

SUMMARY
Gut-associated phages are hypothesized to alter the abundance and activity of their bacterial hosts, contributing to human health and disease. Although temperate phages constitute a significant fraction of the gut virome, the effects of lysogenic infection are underexplored. We report that the temperate phage, Bacteroides phage BV01, broadly alters its host’s transcriptome, the prominent human gut symbiont Bacteroides vulgatus. This alteration occurs through phage-induced repression of a tryptophan-rich sensory protein (TspO) and represses bile acid deconjugation. Because microbially modified bile acids are important signals for the mammalian host, this is a mechanism by which a phage may influence mammalian phenotypes. Furthermore, BV01 and its relatives in the proposed phage family Salyersviridae are ubiquitous in human gut metagenomes, infecting a broad range of Bacteroides hosts. These results demonstrate the complexity of phage-bacteria-mammal relationships and emphasize a need to better understand the role of temperate phages in the gut microbiome.
In Brief
Integrative phages can have myriad effects on bacterial hosts. Campbell et al. find that integration of Bacteroides phage BV01 into Bacteroides vulgatus disrupts expression of TspO, reducing bile acid deconjugation. This is predicted to affect gut colonization and interactions with the human host. BV01 belongs to the Salyersviridae family.
Graphical Abstract
INTRODUCTION
The human gut is colonized by a dense and diverse microbial community comprised of bacterial, archaeal, and fungal cells as well as the viruses that infect them. This gut microbiome is vital for human health and development and is linked to an increasingly long list of disease states (Hills et al., 2019; Schmidt et al., 2018). Recent work has specifically implicated the gut phageome in disease, including inflammatory bowel disease (Norman et al., 2015), malnutrition (Reyes et al., 2015), AIDS (Monaco et al., 2016), colorectal cancers (Hannigan et al., 2018), and hypertension (Han et al., 2018). Broadly, gut phages act as important modulators of bacterial community structure (Hannigan et al., 2018; Khan Mirzaei et al., 2020; Moreno-Gallego et al., 2019) and metabolism (Hsu et al., 2019). Despite their apparent importance, little is known about how most gut-associated phages interact with their bacterial hosts (Mirzaei and Maurice, 2017).
Bacteroides is one of the most common and abundant bacterial genera in the distal human gut. The genus is known to degrade a diversity of complex carbohydrates (El Kaoutari et al., 2013; Salyers et al., 1977) and interact with host immune cells (Hickey et al., 2015; Shen et al., 2012). In a single human host, many Bacteroides species and strains coexist, competing for nutrients under changing environmental conditions caused by host diet (Tuncil et al., 2017), host metabolites (Ridlon et al., 2016), host immune system activities (Cullen et al., 2015; Planer et al., 2016), and phage predation (Porter et al., 2020). Moreover, horizontal gene transfer plays an important role in shaping the evolution and function of Bacteroides genomes (Coyne et al., 2014; Lange et al., 2016). How the diversity of Bacteroides strains in the human gut persists over time in such a dense, dynamic, and competitive environment is likely to have many reasons and is perhaps afforded by their highly plastic genomes.
Phage diversity and phage-host interactions in most commensal gut-associated bacteria, including Bacteroides species, is underexplored, but bioinformatics predictions of Bacteroides phages have been made (Krupovic and Forterre, 2011; Lange et al., 2016). Currently, the most abundant gut-associated phages are crAssphages (Dutilh et al., 2014; Koonin and Yutin, 2020; Yutin et al., 2018), a group of related lytic phages that infect Bacteroides intestinalis and potentially other species. CrAssphages demonstrate how traditional phage techniques (e.g., agar overlay plaque assays) are not reliable for Bacteroides hosts (Shkoporov et al., 2018), likely because of heterogeneity in capsular polysaccharide composition in isogenic cultures (Krinos et al., 2001; Patrick et al., 2010). In fact, deletion of all capsular polysaccharide synthesis loci allows isolation of many phages on the host Bacteroides thetaiotaomicron VPI-5482 (Porter et al., 2020). Most phages isolated against Bacteroides hosts so far exhibit an obligately lytic lifestyle (Jofre et al., 2014; Porter et al., 2020; Shkoporov et al., 2018; Tartera and Jofre, 1987) despite the potentially important role of lysogeny in phage-host interactions in the gut, where at least 17% of the gut phageome is predicted to be temperate (Minot et al., 2011; Ogilvie et al., 2013).
Prophage-host interactions have the potential to cause complex alterations to the host phenotype because of the temperate lifestyle having two distinct phases: lysogeny and lysis (Bondy-Denomy and Davidson, 2014; Howard-Varona et al., 2017). Unlike most strictly lytic phages, temperate phages more readily transfer beneficial genes between hosts, such as antibiotic resistance genes (Abeles et al., 2015) and auxiliary metabolic genes (Anantharaman et al., 2014; Sullivan et al., 2005). Some phage regulatory machinery expressed from prophages can modulate transcription of host genes, resulting in altered phenotypes (Berger et al., 2019; Hernandez-Doria and Sperandio, 2018). Integration of prophages into the host genome may also disrupt or enhance the activity of surrounding chromosomal genes (Carey et al., 2019; Chen et al., 2019; Feiner et al., 2015; Rabinovich et al., 2012). Although much is known about how these prophage-host interactions contribute to virulence in pathogens (Bensing et al., 2001; Jermyn and Boyd, 2002; Nakayama et al., 1999; Sekulovic and Fortier, 2015; Winter et al., 2010), very little is known about how phages modulate the activities of commensals.
Here we identified an active prophage, Bacteroides phage BV01, in a genetically tractable host strain, B. vulgatus ATCC 8482, and characterized its effects on the host’s transcriptome and phenotype. Further, we determine that BV01 represents a larger group of Bacteroides-associated phages comprising the proposed virus family Salyersviridae that common in the human gut phageome. This work provides insight into how Bacteroides react to temperate phage infection and establishes a model system for exploring complex phage-host interactions in an important human symbiont.
RESULTS
Bacteroides Phage BV01 Is a Prophage in B. vulgatus ATCC 8482
Bacteroides phage BV01 was partially predicted previously with the computational tool ProPhinder in the genome of B. vulgatus ATCC 8482 and deposited in a CLAssification of Mobile genetic Elements (ACLAME) database (Leplae et al., 2010; Lima-Mendez et al., 2008). Through comparative genomics and re-annotation of the host genome, we extended the predicted BV01 prophage to 58.9 kb (NCBI RefSeq: NC_009614.1; 3,579,765–3,638,687), which comprises 75 predicted open reading frames (ORFs) (Table S1). This larger predicted region is consistent with an earlier report of a prophage induced from this bacterial strain colonizing gnotobiotic mice (Reyes et al., 2013). BV01 encodes genes suggesting a temperate lifestyle with a putative phage repressor and anti-repressor as well as a holin-lysin-spanin operon for lytic release of phage progeny (Table S1; Figure 1A; Kongari et al., 2018; Young, 2014).
Figure 1. Phage BV01 Is Spontaneously Induced in Culture.

(A) DNA sequencing reads mapped to the BV01 prophage region. Reads from sequencing of B. vulgatus genomic DNA (WT) or isolated phage DNA (free BV01) were normalized to the total number of reads after trimming and are represented as a coverage curve. A cured lysogen (ΔBV01) and integrase deletion mutant (Δint) of B. vulgatus were confirmed by shotgun sequencing of genomic DNA (Tables S2 and S3). The coverage peak at position ~3,588,000 nt in ΔBV01 is attributed to a homologous sequence elsewhere in the B. vulgatus genome. Putative functional categories of BV01 genes are indicated by color; see Table S1 for full BV01 gene annotation.
(B) BV01 is produced at low levels at all stages of host growth. WT cultures were grown in triplicate for 36 h, and BV01 and host abundance were monitored by qPCR. By log phase, cultures reached the limit of detection (LOD) of the spectrophotometer.
(C and D) Lysogeny with phage BV01 does not affect (C) doubling time or (D) terminal optical density (OD) in vitro. Doubling times and terminal ODs were calculated for wild-type (WT), cured lysogen (ΔBV01), and integrase deletion (Δint) strains and averaged across three replicates.
Statistical analyses were performed with one-way ANOVA. N.S., not significant. Phage:host ratio df = 3, F = 4.03, p = 0.051. Doubling time df = 2, F = 2.55, p = 0.157. Percent terminal OD df = 2, F = 6.59, p = 0.101. All error bars represent standard deviation.
BV01 is detectable outside of host cells in the supernatants of in vitro cultures as a DNase-protected, double-stranded DNA (dsDNA) genome by shotgun sequencing (Figure 1A). Assembly of sequencing reads from free BV01 phage DNA results in a circular contig that spans the phage attachment site (attP). BV01 attP is identical to the left and right attachment sites (attL and attR, respectively), a pair of 25-bp direct repeat sequences (5′-GTCTAGTTTAGTTTTTGTGTTGTAA-3′), suggesting that BV01 enters a circular intermediate before replication.
