

Abstract
Previously, we reported that non‐apoptotic cell death was induced in non‐malignant mammary epithelial cells (HMECs) upon loss of anchorage during 48 h incubation in suspension. In this study, we examined HMECs in suspension at an earlier time point and found that most of them lost attachment ability to substrata when replated, although >80% were alive. This suggested that HMECs lost reattachment ability (RA) prior to cell death upon detachment. Concomitant with the loss of RA, a decrease in the levels of β1 and β4 integrin was observed. In sharp contrast, breast cancer cells retained integrin levels, reattached to substrata, and formed colonies after exposure to anchorage loss as efficiently as those maintained under adherent conditions. Such RA of cancer cells is essential for the metastatic process, especially for establishing adhesion contact with ECM in the secondary organ after systemic circulation. Further analysis suggested that sustained levels of β4 integrin, which was mediated by Rac1, was critical for RA after anchorage loss and lung metastasis of breast cancer cells. In the cancer cells, persistent Rac1 activity enhanced escape of β4 integrin from lysosomal degradation depending on actin‐related protein 2/3 and TBC1D2, a GTPase‐activating protein of Rab7 GTPase. Notably, simultaneous high expression of ITGB4 and RAC1 was associated with poor prognosis in patients with breast cancer. Therefore, β4 integrin and Rac1 are attractive therapeutic targets to eliminate RA in cancer cells, thereby preventing the initial step of colonization at the secondary organ during metastasis.
Keywords: beta4 integrin, breast cancer, cell adhesion, neoplasm metastasis, Rac1
Anchorage‐independent reattachment ability (RA) of cells is as important as anchorage‐independent survival or growth for cancer metastasis. The Rac1/beta4 integrin axis has emerged as a critical mediator of RA of breast cancer cells in this study. Notably, simultaneous high expression of RAC1 and ITGB4 was associated with poor prognosis in patients with breast cancer.

Abbreviations
- Arp2/3
actin‐related protein 2/3
- CA
constitutive active
- CFA
colony forming ability
- CRIB
Cdc42‐ and Rac‐interactive binding
- DN
dominant negative
- DOX
doxycycline
- FAK
focal adhesion kinase
- GAP
GTPase‐activating protein
- GEF
guanine nucleotide exchange factor
- HMEC
human mammary epithelial cell
- RA
reattachment ability
- RBD
Rho binding domain
- Tet
tetracycline
1. INTRODUCTION
To accomplish metastasis ending with outgrowth at a distant organ, cancer cells must invade, survive, and proliferate in ectopic environments, where they encounter overall irrelevant and inadequate contact with ectopic ECM, even in the absence of ECM.1 In particular, in the bloodstream, circulating cancer cells that must survive without anchorage long enough to settle in a secondary organ.2 Therefore, cancer cells frequently exhibit anchorage‐independent cell survival and growth or resistance to cell death and growth arrest that are induced in the absence of adhesion, in addition to tolerance to mechanical stress exerted by blood flow and robustness to the attacks of the immune system.
However, even if cancer cells survive adverse conditions in circulation and reach the secondary site, they fail to colonize the site unless they adhere to the ECM or basement membrane underlying the vessel endothelium, establish adhesion contact with substrata, such as laminin, and successfully extravasate into the surrounding tissue.2, 3 Millions of cells are possibly released from a primary tumor every day, and most cells are likely to reach the secondary site shortly after entering the bloodstream; however, only a small minority colonizes a distant organ. Most cells remain trapped in a capillary bed and eventually die intravascularly by apoptosis without forming new anchorage within 1‐2 d.4 Therefore, for metastasis, the ability to reattach and establish adhesion contact with the substrata is considered as important as anchorage‐independent survival and growth. Anchorage‐independent behaviors have been extensively studied as a hallmark of cancer.5 However, the RA of cells has received less attention.
In this study, we investigated the RA of mammary epithelial cells and found that the RA of breast cancer cells was resistant to anchorage loss, similar to survival and growth. They reattached to substrata and formed colonies after suffering anchorage loss with the same efficiency as before. By contrast, in non‐malignant cells, RA was lost after detachment concomitantly with the loss of β1 and β4 integrins. In cancer cells, β1 and β4 integrins persistently existed after detachment, which contributed to RA of cancer cells. Further analysis revealed that Rac1 activity remained in the absence of adhesion and was reactivated in response to reattachment in cancer cells, which mediated the anchorage‐independent β4 integrin existence and RA, resulting in eventual metastasis to the lung. In conclusion, the Rac1/β4 integrin axis has emerged as a critical mediator of extravasation including the establishment of adhesion contact with ECM after systemic circulation, an initial step for colonization at the secondary organ during metastasis.
2. MATERIALS AND METHODS
2.1. Cell lines and culture conditions
All cancer cell lines were obtained from the ATCC and maintained in the appropriate medium, as shown in Table S1. Subtypes of cell lines were categorized as described.6 HMECs were cultured as described.7
For adherent and non‐adherent or suspension cultures, denoted as A0 and S0, respectively (Figure 1A), cells were detached from the culture dish using TrypLE Express (Thermo Fisher Scientific) to obtain a single‐cell suspension in the growth medium supplemented with 0.5% methylcellulose, divided equally, and plated onto culture plates coated with or without polyHEMA for non‐adherent and adherent cultures, respectively. After incubation for 24 h in a CO2 incubator at 37℃, the cells were used for experiments. The culture conditions following A0 and S0 for each experiment are illustrated in Figure 1A, and the representative images of cells under these conditions are shown in Figure S1A.
FIGURE 1.
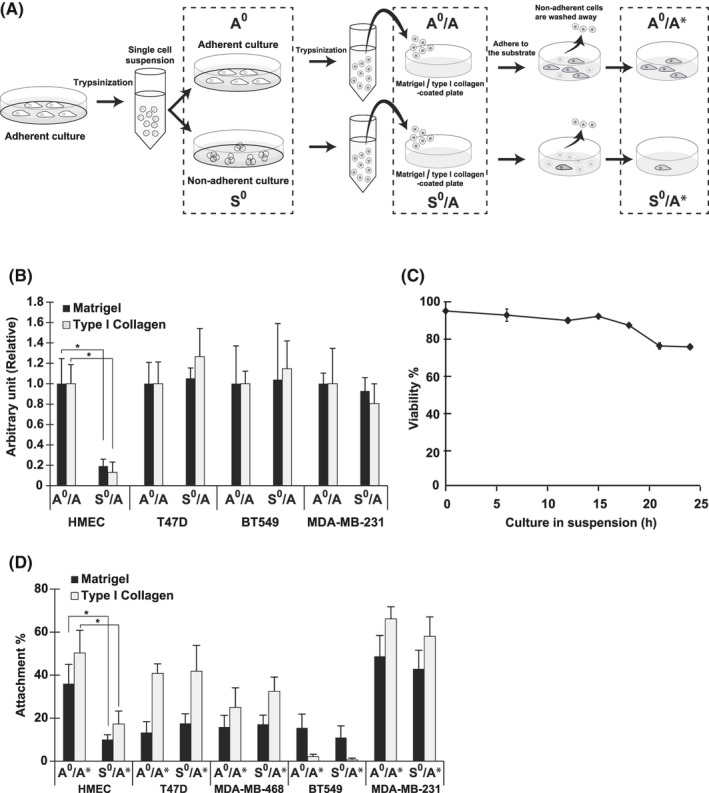
The colony forming ability (CFA) and reattachment ability (RA) of malignant and non‐malignant mammary epithelial cells under adherent and non‐adherent conditions. A, Schematic representation of the experimental procedure used in assays for attachment‐related properties of cells. Cells categorized into groups, adherent (A0) and non‐adherent (S0), and incubated for 24 h under A0 and S0 conditions, respectively, as described in Materials and Methods (cell lines and culture conditions), were used for western blotting. For colony formation and attachment assays, cells (A0 and S0) were dissociated and seeded onto plates coated with Matrigel or type I collagen (A0/A and S0/A). After 14 d, colony formation was quantified. The cell attachment assay was performed with cells (A0 and S0) dissociated and stained with the dye, as described in Materials and Methods. The cells were then replated and incubated for 30 min at A0/A and S0/A. After removal of unattached cells, the attached cells were quantified (A0/A* and S0/A*). Pull‐down assay was conducted following the steps for A0 and S0 or A0/A* and S0/A*. Representative images of cells (HMECs) cultured under each condition are shown in Figure S1A. Details for each assay are described in Materials and Methods. B, CFA of HMECs and cells from breast cancer cell lines (Table S1). Ratio to the control, the value from cells on Matrigel‐coated plates (A0/A; Matrigel) is shown. C, Viability of HMECs in suspension. HMECs were cultured under non‐adherent conditions (S0), and cell viability was determined by trypan blue exclusion at the time indicated. D, RAs of HMECs and cells from breast cancer cell lines assessed by cell attachment assays. Ratio of attached cells to the initial input is shown as attachment percentage. Values represent mean ± SD from at least 3 independent experiments. *P < .01
Other methods are described in Supplementary material Doc. S1.
