

SUMMARY
FXR agonists are used to treat non-alcoholic fatty liver disease (NAFLD), in part because they reduce hepatic lipids. Here, we show that FXR activation with the FXR agonist GSK2324 controls hepatic lipids via reduced absorption and selective decreases in fatty acid synthesis. Using comprehensive lipidomic analyses, we show that FXR activation in mice or humans specifically reduces hepatic levels of mono- and polyunsaturated fatty acids (MUFA and PUFA). Decreases in MUFA are due to FXR-dependent repression of Scd1, Dgat2, and Lpin1 expression, which is independent of SHP and SREBP1c. FXR-dependent decreases in PUFAs are mediated by decreases in lipid absorption. Replenishing bile acids in the diet prevented decreased lipid absorption in GSK2324-treated mice, suggesting that FXR reduces absorption via decreased bile acids. We used tissue-specific FXR KO mice to show that hepatic FXR controls lipogenic genes, whereas intestinal FXR controls lipid absorption. Together, our studies establish two distinct pathways by which FXR regulates hepatic lipids.
In brief
The nuclear receptor FXR lowers hepatic triglycerides to protect against the onset of NAFLD. Clifford et al. demonstrate that activation of FXR decreases hepatic triglycerides through two distinct mechanisms. First, via bile-acid-dependent decreases in intestinal lipid absorption and second, through selective changes in lipogenesis.
Graphical Abstract
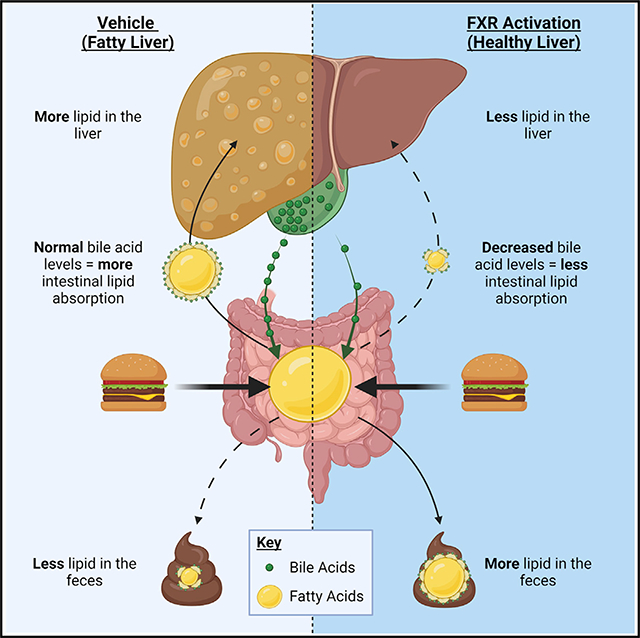
INTRODUCTION
The liver is the nexus for many metabolic pathways, including those that regulate bile acids, fatty acids, cholesterol, and glucose. In healthy humans, metabolized nutrients are distributed to various tissues without long-term accumulation in the liver. In disease states, however, metabolites may accrue in the liver, impairing hepatic function (Alves-Bezerra and Cohen, 2017; Birkenfeld and Shulman, 2014). Owing to a drastic increase in obesity, dysregulation of hepatic lipid metabolism is commonplace and non-alcoholic fatty liver disease (NAFLD) has become the most prevalent liver disease globally (Targher et al., 2018; Younossi et al., 2018). NAFLD comprises a spectrum of liver disorders with the initial accumulation of ectopic hepatic triglycerides (TAGs) progressing to the more serious non-alcoholic steatohepatitis (NASH) in a sub-group of individuals. Patients with NASH exhibit not only elevated hepatic TAG levels, but also fibrosis, hepatocellular ballooning, and lobular inflammation, which may progress further to cirrhosis or liver failure (Cohen et al., 2011; Kleiner and Makhlouf, 2016). Currently, there are limited therapeutic options available to patients diagnosed with NALFD/NASH, but agonists of the nuclear receptor farnesoid X receptor (FXR) have shown considerable promise. FXR agonists, including obeticholic acid (OCA), a synthetic bile acid conjugate, as well as non-steroidal agonists, are currently in clinical trials as potential treatments for NAFLD/NASH (Schumacher and Guo, 2019; Shah and Kowdley, 2020).
FXR is endogenously activated by bile acids and is highly expressed in both hepatocytes and intestinal enterocytes where it controls all aspects of bile acid metabolism including synthesis, export into the bile ducts, and resorption from the intestine (Dawson and Karpen, 2015). Classically, FXR controls the inhibitory arm of the bile acid synthesis pathway whereby elevated bile acid levels activate FXR leading to repression of hepatic genes involved in bile acid synthesis in a negative feedback loop (de Aguiar Vallim et al., 2013a).
Studies in mice and humans have shown that activation of FXR results in a number of clinically relevant changes such as reduced levels of hepatic and plasma TAGs, reduced inflammation, increased insulin sensitivity, and protection from certain toxic drugs (Ahmad and Haeusler, 2019). The mechanism by which FXR is proposed to reduce liver TAG is through reduced hepatic de novo lipogenesis via an FXR-SHP-SREBP1c pathway (Watanabe et al., 2004). More recent paradoxical studies have suggested that specifically activating (Fang et al., 2015) or repressing (Gonzalez et al., 2016; Jiang et al., 2015) FXR, particularly intestinal FXR, is key to the beneficial reduction in hepatic TAG levels.
In this study, we utilize GSK2324, a potent and specific synthetic FXR agonist (de Aguiar Vallim et al., 2015; Bass et al., 2011; Tarling et al., 2017) together with multiple knockout mouse models to identify two distinct pathways that FXR utilizes to reduce hepatic TAG. We show that FXR activation selectively alters lipogenesis by repressing only three of the 24 genes involved in TAG synthesis and that these decreases are independent of SHP and SREBP1c. We use three independent absorption assays to demonstrate that GSK2324 treatment of mice reduces lipid absorption in both low- and high-fat-fed animals. By feeding animals a diet containing bile acids, we show that the FXR-dependent decrease in lipid absorption can be abolished when bile acids are replenished. Lastly, we utilized mice with tissue-specific loss of FXR to demonstrate that hepatic FXR controls the GSK2324-dependent decreases in lipogenic genes, whereas intestinal FXR largely controls changes in lipid absorption. Together, our data present a new understanding of how FXR activation reduces hepatic TAG levels.
RESULTS
Humans and mice treated with FXR agonists have selective reductions in hepatic TAGs containing MUFAs and PUFAs
To study how FXR activation alters hepatic TAG levels, we obtained liver biopsies from human patients awaiting gallstone surgery (n = 11) (Al-Dury et al., 2019). Patients were randomized and treated with either placebo (n = 6) or the FXR agonist OCA (n = 5) for 3 weeks (Figure 1A). Hepatic FXR activation was confirmed in patients receiving OCA by induction of the FXR target gene BSEP and a decrease in the bile acid synthetic enzyme CYP7A1 (Figures S1A and S1B). Serum and gall bladder bile acid composition of the same OCA-treated patients was previously shown to be altered as compared with placebo-treated controls (Al-Dury et al., 2019), consistent with FXR activation. We next used state-of-the-art lipidomics to measure 13 lipid classes and over 700 individual lipid species. Analysis of lipid classes revealed that FXR activation specifically reduced hepatic TAG, which was the most abundant lipid class in these livers but did not affect other lipid classes (Figure 1B). Detailed analysis of individual TAG species showed that OCA treatment decreased most of the highly abundant TAG species, which were predominantly TAG containing fatty acids with one double bond (MUFAs; Figure 1C), and TAG containing two or more double bonds (PUFAs) to different extents, suggesting FXR agonism affects TAG metabolism in complex ways (Figure 1C).
Figure 1. Humans and mice treated with FXR agonists have selective reductions in hepatic TAG containing MUFAs and PUFAs.

(A) Experiment schematic of double-blinded placebo-controlled randomized trial for obeticholic acid (OCA) in human patients.
(B and C) Lipidomic analyses of (B) hepatic lipid classes (nmol/mg liver) and (C) hepatic TAG species (nmol/mg liver) in human patients treated as in (A).
(D) Experiment schematic: wild-type or Fxr−/− mice (n = 11–12/group) fed a standard rodent diet were treated for 3 days with vehicle or FXR agonist GSK2324.
(E) Hepatic mRNA expression of FXR target genes Shp and MafG in mice from (D).
(F) Total hepatic TAG in mice from D.
(G and H) Lipidomic analyses of (G) hepatic lipid classes (nmol/mg liver) and (H) hepatic TAG species (nmol/mg liver) in mice from (D).
(I and J) Bar graphs of individual TAG species containing (I) monounsaturated and (J) polyunsaturated fatty acids in livers of mice from (D).
Data are represented as mean ± SEM with individual animals noted as dots. *p < 0.05, **p < 0.01, ***p < 0.001. See also Figure S1.
To interrogate the molecular mechanism underlying the FXR-mediated decreases in hepatic TAG, we treated wild-type and Fxr−/− mice with the synthetic, water-soluble, and specific FXR agonist GSK2324 (Bass et al., 2011) (Figure 1D). FXR activation increased the expression of two well-characterized FXR target genes Shp and MafG (de Aguiar Vallim et al., 2015; Goodwin et al., 2000) specifically in wild-type, but not in Fxr−/− mice (Figure 1E). As with our human data (Figure 1B), the most pronounced effect of FXR activation on liver lipids was decreased hepatic TAG levels in wild-type, but not Fxr−/− mice (Figures 1F and 1G). In addition to TAG, FXR activation only significantly reduced the levels of phosphatidylethanolamine (PE; Figure 1G) and did not alter hepatic cholesterol (Figure S1C). Closer analysis of distinct TAG species showed that most of the highly abundant TAG species were decreased in wild type, but not Fxr−/− mice treated with GSK2324 (Figure 1H). FXR activation reduced the levels of MUFA-containing (Figure 1I) and PUFA-containing TAGs (Figure 1J), In contrast, TAG species containing only saturated fatty acids were unaltered following GSK2324 treatment (Figure S1D). Taken together, these analyses demonstrate that FXR activation selectively alters specific lipid species, primarily those containing fatty acids with one or more double bonds.