Routine detection of phage BV01 in DNase-treated, cell-free supernatants by PCR suggests that it is spontaneously induced. Furthermore, examination of the phage-to-host ratio by qPCR revealed that it remained approximately 1 throughout log and stationary phase host growth (Figure 1B), which is expected for phages that are spontaneously induced (Baugher et al., 2014; Bonanno et al., 2016). We have yet to identify in vitro induction conditions that increase phage BV01 production above this background level detectable by qPCR. Subinhibitory concentrations of the antibiotics norfloxacin, mitomycin C, tetracycline, and erythromycin; UV irradiation; or co-culture with gut-associated microbes do not increase BV01 production. Furthermore, attempts to visualize BV01 by transmission electron microscopy were not successful, likely because of the small number of phage particles produced.
An isogenic cured lysogen (ΔBV01) strain was constructed by allelic exchange with the corresponding chromosomal attB region of an uninfected B. vulgatus strain (Figure 1A). No apparent fitness effect of BV01 infection was detected in vitro because growth of the lysogen (wild-type [WT]) and cured lysogen strains (ΔBV01) are not significantly different (Figures 1C and 1D). This further supports the conclusion that spontaneous induction of BV01 occurs at a low rate and that sporadic cell lysis events do not have an effect on cell density.
Despite sequence evidence of BV01 phage particles in the culture supernatants, BV01-containing supernatants were never found to produce new infection. No lytic infection was detected using plaque assays (Kropinski et al., 2009) with 10 B. vulgatus isolates (Table S4), the B. vulgatus cured lysogen (ΔBV01), B. thetaiotaomicron, and 4 B. dorei isolates on four different growth media: tryptone-yeast extract-glucose broth (TYG), brain heart infusion (BHI) agar supplemented with 10% defibrinated horse blood (HB), BHI supplemented with hemin and menadione (BHI-HM) (Thornton et al., 2012), and supplemented TYG (TYGS) (Goodman et al., 2009). Enrichment of infectious BV01 particles was attempted by passaging BV01-containing supernatants on B. vulgatus cured lysogen (ΔBV01) every day for 10 days (Shkoporov et al., 2018). However, enrichment was unsuccessful because subsequent plaque assays on naive cured lysogen did not yield any plaques. The same panel of 15 Bacteroides strains was tested for formation of lysogenic infection using non-concentrated or 200× concentrated BV01-containing supernatants. However, no novel infection was detected by PCR with primers designed to span the attachment site. To further test for new lysogenic infection, BV01 was tagged with a copy of tetQ, conferring tetracycline resistance, using allelic exchange. Five recipient strains, 4 B. vulgatus hosts and 1 B. dorei host, were tagged with erythromycin resistance with pNBU2-bla-ermG (Table S4). Double antibiotic selection found no new lysogens after attempted infection assays using BV01-tetQ supernatants or 200× concentrated BV01-tetQ supernatants or after 24-h co-culture of donor and recipient strains. We conclude that BV01 is a latent prophage in vitro, which is further supported by transcriptional data showing that BV01 exists in a largely repressed state (Figure S1A), a state likely maintained by the predicted phage repressor (BVU_RS14475), the most highly transcribed gene in the BV01 prophage (Figure S1A).
The only infectious conditions that were identified for BV01 are in a gnotobiotic mouse model (Figure 2A). Within 1 day of gavage, BV01-tetQ transductants were identified on doubly selective medium from mouse pellets from 4 of the 7 mice. Over the course of the 11-day experiment, transductants were eventually observed in all animals in all cages. The average frequency of transduction ranged from 1.9 × 10−6 to 3.6 × 10−9 per animal. Of 26 transductant isolates that were colony purified, all were confirmed by PCR to have gained BV01 integrated at the known attB. Further, a representative transductant isolate was confirmed by whole-genome sequencing (Tables S2 and S3). These results support our hypothesis that an unknown mammalian host factor is required for novel BV01 infection.
Figure 2. Transduction, Excision, and Circularization of Phage BV01.

(A) BV01 can transduce uninfected hosts in gnotobiotic mice (n = 7). Mice were gavaged with an equal mixture of BV01-tetQ lysogen and erythromycin-tagged cured lysogen (day 0). Recipient, donor, and transductant cells were identified by plating on medium with erythromycin (Erm) or tetracycline (Tet). Calculated means and standard errors (SE) are shown in black.
(B) Excision and circularization activities of the BV01 integrase are confirmed by PCR from late stationary phase cultures of four strains: WT, Δint, ΔBV01, and the int complement strain using a pNBU2 integrative plasmid (Δint + pNBU2-int). The presence of phage DNA was detected by amplification of a phage marker gene (BVU_RS14350). Amplification across the phage attachment site (attP) indicates circularization of the BV01 genome. Note that attP amplicons from Δint + pNBU2-int are ~1.2 kb shorter than WT amplicons because of deletion of the integrase gene. Supernatant fractions were DNase treated, eliminating all contaminating host genomic DNA, as demonstrated by lack of amplification of a host marker gene (16S rRNA). Despite an apparent size shift of BVU_RS14350 amplicons from the pellets and supernatants, Sanger sequencing validated that the products have no apparent insertions (Figure S1B). PCR amplicons were visualized by agarose gel electrophoresis alongside the New England Biolabs (NEB) 1-kb DNA ladder (BVU_RS14350, attP) or GeneRuler Express DNA ladder (16S rRNA); numbers are in kilobases.
To confirm that the release of DNase-protected BV01 genomes from host cells is a phage-encoded process, we sought to identify the phage integrase. BV01 encodes three genes with integrase domains (PF00589). We hypothesized that the gene BVU_RS14130, adjacent to the phage attachment site, was the most likely candidate for catalyzing integration and excision of BV01, as is common in phages (Casjens, 2003). A BVU_RS14130 deletion mutant (Δint) was constructed (Figure 1A), and its activity was assayed by PCR of paired cell pellets and culture supernatants (Figure 2B). Phage DNA was not detected in the supernatants of the Δint strain. Furthermore, the Δint mutant did not yield an amplicon for circularization of BV01. Expression of the int gene in trans complements the circularization and release phenotypes (Figure 2B). These results demonstrate that the integrase encoded by BVU_RS14130 is necessary for phage excision, circularization, and release from the host. Furthermore, they suggest that BV01 is an intact prophage that directs its own mobilization.
Lysogeny with BV01 Alters the Host Transcriptome
We hypothesized that lysogeny with a prophage such as BV01 could alter the activities of the B. vulgatus host. RNA sequencing (RNA-seq) of the B. vulgatus WT lysogen and the isogenic cured lysogen was performed to identify transcripts differentially regulated in response to lysogeny (Figure 3). Our expectation was that if prophage BV01 modulates bacterial activities, then some of those changes would be apparent as changes in host gene expression.
Figure 3. Differential Regulation of the Host Transcriptome in Response to BV01 Lysogeny.

RNA-seq was performed in triplicate on the B. vulgatus WT lysogen, cured lysogen (ΔBV01), and tspO deletion cured lysogen (ΔBV01ΔtspO) strains at early log phase.
(A) Count of differentially regulated transcripts compared with the WT B. vulgatus lysogen (fold change ≥ 2, q ≤ 0.01).
(B) Chromosomal location of the differentially expressed genes (q ≤ 0.01). Each dot represents a differentially expressed transcript on a log2 scale; genes below the 2-fold change cutoff (yellow) and within the BV01 prophage (blue) are not shown. Positive fold change values correspond to ratios above 1 for each comparison.
(C) Functional assignment of 115 genes differentially expressed between WT and cured lysogen strains (fold change ≥ 2, q ≤ 0.01). The locations of tspO and two bile salt hydrolases (BSHs) are indicated. General functions were assigned using the Clusters of Orthologous Groups (COGs); full functional categories are listed in Table S5. Transcripts that are not differentially expressed in other strain comparisons are shown in black. tspO-dependent transcripts are marked with green circles on the right.
Analysis of the RNA-seq data revealed 115 host transcripts differentially regulated in response to lysogeny with BV01 (Figure 3A), 103 of which (89%) were upregulated in the cured lysogen (Table S5). Resequencing of mutant genomes did not reveal any secondary mutations likely to contribute to this global transcriptional response (Tables S2 and S3). These transcriptional changes occur across the host genome (Figure 3B). Functional analysis of these transcripts revealed that most function in metabolism and cellular processes and signaling (Figure 3C). However, pathway analysis using the Kyoto Encyclopedia of Genes and Genomes Pathway Database (Kanehisa and Goto, 2000) failed to yield pathway-level differences, which may be reflective of the level of annotation of the B. vulgatus genome. Taken together, these results indicate broad repression of a diverse array of host metabolic activities in response to prophage BV01, suggesting that it acts through one or more transcriptional regulators.