3. RESULTS
3.1. RA and CFA of malignant and non‐malignant mammary epithelial cells
CFA, an indicator of the proliferative potential of cells, is an in vitro surrogate marker of the aggressiveness or stemness of cancer cells in vivo, and is frequently anchorage independent in cancer cells.5, 8 In this study, we examined the CFA of malignant and non‐malignant mammary epithelial cells in accordance with the experimental procedures as illustrated in Figure 1A. We used Matrigel and type I collagen as in vitro substrates as alternatives to basement membrane under the vascular wall and interstitial matrix in perivascular tissue in vivo, respectively. As shown in Figure 1B, CFAs of breast cancer cells in 2 populations incubated under adherent (A0/A) and non‐adherent conditions (S0/A) were almost equal, confirming that CFA was anchorage independent. We studied 5 breast cancer cell lines as representatives of 4 subtypes (Table S1)6 and obtained the same results.
However, CFA was anchorage dependent in non‐malignant mammary epithelial cells and compromised upon loss of anchorage, usually with the concomitant induction of cell death, known as anoikis.9 Therefore, the CFA of HMECs kept in suspension for 24 h was <20% compared with that of their counterpart (Figures 1B and S1B; HMEC, A0/A vs. S0/A). However, results from parallel experiments revealed that cell viabilities were equally high under both adherent (A0) and non‐adherent (S0) conditions within the span of incubation. Notably, 80.6% of cells were alive at 24 h after detachment in suspension (Figure 1C): cell death became evident only after 48 h, as described.7 This apparent discrepancy between low CFA and high viability of HMECs at an earlier time point indicates that non‐malignant cells suffering anchorage loss lose CFA within 24 h without losing viability.
Loss of CFA in HMECs during early periods after detachment prompted us to examine the attachment properties of HMECs in suspension. We examined RA following the procedure used for the assessment of CFA (Figure 1A). Briefly, cells preincubated under adherent (A0) or non‐adherent conditions (S0) for 24 h, the viabilities of which were >80% as described, were plated on substrata and allowed to adhere for 30 min. The reattachment of cells preincubated in suspension reached a plateau by this time (Figure S1C; S0/A*). After removing unattached cells, the number of attached cells was quantified. Interestingly, the number derived from non‐adherent preculture (Figure 1D; HMEC, S0/A*) was significantly smaller compared with that from the adherent preculture (HMEC, A0/A*). In short, most cells in the non‐adherent culture (S0) were alive but not reattached and were removed as unattached cells, demonstrating that cellular adhesiveness to substrata was selectively impaired within 24 h in cells kept in suspension. Accordingly, it was most likely that the low CFA of HMECs undergoing anchorage loss was primarily due to the impaired adhesiveness to substrata when replated.
By contrast, RA was not compromised in breast cancer cells during incubation in suspension. The number of reattached cells was almost the same between populations preincubated under adherent (A0/A*) and non‐adherent (S0/A*) conditions (Figure 1D) in agreement with the CFA (Figure 1B). Results were the same among breast cancer cell lines studied, although their inherent attachment rates varied. Therefore, unlike HMECs, breast cancer cells retained RA during loss of anchorage, thereby adhering and forming colonies when in contact with substrate. Such persistent RA of cancer cells after anchorage loss presumably plays an important role in accomplishing metastasis, including establishment of adhesion contact with basement membrane under a vascular wall after systemic circulation, followed by extravasation into the parenchyma of a secondary organ.3
3.2. Anchorage dependence and independence of β1 and β4 integrin levels responsible for RA and metastatic potential of mammary epithelial cells
The ability of cells to attach to ECM depends mostly on integrins, a group of cell adhesion receptors that recognize the ECM.3 In this study, we found that protein levels of β1 and β4 integrin were markedly reduced in HMECs after detachment (Figure 2A; HMEC, A0 vs. S0); 2 forms of β1 integrin were detected, which were probably differentially glycosylated.10 Since mRNA levels were unchanged (Figure 2B; HMEC, A0 vs. S0), the decrease in integrins was mainly regulated at a protein level. In contrast with HMECs, cancer cells retained the 2 integrins at comparable levels at both protein and mRNA levels under the 2 culture conditions (Figure 2A,B). Given that β4‐ as well as β1‐containing integrins play fundamental roles in the adhesion of epithelial, including mammary epithelial, cells,11, 12, 13 their decreased level in HMECs in suspension was the most probable cause of the deteriorated RA of cells, and their persistent level in cancer cells potentially underpinned the RA of cells. Analysis of protein stability by cycloheximide chase methods showed that the β4 integrin protein was less stable in HMECs compared with in cancer cells under non‐adherent conditions (Figure 2C).
FIGURE 2.
