

Abstract
Antibody (Ab) crosslinking of HLA I molecules on the surface of endothelial cells (EC) triggers proliferative and pro-survival intracellular signaling, which is implicated in the process of chronic allograft rejection, also known as transplant vasculopathy (TV). Despite the importance of antibody-mediated rejection (AMR) in transplantation, the mechanisms involved remain incompletely understood. Here, we examined the regulation of Yes-Associated Protein (YAP) localization, phosphorylation and transcriptional activity in human endothelial cells (ECs) challenged with Abs that bind HLA I. In unstimulated ECs, YAP localized mainly in the cytoplasm. Stimulation of these cells with Ab W6/32 induced marked translocation of YAP to the nucleus. The nuclear import of YAP was associated with a rapid decrease in YAP phosphorylation at Ser127 and Ser397, sites targeted by LATS1/2 and with the expression of YAP-regulated genes, including Connective Tissue Growth Factor (CTGF), and Cysteine-rich angiogenic inducer 61 (CYR61). Transfection of siRNAs targeting YAP/TAZ blocked the migration of ECs stimulated by ligation of HLA I, indicating that YAP mediates the increase in EC migration induced by HLA I ligation. Treatment of intact ECs with Src family inhibitors induced cytoplasmic localization of YAP in unstimulated ECs and strikingly blocked the nuclear import of YAP induced by Ab-induced HLA I activation in these cells and the increase in the expression of the YAP-regulated genes CTGF and CYR61 induced by HLA I stimulation. Our results identify the Src/YAP axis as a key player in promoting the proliferation and migration of ECs that are critical in the pathogenesis of TV.
Keywords: Endothelial cells, anti-HLA I antibodies, signal transduction, migration, YAP localization
Introduction
Antibody-mediated rejection (AMR) remains as a major subject limiting long-term patient and allograft survival. In many cases, AMR is mediated by the binding of human leukocyte antigen (HLA) antibody (HLA Ab) to the mismatched donor HLA class I (HLA I) and/or class II (HLA II) antigens expressed on the graft endothelial cells (ECs), resulting in microvascular inflammation and intravascular activated mononuclear cells, with or without complement deposition (1–3). Chronic exposure of heart, kidney and lung allografts to HLA Ab can lead to transplant allograft vasculopathy (TAV) resulting in graft dysfunction, loss and patient death (4–6). TV-induced graft failure occurs in up to 50% of renal, heart, lung, small bowel and liver transplants by 10 years post-transplant. Consequently, the elucidation of the pathways that mediate EC responses to Ab-induced ligation of HLA Class I are of major importance and a first step in identifying novel targets to prevent graft failure.
Since HLA molecules do not have endogenous protein kinase activity, they are likely to associate with co-receptors to elicit endothelial cell signaling. We identified integrin β4 to form a molecular complex with HLA I to transduce EC proliferation and migration (7). Previously, we showed that Ab-induced ligation of HLA Class I on the surface of ECs triggers multiple signaling events, including activation of focal adhesion kinase (FAK), Src non-receptor tyrosine kinases (8, 9), phosphatidylinositol 3-kinase (PI3K)/AKT, mechanistic target of rapamycin (mTOR) complex 1 (mTORC1) and mTORC2, p70 S6 Kinase (S6K) and stimulation of extracellular-signal-regulated kinases (ERK1/2) (10, 11). These signaling pathways, mediated in part by engagement of integrin β4 (7), promote cytoskeleton reorganization, stress fiber formation, (11–13), migration and cell proliferation in ECs (10, 14–16). Despite the fundamental significance and translational potential, the transcriptional programs that operate downstream of these signaling networks remain poorly understood.
The highly conserved Hippo pathway, originally identified in Drosophila, is attracting intense attention as a key regulator of development, organ-size, tissue regeneration, tumorigenesis and a central pathway in Rho, mTORC1/2 and PI3K signaling (17–21). Canonical Hippo signals are transduced through a serine/threonine kinase cascade wherein Mst1/2 kinases phosphorylate and activate LATS1/2 (22). In turn, LATS1/2 phosphorylates the transcriptional co-activators Yes-Associated Protein (YAP) and WW-domain-containing Transcriptional co-Activator with PDZ-binding motif (TAZ), two central effectors of the Hippo pathway and novel sensors of mechanical cues (23–25) and diffusible growth-promoting stimuli, including growth factors and G protein-coupled receptor (GPCR) agonists (26). The phosphorylation of YAP and TAZ at multiple serine residues by LATS1/2 restricts their activity, cellular localization and stability (27). In the absence of phosphorylation, YAP localizes to the nucleus where it binds and activates predominantly the TEA-domain DNA-binding transcription factors (TEAD 1–4) thereby stimulating the expression of multiple genes, including Connective Tissue Growth Factor (CTGF) and Cysteine-rich angiogenic inducer 61 (Cyr61). Several recent reports indicate that YAP plays a critical role in the regulation of angiogenesis and vascular development (28, 29). Indeed, loss of YAP/TAZ in the mouse leads to scarcity of ECs, branching irregularities and junction defects (30). Consequently, we hypothesized that the products of YAP/TEAD-regulated genes have a major impact on critical cell processes implicated in chronic AMR, including EC signaling and migration. To examine this hypothesis, we determined the impact of Ab-mediated crosslinking of HLA I molecules on YAP localization, phosphorylation and transcriptional co-activator activity in human ECs and ascertained the role of YAP in the proliferation and migration of these cells.
Here, we report that Ab-induced HLA I activation induces robust YAP nuclear localization and dephosphorylation at residues targeted by LATS1/2 in human arterial ECs. Concomitantly, HLA I activation enhanced YAP co-activator transcriptional activity, leading to increase expression of CTGF and CYR61. Mechanistically, we show that Src family kinases (SFK) play a critical role in mediating YAP localization and activation in response to Ab-induced HLA I signaling in ECs. Our results identify a Src/YAP axis as a putative new target in ECs for therapies to prevent cAMR.
Materials and Methods
Antibodies and Chemicals
Cell culture reagents, transfection reagents, Alexa Fluor 488 conjugated goat anti-mouse antibody (A-1100) and phospho-FAK Tyr576/577 (PA5–37706) were from Invitrogen Life Technologies (Carlsbad, CA). Mouse monoclonal anti–human HLA I Ab (clone W6/32, mIgG2a) was purchased from BioXCell, anti-CD105 mouse monoclonal (clone 43A3, mIgG1) was from BioLegend (San Diego, CA). Human monoclonal allele-specific Ab against HLA-A2 (IgG1, clone SN607D8, was a gift from Dr. Arend Mulder, Leiden University Medical Center, Leiden, Netherlands). Mouse and human IgG isotype control, PP2 and Dasatinib were purchased from Ab Sigma-Aldrich (St. Louis, MO) and Saracatinib (AZD0530) from Selleckchem (Houston, TX). Phospho-YAP Ser127 (D9W2I, 13008), phospho-YAP Ser397 (D1E7Y, 13619), YAP/TAZ (D24E4, 8418), phospho LATS Thr1079 (8654) and LATS2 (5888) were purchased from Cell Signaling Technology (Danvers, MA). YAP (63.1, sc-101199) and GAPDH (sc-365062) were from Santa Cruz Biotechnology. Horseradish peroxidase–conjugated anti-rabbit IgG and anti-mouse IgG were from GE Healthcare Bio-Sciences Corp (Piscataway, NJ). All other reagents were of the highest grade available.