Treatment with GSK2324 selectively reduces the expression of fatty acid and triglyceride synthesis genes
Having determined that FXR activation selectively reduces MUFA- and PUFA-containing hepatic TAGs in humans and mice, we next sought to establish whether genes involved in TAG or fatty acid production were regulated in response to FXR activation. Hepatic de novo TAG synthesis involves the coordinated actions of several enzymes that synthesize fatty acids from two-carbon precursors (Alves-Bezerra and Cohen, 2017) and then conjugate these fatty acids to a glycerol backbone to generate TAGs containing various acyl chains (Takeuchi and Reue, 2009). The enzymes and metabolites of this complex pathway are shown in Figure 2A. We measured hepatic mRNA levels of genes encoding all 24 enzymes in both wild-type and Fxr−/− mice treated with either vehicle or GSK2324 (Figures 2B–2H). There were no significant reductions in mRNAs encoding enzymes of saturated fatty acid synthesis or fatty acid elongation in response to FXR activation (Figures 2B and 2C). Indeed, Fasn mRNA levels were increased (Figure 2B), consistent with an earlier report showing that Fasn is a direct FXR target gene (Matsukuma et al., 2006).
Figure 2. Treatment with GSK2324 selectively reduces the expression of fatty acid and triglyceride synthesis genes.

(A) Schematic of the hepatic de novo lipogenesis pathway, where genes are represented in yellow and metabolites in green.
(B–H) Hepatic mRNA expression for 24 genes encoding major enzymes in hepatic lipogenesis from livers of wild-type or Fxr−/− mice fed a standard rodent diet and treated with vehicle or GSK2324 with most significantly changing genes denoted with red asterisks in (A).
(I) Experimental schematic outlining de novo lipogenesis measurements using deuterium oxide (D2O) in mice treated for 3 days with FXR agonist GSK2324 (n = 8/group).
(J) Fatty acid methyl ester analysis for newly synthesized neutral lipids in livers of mice from (I).
Data are represented as mean ± SEM with individual animals noted as dots. *p < 0.05, **p < 0.01, ***p < 0.001. See also Figure S2.
Importantly, of the 24 mRNAs encoding enzymes involved in hepatic TAG synthesis (Figures 2B–2H), only three genes were reduced in an FXR-dependent fashion following treatment with GSK2324, Scd1 (Figure 2D), Lpin1 (Figure 2G), and Dgat2 (Figure 2H). These three mRNAs were not decreased following GSK2324 treatment of Fxr−/− mice (Figures 2D, 2G, and 2H), consistent with a requirement for FXR. Together, our data suggest that FXR activation does not promote a generalized decrease in the expression of fatty acid synthesis genes, but instead, results in selective changes in fatty acid and TAG metabolism. For example, FXR activation decreases Scd1 expression, the gene encoding the enzyme steroyl CoA desaturase (SCD1), which converts saturated fatty acids (mainly C16:0 and C18:0) to monounsaturated fatty acids (C16:1 and C18:1) (Flowers and Ntambi, 2008). Thus, the FXR-dependent decrease in MUFA-containing TAGs is likely due to decreased expression of SCD1.
We next determined whether FXR activation reduced de novo fatty acid synthesis. To measure lipogenesis, we added deuterium oxide (D2O) to the drinking water of mice, and then treated them with either GSK2324 or vehicle as we have done previously (Parker et al., 2019) (Figure 2I). Hepatic FXR activation was confirmed by induction of FXR target genes Shp and MafG (Figure S2A), and decreased expression of key lipogenic genes Srebp1c, Scd1, Dgat2, and Lpin1 (Figure S2B). To determine which fatty acids were newly synthesized, we extracted neutral lipids to deplete most of the phospholipid classes but retain most of the TAGs, DAGs, CEs and FFAs. The synthesis of MUFAs (C16:1 and C18:1) was dramatically decreased following GSK2324 treatment (Figure 2J), whereas there were only modest changes in the levels of newly synthesized saturated fatty acids (Figure 2J). Notably, the total levels of saturated fatty acids in the neutral lipid fraction were largely unaffected, whereas total MUFA levels were significantly decreased (Figure S2C). Based on these studies, we conclude that FXR activation results in a preferential decrease in the synthesis of MUFA that are being incorporated into hepatic TAGs, leading to the observed changes in TAG-fatty acid composition.
FXR activation reduces hepatic TAG levels independently of SHP and SREBP1c
It has been proposed that FXR regulates hepatic TAGs via an SHP-dependent reduction in SREBP1c (Watanabe et al., 2004), the master transcriptional activator of numerous lipogenic genes (Horton et al., 2002). To determine if the FXR-dependent reductions in TAG were elicited through the transcriptional repressor SHP, we treated wild-type and Shp−/− mice with GSK2324 for 3 days (Figure 3A). FXR activation induced the FXR target gene MafG in wild-type and Shp−/− mice, whereas Shp was induced in wild-type mice but, as expected, was undetectable in Shp−/− mice (Figure 3B). GSK2324 treatment of both wild-type and Shp−/− mice also repressed hepatic levels of Srebp1c, Scd1, Lipin1, and Dgat2 (Figure 3C), indicating that the repression of these genes was independent of SHP. Further, treatment with GSK2324 decreased hepatic TAG in both wild-type and Shp−/− mice equivalently, suggesting SHP is not required for FXR-mediated decreases in hepatic TAGs (Figure 3D), including TAGs containing MUFAs and PUFAs (Figures 3E–3G). Thus, we concluded that the FXR-dependent regulation of hepatic TAG was largely independent of SHP.
Figure 3. FXR activation reduces hepatic TAG levels independently of SHP and SREBP1c.

(A and H) Experiment schematics: wild-type mice and (A) Shp−/− or (H) Srebp1c−/− mice (n = 9–12/group) fed a standard rodent diet were treated for 3 days with vehicle or FXR agonist GSK2324.
(B and I) Hepatic mRNA expression of FXR target genes Shp and MafG in wild-type and (B) Shp−/− or (H) Srebp1c−/− mice.
(C and J) Hepatic mRNA expression of select lipogenic genes in wild-type and (C) Shp−/− or (J) Srebp1c−/− mice.
(D and K) Hepatic lipid classes (nmol/mg liver) in wild-type and (D) Shp−/− or (K) Srebp1c−/− mice.
(E and L) Hepatic TAG species (nmol/mg liver) in wild-type and (E) Shp−/− or (L) Srebp1c−/− mice.
(F, G, M, and N) Individual TAG species containing MUFA and PUFA acyl tails in wild-type and (F and G) Shp−/− or (M and N) Srebp1c−/− mice.
Data are represented as mean ± SEM with individual animals noted as dots. *p < 0.05, **p < 0.01, ***p < 0.001. See also Figure S3.
Next, to address whether FXR activation requires SREBP1c to mediate the changes in hepatic lipids, we treated wild-type and Srebp1c−/− mice with GSK2324 (Figure 3H). The induction of FXR target genes Shp and MafG was identical in both wild-type and Srebp1c−/− mice (Figure 3I), demonstrating robust FXR activation. Hepatic Scd1, Lpin1, and Dgat2 were still decreased in Srebp1c−/− mice treated with GSK2324 (Figure 3J), suggesting the mechanism by which FXR reduces the expression of these genes is also independent of SREBP1c. Lipidomic analysis confirmed decreased hepatic TAG in wild-type mice treated with GSK2324 (Figure 3K), which was especially evident in MUFA- and PUFA-containing TAGs (Figures 3L–3N). Basal hepatic TAG levels in Srebp1c−/− mice were significantly lower than in wild-type littermates (Figures 3K and S3A), likely due to a lifelong deficit in lipogenesis (Horton et al., 2002). Notably, however, treatment with GSK2324 still significantly reduced total hepatic TAG and numerous TAG species in these animals despite significantly lower basal hepatic lipid levels (Figures 3L, 3N, and S3A–S3D), demonstrating that SREBP1c is not required for FXR-mediated reductions in hepatic TAG.
To dissect the molecular mechanism by which FXR activation reduces the expression of Scd1, Lpin1, and Dgat2, we considered direct repression, where FXR may bind to the promoters of these genes and block their transcription by competing with the transcriptional activator PPARα (Lee et al., 2014). To test this hypothesis, we treated wild-type and Pparα−/− mice with GSK2324 (Figure S3E) and confirmed FXR activation through induction of FXR target genes (Figure S3F). In both wild-type and Pparα−/− mice hepatic TAG and TAG species were significantly decreased in response to GSK2324 treatment (Figures S3G–S3I), as was Scd1, Lpin1, and Dgat2 expression (Figure S3J). These data suggest that direct repression by FXR via competition with PPARα at response elements in the three repressed genes is unlikely to be the mechanism for the FXR-dependent repression of specific lipogenic genes. Further, ChIP-seq analysis (Thomas et al., 2010) did not identify proximal FXR-binding sites in these genes compared with known FXR targets Shp (Nr0b2) and Fasn (Figures S3K–S3O). Together, these data show that FXR activation via GSK2324 reduces hepatic TAGs independently of SHP, SREBP1c, and PPARα, suggesting that FXR reduces the expression of these genes via an unknown mechanism.