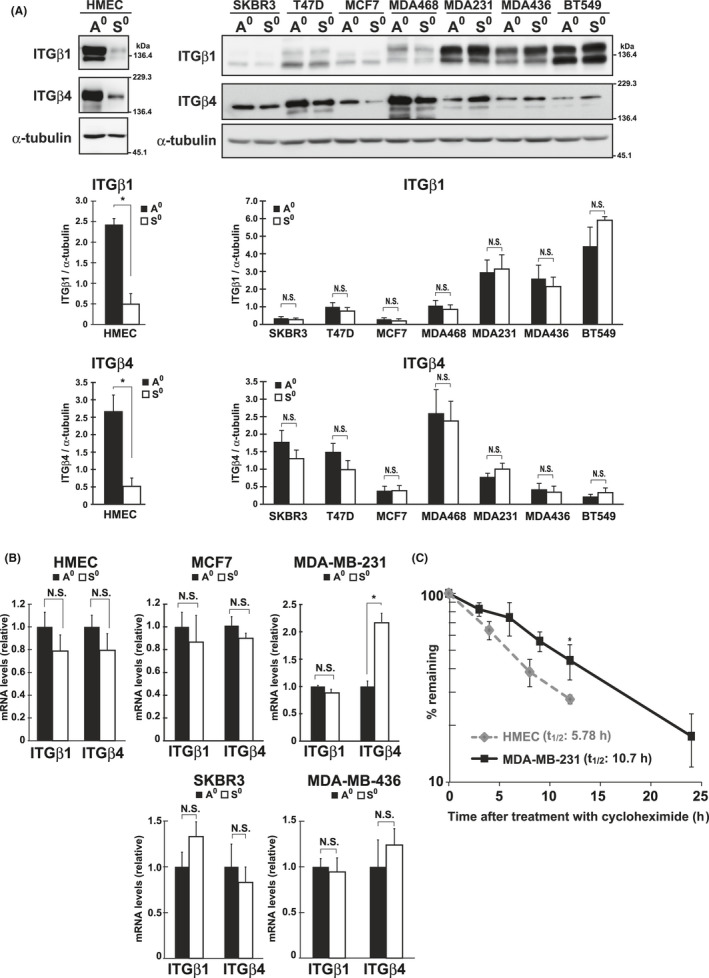
β1 and β4 integrin (ITG) levels under adherent and non‐adherent conditions. A, β1 and β4 integrin levels in HMECs and breast cancer cells under adherent (A0) and non‐adherent (S0) conditions were examined using western blotting with the indicated antibodies. α‐Tubulin was used as the loading control. The band intensities were quantified using ImageJ software, normalized against those of α‐tubulin, and graphed. B, The mRNA levels of β1 and β4 integrin in HMECs and cancer cells cultured as in A were examined by qPCR. The values were normalized with those of the TATA‐box binding protein and shown as relative to the control (A0). C, HMECs and MDA‐MB‐231 cells were incubated under non‐adherent conditions in the presence of cycloheximide (100 µg/mL) for the indicated times, and at each time point, the amount of β4 integrin was quantified by western blotting using ImageJ software. After normalization to values of Coomassie‐stained total protein, the percentages of the remaining amounts of β4 integrin were plotted. The half‐life (t1/2) of the protein was calculated from the slope between the points on the graph and averaged. Values are represented as mean ± SD from at least 3 independent experiments. *P < .01. NS, not significant
To demonstrate the impact of integrin level on RA, we established MDA‐MB‐231 cells expressing shRNAs for β1 or β4 integrins with 2 unrelated sequences (shITG β1#2 and #3, and shITG β4#1 and #5) along with those expressing non‐targeted shRNA (shNT). Successful knockdown of each integrin was confirmed by western blotting and flow cytometry (Figure 3A,B). Knockdown with the shRNAs revealed that the RA of cancer cells deteriorated considerably (Figure 3C); shRNAs for β1 and β4 integrins were comparably effective. Therefore, both β1 and β4 integrins were required for RA in mammary epithelial cells, suggesting that the difference of RA between HMECs and cancer cells could be attributed to the difference in the dependency of their integrin levels on anchorage.
FIGURE 3.
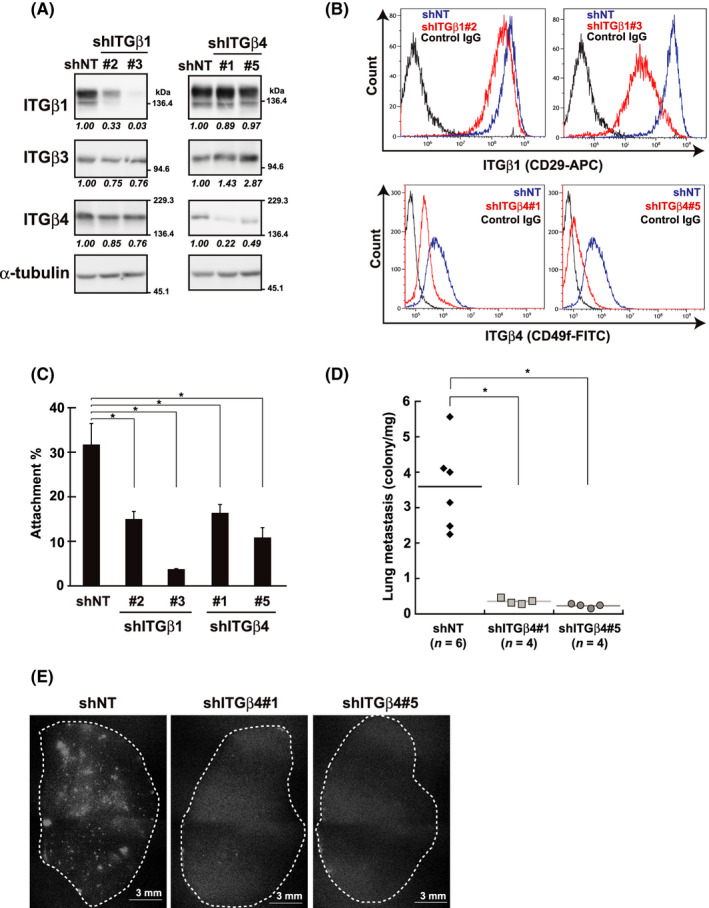
Knockdown of β1 and β4 integrin (ITG) expression and its impact on RA and lung metastasis. shRNA [shITGβ1#2, #3, shITGβ4#1, #5 and control (shNT)]‐expressing cells were established from non‐ (A‐C) or tdTomato‐labeled MDA‐MB‐231 cells (D, E) (see Materials and Methods). Knockdown effects were verified by western blotting (A) and flow cytometry (B) using the indicated antibodies. A, α‐Tubulin was used as the loading control. The band intensities were quantified, normalized with those of α‐tubulin, and shown as relative to the control (shNT) below the panels. B, Representative histograms are shown. C, RA of shRNA‐expressing cells was evaluated using the cell attachment assay on Matrigel‐coated dishes as A0/A* in Figure 1D. Values represent mean ± SD from at least 3 independent experiments. *P < .01. D, E, Cells were inoculated into the mammary fat pads of female NOD/SCID mice. When the tumor volume reached approximately 1.0 cm3, the metastatic nodules in the lobes of the lungs—representative images are shown (E)—were quantified and plotted as described in the Materials and Methods (D). Horizontal lines indicate the means from the indicated number of mice. *P < .01
Notably, the knockdown of β4 integrin dramatically suppressed the lung metastasis of MDA‐MB‐231 cells that were inoculated orthotopically and growing in mammary fat pads (Figure 3D,E). Of the properties underlying the metastatic potential of cells, migratory ability was impaired by this knockdown, while doubling time was not significantly affected (Figure S2A,B). Collectively, it is likely that the decrease in β4 integrin levels led to defects in the RA and migratory ability of cancer cells, thereby interfering with metastasis. Depletion of β1 integrin can also reportedly interfere with lung metastasis of breast cancer cells.14
3.3. Anchorage‐dependent change of Rac1 activity in mammary epithelial cells
Expression levels of integrin and cell adhesion are regulated by the Rho family of small GTPases; representative members are Rac1, Cdc42, and RhoA.15, 16 To understand the anchorage dependence and independence of integrin levels and RA in mammary epithelial cells, we examined the activities of GTPases in cells under the conditions used for the reattachment assay (Figure 1A). Pull‐down experiments detected the active form of Rac1 in samples from HMECs under adherent conditions (Figure 4A; Rac1/GST‐CRIB, A0). However, in cells cultured in suspension, this form was undetectable (Rac1/GST‐CRIB, S0), suggesting that Rac1 activity was anchorage dependent, which was similar to integrin level and RA in HMECs. The active form of Cdc42 was barely detectable under the experimental conditions, and its amount visualized under an extended detection setting was unchanged between culture conditions (Figure 4A; Cdc42/GST‐CRIB, A0 and S0). In addition, the active form of RhoA was unchanged (RhoA/GST‐RBD).
FIGURE 4.