Cell Culture
Primary human aortic endothelial cells were isolated from the aortic rings of explanted donor hearts, as described previously (31) or commercial ECs (lot no. EC5555) were obtained from Lonza/Clonetics (Walkersville, MD). Most experiments were performed using ECs from Lonza/Clonetics. Selected experiments were confirmed using primary human aortic endothelial cells isolated from the aortic rings, as indicated in Results. The cells were cultured in M199 medium (Mediatech, Manassas, VA) supplemented with 20% (v/v) FBS (HyClone), 90 mg/ml Heparin (Sigma-Aldrich), 20 mg/ml Endothelial Cell Growth Supplement (BD Biosciences), 100 U/mL penicillin, 100 µg/mL streptomycin, sodium pyruvate (1 mmol/l) at 37°C with 5% CO2 in a humidified incubator. Cells were cultured in flasks or dishes coated with 0.1% Gelatin (Sigma-Aldrich). For experiments, cells from passage 6 to 8 were used at a confluence of 80–90% and were transferred to medium M199 without serum for 4 h prior to use in experiments, unless otherwise indicated.
Immunofluorescence
Adherent endothelial cells (ECs) were fixed with 4% paraformaldehyde in PBS and then permeabilized with 0.4% Triton X-100 in PBS. After washing with PBS, fixed cells were incubated in blocking solution (3% BSA in PBS) for one hour at room temperature. Cells were then probed with YAP antibody (63.1, sc-101199) (1:200 diluted in 3% BSA in PBS) and incubated at 4°C overnight. Subsequently, cells were washed with PBS and incubated with Alexa Fluor 488– conjugated goat-anti mouse antibody diluted in blocking solution (1:1000 dilution) for one hour at room temperature. The cells were washed with PBS and the nuclei were stained using a Hoechst33342 stain (1:10,000).
Image analysis
To determine the nuclear/cytoplasmic ratios for YAP localization, the average fluorescence intensity in the nucleus and just outside the nucleus (cytoplasm) was measured in individual cells. The Image analysis was performed using Zeiss analysis imaging software. The cells displayed in the appropriate figures were representative of 80% of the population.
Western blot analysis
Confluent cultures of endothelial cells, grown on 35-mm tissue culture dishes, were incubated in serum-free medium for 4 h and then treated as described in individual experiments. The cultures were then washed with ice-cold PBS and directly lysed in 2X SDS-PAGE sample buffer [200 mmol/L Tris-HCl (pH 6.8), 2 mmol/L EDTA, 0.1 mol/L Na3VO4, 6% SDS, 10% glycerol, and 4% 2-mercaptoethanol]. The proteins were separated on 4% to 15% SDS–polyacrylamide and then transferred to Immobilon-P membranes (Millipore). For detection of proteins, membranes were blocked in 5% nonfat dried milk in PBS and incubated overnight with the respective primary antibodies diluted in PBS containing 0.1% Tween. Primary antibodies bound to immunoreactive bands were visualized by enhanced chemiluminescence detection system with horseradish peroxidase (HRP)-conjugated anti-mouse or anti-rabbit antibody, and a FUJI LAS-4000 mini luminescent image analyzer. Quantification of the bands was performed using the FUJI Multi Gauge V3.0 analysis program.
RNA extraction and quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
RNA was isolated from the endothelial cells by using PureLinkTM RNA Mini Kit (Invitrogen) according to manufacturer’s protocol. 1 µg of the isolated RNA was taken for cDNA preparation using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Quantitative RT-PCR was performed using the Applied Biosystems StepOne system and the amplifications were done using the TaqMan Fast Advanced Master Mix. Relative fold change was calculated using the formula 2−ΔΔCt. Gene-specific Homo sapiens oligonucleotide primers for CTGF (assay ID: Hs00170014_m1), CYR61 (assay ID: Hs00155479_m1), and 18S (assay ID: Hs03928990_g1) were obtained from Life Technologies.
Knockdown of YAP/TAZ levels via siRNA transfection
Silencer select siRNAs were designed to target human YAP (Life Technologies #4427037, siRNA_id: s20367) and TAZ (Santa Cruz, sc-38568). Cells were transfected using the reverse transfection method. Either silencer select nontargeting negative control (25 nmol/L) or a target siRNA (25 nmol/L) was mixed with Lipofectamine RNAiMAX according to the manufacturer’s protocol and added to 35-mm tissue culture plates. Two to three days after transfection, cells were used for experiments. Immunoblotting with anti-YAP/TAZ antibody (D24E4) was performed to monitor the efficiency of siRNA knockdown.
Cell proliferation assays by BrdU incorporation
DNA synthesis was measured by BrdU incorporation using BD Pharmingen BrdU flow kits (BD Biosciences) according to the manufacturer’s protocol. Briefly, ECs grown to 70% confluence in 35-mm culture dishes coated with 0.1% gelatin were transfected with YAP/TAZ siRNA or non-targeted negative control (Non-Targ). Two days after transfection 10 µM BrdU was added to the cell culture for 2 hours. The cells were then detached with Accutase (Innovative Cell Technologies, San Diego, CA), fixed and permeabilized at room temperature. DNA was denatured by incubation with DNase for 60 min at 37°C. The cells were incubated with FITC–anti-BrdU Ab for 20 min at room temperature, and then total DNA was stained with 7-aminoactinomycin D (7-AAD). Thereafter, the cells were analyzed for simultaneous green (FL1) and red (FL3) fluorescence emission on a FACSCalibur flow cytometer (Becton Dickinson).
Cell migration assays by wound healing
Confluent Non-Targ control and YAP/TAZ siRNA transfected ECs grown in 35-mm culture dishes were starved with 5% FBS for 4 hours. Starved cells were then treated with 10 µg/ml mitomycin C for 2 hours to inhibit cell proliferation. A scratch wound was created with a sterile 200-µl pipette tip and dishes were rinsed twice with M199 to remove detached cells. Cells were treated with anti-HLA I mAb W6/32 for 16 h. The cells were then fixed with 4% paraformaldehyde, stained with Wright–Giemsa (Sigma-Aldrich) and wound closure was monitored by microscopy. The cell number between two initiated front edges was counted (10 fields). Migration rate was determined as relative fold of wound healing as compared to unstimulated Non-Targ siRNA transfected ECs.
Statistical analysis
Each experiment was repeated three times independently. Unless otherwise noted, data are presented as mean ± SEM. Differences in protein phosphorylation, cell proliferation, or cell migration were determined using Student’s t-test and were considered significant if p < 0.05.
Results
Growing ECs exhibit YAP localization and require YAP for their proliferation.
Because cell density regulates YAP localization through activation of the Hippo protein kinase pathway in a variety of cell types, we initially examined the localization of endogenous YAP in human aortic ECs after various times of plating. YAP predominantly localized in the nuclei of ECs growing at low cell density (2 days after plating). In contrast, YAP localization shifted to the cytoplasm in dense cultures of ECs, i.e. 6 days after plating (Fig. 1A, Left). Quantification of YAP nuclear/cytoplasmic ratio in multiple individual cells corroborated that cell density regulates YAP localization in ECs (Fig. 1B).
