

Abstract
α-Synuclein (α-syn)-induced neurotoxicity has been generally accepted as a key step in the pathogenesis of Parkinson’s disease (PD). Microtubule-associated protein tau, which is considered second only to α-syn, has been repeatedly linked with PD in association studies. However, the underlying interaction between these two PD-related proteins in vivo remains unclear. To investigate how the expression of tau affects α-syn-induced neurodegeneration in vivo, we generated triple transgenic mice that overexpressed α-syn A53T mutation in the midbrain dopaminergic neurons (mDANs) with different expression levels of tau. Here we found that tau had no significant effect on the A53T α-syn-mediated mDANs degeneration. However, tau knockout could modestly promote the formation of α-syn aggregates, accelerate severe and progressive degeneration of parvalbumin-positive (PV+) neurons in substantia nigra pars reticulata (SNR), accompanied with anxiety-like behavior in aged PD-related α-syn A53T mice. The mechanisms may be associated with A53T α-syn-mediated specifically successive impairment of N-methyl-D-aspartate receptor subunit 2B (NR2B), postsynaptic density-95 (PSD-95) and microtubule-associated protein 1A (MAP1A) in PV+ neurons. Our study indicates that MAP1A may play a beneficial role in preserving the survival of PV+ neurons, and that inhibition of the impairment of NR2B/PSD-95/MAP1A pathway, which may be a novel and preferential option to ameliorate α-syn-induced neurodegeneration.
Keywords: Parvalbumin, Synuclein, SNR, Neurodegeneration
INTRODUCTION
Parkinson’s disease (PD) is characterized by relatively selective loss of midbrain dopaminergic neurons (mDANs) and the presence of α-synuclein (α-syn)-positive intracytoplasmic inclusions named Lewy bodies and Lewy neurites (1). Early-onset autosomal dominant familial form of PD can be caused by certain mutations in α-syn gene (SNCA) (A53T, A30P and E46K) and gene duplication/triplication (2). Many studies have confirmed that overexpression of wild-type or mutant α-syn can form fibrillar aggregates (3), which can cause various forms of cytotoxicity, including inhibition of synaptic transmission (4), impairment of proteasome and lysosome activities (5), perturbation of the mitochondrial function (6), and disruption of endoplasmic reticulum (ER)/Golgi transport (7). These pathological and genetic studies clearly point out an important role of α-syn in the pathogenesis of PD.
Microtubule-associated protein tau (MAPT) is involved in the assembly and the stabilization of the microtubule network, and plays an important role in maintaining neuronal integrity and axonal transport (8). The dysfunction and aggregation of tau mediate a number of neurodegenerative disorders, including PD (9). Genome-wide association studies (GWAS) identified that the population attributable risk percent (PAR%) of MAPT region (PAR% = 8%) is second only to that of SNCA (PAR% = 12%), highlighting a significant association between tau and PD (10).
Co-occurrence of α-syn and tau pathology has been detected in postmortem PD brains (11). In vitro, overexpressed α-syn can promote the hyperphosphorylation of tau (12). Conversely, overexpressed tau also can increase phospho-α-syn and aggregated α-syn levels (13). All these observations highly suggest that α-syn and tau can interact with each other, and the synergistic interaction may be essential for the development and spreading of neurodegeneration in PD. However, it is still not fully elucidated how this interaction occurs in vivo and how it affects neurodegenerative processes.
In order to investigate how the expression of tau affects α-syn-induced neurodegeneration in vivo, we generated four lines of triple transgenic mice that overexpressed PD-related α-syn A53T missense mutation in the mDANs with different tau gene dosage, including overexpressed human tau (hTau), tau wild-type (Tau+/+), tau heterozygote (Tau+/−) and tau knockout (Tau−/−), which were named as Pitx3-A53T α-Syn × hTau, Pitx3-A53T α-Syn × Tau+/+, Pitx3-A53T α-Syn × Tau+/− and Pitx3-A53T α-Syn × Tau−/− mice. Our findings suggest that tau knockout can exacerbate A53T α-syn-induced neuropathology and specifically induce massive loss of parvalbumin-positive (PV+) neurons in substantia nigra pars reticulata (SNR), eventually causing the Pitx3-A53T α-Syn × Tau−/− mice to exhibit anxiety-like behavior at 12-month-old.
MATERIALS AND METHODS
Generation of triple-transgenic mice
Tau−/− mice (14) and human wild-type Tau inducible transgenic mice (tetO-hTau) (15) were obtained from Jackson laboratory and maintained on C57BL/6J background. As described previously (7, 16), the line of pituitary homeobox 3 (Pitx3) promoter-controlled tetracycline transactivator (tTA) (Pitx3-tTA) mice, the lines of human α-syn A53T inducible transgenic mice (tetO-A53T) and tetO-hTau, in which the expression of human α-syn A53T and human wild-type Tau were under the transcriptional control of tetracycline operator (tetO), and the line of Tau−/− mice were used to generate triple transgenic mice. All the transgenic mice were maintained on C57BL/6J background. Pitx3-tTA mice were crossbred with tetO-hTau and Tau−/− mice to obtain Pitx3-tTA/tetO-hTau (Pitx3 × hTau) and Pitx3-tTA × Tau+/− mice respectively. Meanwhile, tetO-A53T mice were crossbred with Tau−/− mice to obtain tetO-A53T × Tau+/− mice. Then, Pitx3 × hTau mice were crossbred with tetO-A53T mice to generate the triple transgenic mice: Pitx3-tTA/tetO-A53T/tetO × hTau (Pitx3-A53T α-Syn × hTau). And Pitx3-tTA × Tau+/− mice were crossbred with tetO-A53T × Tau+/− mice to generate the triple transgenic mice: Pitx3-tTA/tetO-A53T × Tau+/+ (Pitx3-A53T α-Syn × Tau+/+), Pitx3-tTA/tetO-A53T × Tau+/− (Pitx3-A53T α-Syn × Tau+/−) and Pitx3-tTA/tetO-A53T × Tau−/− (Pitx3-A53T α-Syn × Tau−/−). The mice were housed in a 12-h light/dark cycle in specific-pathogen-free (SPF) conditions and fed a sterile regular diet ad libitum. All experimental procedures were approved by the Institutional Animal Care and Use Committee of Sun Yat-sen University.
Genotyping
Genomic DNA was extracted from tail biopsy using DirectPCR Lysis Reagent (Viagen Biotech) and subjected to polymerase chain reaction (PCR) amplification using specific sets of PCR primers for each genotype, including Pitx3-tTA transgenic mice (Pitx3-F, GACTGGCTTGCCCTCGTCCCA; Pitx3-R, GTGCACCGAGGCCCCAGATCA), tetO-A53T transgenic mice (PrpEx2-F, TACTGCTCCATTTTGCGTGA; SNCA-R, TCCAGAATTCCTTCCTGTGG), tetO-hTau transgenic mice (IMR1369, GGGAGTTCGAAGTGATGGAA; IMR1370, TCTCCAATGCCTGCTTCTTC), and Tau−/− mice (IMR7415, GCCAGAGGCCACTTGTGTAG; IMR7824 AATGGAAGACCATGCTGGAG; IMR7825 ATTCAACCCCCTCGAATTTT).
Behavior Tests
More than 10 male mice for each genotype were used in the behavior tests. Each test was repeated at 2-, 6-, 12- and 18-month-old.
Body weight:
Mice were weighed at same time point every month.
Rotarod:
As described previously (17), mice were placed onto a rotating rod (YLS-4C, Yi Yan Technology Co. Ltd, China) with auto acceleration from 5 rpm to 40 rpm in 2 min. The length of time the mice was able to stay on the rotating rod was recorded. Before the test, mice were trained on the rotating rod at a low rotational speed for 2 days (1st day: 10 rpm, 10 min; 2nd day: 15 rpm, 10 min), which gave the mice sufficient familiarity and practice with the task to avoid hanging on or immediately falling off. For the motor learning test, four trials were run for each mouse in a 2-h interval per day for three sequential days.
Open field:
As described previously (17), mice were tested in a 40 cm × 40 cm × 38 cm acrylic box (San Diego Instruments, U.S.A). A single light source placed in a dark room opposite of the testing area provided a diffuse source of light. Mice were habituated to the testing room and light level for 30 min prior to testing. The acrylic box was cleaned with 75% ethanol between each test. Photobeam Activity System (San Diego Instruments, U.S.A) was used to trace and quantify the movement of the mouse in the unit as the number of beam breaks per 30 min. Four major criteria were assessed in the test: (1) Horizontal movement: ambulation; (2) Fine movement; (3) Vertical movement: rearing or hole-pokes; (4) Whole movement.
Immunohistochemistry and light microscopy
For each genotype, 5 female mice at 2-, 6- and 12-month-old, and 5 male mice at 18-month-old were used for each immunohistochemistry and biochemical experiment. Mice were perfused via cardiac infusion with cold phosphate-buffered saline (PBS), and brains were dissected out immediately. The right hemisphere was snap frozen in a pre-cooling vial in liquid nitrogen, then transferred to −80°C refrigerator for western blot analysis. The right hemisphere, which were used for immunohistochemistry analysis, was fixed in 4% paraformaldehyde for 48 h, then submerged in 30% sucrose for 24–48 h until drop on the bottom of tube. Sequentially, brain tissues were sectioned at 40 μm thickness using a cryostat (Leica SM 2010R). Antibodies specific to tryosine hydroxylase (TH) (Pel-Freez, P40101, 1:1000), human/mouse- α-syn (Santa Cruz Biotechnology, sc-7011-R, 1:1000), human- α-syn (Santa Cruz Biotechnology, sc-12767, 1:1000), glial fibrillary acidic protein (GFAP; Sigma-Aldrich, G9269, 1:500), ionized calcium-binding adaptor molecule-1 (Iba1; Wako Chemicals, 019-19741, 1:500), PV (Sigma-Aldrich, P3088, 1:500; Abcam, ab11427, 1:1000; ab32895, 1:2000), N-methyl-D-aspartate (NMDA) receptor subunit 2B (NR2B; Abcam, ab65783, 1:200; Thermo Fisher Scientific, MA1-2014, 1:250), postsynaptic density-95 (PSD-95; Sigma-Aldrich, P246, 1:200) and microtubule-associated protein 1A (MAP1A; Abcam, ab11264, 1:250; Sigma-Aldrich, HPA039064, 1:1000) were used as suggested by manufacturers. Alexa 488-, Alexa 546-, Alexa 555- or Alexa 647- conjugated secondary antibody (Invitrogen, 1:500) was use to visualize the staining.