One possible explanation for the widespread transcriptomic response to BV01 lysogeny (Figure 3B) is that a phage product directly alters the transcriptional activity of host genes. BV01 encodes three candidate genes that might act in this way: a predicted phage repressor (BVU_RS14475), anti-repressor (BVU_RS14135), and sigma factor-like protein (BVU_RS14235) (Figure S1A). The putative phage repressor encoded by BVU_RS14475 is the most highly transcribed gene in the BV01 prophage, which might also interact with host promoters. Transcription of BVU_RS14135 and BVU_RS14235 is very low, so they are less likely to play a major role in the observed transcriptional response (Figure S1A). It is also possible that the observed transcriptional response to BV01 is the result of a host response to infection. A host-encoded, universal stress protein (uspA) homolog (BVU_RS16570) is upregulated in the BV01 lysogen (Table S5), but it is unknown whether that is a direct or indirect effect of infection.
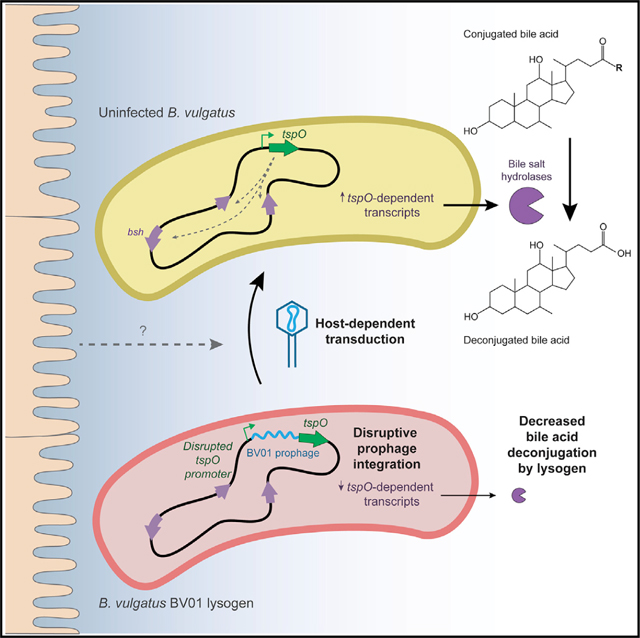
BV01 Disruption of the tspO Promoter Alters Bile Acid Metabolism
Notably, integration of BV01 at attB is associated with a 23-fold downregulation of the adjacent downstream transcript (BVU_RS14490) encoding a predicted tryptophan-rich sensory protein (TspO). We hypothesized that this downregulation is caused by disruption of the tspO promoter because BV01 integrates between the predicted promoter and the ORF (Figure 4A). A low level of expression of this gene is observed in the WT lysogen, perhaps a result of readthrough from phage transcripts. TspO is an intramembrane protein whose endogenous ligand is unknown but that is broadly implicated in metabolic regulation and stress response in other bacteria (Balsemão-Pires et al., 2011; Davey and de Bruijn, 2000; Yeliseev and Kaplan, 1999). Sequence conservation specifically in the unstructured loop (LP1) and the cholesterol recognition/interaction amino acid consensus (CRAC) sequence of TspO suggest that the B. vulgatus copy may function similar to other bacterial copies (Guo et al., 2015; Li et al., 2015a; Zeno et al., 2012; Figure 4B). Although TspO is conserved in many bacteria, archaea, and eukaryotes, it is considered an accessory protein. Indeed, not all gut-associated members of the family Bacteroidales or the genus Bacteroides encode tspO (Figure 4C). Within Bacteroides, tspO is restricted to the clade that includes B. vulgatus, and among B. vulgatus strains, TspO is highly conserved (Figure 4D). Together, this suggests that TspO might play a specialized role in the lifestyle of B. vulgatus.
Figure 4. BV01 Integration Disrupts Transcription of tspO.

(A) Transcriptional activity of the tspO gene region as it exists in the cured lysogen background. Locations of tspO, its predicted promoter elements (Bayley et al., 2000), and the empty BV01 attB are shown. RNA-seq read mapping from WT and ΔBV01 B. vulgatus was normalized to the total number of reads mapping to the genome. Three replicates are shown.
(B) Amino acid alignment of B. vulgatus TspO with known TspO sequences was generated with MUSCLE (Edgar, 2004). Identical and similar residues are colored blue and gray, respectively. Shown are TspO protein sequences from B. vulgatus (Refseq: WP_005843416.1), Bacillus cereus (GenBank: GCF80909.1), Rhodobacter sphaeroides (GenBank: AAF24291.1), Sinorhizobium meliloti (GenBank: AAF01195.1), human (RefSeq: NP_001243460.1), and rat (RefSeq: NP_036647.1). Transmembrane (TM) domains, the unstructured loop (LP), and the cholesterol recognition/interaction amino acid consensus (CRAC) sequence from R. sphaeroides TspO crystal structure are shown (Li et al., 2015a).
(C) The search for TspO homologs in the family Bacteroidales was accomplished with a BLAST-based approach using the B. cereus TspO (GenBank: GCF80909.1) as a query against a database of 154 gut-associated Bacteroidales genomes, 122 of which are in the genus Bacteroides. Genome counts are indicated.
(D) Gene tree estimated from TspO sequences across the Bacteroidales. All B. vulgatus and B. dorei genomes included in the search encode tspO. The clade for B. vulgatus TspO sequences is displayed as a polytomy; all B. vulgatus TspO sequences are at least 98% identical to each other. Numbers above branches represent bootstrap values; bootstraps over 50 are shown. The gene tree was estimated with FastTree (Price et al., 2010).
Given TspO’s important role in regulating cellular activities in other bacterial systems, we hypothesized that it may be responsible for some of the differential regulation observed in response to BV01 lysogeny. A tspO deletion mutant was constructed in the cured lysogen background (ΔBV01ΔtspO), and its transcriptome was sequenced alongside that of the WT and cured lysogen strains (Figure 3). The predicted tspO regulon extends beyond the differential expression observed in the cured lysogen (Figures 3A and 3B), suggesting that the small amount of tspO transcription in the BV01 lysogen exerts effects on the rest of the transcriptome (Table S5).
Transcripts differentially regulated between the BV01 WT lysogen and cured lysogen that returned to WT-like levels upon further deletion of tspO were classified as tspO-dependent transcripts (Figure 3C). TspO-dependent transcripts are not significantly different in a DBV01DtspO/WT comparison because tspO is absent or minimally active under both of these conditions. Of the 115 transcripts differentially regulated in response to BV01, 69 (60%) are tspO-dependent. Of these 69 tspO-dependent genes, 67 are downregulated by BV01 integration (i.e., positive fold change for ΔBV01/WT) and downregulated by tspO deletion (i.e., negative fold change for ΔBV01ΔtspO/ΔBV01), suggesting that tspO is largely involved in activation of downstream transcripts.
How TspO, a membrane protein, affects downstream transcription is unknown (Davey and de Bruijn, 2000; Liu et al., 2006; Yeliseev and Kaplan, 1999). However, we hypothesize that, in B. vulgatus, like in other bacterial models, TspO is involved in a signaling cascade, possibly by transporting unknown ligands across the membrane or by interacting with other membrane proteins (Davey and de Bruijn, 2000; Liu et al., 2006; Yeliseev and Kaplan, 1999). Consistent with TspO’s role in regulating stress, many tspO-dependent transcripts fall into the Clusters of Orthologous Groups (COG) category for post-translational modification, protein turnover, and chaperones, including several thioredoxins, peroxidases, and protein chaperones. TspO-dependent transcripts also account for the majority of metabolic genes differentially regulated in response to BV01 (Table S5).
Two tspO-dependent transcripts that are downregulated in response to lysogeny with BV01 encode putative bile salt hydrolases (BSHs; BVU_RS13575, 5.38-fold change, q < 10−100; BVU_RS20010, 6.58-fold change, q < 10−200) (Table S5). It was hypothesized that these transcriptional differences would reflect enzyme activities. To this end, B. vulgatus strains were grown in the presence of bile acids, and the deconjugation of those bile acids was measured by liquid chromatography-mass spectrometry (LC-MS) (Figure 5).
Figure 5. BV01 Alters Host Interactions with Bile Acids in a tspO-Dependent Manner.

A) Representative LC-MS traces showing that B. vulgatus deconjugates taurocholic acid (TCA) to cholic acid (CA) in the cured lysogen background (ΔBV01), but little or no CA is detectable in the WT or cured lysogen tspO deletion (ΔBV01ΔtspO) backgrounds. B. vulgatus cultures were incubated with 50 μM TCA for 16 h prior to bile acid extraction.
(B) Representative LC-MS traces showing that B. vulgatus deconjugates glycocholic acid (GCA) to CA in the ΔBV01 background but not in the WT or ΔBV01ΔtspO backgrounds. B. vulgatus cultures were incubated with 50 μM GCA for 48 h prior to bile acid extraction. Nordeoxycholic acid (norDCA) was added to a final concentration of 15 μM as an internal standard after incubation. Peaks were labeled for their metabolites based on m/z; TCA, 514.29; GCA, 464.30; CA, 407.28; norDCA, 377.27.