Fig. 1. Cell density regulates YAP localization in ECs.
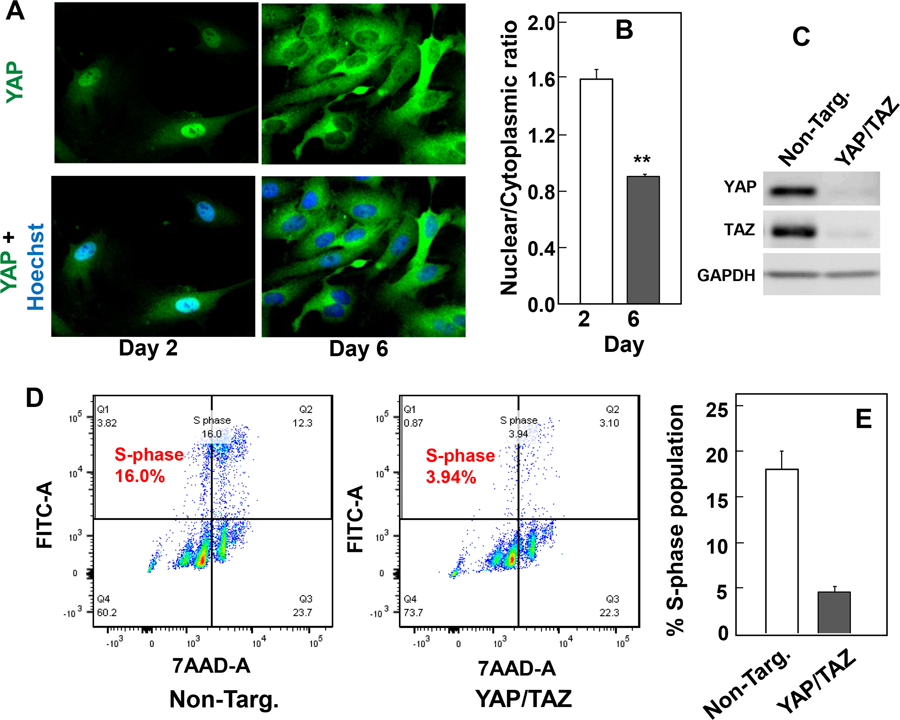
A, Human aortic endothelial cells (ECs) were plated at low density (50 x 104), cells were fixed with 4% paraformaldehyde either at day 2 (low density) or day 6 (high density) after plating. The cultures were then stained with an antibody that detects total YAP and with Hoechst 33342 to visualize the cell nuclei. B, Bars represent the ratio of nuclear/cytoplasm (200–300 cells) mean ± S.E. with similar results obtained in three independent experiments, **p<0.01. Requirement of YAP and TAZ for entry into S phase in growing EC cells. C and D, Endothelial cells, plated at low density, were transfected with non-target siRNA (Non-Targ.) or siRNAs targeting YAP and TAZ for 48 h. C, ECs were then lysed with 2 X SDS-PAGE sample buffer and analyzed by immunoblotting with antibodies that detect total YAP and TAZ. GAPDH was used as loading control. D, Two days after transfection, BrdU was added to the cell culture and ECs proliferation was measured by flow cytometry. E, Bars mean ± S.E., represent the percentage of S-phase population in control non-target (Non-Targ.) and YAP/TAZ siRNA (YAP/TAZ) transfected EC. Similar results were obtained in two independent experiments.
To determine whether YAP/TAZ plays a role in the proliferation EC cells, low density ECs (nuclear YAP) were transfected with non-targeting oligonucleotides or with siRNAs targeting YAP and TAZ. We verified that the transfection of siRNAs effectively decreased the level of YAP and TAZ proteins in ECs (Fig. 1C). The proportion of ECs engaged in DNA synthesis was assessed by flow cytometry after a 2 h pulse with BrdU. Knock down of YAP/TAZ caused 80% decrease in the proportion of ECs engaged in DNA synthesis (Fig. 1D). These results indicate that YAP/TAZ play a critical role in promoting proliferation in ECs.
Class I HLA antibody induces YAP nuclear localization and activity in confluent cultures of human ECs.
In subsequent experiments, we used confluent ECs to examine whether Ab-mediated cross-linking of HLA I molecules on the surface of these cells regulates YAP localization, phosphorylation and transcriptional co-activator activity. To this end, dense cultures of ECs transferred to serum-free medium for 4 h, were stimulated with W6/32, a monoclonal antibody against HLA-I, at 0.1 or 1 μg/ml for 60 min, fixed and stained with an Ab that detects endogenous YAP. In unstimulated cells ECs and in agreement with the results shown in Fig. 1 A, B, YAP localized mainly in the cytoplasm. Stimulation of these cells with Ab W6/32 at either 0.1 or 1 μg/ml induced a significant translocation of YAP to the nucleus (Fig. 2 A). Similar results were obtained with a different human HLA I Ab (HLA-A2, Fig. 2 A) and in other experiments using primary human aortic endothelial cells isolated from the aortic rings (Results not shown). Image analysis of multiple individual cells corroborated that exposure of ECs to W6/32 promoted YAP nuclear localization (Fig. 2 B). The nuclear localization of YAP in response to Ab-induced HLA I crosslinking was comparable to that induced by thrombin, an agonist that activates PAR1, a protease-activated GPCR known to stimulate YAP nuclear import in other cell types (32). In contrast, exposure of ECs to IgG (a negative control) or to Ab CD105 directed against endoglin, did not produce any significant effect on YAP localization (Fig. 2 A; quantification in panel B). Image analysis also demonstrated that the ratio of nuclear to cytoplasmic YAP in the unstimulated cells follows a normal distribution (Fig. 2 C, open bars). Treatment of ECs with W6/32 shifted the distribution to the right (Fig. 2 C, closed bars), in line with YAP translocation from the cytoplasm to the nucleus. These results demonstrate that Ab-mediated crosslinking of HLA I induces nuclear import of YAP in confluent cultures of human ECs.
Fig. 2. Class I HLA antibody induces YAP nuclear localization in confluent cultures of human ECs.

A, Confluent ECs were stimulated with mouse IgG (m-IgG, 0.1 µg/ml), human IgG (h-IgG, 0.1 µg/ml), anti-CD105 mAb (0.1 µg/ml), anti-W6/32 HLA-I mAb (0.1 and 1 µg/ml), human mAb anti-HLA A2 (0.1 µg/ml) or thrombin (1U/ml) for 60 min. The cultures were fixed with 4% paraformaldehyde and stained for YAP and Hoechst 33342 to visualize the cell nuclei. B, Bars represent the ratio of nuclear/cytoplasm of unstimulated and stimulated ECs mean ± S.E n = 250− to 300, *p<0.01. Similar results were obtained in three independent experiments. C, Histogram represents the distribution of unstimulated (control, open bars) and anti-W6/32 HLA-I mAb (0.1 µg/ml) (closed bars) stimulated ECs as a function of nuclear/cytoplasmic ratio of YAP immunofluorescence based on the analysis of 300 cells for each condition.