Fluorescence images were captured using a laser-scanning confocal microscope (LSM 710; Zeiss). The paired images in all of the figures were collected at the same gain and offset settings, and were processed uniformly after collection. The images were presented as either a single optic layer after being acquired in z-series stack scans at the optimal intervals from individual fields or displayed as maximum-intensity projections to represent confocal stacks.
Image analysis
In order to quantify the distribution and expression of GFAP and Iba1 in the SNR, images were taken using identical settings and exported to ImageJ (NIH) for imaging analyses. All the images were converted to an 8-bit color scale (fluorescence intensity from 0 to 255) using ImageJ. Areas of interest were selected by the freehand selection tools and then subjected to measurement by area fractions or polygon areas. The background area was subtracted from the selected area.
Fluoro-Jade C staining
Cell death was detected by Fluoro-Jade C (Chemicon, AG325) staining according to the manufacturer’s protocol. The sections were mounted onto the gelatin coated slide and then air-dried for at least 30 min on a slide warmer at 50°C. Slides bearing frozen cut tissue sections were first immersed in a basic alcohol solution consisting of 1% sodium hydroxide in 80% ethanol for 5 min. They were then rinsed for 1 min × 2 times in 70% ethanol, for 1 min × 3 times in deionized distilled water (ddH2O), and then incubated in 0.06% potassium permanganate solution for 10 min. After being rinsed for 1 min × 3 times in ddH2O, the slides were then transferred for 10 min to a 0.0001% solution of Fluoro-Jade C dissovled in 0.1% acetic acid vehicle. The slides were then rinsed in ddH2O for 1 min × 3 times, and air-dried on a slide warmer at 50°C for at least 5 min. The air-dried slides were then cleared in xylene and coverslipped with nonfluorescent mounting media.
TdT-mediated dUTP nick end labeling (TUNEL) staining
Apoptosis was detected by in situ cell death detection kit (Roche, 11684795910) according to the manufacturer’s protocol. The sections were mounted onto the slides and then air-dried for at least 30 min on a slide warmer at 50°C. After cooling down, the slides were washed with PBS for 5min × 4 times and then incubated in permeabilization solution for 2 min on ice (2–8°C). The slides were rinsed for 5 min × 4 times with PBS. The areas around the samples were dried. TUNEL reaction mixture was added on the samples. The slides were incubated in a humidified atmosphere for 60 min at 37°C in the dark. The slides were then rinsed with PBS for 5 min × 4 times and rinsed in ddH2O for 5 min × 3 times. After being air-dried at 37°C in the dark, the slides were mounted in ProLong® Gold antifade reagent (Invitrogen) prior to analysis.
Nissl staining
Neurons were detected by NeuroTrace Fluorescent Nissl (Invitrogen, N21482) staining according to the manufacturer’s protocol. The cryosections were placed on a slide and then the sections were rehydrated for at least 40 min in 0.1 M PBS. The sections were washed for 10 min in PBS plus 0.1% TritonX-100. Then the sections were washed in PBS for 5 min × 4 times. After that, the sections were incubated with the diluted NeuroTrace stain (Invitrogen, 1:200) for 20 min in dark. The slides were then rinsed with PBS for 5 min × 4 times and rinsed in ddH2O for 5 min × 3 times. After being air-dried at 37°C in the dark, the slides were mounted in ProLong® Gold antifade reagent (Invitrogen, 1724814) prior to analysis.
Stereology
According to the mouse brain in stereotaxic coordinates (18), a series of coronal sections across the striatum (every sixth from bregma, 1.10 to −2.16 mm) and midbrain (every fourth section from bregma, −2.46 to −3.88 mm) (19)were processed for TH staining and visualized using the Vectastain ABC kit (Vector Laboratories, PK6100). Series of coronal sections across the striatum and midbrain were also processed for Fluoro-Jade C and TUNEL staining. Series of coronal sections across the SNR (every fourth from bregma, −3.28 to −4.04 mm) were processed for Nissl and PV staining, and the PV staining was visualized using Alexa 594- conjugated secondary antibody (Invitrogen, 1:500). Unbiased stereological estimation of the total numbers of TH+, Fluoro-Jade C+, TUNEL+, Nissl+ and PV+ neurons in the midbrain were performed using the Optical Fractionator method of Stereo Investigator 8 (MicroBrightField), as described previously (16). Five mice were used per genotype at each time point. Counters were blinded to the genotypes of the samples. The sampling scheme was designed to have a coefficient of error of <10% to get reliable results. The stereological analyses of TH+ and PV+ neuron numbers were performed under the 100× objective of a Zeiss Axio microscope (Imager A1).
Tissue homogenization and western blot
Brain tissues of mice were homogenized in radio immunoprecipitation assay buffer (Sigma-Aldrich) supplemented with protease and phosphatase inhibitors (Roche Applied Science, 04693124001, 04906837001). After a 15-min incubation on ice, protein extracts were centrifuged at 18,000 × g for 30 min at 4°C. The supernatants were quantified for protein content using Pierce BCA protein assay kit (Thermo Fisher Scientific, 23227) and separated by 8–12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). After being transferred to polyvinylidene difluoride (PVDF) membranes, the membranes were blocked with 5% non-fat milk or 10% bovine serum albumin (BSA) in Tris-buffered saline, and were immunoblotted with appropriate primary antibodies: TH (Pel-Freez, P40101, 1:500), human/mouse-α-syn (Santa Cruz Biotechnology, sc-7011-R, 1:1000; 3H2897, 1:50), human-α-syn (Santa Cruz Biotechnology, sc-12767, 1:1000), human-tau (T14, Invitrogen, 13–1400, 1:1000), MAP1A (Abcam, ab11264, 1:500), PSD-95 (Sigma-Aldrich, P264, 1:500), NR2B (Abcam, ab65783, 1:1000), β-actin (Sigma-Aldrich, A5441, 1:5000) and β-tubulin (Sigma-Aldrich, T8578, 1:2000). Signals were visualized by Super Signal west pico chemiluminescent substrate (Thermo Fisher Scientific, 34087) and exposed to autoradiographic film (Kodak). Protein bands were quantified using ImageJ software (NIH) and normalized to β-actin or β-tubulin.
Co-immunoprecipitation (co-IP)
Brain tissues of 2-month-old nTg and Pitx3-A53T α-Syn × Tau+/+ mice were homogenized in lysis buffer (pH 8.0, 50 mM Tris/HCl, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.25% sodiumdeoxycholate). The tissue extracts were clarified by centrifugation at 10,000 g for 15 min at 4°C. The supernatants were incubated with the antibodies for 2 h at 4°C. Then the lysates were incubated with protein A/G beads (Santa Cruz Biotechnology, sc-2003) for 4 h at 4°C and centrifuged at 10,000 × g for 30 min at 4°C. Samples were resolved on 8–12% SDS-PAGE gels and transferred onto PVDF membranes respectively. Mouse or rabbit immunoglobulin G was used as a negative control for the immunoprecipitation of either α-syn or NR2B, PSD-95 and MAP1A. The bound proteins were determined by immunoblotting with the indicated antibodies.
Statistical analysis
The experimenter was blind to the genotype of the mice. Statistical analysis was carried out with GraphPad Prism 5 (GraphPad Software). Data were presented as mean ± SEM. Statistical significance was determined by comparing means of different groups with ANOVA followed by Tukey’s honestly significant difference test post hoc with the level of significance set at P = 0.05.
RESULTS
Pitx3-A53T α-Syn × Tau−/− mice appeared to be anxious at 12-month-old
As described previously (16), Pitx3-tTA mice provide a useful tool to deliver transgenes selectively into the mDANs. We crossbred Pitx3-tTA mice with inducible responder tetO-A53T, tetO × hTau mice or Tau−/− mice to generate the triple transgenic mice, achieving high-level expression of α-syn A53T in the mDANs with different tau gene dosage, including overexpressed human tau (Pitx3-A53T α-Syn × hTau), tau wild-type (Pitx3-A53T α-Syn × Tau+/+), tau heterozygote (Pitx3-A53T α-Syn × Tau+/−) and tau knockout (Pitx3-A53T α-Syn × Tau−/−). While the single transgenic mice were performed normally as non-transgenic (nTg) mice, as shown in our previous studies (16, 20), nTg littermates were used as controls in this study for simplicity.
All lines of triple transgenic mice were viable, developed normally, and could survive their full expected life span. But they were significantly smaller than nTg littermates, and displayed profound posture and motor abnormalities during both resting and moving states. The triple transgenic mice gained significantly less body weight compared with nTg mice since 1-month-old (P < 0.001, Fig. 1A). Since our previous study have demonstrated that variations in tau expression levels did not affect the body weight in mice at different age stages (20), these results suggest that α-syn A53T mutation is the key gene that gives rise to a significant decrease in body weight in transgenic mice. In the rotarod test, compared to nTg mice, all triple transgenic mice exhibited significant impairment in motor coordination and balance from 2-month-old, while Pitx3-A53T α-Syn × Tau+/− mice seemed to perform worse than the other triple transgenic mice at 6-month-old (Fig. 1B and Supplementary Fig. 1A). In the open-field test, triple transgenic mice displayed an obvious decrease in horizontal and vertical movements compared to nTg mice at 2-month-old (Fig. 1C and D). Pitx3-A53T α-Syn × Tau+/− mice became less active than the other triple transgenic mice at 6-month-old (Fig. 1C, D). However, Pitx3-A53T α-Syn × Tau−/− mice displayed a dramatic increase in the movements at 12-month-old, and became even more active than nTg mice (Fig. 1C and Supplemental Fig. S1B). Furthermore, the pathways traced by all mice showed that Pitx3-A53T α-Syn × Tau−/− mice exhibited anxiety-like behavior, as they spent significant shorter time in central zone during habituation compared with nTg mice (the ratio of time in central zone was 5.8% and 15.9%, respectively), while Tau−/− mice showed normal pathways compared with nTg mice (Fig. 1E, F and Supplemental Fig. S1B). Additionally, there were no obvious differences among Pitx3 × hTau, Tau+/−, Tau−/− and nTg mice in the rotarod and open-field tests (Supplemental Fig. S1), indicating that different expression levels of tau could not independently induce the motor abnormalities (20). Taken together, these results suggested that tau deficiency could exacerbate movement impairment in the α-syn A53T conditional transgenic mice. Moreover, at 18-month-old, all mice maintained a low level of activity and there were no significant differences in movement among genotypes. This might be due to the decline in activity, which comes with old age.
Figure. 1.
