

Parkinson’s disease (PD) is the second most common neurodegenerative disorder characterized by multiple motor and non-motor symptoms, which include, among others, constipation, sleep disturbance, bradykinesia, gait and balance abnormalities, muscle stiffness and resting tremor. The motor symptoms are caused by progressive age-related death of dopaminergic neurons and in the vast majority of patients suffering from age-related idiopathic PD the cause of dopaminergic neurodegeneration is unknown. Even in the familial early-onset PD where genetic mutations have been identified, the molecular mechanisms driving degeneration of dopaminergic neurons are far from clear. Consequently, there is no clinically approved disease-modifying therapy capable of stopping or at least slowing down the disease progression.
Intracellular accumulation of insoluble protein deposits emerged as a common element of major age-related neurodegenerative disorders. Such deposits are commonly observed in histopathological examination of brains from patients with Alzheimer’s disease (AD) (α-synuclein (αSyn), β-amyloid peptide and Tau deposits); amyotrophic lateral sclerosis (SOD1, TDP43 and FUS deposits); multiple systems atrophy (MSA), dementia with Lewy bodies (DLB) and PD (αSyn deposits in all three). Moreover, protein aggregation pathology is not limited to neurodegenerative diseases, but can also occur in peripheral organs, such as the gastrointestinal tract and kidneys, for example, in various amyloidoses. Despite being common denominator of most prevalent neurodegenerative disorders, the role of pathological protein aggregation in the etiology of AD, amyotrophic lateral sclerosis, MSA, DLB, and PD remains unclear, with very few molecular mechanisms confirmed in patients. Moreover, several clinical trials have targeted protein aggregates in neurodegenerative disorders (e.g. treatments targeting β-amyloid peptide in AD) with limited success, leading to a view that protein aggregation might be just epiphenomenon accompanying neurodegeneration. However, in our opinion it is rather the lack of mechanistic understanding of pathological protein misfolding, spread and disturbances it causes to cells that hindered successful development of disease-modifying therapies (Chmielarz and Saarma, 2020). Recently developed models utilizing αSyn pre-formed fibrils (αSyn PFFs) have rapidly progressed our understanding of protein aggregation in PD, aiming for the development of more evidence based and ultimately effective treatments.
αSyn is one of the main components of proteinaceous neuronal inclusions called Lewy Bodies characteristic for DLB and PD (Fouka et al., 2020). Lewy Bodies contain αSyn crowded together with fragments of membranous organelles and vesicles (Shahmoradian et al., 2019) and their formation has been recently proposed to drive neuronal pathology (Mahul-Mellier et al., 2020; Figure 1). Mutations and polymorphisms in the gene encoding αSyn are known risk factors and, in some cases, even direct causes of familial forms of PD. During PD progression, insoluble aggregates are increasingly observed in multiple brain regions, suggesting that misfolded αSyn spreading through interconnected neuronal networks possesses prion-like properties. Supporting thе hypothesis, host-to-graft spread of Lewy Body pathology was also observed in fetal midbrain-derived neurons transplanted to the brains of PD patients. Yet, despite that Lewy Bodies in PD have been described more than 100 years ago and for more than 