

Abstract
Starting from the MLPCN probe compound ML300 a structure-based optimization campaign was initiated against the recent severe acute respiratory syndrome coronavirus (SARS-CoV-2) main protease (3CLpro). X-ray structures of SARS-CoV-1 and CoV-2 3CLpro enzymes in complex with multiple ML300 based inhibitors, including the original probe ML300, were obtained and proved instrumental in guiding chemistry towards probe compound 41 (CCF0058981). The inhibitors disclosed utilize a non-covalent mode of action and complex in a non-canonical binding mode not observed by peptidic 3CLpro inhibitors. In vitro DMPK profiling highlight key areas where further optimization in the series is required to obtain useful in vivo probes. Anti-viral activity was established using a SARS-CoV-2 infected Vero E6 cell viability assay, and in a plaque formation assay. Compound 41 demonstrates nanomolar activity in these respective assays, comparable in potency to remdesivir. These findings have implications towards antiviral development to combat current and future SARS-like zoonotic coronavirus outbreaks.
Keywords: 3CLpro, Mpro, SARS-CoV-2, COVID-19, coronavirus
Graphical Abstract
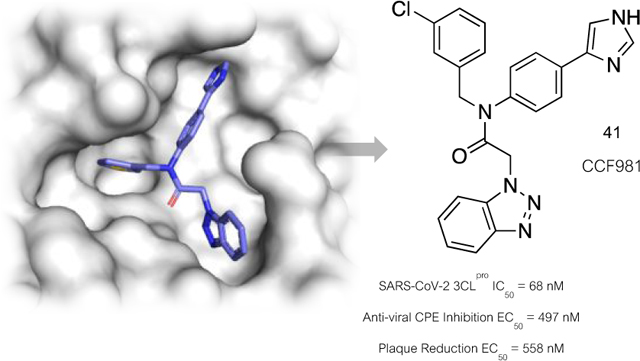
Introduction
Coronaviruses (CoV) are a family of enveloped positive-strand RNA pathogenic viruses that can cause acute and chronic conditions including central nervous system disorders, the common cold, lower respiratory tract infections, and diarrhea.1 The 229E and OC43 strains were among the first characterized human CoV strains starting in 1965.2 The novel severe acute respiratory syndrome CoV reported in 2003,3,4 that is now identified as SARS-CoV-1, became the first global human CoV pandemic leading to progressive respiratory failure in over 8,000 individuals and 916 deaths (fatality rate of 10–15%).5 In the eight years that followed, significantly less lethal human coronaviruses NL64 and HKU1 were identified and characterized.6,7 Subsequently in 2012 the SARS-like MERS (Middle East Respiratory Syndrome) was identified and found to have a low transmission rate, but significant lethality with a total of 2567 patients with confirmed infection worldwide, of which 882 (34% fatality) died from 2012 through February 2nd, 2021. To date, most confirmed MERS cases have been reported from Saudi Arabia.8 Like the SARS-CoV of 2003 and MERS of 2012, the ongoing novel SARS-CoV pandemic of 2020, known as SARS-CoV-2, the causative agent of COVID-19, presents a worldwide threat due to its ability to rapidly spread person-to-person via respiratory droplets and its remarkable capacity to suppress human immune surveillance.9 Unfortunately SARS CoV-2 has been much more extensive than that of MERS and SARS CoV-1 in its spread, with current worldwide infections, as of March 29, 2021 exceeding 126 million confirmed cases of COVID-19 and 2,778,619 confirmed deaths (fatality rate of ~2%) according to the World Health Organization (WHO).10
The coronavirus SARS-CoV-2,11 encodes multiple enzymes that are essential for viral replication.12,13 As potential therapeutic antiviral targets, the two cysteine proteases – the chymotrypsin-like or main protease (3CLpro or Mpro), and the papain-like protease (PLpro) – have garnered significant attention.14–16 Both SARS-CoV-1 and CoV-2 genomes encode a large polyprotein that is proteolytically processed by these respective cysteine proteases. In solution, 3CLpro exists primarily as a dimer and has been confirmed to be the catalytically active species.17 3CLpro is responsible for processing at 11 different cleavage sites within the coronavirus polyprotein and PLpro is responsible for cleavage at three other unique sites. Without these essential proteases replication is impaired and shuts down the viral life cycle.18 CoV 3CLpro enzymes contain three structural domains connected by flexible loops. Domains I and II are β-barrel domains and contain the catalytic active site region, whereas domain III is an α-helical domain shown to be critical for dimerization.19 The active site contains a catalytic dyad consisting of a cysteine residue (Cys-145) that acts as a nucleophile and a histidine residue (His-41) that acts as the general acid-base.
Many published inhibitors of SARS-CoV-1 3CLpro, and more recently for SARS-CoV-2 3CLpro have been peptide-like, and often include a reactive center targeted toward a covalent interaction with the catalytic cysteine, Cys145. In collaboration with Mesecar and co-workers, and other member teams from NIH MLPCN, including the Scripps Research Institute Molecular Screening Center (SRIMSC) and the Vanderbilt Specialized Chemistry Center (VSCC), we participated in efforts to develop inhibitors of a coronavirus 3CLpro in the aftermath of the SARS-CoV-1 outbreak, with a particular focus on the development of non-covalent inhibitors, leading to ML300 and ML188 (1 and 2, Figure 1).20–21 Examples of de novo, non-covalent inhibitors designed towards SARS-CoV-2 3CLpro include the recently disclosed pyridone, 3.22 In the course of the COVID-19 pandemic, two covalent inhibitors previously discovered, 4 (GC-376)23 and 5 (PF-00835231)24 have advanced into clinical trials in 2020. The phosphate pro-drug of 5, PF-07304814 is also under clinical evaluation.25
Figure 1.

Exemplary structures of reported 3CLpro inhibitors for SARS-CoV-1 and SARS-CoV-2 using either a non-covalent (top) or covalent (bottom) mechanism of action.
Among human novel CoV’s the highest sequence homology for the 3CLpro protease exists between SARS-CoV-1 and SARS-CoV-2, with an overall 96% sequence identity and 100% identity in the active site, giving rise to similar substrate specificity,26 thus providing the field with an advanced starting point for the development of SARS-CoV-2 3CLpro inhibitors. Structural biology has played a critical role in 3CLpro protease inhibitor development, beginning with the first X-ray structure in 2003 with a hexapeptidyl chloromethyl ketone inhibitor bound to TGEV 3CLpro (PDB: 1P9U)27 and SARS-CoV-1 3CLpro (PDB: 1UK4).28 Today, a multitude of X-ray crystal structures have emerged comparing CoV 3CLpro’s including SARS-CoV-2 to aid and galvanize inhibitor design efforts. High impact early contributions in the context of the ongoing pandemic include those from the Diamond Light Source XChem X-ray fragment screening program,29 Shanghai Tech University N3-peptide,30 University of Lübeck dicarbonyl compound 13b,26 peptide based aldehydes similar to 5 reported by Dai and co-workers at the Chinese Academy of Sciences,31 and baicalein and related natural products from University of Chinese Academy of Sciences.32 More recent developments include evidence from a March 2020 Science report from Qiao and co-workers33 at Sichuan University demonstrating preclinical in vivo efficacy in a human angiotensin converting enzyme 2 (hACE2) transgenic SARS-CoV-2 murine model. Using a moderate infection threshold the Sichuan team developed a peptidyl aldehyde based upon a Boceprevir design. One of the lead orally bioavailable inhibitors, MI-09, was administered at 100 mg/kg BID 1h prior to virus inoculation and then continuing for 5 days post infection. In this study compound MI-09 demonstrated significant viral RNA load reduction within 3 days, with almost no virus detected by day 5, thus setting the stage as a comparator for emerging tool compounds to be evaluated and assessed against for their efficacy and overall PK-PD.
Our focus was directed to the synthesis and further optimization of non-peptidic, non-covalent inhibitors of SARS-CoV-2 3CLpro that are derived from ML300.21 We were intrigued to investigate the ML300 series against SARS-CoV-2 for several reasons. Notably, in contrast to ML188 and prior peptidomimetics bound to 3CLpro in a canonical binding mode wherein the inhibitor accommodates substrate sub-pockets in the enzyme active-site, the SARS-CoV-1 3CLpro X-ray structure of the ML300 series HTS hit, 6 (Figure 2A) favors an induced-fit complex where key flexible amino acid side chains in the binding pocket, specifically Gln189 and Met49, take on distinct conformations and in tandem reorganize the S4 and S2 binding surface. The observed induced-fit allows the bis-aniline to occupy a newly formed channel proximal between what is traditionally the canonical S1’ and S2 pockets (S2c = S2 channel). Conversely, the pyrrole ring of 6 exists in a deeper sub-pocket of S2 (S2sp) that is more proximal to the traditional S4 pocket.34 Secondly, while the original ML300 series achieved nM inhibition against SARS-CoV-1, leading to compounds such as 7 and 8 (Figure 2C), the series was far from optimized. For example, only a handful of amide replacements presumed to occupy the upper rim of the S2c were explored. In the S2sp region, which the ML300 3-thienyl ring was presumed to act as an P2sp group (Figure 2B), only these two π-excessive heterocycles were examined during hit expansion. Moreover, cellular antiviral structure-activity relationships (SAR) of the ML300 series against SARS CoV’s remained largely unknown, although the potential for broad spectrum utility was precedent based upon studies examining ML300 series analogs targeting HKU4, a suspected reservoir host for MERS.35 Lastly, in vitro ADME properties of ML300 revealed significant hurdles including metabolic instability with no knowledge of the mechanism or site of metabolism. Based on these fundamental gaps and our interest to contribute to understanding if the benzotriazole based ML300 series may have potential as a lead for COVID-19 or future broad spectrum 3CLpro-based CoV antiviral therapies we assembled a team highly focused on the above goals.
Figure 2.

Non-canonical binding mode of MLPCN HTS compound 6 (SID 24808289) and evolution to ML300 and related bi-phenyl analogs 7 and 8 originally developed for SARS-CoV-1 3CLpro. A) Schematic of binding mode of 6 orienting the binding site; B) Solvent accessible surface of SC1 3CLpro:6 complex, illustrating flexibility and reorganization of the binding pocket (PDB: 4MDS). C) Profile of 1 (ML300) and biaryl analogs 7 and 8.
The compounds disclosed herein provide significantly improved characteristics in several areas from the original ML300 report, achieving robust nanomolar biochemical inhibition against SARS-CoV-2 3CLpro, sub-micromolar antiviral and plaque formation inhibition against the SARS-CoV-2 live virus, and a systematic DMPK and metabolite profile to support directions for future optimization. In addition, we compare and contrast several new X-ray structures of inhibitors bound to SARS-CoV-1 and CoV-2 3CLpro, including ML300 and a number of analogs generated using structure-based design.
Results and Discussion
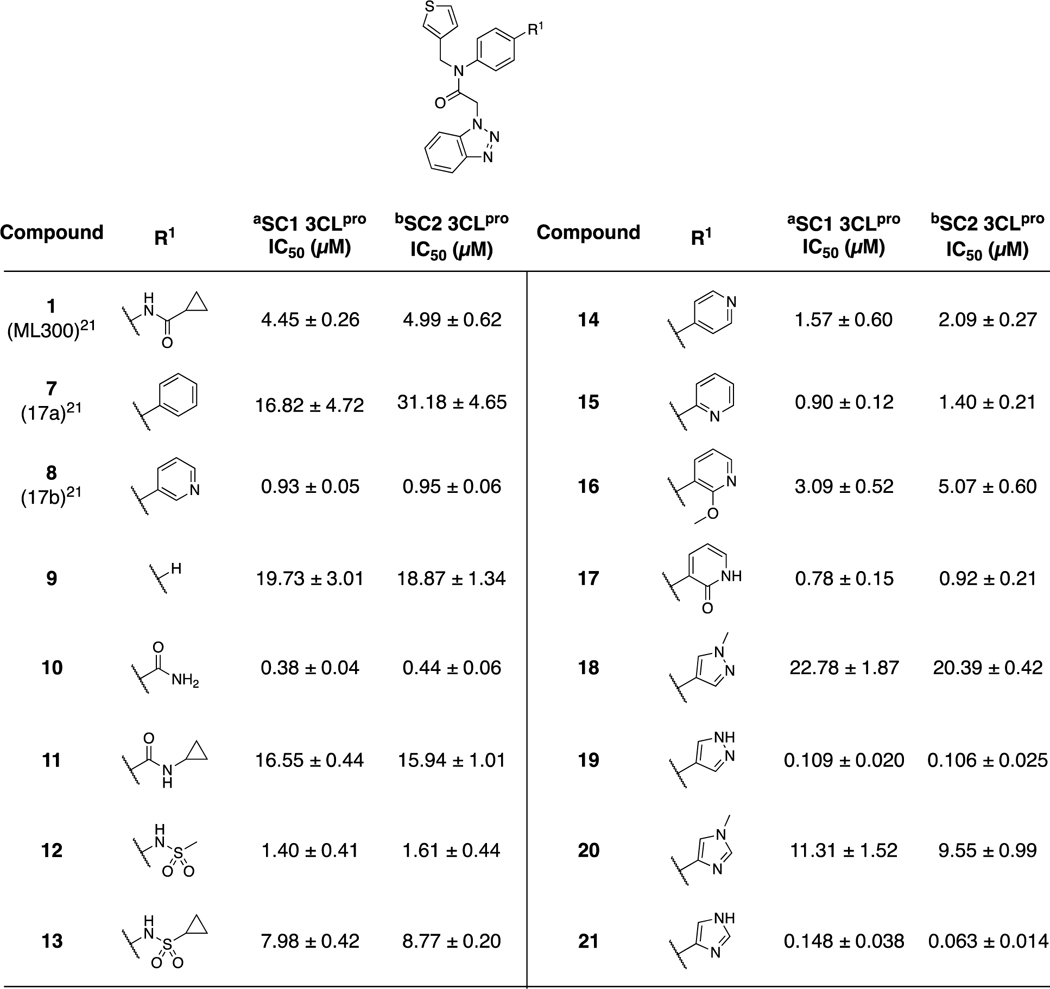
Our efforts began by initially synthesizing 1 (ML300), 7 and 8 and re-screening against SARS-CoV-1 3CLpro (SC1), obtaining for 1 an IC50 of 4.45 μM (Table 1), which was in agreement with reported data; a similar IC50 value against SARS-CoV-2 3CLpro (SC2) was also obtained. However, to our surprise, the previously published biphenyl derivative, 721 did not reconfirm it’s prior published value of 51 nM, and in fact was drastically less potent. Based on the concentration-response-curves (CRC) of compound 7 an SC1 IC50 of 16.8 μM, and SC2 IC50 of 31.2 μM was calculated, which represents potency more similar to the truncated compound, 9 (Table 1). In contrast, the 3-pyridyl congener 8 maintained an IC50 between 930–950 nM for both SC1 and SC2 enzymes that mirrored the initial report from 2013. Relative to 1 the pyridyl biaryl 8 results in ligand efficiency (LE) enhancement from 0.23 to 0.27 respectively based upon SC2 mean IC50 values. Since our findings regarding biphenyl 7 stand in contrast to the original work, we carefully examined our assay reagents, protocols, and compound fidelity. For each 3CLpro construct we utilized the native sequence of the enzyme that represents the in vivo, post-proteolytic form using the protocol and conditions reported by Mesecar and co-workers.36 We also utilized the same FRET-based 3CLpro peptide substrate as reported and achieved excellent plate uniformity using a 384-well plate format, Z-prime > 0.8. Compound CRC’s were generated and tested from at least three experiments using 384-well plate format with technical replicates on each plate. SC1 and SC2 reported IC50 values reported in Tables 1–3 reflect n = 3, with further repeats as required until CV ≤ 0.3 was achieved. The standard deviation is reported and shown for reference. As per published protocols, 0.01% Triton X-100 was included in the assay buffer to remove potential promiscuous inhibition caused by aggregation. At this time the >300-fold SC1 IC50 discrepancy for biphenyl 7 cannot be fully explained; however, it is clear that inherent high lipophilicity of this molecule may have been a contributing factor based on its calculated physicochemical properties (7 cLogP = 5.61, LogS = −6.85, ChemDraw 19.0) and behavior in FRET CRC measurements. For example, Figure 3 shows a comparison of the SC1 and SC2 3CLpro FRET inhibition curves for 1, 7, and 8. Incomplete inhibition is observed for compound 7 based on the curve profile when tested up to 100 μM maximum (Figure 3, 4-fold serial dilution). In addition, there is a clear plateau of inhibition that appears to be begin at concentrations from 25 μM onward, suggesting a likely solubility limit. In contrast, compound 8 behaves well up to a top assay concentration of 100 μM, achieving complete inhibition.
Table 1.
SARS-CoV-1 and SARS-CoV-2 3CLpro inhibition data, exploring P2c SAR
|
IC50 values are average of at least 3 independent assays, each run as technical duplicates;
SARS-CoV-1 3CLpro
SARS-CoV-2 3CLpro.
Table 3.
3CLpro inhibition data, analogs exploring aniline-biaryl P2c combinations
|
|||
|---|---|---|---|
| Compound | R1 | aSC1 3CLpro IC50 (μM) | bSC2 3CLpro IC50 (μM) |
|
| |||
| 35 |
|
0.657 ± 0.405 | 0.746 ± 0.098 |
| 36 |
|
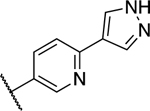
0.334 ±0.126 | 0.270 ± 0.067 |
| 37 |
|
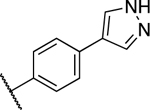
0.206 ± 0.052 | 0.328 ± 0.027 |
| 38 |
|
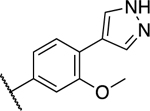
0.208 ± 0.077 | 0.197 ±0.050 |
| 39 |
|
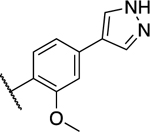
0.336 ± 0.065 | 0.333 ± 0.093 |
| 40 |
|
0.214 ±0.063 | 0.171 ±0.029 |
| 41 |
|
0.019 ±0.005 | 0.068 ± 0.023 |
IC50 values are average of at least 3 independent assays, each run as technical duplicates
SARSCoV-1 3CLpro
SARS-CoV-2 3CLpro
Figure 3.

3CLpro concentration response curves for compounds 1, 7 and 8, against SC1 (A) and SC2 (B) enzymes.
Encouraged by the sub-micromolar SC1 and SC2 inhibitory activity of pyridyl 8 we pursued a series of X-ray crystal studies and obtained high resolution structures of 8 bound to SC1 3CLpro at 1.85 Å via soaking method, in addition to a co-crystal of 1 (ML300) bound to SC2 3CLpro at 2.1 Å resolution via co-crystallization. Shown in Figure 4 is a depiction of key residues and the binding poses of 1 – SC2 3CLpro (panel A) and 8 – SC1 3CLpro (panel B).
Figure 4.

A) X-ray co-crystal structure of 1 in complex with SC2 3CLpro (PDB: 7LME) and B) 8 in complex with SC1 3CLpro (PDB: 7LMH). Left panel: key residues are highlighted as sticks and interatomic distances depicted as dashes; right panel: solvent accessible surface with occupied and neighboring pockets labeled. Binding orientation of each inhibitor is the same in the active site of each monomer of the dimer in the asymmetric unit, only one active site is shown for clarity.
Comparison of 6 – SC1 3CLpro, 1 – SC2 3CLpro, and 5 – SC2 3CLpro X-ray structures
Overall the binding orientation of 1 in the SC2 3CLpro binding pocket (Figure 4A) retains many of the key interactions found in the 6 – SC1 3CLpro complex (Figure 2B); however, key distinctions are observed within the binding site which illuminate prior SAR trends.21 In general, removal of the P3 amide of 6 as the major difference versus inhibitor 1 does not grossly effect the ligand bioactive conformation, leaving the solvent exposed S3 pocket unoccupied. In the S1 pocket, identical to 6 – SC1 3CLpro, the benzotriazole of 1 forms neighboring putative H-bonding interactions with His163 and Cys145, at 2.8 and 3.6 Å, respectively. Similarly, the amide carbonyl oxygen retains a key anchoring hydrogen bond with the backbone NH of Glu166. Within the S2 pocket, as seen previously within the compound 6 – SC1 3CLpro complex, the dynamic nature of the Gln189 loop allows a new S2sp to form to accommodate the thiophene ring, resulting in the thiophene sulfur being sandwiched between Met165 and Met49. Interestingly, the N-methyl pyrrole and thiophene conformationally present distinctly within the S2sp. For example, the bulkier N-methyl pyrrole adopts an 18° torsion to reach into the S2sp pocket, while the thiophene side chain of 6 adopts a 98° torsion with respect to the aniline ring. In addition, there is a slight frame shift of the N-benzyl amide backbone resulting in rotational shift of the aniline vector towards the S2c upper rim that appears to favorably accommodate the larger cyclopropyl amide. Relative to 5 – SC2 3CLpro peptide 5 complex (see Supplemental Figure S1, PDB: 6XHM); 5 and 1 share common H-bond interactions with Glu166 and His163 in the S1 region; however, 5 forms a covalent interaction with the catalytic Cys145 and occupies the solvent exposed S3 pocket. In contrast to 6 – SC1 3CLpro complex, for ligand 1 in chain A of the asymmetric unit the cyclopropyl anilido amide moiety maintains a rotamer in the S2c channel which positions the carbonyl at a 50° torsion from co-planarity relative to the central aniline ring, giving rise to a unique hydrogen bond with the side-chain of Ser46 (Figure 4, left). In chain B in the asymmetric unit the cyclopropyl moiety points further from the pocket towards solvent; however, for chain B, the B-factor values indicate significant motion versus the chain A inhibitor. Interestingly, this observed hydrogen bonding interaction represents an amino acid difference between the SC1 and SC2 3CLpro enzymes, namely an Ala46 in SC1 for Ser46 in SC2. Residue 46 defines part of a short helix-loop-helix motif spanning Arg40 through Lue50. Peptidomimetics efforts to date with a canonical mode of inhibition acknowledge that amino acids within the active-site between SC1 and SC2 3CLpro are 100% sequence identical. In regard to a tailored inhibitor design versus a broad spectrum inhibitor strategy, the observed interactions and residue differences noted within and near S2c for this ML300-based series may present unique challenges as well as opportunities not available to traditional inhibitor scaffolds.
Comparison of 1 – SC2 3CLpro and 8 –SC1 3CLpro- 8 X-ray structures
Relative to 1 – SC2 3CLpro complex, the 8 – SC1 3CLpro binding pose and interactions are otherwise identical with the exception of distinctions within the S2c upper rim region (Figure 4, right versus left). In regard to nearby polar residues that might engage the pyridyl nitrogen, Thr25 side chain is the closest residue in proximity; however, the distance measured at 3.8 angstroms would indicate this is less than favorable. Furthermore, no apparent water mediated interactions were observed with the ligand. In terms of π-interactions with the pyridyl ring of 8, a Ala46 backbone amide- π interaction is apparent based upon proximity and distances from the backbone carbonyl carbon and the pyridyl centroid (3.5–3.8 Å). This stacking interaction may contribute to the beneficial boost in potency of 8 versus biphenyl 7.
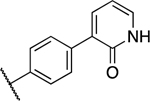
With the above new structural information and encouraging potency for compound 8 in hand we investigated more extensive amide variants and heterocyclic modifications (Table 1). The reverse amide of ML300, 11 had a 3-fold reduction in activity versus the parent, while the primary amide, 10, displayed nanomolar inhibition against both proteases. Sulfonamides 12–13 both measured low micromolar inhibition. Reviewing the SC1 3CLpro X-ray structure of 8, while we did not observe any obvious direct hydrogen bonding interactions between the pyridyl group and the protein, it was clear that the orientation of this compound series was directed toward multiple residues for potential interaction. Additional isomeric analogs such as 4-pyridyl 14 and 2-pyridyl 15 were within 2-fold of the activity of 8 for both SC1 and SC2 3CLpro. 2-Methoxy-pyridyl 16 lost 3–5 fold for both enzymes, while pyridone 17 was essentially equipotent to compound 8. Encouraged by 17 and the observed interactions of 1 within the S2c upper rim we continued to design and pursue modifications in the S2c exploring various H-bond donors both neutral and potentially charged in nature. To our delight a significant enhancement was found with the NH-pyrazole and imidazole derivatives, 19 and 21, reaching an IC50 of 60 nM versus SC2 3CLpro in the case of imidazole 21. Potency of N-methyl analogs 18 and 20 was drastically diminished, between 160–185-fold verses their non-methylated analogs, underscoring the importance of the presence and nature of a H-bond donor in the P2c group
Obtained X-ray co-crystal structures of 19 and 21 in SC2 3CLpro indeed demonstrate the potential for multiple H-bonding interactions from these heterocyclic azole nitrogens (Figure 5, A and B). There are prospective H-bonds with both the hydroxyl side chain of Thr25 and backbone carbonyl of Cys44. The bound pose of the azole heterocycle is co-planar with the central phenyl in both cases and juxtaposed between the residues with distances from the closest nitrogen within acceptable inter-heteroatom H-bonding distances of 2.8 to 3.0 angstroms. The hydrogen bonding array revealed in both structures would suggest the azole is acting as hydrogen bond donor to the carbonyl of Cys44 and then an acceptor from Thr25. We also obtained X-ray structures of these same two compounds in SC1 3CLpro, and in both cases we observed an almost identical binding pose – and binding site orientation – with the exception of a subtle difference in the S2sp region (Supplemental Figure S1A). Here, there is movement of the unstructured loop, and rotation of Gln189, which leads to a small difference in the thiophene ring torsion (86° vs. 64°). This observation is presumed to be driven by different crystallization conditions used for the two proteins; SC2 was co-crystallized, while compounds for SC1 were soaked into apo-3CLpro with increasing amounts of glycerol in the buffer solution. Indeed, density for a glycerol unit is found in the nearby S4 region for all our solved SC1 3CLpro structures.