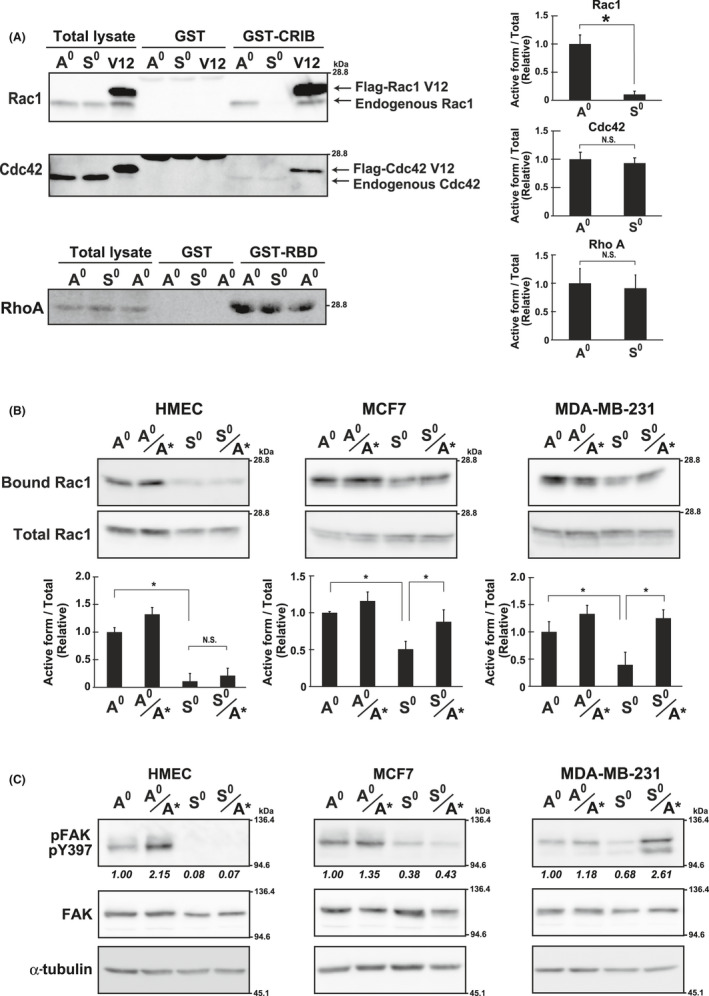
Rac1 activity under adherent and non‐adherent conditions. A, Rac1 and Cdc42, were precipitated with GST‐CRIB and RhoA with GST‐RBD, and analyzed by western blotting in HMECs cultured under adherent (A0) or non‐adherent (S0) conditions (Figure 1A). The Flag‐tagged constitutively active mutant of Rac1 and Cdc42 (Rac1 V12 and Cdc42 V12, respectively) were expressed in HMECs and used as the positive control (V12). The band intensities of the active forms (GST‐CRIB or GST‐RBD) were quantified using ImageJ software, normalized against those of the total lysate, and shown as relative to the control (A0) in graphs. B, Cells cultured as A0 and S0 or reattached as A0/A* and S0/S* (Figure 1A) for 1 h on Matrigel‐coated plates were used for pull‐down assays and analyzed as in (A). Values represent mean ± SD from at least 3 independent experiments. *P < .01. NS, not significant. C, Cells, cultured as in (B), were examined by western blotting using the indicated antibodies. α‐Tubulin was used as the loading control. The band intensities of pFAK were quantified, normalized with those of FAK, and shown as relative to the control (A0) below the panel of pFAK
The active form of Rac1 was also detected under adherent conditions in MCF7 and MDA‐MB‐231 cells, whereas its amount was reduced under non‐adherent conditions (Figure 4B; Bound Rac1/MCF7 and MDA‐MB‐231, A0 vs. S0). Therefore, Rac1 was similarly inactivated upon detachment in HMECs and cancer cells. In cancer cells, however, Rac1 remained active to some extent (MCF7 and MDA‐MB‐231, S0). In addition, Rac1 was reactivated in cancer cells when cells in suspension were replated onto the substratum (S0 vs. S0/A*). Similar to the changes in Rac1 activity, activation or inactivation of FAK, an upstream regulator of Rac1, was observed [Figure 4C; pFAK(Y397)]. In particular, FAK remained partially activated in cancer cells cultured in suspension (MCF7 and MDA‐MB‐231, S0), possibly contributing to the aberrant Rac1 activation in cancer cells. This was in striking contrast with that for HMECs, in which Rac1 and FAK remained off or inactivated after detachment even if the cells were transferred to adherent culture (Figure 4B,C; HMEC, S0 and S0/A*).
In summary, in cancer cells, Rac1 remained activated during loss of anchorage, albeit at a lower level, downstream of FAK, and was reactivated in response to reattachment to the substrate. Therefore, reactivation and/or remaining activity of Rac1 potentially contribute to sustained integrin level and/or RA in cancer cells after anchorage loss.
3.4. Rac1 activity regulates integrin level and RA in mammary epithelial cells
To substantiate the potential involvement of Rac1 activity in sustained integrin level and persistent RA in cancer cells, we interfered with Rac1 activity in cells and observed the effects on their phenotypes. We virally transduced an expression system of the DN form of Rac1 and those of the other members of the family, namely Cdc42 and RhoA, based on the Tet‐Off system in MDA‐MB‐231 cancer cells. After establishing cell lines expressing the DNs in the absence of DOX, we first examined their effects on β1 and β4 integrin level. Except for a slight decrease in response to detachment, which was observed in most cell lines irrespective of the DN expression (Figure 5A; ITGβ1 and ITGβ4, A0 vs. S0), β4 level was significantly affected by Rac1 DN expression and decreased in response to detachment as HMECs in Figure 2A (Figure 5A; ITGβ4/Rac N17, DOX+ vs. DOX−, A0 vs. S0). Conversely, β1 level was almost unchanged in the presence and absence of DOX (ITGβ1/Rac N17, DOX+ vs. DOX−). Cdc42 and RhoA DN expressions had no significant effect on both integrin levels (Cdc42 N17 and RhoA N19, DOX+ vs. DOX−). Therefore, Rac1 activity regulates β4 level in cancer cells. The level of β1 appeared constitutive and independent of Rac1 activity. Consistent with our results, a study reported that activated Rac1 selectively upregulates the expression level of β4 integrin in colon cancer cells.17
FIGURE 5.
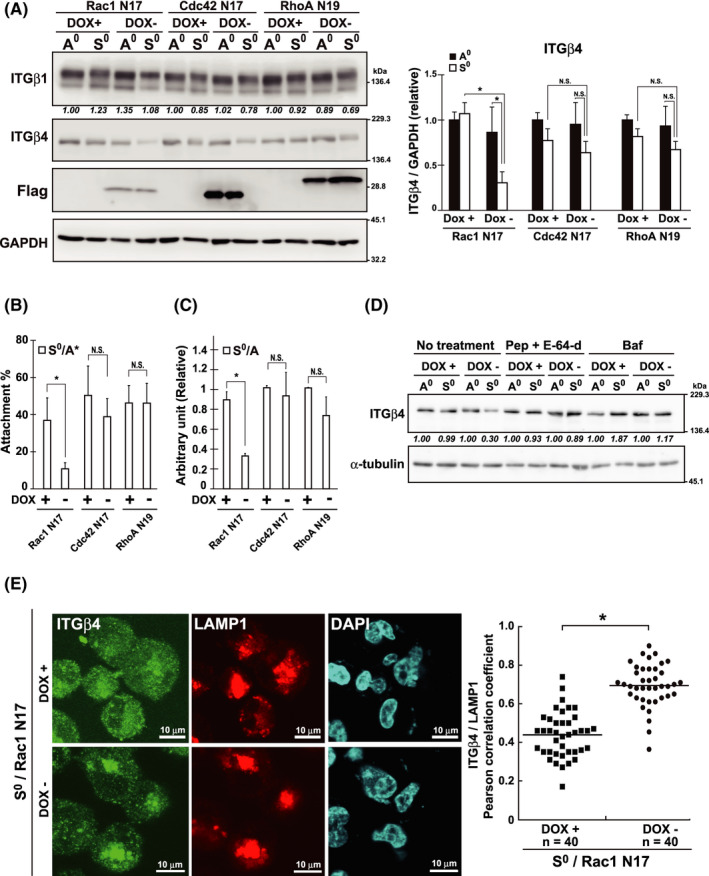
Rac1 activity underlying reattachment ability (RA) and β4 integrin level in breast cancer cells. A‐E, MDA‐MB‐231 cells were infected with Tet‐Off lentiviral expression vectors encoding Flag‐tagged DN mutants of GTPases (Rac1 N17, Cdc42 N17, and Rho N19) and selected with 1 µg/mL puromycin. The resistant cells were incubated for 24 h in the presence (DOX+) or absence (DOX−) of doxycycline (DOX) (2 ng/mL) and then cultured for 24 h under adherent (A0) or non‐adherent (S0) conditions. Total cell lysates were examined by western blotting using the indicated antibodies (A). GAPDH was used as the loading control. The band intensities were quantified using ImageJ software, normalized against those of GAPDH, and shown as relative to the control (DOX+/A0) in graphs. B , C, RA (B) and colony forming ability (CFA) (C) on Matrigel‐coated plates, respectively, were examined. After incubation under non‐adherent conditions for 24 h, cells were assayed for RA (S0/A*) and CFA (S0/A). Values represent mean ± SD from at least 3 independent experiments. *P < .01. NS, not significant. D, Cells expressing Rac1 N17 were incubated under DOX+ or DOX− as in (A) and cultured (A0 and S0) in the presence of bafilomycin A1 (Baf) (100 nM), or pepstatin A (Pep) (10 µg/mL) and E‐64‐d (10 µg/mL) in combination for 24 h, and examined using western blotting. The band intensities of ITGβ4 were quantified, normalized with those of α‐tubulin, and shown as relative to the control (A0) below the panel of ITGβ4. E, Cells expressing Rac1 N17 incubated under DOX+ or DOX− were cultured (S0) as above, and examined by immunocytochemistry using the antibodies forβ4 integrin (ITGβ4) and LAMP1. Representative images at high magnification are shown (low magnification images are shown in Figure S3A). Nuclei were labeled with DAPI. Pearson correlation coefficients for the colocalization of β4 integrin with LAMP1 was computed using the Coloc 2 plugin of the Fiji software and plotted. Horizontal lines indicate the means from the indicated number of cells. *P < .01
Concomitant with β4 integrin levels, RA levels were affected by the expression of Rac1 DN; the number of reattached cells from the suspension culture markedly declined upon Rac1 DN expression in cancer cells (Figure 5B; Rac N17). Consequently, CFA decreased (Figure 5C; Rac N17). The expression of Cdc42 and RhoA DNs had little impact on RA and CFA.