(C) Areas under the curve from extracted ion chromatograms were used to calculate relative intensity and averaged for CA:TCA (n = 3) and CA:GCA (n = 4) ratios. Statistical analyses were performed with one-way ANOVA; CA:TCA df = 2, F = 6.59, p = 0.03; CA:GCA df = 3, F = 103.46, p < 0.0001. Asterisks illustrate the Tukey’s honestly significant difference test p value: *p < 0.05, **p < 0.01.
The LC-MS results show that the WT B. vulgatus lysogen does not significantly deconjugate glycocholic acid (GCA) to cholic acid (CA) and may exhibit modest deconjugation of taurocholic acid (TCA) (Figure 5; Figure S2). This agrees with a previous study that showed that B. vulgatus ATCC 8482 can deconjugate TCA but not GCA over a 48-h incubation (Yao et al., 2018). Importantly, CA is clearly detectable only in the cured lysogen background, consistent with the RNA-seq data (Figures 5A and 5B). This bile acid deconjugation phenotype is ablated with deletion of tspO, further supporting our hypothesis that tspO activates transcription of BSHs via an unknown mechanism, resulting in increased enzymatic activity (Figure 5C). Finally, resequencing of mutant genomes revealed no additional mutations likely to cause the restoration of BSH activity (Tables S2 and S3).
To see whether tspO-disrupted lysogens occur in natural human gut microbiomes, read mapping from 246 healthy human gut metagenomes was performed. Starting with reads that mapped to tspO in the reverse orientation, read mates were checked for mapping to phage BV01 and its relatives (Figure 6). All samples had reads mapping to tspO, with an average of 0.0004% of metagenome reads mapping, indicating that the corresponding population of B. vulgatus and B. dorei encoding tspO is relatively abundant (Figure S3A). Incidence of tspO associated with BV01 or a related phage is also common; 13.3% of samples (n = 34) contained read pairs mapping to tspO and an adjacent prophage. Within an individual microbiome, incidence of tspO-disrupted lysogens appears to be rare, usually comprising 3% or less of the combined B. vulgatus and B. dorei population, although, for some individuals, this incidence rate can be more than 10% (Figure S3B). Together, these data suggest that BV01 and other phages’ effects on downstream phenotypes via tspO are likely quite common among humans, although these lysogens comprise the minority of the overall microbiome.
Figure 6. Salyersviridae Occur throughout the Bacteroides Genus.

(A) Bacteroides phylogeny and occurrence of the Salyersviridae att site. All duplications of the att site are associated with a putative integrated prophage. Host phylogeny was estimated by maximum likelihood from concatenated alignment of 13 core genes.
(B) Consensus att site for Salyersvirinae. The attP is duplicated upon integration of a Salyersvirinae prophage, resulting in direct repeats. The image was made with the WebLogo online tool.
(C) Phylogenomic genome-BLAST distance phylogeny implemented with the VICTOR online tool (Meier-Kolthoff and Göker, 2017) using amino acid data from all phage ORFs. For consistency, all phage genomes were annotated with MetaGeneAnnotator (Noguchi et al., 2008) implemented via VirSorter (Roux et al., 2015). Support values above branches are Genome Blast Distance Phylogeny (GBDP) pseudo-bootstrap values from 100 replications. Family (F), subfamily (Subf), genus (G), and species (Sp) were assigned by OPTSIL clustering (Gker et al., 2009; Table 1). Each leaf of the tree represents a unique phage species, except where indicated by colored boxes. Active prophages were confirmed by sequencing and/or PCR, indicated by black dots; prophages confirmed to have been inactivated by genome rearrangement are indicated by an X; prophages that were tested for activity with inconclusive results are indicated by asterisks (Figure S4).
BV01 Represents the Proposed Virus Family Salyersviridae
While searching for potential new hosts for BV01, its predicted 25-bp attB was queried against 154 gut-associated Bacteroidales genomes. It was found that all Paraprevotella and Bacteroides genomes had at least the first 21 bases of the attachment site conserved (Figure 6A). In 20 genomes, two copies of the att site were found, and all were associated with putative prophages. In all instances, the putative attL and attR sites are direct repeats, as is true for BV01. Importantly, only B. vulgatus lysogens encode tspO (Figure 4); other prophages and attB sites occur in alternate genomic contexts. Alignment of these putative prophage-associated att sites finds that the first 22 bp are always conserved and that an additional 3 bp are variable among lysogen genomes (Figure 6B).
Integration at the same att site suggests that these prophages are genetically related. To assess relatedness, the Virus Classification and Tree Building Online Resource (VICTOR) (Meier-Kolthoff and Göker, 2017) was used to build a genome tree from all of the identified prophages (Figure 6C), and OPTSIL clustering (Göker et al., 2009) was used to predict taxonomic groups, which we named Salyersviridae, Salyersvirinae, and Salyersvirus (Table 1). Taxonomic clustering also defined phage species, four of which have more than one member. The species containing BV01 also contains BV02, a prophage 99.8% nucleotide identical to BV01. BV02 occurs in B. vulgatus VPI-4245, a strain cross-listed as ATCC 8482, suggesting that the BV01 prophage is ancestral to the strain lineage. Interestingly, the phage species BX01 has representatives in two distantly related hosts, Bacteroides xylanisolvens XB1A (phage BX01) and B. thetaiotaomicron 1_1_6 (phage BT01), suggesting that at least some salyersviruses have broad host ranges. Phage BC01 was placed outside of the subfamily Salyersvirinae, which is consistent with its considerable sequence divergence (Figure S4). Further examination of the BC01 attL and attR sites found that the 22-bp consensus sequence is only a portion of the full 69-bp repeat flanking the BC01 prophage, which further supports placement of BC01 outside of Salyersvirinae. Also included in the analysis were three known Bacteroides-infecting lytic phages: B124–14, B40–8, and crAssphage. OPTSIL taxonomic clustering placed phages B124–14 and B40–8 in the family Salyersviridae. This is consistent with an independent network analysis using vConTACT 2.0 (Bin Jang et al., 2019) and existing NCBI RefSeq viral genomes, which creates a cluster consisting of only BV01, B124–14, and B40–8. Similarities between Salyersviridae prophages and the two lytic phages are not detectable at the nucleotide sequence level (Figure S4A) but is likely driven by similarities in several proteins, including homologs to the predicted lysin, excisionase, and single-stranded DNA (ssDNA) binding protein from BV01 (Figure S4B).
Table 1.
Bacteroides Phage Taxonomy as Determined by Whole-Genome Clustering
| Phage | Host | Family | Subfamily | Genus | Species |
|---|---|---|---|---|---|
|
| |||||
| BV01 | B. vulgatus ATCC 8482 | Salyersviridae | Salyersvirinae | Salyersvirus | BV01 |
| BV02 | B. vulgatus VPI-4245 | Salyersviridae | Salyersvirinae | Salyersvirus | BV01 |
| BV03 | B. vulgatus VPI-2365 | Salyersviridae | Salyersvirinae | Salyersvirus | BV03 |
| BV04 | B. vulgatus VPI-6186 | Salyersviridae | Salyersvirinae | Salyersvirus | BV04 |
| BV05 | B. vulgatus VPI-5710 | Salyersviridae | Salyersvirinae | Salyersvirus | BV05 |
| BV06 | B. vulgatus DH4096S | Salyersviridae | Salyersvirinae | Salyersvirus | BV06 |
| BF01 | B. finegoldii CL09T03C10 | Salyersviridae | Salyersvirinae | Salyersvirus | BF01 |
| BT01 | Bacteroides sp. 1_1_6 | Salyersviridae | Salyersvirinae | Salyersvirus | BX01 |
| BO01 | B. ovatus ATCC 8483 | Salyersviridae | Salyersvirinae | Salyersvirus | BO01 |
| BO02 | B. ovatus CL02T12C04 | Salyersviridae | Salyersvirinae | Salyersvirus | BO02 |
| BO03 | Bacteroides sp. 2_2_4 | Salyersviridae | Salyersvirinae | Salyersvirus | BO03 |
| BO04 | Bacteroides sp. D2 | Salyersviridae | Salyersvirinae | Salyersvirus | BO04 |
| BX01 | B. xylanisolvens XB1A | Salyersviridae | Salyersvirinae | Salyersvirus | BX01 |
| BX02 | Bacteroides sp. D1 | Salyersviridae | Salyersvirinae | Salyersvirus | BX02 |
| BX03 | Bacteroides sp. 2_1_22 | Salyersviridae | Salyersvirinae | Salyersvirus | BX02 |
| BX04 | B. ovatus SD. CC 2a | Salyersviridae | Salyersvirinae | Salyersvirus | BX02 |
| BX05 | B. xylanisolvens SD. CC 1b | Salyersviridae | Salyersvirinae | Salyersvirus | BX02 |
| BX06 | Bacteroides sp. D22 | Salyersviridae | Salyersvirinae | Salyersvirus | BX06 |
| BX07 | B. ovatus SD. CMC 3f | Salyersviridae | Salyersvirinae | Salyersvirus | BX07 |
| BC01 | B. clarus YIT 12056 | Salyersviridae | ? | ? | BC01 |
| B124–14 | B. fragilis | Salyersviridae | ? | ? | B124–14 |
| B40–8 | B. fragilis | Salyersviridae | ? | ? | B40–8 |
| crAssphage | B. intestinalis | ? | ? | ? | ? |
To check that the proposed Salyersviridae clade was comprised of active phages, we searched for evidence of activity in culture and in wastewater samples. Sequencing and PCR were used to test the activity of six additional salyersviruses (Figure S5). Phages BV02, BV05, and BV06 were found to be released from host cells in culture, whereas BV03 is likely inactivated by genomic rearrangement (Figure S5D). We did not observe activity for BV04 or BO03 from B. ovatus ATCC 8483, both of which appear complete based on synteny with intact salyersviruses. This may be the result of inactivating mutations or not being induced under the growth conditions tested (Figure S5D).