20 years we know that αSyn is their main component, the causative role of αSyn in PD progression and degeneration of dopaminergic neurons has not been conclusively proven. For example, it is still not clear which conformation(s) and oligomeric state(s) (e.g. soluble oligomers or short fibrils) of misfolded αSyn are the most active in propagation and seeding aggregation in neurons and glial cells in the human brain. The molecular mechanisms for uptake of misfolded αSyn are also not fully elucidated. In addition to genetic and pathological evidence, application of sensitive protein misfolding cyclic amplification or real-time quaking-induced conversion assays demonstrated the presence of misfolded protein species capable of seeding αSyn aggregation in cerebrospinal fluid (Shahnawaz et al., 2020) and even in skin samples of PD patients (Manne et al., 2020). Inoculation with Lewy Body extracts collected post-mortem from PD patients’ brains induced the development and spread of αSyn pathology not only in rodents, but also in non-human primates (Arotcarena et al., 2020). Supported by such data, αSyn has emerged as a promising target for disease-modifying therapy in PD, MSA and, possibly, DLB. Proposed therapeutic approaches targeting αSyn include interfering with initial aggregation by small molecules stabilizing correctly folded protein conformation, directly binding misfolded fibrils and promoting either their dissociation or inhibiting their growth; enhancing degradation of intracellular αSyn by stimulating lysosomal activity or by small molecules – proteolysis targeting chimeras (PROTACs) – promoting preoteasomal degradation; lowering cellular αSyn levels by anti-sense oligonucleotides (ASO) and cell-penetrant nanobodies; inhibiting uptake and targeting transmission of extracellular αSyn with active and passive immunization. Many of such treatments are currently in clinical trials (see https://clinicaltrials.gov/ for a regularly updated list) and some have shown good safety profiles (e.g. anle138b, NPT200-11, ENT-01, nilotinib, memantine and several other small molecules and active and passive immunotherapies targeting αSyn misfolding and spread), however, conclusive evidence of their efficacy is still to be demonstrated. Interestingly, encouraging results have recently been reported from PASADENA phase II clinical trial of monoclonal humanized antibody designed to stop misfolded αSyn transmission. While not meeting its primary endpoint, this trial demonstrated clear improvement in both motor and cognitive symptoms, convincing enough to prompt Roche and Prothena to support moving into the next phase of clinical development (see https://ir.prothena.com/news-releases/news-release-details/roche-and-prothena-will-advance-prasinezumab-late-stage-clinical). Further development of successful αSyn targeting therapies would benefit immensely from a comprehensive understanding of the entire process of the pathology transmission, Lewy Body formation and resulting cellular damage. Significant progress in our understanding of these questions has been made in preclinical studies, thanks to multiple αSyn transgenic models (Airavaara et al., 2020) and the development of αSyn PFFs able to seed aggregation of endogenous αSyn in cultured cells and in vivo (Hijaz and Volpicelli-Daley, 2020).
Figure 1.
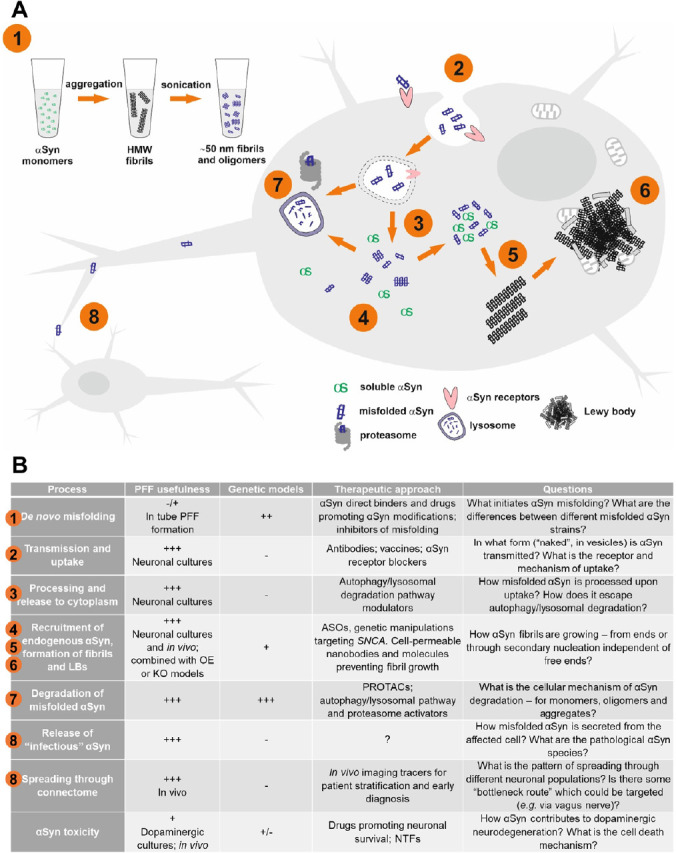
Preparation and application of α-synuclein preformed fibrils to reconstruct multiple steps of Lewy body formation and pathological protein transmission.