In conclusion, Rac1 activity was essential for maintaining β4 integrin levels in cancer cells during anchorage loss, thereby promoting RA in cooperation with β1 integrin, which was also required for RA (Figure 3C). In cancer cells, the expression level of β1 integrin was uncoupled from Rac1 activity potentially through the activation of oncogenic signaling upstream. In HMECs, the β1 integrin level was also regulated by Rac1, as described below.
Integrins on the cell surface are constantly subjected to endocytosis followed by endosomal sorting, which determines the recycling or degradation of these molecules.18 To obtain insight into the mechanisms by which Rac1 regulates β4 integrin levels, we focused on the degradation process, considering that the above observations suggested the involvement of protein stability regulation for the integrin (Figure 2C). Here, we tested the effects of inhibitors targeting lysosomal enzymatic activities, such as bafilomycin A1, pepstatin A, and E‐64‐d, and found that they effectively prevented a decrease in β4 integrin levels in suspended MDA‐MB‐231 cells under Rac1 DN expression. This suggested that integrins were preferably degraded in the lysosomes when Rac1 was inhibited (Figure 5D; No treatment vs. Pep+E‐64‐d and Baf, DOX−/S0). In fact, in the cells expressing Rac1 DN, a significant amount of β4 integrin was apparently colocalized with LAMP1,19 a lysosomal marker, suggesting the accumulation of integrins in the lysosomes under Rac1 inhibition (Figure 5E; DOX−). Under the control (Figures 5E and S3A; S0/DOX+) or adherent conditions (Figure S3A,B; A0/DOX+ and DOX−), the majority of β4 integrin was distributed diffusely over the entire cells. Therefore, it was possible that Rac1 functions to promote the escape of early endosomes containing β4 integrin from lysosomal degradation.
On the basis of the above observations, it was hypothesized that HMECs with constitutively active Rac1 would retain integrins and consequently acquire RA even after anchorage loss, similar to cancer cells. To test this hypothesis, we transduced constitutive active forms (CAs) of the 3 GTPases into HMECs and examined the RA of cells. As expected, in HMECs expressing Rac1 CA, but not Cdc42 and RhoA CA, β1 and β4 integrin levels were maintained during anchorage loss, leading to complete retention of RA and CFA after being kept in suspension, similar to that under adherent conditions (Figure 6A‐D; Mock vs. Rac1 V12).
FIGURE 6.
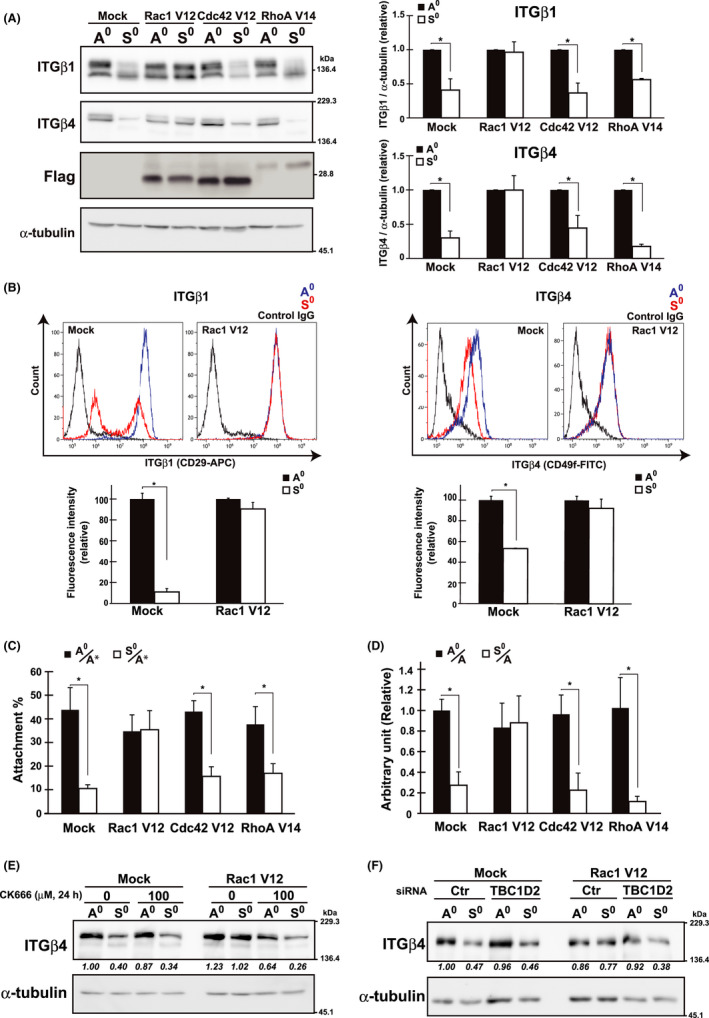
Rac1‐regulated integrin level and reattachment ability (RA) in HMECs. A, HMECs were infected with lentivirus constructs expressing control (Mock) and Flag‐tagged CA mutants of GTPases (Rac1 V12, Cdc42 V12, and RhoA V14). After selection with 10 µg/mL blasticidin, cells stably expressing proteins were cultured and examined using western blotting with the indicated antibodies as in Figure 2A. α‐Tubulin was used as the loading control. The band intensities were quantified using ImageJ software, normalized against those of α‐tubulin, and shown as relative to the control (A0) in graphs. *P < .01. B, HMECs expressing Rac1 V12 were cultured as in (A), and cell surface expression of β1 and β4 integrins was evaluated by flow cytometry. Representative histograms are shown. Fluorescence intensities were quantified using Kaluza Analysis software, and shown as relative to the control (A0) in graphs. *P < .01. C and D, RA (C) and colony forming ability (CFA) (D) of cells plated on Matrigel‐coated dish were examined as in Figure 1D,B, respectively. Values represent mean ± SD from at least 3 independent experiments. *P < .01. E, F, HMECs expressing control (Mock) or Rac1 V12 were cultured under the A0 or S0 conditions for 24 h with or without CK‐666 (E) or after pretreatment with siRNA for control (Ctr) or TBC1D2 for 48 h (F). Total cell lysates were examined by western blotting using the indicated antibodies. α‐Tubulin was used as the loading control. The band intensities of ITGβ4 were quantified, normalized with those of α‐tubulin, and shown as relative to the control (Mock, 0/A0 or Ctr/A0) below the panel of ITGβ4
From these results, the important role of Rac1 in the regulation of integrin levels, specifically at the turnover processes, has emerged (Figure 5D,E). Mechanistically, Rac1 appeared to mobilize actin dynamics or actin‐related protein (Arp)2/3‐dependent actin polymerization to promote integrin trafficking to recycling endosomes.20, 21 A Rac1 effector, TBC1D2, which is a GAP of Rab7 GTPase,22, 23 was also involved; under the treatment with a pharmacological inhibitor of Arp2/3, CK‐666 (Figure 6E), or siRNA for TBC1D2 (Figure 6F), active Rac1 was unable to sustain β4 integrin levels upon detachment (Figure 6E; Rac1 V12, 0/S0 vs. 100/S0) (Figure 6F; Rac1 V12, Ctr/ S0 vs. TBC1D2/S0).