Furthermore, we used read mapping to search for evidence of all 20 Salyersviridae phages in a wastewater metavirome (Figure S5E). Reads mapped to all Salyersviridae genomes; however, it may be the result of sequence conservation within the family because reads often accumulate at the most conserved regions of each phage genome. Further, very little read mapping occurred in the distal portion of the inactivated prophage BV04, which resembles the host chromosome more than the phage sequence, indicating that virome processing removed most cellular DNA prior to sequencing (Figure S5E). De novo assembly of the individual wastewater viromes finds contigs that align with high identity but imperfectly to each Salyersviridae phage, supporting the findings seen by read mapping and suggesting that the real diversity of the family Salyersviridae is far greater than what has been observed to be integrated in cultured host genomes so far. This analysis found that phages infecting B. vulgatus are more abundant than other Salyersviridae phages based on maximum normalized read coverages. A similar comparison concludes that most individual temperate Salyersviridae phages are approximately equal in abundance to the lytic Salyersviridae phages B124–14 and B40–8 and approximately 10-fold less abundant than crAssphage in these wastewater viromes (Figure S5E). Searches for Salyersviridae phages in individual healthy human fecal metagenomes refine this conclusion, showing that, in most human samples, Salyersviridae phages are at least as abundant as crAssphage, although crAssphage can reach very high abundance in a subset of individuals (Figure S5F; Table S6). Although confirmation of most Salyersviridae activities and functions will require better sampling and in vitro testing, these results indicate that the phage family is active in human-associated communities.
DISCUSSION
Here we characterize a unique phage-host interaction between Bacteroides phage BV01 and its host B. vulgatus. We first demonstrate that BV01 is an intact prophage capable of directing its own excision and that it is transducible in vivo in a gnotobiotic mouse model. Using a combination of genetics, RNA-seq, and analytical chemistry, we show that BV01 decreases its host’s ability to deconjugate bile acids by disrupting transcription of the gene adjacent to the attB encoding a TspO homolog. Furthermore, we show that tspO disruption by phage integration is common among, but rare within, healthy human gut microbiomes and can be mediated by BV01 or its relatives. Together, these findings elucidate a complex mechanism by which a phage alters its host’s activities.
Repression of bile acid deconjugation as a consequence of BV01 integration is particularly relevant in the context of the mammalian gut. Mammals secrete conjugated primary bile acids into the small intestine, where they reach concentrations as high as 1 mM (Northfield and McColl, 1973); although the majority of bile acids secreted into the small intestine are reabsorbed, they can still accumulate to concentrations of 0.2–1 mM in the colon (Hamilton et al., 2007). Although bile acids are broadly capable of damaging lipid membranes, Bacteroides species are generally considered bile resistant (Citron et al., 1990), the mechanism of which is unknown. Bile acid deconjugation is a common activity encoded by gut-associated microbes, although its direct benefit to those microbes is unclear. Microbial modification of the bile acid pool can be linked to beneficial changes in the human host metabolism (Joyce et al., 2014; Yao et al., 2018) and varied epithelial susceptibility to viral pathogens (Grau et al., 2020). The link between BV01 and bile acid metabolism suggests a so far undescribed mechanism by which gut phages might influence mammalian host phenotypes.
Here, bile acid deconjugation in B. vulgatus is dependent on a putative TspO. Bacterial TspOs are important for regulating metabolic switches and stress regulation in at least three diverse systems (Balsemão-Pires et al., 2011; Davey and de Bruijn, 2000; Yeliseev and Kaplan, 1999), but the mechanism of action for the protein is unknown. The crystal structure of TspO shows a periplasm-facing binding pocket distinct from the intramembrane cholesterol recognition consensus sequence, which may bind or degrade porphyrins (Guo et al., 2015; Li et al., 2015a; Zeno et al., 2012). The porphyrin degrading and cholesterol transporting functions of TspO, however, have been disputed (Li et al., 2015b; Rone et al., 2012). Despite this, it is notable that cholesterol is structurally similar to bile acids, being their biosynthetic precursor. In at least one other gut-associated microbe, tspO is upregulated by bile acids, suggesting that TspO may be involved more broadly in bile acid metabolism in gut microbes (Devendran et al., 2019). The regulatory link described here between bile acid hydrolysis and TspO suggests a hypothesis where the B. vulgatus TspO might be a sensor and regulator of bile acid interactions.
Induction of BV01 from its integrated state and infection of new hosts remains enigmatic. Prophage induction is canonically linked to stress-dependent pathways, as in lambdoid phages that respond to DNA damage via RecA-dependent cleavage of the CI repressor protein (Roberts et al., 1978). It is possible that prophages in Bacteroides hosts respond to alternative stimuli, as is the case for CTnDOT, a well-studied Bacteroides conjugative transposon whose excision is inducible only by tetracycline (Moon et al., 2005). Neither DNA damage nor antibiotics induce prophage BV01 in vitro, so all experiments here relied on an apparently low rate of spontaneous prophage induction. Similarly, no infection conditions or susceptible hosts have been identified for BV01 in vitro. We demonstrate that BV01 is transducible in a gnotobiotic mouse model, suggesting that an unknown mammalian host factor is required for novel BV01 infection. Enigmatic infection dynamics may be a result of the phase-variable polysaccharide capsule; recent work suggests that heterogeneity in capsule composition hinders phage infection on population scales (Porter et al., 2020). Indeed, it has long been observed that finding phages in Bacteroides using traditional techniques is difficult or impossible for most host strains (Puig and Gironés, 1999; Salyers et al., 2000), making the host-first approach to phage discovery used here especially appealing.
Finally, phage BV01 is the first explored representative of the broad family Salyersviridae, which spans an entire host genus and includes lytic and temperate members. Salyersviridae is common and diverse among natural human samples but rare within individuals, suggesting that lysogenization may confer frequency-dependent advantages to the bacterial host. The genetic context of non-B. vulgatus Salyersviridae lysogens remains unexplored and likely underlies different phage-host interactions. The absence of tspO in these other host systems may provide the ideal background for studying more direct effects on the bacterial host. Certainly, other interactions between BV01 and its host remain to be studied, although they were overshadowed here by the enormous effects of tspO. Future studies should also examine the role of Salyersviridae phages on bacterial host fitness and evolution (Oh et al., 2019; Zhao et al., 2019) because these phages likely have important ripple effects throughout the microbiome and on the mammalian host that remain to be elucidated.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Patrick Degnan (patrick.degnan@ucr.edu).
Materials Availability
Bacterial strains and plasmids generated in this study are available upon request to the Lead Contact, Dr. Patrick Degnan (patrick.degnan@ucr.edu).