Class I HLA antibody induces YAP dephosphorylation in confluent cultures of human ECs.
The phosphorylation of YAP by LATS1/2 plays a major role in restricting its cellular localization to the cytoplasm (Ser127) and in promoting its degradation by the ubiquitin–proteasome system (Ser397) in many cell types (17, 22) but these modifications have not been examined in ECs challenged with Ab that crosslink HLA I. Given that Ab-induced ligation of HLA I promotes YAP nuclear localization, we determined whether HLA I ligation regulates YAP phosphorylation at Ser127 and Ser397, highly conserved residues located within a consensus sequence phosphorylated by the Hippo kinases LATS1/2 (HxRxxS). ECs were stimulated with Ab W6/32 at either 0.1 μg/ml or 1 μg/ml for various times (15, 30 or 60 min) and lysed. Cell lysates were analyzed by Western blotting with antibodies that detect the phosphorylated state of YAP at Ser127 and Ser397. As shown in Fig. 3 A, treatment of ECs with Ab W6/32 induced a rapid decrease in YAP phosphorylation at Ser127. Engagement of HLA I produced an even more pronounced decrease in YAP phosphorylation at Ser397. (Fig 3 B). Stimulation of parallel cultures of ECs with thrombin induced a similar decrease in YAP phosphorylation at Ser127 and Ser397. We also detected a marked decrease in YAP phosphorylation at Ser397 in primary cultures of human aortic endothelial cells challenged with W6/32 (Results not shown). In contrast, exposure of ECs to IgG (a negative control) did not produce any detectable effect on YAP phosphorylation at either Ser127 or Ser397. Collectively, the findings indicate that crosslinking of HLA I rapidly stimulates YAP nuclear localization and concomitantly decreases YAP phosphorylation at residues regulated by LATS1/2 in human ECs.
Fig. 3. Class I HLA antibody induces YAP dephosphorylation in confluent cultures of human ECs.

A and C, A, Confluent ECs were treated with either mouse IgG (IgG, 1 µg/ml, for 60 min) or anti-W6/32 HLA-I mAb (W6/32, 0.1 and 1 µg/ml) or thrombin (1U/ml) for the indicated times. Cultures were then lysed with 2X SDS-PAGE sample buffer and analyzed by immunoblotting with antibodies that detect YAP phosphorylated at Ser127 and Ser397, total YAP and GADPH as a loading control were. B and D, Quantification of phosphorylated at Ser127 and Ser397 using total YAP to normalize. The results represent the mean ± SE; n=3 (independent experiments) and are expressed as ratio of YAP phosphorylated at Ser127 or Ser397 and total YAP. Anti-W6/32 HLA-I mAb significantly decreased YAP phosphorylation at Ser127 and Ser397 as compared with IgG controls (*p<0.05).
Class I HLA antibody induces YAP activity and YAP-dependent migration in confluent cultures of human ECs.
YAP localization may not always be a consistent marker of YAP co-activator activity (33). Consequently, we next determined whether the rapid nuclear import and dephosphorylation of YAP in response to crosslinking of HLA I promotes its coupling to the transcription factors of the TEA domain–containing transcription factors (TEAD1–4) and thereby stimulate the expression of YAP/TEAD-regulated genes, including CTGF and CYR61. CTGF is one of the best-characterized direct target gene of YAP that contains three putative YAP-TEAD binding sites (GGAATG) in its promoter region. As shown in Fig. 4 A, Ab W6/32-mediated crosslinking of HLA I in ECs enhanced the level of CTGF and CYR61 transcripts, as determined by qRT-PCR. The stimulatory effect of Ab W6/32 at 1μg/ml was comparable to that induced by a high concentration of thrombin (I U/ml). In contrast, exposure of ECs to IgG (a negative control) did not produce any detectable effect on CTGF or CYR61 expression (Fig. 4 A). Collectively, these results indicate that YAP activation is a novel early point of transcriptional convergence in human aortic ECs stimulated by Ab-mediated crosslinking of HLA I.
Fig. 4. A, Class I HLA antibody induces YAP activity and YAP-dependent migration in confluent cultures of human ECs.
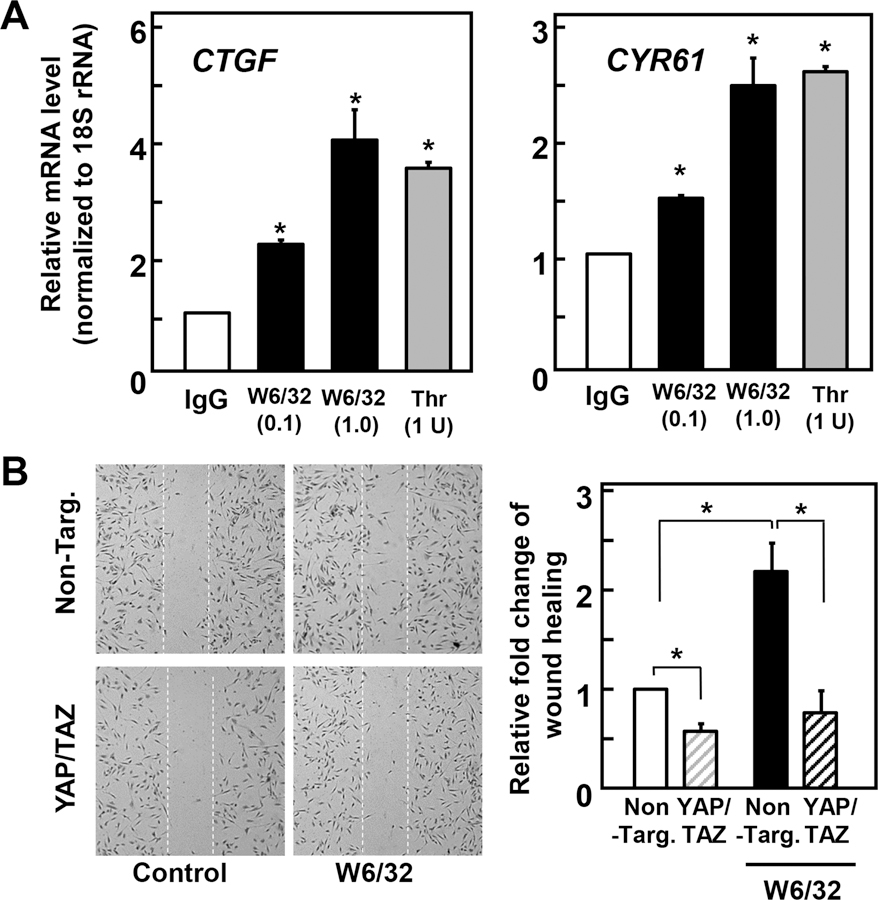
A, Confluent ECs were treated with mouse IgG (IgG, 1 µg/ml), anti-W6/32 HLA-I mAb (W6/32, 0.1 and 1 µg/ml) or thrombin (Thr, 1U/ml) for 60 min, as indicated. RNA was isolated and relative levels (n=3) of CTGF or CYR61 mRNA compared with 18S mRNA were measured by RT-qPCR. Data are presented as mean ± SEM n=3, *p<0.05, compared to IgG control. B, Confluent ECs transfected with either non-targeted (Non-Targ.) or with siRNAs targeting YAP and TAZ (YAP/TAZ) were pretreated with 10 mg/ml mitomycin C for 2 h to inhibit cell proliferation. A scratch wound was then created with a sterile 200-ml pipette tip. After washing, wounded cells were stimulated with or without 1 µg/ml HLA-I mAb W6/32 for 16 h. The cultures were then fixed with 4% paraformaldehyde and stained with Giemsa stain. Representative microscopy fields are shown. C, Bars represents relative migration (average of 10 fields/per experiment) of ECs transfected with non targeted (Non-Targ.) or with siRNAs targeting YAP and TAZ (YAP/TAZ) with or without W6/32 stimulation. Data are presented as mean ± S.E of three independent experiments (*p<0.05).