(A) Major steps in PFF-induced αSyn aggregation model: (1) formation of αSyn fibrils in vitro; (2) cellular uptake of αSyn PFFs; (3) processing and release to cytoplasm; (4) recruitment of endogenous αSyn; (5) seeding of endogenous αSyn misfolding, formation of oligomers and insoluble αSyn fibrils; (6) maturation of fibrils and formation of Levy Bodies containing fragment of lipid membranes and mitochondria; (7) lysosomal and proteasomal degradation of misfolded αSyn oligomers and fibrils; (8) release and propagation of misfolded αSyn. (B) Application of αSyn PFFs and genetic models to study molecular mechanisms and develop therapies targeting pathological αSyn aggregation, and the remaining questions. Numbers correspond to major PFF-induced αSyn aggregation steps depicted in Figure 1. ASOs: Anti-sense oligonucleotides; HMW: high molecular weight; KO: knockout; LBs: Lewy bodies; NTFs: neurothrophic factors; OE: overexpression; PROTACs: proteolysis targeting chimeras.
αSyn PFFs model allows studying multiple steps of Lewy Body pathology development: uptake of pathogenic fibrils, their internalization, transport, processing and recruitment of endogenous αSyn, its post-translational modifications (e.g. phosphorylation of Ser129), formation and maturation of Lewy Body-like inclusions (Figure 1A) and, finally, effects of these processes on cell function and survival. Furthermore, with αSyn PFFs we can study the mechanism of pathology transmission between neurons and in vivo spreading through neuronal connectome as well as motor and behavioral outcomes of accumulation of αSyn deposits in different brain structures (Hijaz and Volpicelli-Daley, 2020). Application of αSyn PFFs as well as genetic models allowed pre-clinical testing of multiple approaches aiming to develop disease-modifying PD therapy (Figure 1B). Additionally, the formation of fibrils may be templated with pathological αSyn species from the brain and/or cerebrospinal fluid samples of PD and MSA patients. In such case the fibrils assume pathological conformation of templating patient material, allowing studying the differences and discriminating pathological αSyn aggregates in PD and MSA (Shahnawaz et al., 2020).
αSyn PFFs are purified prion-like, self-templating and transmittable aggregates of misfolded αSyn, which can induce formation of Lewy Body-like inclusions and cellular dysfunction both in in vitro and in vivo models (Er et al., 2020; Hijaz and Volpicelli-Daley, 2020). αSyn PFFs are formed by bacteria-expressed purified recombinant monomeric αSyn incubated at high concentration at 37°C with shaking for up to seven days, during which time it folds into elongated fibrils, exhibiting amyloid and self templating properties. Obtained fibrils are subsequently sonicated to form short, about 50 nm in length, fragments, most efficient at seeding pathology (Hijaz and Volpicelli-Daley, 2020; Figure 1A).
Formation of fibrils in vitro can be easily monitored to investigate initial steps in this putatively nucleation-dependent process. For example, conversion of monomeric αSyn into pathogenic αSyn PFFs involves the preceding step of liquid-liquid phase separation, which can be fostered by PD linked mutations or heavy metal interaction (Ray et al., 2020). Understanding of the de novo αSyn PFFs formation and ability to monitor it is of obvious value for the development of drugs targeting this process. In vitro PFF model is also useful for testing compounds aiming to dissociate fibrils or inhibit their growth to avoid formation of putatively more toxic αSyn oligomers. Several such compounds (e.g. anle138b, NPT200-11, ENT-01, memantine and others) are currently in different phases of clinical trials.