FXR activation decreases intestinal lipid absorption
In addition to significant decreases in MUFAs, our lipidomic analysis of mouse livers treated with GSK2324 also showed consistent reductions in PUFA-containing TAGs, such as those containing C18:2 (linoleic acid) fatty acyl chains (Figures 1F, 3E, and 3L). Moreover, GC-MS analysis of hepatic fatty acids showed significant reduction in total C18:2 levels (Figure 4A). Linoleic acid is an essential lipid, which cannot be synthesized in mammalian cells and is exclusively acquired from the diet (Burr and Burr, 1930; Spector and Kim, 2015). We therefore hypothesized that FXR activation may decrease absorption of dietary lipids in the intestine.
Figure 4. FXR activation decreases intestinal lipid absorption.

(A) Total hepatic C18:2 in wild-type mice treated with vehicle or GSK2324.
(B, E, and H) Experiment schematics for three different lipid absorption methods. Wild-type mice fed a standard rodent diet were treated with either vehicle or GSK2324 and (B) gavaged with 14C triolein, (E) gavaged with fatty acid conjugated to BODIPY fluorophore, or (H) fed a diet containing non-absorbable lipid standard sucrose polybehenate (C22:0, n = 10/group).
(C and D) Total plasma TAG (C) and plasma radioactive counts (D) from 14C triolein from mice as in (B).
(F and G) (F) Representative images and (G) fluorescence quantification from intestinal sections of mice treated as in (E).
(I) Fatty acid methyl ester analysis of fecal lipids normalized to C22:0 from mice treated as in (H).
Data are represented as mean ± SEM with individual animals noted as dots. **p < 0.01, ***p < 0.001. See also Figure S4.
To investigate the effect of FXR activation on lipid absorption, we utilized three separate absorption assays. First, we treated wild-type mice for 3 days with either vehicle or GSK2324 and then gavaged them with a bolus of olive oil spiked with radiolabeled 14C-triolein (Figure 4B). FXR activation not only lowered plasma total TAG levels after the bolus of oil (Figure 4C) but also lowered the levels of 14C fatty acids in plasma (Figure 4D), consistent with a decrease in lipid absorption.
Second, we treated mice with either vehicle or GSK2324 as before but instead gavaged the mice with a bolus of olive oil containing C12-BODIPY fluorescent fatty acid (Figure 4E). By imaging the intestinal villi, quantification of mean fluorescence intensity revealed reduced signal in the jejunum of mice treated with GSK2324 compared with controls (Figures 4F, 4G, and S4A).
Lastly, we synthesized modified diets containing a non-absorbable lipid, sucrose polybehenate (SPB; C22:0/behenic acid conjugated to a sucrose backbone) (Moser and McLachlan, 1999) to act as an internal control for the measurement of fecal fatty acids (Jandacek et al., 2004). To determine whether GSK2324 altered fatty acid absorption, we individually housed wild-type mice and fed them a diet containing SPB (Figure S4B) for 1 week. During the last 3 days of feeding, the mice were treated daily with either vehicle or GSK2324 (Figure 4H). GSK2324 treatment significantly increased fecal fatty acids (Figures 4I and S4C), consistent with reduced lipid absorption. As expected, fecal fatty acid levels were not different between the groups prior to GSK2324 treatment (Figures S4D–S4F), and the levels of the internal control C22:0 were not altered (Figures S4F and S4G). In summary, we show that FXR activation following GSK2324 treatment decreases the absorption of dietary fatty acids in the intestine.
Decreased intestinal lipid absorption requires FXR-dependent changes in bile acids
Absorption of dietary lipids in the intestine requires that TAGs are dispersed in micelles containing bile acids and phospholipids so that pancreatic-derived lipases can hydrolyze them to release fatty acids and monoacylglycerides (MAGs). These fatty acids and MAGs then enter enterocytes where TAGs are resynthesized and secreted into the lymph via chylomicrons (Hofmann and Borgstroem, 1964; Ko et al., 2020). Bile acids are detergents and play a key role in the formation of micelles in the intestinal lumen. Since FXR is the master regulator of bile acid metabolism (de Aguiar Vallim et al., 2013a, 2013b), we hypothesized that FXR activation may affect intestinal lipid absorption through a reduction in bile acid levels and/or a shift in bile acid composition.
First, we demonstrated that the GSK2324-dependent decreases in absorption required FXR after treatment of wild-type and Fxr−/− mice with GSK2324 and fecal lipid analysis (Figures 5A and 5B). To investigate how GSK2324 treatment alters the bile acid pool, we isolated liver, intestine, and gallbladder, the major organs that contain bile acids (Hofmann, 2009; Rizzolo et al., 2019) to quantify different bile acid species by UPLC-MS/MS (Figures 5C, 5D, and S5A–S5E). FXR activation with GSK2324 significantly reduced total biliary (Figure 5C) and liver (Figure S5A) bile acids in wild-type mice but had no effect on intestinal bile acid levels (Figure S5C). In contrast, no decrease in bile acid levels was noted following treatment of Fxr−/− mice with GSK2324 (Figures 5C, S5A, and S5C). Notably, treatment with GSK2324 altered the bile acid composition irrespective of compartment, with specific reductions in taurocholic acid (TCA) and cholic acid (CA) in wild-type, but not Fxr−/− mice in gallbladder (Figure 5D), liver (Figure S5B), and intestine (Figure S5D). These changes were specific, as other abundant bile acid species, such as muricholic acids (MCAs), were unaffected by GSK2324 treatment (Figures 5D, S5B, and S5D). We propose that biliary bile acids are most indicative of the absorptive properties of bile acid pool, as biliary bile acids are the first detergents to mix with dietary lipids to facilitate absorption (Dawson and Karpen, 2015). Thus, our data suggest that GSK2324 decreases intestinal lipid absorption in an FXR-dependent manner, and this is likely the result of changes in bile acid levels and/or composition.
Figure 5. Decreased intestinal lipid absorption requires FXR-dependent changes in bile acids.

(A) Experiment schematic: wild-type or Fxr−/− mice (n = 11–12/group) fed a standard rodent diet were treated for 3 days with vehicle or FXR agonist GSK2324.
(B) Fecal fatty acids in mice from (A).
(C and D) Total biliary bile acid concentration (C) and biliary bile acid composition (D) in mice from (A).
(E) Experiment schematic: wild-type mice fed either a standard rodent diet or a diet supplemented with cholic acid were treated with vehicle or GSK2324 for3 days (n = 10/group).
(F) Hepatic mRNA expression of Shp and MafG in mice from (E).
(G–J) Total biliary bile acid concentration (G) and biliary bile acid composition (H) in mice from (E). Lipidomic analyses of (I) total hepatic TAG and (J) individual hepatic TAG species (nmol/mg liver) in mice from (E).
(K) Fecal fatty acids in mice from (E).
Data are represented as mean ± SEM with individual animals noted as dots. *p < 0.05, ***p < 0.001. See also Figure S5.
The FXR-dependent changes in bile acids suggest that changes in CA and its derivatives likely play a role in FXR-mediated changes in intestinal lipid absorption (Figures 5A–5D). To test this hypothesis, we performed a rescue experiment to establish if replenishing the bile acid pool would prevent changes in lipid absorption. We fed wild-type mice either a standard diet or a diet supplemented with 0.5% CA for 4 days and then treated each group with either vehicle or GSK2324 for the final three days of the feeding period (Figure 5E). As expected, the CA diet alone increased the expression of hepatic FXR target genes (Figure 5F). However, GSK2324 treatment still resulted in robust induction of FXR target genes Shp and MafG (Figure 5F). GSK2324 significantly decreased total biliary bile acids (and specifically decreased CA bile acids) in mice fed the standard diet, but not in mice fed the CA-containing diet (Figures 5G and 5H). Feeding with 0.5% CA was sufficient to restore the contracted bile acid pool size observed in standard-diet-fed mice as has been shown before (Jones et al., 2012). While GSK2324 did not significantly reduce total amounts of bile acid in standard-diet- or CA-diet-fed mice, the FXR agonists only decreased bile acid pool CA levels in control-fed mice, and this effect was completely ablated by CA feeding (Figures S5F–S5J).
We then measured hepatic TAG using lipidomics and showed that while FXR activation in standard-diet-fed mice decreased both total TAG (Figure 5I) and numerous TAG species (Figure 5J), GSK2324 treatment following CA supplementation in fact resulted in an increase in hepatic TAG (Figures 5I and 5J). Expression of key lipogenic genes Srebp1c, Scd1, Lpin1, and Dgat2 were significantly reduced following GSK2324 treatment in both standard- and CA-fed mice (Figure S5K). Importantly, we show that CA feeding abolished the increase in fecal fatty acids seen after treatment with GSK2324 (Figure 5K). These data demonstrate that FXR activation decreases intestinal lipid absorption and that this decrease in absorption can be rescued with bile acid supplementation, suggesting that the FXR-dependent decreases in lipid absorption are predominantly mediated by changes in the composition of the bile acid pool.