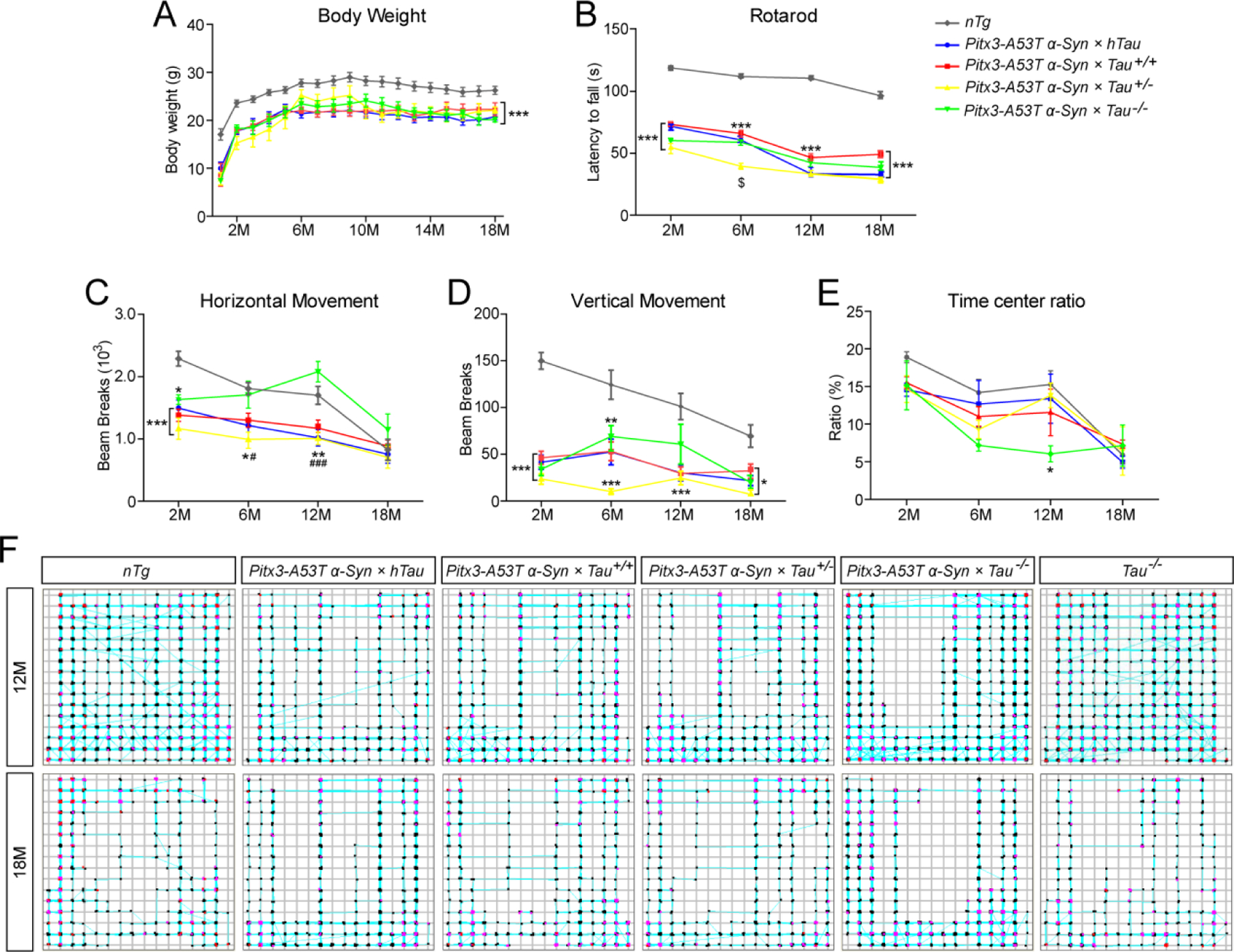
Pitx3-A53T α-Syn × Tau−/− mice showed anxious behavior at 12-month-old. (A) All the mice were weighed monthly for 18 months (M). (B) The latency to fall was quantified by the rotarod test. The horizontal movement (C) and vertical movement (D) were measured using the open-field test. (E) The ratio of time in central zone over total time with any events of mice in the open-field test was measured. (F) The pathways traced by 12- and 18-month-old mice in the open-field test. n ≥ 10 male mice per genotype per time point. Values are mean±SEM. *P < 0.05, **P < 0.01, ***P < 0.001 (Triple transgenic versus age-matched nTg); $P < 0.05 (Pitx3-A53T α-Syn × Tau+/− versus age-matched Pitx3-A53T α-Syn × hTau, Pitx3-A53T α-Syn × Tau+/+ and Pitx3-A53T α-Syn × Tau−/−); #P < 0.05, ###P < 0.001 (Pitx3-A53T α-Syn × hTau, Pitx3-A53T α-Syn × Tau+/+ and Pitx3-A53T α-Syn × Tau+/− versus age-matched Pitx3-A53T α-Syn × Tau−/−).
Tau may have no effect on the A53T α-syn-mediated mDANs degeneration
To determine whether different tau expression levels affect A53T α-syn-mediated degeneration of mDANs (16), we counted the numbers of TH+ mDANs in substantia nigra pars compacta (SNC), ventral tegmental area (VTA), and retro-rubral field (RRF) of mice at 2-, 6-, 12-, and 18-month-old (Fig. 2A–D). Compared to nTg mice, triple transgenic mice developed a robust and progressive loss of mDANs in the SNC and VTA from 2-month-old (P < 0.001, Fig. 2B, C), and in the RRF from 12-month-old (P < 0.001, Fig. 2D), but there were no significant differences among themselves. The costaining of TH and Nissl revealed that the percentage of TH and Nissl double-positive neurons to the total number of TH+ cells in the midbrains of the triple transgenic mice had no significant change (Supplemental Fig. S2C). These results confirm that triple transgenic mice develop a physical mDANs loss. Besides neuronal loss, we found that TH+ fibers were significantly reduced in the striatum of triple transgenic mice, as compared to nTg mice (Supplemental Fig. S2A and 2B). In addition, no significant loss of mDANs was found in the SNC, VTA and RRF of 18-month-old Pitx3 × hTau, Tau+/− and Tau−/− mice, as compared with nTg mice, indicating that different expression levels of tau might not affect the numbers of mDANs (Fig. 2B–D). These results suggested that A53T α-syn should be the key factor causing the degeneration of mDANs in triple transgenic mice, and tau might have no effect on this process.
Figure. 2.
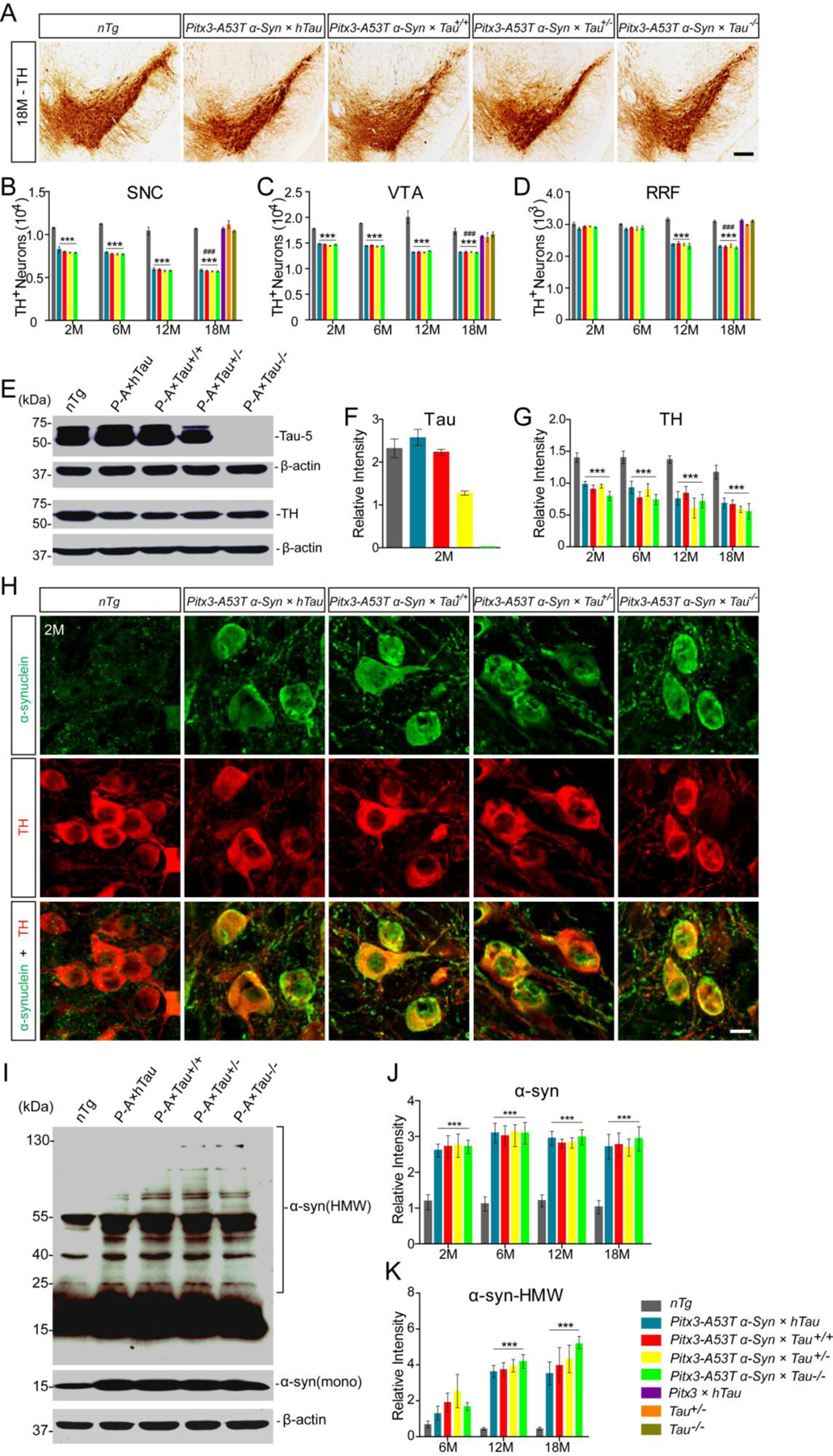
The effects of tau on A53T α-syn-mediated mDANs degeneration and α-syn aggregation. (A) TH immunohistochemistry staining of the midbrain coronal sections of mice. Scale bar: 100 μm. (B-D) The numbers of TH+ neurons in the SNC (B), VTA (C), and RRF (D) of mice. (E) Western blot showed the expression levels of tau and TH in midbrains of 2-month-old mice. (F,G) Bar graphs quantified the levels of tau and TH in midbrain homogenates, respectively. (H) Immunofluorescent images showed α-syn staining (green) in the mDANs (red) at SNC regions of 2-month-old mice. Scale bar: 10 μm. (I) Western blot showed the expression levels of α-syn-positive HMW aggregates in midbrains of 12-month-old mice. (J) Bar graph quantified the levels of total α-syn in midbrain homogenates. (K) Bar graph quantified the levels of α-syn-positive HMW aggregates in midbrain homogenates. n = 5 per genotype per time point. Values are mean±SEM. ***P < 0.001 (Triple transgenic versus age-matched nTg); ###P < 0.001 (Triple transgenic versus age-matched Pitx3 × hTau, Tau+/− and Tau−/−).