αSyn PFFs were also used to study neuronal uptake of misfolded αSyn, which was shown to be receptor-mediated, albeit alternative hypotheses have also been proposed, such as direct penetration of membrane, adsorptive-mediated endocytosis or transmission through tunneling nanotubes or exosomes (Fouka et al., 2020). After uptake, αSyn PFFs localize in lysosomal compartment and partial lysosomal processing might actually enhance or even be required for αSyn PFFs pathogenicity in cells (Hijaz and Volpicelli-Daley, 2020; Figure 1). However, it is still not clear how exactly αSyn PFFs are processed and how they are released to cytoplasm and, possibly, also to extracellular space. Several studies highlight the role of extracellular exosomes in promoting the spread of misfolded αSyn; moreover, elevated levels of αSyn in neuronal exosomes isolated from patients’ serum emerge as a promising diagnostic biomarker of early PD (Hijaz and Volpicelli-Daley, 2020; Jiang et al., 2020). However, additional studies are needed to resolve what type of autophagy/lysosomal pathway modulation will be beneficial and which might be ineffective or even harmful. We have recently utilized αSyn PFFs in vitro in primary dopaminergic neurons and in vivo in mouse to demonstrate protection against formation of αSyn deposits by stimulation of GDNF/RET signaling at early time points after αSyn PFF uptake, presumably through modulation of endo-lysosomal pathway (Chmielarz et al., 2020). Indeed, several molecules, such as ambroxol and venglustat, modulating lysosomal function by affecting levels and activity of lysosomal glucocerebrosidase are currently being tested in clinical trials for PD treatment.
When applied to neuronal cultures, including primary and human induced pluripotent stem cell derived neurons, αSyn PFFs induce accumulation of insoluble intracellular deposits which maturate over time, increasing their resemblance to Lewy Bodies (Mahul-Mellier et al., 2020). Application of αSyn PFFs to neurons cultured in microfluidic chambers allows to dissect cell-to-cell transmission of pathological aggregates and to demonstrate the effectiveness of antibodies targeting extracellular αSyn or putative αSyn receptors in blocking this process. When αSyn PFFs are injected into the brain parenchyma, similar deposits are observed in anatomically connected areas (Arotcarena et al., 2020; Hijaz and Volpicelli-Daley, 2020), resembling the spreading behavior of pathology in PD patients (Horsager et al., 2020). Importantly, formation and spreading of αSyn PFFs induced deposits through neuronal connections is dependent on endogenous αSyn, and the extent of pathology varies between neuronal types (Hijaz and Volpicelli-Daley, 2020). Application of αSyn PFFs to primary mouse midbrain neuronal cultures or human stem cell-derived dopaminergic neurons allows monitoring accumulation of αSyn deposits and their effects on survival of PD-relevant dopaminergic neurons in vitro and in humanized rats (Er et al., 2020; Hoban et al., 2020). Sensitivity of αSyn PFF induced pathology to endogenous αSyn levels makes αSyn PFF based models ideally suited to test the effectiveness of ASOs, PROTACs or gene therapy aimed at lowering expression or promoting degradation of αSyn.
The biggest limitation of αSyn PFFs model is that seeded deposits only modestly impair neuronal function and survival, the latter only after prolonged period of time (Mahul-Mellier et al., 2020), complicating testing of survival-promoting agents. Different research groups have reported variable effects of PFFs on cell survival. This could be partially because of technical difficulties in standardizing PFF preparations and batch-to-batch variations. However, delayed and modest effects on cell survival seem to be more in line with actual slow PD progression in humans. Actually, the mechanism by which pathological αSyn cause cell death remains elusive, although recent data suggest that this process is driven by formation of pathological deposits resembling Lewy Bodies containing not only αSyn but also fragments of membranous organelles (Mahul-Mellier et al., 2020). Understanding how αSyn pathology contributes to neuronal death could allow to design therapies aimed at increasing survival of cells already afflicted with αSyn pathology, such as neurotrophic factors or small molecules mimetics (Chmielarz et al., 2020). From the perspective of drug discovery, αSyn PFF models have advantages of being adaptable to multi-well plate formats with automatized quantification in dopaminergic neurons (Er et al., 2020). While still not feasible for use in high throughput screening campaigns, they can allow for functional validation of promising therapeutics in a physiologically relevant model allowing for monitoring αSyn aggregation and dopaminergic cell survival. Subsequently, therapies can be further validated in αSyn PFF based models in stem cell-derived human dopaminergic neurons and in vivo for long term effectiveness at slowing the spread of pathology and for behavioral outcomes (Hoban et al., 2020). Thanks to progressive nature of αSyn PFF in vivo model, it could also be used to identify a time window for effective treatment. Moreover, by varying the site of αSyn PFF injection (e.g. brain versus duodenum) (Arotcarena et al., 2020), we can verify the effectiveness of treatments in putative brain-first or body-first subtypes of PD (Horsager et al., 2020).