In previous studies, we demonstrated that HLA I signaling induces migration of ECs into a denuded area of the monolayer. Given that the role of YAP/TAZ in cell migration depends on cell context (34, 35), we determined whether YAP/TAZ plays a role in mediating HLA I-stimulated migration in ECs. Because knockdown of YAP/TAZ could reduce the number of cells in the denuded area of the wound by inhibiting cell proliferation rather than migration, we examined migration of ECs pretreated with mitomycin C, a DNA cross-linking agent, to prevent cell proliferation. As shown in Fig. 4 B, transfection of siRNAs targeting YAP/TAZ completely blocked the migration of ECs into the denuded area of the monolayer stimulated by ligation of HLA I, as shown using a scratch wound assay. These results indicate that endogenous YAP mediates the increase in cell migration induced by HLA I ligation in ECs.
Role of Src family kinases (SFKs) in mediating YAP activation by Ab W6/32-induced crosslinking of HLA I in ECs
We next explored the mechanism by which Ab W6/32-mediated crosslinking of HLA I induces YAP nuclear localization and stimulates YAP/TEAD activity in ECs. One of earliest events elicited by HLA I ligation in ECs is the activation of Src kinases (36, 37). Src family kinases (SFK) have been implicated in the regulation of YAP localization and function in other cell types (38) but the mechanisms are cell-context dependent and the role of SFKs in YAP regulation by HLA I in ECs was not examined before. To determine whether Src is required for YAP activation in ECs challenged with HLA I Ab, we treated confluent cultures of ECs with or without the potent SFK inhibitor dasatinib (39) prior to stimulation with Ab W6/32. Exposure to dasatinib induced cytoplasmic localization of YAP in unstimulated ECs and blocked the nuclear import of YAP induced by W6/32 stimulation in these cells (Fig. 5). Similar to dasatinib, the SFK inhibitors sacaratinib and PP2 prevented YAP nuclear localization induced by crosslinking of HLA I. Furthermore, dasatinib and PP2 also prevented the increase in CTGF and CYR61 mRNA levels induced by W6/32 stimulation. These results indicated that SFKs mediate the nuclear localization and transcriptional co-activator activity of YAP induced by HLA I signaling promoted by AbW6/32 in ECs.
Fig. 5. Src family kinase (SFK) inhibitors suppresses YAP nuclear localization and YAP/TAZ–regulated gene expression in ECs.

A, Confluent ECs were treated without (−) or with the SFK inhibitors Dasatinib (Das, 1 μM), Saracatinib (Sar, 10 μM) or PP2 (5 μM), as indicated for 60 min. ECs were then stimulated with W6/32 (0.1 μg/ml) for 60 min. ECs were fixed with 4% paraformaldehyde and stained with an antibody that detects total YAP (green) and Hoechst 33342 to visualize the cell nuclei. B, Bar graphs represent ratio of nuclear/cytoplasmic ratio of unstimulated and stimulated ECs (300 cells) with or without pretreatment of SFK inhibitors. Data are presented as mean ± S.E of three independent experiments (*p<0.05). C, Confluent ECs were treated without (−) or with SFK inhibitors Dasatinib (Das,1 μM) or PP2 (5 μM) as indicated for 60 min. ECs were then stimulated with W6/32 (0.1 μg/ml) for 60 min. RNA was isolated and relative levels (n=3) of CTGF and CYR61 mRNAs compared with 18S mRNA was measured by RT-qPCR. Data are presented as mean ± SEM; n= 3 (*p<0.05).
As Src mediates YAP activation via different mechanisms in different cell types, we next determined whether Src mediates YAP activation through inhibition of the Hippo pathway. In support of this possibility, treatment with increasing concentrations of dasatinib (0.1–1μM) produced a striking increase in YAP phosphorylation at Ser397, a site targeted by LATS (Fig. 6). We verified that dasatinib, at concentrations that promoted robust YAP phosphorylation, completely inhibited SFKs, as judged by the phosphorylation of FAK at Tyr577, a residue within the kinase activation loop phosphorylated by SFKs and a useful biomarker of SFK inhibition within intact cells (40). Treatment with PP2 (5–10 μM) also induced YAP phosphorylation at Ser397 and attenuated FAK phosphorylation at Tyr577 though less effectively than dasatinib (Fig. 6).
Fig. 6. A, Src inhibition induces phosphorylation of YAP at Ser397.
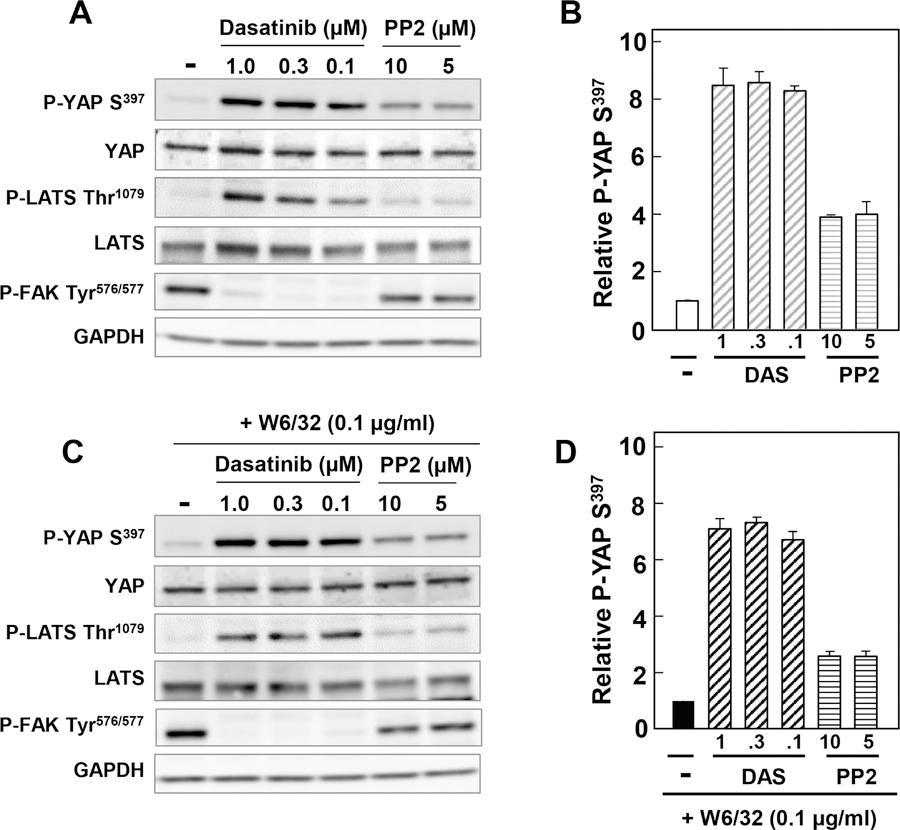
A to D, Confluent ECs were treated without (−) or with the SFK inhibitors Dasatinib (1, 0.3 and 0.1 μM) or PP2 (10 and 5 μM) for 2 h and treated without (A, B) or with 0.1 μg/ml W6/32 (C, D) for an additional 1h. Cultures were then lysed with 2X SDS-PAGE sample buffer and analyzed by immunoblotting with antibodies that detect YAP Ser397, total YAP, phospho LATS Thr1079, LATS2, P-FAK Tyr576/577and GAPDH. A, C Representative western blots in the absence (upper) or presence (lower) of W6/32 stimulation. B, D Bars are the relative fold increases compared to control, mean ± n = 3.