Figure 5.

X-ray co-crystal structures of compounds 19 and 21, select residues are shown as sticks with key hydrogen bonds shown as dashes. A) 19 in complex with SC2 3CLpro (PDB: 7LMD); B) 21 in complex with SC2 3CLpro (PDB: 7LMF)
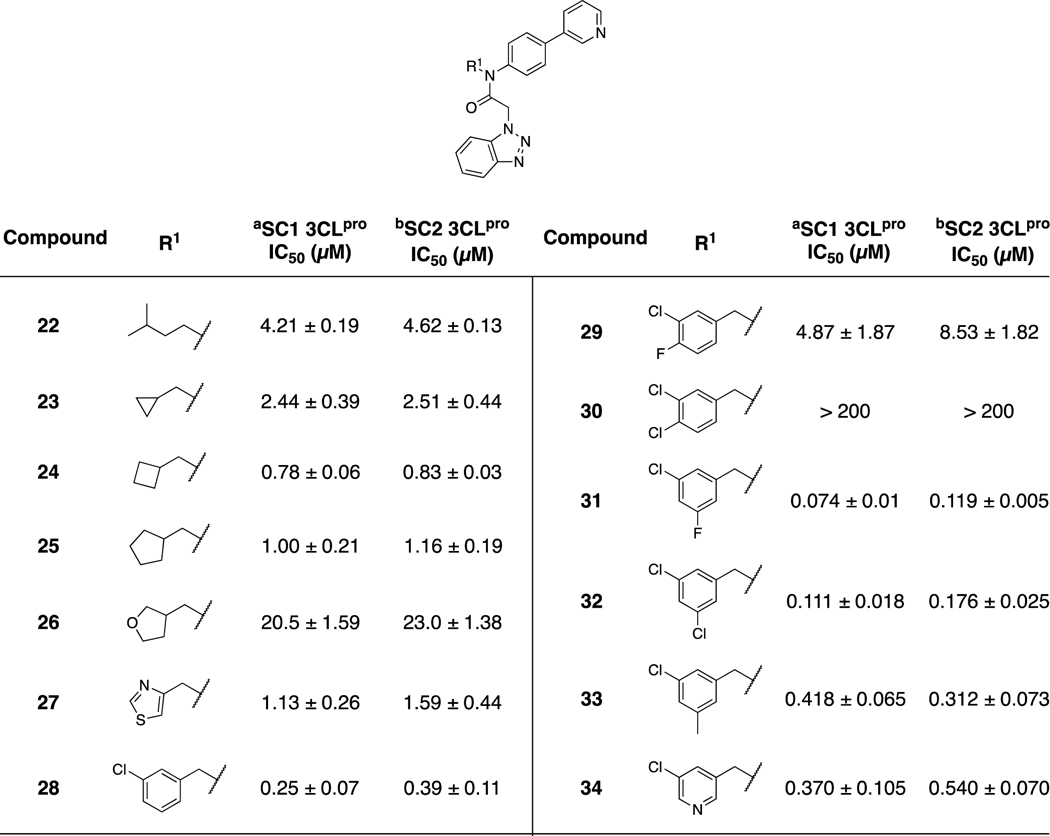
Simultaneously to the synthesis of our first series expanding the scope of the biaryl 8 SAR, we explored thiophene replacements at P2sp (Table 2) holding the P2c group constant as 3-pyridyl. A range of acyclic and cyclic aliphatic groups were targeted, including 22–26, with the cyclobutyl derivative 24 affording a slight improvement relative to the parent 8. Introduction of an ether oxygen via tetrahydrofuran 26 was highly deleterious. Thiazole analog, 27, offered similar inhibition within two-fold of 8 and 24 with early evidence of an improvement in microsomal stability compared to 8 (vide infra). It was quickly established that a 3-chlorophenyl group led to a significant potency increase against both SC1 and SC2, namely compound 28, and subsequent analogs explored SAR of this ring in more depth, 29–34. A 3,4-substitution pattern was clearly not tolerated (29, 30), an observation that was consistent with our emerging X-ray structures and docking studies suggesting a limited steric bulk near the loop residue Arg188. This observation is in contrast to mobility in the loop region near Asn189. A 3,5-substitution afforded examples with inhibition below 200 nM, including analogs 31 and 32, with the 5-fluoro derivative 31 among the most potent in this subseries with IC50 values ranging 74–119 nM. The addition of a pyridine nitrogen to improve the physicochemical properties of this sub-series was tolerated, but with a reduction in SC2 inhibition to 540 nM.
Table 2.
3CLpro inhibition data, exploring P2sp SAR
|
IC50 values are average of at least 3 independent assays, each run as technical duplicates;
SARS-CoV-1 3CLpro
SARS-CoV-2 3CLpro
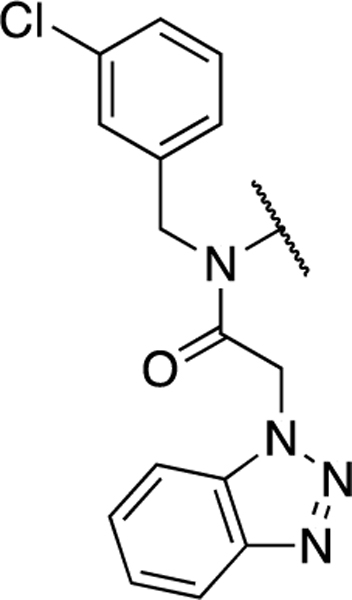
Based on the above data, we elected to hold the P2sp 3-chlorophenyl and P1 benzotriazole constant while exploring combinations P2c pendant biaryls with a handful of aniline modifications (Table 3). Interestingly, a number of the prior P2c favorable modifications, such as pyridone 17 and pyrazole 19, led to active compounds 35 and 36, however, differential SAR was noted. For example, within the pyridone sub-series 3-chlorophenyl (35) versus 3-thienyl (17) 3CLpro inhibition is nearly identical. Within the more active pyrazole comparators (e.g. 36 versus 19) there is a three-fold preference for the 3-thienyl sub-series which is in the opposing direction from that initially described in Table 1. This differential SAR is observed in both SC1 and SC2 3CLpro data, suggesting a real phenomenon, highlighting dynamic SAR presumably resulting from the P2c upper rim interactions identified. Relative to 36 modifications within aniline core such as methoxy analogs 37 and 38 are tolerated with 2-methoxy being slightly improved relative to 36 with an IC50 = 197 nM for SC2 3CLpro. A similar trend was noted for the central pyridyl congeners 39 and 40 with 2-pyridyl displaying identical 1.6-fold increases in potency for both SC1 and SC2 enzymes. Investigation of the 3-chlorophenyl P2sp analog imidazole 21 afforded compound 41. Once again like the pyrazole matched pairs described above, more potent P2c modifications appear to engender differential SAR in context of the P2sp group. In the case of SC1 3CLpro an 8-fold boost in IC50 is noted substituting 3-thienyl for 3-chlorophenyl (21, IC50 = 148 nM versus 41, IC50 = 19 nM), respectively. However, in the case of SC2 3CLpro we observe flat SAR with no real improvement in inhibition when the P2c substituent is C4-imidazole.
As part of the ongoing optimization efforts we spot-checked compounds for DMPK properties throughout, including human and rat intrinsic clearance, plasma protein binding, MDCK passive permeability, and P450 isozyme reversible inhibition at 10 μM. A complete summary is presented in Supplemental Table S1. Beginning with 1 (ML300) and throughout most of the SAR presented, exceptionally high microsomal clearance has been observed, with hepatic clearance equivalent to hepatic blood-flow in both human and rat. We hypothesized that the P1-benzotriazole group may be a source of significant metabolism and indeed, the carbon-linked 3-pyridyl and 5-pyrimidyl analogs 42 and 43 (Figure 6) do show a noteworthy improvement in CLint, with a 2- and 4-fold decrease compared to 36 in microsomes (human, rat) and in human S9 fraction. Unfortunately, these groups afforded a drop in primary biochemical potency back to micromolar levels, demonstrating further P1 optimization is required. The monodirectional cellular permeability is high, as measured in MDCK-MDR1 cells, across the series, and multiple examples have modest free fraction at or above 1% unbound in human (e.g. 21 and 41, Supplemental Table S1). We have also assessed an initial CYP inhibition profile for this series of compounds, and relatively high inhibition at 10 μM was noted in general with few exceptions (3A4, 2D6 >> 50%). Given the structural features of these molecules, this finding was not unexpected, and further optimization is ongoing to mitigate P450 inhibition.
Figure 6.

SC2 3CLpro potency and DMPK profiles for lead benzotriazole 36, 42, and 43.
Subsequently, we initiated soft-spot metabolite ID analysis of a representative benzotriazole, namely 3-chlorobenzyl derivative 36, to further understand the generally high hepatic oxidative metabolism observed and guide future target design. This analysis demonstrated that while there is NADPH-dependent oxidative metabolism of the benzotriazole moiety, this is a secondary metabolite in comparison to oxidation of the benzylic 3-chlorophenyl ring (Figure 7). The M1-metabolite is estimated at 60% based on UV total peak AUC, while M2 is 33%. We note that sample depletion was not observed in the absence of NADPH (data not shown), indicative of oxidative metabolism mediated by P450. A third, minor metabolite (M3, 7%) resulting from cleavage of the 3-chlorobenzyl group is also observed. Additional profiling of related compounds is ongoing to support this observation that benzylic P2sp is the major soft spot in human hepatic S9 fractions. Strategies are underway to reduce oxidative metabolism and increase in vitro half-life.
Figure 7.

Soft-spot metabolite identification studies of 36 using human hepatic S9 fractions. Metabolite formation (%) is based on the total peak area of the three principle metabolites, M1, M2 and M3; see also Supplemental Table S2.
We next turned to evaluation of promising compounds for their antiviral activity. The antiviral efficacy of compounds against infectious SARS-CoV-2 virus in Vero E6 cells was evaluated by cytopathic effect (CPE) inhibition and in a plaque reduction assay. The results are summarized in Table 4. The CPE inhibition assay was performed with 6-point dilution with three independent experiments for each compound, these results were then validated via a plaque reduction assay performed with between 4–6 concentrations of compound, dependent on the measured CPE inhibition.
Table 4.
Assessing antiviral efficacy of compounds, measuring in vitro viral replication.
| Compound | CPE Inhibition Assay EC50 (μM) | Plaque Reduction Assay EC50 (μM) |
|---|---|---|
|
| ||
| Remdesivir | 0.34 ± 0.01 | 0.43 ± 0.06 |
| 1 (ML300) | 19.90 ± 6.32 | 28.15 ± 1.30 |
| 8 | 12.07 ± 0.31 | 14.55 ± 1.22 |
| 19 | 5.76 ± 1.96 | 7.63 ± 1.65 |
| 21 | 1.74 ± 0.19 | 1.75 ± 0.25 |
| 28 | 7.61 ± 1.43 | 11.14 ± 0.37 |
| 35 | 8.77 ± 1.14 | 16.69 ±. 2.44 |
| 36 | 1.98 ± 0.24 | 3.31 ± 0.10 |
| 37 | 2.93 ± 0.28 | 3.24 ± 0.27 |
| 38 | 2.30 ± 0.33 | 2.01 ± 0.49 |
| 39 | 3.81 ± 0.18 | 4.65 ± 0.11 |
| 40 | 1.91 ± 0.30 | 3.39 ± 0.21 |
| 41 | 0.497 ± 0.009 | 0.558 ± 0.041 |
The original declared MLPCN probe ML300 displayed weak activity in the high micromolar range (Table 4, CPE EC50 = 19.9 μM, Plaque reduction EC50 = 28 μM). The pyrazole and imidazole P2c containing ML300 derivatives, 19 and 21, which are among the more biochemically potent analogs, showed improved EC50 values between 1.7–8 μM in both assays. Interestingly, potency of the matched pair analogs 36 and 41 that substitute the P2sp thienyl to 3-chlorophenyl are greatly improved with the EC50 of 41 sub-micromolar in both assays. In addition, the 50% cytotoxic concentration of 41 (CC50) in the CPE antiviral assay was found > 50 μM, indicating an excellent selectivity index (SI > 100, see Supplemental Figure S4 and Table S5). Comparison of EC50/IC50 ratios of the non-covalent compounds can help guide compound design in terms of estimating the required target IC50 in order to achieve a desired pharmacological effect. For example, for 19 and 21 the respective EC50/IC50 ratios are 52 and 29 respectively. These are far from a desired profile. In contrast, the 3-chlorophenyl analogs 36 and 41 have EC50/IC50 ratios of 7.3 and 7.4 respectively. To our knowledge, these values are equivalent to the best reported to-date non-covalent inhibitors of SC2 3CLpro. In addition, the efficacy of 41 was shown in our assays to be comparable to that of the clinically utilized polymerase inhibitor, remdesivir (Figure 8).37, 38
Figure 8.

Summary of data for compound 41 (CCF0058981). Images show antiviral activity of 41 and remdesivir against SARS-CoV-2 by plaque reduction assay. Vero E6 ACE2 cells were infected with approximately 50 plaque forming units per well of SARS-CoV-2. After 1 h incubation, the viral inoculum was removed, and the cells were overlaid with DMEM containing 1% low-melting agarose and indicated concentration of test compounds. At 3 days post-infection, the cells were fixed with 4% formaldehyde for at least 1 h. Overlay was then removed and cells were stained with 0.2% crystal violet containing 20% ethanol. The images are representatives of two repeats. Tier 1 DMPK data were obtained at Q2 Solutions.
Chemistry
The nature of our starting point, 1 (ML300), led to the possibility for modular and systematic exploration of SAR in different vectors of the 3CLpro binding pocket. Our initial focus was on optimization of the groups at P2c, which was previously found to be a source of varied SAR.21 Here, the P2sp group was fixed and thiophene-3-carbalydehyde, 44, was reacted with various anilines via reductive amination (Scheme 1). Amides 10 and 11, the reverse amide relative to 1 were synthesized via benzoic acid intermediate 45; while sulfonamides 12 and 13 were synthesized via the 1,4-dianiline intermediate, 46. The aryl bromide, 47, afforded a versatile intermediate that afforded bi-(hetero)aryl analogs 8, 14–21 using Suzuki-Miyaura cross-coupling conditions. The pyridone example, 17, was synthesized by hydrolysis of the fluoropyridine intermediate 48. The NH-pyrazole could be introduced directly, without protection to afford 19; however, reaction with the analogous NH-imidazole was unsuccessful. In this instance, we utilized 4-bromo-1-trityl-imidazole and the corresponding pinacol ester of 47 to furnish the desired product 21 after deprotection.
Scheme 1.

Synthesis of compounds 8–21, from thiophene-3-carbaldehyde, 44. Reagents and conditions: i) NaBH(OAc)3, AcOH, DCE, rt; ii) benzotriazole-1-acetic acid (50), T3P, EtOAc, rt; iii) LiOH, THF, rt; iv) NH4Cl, EDC, HOBt, Et3N, THF, rt; v) cyclopropylamine, COMU, DIPEA, DCM, rt; vi) TFA, DCM, rt; vii) R-SO2Cl, Et3N, DCM, rt; viii) 50, HATU, Et3N, DCM, rt; ix) ArB(OH)2, Pd(OAc)2, dppf, CuCl, Cs2CO3, DMF, 100 °C; x) Ar-B(OH)2, Pd(PPh3)4, THF:H2O, 100 °C; xi) aq. HCl, dioxane, 80 °C; xii) Ar-Bpin, Pd(dppf)Cl2.DCM, K2CO3, dioxane, 100 °C; xiii) B2pin2, Pd(dppf)Cl2.DCM, KOAc, dioxane, 80 °C; xiv) 4-bromo-1-trityl-imidazole, Pd(dppf)Cl2.DCM, K2CO3, dioxane, 100 °C; xv) AcOH, MeOH, 65 °C.
We simultaneously explored the nature of the P2sp-substituent, with a desire to replace the thiophene moiety of ML-300 (Scheme 2). Here, various aldehydes were reacted with the biaryl aniline 51 via reductive amination, then amide bond formation with benzotriazole-1-acetic acid (50) in the presence of propylphosphonic anhydride (T3P) afforded compounds 22–34.
Scheme 2.

Synthesis of compounds 22–34, varying the P2sp substituent. Reagents and conditions: i) NaBH(OAc)3, AcOH, DCE, rt; ii) T3P, pyridine, DMF, rt
Similar to our initial library, for compounds 35–43 the S2 3-chlorophenyl moiety was fixed as a thiophene replacement (Scheme 3). Reductive amination of 3-chlorophenylbenzaldehyde, 53, with anilines afforded intermediates that follow a similar synthetic sequence as above. The bromo intermediate 58 provided synthetic flexibility, as subsequent Suzuki-Miyaura reaction, followed by T3P amide coupling, led to products 35, 36, 42 and 43. Alternatively, the 1,2,3-benzotriazole moiety could be introduced first to give bromide 61, which was then subjected to Miyaura-Ishiyama-Hartwig borylation and subsequent Suzuki-Miyaura cross-coupling conditions to afford 41 in acceptable yield.
Scheme 3.

Synthesis of compounds 35–43. Reagents and conditions: i) Aryl-amine, NaBH(OAc)3, AcOH, DCE, rt; ii) aryl acetic acid, T3P, pyridine, DMF, rt or 60 °C; iv) 2-fluoropyridine-3-boronic acid, Pd(dppf)Cl2.DCM, K2CO3, dioxane:H2O, 100 °C; v) aq. HCl, dioxane, 80 °C; vi) 1-THP-4-Bpin pyrazole, Pd(dppf)Cl2.DCM, K2CO3, dioxane:H2O, 100 °C; vii) 4M HCl, Dioxane, 60 °C; viii) B2pin2, Pd(dppf)Cl2.DCM, KOAc, dioxane, 100 °C; ix) 4-bromo-1-trityl-imidazole, Pd(dppf)Cl2.DCM, K2CO3, dioxane:H2O, 100 °C; x) AcOH, MeOH, rt; xi) 1-THP-4-Bpin pyrazole, S/XPhos Pd G2, K2CO3, 1-BuOH, 100 °C; xii) 50, cyanuric fluoride, pyridine, DCM, rt then 66 or 67, Et3N, THF 60 °C.
Conclusions
As for many scientists around the globe, the huge effect on our lives caused by the COVID-19 pandemic instilled us to assess how we could have an impact, however large or small, in the study of this disease. Having prior experience targeting the 3CL-protease of SARS-CoV-1 working with leaders in the field we were compelled to further investigate ML300 as a starting point employing all disciplines within our newly formed Center and partnering with our CCF Institute virologist colleagues and experts in the field. After initial disappointment and intrigue, we found that the addition of H-bond donating azole heterocycles, in particular pyrazole and imidazole containing analogs, including 19 and 21, directed to S2c afforded an appreciable increase in potency. This discovery combined with the 3-chlorobenzyl modification in S2sp advanced our optimization efforts to a series of compounds routinely reaching primary IC50 SC2 3CLpro inhibition values below 500 nM. As such the ML300 amide series continues to display an interesting mode of action with differential SAR worthy of further investigation.
Historically the ML300 series has demonstrated basic DMPK properties that were not attractive for further development beyond in vitro studies. While we have observed improvements, the compounds disclosed still lack the properties needed for a full in vivo evaluation in animal models of SARS-CoV-2 infection. The addition of metabolite ID studies have highlighted key areas of the molecule for further optimization to improve the high clearance and metabolic turnover in the series. Similarly, while the unbound fraction and cellular permeability of these compounds is promising, a further hurdle these compounds must overcome is their high inhibition of CYP enzymes which is currently a major area of focus.
Non-covalent small molecule SARS-CoV-2 inhibitor 41 (CCF0058981) provides a significant advance from the original SARS-CoV-1 ML300-derived inhibitor, with low nanomolar biochemical inhibition and efficacy in cellular models comparable to the FDA-approved RNA polymerase inhibitor remdesivir. Optimization of the series of compounds is ongoing, with a particular focus on improving the DMPK profile, as well as further improving the biochemical and cellular efficacy. Further, we look forward to future opportunities to profile the series and compounds herein against a wider panel of coronaviruses, including MERS. We anticipate that investigators targeting SC1 and SC2 3CLpro will benefit from the disclosed SAR and X-ray crystal structures. Furthermore, these findings have implications towards antiviral development to combat future SARS-like zoonotic coronavirus outbreaks.
Experimental Section
SARS-CoV-1/2 3CLpro Protein Expression & Purification
SARS-CoV-1/2 3CLpro enzymes were cloned using previously published methods.30,39 Briefly, genes encoding each protein were codon optimized for E.coli, synthesized, and inserted into a pGEX-6P-1 plasmid between the BamHI and XhoI cut sites (note that additional residues “AVLQ” and “GPHHHHHH-stop” were added to the N and C termini, respectively, of the proteins as previously described)30,39. The resulting expression constructs yield unscarred, native enzymes following protein purification.
Both enzymes were recombinantly expressed using New England Biolabs T7 Express lysY/Iq cells transformed with 3CLpro expression plasmids. The enzymes were expressed and purified using identical methods derived from previously published work.30 Inoculated cultures of Lauri Broth media supplemented with ampicillin were grown at 37 °C with shaking to a density of 0.6–0.8 OD600nm. The incubator/shaker temperature was then reduced to 16 °C and the cultures were induced with 0.5 mM IPTG. Following overnight induction, cells were harvested via centrifugation at 3900 RPM (Eppendorf 5810R, S-4–104 rotor) for 25 minutes and resuspended in 20 mM TRIS, 300 mM NaCl pH 8.0 buffer (Buffer A). Resuspended cells were lysed via sonication, centrifuged at 10,500 RPM (Eppendorf 5810R, FA45–6-30 rotor) for 30 minutes, and the clarified lysate loaded onto a 5 mL Ni-charged Nuvia IMAC column equilibrated with Buffer A. The column was washed with 10 column volumes (CV) of Buffer A and eluted with a 7 CV gradient of 0–100 % Buffer B (Buffer A + 500 mM imidazole). Fractions containing pure 3CLpro were pooled, diluted in half with Buffer A, and dialyzed overnight at 4 °C with PreScission Protease against a 50 mM TRIS, 150 mM NaCl, 1 mM DTT, pH 7.5 buffer. Cleaved protein was passed through a 5 mL Ni-charged Nuvia IMAC column equilibrated with a 50 mM TRIS, 150 mM NaCl pH 7.5 buffer. Collected flow through was concentrated to 5 mL using an Amicon Ultra 10k centrifugal filter and diluted to 25 mL with 50 mM TRIS pH 7.5. Diluted protein was passed through a 5 mL HiTrap Q FF anion exchange column equilibrated with 50 mM TRIS, 25 mM NaCl buffer pH 7.5 (Buffer C). The Q FF column was washed with 10 CV of Buffer C and bound proteins eluted with a 5 CV 0–100 % gradient of Buffer D (Buffer C + 1 M NaCl). Pure 3CLpro was found to be in the non-bound and wash fractions from this chromatographic step. Final purity was assessed via Coomassie stained SDS-PAGE. The purified protein in a 50 mM TRIS and 25 mM NaCl pH 7.5 buffer was pooled, concentrated, aliquoted and frozen at −80 °C for biochemical assays, or used fresh for protein crystallography studies.
SARS-CoV-1/2 3CLpro Biochemical Assay
Protease activity and subsequent 10-point IC50 curves were spectroscopically determined using a scaled down, endpoint assay adapted from a previously described peptide-based Forster Resonance Energy Transfer (FRET) assay. 20,40 Compounds (as 10 mM DMSO stock) were serial diluted 4-fold using 100% DMSO in a LabCyte 384-well LDV plate and acoustically transferred using a LabCyte ECHO 550 into Corning 384-well black NBS plates. Standard 10-point IC50 384-well plate layout is as follows: 100 μM of 8 was stamped into columns 1 and 24 (low control), DMSO was stamped into columns 2 and 23 (high control), and serial diluted compounds were stamped from high (100 μM) to low (0.38 nM) concentrations in columns 3–12 (replicate 1) and 13–22 (replicate 2). Protocol for running the assay is as follows: assay wells stamped with 0.25 μL of compound or DMSO were filled via a ThermoFisher Multidrop Combi liquid dispenser with 14.5 μL of 150 nM or 200 nM (concentration for 25 μL final reaction volume) of SARS-CoV-1 or SARS-CoV-2 3CLpro, respectively, in assay buffer (50 mM HEPES, 0.1 mg/ml BSA, 0.01% v/v TRITON X100, 2 mM DTT, pH 7.5). Assay plates were then centrifuged at 1,000 RPM (Eppendorf 5810R, S-4–104 rotor) for 1 minute, covered, and incubated at room temperature for 15 minutes. Reactions were initiated using the Multidrop Combi liquid dispenser to titrate 10 μL of 2 μM (concentration for 25 μL final reaction volume) of fluorophore-quencher peptide substrate (HiyteFluor™-488ESATLQSGLRKAK-(QXL™)-NH2 from AnaSpec, Inc. Catalog No. AS-65599) solubilized in assay buffer into each well. Assay plates were again centrifuged at 1,000 RPM for 1 minute, covered, and incubated at room temperature for 30 minutes. Biochemical assays were quenched through the addition of 5 μL of 500 mM acetic acid via Multidrop Combi liquid dispenser. Assay plates were then centrifuged at 1,000 RPM for 1 minute and resulting fluoresce intensity measured on a BioTek Cytation 5 multimode plate reader (λex = 485nm, λem = 528nm).