In an NAFLD model, FXR activation dramatically decreases hepatic TAG and intestinal lipid absorption
The standard rodent diet contains relatively little dietary fat, and mice fed this diet typically do not develop fatty liver disease. In contrast, when rodents are fed a diet rich in fat (Western-type diet; WD), they accumulate significantly more TAGs in the liver, reminiscent of NAFLD (Zhang et al., 2020). To better understand the role of FXR in controlling elevated hepatic lipid levels, we fed wild-type and Fxr−/− mice WD for 8 weeks to promote hepatic lipid accumulation. We then treated these mice with either vehicle or GSK2324 for the final 3 days of the experiment (Figure 6A). GSK2324 treatment induced FXR target genes Shp and MafG in an FXR-dependent manner (Figure 6B). WD feeding shifted the hepatic lipidome such that TAG became the predominant type of lipid in the liver (Figure 6C). However, GSK2324 treatment significantly reduced the large amount of hepatic TAG in wild-type, but not Fxr−/− mice (Figure 6C). Fxr−/− mice were protected from diet-induced NAFLD when fed WD, likely a consequence of reduced body weight compared with wild-type mice (Figure S6A), which has been previously reported (Zhang et al., 2010, 2012). There were no signs of liver toxicity as plasma ALT and AST levels were not altered following GSK2324 treatment (Figure S6B). In WD-fed mice, GSK2324 treatment decreased numerous TAG species irrespective of saturation of the acyl tails (Figure 6D). To determine whether GSK2324 altered the bile acid pool in WD-fed mice, we measured bile acids in gall bladder, liver, and intestine by UPLC-MS/MS. The total bile acid pool was significantly decreased in wild-type, but not Fxr−/− mice (Figure 6E), in contrast to animals fed the standard rodent diet where the pool was not significantly altered (Figure S5E). In WD-fed mice, GSK2324 treatment in wild-type, but not Fxr−/−, mice significantly decreased the amount of TCA in the bile acid pool (Figure S6C), whereas T-βMCA levels were unchanged (Figure S6D).
Figure 6. In an NAFLD model, FXR activation dramatically decreases hepatic TAG and intestinal lipid absorption.

(A) Experiment schematic of diet-induced NAFLD model. Wild-type or Fxr−/− mice fed WD for 8 weeks were treated for 3 days with vehicle or FXR agonist GSK2324 (n = 8–10/group).
(B) Hepatic mRNA expression of FXR target genes Shp and MafG in mice from (A).
(C and D) Lipidomic analyses of (C) hepatic lipid classes (nmol/mg liver) and (D) hepatic TAG species (nmol/mg liver) in mice from (A).
(E) Total bile acid pool amount from mice in (A).
(F and J) Experiment schematic for absorption methods. WD-fed wild-type mice were treated with either vehicle or GSK2324 and (F) gavaged with 14C triolein or with fatty acid conjugated to BODIPY fluorophore, or (J) fed a diet containing non-absorbable lipid standard sucrose polybehenate (C22:0, n = 10/group).
(G and H) Representative images (G) and fluorescence quantification (H) from intestinal sections of BODIPY-gavaged mice treated as in (F).
(I) Liver radioactive counts from 14C triolein-gavaged mice as in (F).
(K) Fecal fatty acids after treatment with either vehicle or GSK2324 in mice as in (J).
Data are represented as mean ± SEM with individual animals noted as dots. **p < 0.01, ***p < 0.001. See also Figure S6.
FXR activation resulted in more pronounced decreases in hepatic TAGs after WD feeding, suggesting that changes in lipid absorption may have been enhanced. We therefore repeated our three absorption assays in wild-type mice fed WD for 8 weeks. First, WD-fed mice were gavaged with FA-BODIPY (Figure 6F). Fluorescent signal in the intestinal villi of the jejunum in GSK2324-treated mice was markedly reduced compared with vehicle-treated animals (Figures 6G, 6H, and S6E). In mice gavaged with radiolabeled 14C triolein (Figure 6F) we observed a significant decrease in 14C label in the liver (Figure 6I). Finally, we also fed wild-type mice WD for 7 weeks, and in the final week fed a custom WD containing sucrose polybehenate (Figures 6J and S6F) prior to measuring fecal fatty acids. GSK2324 treatment significantly increased fecal fatty acids (Figures 6K and S6G), consistent with decreases in intestinal lipid absorption. Fecal lipids, including the C22:0 internal standard, were unaltered before GSK2324 treatment (Figures S6H–S6K). Together, our data demonstrate that in WD-fed mice, FXR activation results in pronounced decreases in hepatic TAG and intestinal lipid absorption.
Intestinal FXR mediates changes in lipid absorption, whereas hepatic FXR controls lipid synthesis
To determine whether the FXR-dependent reduction in hepatic lipids required SHP in a NAFLD model, we repeated the WD-feeding experiments with GSK2324 treatment in Shp−/− mice (Figures 7A and S7A–S7E). WD-fed Shp−/− mice had reduced hepatic TAGs, indicating these animals are protected from diet-induced NAFLD (Figure 7B). However, consistent with our findings in low-fat-fed animals, SHP was not required for the GSK2324-dependent reduction in hepatic TAGs (Figure 7B). Similarly, intestinal lipid absorption (Figure S7B) and total bile acids in the pool (Figure S7C) were decreased in both wild-type and Shp−/− mice treated with GSK2324. Importantly, the repression of lipogenic genes by FXR activation was not altered by loss of SHP (Figure S7D), demonstrating the regulation of these genes by FXR in a NAFLD model is independent of SHP. Plasma ALT and AST levels were not significantly elevated irrespective of genotype or treatment (Figure S7E). Next, we repeated the WD-feeding experiment in Srebp1c−/− mice (Figure 7C) and showed that FXR activation in wild-type and Srebp1c−/− mice (Figure S7F) significantly reduced hepatic TAGs in both wild-type and Srebp1c−/− mice (Figure 7D), despite lower lipid levels in Srebp1c−/− mice. The repression of key lipogenic genes after FXR activation was equivalent in both wild-type and Srebp1c−/− mice (Figure S7G).
Figure 7. Intestinal FXR mediates changes in lipid absorption, whereas hepatic FXR controls lipid synthesis.

(A, C, and E) Experiment schematics: wild-type or (A) Shp−/− mice, (C) Srebp1c−/− mice, or (E) Fxrfl/fl, FxrLiv-KO, and FxrInt-KO mice were fed a Western diet for 8 weeks and treated with either vehicle or GSK2324.
(B and D) Lipidomic analysis of hepatic lipid classes in WD-fed (B) Shp−/− mice and (D) Srebp1c−/− mice.
(F) Hepatic mRNA expression of FXR target gene Shp in mice from (E).
(G) Ileal mRNA expression of FXR target gene FGF15 in mice from (E).
(H) Lipidomic analysis of hepatic lipid classes in mice from (E).
(I) Hepatic mRNA expression of select lipogenic genes in mice from (E).
(J) Fecal fatty acids after treatment with either vehicle or GSK2324 in mice as in (E).
(K) Total bile acid pool amount in mice from (E).
Data are represented as mean ± SEM with individual animals noted as dots. *p < 0.05, **p < 0.01, ***p < 0.001. See also Figure S7.
Lastly, we wanted to determine whether hepatic or intestinal FXR mediated the GSK2324-dependent changes in hepatic lipids and absorption. We generated cohorts of mice where FXR was absent in hepatocytes (Fxrfl/fl bred to an albumin-Cre; FxrLiv-KO) or enterocytes (Fxrfl/fl bred to a villin-Cre; FxrInt-KO). We then fed these animals WD for 8 weeks and treated them with GSK2324 for 3 days as before (Figure 7E). To confirm tissue-specific FXR deletion and FXR activation with GSK2324, we measured hepatic FXR target genes Shp and MafG, which were induced in both Fxrfl/fl and FxrInt-KO mice, but not FxrLiv-KO mice (Figures 7F and S7H). Conversely, the intestinal FXR target genes Fgf15 and Fabp6 were induced in the ileum in both Fxrfl/fl and FxrLiv-KO mice, but not FxrInt-KO mice (Figures 7G and S7I), confirming tissue-specific knockout in each of our models.
To determine how GSK2324 altered hepatic lipids in tissue-specific FXR knockout mice, we performed lipidomics. FXR activation significantly reduced hepatic TAG in Fxrfl/fl mice; however, neither FxrLiv-KO nor FxrInt-KO mice had significant reductions in TAG levels after treatment with GSK2324 (Figure 7H), suggesting that both are required for this effect. Detailed analysis of TAG species revealed consistent and robust decreases in TAGs regardless of acyl tail in Fxrfl/fl mice but not in FxrLiv-KO mice or FxrInt-KO mice (Figure S7J). Hepatic expression of lipogenic genes after treatment with GSK2324 showed reduced expression of Srebp1c, Scd1, Dgat2, and to a lesser extent Lpin1 in Fxrfl/fl mice and FxrInt-KO mice, but not FxrLiv-KO mice (Figures 7I, S7K, and S7L). To determine whether hepatic or intestinal FXR mediate changes in lipid absorption, we analyzed fecal lipids and found that FXR activation significantly increased fecal lipids in both Fxrfl/fl and FxrLiv-KO mice, but not FxrInt-KO mice (Figure 7J). These data indicate that a change in intestinal lipid absorption is largely dependent on activation of intestinal FXR. Finally, we measured the bile acid pool of these tissue-specific FXR knockout mice and found that treatment with GSK2324 significantly decreased the bile acid pool in Fxrfl/fl and FxrLiv-KO mice, but not FxrInt-KO mice (Figure 7K). Together, these data suggest that both hepatic and intestinal FXR are required for GSK2324-mediated reductions in hepatic TAG, but through two distinct pathways. Hepatic FXR controls selective changes in de novo lipogenesis, whereas intestinal FXR decreases intestinal lipid absorption.
DISCUSSION
Agonists for the nuclear receptor FXR are currently under investigation for the treatment of NAFLD; however, the mechanisms for the beneficial effects of FXR agonism have not been fully elucidated. Numerous studies implicate various tissue-specific pathways as the mechanisms for the beneficial lipid-lowering effect of FXR activation (Fang et al., 2015; Jiang et al., 2015; Watanabe et al., 2004). Our studies implicate both hepatic and intestinal FXR as required for FXR-mediated reductions in hepatic TAG. These findings may have important clinical significance as our data suggest combined activation of FXR in liver and intestine are required for maximal hepatic TAG lowering.