Data and Code Availability
Trimmed DNA sequencing reads from mutant genomes generated in this study are deposited in the NCBI SRA under PRJNA655911. Trimmed RNA sequencing reads from this study are deposited in the NCBI SRA under PRJNA622597. Trimmed wastewater virome DNA sequencing reads are deposited in the NCBI SRA under PRJNA622299. Ten new assembled B. vulgatus genomes are deposited in NCBI GenBank under PRJNA622758. Untrimmed phage DNA sequencing reads from culture supernatants are deposited in the NCBI SRA under PRJNA655697. All NCBI BioSample Accession numbers are listed in Table S3. The code for annotation of bacterial genomes is available at https://github.com/phdegnan/MICROBIALOMICS.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacterial Strains
A list of bacterial strains used in this work can be found in the Key Resources Table and Table S4. Escherichia coli S17–1 λ pir was grown aerobically in Lysogeny Broth (LB; 10 g/L tryptone, 5 g/L yeast extract, 5 g/L NaCl) at 37°C under agitation. Bacteroides strains were cultured anaerobically in a vinyl anaerobic chamber using 70% N2, 20% CO2, and 10% H2 gas mixture (Coy Laboratory Products, Grass Lake, MI). All Bacteroides cultures were grown on Difco Brain Heart Infusion (BHI) agar supplemented with 10% defibrinated horse blood (BHI-HB; Quad Five, Ryegate, MT), or in tryptone-yeast extract-glucose broth (TYG; 10 g/L tryptone, 5 g/L yeast extract, 2 g/L glucose, 500 mg/L cysteine free base, 100 mM pH 7.2 potassium phosphate, 20 mg/L MgSO4 ∙ 7H2O, 400 mg/L NaHCO3, 80 mg/L NaCl, 100 mg/L resazurin, 0.0008% CaCl2, 0.4 μg/L FeSO4,1 μg/L menadione, 0.2 mM histidine, 1.9 μM hematin) at 37°C. When necessary, ampicillin (100 μg/mL), gentamicin (200 μg/mL), erythromycin (25 μg/mL), 5′-flourodeoxyuridine (FUdR; 20 μg/mL), or tetracycline (2 μg/mL) were supplemented in the media.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Bacterial and Virus Strains | ||
|
| ||
| Bacteroides vulgatus ATCC 8482 | ATCC | ATCC 8482 |
| B. vulgatus Δtdk | This paper | N/A |
| B. vulgatus Δint | This paper | N/A |
| B. vulgatus ΔBV01 | This paper | N/A |
| B. vulgatus ΔBV01 ΔtspO | This paper | N/A |
| B. vulgatus ΔBV01, attN::pNBU2-bla-ermGb | This paper | N/A |
| B. vulgatus BV01-tetQ, attN::pNBU2-bla-cfx | This paper | N/A |
| B. vulgatus ΔBV01, attB::BV01-tetQ, attN::pNBU2-bla-ermGb | This paper | N/A |
| B. vulgatus VPI-4245 | Abigail Salyers | N/A |
| B. vulgatus VPI-2365 | Abigail Salyers | N/A |
| B. vulgatus VPI-6186 | Abigail Salyers | N/A |
| B. vulgatus VPI-5710 | Abigail Salyers | N/A |
| B. vulgatus DH4096S | Abigail Salyers | N/A |
| B. ovatus ATCC 8483 | ATCC | ATCC 8483 |
|
| ||
| Chemicals, Peptides, and Recombinant Proteins | ||
|
| ||
| Taurocholic acid sodium salt hydrate, ≥ 95% | Sigma-Aldrich | CAS #345909–26-4 |
| Glycocholic acid hydrate, ≥ 97% | Sigma-Aldrich | CAS #1192657–83-2 |
|
| ||
| Critical Commercial Assays | ||
|
| ||
| Nextera XT Library Preparation Kit | Illumina | Cat. #FC-131–1096 |
| Nextera XT Index Kit | Illumina | Cat. #FC-131–1002 |
| NEBNext Ultra II FS Kit for Illumina | New England Biolabs | E7805L |
| RNeasy Kit | QIAGEN | Cat. #74104 |
|
| ||
| Deposited Data | ||
|
| ||
| Mutant genome DNA sequencing, see Table S3 | This paper | PRJNA655911 |
| RNA sequencing, see Table S3 | This paper | PRJNA622597 |
| Wastewater virome DNA sequencing, see Table S3 | This paper | PRJNA622299 |
| Bacterial genomes, see Table S3 | This paper | PRJNA622758 |
| Phage DNA sequencing, see Table S3 | This paper | PRJNA655697 |
|
| ||
| Experimental Models: Organisms/Strains | ||
|
| ||
| Germfree C57BL/6J mice | UCR Gnotobiotic Facility | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| Primers for strain construction, see Table S4 | This paper | N/A |
| Primers for integrase activity assays, see Table S4 | This paper | N/A |
| Primers for detection of Salyersviridae phages, see Table S4 | This paper | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pKNOCK-bla-ermGb | Koropatkin et al., 2008 | N/A |
| pExchange-tdkBV | This paper | N/A |
| pNBU2-bla-ermGb | Koropatkin et al., 2008 | N/A |
| pNBU2-int | This paper | N/A |
| pNBU2-bla-cfx | This paper | N/A |
|
| ||
| Software and Algorithms | ||
|
| ||
| Breseq | Deatherage and Barrick, 2014 | https://github.com/barricklab/breseq |
| A5ud | Riley et al., 2017 | https://github.com/phdegnan/A5ud |
| bwa | Li and Durbin, 2009 | https://github.com/lh3/bwa |
| Microbialomics | This paper | https://github.com/phdegnan/MICROBIALOMICS |
| Rockhopper | McClure et al., 2013 | https://cs.wellesley.edu/~btjaden/Rockhopper/ |
| Trimmomatic 0.38 | Bolger et al., 2014 | http://www.usadellab.org/cms/?page=trimmomatic |
| metaSPAdes v3.13.0 | Nurk etal., 2017 | https://github.com/ablab/spades |
|
| ||
| Other | ||
|
| ||
| 30,000 MWCO Corning Spin-X UF 20 Concentrators | Sigma-Aldrich | Cat. #CLS431489 |
| Vivaflow 50R 30,000 MWCO Hydrosart Filter | Sartorius | Cat. #VF05H2 |
| Sep-Pak tC18 500 mg cartridges | Waters Corp | Cat. #WAT036790 |
Mice
Germfree C57BL/6J mice were used in this study. With no a priori reason to expect age to influence transduction rates, animals ranged from 7 weeks to nearly 12 months old. Animals were maintained in flexible plastic gnotobiotic isolators with a 12-hr light/dark cycle. Animals caged individually (n = 1, female) or in pairs of litter mates (n = 6, males) were provided with standard, autoclaved mouse chow (5K67 LabDiet, Purina) ad libitum. All experiments using mice were performed using protocols approved by the University of California Riverside Institutional Animal Care and Use Committee.
METHOD DETAILS
Cloning and Mutagenesis
All primers used to construct genetic mutants are listed in Table S4. Markerless deletion mutants in B. vulgatus were achieved by allelic exchange using a system analogous to that developed in B. thetaiotaomicron, (Koropatkin et al., 2008) and confirmed by PCR and whole genome sequencing. The tdk gene (BVU_RS09305), encoding thymidine kinase, was deleted from B. vulgatus ATCC 8482 by allelic exchange, conferring resistance to the toxic nucleotide analog FUdR. Cloning was performed as described by (Degnan et al., 2014). Briefly, the 3.5 Kb regions flanking either side of tdk were amplified with Kapa HiFi Taq MasterMix (Kapa Biosystems, Wilmington, MA) and joined by splicing overlap exchange (SOE) PCR. The SOE product was purified, restriction digested, and ligated into the suicide vector pKNOCK-bla-ermGb in E. coli, and conjugated into B. vulgatus. Single recombinant merodiploids were selected for on BHI-HB supplemented with gentamicin and erythromycin, and double recombinant deletion mutants subsequently selected for on BHI-HB with FUdR. The counterselectable suicide vector pExchange-tdkBV was constructed by amplifying tdk from B. vulgatus and cloning the tdk amplicon into pKNOCK-bla-ermGb by the same methods used to clone the SOE product above.
Subsequent deletions were accomplished similarly as described for tdk, except using pExchange-tdkBV and flanking regions of ~1 Kb to create the deletion alleles (tspO, int). For deletion of the entire BV01 prophage, an empty attachment site (attB) and the flanking 800 bp were cloned from B. vulgatus VPI-4506, which is 99.9% nucleotide identical to the analogous regions flanking BV01 in B. vulgatus ATCC 8482.
Complementation of the BV01 integrase (BVU_RS14130) was accomplished by cloning the gene and its native promoter into the integrative plasmid pNBU2-bla-ermGb, which has a single integration site in the B. vulgatus genome (attN; RefSeq: NC_009614.1; 2,710,334–2,710,346). This construct was conjugated into B. vulgatus and transconjugants selected for on BHI-HB supplemented with gentamicin and erythromycin (Degnan et al., 2014).
The BV01-tetQ strain was constructed by inserting tetQ from pNBU2-bla-tetQ immediately downstream of the stop codon of BVU_RS14265, upstream of a predicted transcriptional terminator. As was done for deletion constructs, the desired region was constructed on the pExchange-tdkBV plasmid and moved into the wild-type B. vulgatus strain by allelic exchange. First, a ~2 Kb region surrounding the BVU_RS14265 stop codon was amplified in two pieces with SOE primers designed to insert adjacent SpeI and BamHI cut sites downstream of the stop codon and ligated into pExchange-tdkBV. This construct was confirmed by Sanger sequencing before tetQ and its promoter were amplified from CTnDOT, and ligated into the SpeI and BamHI cut sites. Tetracycline was used to select for mutants, and release of BV01-tetQ phages confirmed by PCR.