Data analyses:
Raw fluorescence values were normalized (RFnorm) by dividing each value by the average of DMSO control wells which represents the maximal fluorescence signal (RFnorm = RFsample/Ave RFDMSO control). Dose response curve fitting was performed using Dotmatics Studies (software version 5.4.2), which computes IC50 values utilizing a four-parameter logistical fit. Reported values are average IC50 from at least 3 independent experiments.
Protein Crystallization, Data Collection, and Structure Refinement
SARS-CoV-2 3CLpro was concentrated to 6.5 mg/ml and titrated with inhibitor to give a final concentration of 3% v/v inhibitor (10 mM DMSO stocks of 19 and 21, and 20 mM DMSO stock of 1). Protein-inhibitor complexes were left at room temp for 1 hour prior to setting up crystallization drops. Diffraction quality crystals were obtained by hanging drop vapor diffusion experiments comprised of 1 μL protein/inhibitor mix and 1 μL crystallization solution drops incubated at 16 °C. The Hampton Research Index Screen (HR2–144) yielded a high rate of crystal formation, with optimal crystals forming in either 0.2 M lithium or ammonium sulfate, 0.1 M Bis-TRIS or HEPES pH 6.5–7.5, and 25 % polyethylene glycol 3350 conditions. Protein crystals were directly looped, flash frozen in liquid nitrogen, and shipped for remote X-ray diffraction data collection at the Advanced Photon Source LS-CAT beamline 21-ID-F. Diffraction data was indexed and scaled using HKL2000 and phased (Phaser-MR) using molecular replacement with the search molecule PDB: 6WQF.41–43 Inhibitor coordinates were generating using ELBOW and the resulting protein-inhibitor structures were refined and built using iterative rounds of refinement and manual manipulations with PHENIX Refine and COOT.44–46 X-ray diffraction and model refinement statistics are given in Supplemental Table 1. The final coordinates and electron density maps were deposited into the Protein Data Bank.
SARS-CoV-1 3CLpro was concentrated to 6.0 mg/ml and titrated with inhibitor to give a final concentration of 4% v/v inhibitor (10 mM DMSO stocks of 35 and 25 mM DMSO stock of 1). Protein-inhibitor complexes were left at room temp for 30 min prior to setting up crystallization drops. Diffraction quality crystals were obtained by hanging drop vapor diffusion experiments comprised of 1 μL protein/inhibitor mix and 1 μL crystallization solution drops incubated at 16 °C. The Hampton Research Index Screen (HR2–144) yielded a high rate of crystal formation, with optimal crystals forming in ammonium sulfate, HEPES pH 7.5, and 25 % polyethylene glycol 3350 conditions. For collecting X-ray diffraction data, the crystals were flash-cooled in liquid nitrogen followed by dragging the crystals through the crystallization solution. In another method, SARS-CoV-1 3CLpro was crystallized at 20 °C using the hanging-drop vapor-diffusion method by mixing 6 mg/mL 3CLpro in 1:1 ratio with 100 mM MES buffer in pH 5.5, 12–18% (w/v) polyethylene glycol 6000, 10% (v/v) glycerol, 2% (v/v) DMSO. Single crystals were transferred to 5 μL of the mother liquor with 400 μM inhibitor concentration (10 mM DMSO stocks of 8, 19 and 21). After incubation at 16 °C for 5 min, the crystals were looped and dragged out through the crystallization solution cryoprotected by 20% (v/v) glycerol. The crystals were then shipped for remote X-ray diffraction data collection at the Advanced Photon Source LS-CAT beamline 21-ID-F. Diffraction data was indexed and scaled using HKL2000 and phased with CCP4i molecular replacement program with the search molecule PDB: 3VB3.47 Inhibitor coordinates were generating using ELBOW and the resulting protein-inhibitor structures were refined and built using iterative rounds of refinement and manual manipulations with Refmac (ccp4i)48 and COOT. X-ray diffraction and model refinement statistics are given in Supplemental Table 2. The final coordinates and electron density maps were deposited into the Protein Data Bank.
Virus propagation and titration
The following reagent was deposited by the Centers for Disease Control and Prevention and obtained through BEI Resources, NIAID, NIH: SARS-Related Coronavirus 2, Isolate USA-WA1/2020, NR-52281. The virus was propagated in Vero E6 expressing ACE2 receptor (a gift from Dr. Younho Choi, Cleveland Clinic Lerner Research Institute) in a DMEM media supplemented with 1X penicillin-streptomycin (Gibco) and 0.5μg/ml TPCK-treated trypsin (Worthington Biochemical) at 37 °C in a humidified incubator with 5% CO2. Propagated virus was aliquoted and stored at −80 °C until further use.
The virus titer was determined by plaque assay as previously described with a little modification.49 Briefly, confluent monolayer Vero E6 ACE2 cells cultured in a 6-well plate were infected with 10-fold serial diluted virus inoculum incubated on a rocker for 45 min in 37 °C for virus adsorption. After the removal of virus solution, cells were overlaid with DMEM containing 1% low melting agarose and incubated in a humidified incubator at 37 °C and 5% CO2 for 4 days. To visualize the plaques, the cells were fixed with 4% formaldehyde and stained with 0.2% crystal violet solution containing 20% ethanol.
Virus inhibition assays
Initial antiviral screening was done by using CPE inhibition assay.50 Briefly, Vero E6 ACE2 cells were cultured in a 96-well flat-bottom plates at a density of 2 × 104 cells per well. Following infection of the cells with a 100 TCID50 of SARS-CoV-2, the plates were incubated on a rocker in 37 °C for 45 min for virus adsorption. The cells were then washed with DMEM and added the medium containing the test compounds in the desired concentration. Both the uninfected cells and infected cells treated with 10 μM of Remdesivir were used as controls. The antiviral efficacy of test compounds was determined by the uptake and subsequent extraction of neutral red dye. After infection (68 h), cells were incubated with 0.034% neutral red dye for 3 h at 37 °C. Free dye was washed from the wells and the uptake dye was quantified using a microplate reader with absorbance at 540 nm. Absorbance values were expressed as percentages of uninfected control cells, and EC50 values of the test compounds were determined using Prism software (GraphPad).
The validation of the initial CPE inhibition results was done by testing the compounds by plaque reduction assay.51 Confluent monolayers of Vero E6 ACE2 cells in 12-well plates were washed once with DMEM and infected with approximately 50 plaque forming units (PFUs) of SARS-CoV-2 in each well. The plates were incubated on a rocker in 37 °C for 45 min for virus adsorption. The virus inoculum was removed and replaced by overlay media (DMEM containing 1% low-melting agarose without serum) containing 3-fold serial dilutions of the test compounds and placed in 37 °C CO2 incubator until plaques can be visualized under light. The cells were then fixed with 4% formaldehyde solution for at least 30 min and the overlaid agarose was removed and stained with 0.2% (w/v) crystal violet solution. The plaques were counted by visual examination and the required concentration to reduce 50% plaque number (EC50) was calculated as relative to the control without test compounds.
Hepatic S9 Metabolic Stability Assessment of Compounds
The metabolic stability compounds were investigated in human hepatic S9 fractions pooled from 20 mixed gender donors (Xenotech, catalog # H0606.S9(AX) / lot 1710129) using substrate depletion methodology (percentage of parent compound remaining). Potassium phosphate-buffered (0.1 M, pH 7.4) solutions of test article (1 μM) and S9 (2.5 mg/ml) were incubated at 37°C under ambient oxygenation in the presence or absence of NADPH (2 mM). Total incubation volumes were 0.4 mL with a final organic concentration of 0.5%. Mixtures lacking substrate were pre-incubated at 37 °C for 5 minutes. Reactions were initiated with addition of substrate, and at the designated times (t = 0, 3, 7, 15, 30, and 45 min), a 25 μL aliquot of the incubation mixture was removed and precipitated by the addition of 4 volumes of ice-cold MeCN containing carbamazepine as an internal standard (50 nM). The mixtures were centrifuged at 3500 rcf (4 °C) for 10 min. The resulting supernatants were diluted 1:3 (supernatant-water) into 96-well plates in preparation for LC-MS/MS analysis. All samples were analyzed via electrospray ionization on an AB Sciex API-6500 QTrap (Applied Biosystems, Foster City, CA) instrument that was coupled with LC-20ADXR pumps (Shimadzu, Columbia, MD) and a CTC PAL autosampler (Leap Technologies, Carrboro, NC). Analytes were separated by gradient elution using a Phenomenex Kinetix C18 column (2.1 × 50 mm, 1.7 μm; Phenomenex, Torrance, CA) thermostated at 40 °C. HPLC mobile phase A was 0.1% formic acid in water (pH unadjusted); mobile phase B was 0.1% formic acid in acetonitrile (pH unadjusted). The gradient started at 5% B after a 0.2-min hold and was linearly increased to 90% B over 1.0 min, held at 90% B for 1.0 min, and returned to 5% B in 0.1 min followed by a re-equilibration (0.5 min). The total run time was 2.5 min, and the HPLC flow rate was 0.5 ml/min. The source temperature was set at 500 °C, and mass spectral analyses were performed using multiple reaction monitoring, with transitions and voltages specific for each compound using a Turbo Ion Spray source in positive ionization mode (4.5 kV spray voltage): 36, m/z 443>124.9 CE 38; 40, m/z 444>124.9 CE46; 42, m/z 403>120 CE 34; 43, m/z 404>125 CE 36; Verapamil, m/z 455.1>165.1 CE 40; Carbamazepine, m/z 237.1>194.0 CE 25. All data were analyzed using AB Sciex Analyst 1.5.1 software. Each compound was assayed in triplicate within the same 96-well plate. Hepatic intrinsic clearance (, mL/min/kg) was extrapolated from hepatic S9 using the substrate depletion method and Equation 1:52
| (1) |
Hepatic clearance (, mL/min/kg) was estimated using Equation 2, according to the well-stirred model, uncorrected for fraction unbound in plasma:53
| (2) |
where represents human hepatic blood flow (21 mL/min/kg) and represents the intrinsic clearance calculated from Equation 1.
In Vitro Biotransformation of Compounds in Hepatic S9 Fractions.
The in vitro metabolism of test articles was investigated using hepatic S9 fractions from Sprague-Dawley rats (62 males, pooled) and humans (150-donor UltraPool, BD Biosciences). A potassium phosphate-buffered reaction (0.1 M, pH 7.4) of test article (25 μM), hepatic S9 fractions (5 mg/ml), and MgCl2 (3 mM) was incubated at 37 °C in borosilicate glass test tubes under ambient oxygenation for 1 h with select experiments being fortified with NADPH (2 mM). All S9 reactions were initiated by the addition of NADHP to the in vitro milieu. Protein was precipitated by the addition of 2 volumes of MeCN with subsequent centrifugation (3000 rcf, 10 min). The supernatant was dried under a stream of nitrogen and reconstituted in 85:15 (v/v) ammonium formate (10 mM, pH 4.1)-MeCN in preparation for LC-MS analysis.
Liquid Chromatography-UV-Mass Spectrometry Analysis of Metabolites.
The LC-MS/MS system described in the hepatic S9 metabolic stability experiment was coupled to an Acquity BEH C18 column (2.1 μm, 2.1 × 100 mm; Waters, Billerica, MA). Solvent A was 10 mM (pH 4.1) ammonium formate, and solvent B was MeCN. The initial mobile phase was 85:15 A-B (v/v) and held for 5 min, and by linear gradient was transitioned to 20:80 A-B over 20 min. The flow rate was 0.400 ml/min. The HPLC eluent was first introduced into Shimadzu SPD-20A UV detector (single wavelength selected, 254 nm) followed by electrospray ionization-assisted introduction the 6500 QTrap mass spectrometer operated in the positive ion mode. The electrospray voltage was set at 4.5 kV with a source temperature of 500 °C. The collision energy was 25 when the mass spectrometer was operated in the MS/MS mode.
Compound Synthesis
General Experimental.
All chemical reagents and reaction solvents were purchased from commercial suppliers and used as received. Normal phase chromatography was performed on a Teledyne ISCO CombiFlash NextGen300 system using Teledyne RediSep® normal phase silica cartridges, with average particle size 35–70 micron. Preparative reversed-phase HPLC was performed using a Teledyne ACCQ-Prep HP150 equipped with Phenomenex Kinetex C18 columns, using gradients of MeCN in H2O with 0.1% TFA additive. Compounds that are obtained as a TFA salt after purification were afforded as free base, by dissolving the salt in EtOAc and washing with sat. aq. K2CO3, or by elution through a Biotage ISOLUTE® SCX-II cartridge, loading and washing with MeOH and eluting with 2N NH3 in MeOH. Proton nuclear magnetic resonance (1H NMR) spectra were recorded 400 MHz on a Bruker spectrometer. For 1H NMR spectra, chemical shifts are reported in parts per million (ppm) and are reported relative to residual non-deuterated solvent signals. Coupling constants are reported in hertz (Hz). The following abbreviations (or a combination, thereof) are used to describe splitting patterns: s, singlet; d, doublet; t, triplet; q, quartet; pent, pentet; m, multiplet; br, broad. Analytical thin layer chromatography (TLC) was performed on Kieselgel 60 F254 glass plates precoated with a 0.25 mm thickness of silica gel. TLC plates were visualized with UV light and iodine.
All compounds were of 95% purity or higher, unless otherwise noted, as measured by analytical reversed-phase HPLC. Mass spectra were obtained on an Agilent 1290 series 6230 TOF. Detection methods are diode array (DAD) at 210, 254 nM and positive/negative electrospray ionization (ESI), mass range 100–1200 m/z. All methods use an Agilent InfinityLab Poroshell 120 EC-C18 column, dimensions 4.6 × 50 mm, 2.7 μM, fitted with Poroshell 120 EC-C18, 2.1 mm, 1.9 μM guard. Mobile phase A was 0.1% TFA in H2O, mobile phase B was 0.1% TFA in MeCN.
Method A:
The mobile phase B was 5% for 0.2 min, then a gradient of 5–95% B over 2.0 min, then hold 0.45 min (0.4 mL/min flow rate), using positive ESI.
Method B:
The gradient was 40–95% B for 2.5 min, then hold 0.5 min (0.4 mL/min flow rate), using positive ESI.
2-(Benzotriazol-1-yl)-N-(4-phenylphenyl)-N-(3-thienylmethyl)acetamide (6).
Synthesized according to published procedure.21,54 1H NMR (400 MHz, DMSO-d6) δ 8.03 (d, J = 8.4 Hz, 1H), 7.84 – 7.73 (m, 3H), 7.70 (d, J = 7.6 Hz, 2H), 7.60 – 7.51 (m, 4H), 7.48 (t, J = 7.6 Hz, 2H), 7.44 – 7.36 (m, 2H), 7.33 (s, 1H), 7.05 – 6.99 (m, 1H), 5.47 (s, 2H), 4.92 (s, 2H); 13C NMR (101 MHz, DMSO) δ 165.6, 145.5, 140.6, 140.0, 139.4, 138.1, 134.3, 129.5, 129.2, 128.4, 128.3, 128.2, 127.6, 127.3, 127.1, 124.3, 123.8, 119.4, 111.6, 49.9, 48.7; Purity ≥95% by LCMS (Method A) tR = 2.44 min, m/z = 425.14 [M+H]+; HRMS calculated for C25H21N4OS [M+H]+ 425.1431, found 425.1450.
2-(1H-Benzo[d][1,2,3]triazol-1-yl)-N-(4-(pyridin-3-yl)phenyl)-N-(thiophen-3-ylmethyl)acetamide (7).
Step 1. 4-Bromo-N-(3-thienylmethyl)aniline.
To a solution of 44 (493 mg, 4.4 mmol) and 4-bromoaniline (929 mg, 5.4 mmol) in DCE (44 mL) was added NaBH(OAc)3 (1.42 g, 6.7 mmol) and the mixture stirred for 1 h at rt Sat. aq. NH4Cl (50 mL) was added and DCE layer separated. The aqueous layer was extracted with EtOAc (3 × 40 mL) and concentrated, purified by flash chromatography to afford a product (1.18 g, 4.4 mmol, 99%). LCMS (Method A) tR = 2.28 min, m/z = 269.98 [M+H]+.
Step 2. 2-(benzotriazol-1-yl)-N-(4-bromophenyl)-N-(3-thienylmethyl)acetamide.
To an ice-cold solution of 50 (1.27 g, 7.16 mmol) and Et3N (2.00 mL, 14.32 mmol) in DCM (25 mL) was added HATU (2.72 g, 7.16 mmol) and the mixture stirred for 30 min before the addition of 4-bromo-N-(3-thienylmethyl)aniline (1.28 g, 4.77 mmol) in a single portion. The mixture was stirred for 20 h at rt then washed with water, brine, and concentrated. Purified by flash chromatography to afford a pale brown solid (1.13 g, 2.64 mmol, 55%). LCMS (Method A) tR = 2.34 min, m/z = 429.02 [M+H]+; 1H NMR (400 MHz, CDCl3) δ 8.05 (d, J = 8.4 Hz, 1H), 7.56 (d, J = 8.6 Hz, 2H), 7.53 – 7.45 (m, 2H), 7.42 – 7.33 (m, 1H), 7.30 – 7.27 (m, 1H), 7.03 (s, 1H), 7.00 – 6.93 (m, 3H), 5.15 (s, 2H), 4.86 (s, 2H).
Step 3. 2-(1H-Benzo[d][1,2,3]triazol-1-yl)-N-(4-(pyridin-3-yl)phenyl)-N-(thiophen-3-ylmethyl)acetamide.
A vial was charged with 47 (128 mg, 0.30 mmol), pyridine-3-boronic acid (59 mg, 0.48 mmol) and Pd(PPh3)4 (35 mg, 0.03 mmol). THF (3 mL) and water (0.6 mL) was added into the vial. The mixture was stirred and purged with Ar. The vial was capped and heated at 100°C for 16 h. The reaction was allowed to cool to ambient temperature, diluted with EtOAc, filtered with celite, purified by preparative RP-HPLC to afford a colorless solid (100 mg, 0.24 mmol, 78%). 1H NMR (400 MHz, CD3OD) δ 8.80 (d, J = 2.3 Hz, 1H), 8.55 (dd, J = 4.9, 1.6 Hz, 1H), 8.10 (dt, J = 8.2, 1.9 Hz, 1H), 7.97 (d, J = 8.4 Hz, 1H), 7.74 (d, J = 8.5 Hz, 2H), 7.66 (d, J = 8.4 Hz, 1H), 7.58 – 7.50 (m, 2H), 7.46 – 7.35 (m, 4H), 7.18 (s, 1H), 7.04 (d, J = 5.0 Hz, 1H), 5.45 (s, 2H), 4.98 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 165.08, 149.46, 148.49, 146.25, 140.41, 139.05, 137.06, 135.43, 134.78, 133.98, 129.31, 129.20, 128.55, 128.00, 126.66, 124.87, 124.27, 124.05, 120.37, 110.05, 50.25, 48.84; Purity ≥95% by LCMS (Method A) tR = 1.68 min, m/z = 426.14 [M+H]+; HRMS calculated for C24H19N5OS [M+H]+ 426.1394, found 426.1396.
2-(1H-benzo[d][1,2,3]triazol-1-yl)-N-phenyl-N-(thiophen-3-ylmethyl)acetamide (9).
Step 1. N-(thiophen-3-ylmethyl)aniline.
To a stirring solution of thiophene-3-carbaldehyde, 44 (88 μL, 1.0 mmol) and aniline (112 μL, 1.23 mmol) in DCE (10 mL) was added sodium triacetoxyborohydride (323 mg, 1.52 mmol). The mixture was stirred at rt for 2h, then sat. aq. NH4Cl (20 mL) added and the DCE layer separated. The aqueous was extracted with DCM (3 × 30 mL), combined organics were dried (Na2SO4), concentrated and purified by flash chromatography to afford a colorless solid (188 mg, 1.1 mmol, 99%). LCMS (Method A) tR = 1.57 min, m/z = 190.06 [M+H]+; 1H NMR (400 MHz, CDCl3) δ 7.34 – 7.28 (m, 1H), 7.23 – 7.15 (m, 3H), 7.12 – 7.06 (m, 1H), 6.74 (t, J = 7.4 Hz, 1H), 6.67 (d, J = 7.9 Hz, 2H), 4.34 (s, 2H), 4.20 (d, J = 20.9 Hz, 1H).
Step 2. 2-(1H-benzo[d][1,2,3]triazol-1-yl)-N-phenyl-N-(thiophen-3-ylmethyl)acetamide.
To a stirred solution of N-(thiophen-3-ylmethyl)aniline (208 mg, 1.1 mmol) and benzotriazol-1-ylacetic acid (195 mg, 1.1 mmol) in DMF (5.5 mL) was added T3P (50% in EtOAc) (1.31 mL, 2.2 mmol), followed by pyridine (266 μL, 3.3 mmol). The mixture was stirred at rt for 18 h, then sat. aq. NaHCO3 (20 mL) added. The aqueous was extracted with DCM (3 × 30 mL), combined organics were dried (Na2SO4), concentrated and purified by flash chromatography to afford a colorless solid (325 mg, 1.1 mmol, 85%). 1H NMR (400 MHz, CD3OD) δ 7.98 (d, J = 8.4 Hz, 1H), 7.66 (d, J = 8.4 Hz, 1H), 7.60 – 7.52 (m, 1H), 7.52 – 7.40 (m, 4H), 7.36 (dd, J = 5.0, 2.9 Hz, 1H), 7.30 (d, J = 6.7 Hz, 2H), 7.13 (s, 1H), 6.99 (d, J = 4.9 Hz, 1H), 5.35 (s, 2H), 4.93 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 165.09, 146.25, 140.56, 137.18, 134.04, 130.62, 129.53, 128.68, 128.64, 128.01, 126.50, 124.84, 124.27, 120.38, 110.10, 50.23, 48.78; Purity ≥95% by LCMS (Method A) tR = 2.18 min, m/z = 349.11 [M+H]+; HRMS calculated for C19H16N4OS [M+H]+ 349.1129, found 349.1139.
4-[[2-(Benzotriazol-1-yl)acetyl]-(3-thienylmethyl)amino]benzamide (10).
Step 1. Ethyl 4-(3-thienylmethylamino)benzoate.
To a stirring solution of 44 (1.75 mL, 20 mmol) and ethyl 4-aminobenzoate (3.63 g, 22 mmol) in DCE (100 mL) was added sodium triacetoxyborohydride (5.09 g, 24 mmol). The mixture was stirred at rt for 12 h, then sat. aq. NaHCO3 (100 mL) added and the DCE layer separated. The aqueous was extracted with DCM (3 × 50 mL), combined organics were dried (MgSO4), concentrated and purified by flash chromatography to afford a colorless solid (1.67 g, 6.4 mmol, 32%). LCMS (Method A) tR = 2.02 min, m/z = 262.09 [M+H]+; 1H NMR (400 MHz, CDCl3) δ 7.89 (d, J = 8.3 Hz, 1H), 7.32 (dd, J = 5.0, 2.9 Hz, 1H), 7.22 – 7.18 (m, 1H), 7.07 (dd, J = 5.0, 1.4 Hz, 1H), 6.65 (d, J = 8.3 Hz, 1H), 4.40 (s, 1H), 4.32 (q, J = 7.1 Hz, 1H), 1.36 (t, J = 7.1 Hz, 2H).
Step 2. Ethyl 4-[[2-(benzotriazol-1-yl)acetyl]-(3-thienylmethyl)amino]benzoate.
To a stirred solution of ethyl 4-(3-thienylmethylamino)benzoate (1.67 g, 6.4 mmol) and benzotriazol-1-yl-acetic acid (1.24 g, 7.0 mmol) in EtOAc (12.75 mL) was added T3P (50% in EtOAc) (7.6 mL, 12.75 mmol), followed by pyridine (1.54 mL, 19.12 mmol). The mixture was stirred at rt for 16 h, then washed with water (20 mL) and brine (20 mL). Purification by flash column chromatography afforded a cream solid (2.52 g, 6.0 mmol, 94%). LCMS (Method A) tR = 2.00 min, m/z = 421.04 [M+H]+.
Step 3. 4-[[2-(benzotriazol-1-yl)acetyl]-(3-thienylmethyl)amino]benzoic acid, (45).
To a solution of ethyl 4-[[2-(benzotriazol-1-yl)acetyl]-(3-thienylmethyl)amino]benzoate (2.52 g, 6.0 mmol) in THF (30 mL) was added 2M LiOH (15 mL, 30 mmol) and the mixture stirred at rt for 18 h. The mixture was acidified with 2M HCl (20 mL) and extracted with EtOAc, washed with brine, and concentrated. The crude material was purified by preparative RP-HPLC to afford a colorless solid (1.11 g, 2.83 mmol, 47%). LCMS (Method A) tR = 1.89 min, m/z = 393.10 [M+H]+.
Step 4. 4-[[2-(Benzotriazol-1-yl)acetyl]-(3-thienylmethyl)amino]benzamide.