Western blot analyses of midbrain homogenates confirmed that the expression of human tau was only detected in Pitx3-A53T α-Syn × hTau mice (Supplemental Fig. S2D). In addition, the mice displayed the corresponding gradient levels of tau expression, which were at highest levels in Pitx3-A53T α-Syn × hTau mice, at normal levels in Pitx3-A53T α-Syn × Tau+/+ and nTg mice, and decreased to half levels in Pitx3-A53T α-Syn × Tau+/− mice, but not expressed in Pitx3-A53T α-Syn × Tau−/− mice (Fig. 2E, F). Consistent with the immunostaining results, western blot analyses also revealed that the expression levels of TH in midbrains of triple transgenic mice were much lower than nTg mice, while there were no obvious differences among themselves (Fig. 2E, G). Together, these results indicated that different expression levels of tau had no significant effect on the A53T α-syn-mediated mDANs degeneration.
Tau knockout modestly promoted the formation of α-syn aggregates
Immunostaining of α-syn and TH revealed that, compared with nTg mice, somatic α-syn signals were increased dramatically in the mDANs of triple transgenic mice (Fig. 2H and Supplemental Fig. S2F). Similarly, western blot analyses showed that human specific α-syn was only expressed in triple transgenic mice, but not nTg mice (Supplemental Fig. S2E). Moreover, the expression levels of total α-syn in the midbrain homogenates of triple transgenic mice were significantly increased at 2- to 18-month-old compared with nTg mice, while there were no obvious differences among themselves (Fig. 2I, J). Furthermore, α-syn-positive high molecular weight (HMW) aggregates in the midbrain homogenates of triple transgenic mice were significantly increased at 12- and 18-month-old compared with nTg mice. Interestingly, though no statistical differences were found among the triple transgenic mice, the intensity of α-syn-positive HMW bands was modestly enhanced in the midbrain homogenates of Pitx3-A53T α-Syn × Tau−/− mice, compared to the other triple transgenic mice from 12-month-old (Fig. 2I, K). These results indicated that tau knockout could modestly promote the formation of α-syn aggregates.
Tau knockout accelerated the progression of A53T α-syn-mediated neurodegeneration in SNR
To test whether different expression levels of tau affect A53T α-syn-mediated neurodegeneration, we examined the brain sections for the neuropathology by Fluoro-Jade C, TUNEL, GFAP and Iba1 staining. Both Fluoro-Jade C and TUNEL staining were negative in the brain sections derived from nTg, Pitx3 × hTau, Tau+/− and Tau−/− mice (Fig. 3A and Supplemental Fig. S3), but were positive specifically in the SNR of the triple transgenic mice at 12- and 18-month-old (Fig. 3A). Interestingly, a significant increase in the numbers of Fluoro-Jade C-positive (Fluoro-Jade C+) and TUNEL-positive (TUNEL+) cells was observed in the SNR of Pitx3-A53T α-Syn × Tau−/− mice compared with the other triple transgenic mice (P < 0.05 or P < 0.01, Fig. 3B, C). However, we did not observe any Fluoro-Jade C+ cells in the striatum of triple transgenic mice (Supplemental Fig. S4). Moreover, costaining of TUNEL and Nissl revealed that the apoptotic cells in the SNR of triple transgenic mice were neurons (Fig. 3D, E). Furthermore, Iba1 and GFAP staining showed that the activated microgliosis and astrocytosis were elevated significantly in the SNR of triple transgenic mice compared with nTg mice from 2-month-old. In addition, more Iba1-positive microglia and GFAP-positive astrocytes were observed in the SNR of Pitx3-A53T α-Syn × Tau−/−mice than the other triple transgenic mice (Supplemental Fig. S5A–E). Together, these observations suggested that tau knockout could accelerate the progression of A53T α-syn-mediated neuropathology in the SNR.
Figure. 3.
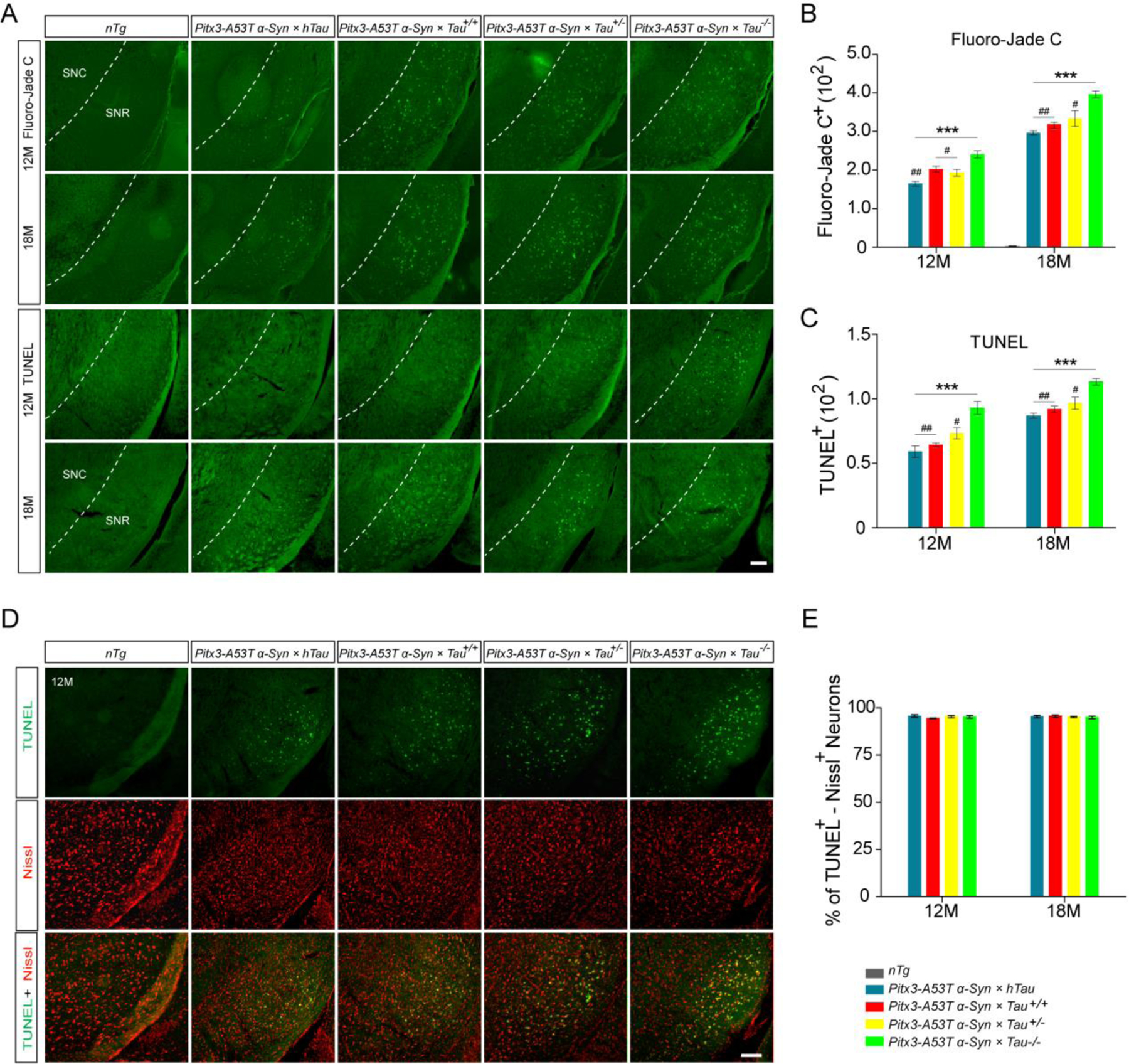
Tau knockout accelerated the progression of A53T α-syn-mediated neurodegeneration in SNR. (A) Representative images for Fluoro-Jade C and TUNEL staining in the SNR of 12- and 18-month-old mice. Scale bar: 100 μm. (B,C) The numbers of Fluoro-Jade C+ and TUNEL+ cells in SNR. (D) TUNEL (green) was highly costained with Nissl (red) in the SNR of the triple transgenic mice at 12-month-old. Scale bar: 100 μm. (E) Percentage of TUNEL and Nissl double-positive neurons to the total number of TUNEL+ cells in the SNR of the triple transgenic mice at 12- and 18-month-old. n=5 per genotype per time point. Values are mean±SEM. **P < 0.01, ***P < 0.001 (Triple transgenic versus age-matched nTg); #P < 0.05, ##P < 0.01 (Pitx3-A53T α-Syn × hTau, Pitx3-A53T α-Syn × Tau+/+ and Pitx3-A53T α-Syn × Tau+/− versus age-matched Pitx3-A53T α-Syn × Tau−/−).
Pitx3-A53T α-Syn × Tau−/− mice developed severe and progressive degeneration of PV+ neurons in SNR
Parvalbumin (PV) is present in GABAergic interneurons and its immunoreactivity can be used to divide the SNR region from midbrain and reflect SNR activity (21, 22). To determine whether A53T α-syn causes the loss of PV+ neurons, we counted the numbers of PV+ neurons in the SNR of the mice. Compared to nTg mice, triple transgenic mice have developed less numbers of PV+ neurons in the SNR from 6-month-old, but there were no significant differences among themselves (Fig. 4A, B). However, the numbers of PV+ neurons in the SNR of Pitx3-A53T α-Syn × Tau−/− mice were decreased significantly at 12- and 18-month-old compared with nTg mice (P < 0.001) and the other triple transgenic mice (P < 0.05) (Fig. 4A, B). Approximately 40% PV+ neurons were lost in the SNR of Pitx3-A53T α-Syn × Tau−/− mice at 6-month-old, while approximately 70% were lost at 18-month-old (Fig. 4B). Meanwhile, compared to nTg mice, the soma size of PV+ neurons in the SNR of triple transgenic mice was smaller, especially in Pitx3-A53T α-Syn × Tau−/− mice (Fig. 4C).We also detected PV expression in the SNR of 18-month-old Tau−/− mice to eliminate the monogenic effect, and the results showed that there were no differences in the number and soma size of PV+ neurons between Tau−/− and nTg mice (Fig. 4B). Furthermore, we found that α-syn staining was mainly presented as small puncta, mainly surrounding the PV+ neuron bodies in the SNR of triple transgenic mice (Fig. 4D). The overexpression of human specific α-syn protein in SNR may reflect the innervation from the mDANs in SNC to this region, because of the enrichment of α-syn protein in the TH+ axon terminals (Supplemental Fig. S6). Intriguingly, in the SNR of Pitx3-A53T α-Syn × Tau−/− mice, along with the increased α-syn accumulation, PV+ neurons were decreased significantly (Fig. 4D). Together, these observations suggested that tau knockout could exacerbate A53T α-syn-induced PV+ neuron degeneration in the SNR.