Select mutant strains were confirmed by whole genome sequencing and analyzed with Breseq (Deatherage and Barrick, 2014) aligned to the wild-type B. vulgatus ATCC 8482 genome (RefSeq: NC_009614.1), and summarized in Table S2.
Bacterial Growth Rate
Triplicate 200 μL cultures of B. vulgatus strains were grown in TYG medium and their optical densities (OD) monitored at a wavelength of 600 nm at 30 min intervals. Doubling times were calculated from cells in early to mid-log phase using the least-squares fitting method on http://www.doubling-time.com/compute.php.
Genome Sequencing
Cells were pelleted from 5 mL overnight culture in TYG by centrifugation at 4,000 × g for 5 min at 4°C, resuspended in 0.5 mL TE buffer (10 mM Tris, 1 mM EDTA), and lysed by adding sodium dodecyl sulfate (SDS) and proteinase K (GoldBio, Olivette, MO) to final concentrations of 0.07% and 300 μg/mL, respectively, and incubating for 2 hr at 55°C. Cellular material was removed by washing twice in an equal volume of buffered phenol, phenol-chloroform-isoamyl alcohol (VWR, Radnor, PA), and DNA precipitated with 100% ethanol in the presence of 0.3M sodium acetate at −20°C overnight. DNA pellets were washed with 70% ethanol, dried, and resuspended in TE buffer.
Phage DNA was prepared from overnight TYG culture supernatants collected after centrifugation and filtration with a 0.22 μm filter and concentrated by centrifugation with 30,000 MWCO Corning Spin-X UF 20 Concentrators (Corning, NY) or by tangential flow filtration with a Vivaflow 50R 30,000 MWCO Hydrosart filter (Sartorius, Gottingen, Germany). Supernatants were treated with 200 μg/mL DNase I and 1 μg/mL RNase A for 1 hr at room temperature to remove unprotected DNA and RNA. Virions were disrupted with 1% SDS and 1 mg/mL proteinase K for 2 hr at 55°C. DNA was further isolated using the same phenol-chloroform-isoamyl alcohol extraction and ethanol precipitation procedures as for cellular DNA.
DNA libraries were constructed with the Nextera XT Library Preparation Kit and Index Kit (Illumina, San Diego, CA) or the NEBNext Ultra II FS kit for Illumina (New England Biolabs, Ipswich, MA). DNA libraries were pooled and sequenced on an Illumina HiSeq5000, HiSeq2500 and MiSeq instruments. Fastq files were generated from demultiplexed reads with bcl2fastq Conversion Software (Illumina, San Diego, CA). Reads were trimmed and assembled using the A5ud pipeline (Riley et al., 2017). Sequencing methods and assembly data are summarized in Table S3. Reads were mapped to the B. vulgatus ATCC 8482 reference genome (RefSeq: NC_009614.1) using bwa in paired end mode (Li and Durbin, 2009) with default parameters to generate Figure 1A.
Genome Annotation
Annotation of cellular genomes was accomplished with a custom Perl script (https://github.com/phdegnan/MICROBIALOMICS) that calls protein coding genes with Prodigal (Hyatt et al., 2010) and RNA coding genes with tRNAscan-SE (Lowe and Eddy, 1997), Rnammer (Lagesen et al., 2007) and Infernal (Nawrocki and Eddy, 2013). Functional predictions are assigned by searching against Kyoto Encyclopedia of Genes and Genomes (Kanehisa and Goto, 2000), Cluster of Orthologous Genes (Tatusov et al., 2003), Pfam (Bateman et al., 2004), and TIGRFAM (Haft et al., 2003) databases, and subCELlular LOcalization predictor (Yu et al., 2014) is used to predict cellular localization.
For phage genomes, open reading frames were called by Prodigal and the gene calling tool within Artemis (Carver et al., 2012). Functional predictions were made as above except with relaxed search parameters (cut_nc in hmmscan), plus using Basic Local Search Alignment Tool (Mount, 2007) with the GenBank virus database (Benson et al., 2009), Phyre2 (Kelley et al., 2015) to identify conserved protein folds, SignalP 5.0 (Almagro Armenteros et al., 2019) to predict signal peptides, and iVireons (Seguritan et al., 2012) to predict structural proteins, and manually comparing and combining results.
In Vivo Transduction
Individually antibiotic resistance marked bacterial strains were grown for ~20h in TYG medium with appropriate antibiotics and frozen at −80°C in anaerobic cryovials. Cell viability was tested by plating and viable CFU counts were used to combine equal parts of the B. vulgatus cured lysogen tagged with pNBU2-bla-ermGb and B. vulgatus BV01-tetQ tagged with pNBU2-bla-cfx. Approximately 4 × 106 CFUs of the combined strains were administered to each animal by oral gavage. Fecal samples were collected on days 1, 3, 7 and 11 from each animal. Fecal pellets were processed by adding 500μl of TYG+20% glycerol to each tube and vigorously shaking in a beadbeater without beads for 1 m 30 s. Fecal slurries were spun down at 2,000 x g for 1 s, followed by serial dilution on selective media (BHI+Tet+Gn, BHI+Erm+Gn, BHI+Erm+Tet+Gn) to determine CFUs. Animals were sacrificed on day 11 following final fecal collection.
Integrase Activity Assays
Integrase activity was assayed through PCR of DNase-treated supernatant DNA. Briefly, free phage DNA was prepared as for DNA sequencing, and amplified with Kapa HiFi Taq MasterMix with primers specific to BV01 (BVU_RS14350) or spanning the circularized attP (Table S4). Free phage DNAs were checked for the presence of contaminating cellular DNA by amplifying the 16S rRNA gene with universal primers. Amplicons were cleaned with a QIAGEN PCR Cleanup kit (Hilden, Germany) and run on an agarose gel in 0.5X Tris-borate-EDTA buffer at 70V alongside 1 Kb ladder (New England BioLabs, Ipswich, MA) or GeneRuler Express DNA Ladder (Thermo Scientific, Waltham, MA) and stained with GelRed (VWR, Radnor, PA). Amplicons generated with attP-flanking primers were sequence confirmed by Sanger sequencing performed by ACGT, Inc (Wheeling, IL).
Transcriptomics
B. vulgatus was grown overnight in 5 mL TYG medium. Each culture was pelleted (4,000 × g for 5 min at 4°C), supernatant decanted, and washed in an equal volume of TYG three times. Cells were normalized to an OD600 of ~0.3 (~2.9×109 CFU/mL) and used to inoculate cultures in 10 mL TYG at a final dilution of 1:1000 in biological triplicate. Cell growth was monitored and cells were harvested at an OD600 of ~0.4 (3.9×109 CFU/mL). Total RNA was prepared with a QIAGEN RNeasy kit (Hilden, Germany) and treated on-column with RNase-free DNase (QIAGEN, Hilden, Germany). RNA was quantitated with a Qubit 2.0 fluorometer (Thermo Fisher, Waltham, MA) and stored at −80°C.
RNA was submitted to the W. M. Keck Center for Comparative and Functional Genomics at the University of Illinois at Urbana-Champaign for quality analysis, rRNA depletion with the RiboZero Bacteria kit (Illumina, San Diego, CA), library construction with the TruSeq Stranded mRNAseq Sample Prep kit (Illumina, San Diego, CA), and sequencing on an Illumina NovaSeq 6000 with the NovaSeq S4 reagent kit. Fastq files of demultiplexed reads were prepared with the bcl2fastq v2.20 Conversion Software (Illumina, San Diego, CA).
RNaseq reads were quality filtered and trimmed with trim_galore v0.4.4 (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/). Rockhopper (McClure et al., 2013) was used to calculate individual transcript Expression and pairwise q-values. Fold-change was calculated in Excel as (Expression A)/(Expression B) if A > B, or as –(Expression B)/(Expression A) if B > A. Differentially expressed transcripts between the isogenic mutants were defined as those ≥ 2-fold change and q ≤ 0.01. Trimmed reads were mapped to the B. vulgatus ATCC 8482 reference genome (RefSeq: NC_009614.1) using bwa in single end mode (Li and Durbin, 2009) with default parameters to generate coverage curves.
Bile Salt Deconjugation Assay and LC-MS
B. vulgatus strains were inoculated in TYG liquid supplemented with 50 μM glycocholic acid (GCA) or 50 μM taurocholic acid (TCA) and allowed to grow for 16 or 48 hr, respectively. B. vulgatus ATCC 8482 had previously been shown to completely hydrolyze TCA in 48 hr (Yao et al., 2018); the 16 hr time point was chosen to ensure incomplete hydrolysis to better detect increased activity. B. vulgatus ATCC 8482 was also unable to hydrolyze GCA in 48 hr (Yao et al., 2018); the 48 hr time point was chosen to ensure that any increase in hydrolysis would be apparent. Grown cultures were brought to a pH 2.0–3.0 with 10 N hydrochloric acid, centrifuged for 5 min at 4,000 x g, and the pellets discarded. Bile acids were isolated by solid phase extraction over Sep-Pak tC18 500 mg cartridges (Waters Corp., Milford, MA). Cartridges were preconditioned by serial washes with 6 mL hexane, 3 mL acetone, 6 mL methanol, and 6 mL water (pH = 3.0). Acidified supernatants were loaded before washing with 3 mL 40% methanol. The column was allowed to dry, then bile acids eluted in 3 mL methanol. Samples were evaporated under nitrogen, resuspended in 20 μL methanol, and centrifuged before analysis by liquid chromatography-mass spectroscopy (LC-MS).