To 45 (100 mg, 0.25 mmol) in THF (1.3 mL) was added 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (98 mg, 0.51 mmol) and HOBt (41 mg, 0.31 mmol) and the mixture stirred at rt for 20 min before the addition of NH4Cl (68 mg, 1.27 mmol) and Et3N (0.35 mL, 2.55 mmol). The mixture was stirred for 18 h, then diluted with EtOAc and washed with sat. aq. NaCl. Purification by ISCO automated flash chromatography afforded title compound as a colorless solid (27 mg, 27%). 1H NMR (400 MHz, CD3OD) δ 7.88 (d, J = 8.4 Hz, 1H), 7.84 (d, J = 8.1 Hz, 1H), 7.56 (d, J = 8.4 Hz, 1H), 7.45 (t, J = 7.7 Hz, 1H), 7.34 (d, J = 7.7 Hz, 1H), 7.31–7.24 (m, 3H), 7.05 (br s, 1H), 6.90 (d, J = 4.9 Hz, 1H), 5.30 (s, 2H), 4.87 (s, 2H); 13C NMR (101 MHz, CD3OD) δ 169.59, 165.61, 145.11, 142.97, 136.79, 134.17, 133.89, 129.10, 128.27, 127.53, 125.98, 124.20, 123.85, 118.49, 110.21, 49.45; Purity ≥ 95% by LCMS (Method A) tR = 1.74 min, m/z = 392.12 [M+H]+; HRMS calculated for C20H17N5O2S [M+H]+ 392.1176, found 392.1163.
4-[[2-(Benzotriazol-1-yl)acetyl]-(3-thienylmethyl)amino]-N-cyclopropyl-benzamide (11).
To a stirred solution of 45 (78 mg, 0.2 mmol) and cyclopropylamine (21 μL, 0.3 mmol) in DCM (1 mL) was added COMU (128 mg, 0.3 mmol) and DIPEA (105 μL, 0.6 mmol). The mixture was stirred at rt for 2 h, then washed with water and concentrated. Purification by RP-HPLC affords a colorless solid (15 mg, 35%). 1H NMR (400 MHz, CD3OD) δ 8.00 (d, J = 8.4 Hz, 1H), 7.88 (d, J = 8.2 Hz, 2H), 7.71 – 7.66 (m, 1H), 7.61 – 7.54 (m, 1H), 7.49 – 7.42 (m, 1H), 7.39 (d, J = 8.2 Hz, 2H), 7.15 (s, 1H), 7.01 (d, J = 4.9 Hz, 1H), 5.41 (s, 2H), 4.98 (s, 2H), 2.87 (dq, J = 7.2, 3.7 Hz, 1H), 0.83 (dt, J = 7.1, 3.6 Hz, 2H), 0.74 – 0.58 (m, 2H); 3C NMR (101 MHz, CD3OD) δ 169.08, 165.59, 145.11, 142.73, 136.78, 134.64, 133.88, 128.72, 127.54, 125.97, 124.20, 123.85, 118.50, 110.22, 60.13, 49.44, 22.68, 5.15; Purity ≥ 95% by LCMS (Method A) tR = 1.91 min, m/z = 432.15 [M+H]+; HRMS calculated for C23H21N5O2S [M+H]+ 432.1489, found 432.1506.
2-(1H-Benzo[d][1,2,3]triazol-1-yl)-N-(4-(methylsulfonamido)phenyl)-N-(thiophen-3-ylmethyl)acetamide (12).
Step 1. tert-Butyl N-[4-(3-thienylmethylamino)phenyl]carbamate.
To a solution of 44 (493 mg, 4.4 mmol) and 4-(tert-butoxycarbonylamino)aniline (1125 mg, 5.4 mmol) in DCE (44 mL) was added NaBH(OAc)3 (1.42 g, 6.7 mmol) and the mixture stirred for 1 h at rt Sat. aq. NH4Cl (50 mL) was added and DCE layer separated. The aqueous layer was extracted with EtOAc (3 × 40 mL) and concentrated, purified by flash chromatography to afford a colorless solid (1.23 g, 4.0 mmol, 92%). LCMS (Method A) tR = 1.81 min, m/z = 305.13 [M+H]+.
Step 2. tert-Butyl N-[4-[[2-(benzotriazol-1-yl)acetyl]-(3-thienylmethyl)amino]phenyl]carbamate.
To an ice-cold solution of 2-(benzotriazol-1-yl)acetic acid (700 mg, 3.95 mmol) and Et3N (1.10 mL, 7.90 mmol) in DCM (20 mL) was added HATU (1.50 g, 3.95 mmol) and the mixture stirred for 30 min before the addition of tert-butyl N-[4-(3-thienylmethylamino)phenyl]carbamate (802 mg, 2.63 mmol) in a single portion. The mixture was stirred for 16 h at rt then washed with water, brine, and concentrated. Purification by flash chromatography afforded title compound as a pale brown solid (1.08 g, 2.34 mmol, 89%). LCMS (Method A) tR = 2.34 min, m/z = 464.18 [M+H]+; 1H NMR (400 MHz, CDCl3) δ 8.05 (d, J = 8.4 Hz, 1H), 7.50 – 7.47 (m, 2H), 7.44 (d, J = 8.7 Hz, 2H), 7.36 (ddd, J = 8.1, 5.6, 2.3 Hz, 1H), 7.26 – 7.23 (m, 1H), 7.05 – 6.99 (m, 3H), 6.97 (dd, J = 5.0, 1.3 Hz, 1H), 6.62 (s, 1H), 5.15 (s, 2H), 4.84 (s, 2H), 1.53 (s, 9H).
Step 3. N-(4-Aminophenyl)-2-(benzotriazol-1-yl)-N-(3-thienylmethyl)acetamide,(46).
To a solution of tert-butyl N-[4-[[2-(benzotriazol-1-yl)acetyl]-(3-thienylmethyl)amino]phenyl]carbamate (588 mg, 1.27 mmol) in DCM (3 mL) was added TFA (2 mL) and stirred for 1 h, then diluted with DCM (10 mL) and washed with sat. aq. K2CO3 (20 mL), water and sat. aq. NaCl affording a cream solid that was used without purification (436 mg, 1.20 mmol, 95%). LCMS (Method A) tR = 1.70 min, m/z = 364.14 [M+H]+; 1H NMR (400 MHz, CDCl3) δ 8.05 (d, J = 8.4 Hz, 1H), 7.50 – 7.46 (m, 2H), 7.36 (ddd, J = 8.1, 5.1, 2.8 Hz, 1H), 7.25 – 7.22 (m, 1H), 7.04 (d, J = 2.9 Hz, 1H), 6.98 (dd, J = 5.0, 1.2 Hz, 1H), 6.86 (d, J = 8.6 Hz, 2H), 6.67 (d, J = 8.6 Hz, 2H), 5.17 (s, 2H), 4.82 (s, 2H), 3.84 (s, 2H).
Step 4. 2-(Benzotriazol-1-yl)-N-[4-(methanesulfonamido)phenyl]-N-(3-thienylmethyl)acetamide.
To a solution of 46 (87 mg, 0.24 mmol) in DCM (2 mL) was added methanesulfonyl chloride (28 μL, 0.36 mmol) and Et3N (509 μL, 3.6 mmol) and stirred for 2 h at rt. The mixture was washed with sat. aq. NH4Cl and concentrated, purified by flash chromatography to afford a colorless solid (26 mg, 0.06 mmol, 25%). 1H NMR (400 MHz, CDCl3) δ 8.04 (d, J = 8.4 Hz, 1H), 7.51 (d, J = 3.9 Hz, 2H), 7.41 – 7.35 (m, 1H), 7.22 (d, J = 8.7 Hz, 2H), 7.05 – 6.98 (m, 3H), 6.97 (d, J = 4.9 Hz, 1H), 6.69 (s, 1H), 5.19 (s, 2H), 4.85 (s, 2H), 3.11 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 165.16, 146.24, 137.90, 137.02, 136.94, 133.90, 129.96, 128.51, 128.18, 126.74, 124.91, 124.45, 121.25, 120.40, 110.09, 50.55, 48.88, 40.46; Purity ≥95% by LCMS (Method A) tR = 1.92 min, m/z = 442.12 [M+H]+; HRMS calculated for C20H19N5O3S2 [M+H]+ 442.1013, found 442.1009.
2-(1H-Benzo[d][1,2,3]triazol-1-yl)-N-(4-(cyclopropanesulfonamido)phenyl)-N-(thiophen-3-ylmethyl)acetamide (13).
To a solution of 46 (36 mg, 0.1 mmol) in DCM (1 mL) was added cyclopropanesulfonyl chloride (12 μL, 0.12 mmol) and Et3N (509 μL, 3.6 mmol) and stirred at rt for 1 h. The mixture was washed with sat. aq. NH4Cl and concentrated, purified by preparative RP-HPLC, washed with aq. K2CO3 to remove TFA and concentrated to afford a colorless solid (8 mg, 0.02 mmol, 7%). 1H NMR (400 MHz, CD3OD) δ 7.98 (d, J = 8.4 Hz, 1H), 7.67 (d, J = 8.4 Hz, 1H), 7.56 (t, J = 7.7 Hz, 1H), 7.44 (t, J = 7.6 Hz, 1H), 7.39 – 7.29 (m, 3H), 7.22 (d, J = 8.8 Hz, 2H), 7.15 (s, 1H), 6.99 (d, J = 4.7 Hz, 1H), 5.38 (s, 2H), 4.91 (s, 2H), 2.65 – 2.56 (m, 1H), 1.10 – 1.03 (m, 2H), 1.00 – 0.93 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 165.19, 146.22, 138.22, 136.96 (two peaks overlap), 133.98, 129.77, 128.52, 128.18, 126.69, 124.89, 124.45, 122.32, 120.38, 110.15, 50.40, 48.84, 30.73, 6.20; Purity ≥ 95% by LCMS (Method A) tR = 2.05 min, m/z = 468.12 [M+H]+; HRMS calculated for C22H21N5O3S2 [M+H]+ 468.1170, found 468.1166.
2-(1H-Benzo[d][1,2,3]triazol-1-yl)-N-(4-(pyridin-4-yl)phenyl)-N-(thiophen-3-ylmethyl)acetamide (14).
An argon purged vial was charged with 47 (85 mg, 0.20 mmol), pyridine-4-boronic acid (39 mg, 0.32 mmol) and Pd(PPh3)4 (23 mg, 0.02 mmol). THF (2 mL) and water (0.4 mL) was added into the vial. The vial was capped and heated at 100°C for 16 h. The reaction was allowed to cool to ambient temperature, diluted with EtOAc, filtered with celite, and purified by preparative RP-HPLC to afford title compound as a colorless solid (52 mg, 0.12 mmol, 61%). 1H NMR (400 MHz, CDCl3) δ 8.72 (d, J = 6.3 Hz, 2H), 8.05 (d, J = 8.4 Hz, 1H), 7.69 (d, J = 8.6 Hz, 2H), 7.58 – 7.46 (m, 4H), 7.40 – 7.34 (m, 1H), 7.31 – 7.27 (m, 1H), 7.23 (d, J = 8.0 Hz, 2H), 7.07 (s, 1H), 7.00 (dd, J = 5.0, 1.3 Hz, 1H), 5.22 (s, 2H), 4.93 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 165.05, 150.62, 147.26, 146.29, 141.29, 139.31, 137.00, 133.99, 129.39, 129.18, 128.54, 128.07, 126.75, 124.94, 124.33, 121.99, 120.43, 110.06, 50.29, 48.86; Purity ≥95% by LCMS (Method A) tR = 1.67 min, m/z = 426.13 [M+H]+; HRMS calculated for C24H19N5OS [M+H]+ 426.1394, found 426.1390.
2-(1H-Benzo[d][1,2,3]triazol-1-yl)-N-(4-(pyridin-2-yl)phenyl)-N-(thiophen-3-ylmethyl)acetamide (15).
A vial was charged with 47 (86 mg, 0.20 mmol), 2-pyridinylboronic acid (49 mg, 0.40 mmol), CsCO3 (261 mg, 0.80 mmol), CuCl (20 mg, 0.20 mmol), Pd(OAc)2 (2 mg, 0.01 mmol) and 1,1-bis(diphenylphosphino)ferrocene (11 mg, 0.02 mmol). DMF (2 mL) was added into the vial. The mixture was stirred and purged with Ar. The vial was capped and heated at 100°C for 15 h. The reaction was allowed to cool to ambient temperature, diluted with EtOAc, filtered with celite, and purified by flash chromatography to afford title compound as a colorless solid (55 mg, 0.13 mmol, 65%). 1H NMR (400 MHz, CD3OD) δ 8.64 (d, J = 4.8 Hz, 1H), 8.05 (d, J = 8.1 Hz, 2H), 7.98 (d, J = 8.4 Hz, 1H), 7.95 – 7.85 (m, 2H), 7.68 (d, J = 8.4 Hz, 1H), 7.55 (t, J = 7.7 Hz, 1H), 7.46 – 7.34 (m, 5H), 7.18 (s, 1H), 7.03 (d, J = 4.9 Hz, 1H), 5.44 (s, 2H), 4.98 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 165.05, 156.12, 150.20, 146.25, 140.96, 140.56, 137.37, 137.05, 134.03, 129.06, 128.99, 128.60, 127.97, 126.55, 124.92, 124.21, 123.13, 121.03, 120.34, 110.04, 50.12, 48.71; Purity ≥95% by LCMS (Method A) tR = 1.76 min, m/z = 426.14 [M+H]+; HRMS calculated for C24H19N5OS [M+H]+ 426.1394, found 426.1397.
2-(1H-Benzo[d][1,2,3]triazol-1-yl)-N-(4-(2-methoxypyridin-3-yl)phenyl)-N-(thiophen-3-ylmethyl)acetamide (16).
A vial was charged with 47 (85 mg, 0.20 mmol), (2-methoxy-3-pyridyl)boronic acid (49 mg, 0.32 mmol) and Pd(PPh3)4 (23 mg, 0.02 mmol). THF (2 mL) and water (0.4 mL) was added into the vial. The mixture was stirred and purged with Ar. The vial was capped and heated at 100°C for 16 h. The reaction was allowed to cool to ambient temperature, diluted with EtOAc, filtered with celite, purified by flash chromatography to afford a colorless solid (90 mg, 0.20 mmol, 99%). 1H NMR (400 MHz, CDCl3) δ 8.21 (dd, J = 5.0, 1.9 Hz, 1H), 8.06 (d, J = 8.5 Hz, 1H), 7.69 – 7.60 (m, 3H), 7.53 – 7.47 (m, 2H), 7.41 – 7.33 (m, 1H), 7.31 – 7.27 (m, 1H), 7.18 (d, J = 8.3 Hz, 2H), 7.10 (s, 1H), 7.07 – 6.98 (m, 2H), 5.23 (s, 2H), 4.92 (s, 2H), 4.01 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 164.86, 160.71, 146.50, 145.98, 139.40, 138.66, 137.80, 136.98, 133.75, 130.97, 128.31, 128.03, 127.67, 126.21, 124.48, 123.91, 122.95, 120.07, 117.25, 109.77, 53.65, 49.94, 48.55; Purity ≥95% by LCMS (Method A) tR = 2.23 min, m/z = 456.15 [M+H]+; HRMS calculated for C25H21N5O2S [M+H]+ 445.1489, found 456.1499.
2-(1H-Benzo[d][1,2,3]triazol-1-yl)-N-(4-(2-oxo-1,2-dihydropyridin-3-yl)phenyl)-N-(thiophen-3-ylmethyl)acetamide (17).
Step 1. 2-(benzotriazol-1-yl)-N-[4-(2-fluoro-3-pyridyl)phenyl]-N-(3-thienylmethyl)acetamide (48).
A vial was charged with 47 (85 mg, 0.20 mmol), 2-fluoropyridine-3-boronic acid (45 mg, 0.32 mmol) and Pd(PPh3)4 (23 mg, 0.02 mmol). THF (2 mL) and water (0.4 mL) was added into the vial. The mixture was stirred and purged with Ar. The vial was capped and heated at 100°C for 16 h. The reaction was allowed to cool to ambient temperature, diluted with EtOAc, filtered with celite, purified by RP-HPLC and flash chromatography to afford a colorless solid (80 mg, 0.18 mmol, 90%). LCMS (Method A) tR = 2.16 min, m/z = 444.13 [M+H]+; 1H NMR (400 MHz, CDCl3) δ 8.25 (dt, J = 4.8, 1.6 Hz, 1H), 8.09 – 8.02 (m, 1H), 7.88 (ddd, J = 9.6, 7.5, 1.9 Hz, 1H), 7.67 – 7.60 (m, 2H), 7.53 – 7.47 (m, 2H), 7.41 – 7.31 (m, 2H), 7.30 – 7.27 (m, 1H), 7.21 (d, J = 8.1 Hz, 2H), 7.08 (s, 1H), 7.01 (dd, J = 5.0, 1.3 Hz, 1H), 5.23 (s, 2H), 4.93 (s, 2H).
Step 2. 2-(1H-benzo[d][1,2,3]triazol-1-yl)-N-(4-(2-oxo-1,2-dihydropyridin-3-yl)phenyl)-N-(thiophen-3-ylmethyl)acetamide.
To a solution of 48 (44 mg, 0.10 mmol) in dioxane (1 mL) was added conc. aq. HCl (0.08 mL, 1 mmol) and the mixture stirred for 16 h at 80°C. The reaction was allowed to cool to room temperature, concentrated, and purified by flash chromatography to afford a colorless solid (9.0 mg, 0.02 mmol, 20%). 1H NMR (400 MHz, CDCl3) δ 8.06 (d, J = 8.4 Hz, 1H), 7.83 (d, J = 8.3 Hz, 2H), 7.66 (dd, J = 7.1, 2.0 Hz, 1H), 7.53 – 7.47 (m, 2H), 7.43 (dd, J = 6.5, 2.0 Hz, 1H), 7.40 – 7.33 (m, 1H), 7.30 – 7.26 (m, 1H), 7.20 (d, J = 8.1 Hz, 2H), 7.09 (s, 1H), 7.03 (dd, J = 5.0, 1.3 Hz, 1H), 6.44 (t, J = 6.8 Hz, 1H), 5.22 (s, 2H), 4.91 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 165.20, 163.62, 146.30, 140.61, 140.03, 137.63, 137.30, 134.75, 134.08, 130.61, 130.28, 128.64, 128.46, 128.01, 126.53, 124.81, 124.25, 120.40, 110.11, 107.71, 50.26, 48.90; Purity ≥95% by LCMS (Method A) tR = 1.65 min, m/z = 442.13 [M+H]+; HRMS calculated for C24H19N5O2S [M+H]+ 442.1343, found 442.1342.
2-(Benzotriazol-1-yl)-N-[4-(1-methylpyrazol-4-yl)phenyl]-N-(3-thienylmethyl)acetamide (18).
A vial was charged with 47 (128 mg, 0.30 mmol), 1-methyl-1H-pyrazole-4-boronic acid, pinacol ester (75 mg, 0.36 mmol), 2M aq. K2CO3 (0.3 mL, 0.60 mmol) and Pd(dppf)Cl2.DCM (12 mg, 0.02 mmol). 1,4-Dioxane (1.5 mL) was added and the mixture degassed under a stream of Ar for 15 mins then heated at 100 °C for 16 h. The reaction was diluted with DCM, washed with water, dried (Na2SO4) and concentrated in vacuo. Residue was purified by preparative RP-HPLC (5–95% MeCN in H2O, 0.1% TFA), pure fractions were combined and concentrated in vacuo. The free base was obtained by SCX-II chromatography (load/wash MeOH, elution with 2N NH3 in MeOH) to afford the title compound as a colorless solid (57 mg, 0.13 mmol, 44%). 1H NMR (400 MHz, CD3OD) δ 8.02 – 7.94 (m, 2H), 7.84 (s, 1H), 7.69 – 7.59 (m, 3H), 7.54 (t, J = 7.6 Hz, 1H), 7.46 – 7.39 (m, 1H), 7.37 (dd, J = 5.0, 2.9 Hz, 1H), 7.29 – 7.21 (m, 2H), 7.15 (d, J = 3.0 Hz, 1H), 7.04 – 6.98 (m, 1H), 5.40 (s, 2H), 4.93 (s, 2H), 3.93 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 165.62, 145.46, 138.26, 138.06, 136.79, 134.27, 133.35, 129.17, 128.66, 128.22, 127.62, 127.00, 126.54, 124.25, 123.88, 121.43, 119.41, 111.58, 49.82, 48.48, 39.19; Purity ≥95% by LCMS (Method A) tR = 1.84 min, m/z = 429.15 [M+H]+; HRMS calculated for C23H20N6OS [M+H]+ 429.1492, found 429.1497.
2-(Benzotriazol-1-yl)-N-[4-(1H-pyrazol-4-yl)phenyl]-N-(3-thienylmethyl)acetamide (19).
A vial was charged with 46 (128 mg, 0.30 mmol), 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (70 mg, 0.36 mmol), 2M K2CO3 (0.3 mL, 0.60 mmol) and Pd(dppf)Cl2.DCM (12 mg, 0.02 mmol). 1,4-Dioxane (1.5 mL, 0.2 M) was added and degassed under a stream of Ar for 15 min then heated at 110 °C for 4 h. The reaction mixture was filtered through celite with EtOAc. The filtrate was washed with water, dried (Na2SO4) and concentrated in vacuo. The residue was purified by preparative RP-HPLC (5–95% MeCN in H2O, 0.1% TFA). The pure fractions were combined and concentrated in vacuo. The free base was obtained by SCX-II chromatography (load/wash MeOH, elution with 2N NH3 in MeOH) to afford the title compound as a colorless solid (63 mg, 0.15 mmol, 51%). 1H NMR (400 MHz, CD3OD) δ 8.01 – 7.76 (m, 3H), 7.45 (t, J = 7.6 Hz, 1H), 7.33 (t, J = 7.7 Hz, 1H), 7.27 (dd, J = 5.0, 2.9 Hz, 1H), 7.20 – 7.13 (m, 2H), 7.06 (d, J = 3.0 Hz, 1H), 6.92 (d, J = 4.9 Hz, 1H), 5.31 (s, 2H), 4.84 (s, 2H); 13C NMR (101 MHz, CD3OD) δ 165.91, 153.23, 145.12, 137.87, 137.05, 133.91, 128.58, 127.67, 127.48, 126.61, 125.82, 124.17, 123.80, 121.07, 118.46, 110.23, 49.39; Purity ≥95% by LCMS (Method A) tR = 1.71 min, m/z = 415.13 [M+H]+; HRMS calculated for C22H18N6OS [M+H]+ 415.1336, found 415.1328.
2-(Benzotriazol-1-yl)-N-[4-(1-methylimidazol-4-yl)phenyl]-N-(3-thienylmethyl)acetamide (20).
Step 1. 2-(Benzotriazol-1-yl)-N-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]-N-(3-thienylmethyl)acetamide.
Compound 47 (717 mg, 1.68 mmol), bis(pinacolato)diboron (511 mg, 2.01 mmol), Pd(dppf)Cl2.DCM (68 mg, 0.08 mmol), and KOAc (494 mg, 5.03 mmol) were combined in dioxane (8 mL) and heated to 100 °C for 16 h. Upon cooling, the mixture was filtered, concentrated and purified by ISCO flash chromatography (0–100% EtOAc in hexanes) to afford a pale brown solid (650 mg, 1.37 mmol, 82%). LCMS (Method A) tR = 2.22 min, m/z = 475.09 [M+H]+; 1H NMR (400 MHz, CDCl3) δ 8.05 (d, J = 8.2 Hz, 1H), 7.88 (d, J = 7.2 Hz, 2H), 7.53 – 7.44 (m, 2H), 7.36 (t, J = 7.0 Hz, 1H), 7.30 – 7.21 (m, 1H), 7.14 (d, J = 7.5 Hz, 2H), 7.02 (d, J = 2.9 Hz, 1H), 6.96 (d, J = 4.9 Hz, 1H), 5.14 (s, 2H), 4.89 (s, 2H), 1.36 (s, 12H).
Step 2. 2-(Benzotriazol-1-yl)-N-[4-(1-methylimidazol-4-yl)phenyl]-N-(3-thienylmethyl)acetamide
2-(Benzotriazol-1-yl)-N-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]-N-(3-thienylmethyl)acetamide (100 mg, 0.21 mmol), 4-bromo-1-methyl-1H-pyrazole (41 mg, 0.25 mmol), 2M aq. K2CO3 (0.21 mL, 0.42 mmol) and Pd(dppf)Cl2.DCM (9 mg, 0.01 mmol) were combined in 1,4-dioxane (1 mL), degassed under a stream of Ar for 15 mins then heated at 100 °C for 18 h. The reaction mixture was filtered through celite, washing with EtOAc. The filtrate was washed with water, dried (Na2SO4) and concentrated in vacuo. Residue was purified by preparative RP-HPLC (5–95% MeCN in H2O, 0.1% TFA), pure fractions were combined and concentrated in vacuo. The free base was obtained by SCX-II chromatography (load/wash MeOH, elution with 2N NH3 in MeOH) to afford the title compound as a colorless solid (36 mg, 0.08 mmol, 40%). 1H NMR (400 MHz, CD3OD) δ 7.98 (d, J = 8.4 Hz, 1H), 7.80 (d, J = 8.4 Hz, 2H), 7.71 (s, 1H), 7.67 (d, J = 8.4 Hz, 1H), 7.59 – 7.51 (m, 2H), 7.43 (t, J = 7.7 Hz, 1H), 7.37 (dd, J = 5.0, 2.9 Hz, 1H), 7.28 (d, J = 8.6 Hz, 2H), 7.16 (s, 1H), 7.01 (d, J = 5.0 Hz, 1H), 5.41 (s, 2H), 4.94 (s, 2H), 3.78 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 165.61, 145.47, 139.90, 139.18, 138.51, 138.05, 134.29, 128.90, 128.22, 127.64, 126.99, 125.78, 124.25, 123.88, 119.42, 118.22, 111.59, 49.80, 48.48, 33.68; Purity ≥95% by LCMS (Method A) tR = 1.43 min, m/z = 429.03 [M+H]+; HRMS calculated for C23H20N6OS [M+H]+ 429.1492, found 429.1497.