To test further whether the Hippo pathway mediates the increase in YAP phosphorylation, we examined the impact of SKF inhibitors on LATS1/2 activity in ECs. As shown in Fig. 6, exposure to dasatinib (0.1–1 μM) produced a notable increase in LATS1/2 phosphorylation at Thr1079, a site targeted by MST1/2 in the hydrophobic motif of LATS1/2 leading to LATS1/2 autophosphorylation and activation. These results suggest that SFK inhibition leads to dramatic stimulation of LATS1/2 activity and consequent YAP phosphorylation at inhibitory serine sites that regulate its nuclear/cytoplasmic distribution and transcriptional co-activator activity.
Discussion
Solid organ transplant recipients exhibiting donor specific antibodies most notably against HLA are at a higher risk for graft loss due to chronic antibody-mediated rejection (cAMR) and develop a progressive vascular disease known as transplant vasculopathy (6). Despite the important problem of donor specific antibodies in poorer graft outcomes, the mechanisms mediating antibody-mediated graft injury remain poorly defined. Specifically, a major gap in our current understanding is the identity of the key downstream transcriptional programs in HLA signaling that drive EC proliferation and migration.
The Hippo pathway, originally identified in Drosophila, is attracting intense interest as a key regulator of organ-size, tissue regeneration, tumorigenesis and GPCR signaling (17). Canonical Hippo signals in vertebrate cells proceed through a serine/threonine kinase cascade wherein MST1/2 kinases phosphorylate and activate LATS1/2. In turn, LATS1/2 phosphorylate YAP and TAZ at multiple serine residues. The phosphorylation of YAP by LATS1/2 at Ser127 creates binding sites for 14–3–3 proteins, which localize and anchor YAP in the cytoplasm. In turn, phosphorylation of YAP by LATS1/2 at Ser397 promotes proteolytic degradation. Despite its potential importance in the context of cAMR, the impact of Ab-mediated crosslinking of HLA molecules on YAP localization, phosphorylation and transcriptional co-activator activity has not been examined and the role of YAP in HLA signaling in ECs remained unknown.
Initially, our results demonstrate that growing cultures of ECs display nuclear localization of YAP and that knock down of YAP/TAZ strikingly impairs the entry of ECs into the S phase of the cell cycle. Subsequently, we show that stimulation of confluent cultures of ECs with the monoclonal antibody W6/32 directed against HLA-I induces rapid YAP translocation from the cytoplasm to the nucleus and concomitantly decreases YAP phosphorylation at Ser397 and Ser127, residues phosphorylated by LATS1/2. In line with the stimulation of YAP nuclear import, Ab-induced HLA I activation promotes expression of YAP/TEAD-regulated genes, including CTGF and CYR61. The products of these genes (i.e. CTGF and CYR61) are matricellular proteins that are involved in cell adhesion and migration (41) and our previous studies demonstrated that HLA I signaling induces migration of ECs into a denuded area of the monolayer. Here, we found that knock down of YAP/TAZ markedly inhibits HLA I-stimulated migration in ECs. Our results indicate that activation of the YAP/TEAD axis is a novel early point of transcriptional convergence in HLA signaling in human endothelial cells leading to migration.
Our previous studies demonstrated that crosslinking of HLA-I molecules at the surface of ECs promotes molecular association between HLA-I and the integrin β4 subunit thereby triggering the subsequent activation of intracellular signals involving Src kinases (7), one of the earliest events in HLA I signaling (36, 37). In agreement with this model, dimerization of the cytoplasmic domain of the integrin β4 subunit leads to the activation of SKF signaling (42, 43). Here, we used Src family kinases (SFK) inhibitors, including dasatinib, sacaratinib and PP2 to determine the role of Src kinases in the regulation of YAP in response to HLA I Ab ligation. We found that treatment of intact ECs with SFK inhibitors induced cytoplasmic localization of YAP in unstimulated ECs and strikingly blocked the nuclear import of YAP induced by Ab-induced HLA I activation in these cells. Furthermore, SKF inhibitors also prevented the increase in the expression of the YAP/TEAD-regulated genes CTGF and CYR61 induced by HLA I stimulation. These results indicate that SFKs mediate the nuclear localization and transcriptional co-activator activity of YAP induced by HLA I signaling in ECs.
Several studies have demonstrated that Src promotes YAP/TAZ activity through multiple independent mechanisms in different cell types (44), including repression of LATS1/2 activity (38, 45). Our results show that treatment of intact ECs with dasatinib produced a marked increase in YAP phosphorylation at sites targeted by LATS1/2, including Ser127 and Ser397, and in LATS1/2 phosphorylation of Thr1079, a site targeted by MST1/2 in the hydrophobic motif of LATS1/2 leading to its autophosphorylation and activation. Dasatinib, at concentrations that promoted robust YAP phosphorylation and LATS1/2 activation, profoundly inhibited SFK activity, as judged by the phosphorylation of FAK at Tyr576/577, residues within the activation loop of FAK phosphorylated by SKF, which are recognized biomarkers of SFK inhibition within intact cells (40). These results imply that SFKs repress LATS1/2 activity in ECs and consequently prevent YAP phosphorylation at inhibitory serine sites (Ser397 and Ser127) that regulate its nuclear/cytoplasmic distribution and activity in these cells. Additional experimental work will be required to define whether SKF directly phosphorylate and inhibit LATS1 (45), repress LATS1/2 activity via phosphorylation of proteins that bind to LATS1/2 and regulate their activity (38) or block Hippo kinases through PI3K activation (46, 47). These mechanisms are not mutually exclusive and therefore it is conceivable that HLA I activation induces YAP activation through multiple Src-dependent pathways.
As discussed above, donor specific HLA antibodies are an important problem in poorer graft outcomes. Our results identify the Src/YAP axis as a key player in promoting the proliferation and migration of ECs that are critical in the pathogenesis of TV. While targeting transcriptional co-activators is challenging, a number of FDA-approved inhibitors of SKF have been developed for use in oncology but also proposed for non-malignant disorders (48). Given the findings presented here, these agents could be repurposed for the treatment and prevention of cAMR. To extend further this notion, it will be also important in the future to define the role of the Src/YAP axis in EC signaling as a point of convergence in the action of HLA Class II as well as non-HLA antibodies (3) that also contribute to transplant rejection.
