed a similar role of RRM2 with CDKL3 in glioma: inhibition of cell proliferation and migration, promotion of cell apoptosis, and arrest of cell cycle by gene knockdown. More direct evidence was provided by the RRM2 knockdown‐induced impairment of the promoted cell proliferation by CDKL3 overexpression. One of the most prominent features of malignant tumors is the rapid proliferation of tumor cells, which means that tumor cells need more deoxyribonucleotides than normal cells for DNA replication. Ribonucleotide reductase is the only enzyme that converts intracellular ribonucleotides into deoxyribonucleotides, thus providing support for DNA replication and repair.25 Ribonucleotide reductase plays an important role not only in the proliferation and differentiation of normal cells, but also in the proliferation, metastasis, and drug resistance of tumor cells.26 Human RR total enzyme is a polymer composed of the large subunit RRM1 and small subunit RRM2.25 The latter is the catalytic subunit of RR, which catalyzes the reduction of ribonucleotide diphosphate to deoxyribonucleotide diphosphate.27 Previous studies have revealed that RRM2 was upregulated in various types of human cancer. For example, Lee et al28 utilized GTI‐2040 for the transfection of cancer cells to downregulate the mRNA and protein levels of RRM2, inhibiting the growth of colorectal, prostate, liver, lung, breast, kidney, pancreatic, ovarian, and cervical cancer, and suppressing pulmonary metastasis of melanoma in a mouse model. More importantly, the role of RRM2 in the development and progression of glioma has been proven to some extent. Lee et al revealed that BRCA1‐regulated RRM2 expression could protect glioblastoma cells from endogenous replication stress and promote tumorigenicity.29 A recent study also showed that RRM2 promoted proliferation, migration, and invasiveness but inhibited apoptosis of human glioblastoma cells.30
In this study, the involvement of the JNK/MAPK signaling pathway in CDKL3‐induced promotion of glioma was predicted by KEGG pathway cluster analysis and verified by WB analysis. Further verification was achieved by treatment of cells with JNK activator anisomycin, which could promote glioma cell proliferation and reverse the inhibition induced by CDKL3 knockdown. The suppressed activity of the JNK pathway by RRM2 knockdown further verified the involvement of RRM2 and the JNK pathway in CDKL3‐induced regulation of glioma. The CDKL3 gene sequence contains highly conserved kinase sequences at both ends, endowing it with the activity of MAPKs and CDKs, which are both related to signal transduction, cell cycle, cell proliferation, and migration.22 The MAPK signaling pathway is an important signal transduction system transducing extracellular signals into cells, and it is a key pathway in the regulation of cell proliferation and apoptosis.31 Accumulating evidence has proven that the MAPK signaling pathway is involved in the development and metastasis of various types of human cancers such as colorectal, prostate, and lung cancer.32, 33, 34 In addition, studies have revealed that MAPK signaling pathways such as ERK1/2, JNK, and p38MAPK were activated in glioma tissues and related with the development and prognosis of glioma.35 Recently, studies indicated that ERK1/2 could regulate the development and progression of glioma through several pathways, such as the regulation of MMP expression. For instance, Han et al36 suggested that long noncoding RNA MALAT1 executed its function as tumor suppressor by deactivating ERK/MAPK signaling. The activation of JNK and p38MAPK signaling pathways was critical in the regulation of apoptosis, invasion, and metastasis of tumor cells.37 Recent studies have shown that activation of JNK and p38MAPK signaling pathways can also mediate cell proliferation in a variety of tumors, including gliomas. For example, the miR‐129‐5p/Wnt5a axis‐mediated JNK pathway was reported to have therapeutic potential in GBM treatment.38
In conclusion, our results identified CDKL3 as a tumor promotor for glioma, probably through the regulation of RRM2 and activation of the JNK signaling pathway (Figure 7F). Our data reveal the role of CDKL3 in glioma, highlighting the significance of CDKL3 as a putative therapeutic target.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
Supporting information
Fig S1‐S13
Table S1‐S3
ACKNOWLEDGMENTS
This work was financially supported by National Natural Science Foundation of China (No.81472354).
Cui Y, Yang Z, Wang H, et al. Identification of CDKL3 as a critical regulator in development of glioma through regulating RRM2 and the JNK signaling pathway. Cancer Sci. 2021;112:3150–3162. 10.1111/cas.15010
Cui, Yang, and Wang contributed equally to this work.