2-(Benzotriazol-1-yl)-N-[4-(1H-imidazol-4-yl)phenyl]-N-(3-thienylmethyl)acetamide (21).
Step 1: 2-(Benzotriazol-1-yl)-N-(3-thienylmethyl)-N-[4-(1-tritylimidazol-4-yl)phenyl]acetamide.
Compound 49 2-(benzotriazol-1-yl)-N-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]-N-(3-thienylmethyl)acetamide (122 mg, 0.31 mmol), 4-bromo-1-trityl-imidazole (178 mg, 0.38 mmol), 2M aq. K2CO3 (0.31 mL, 0.63 mmol) and Pd(dppf)Cl2.DCM (13 mg, 0.02 mmol) were combined in 1,4-dioxane (1.6 mL), degassed under a stream of Ar for 15 mins then heated at 100 °C for 18 h. The reaction mixture was filtered through celite, washing with EtOAc. The filtrate was washed with water, dried (Na2SO4) and concentrated in vacuo. Residue was purified by ISCO flash chromatography (24 g, 0–60% EtOAc in hexanes) to afford title compound (117 mg, 0.18 mmol, 57%). Purity = 85% by LCMS (210, 254 nm); tR = 1.78 min, m/z = 657.24 [M+H]+.
Step 2: 2-(Benzotriazol-1-yl)-N-[4-(1H-imidazol-4-yl)phenyl]-N-(3-thienylmethyl)acetamide.
Compound 49 (50 mg, 0.08 mmol) was dissolved in MeOH (380 μL), acetic acid (95 μL, 0.08 mmol) was added and the reaction mixture stirred at 65 °C for 2 h. The mixture was concentrated in vacuo then purified by preparative RP-HPLC (5–40% MeCN in H2O, 0.1% TFA). The free base was obtained by SCX-II chromatography (load/wash MeOH, elution with 2N NH3 in MeOH) to afford the title compound as a colorless solid (11 mg, 0.03 mmol, 35%). 1H NMR (400 MHz, CD3OD) δ 9.04 – 9.00 (m, 1H), 8.01 – 7.94 (m, 2H), 7.80 (d, J = 8.1 Hz, 2H), 7.67 (s, 1H), 7.56 (t, J = 7.7 Hz, 1H), 7.46 – 7.35 (m, 4H), 7.16 (s, 1H), 7.02 (d, J = 4.9 Hz, 1H), 5.44 (s, 2H), 4.98 (s, 2H); 13C NMR (101 MHz, CD3OD) δ 165.61, 145.47, 138.38, 138.04, 136.75, 134.29, 128.90, 128.24, 127.64, 126.99, 125.86, 124.25, 123.92, 119.42, 111.59, 49.80, 48.45; Purity ≥95% by LCMS (Method A) tR = 1.65 min, m/z = 415.13 [M+H]+; HRMS calculated for C22H18N6OS [M+H]+ 415.1336, found 415.1341.
2-(1H-Benzo[d][1,2,3]triazol-1-yl)-N-isopentyl-N-(4-(pyridin-3-yl)phenyl)acetamide (22).
Step 1. 4-(3-Pyridyl)aniline (51).
To a vial containing 4-bromoaniline (1.72 g, 10 mmol), pyridine-3-boronic acid (1.47 g, 12 mmol), Pd(dppf)Cl·DCM (203.66 mg, 0.25 mmol) and K2CO3 (2.76 g, 20 mmol) was added dioxane (25 mL) and water (5 mL). The mixture was heated to 100 °C for 18 h. The mixture was diluted with EtOAc and washed with water, brine, then dried over MgSO4 and concentrated. The crude material was purified by flash chromatography to afford a pale brown solid (1.45 g, 8.54 mmol, 85%). LCMS (Method A) tR = 0.53 min, m/z = 171.03 [M+H]+.
Step 2. N-Isopentyl-4-(3-pyridyl)aniline.
To a solution of isovaleraldehyde (0.05 mL, 0.50 mmol) and 51 (104 mg, 0.61 mmol) in DCE (5 mL) was added NaBH(OAc)3 (161 mg, 0.76 mmol) and the mixture stirred for 2 h at rt. Saturated aq. NH4Cl (30 mL) was added and DCE layer separated. The aqueous layer was extracted with DCM (3 × 10 mL) and concentrated to dryness. Purification by flash chromatography afford a yellow solid (107 mg, 0.45 mmol, 89%). 1H NMR (400 MHz, CDCl3) δ 8.80 (s, 1H), 8.48 (dd, J = 5.0, 1.6 Hz, 1H), 7.82 (dt, J = 8.1, 1.9 Hz, 1H), 7.43 (d, J = 8.7 Hz, 2H), 7.31 (dd, J = 8.0, 4.9 Hz, 1H), 6.69 (d, J = 8.7 Hz, 2H), 3.18 (t, J = 7.4 Hz, 2H), 1.81 – 1.67 (m, 1H), 1.55 (q, J = 7.2 Hz, 2H), 0.97 (d, J = 6.6 Hz, 6H).
Step 3. 2-(1H-Benzo[d][1,2,3]triazol-1-yl)-N-isopentyl-N-(4-(pyridin-3-yl)phenyl)acetamide.
To a stirred solution of N-isopentyl-4-(3-pyridyl)aniline (48 mg, 0.2 mmol) and 50 (35 mg, 0.2 mmol) in DMF (0.5 mL) was added T3P (50% in EtOAc) (238 μL, 0.4 mmol), followed by pyridine (48 μL, 0.6 mmol). The mixture was stirred at rt for 16 h, then directly purified by RP-HPLC to afford a colorless solid (30 mg, 0.08 mmol, 38%). 1H NMR (400 MHz, CDCl3) δ 8.88 (d, J = 2.4 Hz, 1H), 8.66 (dd, J = 4.9, 1.7 Hz, 1H), 8.04 (d, J = 8.4 Hz, 1H), 7.92 (dt, J = 7.9, 2.0 Hz, 1H), 7.71 (d, J = 8.1 Hz, 2H), 7.55 – 7.31 (m, 6H), 5.21 (s, 2H), 3.84 – 3.75 (m, 2H), 1.65 – 1.52 (m, 1H), 1.52 – 1.43 (m, 2H), 0.89 (d, J = 6.6 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ 164.95, 149.43, 148.49, 146.28, 140.92, 138.92, 135.58, 134.90, 134.06, 129.39, 129.25, 128.00, 124.25, 124.12, 120.36, 110.17, 50.34, 49.09, 36.63, 26.37, 22.79; Purity ≥95% by LCMS (Method A) tR = 1.76 min, m/z = 400.21 [M+H]+; HRMS calculated for C24H25N5O [M+H]+ 400.2143, found 400.2131.
2-(1H-Benzo[d][1,2,3]triazol-1-yl)-N-isopentyl-N-(4-(pyridin-3-yl)phenyl)acetamide (23).
Step 1. N-(Cyclopropylmethyl)-4-(3-pyridyl)aniline
To a solution of cyclopropanecarbaldehyde (0.04 mL, 0.50 mmol) and 51 (104 mg, 0.61 mmol) in DCE (5 mL) was added NaBH(OAc)3 (161.4 mg, 0.76 mmol) and the mixture stirred for 1.5 h at rt. Saturated aq. NH4Cl (30 mL) was added and DCE layer separated. The aqueous layer was extracted with EtOAc (3 × 10 mL) and concentrated. Purification by flash chromatography afforded a pale cream solid (100 mg, 0.45 mmol, 89%). 1H NMR (400 MHz, CDCl3) δ 8.80 (s, 1H), 8.52 – 8.46 (m, 1H), 7.81 (dt, J = 7.9, 2.0 Hz, 1H), 7.43 (d, J = 8.6 Hz, 2H), 7.30 (dd, J = 7.9, 4.8 Hz, 1H), 6.70 (d, J = 8.6 Hz, 2H), 4.03 (s, 1H), 3.01 (d, J = 6.9 Hz, 2H), 1.19 – 1.05 (m, 1H), 0.63 – 0.54 (m, 2H), 0.31 – 0.23 (m, 2H).
Step 2. 2-(1H-Benzo[d][1,2,3]triazol-1-yl)-N-isopentyl-N-(4-(pyridin-3-yl)phenyl)acetamide.
To a stirred solution of N-(cyclopropylmethyl)-4-(3-pyridyl)aniline (45 mg, 0.2 mmol) and 50 (35 mg, 0.2 mmol) in DMF (0.5 mL) was added T3P (50% in EtOAc) (238 μL, 0.4 mmol), followed by pyridine (48 μL, 0.6 mmol). The mixture was stirred at rt for 14 h, then directly purified by RP-HPLC to afford a colorless solid (50 mg, 0.13 mmol, 65%). 1H NMR (400 MHz, CDCl3) δ 8.88 (d, J = 2.4 Hz, 1H), 8.66 (dd, J = 4.8, 1.6 Hz, 1H), 8.04 (d, J = 8.4 Hz, 1H), 7.92 (dt, J = 7.9, 2.0 Hz, 1H), 7.70 (d, J = 8.3 Hz, 2H), 7.55 – 7.40 (m, 5H), 7.36 (ddd, J = 8.1, 6.5, 1.4 Hz, 1H), 5.23 (s, 2H), 3.67 (d, J = 7.3 Hz, 2H), 1.08 – 0.96 (m, 1H), 0.54 – 0.45 (m, 2H), 0.23 – 0.14 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 165.02, 149.46, 148.54, 146.28, 141.00, 138.96, 135.57, 134.84, 134.04, 129.54, 129.24, 127.95, 124.23, 124.09, 120.34, 110.19, 54.68, 50.36, 10.04, 4.18; Purity ≥ 95% by LCMS (Method A) tR = 1.59 min, m/z = 384.18 [M+H]+; HRMS calculated for C23H21N5O [M+H]+ 384.1830, found 384.1813.
2-(1H-Benzo[d][1,2,3]triazol-1-yl)-N-(cyclobutylmethyl)-N-(4-(pyridin-3-yl)phenyl)acetamide (24).
Step 1. N-(Cyclobutylmethyl)-4-(3-pyridyl)aniline.
To a solution of cyclobutanecarbaldehyde (0.04 mL, 0.50 mmol) and 51 (104 mg, 0.61 mmol) in DCE (5 mL) was added NaBH(OAc)3 (161 mg, 0.76 mmol) and the mixture stirred for 1 h at rt Sat. aq. NH4Cl (30 mL) was added and DCE layer separated. The aqueous layer was extracted with DCM (3 × 10 mL) and concentrated, purified by flash chromatography to afford as a light-yellow solid (110 mg, 0.46 mmol, 92%). 1H NMR (400 MHz, CDCl3) δ 8.80 (s, 1H), 8.48 (dd, J = 4.9, 1.7 Hz, 1H), 7.82 (dt, J = 8.1, 1.9 Hz, 1H), 7.42 (d, J = 8.7 Hz, 2H), 7.31 (dd, J = 7.9, 4.9 Hz, 1H), 6.69 (d, J = 8.7 Hz, 2H), 3.18 (d, J = 7.3 Hz, 2H), 2.68 – 2.54 (m, 1H), 2.21 – 2.09 (m, 2H), 2.02 – 1.87 (m, 3H), 1.83 – 1.70 (m, 3H).
Step 2. 2-(1H-Benzo[d][1,2,3]triazol-1-yl)-N-(cyclobutylmethyl)-N-(4-(pyridin-3-yl)phenyl)acetamide.
To a stirred solution of N-(cyclobutylmethyl)-4-(3-pyridyl)aniline (48 mg, 0.2 mmol) and 50 (35 mg, 0.2 mmol) in DMF (0.5 mL) was added T3P (50% in EtOAc) (238 μL, 0.4 mmol), followed by pyridine (48 μL, 0.6 mmol). The mixture was stirred at rt for 13 h, then directly purified by RP-HPLC to afford a colorless solid (30 mg, 0.08 mmol, 38%). 1H NMR (400 MHz, MeOD) δ 8.85 (d, J = 2.4 Hz, 1H), 8.60 – 8.54 (m, 1H), 8.15 (dt, J = 8.2, 2.0 Hz, 1H), 7.97 (d, J = 8.4 Hz, 1H), 7.83 (d, J = 8.1 Hz, 2H), 7.64 (d, J = 8.4 Hz, 1H), 7.61 – 7.49 (m, 4H), 7.41 (t, J = 7.7 Hz, 1H), 5.39 (s, 2H), 3.87 (d, J = 7.6 Hz, 2H), 2.64 – 2.51 (m, 1H), 2.10 – 1.97 (m, 2H), 1.97 – 1.81 (m, 2H), 1.78 – 1.67 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 165.20, 149.33, 148.40, 146.29, 140.92, 138.85, 135.62, 134.97, 134.03, 129.28 (two peaks overlap), 127.96, 124.24, 124.15, 120.35, 110.21, 55.14, 50.38, 34.01, 26.54, 18.64; Purity ≥95% by LCMS (Method A) tR = 1.70 min, m/z = 398.20 [M+H]+; HRMS calculated for C24H23N5O [M+H]+ 398.1986, found 398.1969.
2-(1H-Benzo[d][1,2,3]triazol-1-yl)-N-(cyclopentylmethyl)-N-(4-(pyridin-3-yl)phenyl)acetamide (25).
Step 1. N-(Cyclobutylmethyl)-4-(3-pyridyl)aniline
To a solution of cyclopentanecarbaldehyde (0.05 mL, 0.50 mmol) and 51 (104 mg, 0.61 mmol) in DCE (5 mL) was added NaBH(OAc)3 (161 mg, 0.76 mmol) and the mixture stirred for 1 h at rt Sat. aq. NH4Cl (30 mL) was added and DCE layer separated. The aqueous layer was extracted with EtOAc (3 × 10 mL) and concentrated. Purification by flash chromatography afforded a light yellow solid (126 mg, 0.20 mmol, 99%). LCMS (Method A) tR = 1.55 min, m/z = 253.09 [M+H]+.
Step 2. 2-(1H-benzo[d][1,2,3]triazol-1-yl)-N-(cyclopentylmethyl)-N-(4-(pyridin-3-yl)phenyl)acetamide.
To an ice-cold solution of 50 (53 mg, 0.30 mmol) and Et3N (0.08 mL, 0.60 mmol) in DCM (1 mL) was added HATU (114 mg, 0.30 mmol) and the mixture stirred for 30 min before the addition of N-(cyclopentylmethyl)-4-(3-pyridyl)aniline (51 mg, 0.20 mmol) in a single portion. The mixture was stirred for 20 h at rt then washed with water, brine, and concentrated. Purification by flash chromatography afforded title compound as a colorless solid (69.1 mg, 0.17 mmol, 84%). 1H NMR (400 MHz, MeOD) δ 8.85 (d, J = 2.4 Hz, 1H), 8.60 – 8.54 (m, 1H), 8.15 (dt, J = 8.0, 2.0 Hz, 1H), 7.97 (d, J = 8.4 Hz, 1H), 7.84 (d, J = 8.4 Hz, 2H), 7.69 – 7.59 (m, 3H), 7.60 – 7.50 (m, 2H), 7.41 (t, J = 7.7 Hz, 1H), 5.42 (s, 2H), 3.79 (d, J = 7.7 Hz, 2H), 2.19 – 2.06 (m, 1H), 1.82 – 1.71 (m, 2H), 1.69 – 1.50 (m, 4H), 1.35 – 1.24 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 165.29, 149.47, 148.54, 146.25, 140.96, 138.86, 135.54, 134.81, 134.03, 129.30, 129.23, 127.95, 124.22, 124.08, 120.31, 110.19, 54.82, 50.40, 38.25, 30.65, 25.52; Purity ≥95% by LCMS (Method A) tR = 1.81 min, m/z = 412.21 [M+H]+; HRMS calculated for C25H25N5O [M+H]+ 412.2143, found 412.2125.
2-(1H-Benzo[d][1,2,3]triazol-1-yl)-N-(4-(pyridin-3-yl)phenyl)-N-((tetrahydrofuran-3-yl)methyl)acetamide (26).
Step 1. 4-(3-Pyridyl)-N-(tetrahydrofuran-3-ylmethyl)aniline.
To a solution of tetrahydrofuran-3-carbaldehyde (50% aq., 91 μL, 0.50 mmol) and 51 (98 mg, 0.58 mmol) in DCE (5 mL) was added NaBH(OAc)3 (148 mg, 0.70 mmol) and the mixture stirred for 1 h at rt. Saturated aq. NH4Cl (30 mL) was added and DCE layer separated. The aqueous layer was extracted with EtOAc (3 × 10 mL) and concentrated. Purification by flash chromatography afforded a yellow oil (114 mg, 0.45 mmol, 90%). LCMS (Method A) tR = 1.15 min, m/z = 255.08 [M+H]+.
Step 2. 2-(1H-Benzo[d][1,2,3]triazol-1-yl)-N-(4-(pyridin-3-yl)phenyl)-N-((tetrahydrofuran-3-yl)methyl)acetamide.
To a stirred solution of 4-(3-pyridyl)-N-(tetrahydrofuran-3-ylmethyl)aniline (51 mg, 0.2 mmol) and 50 (35 mg, 0.2 mmol) in DMF (0.5 mL) was added T3P (50% in EtOAc) (238 μL, 0.4 mmol), followed by pyridine (48 μL, 0.6 mmol). The mixture was stirred at rt for 16 h, then washed with water (20 mL) and brine (20 mL). Purification by flash chromatography afforded a colorless solid (47 mg, 0.11 mmol, 57%). 1H NMR (400 MHz, CDCl3) δ 8.88 (s, 1H), 8.67 (d, J = 4.8 Hz, 1H), 8.05 (d, J = 8.4 Hz, 1H), 7.95 (d, J = 8.1 Hz, 1H), 7.71 (d, J = 8.3 Hz, 2H), 7.53 – 7.45 (m, 3H), 7.42 (d, J = 8.1 Hz, 2H), 7.40 – 7.33 (m, 1H), 5.29 – 5.17 (m, 2H), 3.99 (dd, J = 13.5, 7.7 Hz, 1H), 3.90 – 3.68 (m, 4H), 3.51 – 3.43 (m, 1H), 2.59 – 2.48 (m, 1H), 2.06 – 1.94 (m, 1H), 1.68 – 1.64 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 165.61, 149.54, 148.48, 146.26, 140.75, 139.18, 135.41, 134.88, 133.98, 129.54, 129.09, 128.06, 124.32, 124.14, 120.40, 110.04, 71.59, 68.00, 52.86, 50.32, 38.45, 30.46; Purity ≥95% by LCMS (Method A) tR = 1.44 min, m/z = 414.19 [M+H]+; HRMS calculated for C24H23N5O2 [M+H]+ 414.1935, found 414.1936.
2-(Benzotriazol-1-yl)-N-[4-(3-pyridyl)phenyl]-N-(thiazol-4-ylmethyl)acetamide (27).
Step 1. 4-(3-Pyridyl)-N-(thiazol-4-ylmethyl)aniline.
To a solution of 51 (34 mg, 0.20 mmol) and thiazole-4-carbaldehyde (25 mg, 0.22 mmol) in DCM (1 mL) was added AcOH (50 μL) and stirred for 30 min before the addition of NaBH(OAc)3 (64 mg, 0.30 mmol). The mixture was stirred for 16 h, then diluted with DCM and washed with sat. aq. NaHCO3, concentrated and purified by ISCO flash chromatography (0–100% EtOAc in hexanes) to afford a cream colored solid (47 mg, 0.18 mmol, 88%). LCMS (Method A) tR = 1.13 min, m/z = 268.01 [M+H]+.
Step 2. 2-(Benzotriazol-1-yl)-N-[4-(3-pyridyl)phenyl]-N-(thiazol-4-ylmethyl)acetamide.
To a solution of 4-(3-pyridyl)-N-(thiazol-4-ylmethyl)aniline (47 mg, 0.18 mmol) and 50 (31 mg, 0.18 mmol) in DMF (1 mL) was added T3P (50% in DMF, 0.21 mL, 0.35 mmol) and pyridine (43 μL, 0.53 mmol). The mixture was stirred at rt for 16 h, then filtered and purified by preparative RP-HPLC. The free base was obtained by SCX-II chromatography (eluant 2N NH3/MeOH) affording a colorless solid (27 mg, 0.06 mmol, 36%). 1H NMR (400 MHz, MeOH-d4) ∂H 8.99 (s, 1H), 8.82 (s, 1H), 8.61 – 8.52 (m, 1H), 8.12 (d, J = 8.0 Hz, 1H), 7.99 (d, J = 8.4 Hz, 1H), 7.77 (d, J = 8.0 Hz, 2H), 7.71 (d, J = 8.4 Hz, 1H), 7.60 – 7.47 (m, 6H), 7.43 (t, J = 7.7 Hz, 1H), 5.50 (s, 2H), 5.18 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 165.61, 154.83, 152.81, 149.31, 148.20, 145.46, 140.95, 134.92, 134.70, 134.19, 129.38, 128.66, 127.61, 124.37, 124.26, 119.42, 117.47, 111.58, 50.00, 49.50; Purity ≥95% by LCMS (Method A) tR = 1.24 min, m/z = 427.13 [M+H]+; HRMS calculated for C23H18N6OS [M+H]+ 427.1336, found 427.1339.
2-(Benzotriazol-1-yl)-N-[(3-chlorophenyl)methyl]-N-[4-(3-pyridyl)phenyl]acetamide (28).
Step 1. N-[(3-Chlorophenyl)methyl]-4-(3-pyridyl)aniline.
To a solution of 51 (34 mg, 0.20 mmol) and 3-chlorobenzaldehyde (25 μL, 0.22 mmol) in DCM (1 mL) was added AcOH (50 μL) and stirred for 30 min before the addition of NaBH(OAc)3 (64 mg, 0.30 mmol). The mixture was stirred for 16 h, then diluted with DCM and washed with sat. aq. NaHCO3. Concentration and purification by ISCO flash chromatography (0–100% EtOAc in hexanes) afforded a cream colored solid (51 mg, 0.17 mmol, 86%). LCMS (Method A) tR = 1.63 min, m/z = 295.02 [M+H]+.
Step 2. 2-(Benzotriazol-1-yl)-N-[(3-chlorophenyl)methyl]-N-[4-(3-pyridyl)phenyl]acetamide.
To a solution of N-[(3-chlorophenyl)methyl]-4-(3-pyridyl)aniline (51 mg, 0.17 mmol) and 50 (30 mg, 0.17 mmol) in DMF (1 mL) was added T3P (50% in DMF, 206 μL, 0.35 mmol) and pyridine (42 μL, 0.52 mmol). The mixture was stirred at rt for 16 h, then filtered and purified by preparative RP-HPLC. The free base was obtained after SCX-II chromatography (eluent 2N NH3/MeOH) affording a colorless solid (48 mg, 0.11 mmol, 61%). 1H NMR (400 MHz, DMSO-d6) δ 8.93 (s, 1H), 8.59 (d, J = 4.8 Hz, 1H), 8.11 (d, J = 8.0 Hz, 1H), 8.04 (d, J = 8.4 Hz, 1H), 7.86 (d, J = 8.1 Hz, 2H), 7.82 (d, J = 8.4 Hz, 1H), 7.64 (d, J = 8.0 Hz, 2H), 7.55 (t, J = 7.7 Hz, 1H), 7.50 (dd, J = 7.9, 4.8 Hz, 1H), 7.44 – 7.31 (m, 4H), 7.26 (d, J = 6.1 Hz, 1H), 5.55 (s, 2H), 4.98 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 166.19, 149.33, 148.20, 145.44, 140.58, 139.92, 137.49, 134.83, 134.69, 134.20, 133.58, 130.80, 129.24, 128.74, 128.17, 127.83, 127.63, 127.07, 124.36, 124.28, 119.43, 111.57, 52.49, 49.98; Purity ≥95% by LCMS (Method A) tR = 1.60 min, m/z = 454.14 [M+H]+; HRMS calculated for C26H20ClN5O [M+H]+ 454.1429, found 454.1445.
2-(Benzotriazol-1-yl)-N-[(3-chloro-4-fluoro-phenyl)methyl]-N-[4-(3-pyridyl)phenyl]acetamide (29).
Step 1: N-[(3-Chloro-4-fluoro-phenyl)methyl]-4-(3-pyridyl)aniline.
3-chloro-4-fluoro-benzaldehyde (35 mg, 0.22 mmol) and 51 (34 mg, 0.20 mmol) were dissolved in DCM:AcOH (0.2 M, 20:1) and stirred for 30 min at 23 °C. To the mixture, NaBH(OAc)3 (63.6 mg, 0.30 mmol) was added and stirred at 23 °C for 18 h. The reaction mixture was diluted with DCM and washed with sat. aq. NaHCO3. The organic layer was dried (Na2SO4), filtered, and concentrated in vacuo. The resulting residue was purified by ISCO automated flash chromatography (4 g, 0–100% EtOAc in hexanes) to afford a cream colored solid (19 mg, 0.06 mmol, 30%). LCMS (Method A), tR = 1.86 min, m/z = 313.09 [M+H]+.