Figure. 4.
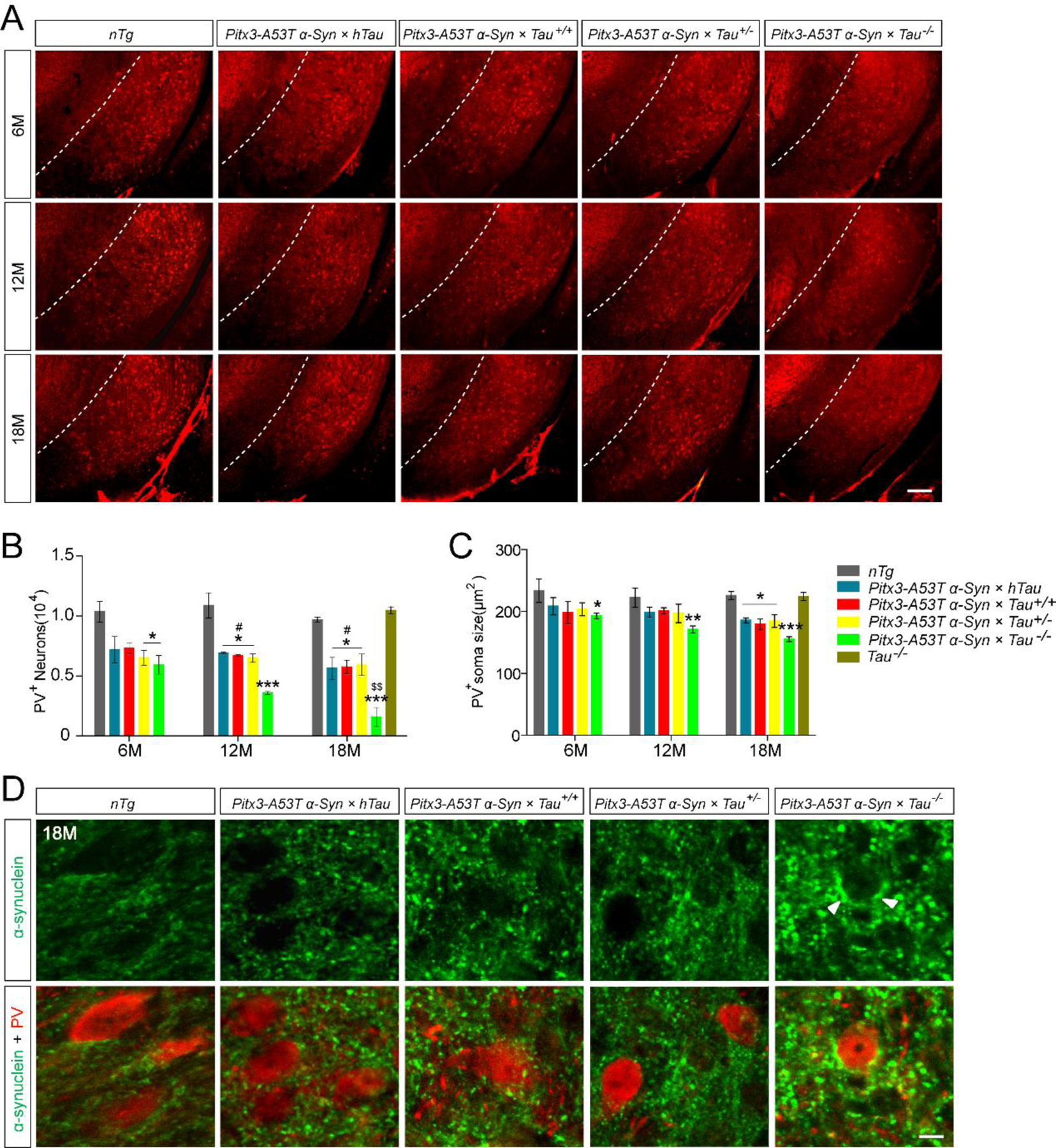
Pitx3-A53T α-Syn × Tau−/− mice developed severe and progressive degeneration of PV+ neurons in the SNR. (A) PV staining in the SNR of mice. Representative coronal sections of midbrain had been shown with white dotted lines demarcating the boundary between SNC and SNR. The ventrolateral area was considered as SNR. Scale bar: 100 μm. (B) The numbers of PV+ neurons in the SNR of mice. (C) The soma size of PV+ neurons in the SNR of mice. Values are mean ±SEM. *P < 0.05, ***P < 0.001 (Triple transgenic versus age-matched nTg); #P < 0.05 (Pitx3-A53T α-Syn × hTau, Pitx3-A53T α-Syn × Tau+/+ and Pitx3-A53T α-Syn × Tau+/− versus age-matched Pitx3-A53T α-Syn × Tau−/−); $$P < 0.01 (18-month-old versus 6-month-old Pitx3-A53T α-Syn × Tau−/−). (D) α-syn (green, C20) and PV (red) costaining in the SNR of 18-month-old mice. The arrowheads pointed to α-syn-positive aggregates. Scale bar: 10 μm. n = 5 per genotype per time point.
Pitx3-A53T α-Syn × Tau−/− mice specifically developed severe impairment of MAP1A in the SNR, which may contribute to the degeneration of PV+ neurons
As reported previously, there has been a compensatory mechanism existing in Tau−/− mice for the lack of tau. The levels of MAP1A, a member of the microtubule-associated proteins (MAPs) family, were increased about 2-fold around birth and 1.3-fold in adulthood, but were declined back to normal at 12-month-old in Tau−/− mice, as compared to nTg mice (14, 21). While the expression levels of other relative molecules, such as microtubule-associated protein 1B, microtubule-associated protein 2, neurofilament proteins, synapsin-1, and various tubulin isoforms in Tau−/− mice were similar to those in nTg mice (14, 21). These data suggested that the increased MAP1A from infancy to adulthood may compensate for the lack of tau in Tau−/− mice. In our study, the expression levels of MAP1A in the midbrains of Pitx3-A53T α-Syn × Tau−/− mice also increased at 2- and 6-month-old (1.2-fold and 1.4-fold, respectively), but then decreased significantly at 12- and 18-month-old (Fig. 5A, B, D). However, the MAP1A expression levels in the midbrains of the other triple transgenic mice and nTg mice were not significantly altered even at 18-month-old (Fig. 5A–C). Additionally, there were no differences in the MAP1A expression levels between Tau−/− mice and nTg mice at 12- and 18-month-old (Fig. 5C, D). These results suggested that before 12-month-old, MAP1A may compensate for loss of tau in the Pitx3-A53T α-Syn × Tau−/− mice; whereas from 12-month-old, tau knockout might cause the loss of this ability.
Figure. 5.
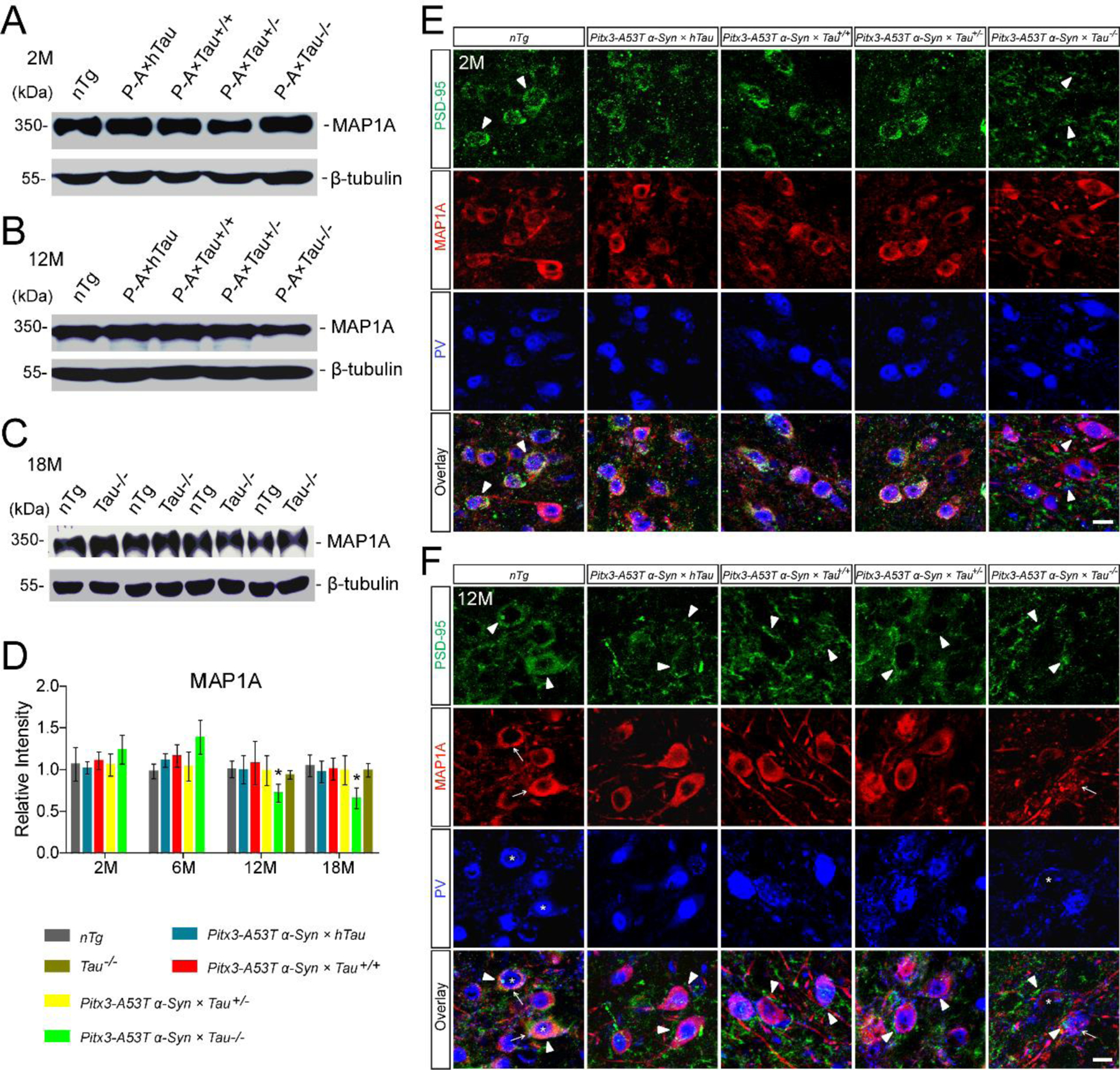
Pitx3-A53T α-Syn × Tau−/− mice specifically developed severe impairment of MAP1A in the SNR. (A,B) Western blot showed the expression levels of MAP1A in the midbrain homogenates of 2- and 12-month-old mice, respectively. (C) Western blot showed the expression levels of MAP1A in the midbrain homogenates of 18-month-old nTg and Tau−/− mice. (D) Bar graph represented the expression levels of MAP1A in the midbrains of 2-, 6-, 12- and 18-month-old mice. Values are mean ± SEM. *P < 0.05 (12- and 18-month-old versus 6-month-old Pitx3-A53T α-Syn × Tau−/−). (E,F) PSD-95 (green), MAP1A (red) and PV (blue) costaining in the SNR of 2- and 12-month-old mice, respectively. Arrowheads marked PSD-95 staining; arrows pointed to MAP1A staining; asterisks indicated PV+ neurons. Scale bar: 10 μm. n = 5 per genotype per time point.