3.5. Poor prognosis of patients with breast cancer with simultaneous high expression of ITGB4 and RAC1
Results from in vitro experiments suggested that cancer cells acquired RA based on persistent Rac1 activity and β4 integrin level, thereby accomplishing metastasis in vivo. To demonstrate the importance of Rac1 and β4 integrin‐mediated RA in cancer progression, we analyzed the correlation of ITGB4 and RAC1 expression with the prognosis of patients with breast cancer using the METABRIC database.24, 25 Notably, survival analysis indicated that the high expression of both ITGB4 and RAC1, but not each alone, significantly correlated with poor prognosis (Figure 7A‐C).
FIGURE 7.
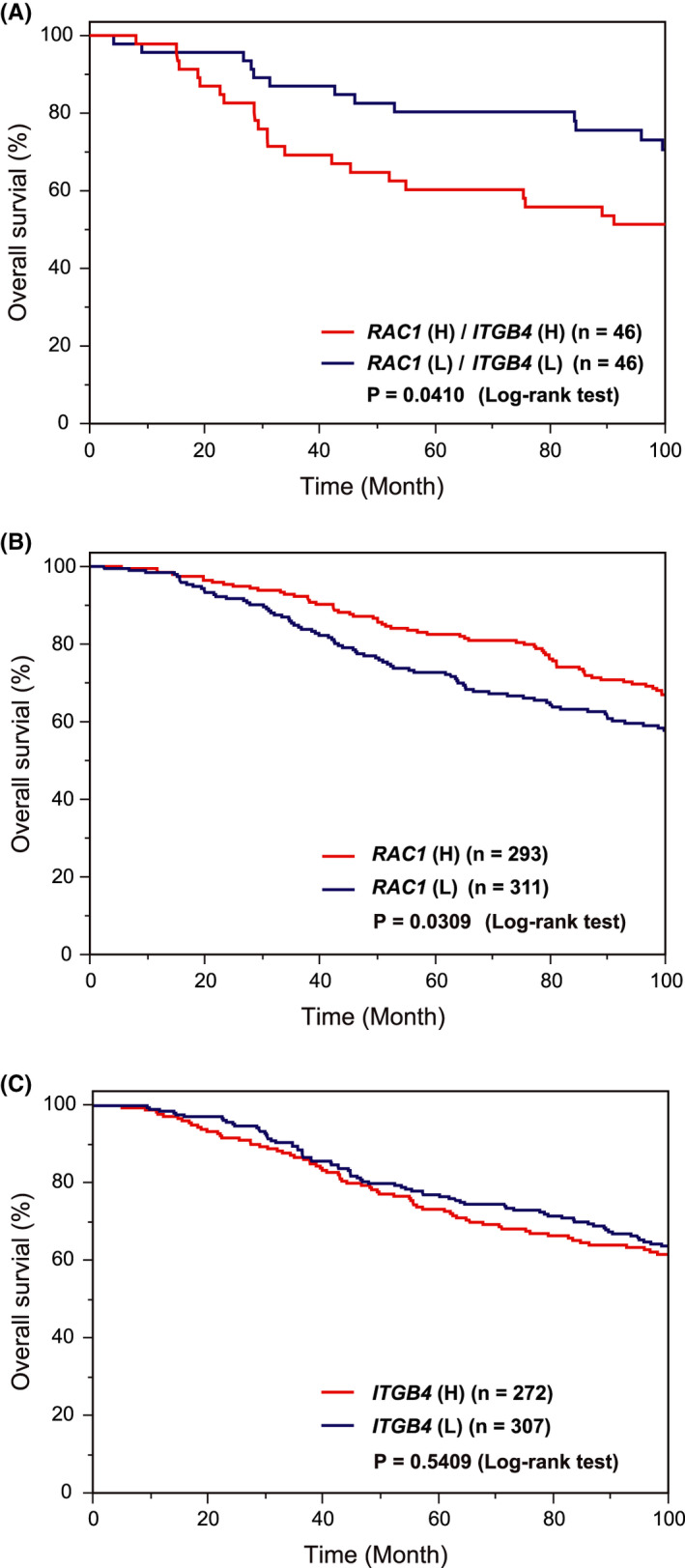
The impact RAC1 and ITGB4 expression on survival outcomes in patient with breast cancer. A‐C, Kaplan‐Meier plots with patients categorized by expression levels [H; high (z‐score >1), and L; low (z‐score < −1)] of RAC1 and ITGB4 were obtained using METABRIC dataset through the cBioPortal, as described in Materials and Methods. A, RAC1 (H)/ITGB4 (H) vs. RAC1 (L)/ITGB4 (L). B, RAC1 (H) vs. (L). C, ITGB4 (H) vs. (L)
4. DISCUSSION
Rac GTPases belongs to the Rho family of small G‐proteins and transduce signals from tyrosine kinases, G protein‐coupled receptors, and integrins to regulate cytoskeletal reorganization, and consequently, cell adhesion and motility.26 Therefore, they engage in cellular responses ranging from morphogenesis to disease, including malignant transformation. Their activities are upregulated and downregulated by guanine nucleotide exchange factors (GEFs) and GAPs, respectively, which are downstream of various upstream stimuli. In cancer cells, due to aberrant upstream signaling, several Rac‐GEFs are upregulated or overexpressed, leading to deregulation of Rac activity.27
In this study, we found Rac1 activity remaining in the absence of anchorage and reactivated in response to recontact with substrate in cancer cells, whereas in non‐malignant cells, the activity was almost completely abolished under conditions without anchorage. The sustained Rac1 activity in cancer cells possibly due to deregulated upstream oncogenic signaling. Notably, we found that Rac1 activity regulated the persistent level of β4 integrin, thereby conferring RA to cancer cells after anchorage loss.
Therefore, distinctive features of Rac1 activity have emerged in the regulation of β4 integrin level and RA in cancer cells. The involvement of other members of the family, such as Cdc42 and RhoA, in this process was less likely, because they are either active or inactive in an anchorage‐independent manner, and manipulation of their activities had little impact on β4 integrin level and RA.
Given the emerging role of Rac1 critical for persistent RA, Rac1 activity would be an attractive target for cancer therapy focusing on the colonizing phase of metastasis. According to our results, the inhibition of Rac1 activity in cancer cells within the vasculature is expected to decrease β4 integrin levels, thereby depriving the cells of RA and preventing establishment of stable adhesion to ECM in a secondary organ.