Step 2: 2-(Benzotriazol-1-yl)-N-[(3-chloro-4-fluoro-phenyl)methyl]-N-[4-(3-pyridyl)phenyl]acetamide.
Pyridine (14 μL, 0.17 mmol) and T3P (50% in DMF) (78 μL, 0.12 mmol) were added to a solution of N-[(3-chloro-4-fluoro-phenyl)methyl]-4-(3-pyridyl)aniline (18 mg, 0.06 mmol) and 50 (11 mg, 0.06 mmol) in DMF (0.5 mL, 0.2 M) and stirred at 23 °C for 18 h. The concentrated reaction mixture was purified by preparative RP-HPLC (5–50% MeCN in H2O, 0.1% TFA), pure fractions were combined and concentrated in vacuo. The free base was obtained by SCX-II chromatography (load/wash MeOH, elution with 2N NH3 in MeOH) to afford the title compound as a colorless solid (7.5 mg, 0.02 mmol, 28%). 1H NMR (400 MHz, CD3OD) δ 9.02 (d, J = 2.3 Hz, 1H), 8.74 (dd, J = 5.5, 1.5 Hz, 1H), 8.61 – 8.54 (m, 1H), 7.98 (d, J = 8.5 Hz, 1H), 7.91 (dd, J = 8.1, 5.5 Hz, 1H), 7.83 (d, J = 8.4 Hz, 2H), 7.69 (d, J = 8.4 Hz, 1H), 7.59 – 7.52 (m, 1H), 7.50 – 7.34 (m, 4H), 7.24 – 7.13 (m, 2H), 5.49 (s, 2H), 4.99 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 165.71, 157.68, 155.23, 148.85, 147.71, 144.97, 139.96, 137.06, 134.86, 134.82, 134.33, 134.21, 133.71, 130.02, 128.81, 128.28, 127.15, 123.88, 119.46, 119.28, 118.95, 116.97, 116.76, 111.09, 51.42, 49.51; 19F NMR (376 MHz, CD3OD δ −112.43; Purity ≥95% by LCMS (Method A) tR = 1.86 min, m/z = 472.13 [M+H]+; HRMS calculated for C26H19ClFN5O [M+H]+ 472.1335, found 472.1345.
2-(Benzotriazol-1-yl)-N-[(3,4-dichlorophenyl)methyl]-N-[4-(3-pyridyl)phenyl]acetamide (30).
Step 1: N-[(3,4-Dichlorophenyl)methyl]-4-(3-pyridyl)aniline.
3,4-Dichlorobenzaldehyde. (39 mg, 0.22 mmol) and 51 (34 mg, 0.20 mmol) were dissolved in DCM:AcOH (0.2 M, 20:1) and stirred for 30 min at rt. To the mixture, NaBH(OAc)3 (63.6 mg, 0.30 mmol) was added and stirred at rt for 18 h. The reaction mixture was diluted with DCM and washed with sat. aq. NaHCO3. The organic layer was dried (Na2SO4), filtered, and concentrated in vacuo. The resulting residue was purified by ISCO flash chromatography (4 g, 0–100% EtOAc in hexanes) to afford N-[(3,4-dichlorophenyl)methyl]-4-(3-pyridyl)aniline as a cream colored solid (40 mg, 0.12 mmol, 61%). LCMS (Method A) tR = 1.93 min, m/z = 329.06 [M+H]+.
Step 2: 2-(Benzotriazol-1-yl)-N-[(3,4-dichlorophenyl)methyl]-N-[4-(3-pyridyl)phenyl]acetamide.
Pyridine (29 μL, 0.36 mmol) and T3P (50% in DMF) (160 μL, 0.24 mmol) were added to a solution of N-[(3,4-dichlorophenyl)methyl]-4-(3-pyridyl)aniline (39 mg, 0.12 mmol) and 50 (23 mg, 0.13 mmol, 1.1 eq.) in DMF (0.6 mL, 0.2 M) and stirred at rt for 18 h. The reaction mixture was purified directly by preparative RP-HPLC (5–50% MeCN in H2O, 0.1% TFA). The product containing fractions were combined and concentrated in vacuo. The free base was obtained by SCX-II chromatography (load/wash MeOH, elution with 2N NH3 in MeOH) to afford the title compound as a colorless solid (36 mg, 0.07 mmol, 62%). 1H NMR (400 MHz, CD3OD) δ 9.02 (d, J = 2.3 Hz, 1H), 8.74 (dd, J = 5.5, 1.5 Hz, 1H), 8.57 (dd, J = 6.1, 4.1 Hz, 1H), 7.98 (d, J = 8.4 Hz, 1H), 7.92 (dd, J = 8.1, 5.4 Hz, 1H), 7.87 – 7.80 (m, 2H), 7.69 (d, J = 8.4 Hz, 1H), 7.58 – 7.53 (m, 1H), 7.51 – 7.39 (m, 5H), 7.20 (d, J = 8.3 Hz, 1H), 5.50 (s, 2H), 5.00 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 166.23, 158.89, 158.53, 146.78, 145.79, 145.45, 140.92, 138.57, 137.69, 135.88, 134.17, 131.54, 131.10, 130.45, 130.41, 129.32, 128.98, 128.82, 127.65, 125.47, 124.30, 119.44, 111.58, 51.91, 50.00; Purity ≥95% by LCMS (Method A) tR = 1.93 min, m/z = 488.10 [M+H]+; HRMS calculated for C26H19Cl2N5O [M+H]+ 488.1039, found 488.1048.
2-(Benzotriazol-1-yl)-N-[(3-chloro-5-fluoro-phenyl)methyl]-N-[4-(3-pyridyl)phenyl]acetamide (31).
Step 1: N-[(3-Chloro-5-fluoro-phenyl)methyl]-4-(3-pyridyl)aniline.
3-Chloro-5-fluoro-benzaldehyde (35 mg, 0.22 mmol) and 51 (34 mg, 0.20 mmol) were dissolved in DCM:AcOH (0.2 M, 20:1) and stirred for 30 min at 23 °C. To the mixture, NaBH(OAc)3 (64 mg, 0.30 mmol) was added and stirred at rt for 18 h. The reaction mixture was diluted with DCM and washed with sat. aq. NaHCO3. The organic layers were dried (Na2SO4), filtered, and concentrated in vacuo. The resulting residue was purified by ISCO automated flash chromatography (4 g, 0–100% EtOAc in hexanes) to afford a colorless solid (56 mg, 0.18 mmol, 90%). LCMS (Method A) tR = 1.87 min, m/z = 313.09 [M+H]+.
Step 2: 2-(Benzotriazol-1-yl)-N-[(3-chloro-5-fluoro-phenyl)methyl]-N-[4-(3-pyridyl)phenyl]acetamide.
Pyridine (40 μL, 0.50 mmol) and T3P (50% in DMF) (224 μL, 0.33 mmol) were added to a solution of N-[(3-chloro-5-fluoro-phenyl)methyl]-4-(3-pyridyl)aniline (52 mg, 0.17 mmol) and 50 (32 mg, 0.18 mmol) in DMF (0.8 mL) and stirred at rt for 18 h. The reaction mixture was purified directly by preparative RP-HPLC (5–50% MeCN in H2O, 0.1% TFA). The product containing fractions were combined and concentrated in vacuo. The free base was obtained by SCX-II chromatography (load/wash MeOH, elution with 2N NH3 in MeOH) to afford title compound as a colorless solid (26 mg, 0.06 mmol, 33%). 1H NMR (400 MHz, CD3OD) δ 8.81 (s, 1H), 8.56 (d, J = 4.8 Hz, 1H), 8.11 (dt, J = 7.9, 2.0 Hz, 1H), 7.98 (d, J = 8.4 Hz, 1H), 7.76 (d, J = 8.4 Hz, 2H), 7.68 (d, J = 8.4 Hz, 1H), 7.54 (dd, J = 8.3, 5.9 Hz, 2H), 7.49 – 7.38 (m, 3H), 7.17 – 7.09 (m, 2H), 7.02 (d, J = 9.3 Hz, 1H), 5.51 (s, 2H), 5.00 (s, 2H); 13C NMR (101 MHz, DMSOd6) δ 166.38, 163.87, 161.41, 149.15, 148.04, 145.45, 142.12, 142.04, 140.55, 137.47, 134.91, 134.47, 134.36, 134.17, 129.17, 128.80, 127.62, 124.44, 124.28, 119.43, 115.53, 115.28, 114.22, 114.00, 111.59, 52.19, 50.01; 19F NMR (376 MHz, CD3OD δ −110.58; Purity ≥95% by LCMS (Method A) tR = 1.87 min, m/z = 472.13 [M+H]+; HRMS calculated for C26H19ClFN5O [M+H]+ 472.1335, found 472.1343.
2-(Benzotriazol-1-yl)-N-[(3,5-dichlorophenyl)methyl]-N-[4-(3-pyridyl)phenyl]acetamide (32).
Step 1: N-[(3,5-Dichlorophenyl)methyl]-4-(3-pyridyl)aniline.
Compound 51 (34 mg, 0.20 mmol) and 3,5-dichlorobenzaldehyde (39 mg, 0.22 mmol) were dissolved in DCM:AcOH (0.2 M, 20:1) and stirred for 30 min at 23 °C. To the mixture, NaHB(OAc)3 (64 mg, 0.30 mmol) was added and stirred at rt for 18 h. The reaction mixture was diluted with DCM and washed with sat. aq. NaHCO3. The organic layers were dried (Na2SO4), filtered, and concentrated in vacuo. The resulting residue was purified by ISCO automated flash chromatography (4 g, 0–100% EtOAc in hexanes) to afford a colorless solid (65 mg, 0.20 mmol, 99%). LCMS (Method A) tR = 1.95 min, m/z = 329.06 [M+H]+.
Step 2: 2-(Benzotriazol-1-yl)-N-[(3,5-dichlorophenyl)methyl]-N-[4-(3-pyridyl)phenyl]acetamide.
Pyridine (46 μL, 0.57 mmol) and T3P (50% in DMF) (258 μL, 0.38 mmol) were added to a solution of N-[(3,5-dichlorophenyl)methyl]-4-(3-pyridyl)aniline (63 mg, 0.19 mmol) and 50 (37 mg, 0.21 mmol) in DMF (1 mL) and stirred at rt for 1 h. The reaction mixture was purified directly by preparative RP-HPLC (5–50% MeCN in H2O, 0.1% TFA). The product containing fractions were combined and concentrated in vacuo. The free base was obtained by SCX-II chromatography (load/wash MeOH, elution with 2N NH3 in MeOH) to afford the title compound as a beige powder (76 mg, 0.16 mmol, 81%). 1H NMR (400 MHz, DMSO-d6) δ 8.93 (d, J = 2.4 Hz, 1H), 8.59 (dd, J = 4.6, 1.7 Hz, 1H), 8.11 (d, J = 7.7 Hz, 1H), 8.03 (d, J = 8.4 Hz, 1H), 7.87 (d, J = 8.0 Hz, 2H), 7.80 (d, J = 8.4 Hz, 1H), 7.67 (d, J = 8.0 Hz, 2H), 7.58 – 7.46 (m, 3H), 7.43 – 7.35 (m, 3H), 5.56 (s, 2H), 4.98 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 166.42, 149.34, 148.21, 145.45, 141.80, 140.49, 134.81, 134.70, 134.57, 134.15, 129.14, 128.79, 127.62, 127.53, 127.05, 124.37, 124.29, 119.44, 111.57, 52.09, 50.03; Purity ≥95% by LCMS (Method A) tR = 1.95 min, m/z = 488.10 [M+H]+; HRMS calculated for C26H19Cl2N5O [M+H]+ 488.1039, found 488.1049.
2-(Benzotriazol-1-yl)-N-[(3-chloro-5-methyl-phenyl)methyl]-N-[4-(3-pyridyl)phenyl]acetamide (33).
Step 1. N-[(3-chloro-5-methyl-phenyl)methyl]-4-(3-pyridyl)aniline.
Compound 51 (34 mg, 0.20 mmol) and 3-chloro-5-methyl-benzaldehyde (28 μL, 0.22 mmol) were taken in DCM (1mL) and acetic acid (0.05 mL) and stirred for 30 min before the addition of NaHB(OAc)3 (64 mg, 0.30 mmol) and stirred for 18 h at rt. The mixture was diluted with DCM and washed with sat. aq. NaHCO3. The organic layers were separated and concentrated to dryness. Purification by ISCO automated flash chromatography (4 g, 0–100% EtOAc in hexanes) afforded a straw-colored oil (54 mg, 0.17 mmol, 87%). LCMS (Method A) tR = 1.94 min, m/z = 309.12 [M+H]+.
Step 2. 2-(Benzotriazol-1-yl)-N-[(3-chloro-5-methyl-phenyl)methyl]-N-[4-(3-pyridyl)phenyl]acetamide.
To a solution of N-[(3-chloro-5-methyl-phenyl)methyl]-4-(3pyridyl)aniline (54 mg, 0.17 mmol) and 50 (31 mg, 0.17 mmol) in DMF (1 mL) was added T3P (50% in EtOAc, 0.21 mL, 0.35 mmol) and pyridine (43 μL, 0.53 mmol). The mixture was stirred at rt for 16 h, then filtered and purified by preparative RP-HPLC. The free base was obtained by SCX-II chromatography (eluant 2N NH3/MeOH) affording a colorless solid (39 mg, 0.08 mmol, 48%). 1H NMR (400 MHz, DMSO-d6) δ 8.93 (s, 1H), 8.60 (d, J = 4.9 Hz, 1H), 8.12 (d, J = 8.0 Hz, 1H), 8.04 (d, J = 8.4 Hz, 1H), 7.86 (d, J = 8.1 Hz, 2H), 7.81 (d, J = 8.4 Hz, 1H), 7.64 (d, J = 8.1 Hz, 2H), 7.55 (t, J = 7.6 Hz, 1H), 7.50 (dd, J = 8.0, 4.8 Hz, 1H), 7.40 (t, J = 7.6 Hz, 1H), 7.16 (d, J = 8.4 Hz, 2H), 7.06 (s, 1H), 5.55 (s, 2H), 4.94 (s, 2H), 2.29 (s, 3H); 13C NMR (101 MHz, DMSO) δ 166.18, 151.21, 149.32, 148.20, 145.45, 140.64, 139.64, 137.47, 134.84, 134.69, 134.16, 133.33, 129.19, 128.73, 128.23, 127.63, 125.14, 124.37, 124.29, 119.44, 111.55, 52.47, 50.00, 21.12; Purity ≥95% by LCMS (Method A) tR = 1.91 min, m/z = 468.16 [M+H]+; HRMS calculated for C27H22ClN5O [M+H]+ 468.1586, found 468.1592.
2-(Benzotriazol-1-yl)-N-[(5-chloro-3-pyridyl)methyl]-N-[4-(3-pyridyl)phenyl]acetamide (34).
Step 1. N-[(5-Chloro-3-pyridyl)methyl]-4-(3-pyridyl)aniline.
Compounds 51 (34 mg, 0.20 mmol) and 5-chloropyridine-3-benzaldehyde (31 mg, 0.22 mmol) were taken in DCM (1mL) and acetic acid (0.05 mL) and stirred for 30 min before the addition of NaHB(OAc)3 (64 mg, 0.30 mmol) and stirred for 18 h at rt. The mixture was diluted with DCM and washed with sat. aq. NaHCO3. The organic layers were separated and concentrated to dryness. Purification by ISCO flash chromatography (4 g, 0–100% EtOAc in hexanes) afforded a cream colored solid (53 mg, 0.18 mmol, 90%). LCMS (Method A) tR = 1.45 min, m/z = 296.10 [M+H]+.
Step 2. 2-(Benzotriazol-1-yl)-N-[(5-chloro-3-pyridyl)methyl]-N-[4-(3-pyridyl)phenyl]acetamide.
To a solution of N-[(5-chloro-3-pyridyl)methyl]-4-(3-pyridyl)aniline (53 mg, 0.18 mmol) and 50 (32 mg, 0.17 mmol) in DMF (1 mL) was added T3P (50% in EtOAc, 0.21 mL, 0.35 mmol) and pyridine (43 μL, 0.53 mmol). The mixture was stirred at rt for 16 h, then filtered and purified by preparative RP-HPLC. The free base was obtained by SCX-II chromatography (eluant 2N NH3/MeOH) affording a colorless solid (42 mg, 0.09 mmol, 52%). 1H NMR (400 MHz, DMSO-d6) δ 8.94 (d, J = 2.5 Hz, 1H), 8.60 (dd, J = 4.8, 1.7 Hz, 1H), 8.55 (d, J = 2.5 Hz, 1H), 8.46 (s, 1H), 8.12 (d, J = 8.0 Hz, 1H), 8.03 (d, J = 8.4 Hz, 1H), 7.91 – 7.85 (m, 3H), 7.82 (d, J = 8.4 Hz, 1H), 7.69 (d, J = 8.0 Hz, 2H), 7.59 – 7.48 (m, 2H), 7.40 (t, J = 7.6 Hz, 1H), 5.54 (s, 2H), 5.03 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 166.36, 149.36, 148.21, 147.97, 147.65, 145.44, 140.39, 137.63, 135.86, 134.86, 134.81, 134.72, 134.18, 131.41, 129.30, 128.84, 127.63, 124.37, 124.29, 119.43, 111.61, 50.22, 50.01; Purity ≥95% by LCMS (Method A) tR = 1.59 min, m/z = 455.14 [M+H]+; HRMS calculated for C25H19ClN6O [M+H]+ 455.1382, found 455.1393.
2-(1H-Benzo[d][1,2,3]triazol-1-yl)-N-(3-chlorobenzyl)-N-(4-(2-oxo-1,2-dihydropyridin-3-yl)phenyl)acetamide (35).
Step 1. 4-Bromo-N-[(3-chlorophenyl)methyl]aniline (58).
To a solution of 3-chlorobenzaldehyde (1.13 mL, 10.0 mmol) and 4-bromoaniline (2.06 g, 12.0 mmol) in DCE (50 mL) was added NaHB(OAc)3 (2.76 g, 13.0 mmol) and the mixture stirred for 2 h at rt. Saturated aq. NH4Cl (50 mL) was added and the DCE layer separated. The aqueous layer was extracted with EtOAc (3 × 30 mL) and concentrated. Purification by ISCO automated flash chromatography afforded a tan oil (2.0 g, 6.74 mmol, 67%). 1H NMR (400 MHz, CDCl3) δ 7.36 (s, 1H), 7.34 – 7.21 (m, 6H), 6.51 (d, J = 8.8 Hz, 2H), 4.32 (s, 2H).
Step 2. N-[(3-Chlorophenyl)methyl]-4-(2-fluoro-3-pyridyl)aniline.
To a vial containing 4-bromo-N-[(3-chlorophenyl)methyl]aniline (415 mg, 1.4 mmol), 2-fluoropyridine-3-boronic acid (237 mg, 1.68 mmol), Pd(dppf)Cl·DCM (57 mg, 0.07 mmol) and K2CO3 (386.99 mg, 2.8 mmol) was added dioxane (3.5 mL) and water (0.7 mL). The mixture was heated to 100 °C for 20 h. The mixture was diluted with EtOAc and washed with water, brine, then dried over Na2SO4 and concentrated. The mixture was purified by ISCO automated flash chromatography to afford title intermediate (157 mg, 0.50 mmol, 36%). 1H NMR (400 MHz, CDCl3) δ 8.10 (dt, J = 4.9, 1.6 Hz, 1H), 7.81 (ddd, J = 9.6, 7.4, 2.0 Hz, 1H), 7.42 (dd, J = 8.6, 1.8 Hz, 2H), 7.38 (s, 1H), 7.27 (d, J = 2.8 Hz, 3H), 7.22 (ddd, J = 6.9, 4.8, 1.8 Hz, 1H), 6.71 (d, J = 8.2 Hz, 2H), 4.38 (s, 2H).
Step 3. 3-[4-[(3-Chlorophenyl)methylamino]phenyl]-1H-pyridin-2-one (59).
To a solution of N-[(3-chlorophenyl)methyl]-4-(2-fluoro-3-pyridyl)aniline (94 mg, 0.30 mmol) in dioxane (1 mL) was added conc. aq. HCl (0.2 mL, 2.4 mmol) and the mixture stirred for 16 h at 80 °C. The reaction was allowed to cool to room temperature, neutralized with sat. aq. NaHCO3, extracted with EtOAc, concentrated, and purified by ISCO automated flash chromatography to afford the title intermediate (87 mg, 0.28 mmol, 93%). LCMS (Method A) tR = 1.90 min, m/z = 311.10 [M+H]+.
Step 4. 2-(1H-Benzo[d][1,2,3]triazol-1-yl)-N-(3-chlorobenzyl)-N-(4-(2-oxo-1,2-dihydropyridin-3-yl)phenyl)acetamide.
To a stirred solution of 59 (49 mg, 0.16 mmol) and 50 (28 mg, 0.16 mmol) in DMF (0.5 mL) was added T3P (50% in EtOAc, 189 μL, 0.32 mmol), followed by pyridine (38 μL, 0.48 mmol). The mixture was stirred at rt for 16 h. Purification by preparative RP-HPLC afforded a colorless solid (36 mg, 0.08 mmol, 49%). 1H NMR (400 MHz, CD3OD) δ 7.99 (d, J = 8.5 Hz, 1H), 7.80 – 7.72 (m, 3H), 7.69 (d, J = 8.4 Hz, 1H), 7.57 (t, 1H), 7.49 – 7.40 (m, 2H), 7.37 (d, J = 8.6 Hz, 2H), 7.33 – 7.25 (m, 3H), 7.19 (s, 1H), 6.50 (t, J = 6.8 Hz, 1H), 5.47 (s, 2H), 4.98 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 165.63, 163.89, 146.29, 140.65, 139.82, 138.83, 137.84, 134.95, 134.75, 134.04, 130.73, 130.27, 130.03, 129.27, 128.42, 128.34, 128.07, 127.46, 124.29, 120.41, 110.06, 107.66, 53.60, 50.22; Purity ≥95% by LCMS (Method A) tR = 2.01 min, m/z = 470.14 [M+H]+; HRMS calculated for C26H20ClN5O2 [M+H]+ 470.1389, found 470.1389.
N-(4-(1H-Pyrazol-4-yl)phenyl)-2-(1H-benzo[d][1,2,3]triazol-1-yl)-N-(3-chlorobenzyl)acetamide (36).
Step 1. N-[(3-chlorophenyl)methyl]-4-(1-tetrahydropyran-2-ylpyrazol-4-yl)aniline (60).
To a vial containing 4-bromo-N-[(3-chlorophenyl)methyl]aniline (138 mg, 0.47 mmol), 1-(tetrahydro-2H-pyran-2-yl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (155 mg, 0.56 mmol), Pd(dppf)Cl·DCM (19 mg, 0.02 mmol) and K2CO3 (129 mg, 0.93 mmol) was added dioxane (1.2 mL) and water (0.12 mL). The mixture was heated to 100 °C for 16 h. The mixture was diluted with EtOAc and washed with water, brine, then dried over Na2SO4 and concentrated top dryness. The resulting crude material was purified by ISCO automated flash chromatography to afford a pale-yellow oil (74 mg, 0.20 mmol, 43%). LCMS (Method B) tR = 1.36 min, m/z = 311.10 [M+H]+.
Step 2. 2-(benzotriazol-1-yl)-N-[(3-chlorophenyl)methyl]-N-[4-(1-tetrahydropyran-2-ylpyrazol-4-yl)phenyl]acetamide.
To a stirred solution of 60 (74 mg, 0.2 mmol) and 50 (35 mg, 0.20 mmol) in DMF (1 mL) was added T3P (50% in EtOAc, 238 μL, 0.40 mmol), followed by pyridine (48 μL, 0.60 mmol). The mixture was stirred at 60 °C for 20 h, then washed with water (20 mL) and extracted with EtOAc. The combined organic layers were purified by ISCO automated flash chromatography to afford the title intermediate (77 mg, 0.15 mmol, 73%). LCMS (Method A) tR = 2.30 min, m/z = 527.20 [M+H]+.
Step 3. N-(4-(1H-pyrazol-4-yl)phenyl)-2-(1H-benzo[d][1,2,3]triazol-1-yl)-N-(3-chlorobenzyl)acetamide.
To a solution of 2-(benzotriazol-1-yl)-N-[(3-chlorophenyl)methyl]-N-[4-(1-tetrahydropyran-2-ylpyrazol-4-yl)phenyl]acetamide (77 mg, 0.15 mmol) in THF (1.5 mL) was added 1N aq. HCl (1.5 mL, 1.5 mmol) and the mixture stirred for 16 h at rt. The reaction was allowed to cool to rt, neutralized with sat. aq. NaHCO3, extracted with EtOAc, concentrated, and purified by ISCO automated flash chromatography to afford title compounds as a colorless solid (41 mg, 0.09 mmol, 63%). 1H NMR (400 MHz, CD3OD) δ 8.07 – 7.89 (m, 3H), 7.71 – 7.64 (m, 3H), 7.55 (t, J = 7.7 Hz, 1H), 7.43 (t, J = 7.7 Hz, 1H), 7.33 – 7.23 (m, 5H), 7.20 – 7.14 (m, 1H), 5.45 (s, 2H), 4.95 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 165.60, 146.27, 138.71, 138.51, 134.76, 134.01, 133.95, 131.76, 130.26, 129.42, 129.09, 128.48, 128.09, 127.78, 127.59, 124.34, 121.70, 120.41, 110.06, 53.57, 50.18; Purity ≥95% by LCMS (Method A) tR = 2.04 min, m/z = 443.14 [M+H]+; HRMS calculated for C24H19ClN6O [M+H]+ 443.1393, found 443.1385.