We demonstrate that activation of FXR decreases both intestinal lipid absorption and bile acids, particularly CA and T-CA. These data highlight that changes in lipid absorption are a key regulatory pathway in mediating hepatic lipid accumulation, and this mechanism may be especially important in determining the efficiency of FXR agonists in clinical practice. Importantly, the decrease in intestinal lipid absorption was abolished when the diet was supplemented with CA, suggesting that changes in bile acids are key mediators in the FXR-dependent changes in lipid absorption. Specific shifts in bile acid composition were recently shown to reduce lipid absorption following vertical sleeve gastrectomy, and decreases in TCA and CA levels were also reported to cause decreased intestinal lipid absorption (Ding et al., 2021). The concept that changes in bile acid composition affect lipid absorption is further supported by Cyp8b1−/− mice, which do not synthesize CA (Li-Hawkins et al., 2002). These animals are protected from diet-induced steatosis, and have reduced lipid absorption (Bonde et al., 2016). Thus, lipid absorption may be regulated not only by decreases in the bile acid pool but also by specific changes to the bile acid pool composition without affecting the overall pool size. Since we show FXR alters lipid absorption, this may also result in decreased uptake of essential fat-soluble vitamins, which should be carefully monitored in patients taking FXR agonists.
Changes in lipid absorption may also explain conflicting studies showing that both inhibiting (Jiang et al., 2015) and activating (Fang et al., 2015) intestinal FXR can have beneficial effects on hepatic TAG levels. In these studies, conclusions were based on the actions of intestinal FXR only. Jiang and colleagues report that intestinal FXR signaling was suppressed through increases in T-b-MCA, a proposed FXR antagonist (Sayin et al., 2013), which was protective against hepatic TAG accumulation (Jiang et al., 2015). We postulate that substantially increasing T-β-MCA could alter absorption independently of its actions on FXR, as β-MCA is a poor detergent, compared with TCA (Montet et al., 1987). Thus, based on the current data, we propose that activation of intestinal FXR by non-bile acid agonists reduces lipid absorption as a result of a decrease in the bile acid pool size and altered composition. In contrast, treatment of mice with the FXR antagonist T-β-MCA alters bile acid composition also resulting in decreased lipid absorption.
A second important finding in these studies is that the activation of hepatic FXR mediates selective changes in hepatic lipogenesis. The current paradigm is that FXR activation reduces hepatic TAGs and lipogenic gene expression through a SHP-SREBP1c pathway (Watanabe et al., 2004). Watanabe and colleagues used a bile acid or GW4064 as FXR agonists in hypertriglyceridemic mice and proposed that FXR reduced triglycerides via a SHP-dependent reduction in fatty acid synthesis by suppressing Srebp1c (Watanabe et al., 2004). However, the Watanabe et al. studies mainly measured plasma TAG, which we have previously demonstrated and has almost no correlation with hepatic lipid burden (Parker et al., 2019), thus highlighting a key difference with our studies that measure liver TAG. We have demonstrated using knockout mice that FXR activation reduces hepatic lipids independently of both SHP and SREBP1c. We also demonstrate that FXR activation in mice fed a WD, which promotes significant hepatic TAG accumulation, still reduces hepatic TAG independently of SHP and SREBP1c. In agreement with previous studies, we found that whole-body knockout of SHP is protective against hepatosteatosis (Akinrotimi et al., 2017; Huang et al., 2007), as was SREBP1c knockout. Yet despite these lower basal TAG levels, treatment with GSK2324 was still able to reduce hepatic TAG in both Shp−/− and Srebp1c−/− mice. Further, we show that rather than a decrease in the expression of all lipogenic enzymes, FXR activation causes selective reductions in the expression of only three hepatic fatty acid synthesis genes, Scd1, Lpin1, and Dgat2. The mechanism for this repression remains to be elucidated.
Taken together, our data demonstrate that intestinal lipid absorption is a novel mechanism by which FXR activation decreases hepatic lipid accumulation. Using a specific, non-steroidal FXR agonist (GSK2324), we demonstrate that FXR activation decreases hepatic TAG levels in mice fed a standard rodent diet and in animals with fatty livers due to prolonged WD feeding. Our studies demonstrate two complex and distinct mechanisms for a clinically relevant class of drug used to treat NAFLD, first through the selective regulation of lipogenic genes and second through decreased intestinal lipid absorption. Importantly, this highlights lipid absorption as a mode of action of FXR agonists, which is likely to be clinically significant.
Limitations of study
We demonstrate that activation of FXR results in significant changes to intestinal lipid absorption as well as selective decreases in lipogenesis. It is difficult to determine the relative contribution of each pathway, however, our tissue-specific FXR knockout mice suggest both pathways play a significant role. Our studies were all carried out using a relatively acute agonist treatment regimen (3 days of agonist treatment), which was sufficient to reduce liver TAG burden in multiple mouse models. However, it remains to be determined how longer-term administration of FXR agonists alters hepatic lipids.
There are different classes of FXR agonists that are currently being evaluated clinically, and these are either bile acid analogs or non-steroidal agonists. This study used mostly a non-steroidal agonist and our data point to a potentially important difference between the two types of FXR agonists. Since we show that bile acids are required for FXR-dependent decreases in lipid absorption, we reason that the use of a bile acid-derived agonist may replenish or alter the bile acid pool and thus may not alter lipid absorption and have reduced efficacy in reducing liver TAGs. Human patients described in this publication were previously shown to have altered biliary bile composition after treatment with the bile acid analog OCA (Al-Dury et al., 2019), suggesting changes in lipid absorption may still have occurred. However, it seems important to carry out studies directly comparing different FXR agonists while monitoring changes in bile acid composition.
Finally, we and others mostly utilize mouse models to determine the effects of FXR activation on liver TAG. However, it is well known that humans and mice have different types of bile acids, for example, mice produce muricholic acids that are absent in humans. While the changes in bile acids were predominantly observed in CA and TCA, a bile acid that is prominent in both mice and humans, these bile acid species differences may also result in additional mechanisms in humans that cannot be elucidated in mice.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Thomas Vallim (tvallim@mednet.ucla.edu).
Materials availability
This study did not generate new unique reagents. GSK2324 was obtained via a material transfer agreement from GlaxoSmithKline.
Data and code availability
This paper analyzes existing, publicly available ChIP-Seq data. The reference for these data is listed in the key resources table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Human liver biopsies | Al-Dury et al., 2019 | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Sucrose polybehenate (SPB) | CarboMer | Cat # 56449-50-4 |
| Deuterium oxide | Sigma-Aldrich | Cat # 151882 |
| Bile acid standards | See Table S3 | N/A |
| Fatty Acid Standards | Nu-Chek-Prep | GLC96, GLC20a, GLC96b, GLC96c |
| Experimental models: Organisms/strains | ||
| Fxr−/− mice (whole-body) | Jackson Laboratory | Cat# JAX 004144 |
| Shp−/− mice | Kind gift from Dr. Bryan Goodwin | N/A |
| Srebp1c−/− mice | Jackson Laboratory | Cat# JAX 004365 |
| Pparα−/− mice | Jackson Laboratory | Cat# JAX 008154 |
| Albumin-Cre mice | Jackson Laboratory | Cat# JAX 003574 |
| Villin-Cre mice | Jackson Laboratory | Cat# JAX 021504 |
| Fxr fl/fl | Jackson Laboratory | Cat# JAX 028393 |
| Oligonucleotides | ||
| Primer sequences for qPCR | See Table S1 | N/A |
| Software and algorithms | ||
| MsAnalyzer (Custom software for GCMS Analysis) | https://github.com/gcalmettes/labUtils | N/A |
| Other | ||
| Standard rodent chow | Purina | Cat# 5053 |
| Western diet | Research Diets | Cat# D12079B |
| Standard rodent chow with 0.8% SPB | Envigo | Cat# TD170975 |
| Standard rodent chow with 0.8% SPB and 0.5% CA | Envigo | Cat# TD170976 |
| Western diet with 1.04% SPB | Research Diets | Cat# D17121803 |
All original code for the custom GC/MS analysis software has been deposited at GitHub and is publicly available at the time of publication. Repository information is listed in the software section of the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice, treatments, and diets
All mice used in the following studies were male and 8 weeks of age at the start of each experiment. Wildtype and Fxr−/− mice were previously described (Sinal et al., 2000), and have been crossed to a C57BL/6J background for over 15 generations and bred as separate colonies. Shp−/− mice were a kind gift from Dr. Bryan Goodwin and were previously described (Hartman et al., 2009) and have been backcrossed to C57BL/6J for over 10 generations. Srebp1c−/− mice (Jax004365) and Pparα−/− mice (Jax008154) were purchased from Jackson Laboratories and backcrossed for 10 generations. FXR floxed mice (Fxrfl/fl) have been previously described (Kim et al., 2007; de Aguiar Vallim et al., 2013b) and have been backcrossed to C57BL/6J for 12 generations, before being crossed to Albumin-Cre or Vilin-Cre (Jackson Laboratories). Cre negative, littermate floxed controls from both Albumin- and Vilin-Cre breeding pairs were combined and analyzed as a single control group. GSK2324 has been extensively described (de Aguiar Vallim et al., 2015; Tarling et al., 2017). In all studies, mice were treated with either vehicle (water) or GSK2324 (dissolved in water) via intraperitoneal injection once daily at 30mg/kg body weight for three days. Animals were fed ad libitum until the final day of treatment, when mice were injected with GSK2324 starting 9am, fasted for 4 hours, and sacrificed starting 1pm to ensure a consistent circadian cycle time. All mice were housed on a 12-hour light/dark cycle. All animals were maintained on a standard laboratory rodent chow (referred to as “standard rodent diet” in the text, Ralston Purina Company, #5053), unless specified.