Similarly, β4 integrin downstream of Rac1 provides an alternative target for cancer therapy against the colonizing process. Previous studies have identified integrin species that were engaged in the extravasation of circulating tumor cells at almost every step.3 Our findings revealed that β4 integrin, which was originally identified as a tumor‐associated antigen overexpressed in several types of metastatic cancers,28, 29 has an important role the in RA of circulating tumor cells. β4 integrin also mediated the interaction of circulating colon carcinoma and breast cancer cells with adhesion molecules on endothelial cells.30, 31 These findings underscore the importance of β4 integrin in the metastasis of cancer cells, specifically at the initial stages of colonization. These features of β4 integrin are probably associated with its exceptionally long cytoplasmic tail containing multiple interacting and signaling domains.32, 33 β4 integrin together with its heterodimerizing partner α6 integrin links intracellular intermediate filaments to extracellular laminins through these domains, and forms aggregates called hemidesmosomes, which anchor the basal layer of epithelial cells to the basement membrane.34
Notably, β4 integrin cooperates with growth factor receptors and other integrins to transduce signals down to major signaling molecules, such as RAS, PI3K, and MAPK, through the signaling domain in the cytoplasmic tail.32, 33, 35, 36, 37 In particular, β4 integrin mediates Rac1 activation in response to upstream inputs.38, 39 Our results showed that Rac1 activity upregulates β4 integrin levels. These findings suggest that Rac1 and β4 integrin mutually potentiate each other to achieve sustainable levels of β4 integrin and Rac1 activity during anchorage loss, consequently leading to RA of cancer cells after anchorage loss. Therefore, RAC1 and ITGB4 are considered to configure a functional axis that operates to promote the aggressiveness of cancer. Therapeutically, the inhibition of either Rac1 activity or β4 integrin expression would be effective to interfere with the function of the axis.
With respect to the mechanisms underlying the regulation of β4 integrin trafficking by Rac1, our results demonstrated the importance of Arp2/3‐dependent actin polymerization, consistent with the prevailing views that the Rho GTPases regulate actin dynamics,40 and that actin dynamics affects the endosomal sorting process.18 According to previous studies, the involvement of vimentin, an intermediate filament, which interacts with β4 integrin through a linker protein,41 was also likely.42 In addition, Rab7, a member of the Rab family of small GTPases, was implicated in regulation in this study. Rab7 is a key regulator of vesicular membrane traffic to late endosomes and lysosomes, including biogenesis of lysosomes.43 Therefore, it is important in targeting surface molecules to lysosome.44, 45 However, when a GAP of Rab7, TBC1D2, is activated downstream of Rac1, Rab7 is inactivated.22 Accordingly, Rac1 activation leads to the suppression of the lysosomal degradation of β4 integrins in a TBC1D2‐dependent manner.
Conversely, Rac1 is regulated by downstream components, such as Rab7, as well as β4 integrin as discussed above. Vimentin also regulates Rac1 activity.46 Moreover, Rab7 interacts with Rac147 and vimentin,48 suggesting interconnected intricate regulation among these molecules, which enables Rac1 to regulate the integrin level in a spatiotemporally precise way by integrating vesicle trafficking and cytoskeleton reorganization.
Despite mounting supportive evidence of the relevance of integrins as targets in cancer therapy and encouraging preclinical data, clinical studies have failed, so far, to demonstrate therapeutic benefits of targeting integrins in patients with cancer. Nevertheless, integrins including β4 remain a valuable target for cancer therapy49 and await reconsiderations, improvements, and innovations in preclinical and clinical approaches, such as the choice of integrin species targeted, and biological processes targeted, and strategies in clinical trials.50 Survival analysis presented in this study suggested the potential to target β4 integrin and Rac1, which would improve outcomes in patients with cancer expressing both genes at high levels.
CONFLICT OF INTEREST
The authors have no conflict of interest to declare.
Supporting information
Fig S1‐4
Table S1‐3
Supplementary Material
ACKNOWLEDGMENTS
We thank Ms S. Ohya, Mr. S. Takagi, Ms M. Torii, Mr. R. Takahashi, Mr. T. Hasegawa, Ms M. Ueno, Ms S. Ito, Ms S. Ikegami, Ms K. Nagashima, and Ms S. Nakahara for contributing to this work as part of their bachelor's degrees. This work was supported in part by JSPS KAKENHI Grant Number 19K07390. The authors would also like to thank Enago (www.enago.jp) for the English language review.
Mori K, Higurashi M, Ishikawa F, Shibanuma M. Rac1‐mediated sustained β4 integrin level develops reattachment ability of breast cancer cells after anchorage loss. Cancer Sci. 2021;112:3205–3217. 10.1111/cas.14985
REFERENCES
- 1.Gupta GP, Massague J. Cancer metastasis: building a framework. Cell. 2006;127:679‐695. [DOI] [PubMed] [Google Scholar]
- 2.Strilic B, Offermanns S. Intravascular survival and extravasation of tumor cells. Cancer Cell. 2017;32:282‐293. [DOI] [PubMed] [Google Scholar]
- 3.Hamidi H, Ivaska J. Every step of the way: integrins in cancer progression and metastasis. Nat Rev Cancer. 2018;18:533‐548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wong CW, Lee A, Shientag L, et al. Apoptosis: an early event in metastatic inefficiency. Cancer Res. 2001;61:333‐338. [PubMed] [Google Scholar]
- 5.Simpson CD, Anyiwe K, Schimmer AD. Anoikis resistance and tumor metastasis. Cancer Lett. 2008;272:177‐185. [DOI] [PubMed] [Google Scholar]
- 6.Dai X, Cheng H, Bai Z, Li J. Breast cancer cell line classification and its relevance with breast tumor subtyping. J Cancer. 2017;8:3131‐3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ishikawa F, Ushida K, Mori K, Shibanuma M. Loss of anchorage primarily induces non‐apoptotic cell death in a human mammary epithelial cell line under atypical focal adhesion kinase signaling. Cell Death Dis. 2015;6(1):e1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rajendran V, Jain MV. In vitro tumorigenic assay: colony forming assay for cancer stem cells. Methods Mol Biol. 2018;1692:89‐95. [DOI] [PubMed] [Google Scholar]
- 9.Guo W, Giancotti FG. Integrin signalling during tumour progression. Nat Rev Mol Cell Biol. 2004;5:816‐826. [DOI] [PubMed] [Google Scholar]
- 10.Meng X, Cheng K, Krohkin O, et al. Evidence for the presence of a low‐mass beta1 integrin on the cell surface. J Cell Sci. 2005;118:4009‐4016. [DOI] [PubMed] [Google Scholar]
- 11.Suzuki K, Takahashi K. Reduced substratum adhesion and decreased expressions of 1 and 4 integrins in human breast cancer cells with a property of anchorage‐independent growth. Int J Oncol. 1999;14:897‐904. [DOI] [PubMed] [Google Scholar]
- 12.Carter WG, Kaur P, Gil SG, Gahr PJ, Wayner EA. Distinct functions for integrins alpha 3 beta 1 in focal adhesions and alpha 6 beta 4/bullous pemphigoid antigen in a new stable anchoring contact (SAC) of keratinocytes: relation to hemidesmosomes. J Cell Biol. 1990;111:3141‐3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sonnenberg A, Linders CJ, Daams JH, Kennel SJ. The alpha 6 beta 1 (VLA‐6) and alpha 6 beta 4 protein complexes: tissue distribution and biochemical properties. J Cell Sci. 1990;96(Pt 2):207‐217. [DOI] [PubMed] [Google Scholar]