REFERENCES
- 1.Furnari FB, Fenton T, Bachoo RM, et al. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Gene Dev. 2007;21:2683‐2710. [DOI] [PubMed] [Google Scholar]
- 2.Minniti G, Muni R, Lanzetta G, Marchetti P, Enrici RM. Chemotherapy for glioblastoma: current treatment and future perspectives for cytotoxic and targeted agents. Anticancer Res. 2009;29:5171‐5184. [PubMed] [Google Scholar]
- 3.Bradley D, Rees J. Updates in the management of high‐grade glioma. J Neurol. 2014;261:651‐654. [DOI] [PubMed] [Google Scholar]
- 4.Bao S, Wu Q, Mclendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756‐760. [DOI] [PubMed] [Google Scholar]
- 5.Cohen AL, Colman H. Glioma biology and molecular markers. Cancer Treat Res. 2015;163:15‐30. [DOI] [PubMed] [Google Scholar]
- 6.Grauer OM, Wesseling P, Adema GJ. Immunotherapy of diffuse gliomas: biological background, current status and future developments. Brain Pathol. 2010;19:674‐693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807‐1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001;411:342‐348. [DOI] [PubMed] [Google Scholar]
- 9.Malumbres M, Harlow E, Hunt T, et al. Cyclin‐dependent kinases: a family portrait. Nat Cell Biol. 2009;11:1275‐1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Canning P, Park K, Gonçalves J, et al. CDKL family kinases have evolved distinct structural features and ciliary function. Cell Rep. 2018;22:885‐894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Midmer M, Haq RJ, Zanke B. Identification of NKIAMRE, the human homologue to the mitogen‐activated protein kinase‐/cyclin‐dependent kinase‐related protein kinase NKIATRE, and its loss in leukemic blasts with chromosome arm 5q deletion. Cancer Res. 1999;59:4069‐4074. [PubMed] [Google Scholar]
- 12.Dubos A, Pannetier S, Hanauer A. Inactivation of the CDKL3 gene at 5q31.1 by a balanced t(X;5) translocation associated with nonspecific mild mental retardation. Am J Med Genet A. 2010;146A:1267‐1279. [DOI] [PubMed] [Google Scholar]
- 13.Liu Z, Tao D. Inactivition of CDKL3 mildly inhibits proliferation of cells at VZ/SVZ in brain. Neurol Sci. 2015;36:297‐302. [DOI] [PubMed] [Google Scholar]
- 14.Thompson MA, Stumph J, Henrickson SE, et al. Differential gene expression in anaplastic lymphoma kinase‐positive and anaplastic lymphoma kinase‐negative anaplastic large cell lymphomas. Hum Pathol. 2005;36:494‐504. [DOI] [PubMed] [Google Scholar]
- 15.Maher EA, Furnari FB, Bachoo RM, et al. Malignant glioma: genetics and biology of a grave matter. Gene Dev. 2001;15:1311‐1333. [DOI] [PubMed] [Google Scholar]
- 16.Nayak L, Reardon DA. High‐grade gliomas. Continuum. 2017;23:1548‐1563. [DOI] [PubMed] [Google Scholar]
- 17.Azambuja JH, Gelsleichter NE, Beckenkamp LR, et al. CD73 downregulation decreases in vitro and in vivo glioblastoma growth. Mol Neurobiol. 2019;56(5):3260‐3279. 10.1007/s12035-018-1240-4 [DOI] [PubMed] [Google Scholar]
- 18.Talamillo A, Grande L, Ruiz‐Ontañon P, et al. ODZ1 allows glioblastoma to sustain invasiveness through a Myc‐dependent transcriptional upregulation of RhoA. Oncogene. 2016;36:1733‐1744. [DOI] [PubMed] [Google Scholar]
- 19.Brandes AA, Franceschi E, Tosoni A, et al. MGMT promoter methylation status can predict the incidence and outcome of pseudoprogression after concomitant radiochemotherapy in newly diagnosed glioblastoma patients. J Clin Oncol. 2008;26:2192‐2197. [DOI] [PubMed] [Google Scholar]
- 20.Omuro AM, Faivre S, Raymond E. Lessons learned in the development of targeted therapy for malignant gliomas. Mol Cancer Ther. 2007;6:1909‐1919. [DOI] [PubMed] [Google Scholar]
- 21.Molinari F, Rio M, Meskenaite V, et al. Truncating neurotrypsin mutation in autosomal recessive nonsyndromic mental retardation. Science. 2002;298:1779‐1781. [DOI] [PubMed] [Google Scholar]