Lastly, in vivo αSyn PFF models will be useful for development of PET tracers for longitudinal assessment of αSyn pathology in PD patients. When developed, such imaging tools would be extremely useful both for monitoring of clinical trials and early diagnosis of the disease.
Overall, αSyn PFFs allow to study both pathophysiology of PD and the effectiveness of therapeutic approaches in physiologically relevant cellular and in vivo models, amenable to both meticulous investigations of mechanisms driving Lewy Body formation and spread, and screening and efficacy validation of compounds blocking these processes. While we still have many questions related to αSyn misfolding and pathology progression (Figure 1B), αSyn PFFs have been and will be very instrumental for helping researchers to find answers to these questions. Not surprisingly, αSyn PFFs elicit great hopes as a reliable and robust tool to model pathological protein aggregation in PD research and have seen extensive adoption in the field.
The present work was supported by grants from the Academy of Finland #293392, #319195; and Päivikki and Sakari Sohlberg Foundation (to AD); and Polish National Science Centre grant 2019/35/D/NZ7/03200 - Sonata 15 (to PC).
Additional file: Open peer review report 1 (86.9KB, pdf) .
Footnotes
Copyright license agreement:The Copyright License Agreement has been signed by both authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open peer reviewer:Johannes Haybaeck, Medical University of Innsbruck, Austria.
P-Reviewer: Haybaeck J; C-Editors: Zhao M, Qiu Y; T-Editor: Jia Y
References
- 1.Airavaara M, Parkkinen I, Konovalova J, Albert K, Chmielarz P, Domanskyi A. Back and to the future: from neurotoxin-induced to human Parkinson’s disease models. Curr Protoc Neurosci. 2020;91:e88. doi: 10.1002/cpns.88. [DOI] [PubMed] [Google Scholar]
- 2.Arotcarena ML, Dovero S, Prigent A, Bourdenx M, Camus S, Porras G, Thiolat ML, Tasselli M, Aubert P, Kruse N, Mollenhauer B, Trigo Damas I, Estrada C, Garcia-Carrillo N, Vaikath NN, El-Agnaf OMA, Herrero MT, Vila M, Obeso JA, Derkinderen P, et al. Bidirectional gut-to-brain and brain-to-gut propagation of synucleinopathy in non-human primates. Brain. 2020;143:1462–1475. doi: 10.1093/brain/awaa096. [DOI] [PubMed] [Google Scholar]
- 3.Chmielarz P, Saarma M. Neurotrophic factors for disease-modifying treatments of Parkinson’s disease: gaps between basic science and clinical studies. Pharmacol Rep. 2020;72:1195–1217. doi: 10.1007/s43440-020-00120-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chmielarz P, Er S, Konovalova J, Bandres L, Hlushchuk I, Albert K, Panhelainen A, Luk K, Airavaara M, Domanskyi A. GDNF/RET signaling pathway activation eliminates Lewy body pathology in midbrain dopamine neurons. Mov Disord. 2020;35:2279–2289. doi: 10.1002/mds.28258. [DOI] [PubMed] [Google Scholar]
- 5.Er S, Hlushchuk I, Airavaara M, Chmielarz P, Domanskyi A. Studying pre-formed fibril induced alpha-synuclein accumulation in primary embryonic mouse midbrain dopamine neurons. J Vis Exp. 2020 doi: 10.3791/61118. doi: 103791/61118. [DOI] [PubMed] [Google Scholar]