We conducted immunofluorescence assay to further identify the distribution of MAP1A in midbrain. Interestingly, MAP1A was preferentially expressed in the SNR and extensively overlapped with PV staining, presenting a ring-like morphology around PV+ neurons, whereas the mDANs in the SNC and VTA expressed MAP1A (Supplemental Fig. S7A–C) at low levels. Moreover, consistent with the western blot analyses, the immunostaining showed that in the SNR of Pitx3-A53T α-Syn × Tau−/− mice at 12-month-old, compared with themselves at 2-month-old, MAP1A expression was decreased, and its normal ring-like morphology seemed to be fragmented, indicating the impairment of MAP1A; meanwhile, PV+ neurons were significantly decreased (Fig. 5E, F). However, in the SNR of the other triple transgenic mice and nTg mice from 2- to 18-month-old, the expression and morphology of MAP1A were not varied, and their PV+ neurons survived (Fig. 5). These results indicated that Pitx3-A53T α-Syn × Tau−/− mice specifically developed severe impairment of MAP1A in the SNR from 12-month-old, which may contribute to the degeneration of PV+ neurons.
A53T α-syn-mediated MAP1A impairment through the NR2B/PSD-95 pathway
Considering that MAP1A expression was decreased in the midbrains of Pitx3-A53T α-Syn × Tau−/− mice, but not altered in Tau−/− mice as same as in nTg mice at 12-month-old, we proposed that A53T α-syn might be the key factor causing MAP1A impairment. Therefore, we conducted the co-IP analyses to test whether α-syn interacted with MAP1A directly. Unexpectedly, the results indicated that there was no direct interaction between α-syn and MAP1A (Supplemental Fig. S8). Previous studies have demonstrated that NR2B could interact with α-syn (23) and PSD-95 (24), a member of the membrane-associated guanylate kinase (MAGUK) family. Furthermore, PSD-95 and MAP1A could bind directly to each other and be highly co-localized in the central synapses (25). Based on the above research reports, we further investigated whether NR2B/PSD-95 was the pathway mediating α-syn neurotoxicity and leading to MAP1A impairment. The co-IP analyses showed that α-syn was present, together with PSD-95 and the PSD-95-interacting protein MAP1A, in the NR2B immunoprecipitated protein extracts from midbrain tissues of 2-month-old nTg mice and Pitx3-A53T α-Syn × Tau+/+ mice (Fig. 6A, B). These results indicated that NR2B/PSD-95 should provide a vital linkage between α-syn and MAP1A. The immunostaining in the SNR revealed that PSD-95-positive puncta were co-localized with MAP1A in the PV+ neurons (Fig. 5D). Interestingly, PSD-95 staining was decreased and appeared to be significantly fragmented in the PV+ neurons of Pitx3-A53T α-Syn × Tau−/− mice from 2-month-old, indicating that PSD-95 impaired earlier than MAP1A (Fig. 5D), while the impairment of PSD-95 was delayed in the other triple transgenic mice until they were 12-month-old (Fig. 5E). Moreover, PSD-95 was also co-localized with NR2B (Fig. 6C), while NR2B was distributed surrounding the PV+ neurons, and partially co-localized with α-syn (Fig. 6D). Western blot analyses further revealed that compared with nTg mice, the expression levels of NR2B and PSD-95 in the midbrain homogenates of Pitx3-A53T α-Syn × Tau−/− mice decreased significantly from 2-month-old (P < 0.001 and P < 0.01, respectively), while these decreased significantly in the other triple transgenic mice from 6-month-old (P < 0.01) (Fig. 6E–J). Taken together, these suggested that A53T α-syn might mediate the successive impairment of NR2B/PSD-95/MAP1A, and tau knockout would exacerbate this process.
Figure. 6.
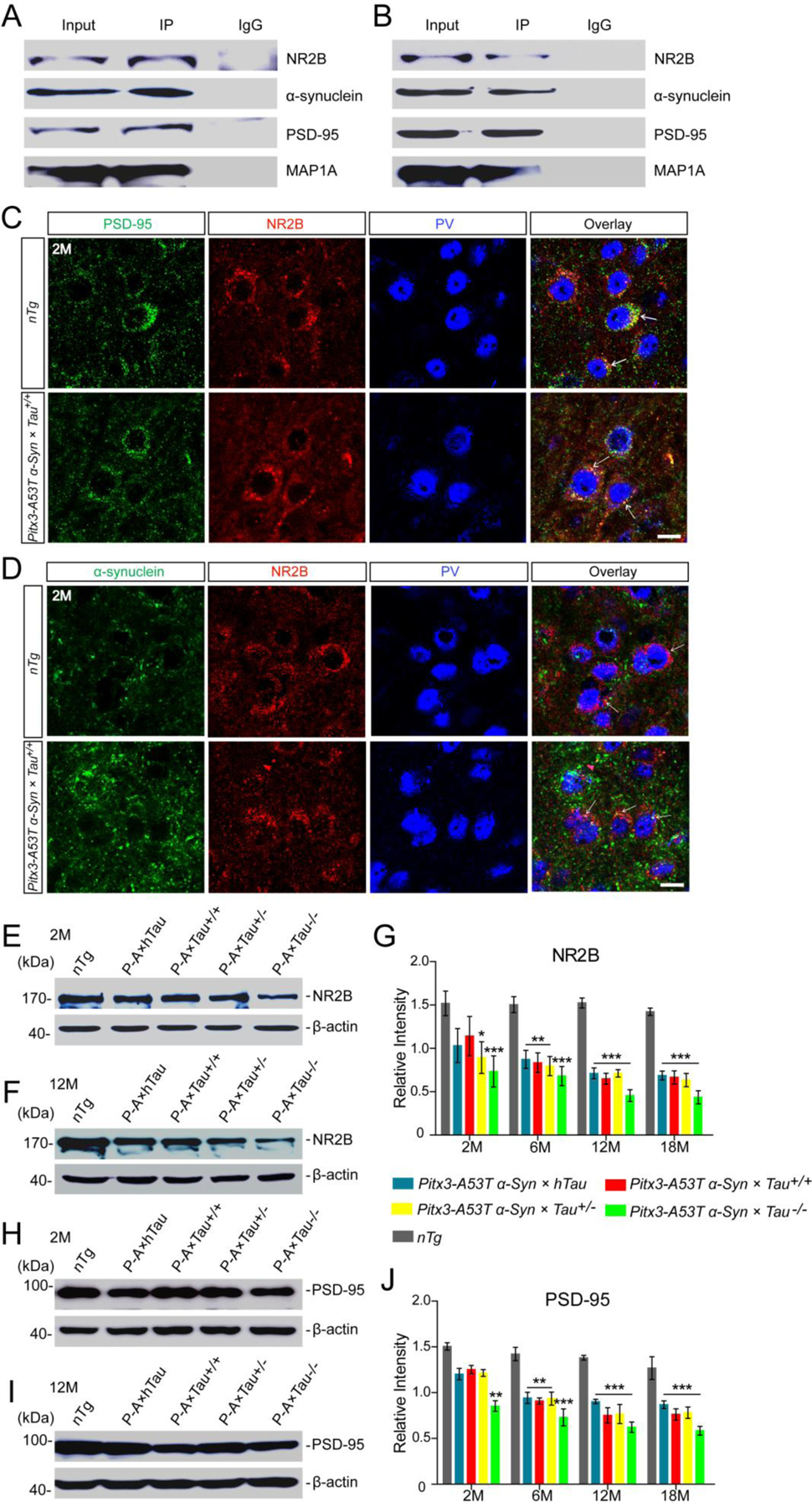
A53T α-syn mediated MAP1A impairment through the NR2B/PSD-95 pathway. (A,B) Representative immunoblotting showed the presence of α-syn, NR2B, PSD-95 and MAP1A positive bands in NR2B-immunoprecipitated protein fractions from the midbrain homogenates of 2 month-old nTg and Pitx3-A53T α-Syn × Tau+/+ mice, respectively. Mouse or rabbit IgG were used as negative controls. (C,D) Costaining images in the SNR of 2-month-old nTg and Pitx3-A53T α-Syn × Tau+/+ mice showed the apparent co-localization of PSD-95 and NR2B (C), α-syn and NR2B (D). Arrows pointed to the costainings. Scale bar: 10 μm. (E,F) Western blot showed the expression levels of NR2B in the midbrains of 2- and 12-month-old mice, respectively. (G) Bar graph illustrated the representative levels of NR2B. (H,I) Western blot showed the expression levels of PSD-95 in the midbrain homogenates of 2- and 12-month-old mice, respectively. (J) Bar graph showed the representative levels of PSD-95. Values are mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 (Triple transgenic versus age-matched nTg). n = 5 per genotype per time point.