2-(Benzotriazol-1-yl)-N-[(3-chlorophenyl)methyl]-N-[3-methoxy-4-(1H-pyrazol-4-yl)phenyl]acetamide (37).
Step 1. 4-Bromo-N-[(3-chlorophenyl)methyl]-3-methoxy-aniline (54).
A mixture of 3-chlorobenzaldehyde (216 μL, 1.91 mmol) and 4-bromo-3-methoxyaniline (350 mg, 1.73 mmol) in DCM (8.25 mL) and AcOH (0.41 mL) was stirred for 30 min at rt before the addition of NaBH(OAc)3 (551 mg, 2.60 mmol). The mixture was stirred for an additional 18 h at rt. Saturated aq. NH4Cl (50 mL) was added and the crude residue extracted with DCM, concentrated and purified by ISCO automated flash chromatography to afford a straw-colored oil (470 mg, 1.44 mmol, 83%). LCMS (Method B) tR = 1.85 min, m/z = 325.99 [M+H]+; 1H NMR (400 MHz, CDCl3) δ 7.37 (s, 1H), 7.33–7.18 (m, 4H), 6.21 (d, J = 2.5 Hz, 1H), 6.15 (dd, J = 8.4, 2.3 Hz, 1H), 4.32 (s, 2H), 3.81 (s, 3H).
Step 2. 2-Benzotriazol-1-yl)-N-(4-bromo-3-methoxy-phenyl)-N-[(3-chlorophenyl)methyl]acetamide (56).
To a stirred solution of 54 (465 mg, 1.42 mmol) and 50 (265 mg, 1.50 mmol) in THF (7 mL) was added T3P (50% in EtOAc, 1.70 mL, 2.85 mmol), followed by pyridine (344 μL, 4.27 mmol). The mixture was stirred at 60 °C for 20 h, then diluted with water (20 mL) and extracted with EtOAc. The combined organic layers were washed with brine, concentrated and purified by flash chromatography to afford a colorless solid (651 mg, 1.34 mmol, 94%). LCMS (Method B) tR = 1.81 min, m/z = 485.03 [M+H]+; 1H NMR (400 MHz, CDCl3) δ 8.06 (d, J = 8.4 Hz, 1H), 7.59 (d, J = 8.2 Hz, 1H), 7.54–7.47 (m, 2H), 7.43–7.35 (m, 1H), 7.32– 7.19 (m, 2H), 7.13–7.06 (m, 1H), 6.64 (dd, J = 8.2, 2.3 Hz, 1H), 6.47 (d, J = 2.3 Hz, 1H), 5.24 (s, 2H), 4.85 (s, 2H), 3.79 (s, 3H).
Step 3. 2-(Benzotriazol-1-yl)-N-[(3-chlorophenyl)methyl]-N-[3-methoxy-4-(1H-pyrazol-4-yl)phenyl]acetamide.
A vial was charged with 2-(benzotriazol-1-yl)-N-(4-bromo-3-methoxy-phenyl)-N-[(3-chlorophenyl)methyl]acetamide (100 mg, 0.21 mmol), 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (48 mg, 0.25 mmol) and Pd(dppf)Cl2·DCM (8.3 mg, 0.01 mmol). Dioxane (1.0 mL) and 2M aq. K2CO3 (0.2 mL) were then added and the resulting solution purged with argon for 15 min. The mixture was then heated to 110 °C and stirred vigorously. After 18 h, the resulting mixture was cooled to rt, filtered through celite, concentrated, and purified by preparative RP-HPLC (5–95% MeCN in H2O, 0.1% TFA) to afford a colorless solid (2.6 mg, 5.5 μmol, 3%). 1H NMR (400 MHz, CD3OD) δ 7.98 (s, 2H), 7.87 (d, J = 8.4 Hz, 1H), 7.55 (t, J = 8.5 Hz, 2H), 7.43 (t, J = 7.7 Hz, 1H), 7.35 – 7.27 (m, 1H), 7.23 – 7.16 (m, 3H), 7.11 – 7.04 (m, 1H), 6.82 – 6.74 (m, 2H), 5.39 (s, 2H), 4.85 (s, 2H), 3.75 (s, 3H); 13C NMR (101 MHz, CD3OD) δ 166.3, 156.8, 139.0, 138.5, 134.0, 133.81 132.8, 129.7, 128.6, 128.1, 127.52, 127.48, 127.1, 124.2, 120.2, 118.4, 111.1, 110.2, 54.7, 52.5, 49.5; Purity ≥95% by LCMS (Method A) tR = 2.10, m/z = 473.15 [M+H]+; HRMS calculated for C25H21ClN6O2 [M+H]+ 473.1487, found 473.1497.
2-(Benzotriazol-1-yl)-N-[(3-chlorophenyl)methyl]-N-[2-methoxy-4-(1H-pyrazol-4-yl)phenyl]acetamide (38).
Step 1. 4-Bromo-N-[(3-chlorophenyl)methyl]-2-methoxy-aniline (55).
To a solution of 3-chlorobenzaldehyde (200 μL, 1.77 mmol) and 4-bromo-2-methoxyaniline (325 mg, 1.61 mmol) in DCM (7.7 mL) and AcOH (0.4 mL) was stirred for 30 min at rt before the addition of NaBH(OAc)3 (511 mg, 2.41 mmol). The mixture was stirred for an additional 18 h at rt. Saturated aq. NH4Cl (50 mL) was added and the crude residue extracted with DCM, concentrated and purified by ISCO automated flash chromatography to afford a straw-colored oil (446 mg, 1.37 mmol, 85%). LCMS (Method B) tR = 2.05 min, m/z = 325.99 [M+H]+; 1H NMR (400 MHz, CDCl3) δ 7.35 (s, 1H), 7.31 – 7.19 (m, 3H), 6.92 (dd, J = 8.4, 2.1 Hz, 1H), 6.89 (d, J = 2.1 Hz, 1H), 6.37 (d, J = 8.4 Hz, 1H), 4.76 (br s, 1H), 4.32 (s, 2H), 3.87 (s, 3H).
Step 2. 2-(Benzotriazol-1-yl)-N-(4-bromo-2-methoxy-phenyl)-N-[(3-chlorophenyl)methyl]acetamide (57).
To a stirred solution of 55 (440 mg, 1.35 mmol) and 50 (251 mg, 1.41 mmol) in THF (7 mL) was added T3P (50% in EtOAc, 1.60 mL, 2.69 mmol), followed by pyridine (326 μL, 4.04 mmol). The mixture was stirred at 60 °C for 20 h, then diluted with water (20 mL) and extracted with EtOAc. The combined organic layers were washed with brine, concentrated and purified by ISCO automated flash chromatography to afford a colorless solid (507 mg, 1.04 mmol, 77%). LCMS (Method B) tR = 1.83 min, m/z = 485.04 [M+H]+; 1H NMR (400 MHz, CDCl3) δ 8.05 (d, J = 8.4 Hz, 1H), 7.56 – 7.45 (m, 2H), 7.42 – 7.33 (m, 1H), 7.29 – 7.01 (m, 6H), 6.84 (d, J = 8.3 Hz, 1H), 5.30 (s, 2H), 5.22 (d, J = 16.6 Hz, 1H), 5.11 (d, J = 16.6 Hz, 1H), 5.01 (d, J = 14.3 Hz, 1H), 4.52 (d, J = 14.3 Hz, 1H), 3.78 (s, 3H).
Step 3. 2-(Benzotriazol-1-yl)-N-[(3-chlorophenyl)methyl]-N-[2-methoxy-4-(1H-pyrazol-4-yl)phenyl]acetamide.
A vial was charged with 57 (100 mg, 0.21 mmol), 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (48 mg, 0.25 mmol) and Pd(dppf)Cl2.DCM (8.3 mg, 0.01 mmol). Dioxane (1.0 mL) and 2M aq. K2CO3 (0.2 mL) were then added and the resulting solution purged with argon for 15 min. The mixture was then heated to 110 °C and stirred vigorously. After 18 h, the resulting mixture was cooled to rt, filtered through celite, concentrated, and purified by preparative RP-HPLC (5–95% MeCN in H2O, 0.1% TFA) to afford a colorless solid (2.5 mg, 5.3 μmol, 3%). 1H NMR (400 MHz, CD3OD) δ 7.96 (s, 2H), 7.88 (d, J = 8.4 Hz, 1H), 7.53 – 7.42 (m, 2H), 7.38 – 7.29 (m, 1H), 7.23 (d, J = 1.8 Hz, 1H), 7.20 – 7.10 (m, 4H), 7.06 (d, J = 8.1 Hz, 1H), 7.04 – 6.99 (m, 1H), 5.34 (d, J = 17.0 Hz, 1H), 5.19 (d, J = 17.0 Hz, 1H), 4.86 (d, J = 14.5 Hz, 1H), 4.62 (d, J = 14.5 Hz, 1H), 3.79 (s, 3H); 13C NMR (101 MHz, CD3OD) δ 167.0, 155.3, 138.9, 138.8, 135.6, 133.7, 129.8, 129.4, 128.7, 127.5, 127.4, 127.2, 125.8, 124.2, 118.5, 118.1, 110.1, 109.4, 55.1, 51.9, 49.3; Purity ≥95% by LCMS (Method A) tR = 2.13, m/z = 473.15 [M+H]+; HRMS calculated for C25H21ClN6O2 [M+H]+ 473.1487, found 473.1476.
2-(Benzotriazol-1-yl)-N-[(3-chlorophenyl)methyl]-N-[6-(1H-pyrazol-4-yl)-3-pyridyl]acetamide (39).
Step 1. 6-(1-Tetrahydropyran-2-ylpyrazol-4-yl)pyridin-3-amine, (64).
1-(Tetrahydro-2H-pyran-2-yl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (560 mg, 2.01 mmol), 6-chloropyridin-3-amine (388 mg, 3.02 mmol), potassium carbonate (3.02 mL, 6.04 mmol) and SPhos Pd G2 (72 mg, 0.10 mmol) were combined in 1-butanol (5.3 mL) in a sealed-tube and degassed under a stream of argon for 20 mins followed by heating at 100 °C for 16 h. The reaction mixture was filtered through celite, washed with EtOAc (10 mL), concentrated under reduced pressure and purified by ISCO automated flash chromatography (24 g, 0–8 % MeOH in DCM) to afford a viscous oil (414 mg, 1.69 mmol, 84%). LCMS (Method A) tR = 1.30 min, m/z = 245.13 [M+H]+; 1H NMR (400 MHz, CD3OD) δ 8.15 (s, 1H), 7.95 (d, J = 2.8 Hz, 1H), 7.92 (s, 1H), 7.42 (d, J = 8.5 Hz, 1H), 7.11 (dd, J = 8.5, 2.8 Hz, 1H), 5.41 (dd, J = 10.0, 2.3 Hz, 1H), 4.10 – 4.00 (m, 1H), 3.74 (td, J = 11.2, 3.1 Hz, 1H), 2.21 – 2.08 (m, 1H), 2.08 – 1.98 (m, 2H), 1.84 – 1.55 (m, 3H).
Step 2. N-[(3-Chlorophenyl)methyl]-6-(1-tetrahydropyran-2-ylpyrazol-4-yl)pyridin-3-amine (66).
Compound 64 (114 mg, 0.47 mmol) and 3-chlorobenzaldehyde (0.05 mL, 0.43 mmol) were taken in DCM (4 mL) and stirred for 15 min. NaHB(OAc)3 (135 mg, 0.64 mmol) was added at rt and stirring continued for 16 h. To the reaction mixture sat. aq. NaHCO3 (5 ml) was added. The resulting residue was extracted with DCM (2 × 10 mL), dried over sodium sulfate, concentrated to dryness and then purified by ISCO automated flash chromatography (12 g, 0–3 % MeOH in DCM) to afford a viscous oil (96 mg, 0.26 mmol, 61%). LCMS (Method A) tR = 1.84 min, m/z = 369.14 [M+H]+; 1H NMR (400 MHz, CD3OD) δ 8.13 (s, 1H), 7.90 (s, 1H), 7.88 (d, J = 2.8 Hz, 1H), 7.42 (d, J = 9.3 Hz, 2H), 7.34 – 7.28 (m, 2H), 7.28 – 7.21 (m, 1H), 7.01 (dd, J = 8.7, 2.8 Hz, 1H), 5.40 (dd, J = 10.0, 2.4 Hz, 1H), 4.38 (s, 2H), 4.04 (d, J = 11.7 Hz, 1H), 3.79 – 3.68 (m, 1H), 2.20 – 2.07 (m, 1H), 2.06 – 1.99 (m, 2H), 1.83 – 1.56 (m, 3H).
Step 3. 2-(Benzotriazol-1-yl)-N-[(3-chlorophenyl)methyl]-N-[6-(1H-pyrazol-4-yl)-3-pyridyl]acetamide (39).
To a stirred solution of 50 (1.10 g, 6.24 mmol) in DCM (20 mL) was added pyridine (0.60 mL, 7.48 mmol) and cyanuric fluoride (0.64 mL, 7.48 mmol) sequentially at rt and continued stirring for 1 h. The precipitated solid was filtered and washed with DCM (10 mL). The filtrate was concentrated to afford crude 2-(benzotriazol-1-yl)acetyl fluoride that was immediately dissolved in anhydrous THF (12 mL). To this was added a solution of 66 (230 mg, 0.62 mmol) and Et3N (0.91 mL, 6.55 mmol) in THF (2 mL). The reaction mixture was stirred at 60 °C for 16 h. Water (20 mL) was added followed by extraction with EtOAc (25 mL x 2). The organic layer was dried over Na2SO4, concentrated under reduced pressure, and passed quickly through a flash column using 0–10 % MeOH in DCM to afford an impure intermediate, 2-(benzotriazol-1-yl)-N-[(3-chlorophenyl)methyl]-N-[6-(1-tetrahydropyran-2-ylpyrazol-4-yl)-3-pyridyl]acetamide (212 mg), which was further treated with 5 mL of 4:1 mixture of MeOH and 4 N HCl in dioxane at rt for 40 min. To the reaction mixture EtOAc (15 mL) was added followed by washing with sat. NaHCO3. The organic layer was dried over Na2SO4, concentrated under reduced pressure and purified by ISCO automated flash chromatography (24 g, 0–6 % MeOH in DCM) to afford desired product as a yellow solid (60 mg, 0.13 mmol, 22 % over 2 steps). 1H NMR (400 MHz, DMSO-d6) δ 13.11 (s, 1H), 8.59 (d, J = 2.6 Hz, 1H), 8.36 (s, 1H), 8.08 (s, 1H), 8.03 (d, J = 8.3 Hz, 1H), 7.92 – 7.85 (m, 1H), 7.81 (dd, J = 14.2, 8.3 Hz, 2H), 7.55 (t, J = 7.7 Hz, 1H), 7.40 (t, J = 7.7 Hz, 1H), 7.33 (d, J = 3.3 Hz, 3H), 7.21 (d, J = 6.7 Hz, 1H), 5.54 (s, 2H), 4.94 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 165.89, 151.92, 148.94, 144.98, 139.11, 137.55, 136.57, 133.71, 133.58, 133.11, 130.36, 128.03, 127.49, 127.17, 126.95, 123.85, 123.82, 121.42, 120.02, 118.96, 111.11, 51.88, 49.48; Purity ≥95% by LCMS (Method A) tR = 1.89, m/z = 443.13 [M+H]+; HRMS calculated for C23H18ClN7O [M+H]+ 443.1334, found 443.1344.
2-(Benzotriazol-1-yl)-N-[(3-chlorophenyl)methyl]-N-[5-(1H-pyrazol-4-yl)-2-pyridyl]acetamide (40).
Step 1. 5-(1-Tetrahydropyran-2-ylpyrazol-4-yl)pyridin-2-amine (65).
1-(Tetrahydro-2H-pyran-2-yl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (560 mg, 2.01 mmol), 2-amino-5-chloropyridine (388 mg, 3.02 mmol), potassium carbonate (2 M, 3.02 mL, 6.04 mmol) and XPhos Pd G2 (79 mg, 0.10 mmol) were combined in dioxane (5.3 mL) in a sealed-tube and degassed under a stream of argon for 20 mins followed by heating at 100 °C for 16 h. The reaction mixture was filtered through celite, washed with EtOAc (10 mL), concentrated under reduced pressure and purified by ISCO automated flash chromatography (24 g, 0–7% MeOH in DCM) to afford an off-white solid (479 mg, 1.96 mmol, 97%). LCMS (Method A) tR = 1.32 min, m/z = 245.13 [M+H]+; 1H NMR (400 MHz, CD3OD) δ 8.12 (d, J = 2.4 Hz, 1H), 8.06 (s, 1H), 7.79 (s, 1H), 7.67 (dd, J = 8.6, 2.4 Hz, 1H), 6.62 (d, J = 8.6 Hz, 1H), 5.40 (dd, J = 10.2, 2.4 Hz, 1H), 4.09 – 4.00 (m, 1H), 3.73 (td, J = 11.2, 3.0 Hz, 1H), 2.21 – 2.09 (m, 1H), 2.09 – 1.98 (m, 2H), 1.84 – 1.57 (m, 3H).
Step 2. N-[(3-Chlorophenyl)methyl]-5-(1-tetrahydropyran-2-ylpyrazol-4-yl)pyridin-2-amine (67).
Intermediate 65 (133 mg, 0.55 mmol) from above and 3-chlorobenzaldehyde (0.06 mL, 0.50 mmol) were taken in DCM (4 mL) and stirred for 15 min. NaHB(OAc)3 (158 mg, 0.75 mmol) was added at rt and stirring was continued for 16 h. The reaction mixture was quenched with sat. aq. NaHCO3 solution (5 ml), the aqueous layer was extracted with DCM (2 × 10 mL), dried over Na2SO4, concentrated under reduced pressure and purified by ISCO automated flash chromatography (12 g, 0–3% MeOH in DCM) to afford a colorless solid (100 mg, 0.27 mmol, 54 %). LCMS (Method A) tR = 1.80 min, m/z = 369.14 [M+H]+; 1H NMR (400 MHz, CD3OD) δ 8.16 (d, J = 2.3 Hz, 1H), 8.04 (s, 1H), 7.78 (s, 1H), 7.65 (dd, J = 8.7, 2.3 Hz, 1H), 7.38 – 7.34 (m, 1H), 7.28 (dd, J = 4.2, 2.0 Hz, 2H), 7.28 – 7.20 (m, 1H), 6.58 (d, J = 8.7 Hz, 1H), 5.40 (dd, J = 10.1, 2.4 Hz, 1H), 4.52 (s, 2H), 4.08–4.01 (m, 1H), 3.77–3.69 (m, 1H), 2.22 – 1.96 (m, 3H), 1.81 – 1.56 (m, 3H).
Step 3. 2-(Benzotriazol-1-yl)-N-[(3-chlorophenyl)methyl]-N-[5-(1H-pyrazol-4-yl)-2-pyridyl]acetamide.
To a stirred solution of 50 (1.44 g, 8.13 mmol) in DCM (20 mL) was added pyridine (0.79 mL, 9.76 mmol) and cyanuric fluoride (0.84 mL, 9.76 mmol) sequentially at room temperature and continued stirring for 1 h. The precipitated solid was filtered and washed with DCM (10 mL). The filtrate was concentrated to get crude 2-(benzotriazol-1-yl)acetyl fluoride that was immediately dissolved in anhydrous THF (12 mL). To this was added a solution of 67 (300 mg, 0.81 mmol) and Et3N (1.19 mL, 8.54 mmol) in THF (2 mL). The reaction mixture was stirred at 60 °C for 16 h. Water (20 mL) was added followed by extraction with EtOAc (25 mL x 2). The organic layer was dried over Na2SO4, concentrated under reduced pressure, and passed quickly through the flash column using 0–10 % MeOH in DCM to afford a impure intermediate 2-(benzotriazol-1-yl)-N-[(3-chlorophenyl)methyl]-N-[5-(1-tetrahydropyran-2-ylpyrazol-4-yl)-2-pyridyl]acetamide (364 mg) which was further treated with 5 mL of 4:1 mixture of MeOH and 4 N HCl in dioxane at room temperature for 40 min. To the reaction mixture EtOAc (15 mL) was added followed by washing with sat. aq. NaHCO3, dried over Na2SO4, concentrated under reduced pressure, and purified by ISCO automated flash chromatography (24 g, 0–4% MeOH in DCM) to afford desired product as a yellow solid (150 mg, 0.33 mmol, 41 % over 2 steps). 1H NMR (400 MHz, CD3OD) δ 8.78 (d, J = 2.4 Hz, 1H), 8.21 – 8.01 (m, 3H), 7.97 (d, J = 8.4 Hz, 1H), 7.69 (d, J = 8.4 Hz, 1H), 7.59 – 7.51 (m, 1H), 7.43 (d, J = 7.6 Hz, 1H), 7.39 (d, J = 8.2 Hz, 1H), 7.34 (s, 1H), 7.32 – 7.18 (m, 3H), 5.73 (s, 2H), 5.11 (s, 2H); 13C NMR (101 MHz, CD3OD) δ 168.24, 152.24, 147.13, 146.52, 140.36, 137.02, 135.49, 135.19, 131.14, 129.10, 128.93, 128.73, 127.51, 125.72, 125.62, 122.08, 119.89, 119.06, 111.64, 111.41, 109.89, 52.27, 45.71; Purity ≥95% by LCMS (Method A) tR = 1.98, m/z = 443.13 [M+H]+; HRMS calculated for C23H18ClN7O [M+H]+ 443.1334, found 443.1323.
N-(4-(1H-Imidazol-4-yl)phenyl)-2-(1H-benzo[d][1,2,3]triazol-1-yl)-N-(3-chlorobenzyl)acetamide (41).
Step 1. 2-(benzotriazol-1-yl)-N-(4-bromophenyl)-N-[(3-chlorophenyl)methyl]acetamide (61).
To a stirred solution of intermediate 58 (1.48 g, 5.0 mmol) and acid 50 (886 mg, 5 mmol) in THF (25 mL) was added T3P (50% in EtOAc, 5.95 mL, 10 mmol), followed by pyridine (1.21 mL, 15 mmol). The mixture was stirred at rt for 16 h, then diluted with water (20 mL) and extracted with EtOAc. The combined organic layers were washed with brine, concentrated, and purified by ISCO automated flash chromatography to afford a colorless solid (1.58 g, 3.46 mmol, 69%). LCMS (Method B) tR = 2.40 min, m/z = 457.03 [M+H]+.
Step 2. 2-(benzotriazol-1-yl)-N-[(3-chlorophenyl)methyl]-N-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]acetamide (62).
To a vial containing 61 (228 mg, 0.50 mmol), bis(pinacolato)diboron (152 mg, 0.60 mmol), Pd(dppf)Cl·DCM (20 mg, 0.03 mmol) and KOAc (221 mg, 2.25 mmol) was added dioxane (2.5 mL). The mixture was heated to 100 °C for 16 h. The mixture was diluted with EtOAc and washed with water, brine, then dried over Na2SO4 and concentrated. The mixture was purified by ISCO automated chromatography to afford a cream colored solid (176 mg, 0.35 mmol, 70%). LCMS (Method A) tR = 2.53 min, m/z = 503.21 [M+H]+.
Step 3. 2-(benzotriazol-1-yl)-N-[(3-chlorophenyl)methyl]-N-[4-(1-tritylimidazol-4-yl)phenyl]acetamide.
To a vial containing 62 (127 mg, 0.25 mmol), 4-bromo-1-trityl-imidazole (90 mg, 0.23 mmol), Pd(PPh3)4 (40 mg, 0.15 mmol) and K2CO3 (64 mg, 0.46 mmol) was added dioxane (1.9 mL) and water (0.4 mL). The mixture was degassed with argon, heated to 100 °C for 16 h. The mixture was diluted with EtOAc and washed with water, brine, then dried over Na2SO4 and concentrated. The mixture was purified by ISCO automated chromatography to afford the desired intermediate (100 mg, 0.15 mmol, 63%). LCMS (Method B) tR = 1.53 min, m/z = 685.25 [M+H]+.
Step 4. N-(4-(1H-imidazol-4-yl)phenyl)-2-(1H-benzo[d][1,2,3]triazol-1-yl)-N-(3-chlorobenzyl)acetamide.
To 2-(Benzotriazol-1-yl)-N-[(3-chlorophenyl)methyl]-N-[4-(1-tritylimidazol-4-yl)phenyl]acetamide (100 mg, 0.15 mmol) in MeOH (1.5 mL) was added AcOH (170 μL, 3.0 mmol) and the mixture stirred for 2 h at 65 °C. The reaction was allowed to cool to rt, concentrated, and purified by preparative RP-HPLC to afford a colorless solid (30 mg, 0.07 mmol, 45%). 1H NMR (400 MHz, DMSO-d6) δ 12.42 (br s, 1H), 8.04 (d, J = 8.4 Hz, 1H), 7.87 (d, J = 8.1 Hz, 2H), 7.82 (d, J = 8.4 Hz, 1H), 7.78 (s, 1H), 7.69 (s, 1H), 7.56 (t, J = 7.7 Hz, 1H), 7.46 (d, J = 8.1 Hz, 2H), 7.40 (t, J = 7.7 Hz, 1H), 7.35–7.29 (m, 3H), 7.21 (d, J = 6.8 Hz, 1H), 5.49 (s, 2H), 4.92 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 165.7, 145.0, 139.5, 137.8, 137.6, 136.3, 134.3, 133.8, 133.0, 130.3, 128.4, 127.9, 127.3, 127.2, 126.8, 125.5, 123.8, 119.0, 115.2, 111.1, 52.0, 49.3; Purity ≥95% by LCMS (Method A) tR = 1.80 min, m/z = 443.14 [M+H]+; HRMS calculated for C24H19ClN6O [M+H]+ 443.1393, found 443.1375.