- 6.Fouka M, Mavroeidi P, Tsaka G, Xilouri M. In search of effective treatments targeting alpha-synuclein toxicity in synucleinopathies: pros and cons. Front Cell Dev Biol. 2020;8:559791. doi: 10.3389/fcell.2020.559791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hijaz BA, Volpicelli-Daley LA. Initiation and propagation of alpha-synuclein aggregation in the nervous system. Mol Neurodegener. 2020;15:19. doi: 10.1186/s13024-020-00368-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoban DB, Shrigley S, Mattsson B, Breger LS, Jarl U, Cardoso T, Nelander Wahlestedt J, Luk KC, Bjorklund A, Parmar M. Impact of alpha-synuclein pathology on transplanted hESC-derived dopaminergic neurons in a humanized alpha-synuclein rat model of PD. Proc Natl Acad Sci U S A. 2020;117:15209–15220. doi: 10.1073/pnas.2001305117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Horsager J, Andersen KB, Knudsen K, Skjærbæk C, Fedorova TD, Okkels N, Schaeffer E, Bonkat SK, Geday J, Otto M, Sommerauer M, Danielsen EH, Bech E, Kraft J, Munk OL, Hansen SD, Pavese N, Göder R, Brooks DJ, Berg D, et al. Brain-first versus body-first Parkinson’s disease: a multimodal imaging case-control study. Brain. 2020;143:3077–3088. doi: 10.1093/brain/awaa238. [DOI] [PubMed] [Google Scholar]
- 10.Jiang C, Hopfner F, Katsikoudi A, Hein R, Catli C, Evetts S, Huang Y, Wang H, Ryder JW, Kuhlenbaeumer G, Deuschl G, Padovani A, Berg D, Borroni B, Hu MT, Davis JJ, Tofaris GK. Serum neuronal exosomes predict and differentiate Parkinson’s disease from atypical parkinsonism. J Neurol Neurosurg Psychiatry. 2020;91:720–729. doi: 10.1136/jnnp-2019-322588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mahul-Mellier AL, Burtscher J, Maharjan N, Weerens L, Croisier M, Kuttler F, Leleu M, Knott GW, Lashuel HA. The process of Lewy body formation, rather than simply alpha-synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc Natl Acad Sci U S A. 2020;117:4971–4982. doi: 10.1073/pnas.1913904117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Manne S, Kondru N, Jin H, Serrano GE, Anantharam V, Kanthasamy A, Adler CH, Beach TG, Kanthasamy AG. Blinded RT-QuIC analysis of alpha-synuclein biomarker in skin tissue from Parkinson’s disease patients. Mov Disord. 2020;35:2230–2239. doi: 10.1002/mds.28242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ray S, Singh N, Kumar R, Patel K, Pandey S, Datta D, Mahato J, Panigrahi R, Navalkar A, Mehra S, Gadhe L, Chatterjee D, Sawner AS, Maiti S, Bhatia S, Gerez JA, Chowdhury A, Kumar A, Padinhateeri R, Riek R, et al. alpha-Synuclein aggregation nucleates through liquid-liquid phase separation. Nat Chem. 2020;12:705–716. doi: 10.1038/s41557-020-0465-9. [DOI] [PubMed] [Google Scholar]
- 14.Shahmoradian SH, Lewis AJ, Genoud C, Hench J, Moors TE, Navarro PP, Castaño-Díez D, Schweighauser G, Graff-Meyer A, Goldie KN, Sütterlin R, Huisman E, Ingrassia A, Gier Y, Rozemuller AJM, Wang J, Paepe A, Erny J, Staempfli A, Hoernschemeyer J, et al. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat Neurosci. 2019;22:1099–1109. doi: 10.1038/s41593-019-0423-2. [DOI] [PubMed] [Google Scholar]
- 15.Shahnawaz M, Mukherjee A, Pritzkow S, Mendez N, Rabadia P, Liu X, Hu B, Schmeichel A, Singer W, Wu G, Tsai AL, Shirani H, Nilsson KPR, Low PA, Soto C. Discriminating alpha-synuclein strains in Parkinson’s disease and multiple system atrophy. Nature. 2020;578:273–277. doi: 10.1038/s41586-020-1984-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.