For Western diet fed studies, mice were fed a typical Western diet for 8 weeks (Research Diets, D12079B; 43% kcal from carbohydrate, 40% kcal from fat, 17%kcal from protein. 0.21% cholesterol). For experiments involving measurement of fecal lipids, mice were fed with modified diets containing the compound sucrose polybehenate (SPB, CarboMer, 56449–50-4) at a concentration of 5% of total dietary fat (Jandacek et al., 2004). These included standard laboratory diet (0.8% total SPB; Envigo, TD170975), Western diet (1.04% total SPB; Research Diets, D17121803), and a standard laboratory diet with 0.5% cholic acid diet (0.8% total SPB, Envigo, TD170976). For absorption studies, mice were singly housed and diet switched to the appropriate custom diet for the final 7 days of the study. In the cholic acid feeding study, mice were fed standard SPB diet for 3 days, and half of the animals switched to the cholic acid SPB diet for the final 4 days. In every experiment, cages were changed after 48 hours on SPB diet to ensure subsequent fecal collections were of SPB-containing samples. Fecal collections were made prior to the first GSK2324 treatment and after the 4-hour fast period following the final GSK2324 treatment, prior to sacrifice. Animals were housed in fresh cages for the 4-hour fast period to ensure post-GSK2324 fecal collections contained only feces excreted after all three GSK treatments.
For deuterium labeling studies, mouse drinking water was supplemented with deuterium oxide at a concentration of 5% for the final 4 days of the study (Sigma-Aldrich 151882).
Human studies
Human liver samples were obtained from a cohort of patients that has been previously described (Al-Dury et al., 2019). Briefly, 11 patients awaiting laparoscopic gallstone surgery were randomized to receive either placebo or obeticholic acid (OCA) at 25mg/day for 3 weeks until the day of surgery (placebo n=6, OCA n=5). All patients had symptomatic gallstone disease but had no significant comorbidities or medications known to affect glucose or lipid metabolism. At the beginning of the operation, approximately 1×1cm liver specimens were obtained by electrocautery. Tissues were frozen immediately in liquid nitrogen. All samples were kept frozen at −80 °C until analysis.
Study approval
All animal experiments were approved by the Office of Animal Research Oversight (OARO) and the Institutional Animal Care and Use Committee (IACUC) at the University of California Los Angeles.
The human study was approved by the Regional Ethical Committee in Gothenburg (Dnr 199/11: 08JUN2011) and the Swedish Medical Products Agency (EudraCT 2011–0008-13–37;07MAY2012). Informed consent was obtained from all patients before participation in the study.
METHOD DETAILS
Gene expression analyses
RNA was extracted from snap frozen liver pieces (approximately 50mg in weight) using QIAZOL (Qiagen), DNAse-I treated (Thermo Scientific) for 30 minutes and re-extracted using acidified Phenol:Chloroform:Isoamyl alcohol (Thermo Scientific). The same extraction process was used for ileum samples. At sacrifice, ileal contents were pushed up the intestinal tract to preserve contents for bile analysis and approximately 2.5cm section of clean ileum was cut from the end of the intestine for RNA isolation. Purified RNA (500ng) was used to make cDNA, which was diluted 1:10 for gene expression analysis. Gene expression was determined using primers specific for each gene (primer sequences can be found in Table S1) using a SYBR-Green master mix (Roche Diagnostics) and relative quantification was determined using an efficiency corrected method (Roche Diagnostics) using a Lightcycler 480 II qPCR machine (Roche Diagnostics). All genes were normalized to either Tbp or 36B4.
Lipidomic analysis
Lipidomic analyses were carried out at the UCLA lipidomics core with one exception (Pparα−/− mice), which was done at the Baker Heart & Diabetes Institute, Melbourne. The UCLA lipidomics core protocol has been previously described (Hsieh et al., 2021). In brief, for tissue, 50–100 mg of frozen liver were placed in a 2mL homogenizer tube pre-loaded with 2.8mm ceramic beads (Omni #19–628). PBS was added to the tube and the sample homogenized in the Omni Bead Ruptor Elite (3 cycles of 10 seconds at 5 m/s with a 10 second dwell time). For lipid extraction, 3–6mg of tissue homogenate were transferred to a glass tube for a lipid extraction using a modified Bligh and Dyer extraction (Bligh and Dyer, 1959). Prior to biphasic extraction, a 13-lipid class Lipidyzer Internal Standard Mix was added to each sample (AB Sciex, 5040156). Following two successive extractions, pooled organic layers were dried down using a Genevac EZ-2 Elite. Lipid samples were resuspended in 1:1 methanol/dichloromethane with 10mM Ammonium Acetate and transferred to robovials (Thermo 10800107) for analysis. Samples were analyzed on the Sciex Lipidyzer Platform for targeted quantitative measurement of 1100 lipid species across 13 classes. Differential Mobility Device on Lipidyzer was tuned with SelexION tuning kit (Sciex 5040141). Instrument settings, tuning settings, and MRM list available upon request. Data analysis was performed on the Lipidyzer software. Quantitative values were normalized to mg of liver. The Baker Institute protocol is an expanded iteration of Huynh et al. (2019) where 636 unique lipid species were analyzed.
Quanitifcation of FAMEs by GC-MS
For liver, 100mg of frozen sample was weighed and homogenized in ice-cold PBS using the OMNI Bead Ruptor (OMNI International), and 5uL of homegenate was used for analysis. Fecal pellets were powdered using liquid nitrogen and 10mg weighed for analysis. Fatty acid methyl esters (FAMEs) were extracted from liver and fecal samples using mild acid methanolysis. Samples were incubated at 45°C overnight in a hydrochloric acid/methanol/toluene mixture supplemented with trinonadecanoin (Nu- chek Prep, T-165) as an internal standard. Resultant FAMEs were extracted in 1mL of hexane and 1–20μL was analyzed by gas chromatography-mass spectrometry (GC-MS) using an Agilent 7890B/5977A with DB-WAX UI column (Agilent, 122–7032 UI). Complete GC-MS configurations and method programs are available upon request. Quantification of all ions was determined using custom Python Tkinter software by comparison of sample data with serial dilutions of Nu-Check-Prep Fatty Acid Standard Mixes (GLC20a for liver, GLC96, GLC96b and GLC96c for feces) that contain a mixture of various long-chain fatty acid species (methyl myristate, palmitate, palmitoleate, stearate, oleate, linoleate, linolenate, arachidonate and behenate). Integration of all ions (samples and standards) was performed on MassHunter Quantitative Analysis Program (Agilent Technologies). De novo lipogenesis analysis was measured by labelled isotopic enrichment of FAMES after correcting for the natural abundance of stable isotopes using the modern least-squares implementation of the skewed matrix correction method as we have published previously (Parker et al., 2019).
Absorption studies
Radioactive lipid absorption studies were performed as previously described (Wang et al., 2016). Mice were fasted for 5h and gavaged with 1μCi of 14C-trioleoylglycerol (Perkin Elmer) in 200μl of olive oil (Sigma-Aldrich). After 2h, mice were anesthetized and bled from the retroorbital capillary bed. 100μl of plasma was added to 3mL of Bio-Safe II scintillation fluid (RPI). Fluorescent fatty acid absorption studies were performed as previously described, with some modifications (Zhang et al., 2019). Mice were fasted for 5h and gavaged with olive oil (10μL/g body weight) containing BODIPY FL C12 (Invitrogen). 30-minutes post gavage, the mice were anesthetized, the chest cavity was opened to expose the heart and 10mL of 4% PFA was injected via syringe through the left ventricle. After perfusion, the entire length of the intestine was removed, extended and measured lengthwise from the stomach using a ruler to collect 5cm past the pyloric sphincter to 15cm, thus approximating the jejunum. This segment was pinned in a silicone-plated dish containing 1X PBS (Gibco #10010–023) and flushed with 1X PBS using a 10mL syringe and a reusable feeding needle (Fine Science Tools #18060–20) to remove feces. Next, ball-tip artery scissors (Fine Science Tools #14080–11) were used to open the intestine. The opened jejunum segment was pinned (Fine Science Tools #26002–20) in 4% PFA for 4 hours. The orientation of each intestine sample was kept consistent to enable comparison of the same region. Tissues were washed with 1X PBS overnight on a digital waving rotator at 4°C. Segments of the jejunum were cut from the same location proximal to the duodenum and single-villus wide slices were isolated using Dumont #5 Fine Forceps (Fine Science Tools #11254–20) and Trabeculum Scissors (Geuder #G-19745) under a Leica MZ95 Stereomicroscope. Each slice was then individually transferred to glass microscope slide. The intestine slices were mounted using ReadiUse microscope mounting solution (AAT Bioquest #20009) and a glass coverslip (Fisher Scientific #12548C). Imaging was performed on a Zeiss LSM 880 or LSM 900 equipped with Plan-Apochromat 20x/0.8 M27 objective and 488nm Argon laser with GaAsP PMT array detector set to acquire 499–552nm. Laser intensity settings were optimized for a vehicle-treated intestine sample and then applied to all other samples. Z-stacks were performed to image 50μm sections of the intestinal villi making sure to capture the villi tips. After acquisition, a maximum intensity projection of the Z-stack was applied to visualize the 50μm sections. To quantify the fluorescence intensity in ZEN Blue, individual villi were traced using ‘Draw Spline Contour’ tool to define an individual region of interest (ROI) and the arithmetic mean intensity was calculated for each villus/ROI. Three individual villi were quantified per mouse for a total of 36 measurements from 12 animals (6 vehicle-treated and 6 GSK2324-treated). For the Western fed animals, seven individual villi were quantified per mouse for a total of 14 measurements from 4 animals (2 vehicle-treated and 2 GSK2324-treated).