N-[(3-Chlorophenyl)methyl]-N-[4-(1H-pyrazol-4-yl)phenyl]-2-(3-pyridyl)acetamide (42).
To a solution of N-[(3-Chlorophenyl)methyl]-4-(1-tetrahydropyran-2-ylpyrazol-4-yl)aniline, 60 (37 mg, 0.1 mmol) in THF (1 mL), was added 3-pyridylacetic acid (14 mg, 0.1 mmol), T3P (50% in EtOAc, 0.18 mL, 0.30 mmol) and DIPEA (52 μL, 0.30 mmol) and the mixture stirred at 65 °C for 16 h. The mixture was concentrated, then re-dissolved in HCl (4.0 M in dioxane, 1 mL) and stirred at 60 °C for 1 h. The mixture was concentrated and purified by RP-HPLC (20–100% MeCN in H2O, 0.1% TFA) and free-base obtained by washing product with sat. aq. K2CO3, affording the title compound as a colorless solid (18 mg, 0.04 mmol, 47%). 1H NMR (400 MHz, DMSO-d6) δ 8.74 (d, J = 5.4 Hz, 1H), 8.67 (s, 1H), 8.27 (d, J = 8.0 Hz, 1H), 8.10 (br s, 2H), 7.88 (dd, J = 8.0, 5.5 Hz, 1H), 7.68 (d, J = 8.0 Hz, 2H), 7.38 – 7.25 (m, 5H), 7.20 (d, J = 6.6 Hz, 1H), 4.91 (s, 2H), 3.77 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 169.49, 158.88, 158.52, 145.80, 144.83, 142.20, 140.38, 139.28, 135.59, 133.46, 133.36, 130.69, 129.20, 128.33, 127.70, 127.26, 126.60, 126.06, 120.68, 117.84, 114.92, 52.26, 37.61; Purity ≥95% by LCMS (Method A) tR = 1.71, m/z = 403.13 [M+H]+; HRMS calculated for C23H19ClN4O [M+H]+ 403.1320, found 403.1340.
N-[(3-Chlorophenyl)methyl]-N-[4-(1H-pyrazol-4-yl)phenyl]-2-pyrimidin-5-yl-acetamide (43).
To a solution of N-[(3-chlorophenyl)methyl]-4-(1-tetrahydropyran-2-ylpyrazol-4-yl)aniline, 60 (37 mg, 0.1 mmol) in THF (1 mL), was added 2-pyrimidin-5-ylacetic acid (14 mg, 0.1 mmol), T3P (50% in EtOAc, 0.18 mL, 0.30 mmol) and DIPEA (52 μL, 0.30 mmol) and the mixture stirred at 65 °C for 16 h. The mixture was concentrated, then re-dissolved in HCl (4.0 M in dioxane, 1 mL) and stirred at 60 °C for 1 h. The mixture was concentrated and purified by RP-HPLC (20–100% MeCN in H2O, 0.1% TFA) and free-base obtained by washing product with sat. aq. K2CO3, affording the title compound as a colorless solid (9 mg, 0.02 mmol, 23%). 1H NMR (400 MHz, CD3OD) δ 9.04 (s, 1H), 8.61 (s, 2H), 8.03 (s, 2H), 7.68 (d, J = 8.2 Hz, 2H), 7.32 – 7.26 (m, 3H), 7.18 (d, J = 8.2 Hz, 3H), 4.95 (s, 2H), 3.66 (s, 2H); 13C NMR (101 MHz, CD3OD) δ 170.15, 157.57, 156.10, 139.32, 139.07, 133.96, 133.48, 130.23, 129.68, 128.75, 128.40, 127.37, 126.84, 126.48, 121.06, 52.38, 35.30; Purity ≥95% by LCMS (Method A) tR = 1.84, m/z = 404.13 [M+H]+; HRMS calculated for C22H18ClN5O [M+H]+ 404.1273, found 404.1289.
Supplementary Material
Acknowledgements
The authors thank J. Scott Daniels, PhD and Matthew J. Vergne, PhD at Lipscomb University College of Pharmacy for their assistance and analytic support for the metabolite ID studies. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817).
Funding
We thank Lerner Research Institute for lab start-up support and Cleveland Clinic Philanthropy and an anonymous donor for a generous donation to support COVID related capital purchases for this research to S.R.S. This work was partly supported by: grant no. CA200422, CA251275, AI140718, AI140705, AI140705S1, AI152190, DE023926, DE027888, DE028521, KGM9942011, and the Betsy B. deWindt endowment to J.U.J
Abbreviations Used
- 3CLpro
3C-like protease
- CoV
coronavirus
- CPE
cytopathic effect
- MERS
Middle East respiratory syndrome
- MLPCN
Molecular Libraries Probe Production Centers Network
- SARS
severe acute respiratory syndrome
- SC1
SARS-CoV-1
- SC2
SARS-CoV-2
Footnotes
Associated Content
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI:
- Supplemental Figures, SARS-CoV-1 3CLpro X-ray structures; Tier 1 DMPK Summary; Human S9 generated metabolites and MS fragmentation; X-ray data collection and refinement statistics; ligand electron density maps.
- Molecular-formula strings
- UV-based HPLC chromatograms of final compounds
- 41 CPE SARS-CoV-2 infected Vero E6 concentration-response-curve and Table
PDB ID Codes
SARS-CoV-2 complexes: 1 PDB: 7LME; 19 PDB: 7LMD; 21 PDB: 7LMF.
SARS-CoV-1 complexes: 8 PDB: 7LMH; 19 PDB: 7LMI; 21 PDB: 7LMG; 35 PDB: 7LMJ.
Authors will release the atomic coordinates and experimental data upon article publication.
References
- (1).Myint SH Human Coronavirus Infections. In The Coronaviridae; Siddell SG, Ed.; Springer US: Boston, MA, 1995; pp 389–401. [Google Scholar]
- (2).McIntosh K; Dees JH; Becker WB; Kapikian AZ; Chanock RM Recovery in Tracheal Organ Cultures of Novel Viruses from Patients with Respiratory Disease. Proc. Natl. Acad. Sci. U. S. A 1967, 57 (4), 933–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Ksiazek TG; Erdman D; Goldsmith CS; Zaki SR; Peret T; Emery S; Tong S; Urbani C; Comer JA; Lim W; Rollin PE; Dowell SF; Ling A; Humphrey CD; Shieh W-J; Guarner J; Paddock CD; Rota P; Fields B; DeRisi J; Yang J; Cox N; Hughes JM; LeDuc JW; Bellini WJ; Anderson LJ A Novel Coronavirus Associated with Severe Acute Respiratory Syndrome. N. Engl. J. Med 2003, 348 (20), 1953–1966. [DOI] [PubMed] [Google Scholar]
- (4).Drosten C; Günther S; Preiser W; van der Werf S; Brodt H-R; Becker S; Rabenau H; Panning M; Kolesnikova L; Fouchier RAM; Berger A; Burguière A-M; Cinatl J; Eickmann M; Escriou N; Grywna K; Kramme S; Manuguerra J-C; Müller S; Rickerts V; Stürmer M; Vieth S; Klenk H-D; Osterhaus ADME; Schmitz H; Doerr HW Identification of a Novel Coronavirus in Patients with Severe Acute Respiratory Syndrome. N. Engl. J. Med 2003, 348 (20), 1967–1976. [DOI] [PubMed] [Google Scholar]
- (5).WHO Emergencies preparedness, response - Update 49 - SARS case fatality ratio, incubation period.
- (6).Pyrc K; Berkhout B; van der Hoek L. The Novel Human Coronaviruses NL63 and HKU1. J. Virol 2007, 81 (7), 3051–3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Fielding BC Human Coronavirus NL63: A Clinically Important Virus? Future Microbiol. 2011, 6 (2), 153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Middle East respiratory syndrome coronavirus (MERS-CoV) - United Arab Emirates. https://www.who.int/emergencies/emergency-events/item/2021-DON314.
- (9).Liu G; Lee J-H; Parker ZM; Acharya D; Chiang JJ; van Gent M; Riedl W; Davis-Gardner ME; Wies E; Chiang C; Gack MU ISG15-Dependent Activation of the Sensor MDA5 Is Antagonized by the SARS-CoV-2 Papain-like Protease to Evade Host Innate Immunity. Nat. Microbiol 2021, 6, 467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).WHO Coronavirus (COVID-19) Data Surveillance Dashboard https://covid19.who.int/(accessed Mar 29, 2021).
- (11).Wu F; Zhao S; Yu B; Chen YM; Wang W; Song ZG; Hu Y; Tao ZW; Tian JH; Pei YY; Yuan ML; Zhang YL; Dai FH; Liu Y; Wang QM; Zheng JJ; Xu L; Holmes EC; Zhang YZ A New Coronavirus Associated with Human Respiratory Disease in China. Nature 2020, 579 (7798), 265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Morse JS; Lalonde T; Xu S; Liu WR Learning from the Past: Possible Urgent Prevention and Treatment Options for Severe Acute Respiratory Infections Caused by 2019-NCoV. ChemBioChem 2020, 21 (5), 730–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Dömling A; Gao L. Chemistry and Biology of SARS-CoV-2. Chem 2020, 6 (6), 1283–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Ghosh AK; Brindisi M; Shahabi D; Chapman ME; Mesecar AD Drug Development and Medicinal Chemistry Efforts toward SARS-Coronavirus and Covid-19 Therapeutics. ChemMedChem 2020, 15 (11), 907–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Cannalire R; Cerchia C; Beccari AR; Di Leva FS; Summa V. Targeting SARS-CoV-2 Proteases and Polymerase for COVID-19 Treatment: State of the Art and Future Opportunities. J. Med. Chem 2020, doi. 10.1021/acs.jmedchem.0c01140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Gil C; Ginex T; Maestro I; Nozal V; Barrado-Gil L; Cuesta-Geijo MÁ; Urquiza J; Ramírez D; Alonso C; Campillo NE; Martinez A. COVID-19: Drug Targets and Potential Treatments. J. Med. Chem 2020, 63 (21), 12359–12386. [DOI] [PubMed] [Google Scholar]
- (17).Fan K; Wei P; Feng Q; Chen S; Huang C; Ma L; Lai B; Pei J; Liu Y; Chen J; Lai L. Biosynthesis, Purification, and Substrate Specificity of Severe Acute Respiratory Syndrome Coronavirus 3C-like Proteinase. J. Biol. Chem 2004, 279 (3), 1637–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Thiel V; Ivanov KA; Putics Á; Hertzig T; Schelle B; Bayer S; Weißbrich B; Snijder EJ; Rabenau H; Doerr HW; Gorbalenya AE; Ziebuhr J. Mechanisms and Enzymes Involved in SARS Coronavirus Genome Expression. J. Gen. Virol 2003, 84 (9), 2305–2315. [DOI] [PubMed] [Google Scholar]
- (19).Shi J; Wei Z; Song J. Dissection Study on the Severe Acute Respiratory Syndrome 3C-like Protease Reveals the Critical Role of the Extra Domain in Dimerization of the Enzyme. J. Biol. Chem 2004, 279, 24765–24773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Jacobs J; Grum-Tokars V; Zhou Y; Turlington M; Saldanha SA; Chase P; Eggler A; Dawson ES; Baez-Santos YM; Tomar S; Mielech AM; Baker SC; Lindsley CW; Hodder P; Mesecar A; Stauffer SR Discovery, Synthesis, and Structure-Based Optimization of a Series of N -(Tert -Butyl)-2-(N -Arylamido)-2-(Pyridin-3-Yl) Acetamides (ML188) as Potent Noncovalent Small Molecule Inhibitors of the Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) 3CL. J. Med. Chem 2013, 56 (2), 534–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Turlington M; Chun A; Tomar S; Eggler A; Grum-Tokars V; Jacobs J; Daniels JS; Dawson E; Saldanha A; Chase P; Baez-Santos YM; Lindsley CW; Hodder P; Mesecar AD; Stauffer SR Discovery of N-(Benzo[1,2,3]Triazol-1-yl)-N-(Benzyl)Acetamido)Phenyl) Carboxamides as Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) 3CLpro Inhibitors: Identification of ML300 and Noncovalent Nanomolar Inhibitors with an Induced-Fit Binding. Bioorganic Med. Chem. Lett 2013, 23 (22), 6172–6177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Zhang C; Stone EA; Deshmukh M; Ippolito JA; Ghahremanpour MM; Tirado-rives J; Spasov KA; Zhang S; Takeo Y; Kudalkar SN; Liang Z; Isaacs F; Lindenbach B; Miller SJ; Anderson KS; Jorgensen WL Potent Noncovalent Inhibitors of the Main Protease of SARS-CoV‑2 from Molecular Sculpting of the Drug Perampanel Guided by Free Energy Perturbation Calculations. ACS Cent. Sci 2021, 7 (3), 467–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Kim Y; Lovell S; Tiew K-C; Mandadapu SR; Alliston KR; Battaile KP; Groutas WC; Chang K-O Broad-Spectrum Antivirals against 3C or 3C-Like Proteases of Picornaviruses, Noroviruses, and Coronaviruses. J. Virol 2012, 86 (21), 11754–11762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Hoffman RL; Kania RS; Brothers MA; Davies JF; Ferre RA; Gajiwala KS; He M; Hogan RJ; Kozminski K; Li LY; Lockner JW; Lou J; Marra MT; Mitchell LJ; Murray BW; Nieman JA; Noell S; Planken SP; Rowe T; Ryan K; Smith GJ; Solowiej JE; Steppan CM; Taggart B. Discovery of Ketone-Based Covalent Inhibitors of Coronavirus 3CL Proteases for the Potential Therapeutic Treatment of COVID-19. J. Med. Chem 2020, 63 (21), 12725–12747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Boras B; Jones RM; Anson BJ; Arenson D; Aschenbrenner L; Bakowski MA; Beutler N; Binder J; Chen E; Eng H; Hammond J; Hoffman R; Kadar EP; Kania R; Kimoto E; Kirkpatrick MG; Lanyon L; Lendy EK; Lillis JR; Luthra SA; Ma C; Noell S; Obach RS; O’Brien MN; O’Connor R; Ogilvie K; Owen D; Pettersson M; Reese MR; Rogers T; Rossulek MI; Sathish JG; Steppan C; Ticehurst M; Updyke LW; Zhu Y; Wang J; Chatterjee AK; Mesecar AD; Anderson AS; Allerton C. Discovery of a Novel Inhibitor of Coronavirus 3CL Protease as a Clinical Candidate for the Potential Treatment of COVID-19. bioRxiv 2020. doi. 10.1101/2020.09.12.293498. [DOI] [Google Scholar]
- (26).Zhang L; Lin D; Sun X; Curth U; Drosten C; Sauerhering L; Becker S; Rox K; Hilgenfeld R. Crystal Structure of SARS-CoV-2 Main Protease Provides a Basis for Design of Improved α-Ketoamide Inhibitors. Science, 2020, 368 (6489), 409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Anand K. Coronavirus Main Proteinase (3CLpro) Structure: Basis for Design of Anti-SARS Drugs. Science, 2003, 300 (5626), 1763–1767. [DOI] [PubMed] [Google Scholar]
- (28).Yang H; Yang M; Ding Y; Liu Y; Lou Z; Zhou Z; Sun L; Mo L; Ye S; Pang H; Gao GF; Anand K; Bartlam M; Hilgenfeld R; Rao Z. The Crystal Structures of Severe Acute Respiratory Syndrome Virus Main Protease and Its Complex with an Inhibitor. Proc. Natl. Acad. Sci 2003, 100 (23), 13190–13195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Douangamath A; Fearon D; Gehrtz P; Krojer T; Lukacik P; Owen CD; Resnick E; Strain-Damerell C; Aimon A; Ábrányi-Balogh P; Brandão-Neto J; Carbery A; Davison G; Dias A; Downes TD; Dunnett L; Fairhead M; Firth JD; Jones SP; Keeley A; Keserü GM; Klein HF; Martin MP; Noble MEM; O’Brien P; Powell A; Reddi RN; Skyner R; Snee M; Waring MJ; Wild C; London N; von Delft F; Walsh MA Crystallographic and Electrophilic Fragment Screening of the SARS-CoV-2 Main Protease. Nat. Commun 2020, 11 (5047), doi. 10.1038/s41467-020-18709-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Jin Z; Du X; Xu Y; Deng Y; Liu M; Zhao Y; Zhang B; Li X; Zhang L; Peng C; Duan Y; Yu J; Wang L; Yang K; Liu F; Jiang R; Yang X; You T; Liu X; Yang X; Bai F; Liu H; Liu X; Guddat LW; Xu W; Xiao G; Qin C; Shi Z; Jiang H; Rao Z; Yang H. Structure of Mpro from SARS-CoV-2 and Discovery of Its Inhibitors. Nature 2020, 582 (7811), 289–293. [DOI] [PubMed] [Google Scholar]
- (31).Dai W; Zhang B; Jiang XM; Su H; Li J; Zhao Y; Xie X; Jin Z; Peng J; Liu F; Li C; Li Y; Bai F; Wang H; Cheng X; Cen X; Hu S; Yang X; Wang J; Liu X; Xiao G; Jiang H; Rao Z; Zhang LK; Xu Y; Yang H; Liu H, Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368 (6497), 1331–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Su HX; Yao S; Zhao WF; Li MJ; Liu J; Shang WJ; Xie H; Ke CQ; Hu HC; Gao MN; Yu KQ; Liu H; Shen JS; Tang W; Zhang LK; Xiao GF; Ni L; Wang DW; Zuo JP; Jiang HL; Bai F; Wu Y; Ye Y; Xu YC, Anti-SARS-CoV-2 activities in vitro of Shuanghuanglian preparations and bioactive ingredients. Acta Pharmacol Sin 2020, 41 (9), 1167–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Qiao J; Li YS; Zeng R; Liu FL; Luo RH; Huang C; Wang YF; Zhang J; Quan B; Shen C; Mao X; Liu X; Sun W; Yang W; Ni X; Wang K; Xu L; Duan ZL; Zou QC; Zhang HL; Qu W; Long YH; Li MH; Yang RC; Liu X; You J; Zhou Y; Yao R; Li WP; Liu JM; Chen P; Liu Y; Lin GF; Yang X; Zou J; Li L; Hu Y; Lu GW; Li WM; Wei YQ; Zheng YT; Lei J; Yang S, SARS-CoV-2 M(pro) inhibitors with antiviral activity in a transgenic mouse model. Science 2021, 371 (6536), 1374–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Lee TW; Cherney MM; Liu J; James KE; Powers JC; Eltis LD; James MNG Crystal Structures Reveal an Induced-Fit Binding of a Substrate-like Aza-Peptide Epoxide to SARS Coronavirus Main Peptidase. J. Mol. Biol 2007, 366 (3), 916–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).St. John SE; Tomar S; Stauffer SR; Mesecar AD Targeting Zoonotic Viruses: Structure-Based Inhibition of the 3C-like Protease from Bat Coronavirus HKU4 - The Likely Reservoir Host to the Human Coronavirus That Causes Middle East Respiratory Syndrome (MERS). Bioorganic Med. Chem 2015, 23 (17), 6036–6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Grum-Tokars V; Ratia K; Begaye A; Baker SC; Mesecar AD Evaluating the 3C-like Protease Activity of SARS-Coronavirus: Recommendations for Standardized Assays for Drug Discovery. Virus Res. 2008, 133 (1), 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Uzunova K; Filipova E; Pavlova V; Vekov T. Insights into Antiviral Mechanisms of Remdesivir, Lopinavir/Ritonavir and Chloroquine/Hydroxychloroquine Affecting the New SARS-CoV-2. Biomed. Pharmacother 2020, 131 (May), 110668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Wang M; Cao R; Zhang L; Yang X; Liu J; Xu M; Shi Z; Hu Z; Zhong W; Xiao G. Remdesivir and Chloroquine Effectively Inhibit the Recently Emerged Novel Coronavirus (2019-NCoV) in Vitro. Cell Res. 2020, 30 (3), 269–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Xue X; Yang H; Shen W; Zhao Q; Li J; Yang K; Chen C; Jin Y; Bartlam M; Rao Z. Production of Authentic SARS-CoV Mpro with Enhanced Activity: Application as a Novel Tag-Cleavage Endopeptidase for Protein Overproduction. J. Mol. Biol 2007, 366 (3), 965–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Tomar S; Johnston ML; John SES; Osswald HL; Nyalapatla PR; Paul LN; Ghosh AK; Denison MR; Mesecar AD Ligand-Induced Dimerization of Middle East Respiratory Syndrome (MERS) Coronavirus Nsp5 Protease (3CLpro): Implications for Nsp5 Regulation and the Development of Antivirals. J. Biol. Chem 2015, 290 (32), 19403–19422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Otwinowski Z; Minor W. Processing of X-Ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997, 276, 307–326. [DOI] [PubMed] [Google Scholar]
- (42).McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ Phaser Crystallographic Software. J. Appl. Crystallogr 2007, 40 (4), 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Kneller DW; Phillips G; O’Neill HM; Jedrzejczak R; Stols L; Langan P; Joachimiak A; Coates L; Kovalevsky A. Structural Plasticity of SARS-CoV-2 3CL Mpro Active Site Cavity Revealed by Room Temperature X-Ray Crystallography. Nat. Commun 2020, 11 (1), 7–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Moriarty NW; Grosse-Kunstleve RW; Adams PD Electronic Ligand Builder and Optimization Workbench (ELBOW): A Tool for Ligand Coordinate and Restraint Generation. Acta Crystallogr. Sect. D Biol. Crystallogr 2009, 65 (10), 1074–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Liebschner D; Afonine PV; Baker ML; Bunkoczi G; Chen VB; Croll TI; Hintze B; Hung LW; Jain S; McCoy AJ; Moriarty NW; Oeffner RD; Poon BK; Prisant MG; Read RJ; Richardson JS; Richardson DC; Sammito MD; Sobolev OV; Stockwell DH; Terwilliger TC; Urzhumtsev AG; Videau LL; Williams CJ; Adams PD Macromolecular Structure Determination Using X-Rays, Neutrons and Electrons: Recent Developments in Phenix. Acta Crystallogr. Sect. D Struct. Biol 2019, 75, 861–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Emsley P; Lohkamp B; Scott WG; Cowtan K. Features and Development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr 2010, 66 (4), 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Chuck CP; Chen C; Ke Z; Chi-Cheong Wan D; Chow HF; Wong KB Design, Synthesis and Crystallographic Analysis of Nitrile-Based Broad-Spectrum Peptidomimetic Inhibitors for Coronavirus 3C-like Proteases. Eur. J. Med. Chem 2013, 59, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Winn MD; Ballard CC; Cowtan KD; Dodson EJ; Emsley P; Evans PR; Keegan RM; Krissinel EB; Leslie AGW; Mccoy A; Mcnicholas SJ; Murshudov GN; Pannu NS; Potterton EA; Powell HR; Read RJ; Vagin A; Wilson KS Overview of the CCP4 Suite and Current Developments. Acta Crystallogr. Sect. D 2011, 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Shin WJ; Hara D; Gbormittah F; Chang H; Chang BS; Jung JU Development of Thermostable Lyophilized Sabin Inactivated Poliovirus Vaccine. MBio 2018, 9 (6), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Shin W-J; Nam K-Y; Kim N-D; Kim S-H; No K-T; Seong B-L Identification of a Small Benzamide Inhibitor of Influenza Virus Using a Cell-Based Screening. Chemotherapy 2016, 61 (3), 159–166. [DOI] [PubMed] [Google Scholar]
- (51).Wang W; Shin WJ; Zhang B; Choi Y; Yoo JS; Zimmerman MI; Frederick TE; Bowman GR; Gross ML; Leung DW; Jung JU; Amarasinghe GK The Cap-Snatching SFTSV Endonuclease Domain Is an Antiviral Target. Cell Rep. 2020, 30 (1), 153–163.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Zientek M; Jiang Y; Youdim K; Obach RS In Vitro-In Vivo Correlation for Intrinsic Clearance for Drugs Metabolized by Human Aldehyde Oxidase. Drug Metab. Dispos 2010, 38 (8), 1322–1327 [DOI] [PubMed] [Google Scholar]
- (53).Obach RS Prediction of Human Clearance of Twenty-Nine Drugs from Hepatic Microsomal Intrinsic Clearance Data: An Examination of in Vitro Half-Life Approach and Nonspecific Binding to Microsomes. Drug Metab. Dispos 1999, 27 (11), 1350–1359. [PubMed] [Google Scholar]
- (54).Turlington M; Chun A; Jacobs J; Dawson E; Daniels JS; Saldanha A; Chase P; Hodder P; Eggler A; Tokars V; Mesecar A; Lindsley CW; Stauffer SR Non-Covalent Triazole-Based Inhibitors of the SARS Main Proteinase 3CLpro; Probe Reports from the NIH Molecular Libraries Program, 2013. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.