DISCUSSION
α-Syn and tau can interact with each other, and their synergistic interaction may play a critical role in the pathogenesis of PD (26). In order to systematically investigate the effect of different tau expression levels on A53T α-syn-mediated neurodegeneration, we generated four lines of triple transgenic mice overexpressing PD-related A53T α-syn mutant in the mDANs with different tau gene dosage (Pitx3-A53T α-Syn × hTau, Pitx3-A53T α-Syn × Tau+/+, Pitx3-A53T α-Syn × Tau+/−, Pitx3-A53T α-Syn × Tau−/−). Here we showed that Pitx3-A53T α-Syn × Tau−/− mice exhibited anxiety-like behavior at 12-month-old, perhaps reflecting a severe degeneration of PV+ neurons in SNR. Indeed, we found that A53T α-syn-mediated PV+ neuron degeneration through the successive impairment of NR2B/PSD-95/MAP1A pathway, and tau knockout could specifically exacerbate this process.
Our previous study had generated a line of α-syn A53T conditional transgenic mice that developed profound motor disabilities as well as robust and progressive mDANs degeneration, resembling some key motor and pathological phenotypes of PD (16). Here our study on triple transgenic mice has revealed that the different expression levels of tau may have no effect on the A53T α-syn-mediated mDANs degeneration. However, tau knockout might modestly promote the formation of α-syn aggregates in the midbrain. It might be attributed to the deficiency of tau, which plays an important role in maintaining the normal organization of Golgi complex (27), while the dysfunction of ER-Golgi trafficking can lead to the formation of α-syn aggregates (7). Interestingly, besides mDANs degeneration, we found that all the triple transgenic mice developed different degrees of neuronal apoptosis in the SNR from 12-month-old. Moreover, tau knockout could exacerbate the A53T α-syn-mediated neurodegeneration in the SNR.
The proportion of neurons in SNR is occupied by a majority of GABAergic neurons, and more than 80% of them express PV (28, 29). Many studies have reported that PV can be used as a specific marker for dividing the SNR region from midbrain and indicating the activity of the SNR (30, 31). Contrary to the effect of ultra-short SNC→SNR direct postsynaptic dopamine exciting PV+ neurons, the effect of presynaptic gamma-aminobutyric acid (GABA) released from striatonigral axon terminals inhibits PV+ neurons. The antagonistical actions could adjust precisely the proper activity of SNR PV+ neurons (32). Moreover, loss of mDANs in the SNC of PD animal models and PD patients may affect PV+ neuron activity, through altering their firing frequency and/or pattern (33). In our study, all the triple transgenic mice developed substantial loss of mDANs in the SNC, this may account for the lesser number of PV+ neurons when compared to nTg mice. Furthermore, Pitx3-A53T α-Syn × Tau−/− mice developed more severe PV+ neuron degeneration after 6-month-old, which could be due to the other pathway and which might have a greater impact, since there were no differences in the number of mDANs in the SNC among the triple transgenic mice.
MAP1A, one of the most abundant MAPs in the adult brain, plays an essential role in neuronal microtubule organization, synaptic protein modulation, and neuronal survival. Loss of MAP1A could cause neuronal death (34). In our study, the expression levels of MAP1A were increased about 1.2- and 1.4-fold in the Pitx3-A53T α-Syn × Tau−/− mice at 2- and 6-month-old, which may compensate for the loss of tau. These results were consistent with the previous studies reporting that the levels of MAP1A were increased about 1.3-fold in adult Tau−/− mice, as compared to nTg mice (14, 21). Interestingly, we observed that MAP1A was preferentially expressed in the SNR PV+ neurons, presenting a ring-like morphology around the PV staining, but not in the SNC and VTA mDANs. Furthermore, Pitx3-A53T α-Syn × Tau−/− mice developed severe and progressive loss of PV+ neurons in the SNR, synchronously with the impairment of MAP1A, from 12-month-old, while the PV+ neurons and the MAP1A expression in the other triple transgenic mice and Tau−/− mice were not varied even at 18-month-old. Together, these indicated that MAP1A might play a beneficial role in promoting the survival of PV+ neurons. Tau knockout could greatly promote the progression of A53T α-syn-mediated MAP1A impairment and then induce the PV+ neuron degeneration in the SNR.
As the co-IP experiments showed that there was no direct interaction between α-syn and MAP1A, we tried to find out the possible indirect connection between them. Previous studies have reported that MAP1A and PSD-95 could bind directly to each other (25). Moreover, NR2B could directly interact with PSD-95 (24) and α-syn (23). Our results suggested that NR2B, PSD-95 and MAP1A may form protein complexes: “NR2B/PSD-95/MAP1A”. These complexes have also been reported in a recent study, which revealed that MAP1A could tether NR2B to the cytoskeleton by binding with PSD-95 scaffold (35). We further found that NR2B/PSD-95 impaired earlier than MAP1A in Pitx3-A53T α-Syn × Tau−/− mice, suggesting that A53T α-syn-mediated NR2B/PSD-95 impairment could be the upstream signal triggering MAP1A impairment. It has been reported that α-syn could act postsynaptically in a prion-like manner, independent of pore formation and membrane leakage, to disturb Ca2+ homeostasis and impair neuronal plasticity via NR2B-mediated mechanism, eventually leading to neural degeneration (36). This strongly indicated our results when there were the most increased postsynaptical α-syn accumulation surrounds the PV+ neuron in the SNR of Pitx3-A53T α-Syn × Tau−/− mice. Together, all these results indicated that A53T α-syn might induce PV+ neuron degeneration in the SNR through the successive impairment of the NR2B/PSD-95/MAP1A pathway and that tau knockout would exacerbate this process.
All the triple transgenic mice in our study developed profound motor disabilities, which resembled some typical motor phenotypes of PD (16). Furthermore, we revealed that tau deficiency may exacerbate movement impairment in the α-syn A53T conditional transgenic mice. The Pitx3-A53T α-Syn × Tau+/− mice displayed more severe movement impairment than the other triple transgenic mice at 6-month-old, especially in terms of movement coordination and rearing activities. It may be attributed to tau heterozygote wherein the compensation effect of MAP1A could not be activated (37). The Pitx3-A53T α-Syn × Tau−/− mice exhibited anxiety-like behavior in the open field at 12-month-old, this should be attributed to the MAP1A impairment-induced substantial loss of PV+ neurons in SNR. PV+ neurons are GABAergic and project largely to the motor thalamus and tectum, their loss can increase cortical activation via decreasing the inhibition of thalamic pathway (31, 38). In line with this notion, the animal models of schizophrenia, which are subjected to NMDA receptor-antagonist, also develop a decrease in the expression of PV in several brain regions, and lead to the emergence of schizophrenia-like behavioral dysfunctions, including hyperactivity and anxiety (39). In addition, increasing studies have revealed that PV+ neurons should be tightly correlated with the anxiety-like behaviors (40). These suggest that the degeneration of PV+ neurons in the SNR of 12-month-old Pitx3-A53T α-Syn × Tau−/− mice may contribute significantly to the emergence of anxiety-like behavior.
In summary, our study has revealed that tau knockout could specifically exacerbate A53T α-syn-induced PV+ neuron degeneration in the SNR, through the successive impairment of the NR2B/PSD-95/MAP1A pathway, and eventually cause anxiety-like behavior in PD-related α-syn A53T mice. We suggest that MAP1A may play a beneficial role in promoting the survival of PV+ neurons and that inhibition of the impairment of the NR2B/PSD-95/MAP1A pathway may provide a potential therapeutic target to ameliorate α-syn-induced neurodegeneration in PD or other related neurodegenerative diseases.
Supplementary Material
Acknowledgments
This project was sponsored in part by the National Natural Science Foundation of China Grants 31271555 and 81171211(to X.L.); Guangdong Province Special Project of Collaborative Innovation and Platform Environment Construction Grant 2016A050502009 (to X.L.); the Natural Science Foundation of Guangdong Province, China Grants 2020A1515010318 (to X.L.); the Scientific Research Foundation for the Returned Overseas Chinese Scholar, State Education Ministry Grant 2012-1707(to X.L.); Science and Technology Plan Foundation of Guangzhou(No:201904010066 and 2010B031600167, to J.H. D.); Natural Science of Foundation of Guangdong Province, China (No:7300973, S2012010009108, to J.H. D.) the Intramural Research Program of the U.S. National Institutes of Health, National Institute on Aging Grant AG000928 (to H.B.C.).
Special Acknowledgment
We thank Dr. Raluca Pana (Department of Neurology, Cleverland Clinic, U.S.A.) for her helpful editing this manuscript in English grammar and word.
ABBREVIATIONS:
- α-syn
α-synuclein
- co-IP
co-immunoprecipitation
- ddH2O
deionized distilled water
- GABA
gamma-aminobutyric acid
- GFAP
glial fibrillary acidic protein
- HMW
high molecular weight
- hTau
human tau
- Iba1
ionized calcium binding adaptor molecule-1
- MAP1A
microtubule-associated protein 1A
- mDANs
midbrain dopaminergic neurons
- NeuN
neuronal nuclei
- NR2B
N-methyl-D-aspartate receptor subunit 2B
- nTg
non-transgenic
- PAR%
population attributable risk percent
- PBS
phosphate-buffered saline
- PCR
polymerase chain reaction
- PD
Parkinson’s disease
- Pitx3
pituitary homeobox 3
- PSD-95
postsynaptic density-95
- PV
parvalbumin
- RRF
retro-rubral field
- SNC
substantia nigra pars compacta
- SNR
substantia nigra pars reticulate
- Tau+/+
tau wild-type
- Tau+/−
tau heterozygote
- Tau−/−
tau knockout
- tetO
tetracycline operator
- TH
tyrosine hydroxylase
- tTA
tetracycline transactivator
- TUNEL
TdT-mediated dUTP nick end labeling
- VTA
ventral tegmental area
Footnotes
Conflict of interest
The authors declare no competing financial interests.