- 22.Yee KW, Moore SJ, Midmer M, et al. NKIAMRE, a novel conserved CDC2‐related kinase with features of both mitogen‐activated protein kinases and cyclin‐dependent kinases. Biochem Biophys Res Commun. 2003;308:784‐792. [DOI] [PubMed] [Google Scholar]
- 23.Rudin CM, Durinck S, Stawiski EW, et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small‐cell lung cancer. Nat Genet. 2012;44:1111‐1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jaluria P, Betenbaugh M, Konstantopoulos K, Shiloach J. Enhancement of cell proliferation in various mammalian cell lines by gene insertion of a cyclin‐dependent kinase homolog. BMC Biotechnol. 2007;7:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shao J, Zhou BB, Yen Y. Ribonucleotide reductase inhibitors and future drug design. Curr Cancer Drug Tar. 2006;6:409‐431. [DOI] [PubMed] [Google Scholar]
- 26.Tanaka H, Arakawa H, Yamaguchi T, et al. A ribonucleotide reductase gene involved in a p53‐dependent cell‐cyclecheckpoint for DNA damage. Nature. 2000;404:42‐49. [DOI] [PubMed] [Google Scholar]
- 27.Rahman MA, Amin ARMR, Wang D, et al. RRM2 regulates Bcl‐2 in head and neck and lung cancers: a potential target for cancer therapy. Clin Cancer Res. 2013;19:3416‐3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee Y, Vassilakos A, Feng N, et al. GTI‐2040, an antisense agent targeting the small subunit component (R2) of human ribonucleotide reductase, shows potent antitumor activity against a variety of tumors. Cancer Res. 2003;63:2802‐2811. [PubMed] [Google Scholar]
- 29.Rasmussen RD, Gajjar MK, Tuckova L, et al. BRCA1‐regulated RRM2 expression protects glioblastoma cells from endogenous replication stress and promotes tumorigenicity. Nat Commun. 2016;7:13398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li C, Zheng J, Chen S, et al. RRM2 promotes the progression of human glioblastoma. J Cell Physiol. 2018;233:6759‐6767. [DOI] [PubMed] [Google Scholar]
- 31.Burotto M, Chiou VL, Lee JM, Kohn EC. The MAPK pathway across different malignancies: a new perspective. Cancer‐Am Cancer Soc. 2015;120:3446‐3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fang JY, Richardson BC. The MAPK signalling pathways and colorectal cancer. Lancet Oncol. 2005;6:322‐327. [DOI] [PubMed] [Google Scholar]
- 33.Caromile LA, Dortche K, Rahman MM, et al. PSMA redirects cell survival signaling from the MAPK to the PI3K‐AKT pathways to promote the progression of prostate cancer. Sci Signal. 2017;10:g3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tang Z, Yu W, Zhang C, et al. CREB‐binding protein regulates lung cancer growth by targeting MAPK and CPSF4 signaling pathway. Mol Oncol. 2016;10:317‐329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang R, Deng D, Shao N, et al. Evodiamine activates cellular apoptosis through suppressing PI3K/AKT and activating MAPK in glioma. Onco Targets Ther. 2018;11:1183‐1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Han Y, Wu Z, Wu T, et al. Tumor‐suppressive function of long noncoding RNA MALAT1 in glioma cells by downregulation of MMP2 and inactivation of ERK/MAPK signaling. Cell Death Dis. 2016;7:e2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wada T, Penninger JM. Mitogen‐activated protein kinases in apoptosis regulation. Oncogene. 2004;23:2838‐2849. [DOI] [PubMed] [Google Scholar]
- 38.Zeng A, Yin J, Li Y, et al. miR‐129‐5p targets Wnt5a to block PKC/ERK/NF‐κB and JNK pathways in glioblastoma. Cell Death Dis. 2018;9:394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang H, Song X, Huang Q, et al. LGALS3 promotes treatment resistance in glioblastoma and is associated with tumor risk and prognosis. Cancer Epidem Biomar. 2019;28:760‐769. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S13
Table S1‐S3