Key Points.
HLA Class I signaling induces robust activation of YAP in human endothelial cells.
Src kinases mediate YAP activation in response to HLA I signaling in ECs.
Src/YAP plays a critical role in HLA I-induced proliferation and migration of ECs.
Acknowledgments
We like to thank the Imaging and Stem Cell Biology Core of the CURE: Digestive Diseases Research Core Center (P30 DK041301).
This work was supported by a grant to EFR and ER of the Connie Frank and Evan Thompson Program for Collaborative Restorative Transplantation Research. EFR is also supported by the National Institute of Allergy and Infectious Diseases Grants RO1 AI135201, NIAID U19 AI128913, P01AI120944 and NIAID U01 AI-124319. ER is also supported by Grants R01 DK100405, P30 DK041301, P01CA236585 and Department of Veterans Affair Merit Award 1I01BX003801.
Footnotes
Disclosures: The authors have no financial conflict of interest.
References
- 1.Abrahimi P, Liu R, and Pober JS 2015. Blood Vessels in Allotransplantation. Am J Transplant 15: 1748–1754. [DOI] [PubMed] [Google Scholar]
- 2.Valenzuela NM, and Reed EF 2015. Antibodies to HLA Molecules Mimic Agonistic Stimulation to Trigger Vascular Cell Changes and Induce Allograft Injury. Curr Transplant Rep 2: 222–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kummer L, Zaradzki M, Vijayan V, Arif R, Weigand MA, Immenschuh S, Wagner AH, and Larmann J 2020. Vascular Signaling in Allogenic Solid Organ Transplantation - The Role of Endothelial Cells. Front Physiol 11: 443–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fishbein GA, and Fishbein MC 2012. Morphologic and immunohistochemical findings in antibody-mediated rejection of the cardiac allograft. Hum Immunol 73: 1213–1217. [DOI] [PubMed] [Google Scholar]
- 5.Loupy A, Toquet C, Rouvier P, Beuscart T, Bories MC, Varnous S, Guillemain R, Pattier S, Suberbielle C, Leprince P, Lefaucheur C, Jouven X, Bruneval P, and Duong Van Huyen JP 2016. Late Failing Heart Allografts: Pathology of Cardiac Allograft Vasculopathy and Association With Antibody-Mediated Rejection. Am J Transplant 16: 111–120. [DOI] [PubMed] [Google Scholar]
- 6.Valenzuela NM, and Reed EF 2017. Antibody-mediated rejection across solid organ transplants: manifestations, mechanisms, and therapies. J Clin Invest 127: 2492–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang X, Rozengurt E, and Reed EF 2010. HLA class I molecules partner with integrin beta4 to stimulate endothelial cell proliferation and migration. Sci Signal 3: ra85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jin YP, Korin Y, Zhang X, Jindra PT, Rozengurt E, and Reed EF 2007. RNA interference elucidates the role of focal adhesion kinase in HLA class I-mediated focal adhesion complex formation and proliferation in human endothelial cells. J Immunol 178: 7911–7922. [DOI] [PubMed] [Google Scholar]
- 9.Jin YP, Singh RP, Du ZY, Rajasekaran AK, Rozengurt E, and Reed EF 2002. Ligation of HLA class I molecules on endothelial cells induces phosphorylation of Src, paxillin, and focal adhesion kinase in an actin-dependent manner. J Immunol 168: 5415–5423. [DOI] [PubMed] [Google Scholar]
- 10.Jindra PT, Jin YP, Jacamo R, Rozengurt E, and Reed EF 2008. MHC class I and integrin ligation induce ERK activation via an mTORC2-dependent pathway. Biochem Biophys Res Commun 369: 781–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ziegler ME, Jin YP, Young SH, Rozengurt E, and Reed EF 2012. HLA class I-mediated stress fiber formation requires ERK1/2 activation in the absence of an increase in intracellular Ca2+ in human aortic endothelial cells. American journal of physiology. Cell physiology 303: C872–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lepin EJ, Zhang Q, Zhang X, Jindra PT, Hong LS, Ayele P, Peralta MV, Gjertson DW, Kobashigawa JA, Wallace WD, Fishbein MC, and Reed EF 2006. Phosphorylated S6 ribosomal protein: a novel biomarker of antibody-mediated rejection in heart allografts. Am J Transplant 6: 1560–1571. [DOI] [PubMed] [Google Scholar]
- 13.Ziegler ME, Souda P, Jin YP, Whitelegge JP, and Reed EF 2012. Characterization of the endothelial cell cytoskeleton following HLA class I ligation. PloS one 7: e29472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jindra PT, Jin YP, Rozengurt E, and Reed EF 2008. HLA Class I Antibody-Mediated Endothelial Cell Proliferation via the mTOR Pathway. J Immunol 180: 2357–2366. [DOI] [PubMed] [Google Scholar]
- 15.Li F, Lai C, Kobashigawa JA, Fishbein MC, and Reed EF 2011. Phosphorylated Signaling Proteins as Biomarkers of Antibody-Mediated Heart Allograft Rejection. J Heart Lung Transpl 30: S41–S41. [Google Scholar]
- 16.Jin YP, Valenzuela NM, Ziegler ME, Rozengurt E, and Reed EF 2014. Everolimus Inhibits Anti-HLA I Antibody-Mediated Endothelial Cell Signaling, Migration and Proliferation More Potently Than Sirolimus. Am J Transplant 14: 806–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu F-X, and Guan K-L 2013. The Hippo pathway: regulators and regulations. Genes Dev 27: 355–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moroishi T, Hansen CG, and Guan K-L 2015. The emerging roles of YAP and TAZ in cancer. Nat Rev Cancer 15: 73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Straßburger K, Tiebe M, Pinna F, Breuhahn K, and Teleman AA 2012. Insulin/IGF signaling drives cell proliferation in part via Yorkie/YAP. Dev Biol 367: 187–196. [DOI] [PubMed] [Google Scholar]
- 20.Wang J, Sinnett-Smith J, Stevens JV, Young SH, and Rozengurt E 2016. Biphasic Regulation of Yes-associated Protein (YAP) Cellular Localization, Phosphorylation, and Activity by G Protein-coupled Receptor Agonists in Intestinal Epithelial Cells: A NOVEL ROLE FOR PROTEIN KINASE D (PKD). J Biol Chem 291: 17988–18005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu F-X, Zhao B, and Guan K-L 2015. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 163: 811–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meng Z, Moroishi T, and Guan K-L 2016. Mechanisms of Hippo pathway regulation. Genes Dev 30: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Enzo E, Santinon G, Pocaterra A, Aragona M, Bresolin S, Forcato M, Grifoni D, Pession A, Zanconato F, Guzzo G, Bicciato S, and Dupont S 2015. Aerobic glycolysis tunes YAP/TAZ transcriptional activity. EMBO J 34: 1349–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Z, Wu Y, Wang H, Zhang Y, Mei L, Fang X, Zhang X, Zhang F, Chen H, Liu Y, Jiang Y, Sun S, Zheng Y, Li N, and Huang L 2014. Interplay of mevalonate and Hippo pathways regulates RHAMM transcription via YAP to modulate breast cancer cell motility. Proc Natl Acad Sci U S A 111: E89–E98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Santinon G, Pocaterra A, and Dupont S 2016. Control of YAP/TAZ Activity by Metabolic and Nutrient-Sensing Pathways. Trends Cell Biol 26: 289–299. [DOI] [PubMed] [Google Scholar]