- 14.Huck L, Pontier SM, Zuo DM, Muller WJ. beta1‐integrin is dispensable for the induction of ErbB2 mammary tumors but plays a critical role in the metastatic phase of tumor progression. Proc Natl Acad Sci USA. 2010;107:15559‐15564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haga RB, Ridley AJ. Rho GTPases: regulation and roles in cancer cell biology. Small GTPases. 2016;7:207‐221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cernuda‐Morollon E, Ridley AJ. Rho GTPases and leukocyte adhesion receptor expression and function in endothelial cells. Circ Res. 2006;98:757‐767. [DOI] [PubMed] [Google Scholar]
- 17.Mohri T, Adachi Y, Ikehara S, Hioki K, Tokunaga R, Taketani S. Activated Rac1 selectively up‐regulates the expression of integrin alpha6beta4 and induces cell adhesion and membrane ruffles of nonadherent colon cancer Colo201 cells. Exp Cell Res. 1999;253:533‐540. [DOI] [PubMed] [Google Scholar]
- 18.De Franceschi N, Hamidi H, Alanko J, Sahgal P, Ivaska J. Integrin traffic ‐ the update. J Cell Sci. 2015;128:839‐852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alessandrini F, Pezze L, Ciribilli Y. LAMPs: shedding light on cancer biology. Semin Oncol. 2017;44:239‐253. [DOI] [PubMed] [Google Scholar]
- 20.Duleh SN, Welch MD. Regulation of integrin trafficking, cell adhesion, and cell migration by WASH and the Arp2/3 complex. Cytoskeleton. 2012;69:1047‐1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zech T, Calaminus SD, Caswell P, et al. The Arp2/3 activator WASH regulates alpha5beta1‐integrin‐mediated invasive migration. J Cell Sci. 2011;124:3753‐3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frasa MA, Maximiano FC, Smolarczyk K, et al. Armus is a Rac1 effector that inactivates Rab7 and regulates E‐cadherin degradation. Curr Biol. 2010;20:198‐208. [DOI] [PubMed] [Google Scholar]
- 23.Carroll B, Mohd‐Naim N, Maximiano F, et al. The TBC/RabGAP Armus coordinates Rac1 and Rab7 functions during autophagy. Dev Cell. 2013;25:15‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pereira B, Chin SF, Rueda OM, et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun. 2016;7:11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Curtis C, Shah SP, Chin SF, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346‐352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Etienne‐Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629‐635. [DOI] [PubMed] [Google Scholar]
- 27.Wertheimer E, Gutierrez‐Uzquiza A, Rosemblit C, Lopez‐Haber C, Sosa MS, Kazanietz MG. Rac signaling in breast cancer: a tale of GEFs and GAPs. Cell Signal. 2012;24:353‐362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Falcioni R, Kennel SJ, Giacomini P, Zupi G, Sacchi A. Expression of tumor antigen correlated with metastatic potential of Lewis lung carcinoma and B16 melanoma clones in mice. Cancer Res. 1986;46:5772‐5778. [PubMed] [Google Scholar]
- 29.Kennel SJ, Foote LJ, Lankford PK. Analysis of surface proteins of mouse lung carcinomas using monoclonal antibodies. Cancer Res. 1981;41:3465‐3470. [PubMed] [Google Scholar]
- 30.Laferriere J, Houle F, Huot J. Adhesion of HT‐29 colon carcinoma cells to endothelial cells requires sequential events involving E‐selectin and integrin beta4. Clin Exp Metastasis. 2004;21:257‐264. [DOI] [PubMed] [Google Scholar]
- 31.Abdel‐Ghany M, Cheng HC, Elble RC, Pauli BU. The breast cancer beta 4 integrin and endothelial human CLCA2 mediate lung metastasis. J Biol Chem. 2001;276:25438‐25446. [DOI] [PubMed] [Google Scholar]
- 32.Stewart RL, O'Connor KL. Clinical significance of the integrin alpha6beta4 in human malignancies. Lab Invest. 2015;95:976‐986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bon G, Folgiero V, Di Carlo S, Sacchi A, Falcioni R. Involvement of alpha6beta4 integrin in the mechanisms that regulate breast cancer progression. Breast Cancer Res. 2007;9:203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rezniczek GA, de Pereda JM , Reipert S, Wiche G. Linking integrin alpha6beta4‐based cell adhesion to the intermediate filament cytoskeleton: direct interaction between the beta4 subunit and plectin at multiple molecular sites. J Cell Biol. 1998;141:209‐225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dans M, Gagnoux‐Palacios L, Blaikie P, Klein S, Mariotti A, Giancotti FG. Tyrosine phosphorylation of the beta 4 integrin cytoplasmic domain mediates Shc signaling to extracellular signal‐regulated kinase and antagonizes formation of hemidesmosomes. J Biol Chem. 2001;276:1494‐1502. [DOI] [PubMed] [Google Scholar]
- 36.Shaw LM, Rabinovitz I, Wang HH, Toker A, Mercurio AM. Activation of phosphoinositide 3‐OH kinase by the alpha6beta4 integrin promotes carcinoma invasion. Cell. 1997;91:949‐960. [DOI] [PubMed] [Google Scholar]
- 37.Yoon SO, Shin S, Lipscomb EA. A novel mechanism for integrin‐mediated ras activation in breast carcinoma cells: the alpha6beta4 integrin regulates ErbB2 translation and transactivates epidermal growth factor receptor/ErbB2 signaling. Cancer Res. 2006;66:2732‐2739. [DOI] [PubMed] [Google Scholar]
- 38.Cruz‐Monserrate Z, O'Connor KL. Integrin alpha 6 beta 4 promotes migration, invasion through Tiam1 upregulation, and subsequent Rac activation. Neoplasia. 2008;10:408‐417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.O'Connor KL, Chen M, Towers LN. Integrin alpha6beta4 cooperates with LPA signaling to stimulate Rac through AKAP‐Lbc‐mediated RhoA activation. Am J Physiol Cell Physiol. 2012;302:C605‐614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ridley AJ. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol. 2006;16:522‐529. [DOI] [PubMed] [Google Scholar]
- 41.Homan SM, Martinez R, Benware A, LaFlamme SE. Regulation of the association of alpha 6 beta 4 with vimentin intermediate filaments in endothelial cells. Exp Cell Res. 2002;281:107‐114. [DOI] [PubMed] [Google Scholar]
- 42.Dmello C, Sawant S, Alam H, et al. Vimentin‐mediated regulation of cell motility through modulation of beta4 integrin protein levels in oral tumor derived cells. Int J Biochem Cell Biol. 2016;70:161‐172. [DOI] [PubMed] [Google Scholar]
- 43.Guerra F, Bucci C. Multiple roles of the small GTPase Rab7. Cells. 2016;5(3):34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vitelli R, Santillo M, Lattero D, et al. Role of the small GTPase Rab7 in the late endocytic pathway. J Biol Chem. 1997;272:4391‐4397. [DOI] [PubMed] [Google Scholar]
- 45.Palacios F, Tushir JS, Fujita Y, D'Souza‐Schorey C. Lysosomal targeting of E‐cadherin: a unique mechanism for the down‐regulation of cell‐cell adhesion during epithelial to mesenchymal transitions. Mol Cell Biol. 2005;25:389‐402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Colburn ZT, Jones JCR. Complexes of alpha6beta4 integrin and vimentin act as signaling hubs to regulate epithelial cell migration. J Cell Sci. 2018;131:jcs.214593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sun Y, Buki KG, Ettala O, Vaaraniemi JP, Vaananen HK. Possible role of direct Rac1‐Rab7 interaction in ruffled border formation of osteoclasts. J Biol Chem. 2005;280:32356‐32361. [DOI] [PubMed] [Google Scholar]
- 48.Cogli L, Progida C, Bramato R, Bucci C. Vimentin phosphorylation and assembly are regulated by the small GTPase Rab7a. Biochim Biophys Acta. 2013;1833:1283‐1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ruan S, Lin M, Zhu Y, et al. Integrin beta4‐targeted cancer immunotherapies inhibit tumor growth and decrease metastasis. Cancer Res. 2020;80:771‐783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alday‐Parejo B, Stupp R, Ruegg C. Are integrins still practicable targets for anti‐cancer therapy? Cancers. 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐4
Table S1‐3
Supplementary Material