References
- 1.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, and Goedert M (1997) Alpha-synuclein in Lewy bodies. NATURE 388, 839–840 [DOI] [PubMed] [Google Scholar]
- 2.Hardy J, Cai H, Cookson MR, Gwinn-Hardy K, and Singleton A (2006) Genetics of Parkinson’s disease and parkinsonism. ANN NEUROL 60, 389–398 [DOI] [PubMed] [Google Scholar]
- 3.Narhi L, Wood SJ, Steavenson S, Jiang Y, Wu GM, Anafi D, Kaufman SA, Martin F, Sitney K, Denis P, Louis JC, Wypych J, Biere AL, and Citron M (1999) Both familial Parkinson’s disease mutations accelerate alpha-synuclein aggregation. J BIOL CHEM 274, 9843–9846 [DOI] [PubMed] [Google Scholar]
- 4.Nemani VM, Lu W, Berge V, Nakamura K, Onoa B, Lee MK, Chaudhry FA, Nicoll RA, and Edwards RH (2010) Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. NEURON 65, 66–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, and Sulzer D (2004) Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. SCIENCE 305, 1292–1295 [DOI] [PubMed] [Google Scholar]
- 6.Hsu LJ, Sagara Y, Arroyo A, Rockenstein E, Sisk A, Mallory M, Wong J, Takenouchi T, Hashimoto M, and Masliah E (2000) alpha-synuclein promotes mitochondrial deficit and oxidative stress. AM J PATHOL 157, 401–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin X, Parisiadou L, Gu XL, Wang L, Shim H, Sun L, Xie C, Long CX, Yang WJ, Ding J, Chen ZZ, Gallant PE, Tao-Cheng JH, Rudow G, Troncoso JC, Liu Z, Li Z, and Cai H (2009) Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson’s-disease-related mutant alpha-synuclein. NEURON 64, 807–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson GV, and Stoothoff WH (2004) Tau phosphorylation in neuronal cell function and dysfunction. J CELL SCI 117, 5721–5729 [DOI] [PubMed] [Google Scholar]
- 9.Lei P, Ayton S, Finkelstein DI, Adlard PA, Masters CL, and Bush AI (2010) Tau protein: relevance to Parkinson’s disease. Int J Biochem Cell Biol 42, 1775–1778 [DOI] [PubMed] [Google Scholar]
- 10.Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG, Kruger R, Federoff M, Klein C, Goate A, Perlmutter J, Bonin M, Nalls MA, Illig T, Gieger C, Houlden H, Steffens M, Okun MS, Racette BA, Cookson MR, Foote KD, Fernandez HH, Traynor BJ, Schreiber S, Arepalli S, Zonozi R, Gwinn K, van der Brug M, Lopez G, Chanock SJ, Schatzkin A, Park Y, Hollenbeck A, Gao J, Huang X, Wood NW, Lorenz D, Deuschl G, Chen H, Riess O, Hardy JA, Singleton AB, and Gasser T (2009) Genome-wide association study reveals genetic risk underlying Parkinson’s disease. NAT GENET 41, 1308–1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wills J, Jones J, Haggerty T, Duka V, Joyce JN, and Sidhu A (2010) Elevated tauopathy and alpha-synuclein pathology in postmortem Parkinson’s disease brains with and without dementia. EXP NEUROL 225, 210–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawakami F, Suzuki M, Shimada N, Kagiya G, Ohta E, Tamura K, Maruyama H, and Ichikawa T (2011) Stimulatory effect of alpha-synuclein on the tau-phosphorylation by GSK-3beta. FEBS J 278, 4895–4904 [DOI] [PubMed] [Google Scholar]
- 13.Badiola N, de Oliveira RM, Herrera F, Guardia-Laguarta C, Goncalves SA, Pera M, Suarez-Calvet M, Clarimon J, Outeiro TF, and Lleo A (2011) Tau enhances alpha-synuclein aggregation and toxicity in cellular models of synucleinopathy. PLOS ONE 6, e26609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dawson HN, Ferreira A, Eyster MV, Ghoshal N, Binder LI, and Vitek MP (2001) Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J CELL SCI 114, 1179–1187 [DOI] [PubMed] [Google Scholar]
- 15.Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, and Ashe KH (2005) Tau suppression in a neurodegenerative mouse model improves memory function. SCIENCE 309, 476–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin X, Parisiadou L, Sgobio C, Liu G, Yu J, Sun L, Shim H, Gu XL, Luo J, Long CX, Ding J, Mateo Y, Sullivan PH, Wu LG, Goldstein DS, Lovinger D, and Cai H (2012) Conditional expression of Parkinson’s disease-related mutant alpha-synuclein in the midbrain dopaminergic neurons causes progressive neurodegeneration and degradation of transcription factor nuclear receptor related 1. J NEUROSCI 32, 9248–9264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chandran JS, Lin X, Zapata A, Hoke A, Shimoji M, Moore SO, Galloway MP, Laird FM, Wong PC, Price DL, Bailey KR, Crawley JN, Shippenberg T, and Cai H (2008) Progressive behavioral deficits in DJ-1-deficient mice are associated with normal nigrostriatal function. NEUROBIOL DIS 29, 505–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paxinos GAFK (2008) The mouse brain in stereotaxic coordinates (Compact third edition) (San Diego, CA: Academic press) Vol. 18
- 19.Fu Y, Yuan Y, Halliday G, Rusznak Z, Watson C, and Paxinos G (2012) A cytoarchitectonic and chemoarchitectonic analysis of the dopamine cell groups in the substantia nigra, ventral tegmental area, and retrorubral field in the mouse. BRAIN STRUCT FUNCT 217, 591–612 [DOI] [PubMed] [Google Scholar]
- 20.Tang X, Jiao L, Zheng M, Yan Y, Nie Q, Wu T, Wan X, Zhang G, Li Y, Wu S, Jiang B, Cai H, Xu P, Duan J, and Lin X (2018) Tau Deficiency Down-Regulated Transcription Factor Orthodenticle Homeobox 2 Expression in the Dopaminergic Neurons in Ventral Tegmental Area and Caused No Obvious Motor Deficits in Mice. NEUROSCIENCE 373, 52–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harada A, Oguchi K, Okabe S, Kuno J, Terada S, Ohshima T, Sato-Yoshitake R, Takei Y, Noda T, and Hirokawa N (1994) Altered microtubule organization in small-calibre axons of mice lacking tau protein. NATURE 369, 488–491 [DOI] [PubMed] [Google Scholar]
- 22.McRitchie DA, Hardman CD, and Halliday GM (1996) Cytoarchitectural distribution of calcium binding proteins in midbrain dopaminergic regions of rats and humans. J COMP NEUROL 364, 121–150 [DOI] [PubMed] [Google Scholar]
- 23.Navarria L, Zaltieri M, Longhena F, Spillantini MG, Missale C, Spano P, and Bellucci A (2015) Alpha-synuclein modulates NR2B-containing NMDA receptors and decreases their levels after rotenone exposure. NEUROCHEM INT 85–86, 14–23 [DOI] [PubMed] [Google Scholar]
- 24.Kornau HC, Schenker LT, Kennedy MB, and Seeburg PH (1995) Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. SCIENCE 269, 1737–1740 [DOI] [PubMed] [Google Scholar]
- 25.Clemmensen C, Aznar S, Knudsen GM, and Klein AB (2012) The microtubule-associated protein 1A (MAP1A) is an early molecular target of soluble Abeta-peptide. CELL MOL NEUROBIOL 32, 561–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Waxman EA, and Giasson BI (2011) Induction of intracellular tau aggregation is promoted by alpha-synuclein seeds and provides novel insights into the hyperphosphorylation of tau. J NEUROSCI 31, 7604–7618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thyberg J, and Moskalewski S (1999) Role of microtubules in the organization of the Golgi complex. EXP CELL RES 246, 263–279 [DOI] [PubMed] [Google Scholar]
- 28.Zhou FW, Jin Y, Matta SG, Xu M, and Zhou FM (2009) An ultra-short dopamine pathway regulates basal ganglia output. J NEUROSCI 29, 10424–10435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gonzalez-Hernandez T, and Rodriguez M (2000) Compartmental organization and chemical profile of dopaminergic and GABAergic neurons in the substantia nigra of the rat. J COMP NEUROL 421, 107–135 [DOI] [PubMed] [Google Scholar]
- 30.Goetz CG, De Long MR, Penn RD, and Bakay RA (1993) Neurosurgical horizons in Parkinson’s disease. NEUROLOGY 43, 1–7 [DOI] [PubMed] [Google Scholar]
- 31.Hardman CD, McRitchie DA, Halliday GM, Cartwright HR, and Morris JG (1996) Substantia nigra pars reticulata neurons in Parkinson’s disease. Neurodegeneration 5, 49–55 [DOI] [PubMed] [Google Scholar]
- 32.Zhou FM, and Lee CR (2011) Intrinsic and integrative properties of substantia nigra pars reticulata neurons. NEUROSCIENCE 198, 69–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nevet A, Morris G, Saban G, Fainstein N, and Bergman H (2004) Discharge rate of substantia nigra pars reticulata neurons is reduced in non-parkinsonian monkeys with apomorphine-induced orofacial dyskinesia. J NEUROPHYSIOL 92, 1973–1981 [DOI] [PubMed] [Google Scholar]
- 34.Liu Y, Lee JW, and Ackerman SL (2015) Mutations in the microtubule-associated protein 1A (Map1a) gene cause Purkinje cell degeneration. J NEUROSCI 35, 4587–4598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takei Y, Kikkawa YS, Atapour N, Hensch TK, and Hirokawa N (2015) Defects in Synaptic Plasticity, Reduced NMDA-Receptor Transport, and Instability of Postsynaptic Density Proteins in Mice Lacking Microtubule-Associated Protein 1A. J NEUROSCI 35, 15539–15554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ferreira DG, Temido-Ferreira M, Vicente MH, Batalha VL, Coelho JE, Szego EM, Marques-Morgado I, Vaz SH, Rhee JS, Schmitz M, Zerr I, Lopes LV, and Outeiro TF (2017) alpha-synuclein interacts with PrP(C) to induce cognitive impairment through mGluR5 and NMDAR2B. NAT NEUROSCI 20, 1569–1579 [DOI] [PubMed] [Google Scholar]
- 37.Zheng M, Jiao L, Tang X, Xiang X, Wan X, Yan Y, Li X, Zhang G, Li Y, Jiang B, Cai H, and Lin X (2017) Tau haploinsufficiency causes prenatal loss of dopaminergic neurons in the ventral tegmental area and reduction of transcription factor orthodenticle homeobox 2 expression. FASEB J 31, 3349–3358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deniau JM, Mailly P, Maurice N, and Charpier S (2007) The pars reticulata of the substantia nigra: a window to basal ganglia output. PROG BRAIN RES 160, 151–172 [DOI] [PubMed] [Google Scholar]
- 39.Powell SB, Sejnowski TJ, and Behrens MM (2012) Behavioral and neurochemical consequences of cortical oxidative stress on parvalbumin-interneuron maturation in rodent models of schizophrenia. NEUROPHARMACOLOGY 62, 1322–1331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liang D, Li G, Liao X, Yu D, Wu J, and Zhang M (2016) Developmental loss of parvalbumin-positive cells in the prefrontal cortex and psychiatric anxiety after intermittent hypoxia exposures in neonatal rats might be mediated by NADPH oxidase-2. BEHAV BRAIN RES 296, 134–140 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.