Bile acid extraction and analysis
Bile obtained from mouse gallbladders was diluted 1 to 100 and 1 to 10,000 in Optima methanol (ThermoFisher) prior to analysis. For livers, 100–200mg of liver tissue was homogenized in 4mL of Optima methanol containing a spike in of 1μL of 10mM (10nmol) GCA and GCDCA as recovery standard. Whole intestine (and contents) were homogenized in 9mL of Optima methanol containing a spike in of 4.5μL of 100mM (450nmol) GCA and GCDCA as recovery standard. Homogenates were vortexed thoroughly and incubated at room temperature overnight. The following day, samples were centrifuged at 8000rpm for 10 minutes, and the supernatant used for bile acid analysis. Intestinal samples were diluted 1 to 10 prior to analysis.
Identification and quantification of bile acids was achieved by LC-MS using an Ultimate3000 UHPLC liquid chromatography system (ThermoScientific) equipped with a C18-PFP 1.7um column (ACE Excel) paired to a TSQ Quantiva Mass Spectrometer (ThermoScientific). 10mM ammonium acetate buffer (eluent A) and 75% acetonitrile, 25% methanol with 10mM ammonium acetate (eluent B) were used with a flow rate of 0.325mL/min. For detection of bile acids, a gradient method was used starting at 26% eluent B for 5 min, and increasing to 98% eluent B by 25 min. A wash step was performed after each run using 100% methanol for 2 min, followed by equilibration in 100% eluent A for 5 minutes. Ionization of the samples was performed with the following settings: spray voltage, 3500 V; vaporizer temperature, 350°C; sheath and auxiliary gas (nitrogen) pressure, 20 and 2 arbitrary units, respectively; ion transfer capillary temperature, 300°C; collision gas (argon) pressure, 1.5 mTorr; collision energy 10–55V; and ion polarity, negative. Selected reaction monitoring (SRM) was conducted using the characteristic precursor-to-product ion transition and retention times listed in Table S2. Sample (1μL) was injected into the LC-MS by autoinjector. Bile acid retention time was determined empirically using pure standards of each bile acid. Bile acid concentrations in samples were determined using standard curves for each bile acid except T-αMCA, T-βMCA and T-ωMCA where TCA was used as a class standard, as shown in Table S3. Bile acid abbreviations, common names and systematic name from LIPIDS MAPS Lipidomics Gateway (https://lipidmaps.org/) are also provided in Table S3.
QUANTIFICATION AND STATISTICAL ANALYSIS
All error bars represent the mean ± SEM unless otherwise stated in the figure legend. For comparison of two groups significance was determined using a two-tailed, paired Student’s t-test. For comparison of more than two groups a one- or two-way ANOVA was performed as appropriate with multiple comparisons performed using Fisher’s Least Squares Difference (LSD) test. A p-value of less than 0.05 was taken to be statistically significant.
Supplementary Material
Highlights.
Non-steroidal agonists of FXR significantly decrease intestinal lipid absorption
FXR decreases hepatic triglycerides independently of SHP and SREBP1C
FXR activation reduces expression of three key lipogenic genes, Scd1, Lpin1, and Dgat2
Intestinal and hepatic FXR are both required to decrease hepatic triglycerides
ACKNOWLEDGMENTS
We thank Michelle Steel, Joan Cheng, and Elizabeth Vanderwall for help with mice. We thank all members of the Tarling-Vallim, Edwards, Tontonoz, and Bensinger labs at UCLA for useful advice and discussion and sharing reagents and resources. We thank David Deaton and Tim Willson for the generous gift of GSK2324. The graphical abstract was created using BioRender.com. B.L.C. was sponsored by an AHA post-doctoral fellowship 19POST34380145. L.R.S. was funded by a diversity supplement to NIH R01 DK112119 and T32EB027629. K.E.J. is sponsored by a UCLA Vascular Biology T32HL069766. A.P.C. is funded by T32DK007180. M.C.B.-S. is funded by an AHA post-doctoral fellowship (836561). H.U.M. is funded by grants from the Swedish Research Council (2013-2569/2016-01125) and the Swedish state under the agreement between the Swedish Government and the county councils and the ALF agreement (ALFGBG-426741/717321). A.T. is financially supported by grants from the Erling-Persson Family Foundation (140604). B.G.D. is funded by an Australian Heart Foundation Future Leader Fellowship (101789). J.J.M. is sponsored by an AHA Career Development award (19CDA34760007). E.J.T. is funded by NIH grants HL118161 and HL136543, E.J.T. and T.Q.d.A.V. are funded by NIH grant DK128952, T.Q.d.A.V. is funded by NIH grants HL122677 and DK119112, and P.A.E. and T.Q.d.A.V. are sponsored by NIH grant DK118064.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper received support from a program designed to increase minority representation in science.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.cmet.2021.06.012.
REFERENCES
- Ahmad TR, and Haeusler RA (2019). Bile acids in glucose metabolism and insulin signalling—mechanisms and research needs. Nat. Rev. Endocrinol. 15, 701–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akinrotimi O, Riessen R, Vanduyne P, Park JE, Lee YK, Wong LJ, Zavacki AM, Schoonjans K, and Anakk S (2017). Small heterodimer partner deletion prevents hepatic steatosis and when combined with farnesoid X receptor loss protects against type 2 diabetes in mice. Hepatology 66, 1854–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Dury S, Wahlström A, Panzitt K, Thorell A, Ståhlman M, Trauner M, Fickert P, Bäckhed F, Fändriks L, Wagner M, et al. (2019). Obeticholic acid may increase the risk of gallstone formation in susceptible patients. J. Hepatol. 71, 986–991. [DOI] [PubMed] [Google Scholar]
- Alves-Bezerra M, and Cohen DE (2017). Triglyceride metabolism in the liver. Compr. Physiol. 8, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass JY, Caravella JA, Chen L, Creech KL, Deaton DN, Madauss KP, Marr HB, McFadyen RB, Miller AB, Mills WY, et al. (2011). Conformationally constrained farnesoid X receptor (FXR) agonists: heteroaryl replacements of the naphthalene. Bioorg. Med. Chem. Lett. 21, 1206–1213. [DOI] [PubMed] [Google Scholar]
- Birkenfeld AL, and Shulman GI (2014). Nonalcoholic fatty liver disease, hepatic insulin resistance, and type 2 diabetes. Hepatology 59, 713–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bligh EG, and Dyer WJ (1959). A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917. [DOI] [PubMed] [Google Scholar]
- Bonde Y, Eggertsen G, and Rudling M (2016). Mice abundant in muricholic bile acids show resistance to dietary induced steatosis, weight gain, and to impaired glucose metabolism. PLoS One 11, e0147772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burr GO, and Burr MM (1930). On the nature and role of the fatty acids essential in nutrition. J. Biol. Chem. 86, 587–621. [Google Scholar]
- Cohen JC, Horton JD, and Hobbs HH (2011). Human fatty liver disease: old questions and new insights. Science 332, 1519–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson PA, and Karpen SJ (2015). Intestinal transport and metabolism of bile acids. J. Lipid Res. 56, 1085–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Aguiar Vallim TQ, Tarling EJ, and Edwards PA (2013a). Pleiotropic roles of bile acids in metabolism. Cell Metab 17, 657–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Aguiar Vallim TQ, Tarling EJ, Kim T, Civelek M, Baldán Á, Esau C, and Edwards PA (2013b). MicroRNA-144 regulates hepatic ABCA1 and plasma HDL following activation of the nuclear receptor FXR. Circ. Res. 112, 1602–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Aguiar Vallim TQ, Tarling EJ, Ahn H, Hagey LR, Romanoski CE, Lee RG, Graham MJ, Motohashi H, Yamamoto M, and Edwards PA (2015). MAFG is a transcriptional repressor of bile acid synthesis and metabolism. Cell Metab 21, 298–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Zhang E, Yang Q, Jin L, Sousa KM, Dong B, Wang Y, Tu J, Ma X, Tian J, et al. (2021). Vertical sleeve gastrectomy confers metabolic improvements by reducing intestinal bile acids and lipid absorption in mice. Proc. Natl. Acad. Sci. U. S. A. 118, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang S, Suh JM, Reilly SM, Yu E, Osborn O, Lackey D, Yoshihara E, Perino A, Jacinto S, Lukasheva Y, et al. (2015). Intestinal FXR agonism promotes adipose tissue browning and reduces obesity and insulin resistance. Nat. Med. 21, 159–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flowers MT, and Ntambi JM (2008). Role of stearoyl-coenzyme A desaturase in regulating lipid metabolism. Curr. Opin. Lipidol. 19, 248–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez FJ, Jiang C, and Patterson AD (2016). An intestinal microbiota-farnesoid X receptor axis modulates metabolic disease. Gastroenterology 151, 845–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, Galardi C, Wilson JG, Lewis MC, Roth ME, et al. (2000). A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol. Cell 6, 517–526. [DOI] [PubMed] [Google Scholar]
- Hartman HB, Lai KD, and Evans MJ (2009). Loss of small heterodimer partner expression in the liver protects against dyslipidemia. J. Lipid Res. 50, 193–203. [DOI] [PubMed] [Google Scholar]
- Hofmann AF (2009). The enterohepatic circulation of bile acids in mammals: form and functions. Front. Biosci. (Landmark Ed.) 14, 2584–2598. [DOI] [PubMed] [Google Scholar]
