

Charlevoix‐Saguenay (ARSACS) is a rare autosomal recessive spastic ataxia.1 The disease is characterized by progressive ataxia, pyramidal involvement, peripheral neuropathy and hypermyelination of the retinal nerve fibers due to the mutation in the SACS gene, SACS that encodes protein sacsin. First mutations in the gene were found in French Canadians from the Saguenay region.2
FIG. 1.
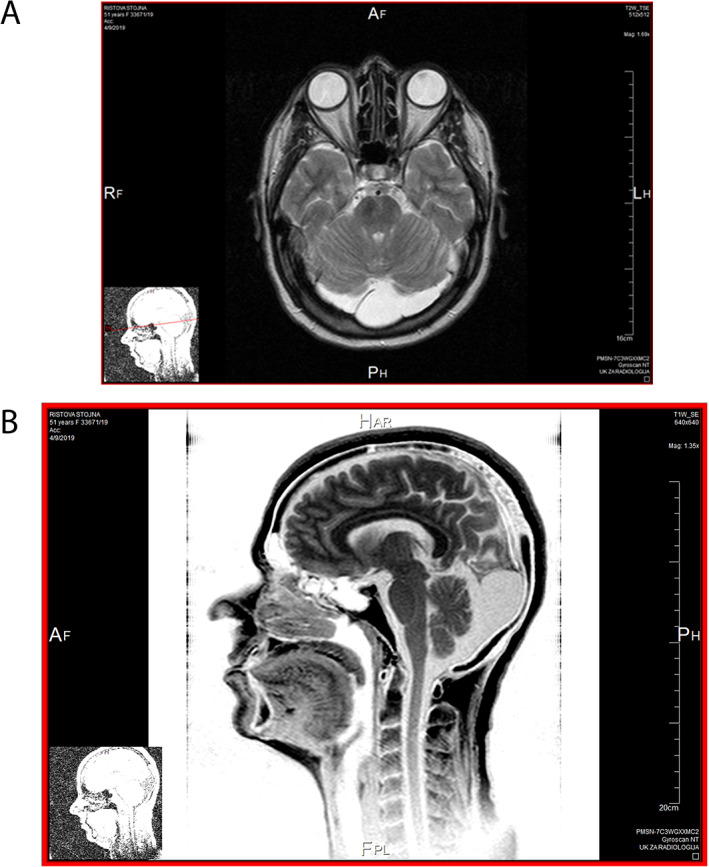
MRI of the brain of the 53 years old woman with autosomal recessive spastic ataxia Charlevoix‐Saguenay. (A) Bilateral cerebellar atrophy and large arachnoidal cyst in posterior cranial fosa exactly in the projection of cisterna magna. (B) Cerebellar atrophy.
A fifty‐three‐year‐old Macedonian right‐handed woman born from non‐consanguineous parents, who was hospitalized at the Movement disorders department in Skopje, was complaining of progressive gait disturbance, walking difficulties, speech problems, changes in her mood, and slowness of performing her daily activities. Her first symptoms of depression began in 2004, at the age of 37 years, when she had an unsuccessful attempt of suicide for which she was hospitalized in psychiatric clinic in Negorci. Her sister also committed suicide. Around one year after this she developed progressive speech and gait difficulties. There were no other medical conditions of note. Her brother had similar symptoms.
Neurological examination revealed severe gait ataxia, and she was unable to walk tandem and had a broad base. Her speech was dysarthric and slurred. There was spasticity in the legs. Romberg sign was positive. Deep tendon reflexes were increased with extended reflexogenic area and Babinski sign was bilaterally positive. This patient had pyramidal signs as well as moderate bradykinesia and hypomimia.3 (Video 1) She had late onset ataxia and the cognitive decline that was shown on the neuropsychological tests is novel in this case.4
Video 1.
53 years old woman with autosomal recessive spastic ataxia Charlevoix‐ Saguenay. Ataxia, dysarthria, bilaterally positive Babinski sign, increase deep tendon reflexes and positive Romberg sign is presented.
MRI of the brain revealed bilateral cerebellar atrophy and a large arachnoidal cyst in the posterior cranial fossa in the projection of the cisterna magna. Occipital bilateral cortical atrophy is also seen (Fig. 1).5 Electromyography (EMG) and nerve conduction studies revealed distal lower limb predominant peripheral axonal neuropathy. Somatosensory evoked potentials (SEP) on the tibial nerve showed prolonged latencies of the cortical waves. Visual evoked potential (VEP) was normal. Electroencephalography (EEG) was normal. Cerebrospinal fluid analysis (CSF) showed slight elevation in the total protein, elevation in the albumin fraction and IgG and the type of electropherogram was transudative suggesting seepage through the blood brain barrier. Routine laboratory tests and routine blood tests were normal.
Genetic testing was performed. Targeted re‐sequencing of 4800 clinically significant genes was made using TruSight One kit, Illumina and bioinformatic processing of the genes associated with the clinical condition of the patient.