- 26.Hansen CG, Moroishi T, and Guan K-L 2015. YAP and TAZ: a nexus for Hippo signaling and beyond. Trends Cell Biol 25: 499–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meng Z, Moroishi T, Mottier-Pavie V, Plouffe SW, Hansen CG, Hong AW, Park HW, Mo J-S, Lu W, Lu S, Flores F, Yu F-X, Halder G, and Guan K-L 2015. MAP4K family kinases act in parallel to MST1/2 to activate LATS1/2 in the Hippo pathway. Nat Commun 6: 8357–8357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim J, Kim YH, Kim J, Park DY, Bae H, Lee D-H, Kim KH, Hong SP, Jang SP, Kubota Y, Kwon Y-G, Lim D-S, and Koh GY 2017. YAP/TAZ regulates sprouting angiogenesis and vascular barrier maturation. J Clin Invest 127: 3441–3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang X, Freire Valls A, Schermann G, Shen Y, Moya IM, Castro L, Urban S, Solecki GM, Winkler F, Riedemann L, Jain RK, Mazzone M, Schmidt T, Fischer T, Halder G, and Ruiz de Almodóvar C 2017. YAP/TAZ Orchestrate VEGF Signaling during Developmental Angiogenesis. Dev Cell 42: 462–478.e467. [DOI] [PubMed] [Google Scholar]
- 30.Neto F, Klaus-Bergmann A, Ong YT, Alt S, Vion A-C, Szymborska A, Carvalho JR, Hollfinger I, Bartels-Klein E, Franco CA, Potente M, and Gerhardt H 2018. YAP and TAZ regulate adherens junction dynamics and endothelial cell distribution during vascular development. eLife 7: e31037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yeh M, Leitinger N, de Martin R, Onai N, Matsushima K, Vora Devendra K, Berliner Judith A, and Reddy Srinivasa T 2001. Increased Transcription of IL-8 in Endothelial Cells Is Differentially Regulated by TNF-α and Oxidized Phospholipids. Arterioscler Thromb Vasc Biol 21: 1585–1591. [DOI] [PubMed] [Google Scholar]
- 32.Mo J-S, Yu F-X, Gong R, Brown JH, and Guan K-L 2012. Regulation of the Hippo-YAP pathway by protease-activated receptors (PARs). Genes Dev 26: 2138–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen Q, Zhang N, Xie R, Wang W, Cai J, Choi K-S, David KK, Huang B, Yabuta N, Nojima H, Anders RA, and Pan D 2015. Homeostatic control of Hippo signaling activity revealed by an endogenous activating mutation in YAP. Genes Dev 29: 1285–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mason DE, Collins JM, Dawahare JH, Nguyen TD, Lin Y, Voytik-Harbin SL, Zorlutuna P, Yoder MC, and Boerckel JD 2019. YAP and TAZ limit cytoskeletal and focal adhesion maturation to enable persistent cell motility. J Cell Biol 218: 1369–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van der Stoel M, Schimmel L, Nawaz K, van Stalborch A-M, de Haan A, Klaus-Bergmann A, Valent ET, Koenis DS, van Nieuw Amerongen GP, de Vries CJ, de Waard V, Gloerich M, van Buul JD, and Huveneers S 2020. DLC1 is a direct target of activated YAP/TAZ that drives collective migration and sprouting angiogenesis. J Cell Sci 133: jcs239947. [DOI] [PubMed] [Google Scholar]
- 36.Jin YP, Singh RP, Du ZY, Rajasekaran AK, Rozengurt E, and Reed EF 2002. Ligation of HLA class I molecules on endothelial cells induces phosphorylation of Src, paxillin, and focal adhesion kinase in an actin-dependent manner. J. Immunol 168: 5415–5423. [DOI] [PubMed] [Google Scholar]
- 37.Jin Y-P, Korin Y, Zhang X, Jindra PT, Rozengurt E, and Reed EF 2007. RNA Interference Elucidates the Role of Focal Adhesion Kinase in HLA Class I-Mediated Focal Adhesion Complex Formation and Proliferation in Human Endothelial Cells. J Immunol 178: 7911–7922. [DOI] [PubMed] [Google Scholar]
- 38.Lamar JM, Xiao Y, Norton E, Jiang Z-G, Gerhard GM, Kooner S, Warren JSA, and Hynes RO 2019. SRC tyrosine kinase activates the YAP/TAZ axis and thereby drives tumor growth and metastasis. J Biol Chem 294: 2302–2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Williams NK, Lucet IS, Klinken SP, Ingley E, and Rossjohn J 2009. Crystal Structures of the Lyn Protein Tyrosine Kinase Domain in Its Apo- and Inhibitor-bound State. J Biol Chem 284: 284–291. [DOI] [PubMed] [Google Scholar]
- 40.Ciccimaro E, Hanks SK, and Blair IA 2009. Quantification of focal adhesion kinase activation loop phosphorylation as a biomarker of Src activity. Mol Pharmacol 75: 658–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen C-C, and Lau LF 2009. Functions and mechanisms of action of CCN matricellular proteins. Int J Biochem Cell Biol 41: 771–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Merdek KD, Yang X, Taglienti CA, Shaw LM, and Mercurio AM 2007. Intrinsic Signaling Functions of the β4 Integrin Intracellular Domain. J Biol Chem 282: 30322–30330. [DOI] [PubMed] [Google Scholar]
- 43.Giancotti FG 2007. Targeting integrin β4 for cancer and anti-angiogenic therapy. Trends Pharmacol Sci 28: 506–511. [DOI] [PubMed] [Google Scholar]
- 44.Warren JSA, Xiao Y, and Lamar JM 2018. YAP/TAZ Activation as a Target for Treating Metastatic Cancer. Cancers (Basel) 10: 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Si Y, Ji X, Cao X, Dai X, Xu L, Zhao H, Guo X, Yan H, Zhang H, Zhu C, Zhou Q, Tang M, Xia Z, Li L, Cong Y-S, Ye S, Liang T, Feng X-H, and Zhao B 2017. Src Inhibits the Hippo Tumor Suppressor Pathway through Tyrosine Phosphorylation of Lats1. Cancer Res 77: 48684880. [DOI] [PubMed] [Google Scholar]
- 46.Kim N-G, and Gumbiner BM 2015. Adhesion to fibronectin regulates Hippo signaling via the FAK–Src–PI3K pathway. J Cell Biol 210: 503–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fan R, Kim N-G, and Gumbiner BM 2013. Regulation of Hippo pathway by mitogenic growth factors via phosphoinositide 3-kinase and phosphoinositide-dependent kinase-1. Proc Natl Acad Sci U S A 110: 2569–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grimminger F, Schermuly RT, and Ghofrani HA 2010. Targeting non-malignant disorders with tyrosine kinase inhibitors. Nat Rev Drug Discov 9: 956–970. [DOI] [PubMed] [Google Scholar]