- Hofmann AF, and Borgstroem B (1964). The intraluminal phase of fat digestion in man: the lipid content of the micellar and oil phases of intestinal content obtained during fat digestion and absorption. J. Clin. Invest. 43, 247–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JD, Goldstein JL, and Brown MS (2002). SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 109, 1125–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh WY, Williams KJ, Su B, and Bensinger SJ (2021). Profiling of mouse macrophage lipidome using direct infusion shotgun mass spectrometry. Star Protoc 2, 100235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Iqbal J, Saha PK, Liu J, Chan L, Hussain MM, Moore DD, and Wang L (2007). Molecular characterization of the role of orphan receptor small heterodimer partner in development of fatty liver. Hepatology 46, 147–157. [DOI] [PubMed] [Google Scholar]
- Huynh K, Barlow CK, Jayawardana KS, Weir JM, Mellett NA, Cinel M, Magliano DJ, Shaw JE, Drew BG, and Meikle PJ (2019). High-throughput plasma lipidomics: detailed mapping of the associations with cardiometabolic risk factors. Cell Chem. Biol. 26, 71–84.e4. [DOI] [PubMed] [Google Scholar]
- Jandacek RJ, Heubi JE, and Tso P (2004). A novel, noninvasive method for the measurement of intestinal fat absorption. Gastroenterology 127, 139–144. [DOI] [PubMed] [Google Scholar]
- Jiang C, Xie C, Li F, Zhang L, Nichols RG, Krausz KW, Cai J, Qi Y, Fang ZZ, Takahashi S, et al. (2015). Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. J. Clin. Invest. 125, 386–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RD, Repa JJ, Russell DW, Dietschy JM, and Turley SD (2012). Delineation of biochemical, molecular, and physiological changes accompanying bile acid pool size restoration in Cyp7a1(−/−) mice fed low levels of cholic acid. Am. J. Physiol. Gastrointest. Liver Physiol. 303, G263–G274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I, Ahn SH, Inagaki T, Choi M, Ito S, Guo GL, Kliewer SA, and Gonzalez FJ (2007). Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. J. Lipid Res. 48, 2664–2672. [DOI] [PubMed] [Google Scholar]
- Kleiner DE, and Makhlouf HR (2016). Histology of NAFLD and NASH in adults and children. Clin. Liver Dis. 20, 293–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko CW, Qu J, Black DD, and Tso P (2020). Regulation of intestinal lipid metabolism: current concepts and relevance to disease. Nat. Rev. Gastroenterol. Hepatol. 17, 169–183. [DOI] [PubMed] [Google Scholar]
- Lee JM, Wagner M, Xiao R, Kim KH, Feng D, Lazar MA, and Moore DD (2014). Nutrient-sensing nuclear receptors coordinate autophagy. Nature 516, 112–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li-Hawkins J, Gåfvels M, Olin M, Lund EG, Andersson U, Schuster G, Björkhem I, Russell DW, and Eggertsen G (2002). Cholic acid mediates negative feedback regulation of bile acid synthesis in mice. J. Clin. Invest. 110, 1191–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsukuma KE, Bennett MK, Huang J, Wang L, Gil G, and Osborne TF (2006). Coordinated control of bile acids and lipogenesis through FXR-dependent regulation of fatty acid synthase. J. Lipid Res. 47, 2754–2761. [DOI] [PubMed] [Google Scholar]
- Montet JC, Parquet M, Sacquet E, Montet AM, Infante R, and Amic J (1987). Beta-muricholic acid; potentiometric and cholesterol-dissolving properties. Biochim. Biophys. Acta 918, 1–10. [DOI] [PubMed] [Google Scholar]
- Moser GA, and McLachlan MS (1999). A non-absorbable dietary fat substitute enhances elimination of persistent lipophilic contaminants in humans. Chemosphere 39, 1513–1521. [DOI] [PubMed] [Google Scholar]
- Parker BL, Calkin AC, Seldin MM, Keating MF, Tarling EJ, Yang P, Moody SC, Liu Y, Zerenturk EJ, Needham EJ, et al. (2019). An integrative systems genetic analysis of mammalian lipid metabolism. Nature 567, 187–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzolo D, Buckley K, Kong B, Zhan L, Shen J, Stofan M, Brinker A, Goedken M, Buckley B, and Guo GL (2019). Bile acid homeostasis in a cholesterol 7α-hydroxylase and sterol 27-hydroxylase double knockout mouse model. Hepatology 70, 389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayin SI, Wahlström A, Felin J, Jäntti S, Marschall HU, Bamberg K, Angelin B, Hyötyläinen T, Orešič M, and Bäckhed F (2013). Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab 17, 225–235. [DOI] [PubMed] [Google Scholar]
- Schumacher JD, and Guo GL (2019). Pharmacologic modulation of bile acid-FXR-FGF15/FGF19 pathway for the treatment of nonalcoholic steatohepatitis. Handb. Exp. Pharmacol. 256, 325–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah RA, and Kowdley KV (2020). Obeticholic acid for the treatment of nonalcoholic steatohepatitis. Expert Rev. Gastroenterol. Hepatol. 14, 311–321. [DOI] [PubMed] [Google Scholar]
- Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, and Gonzalez FJ (2000). Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell 102, 731–744. [DOI] [PubMed] [Google Scholar]
- Spector AA, and Kim HY (2015). Discovery of essential fatty acids. J. Lipid Res. 56, 11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi K, and Reue K (2009). Biochemistry, physiology, and genetics of GPAT, AGPAT, and lipin enzymes in triglyceride synthesis. Am. J. Physiol. Endocrinol. Metab. 296, E1195–E1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Targher G, Lonardo A, and Byrne CD (2018). Nonalcoholic fatty liver disease and chronic vascular complications of diabetes mellitus. Nat. Rev. Endocrinol. 14, 99–114. [DOI] [PubMed] [Google Scholar]
- Tarling EJ, Clifford BL, Cheng J, Morand P, Cheng A, Lester E, Sallam T, Turner M, and De Aguiar Vallim TQ (2017). RNA-binding protein ZFP36L1 maintains posttranscriptional regulation of bile acid metabolism. J. Clin. Invest. 127, 3741–3754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas AM, Hart SN, Kong B, Fang J, Zhong XB, and Guo GL (2010). Genome-wide tissue-specific farnesoid X receptor binding in mouse liver and intestine. Hepatology 51, 1410–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Rong X, Duerr MA, Hermanson DJ, Hedde PN, Wong JS, Vallim TQ, Cravatt BF, Gratton E, Ford DA, et al. (2016). Intestinal phospholipid remodeling is required for dietary-lipid uptake and survival on a high-fat diet. Cell Metab 23, 492–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Houten SM, Wang L, Moschetta A, Mangelsdorf DJ, Heyman RA, Moore DD, and Auwerx J (2004). Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J. Clin. Invest. 113, 1408–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, George J, and Bugianesi E (2018). Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 15, 11–20. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Yin L, Anderson J, Ma H, Gonzalez FJ, Willson TM, and Edwards PA (2010). Identification of novel pathways that control farnesoid X receptor-mediated hypocholesterolemia. J. Biol. Chem. 285, 3035–3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Ge X, Heemstra LA, Chen WD, Xu J, Smith JL, Ma H, Kasim N, Edwards PA, and Novak CM (2012). Loss of FXR protects against diet-induced obesity and accelerates liver carcinogenesis in ob/ob mice. Mol. Endocrinol. 26, 272–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Csaki LS, Ronquillo E, Baufeld LJ, Lin JY, Gutierrez A, Dwyer JR, Brindley DN, Fong LG, Tontonoz P, et al. (2019). Lipin 2/3 phosphatidic acid phosphatases maintain phospholipid homeostasis to regulate chylomicron synthesis. J. Clin. Invest. 129, 281–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Léveillé M, Courty E, Gunes A, N Nguyen B, and Estall JL (2020). Differences in metabolic and liver pathobiology induced by two dietary mouse models of nonalcoholic fatty liver disease. Am. J. Physiol. Endocrinol. Metab. 319, E863–E876. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This paper analyzes existing, publicly available ChIP-Seq data. The reference for these data is listed in the key resources table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Human liver biopsies | Al-Dury et al., 2019 | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Sucrose polybehenate (SPB) | CarboMer | Cat # 56449-50-4 |
| Deuterium oxide | Sigma-Aldrich | Cat # 151882 |
| Bile acid standards | See Table S3 | N/A |
| Fatty Acid Standards | Nu-Chek-Prep | GLC96, GLC20a, GLC96b, GLC96c |
| Experimental models: Organisms/strains | ||
| Fxr−/− mice (whole-body) | Jackson Laboratory | Cat# JAX 004144 |
| Shp−/− mice | Kind gift from Dr. Bryan Goodwin | N/A |
| Srebp1c−/− mice | Jackson Laboratory | Cat# JAX 004365 |
| Pparα−/− mice | Jackson Laboratory | Cat# JAX 008154 |
| Albumin-Cre mice | Jackson Laboratory | Cat# JAX 003574 |
| Villin-Cre mice | Jackson Laboratory | Cat# JAX 021504 |
| Fxr fl/fl | Jackson Laboratory | Cat# JAX 028393 |
| Oligonucleotides | ||
| Primer sequences for qPCR | See Table S1 | N/A |
| Software and algorithms | ||
| MsAnalyzer (Custom software for GCMS Analysis) | https://github.com/gcalmettes/labUtils | N/A |
| Other | ||
| Standard rodent chow | Purina | Cat# 5053 |
| Western diet | Research Diets | Cat# D12079B |
| Standard rodent chow with 0.8% SPB | Envigo | Cat# TD170975 |
| Standard rodent chow with 0.8% SPB and 0.5% CA | Envigo | Cat# TD170976 |
| Western diet with 1.04% SPB | Research Diets | Cat# D17121803 |
All original code for the custom GC/MS analysis software has been deposited at GitHub and is publicly available at the time of publication. Repository information is listed in the software section of the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.