The patient's genotype is NM_014363.5:c.(13721T > G);(13721T > G). Pathogenic change c.13721 T > g;p (Phe 4574Cys) in the SACS gene was found. The patient is homozygous for pathogenic change c.13721 T > G;p. (Phe4574Cys) in SACS gene. This variant in exon 10 of SACS gene is missense variant that on protein level cause change of aminoacid phenylalanine on position 4574 in the aminoacid cystein. This is a novel variant that has not been published in the literature so far, and it is not present in the database of pathogenic variants of SACS gene. The change is in the recessive gene in homozygous form. A similar genetic finding was also detected in a homozygous state in the patient's brother who has a similar clinical manifestation of the disease. This genotype confirmed the diagnosis of autosomal recessive spastic ataxia of Charlevoix‐Saguenay in the patient and her brother. The genetic testing was made in the Macedonian Academy of Science and Art (MANU) in Skopje, Republic of North Macedonia.
The patient was treated with low doses of levodopa carbidopa with minimal effect on her bradykinesia.
The novelty in this article is that it is presented a patient from Republic of North Macedonia in whom a novel missense pathogenic variant c.13721 T > G;p. (Phe4574Cys) in SACS gene was found which has not been published in the literature before.6, 7, 8 The patient had mild cognitive decline, moderate bradykinesia, and hypomimia with ataxia of late onset.
Duquette et al. found that in the clinical picture increased deep tendon reflexes are more common than spasticity, and neuropathy became more apparent in the second decade. Despite a homogeneous genetic background, some patients showed no signs of neuropathy or spasticity by the age of 18 years.9 In the patient described here she had clear spasticity and has been symptomatic at a late age.
Synofzik et al. found on MRI pontine hypointensities, all patients (100%) showed hyperintensities of the lateral pons merging into the (thickened) middle cerebellar peduncles. In addition, 63% exhibited bilateral parietal cerebral atrophy, and 63% a short circumscribed thinning of the posterior midbody of the corpus callosum.5
Hara et al. found that two patients had a unique phenotype characterized by dementia, ophthalmoplegia, and the absence of prominent retinal myelinated fibers.10 In the patient described in the article, VEP was normal and there were no visual difficulties.
Author Roles
(1) Research project: A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript Preparation: A. Writing of the first draft, B. Review and Critique.
I.P.: 3A, 3B
Disclosures
Ethical Compliance Statement
The author confirms that the patient provided written consent, consenting to use for educational purposes, but because this article is a case report, no IRB approval was necessary. I confirm that I have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflict of Interest
No specific funding was received for this work and the author declares that there are no conflicts of interest relevant to this work.
Financial Disclosures for the Previous 12 Months
The author declares that there are no additional disclosures to report.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1.Bouchard JP, Barbeau A, Bouchard R, Bouchard RW. Autosomal recessive spastic ataxia of Charlevoix Saguenay. Can J Neurol Sci 1978;5:61–69. [PubMed] [Google Scholar]
- 2.Engert JC, Berube P, Mercier J, et al. ARSACS, a spastic ataxia common in northeastern Quebec, is caused by mutations in a new gene encoding an 11.5kb ORF. Nat Genet 2000;24:120–125. [DOI] [PubMed] [Google Scholar]
- 3.Bouhlal Y, Amouri R, El Euch FG, Hentati F. Autosomal recessive spastic ataxia of Charlevoix Saguenay: an overview. Parkinsonism Relat Disord 2011;17:418–422. [DOI] [PubMed] [Google Scholar]
- 4.Gazulla J, Benavente I, Vela AC, et al. New findings in the ataxia of Charlevoix Saguenay. J Neurol 2012;259:869–878. [DOI] [PubMed] [Google Scholar]
- 5.Synofzik M, Soehn A, Gburek Augustat J, et al. Autosomal recessive spastic ataxia of Charlevoix Saguenay (ARSACS): expanding the genetic, clinical and imaging spectrum. Orphanet J Rare Dis 2013;8:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Richter A, Ozgul RK, Poisson VC, et al. Private SACS mutations in autosomal recessive spastic ataxia of Charlevoix Saguenay (ARSACS) families from Turkey. Neurogenetics 2004;5:165–170. [DOI] [PubMed] [Google Scholar]
- 7.Criscuolo C, Saccà F, De Michele G, et al. Novel mutation of SACS gene in a Spanish family with autosomal recessive spastic ataxia. Mov Disord 2005;20:1358–1361. [DOI] [PubMed] [Google Scholar]
- 8.Thiffault I, Dicaire MJ, Tetreault M, et al. Diversity of ARSACS mutations in French Canadians. Can J Neurol Sci 2013;40:61–66. [DOI] [PubMed] [Google Scholar]
- 9.Duquette A, Brais B, Bouchard JP, Mathieu J. Clinical presentation and early evolution of spastic ataxia of Charlevoix Saguenay. Mov Disord 2013;28:2011–2014. [DOI] [PubMed] [Google Scholar]
- 10.Hara K, Onodera O, Endo M, et al. Sacsin related autosomal recessive ataxia without prominent retinal myelinated fibers in Japan. Mov Disord 2005;20:380–382. [DOI] [PubMed] [Google Scholar]