LC-MS for all samples was performed on a Waters Aquity UPLC coupled with a Waters Synapt G2-Si ESI MS. Chromatography was performed using a Waters Cortecs UPLC C18 column (1.6 μm particle size) (2.5 mm x 50 mm) with a column temperature of 40°C. Samples were injected at 1 μl. Solvent A consisted of 95% water, 5% acetonitrile, and 0.1% formic acid. Solvent B consisted of 95% acetonitrile, 5% water, and 0.1% formic acid. The initial mobile phase was 90% Solvent A, 10% Solvent B and increased linearly until the gradient reached 50% Solvent A and 50% Solvent B at 7.5 min. Solvent B was increased linearly again until it was briefly 100% at 8.0 min until returning to the initial mobile phase (90% Solvent A, 10% Solvent B) over the next 2 min. The total run was 10 min with a flow rate of 10 μL/min. MS was performed in negative ion mode. Nebulizer gas pressure was maintained at 400°C and gas flow was 800 L/hour. The capillary voltage was set at 2,000 V in negative mode. MassLynx was used to visualize and analyze chromatographs, including calculating areas under the curve (AUC). Ratios of AUCs are presented in Figure 5C.
Taxonomic nomenclature
The family, subfamily, and generic viral taxa names were chosen to honor the microbiologist Abigail A. Salyers, who made significant contributions to understanding the functions and genetics of human gut anaerobes and the importance of their mobile genetic elements.
Wastewater collection, processing, and viromics
From the Urbana and Champaign Sanitary District Northeast Plant (Urbana, IL), 1 L of unprocessed wastewater was collected at each of three time points: May 25, 2016, June 23, 2016, and October 3, 2016.
Wastewater samples were transported on ice, immediately centrifuged at 2,500 × g for 10 min at 4°C and filtered through a 0.4 μm polyethersulfone filter to remove large particulate and cellular matter. The sample was split into three aliquots and processed three ways. One aliquot was not processed further (F). Another aliquot was filtered a second time through a 0.22 μm polyethersulfone filter (DF). The last aliquot was washed three times with an equal volume of chloroform (FC). All aliquots were concentrated 100-fold and virome DNA was isolated from each as described for genome sequencing of phages.
DNA libraries of virome DNA were prepared using the same methods as described for phage genome sequencing and were sequenced on an Illumina HiSeq 2500 sequencer with a HiSeq v4 SBS sequencing kit (Illumina, San Diego, CA) producing 2 × 160-bp paired-end reads. Fastq files of demultiplexed reads were generated with the bcl2fastq v2.17.1.14 Conversion Software (Illumina, San Diego, CA). Reads were trimmed and quality filtered using Trimmomatic 0.38 (Bolger et al., 2014) and assembled with metaSPAdes v3.13.0 using default parameters (Nurk et al., 2017). Sequencing and assembly data for wastewater viromes is summarized in Table S4. Read mapping to phage genomes was performed with bwa in paired end mode (Li and Durbin, 2009).
Human Microbiome Project Healthy Human Subjects Study samples were downloaded with portal_client (Table S6). Read mapping was performed with bwa (Li and Durbin, 2009).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical tests, number of events quantified, standard deviation of the mean, and statistical significance is reported in figure legends. ANOVA and Tukey HSD tests were run on http://vassarstats.net/anova1u.html.
Supplementary Material
Highlights.
Integration of BV01 in the tspO promoter causes genome-wide transcriptome change
B. vulgatus bile salt hydrolase (BSH) activity is repressed by BV01 integration
Transduction of BV01 appears to be host dependent because it is only observed in vivo
Phage BV01 is a member of the Salyersviridae phage family
ACKNOWLEDGMENTS
We thank Nadja Shoemaker and Abigail Salyers for access to an impressive collection of Bacteroides isolates, Ken Ringwald for critical review of the manuscript, Jim Imlay for insightful discussions regarding metabolism and stress, Alvaro Hernandez and Chris Wright for DNA and RNA sequencing, Bruce Rabe for aid with wastewater collection, Jonathan Mitchell for maintenance and animal care at the UCR vivarium, Jericho Ortanez for assistance with construction of pNBU2-bla-cfx, and Nicholas Zanghi for assistance with construction of the Dint mutant. This research was supported by initial complement funding to P.H.D. from the University of Illinois at Urbana-Champaign (UIUC) and the University of California, Riverside (UCR), and D.E.C. was supported by the UIUC Department of Microbiology. L.K.L. was supported by a National Science Foundation graduate research fellowship. Gnotobiotic mouse work and A.H. were supported by National Institute of General Medical Sciences grant R35GM124724. R.J.W. is supported by the Allen Foundation with an Allen Distinguished Investigator Award.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests. Received: April 10, 2020
SUPPORTING CITATIONS
The following references appear in the Supplemental Information: Caporaso et al. (2011), Eggerth and Gagnon (1933), Johnson (1978), Shoemaker et al. (2001), Simon et al. (1983), Whittle et al. (2001).
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108142.
REFERENCES
- Abeles SR, Ly M, Santiago-Rodriguez TM, and Pride DT (2015). Effects of long term antibiotic therapy on human oral and fecal viromes. PLoS ONE 10, e0134941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almagro Armenteros JJ, Tsirigos KD, Sønderby CK, Petersen TN, Winther O, Brunak S, von Heijne G, and Nielsen H. (2019). SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol 37, 420–423. [DOI] [PubMed] [Google Scholar]
- Anantharaman K, Duhaime MB, Breier JA, Wendt KA, Toner BM, and Dick GJ (2014). Sulfur oxidation genes in diverse deep-sea viruses. Science 344, 757–760. [DOI] [PubMed] [Google Scholar]
- Balsemão-Pires E, Jaillais Y, Olson BJ, Andrade LR, Umen JG, Chory J, and Sachetto-Martins G. (2011). The Arabidopsis translocator protein (AtT-SPO) is regulated at multiple levels in response to salt stress and perturbations in tetrapyrrole metabolism. BMC Plant Biol. 11, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman A, Coin L, Durbin R, Finn RD, Hollich V, Griffiths-Jones S, Khanna A, Marshall M, Moxon S, Sonnhammer ELL, et al. (2004). The Pfam protein families database. Nucleic Acids Res. 32, D138–D141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baugher JL, Durmaz E, and Klaenhammer TR (2014). Spontaneously induced prophages in Lactobacillus gasseri contribute to horizontal gene transfer. Appl. Environ. Microbiol 80, 3508–3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayley DP, Rocha ER, and Smith CJ (2000). Analysis of cepA and other Bacteroides fragilis genes reveals a unique promoter structure. FEMS Microbiol. Lett 193, 149–154. [DOI] [PubMed] [Google Scholar]
- Bensing BA, Siboo IR, and Sullam PM (2001). Proteins PblA and PblB of Streptococcus mitis, which promote binding to human platelets, are encoded within a lysogenic bacteriophage. Infect. Immun 69, 6186–6192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, and Sayers EW (2009). GenBank. Nucleic Acids Res. 37, D26–D31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger P, Kouzel IU, Berger M, Haarmann N, Dobrindt U, Koudelka GB, and Mellmann A. (2019). Carriage of Shiga toxin phage profoundly affects Escherichia coli gene expression and carbon source utilization. BMC Genomics 20, 504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bin Jang H, Bolduc B, Zablocki O, Kuhn JH, Roux S, Adriaenssens EM, Brister JR, Kropinski AM, Krupovic M, Lavigne R, et al. (2019). Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat. Biotechnol 37, 632–639. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonanno L, Petit M-A, Loukiadis E, Michel V, and Auvray F. (2016). Heterogeneity in induction level, infection ability, and morphology of Shiga toxin-encoding phages (Stx Phages) from dairy and human Shiga toxin-producing Escherichia coli O26:H11 isolates. Appl. Environ. Microbiol 82, 2177–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondy-Denomy J, and Davidson AR (2014). When a virus is not a parasite: the beneficial effects of prophages on bacterial fitness. J. Microbiol 52, 235–242. [DOI] [PubMed] [Google Scholar]
- Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, and Knight R. (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 108 (Suppl 1), 4516–4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey JN, Mettert EL, Fishman-Engel DR, Roggiani M, Kiley PJ, and Goulian M. (2019). Phage integration alters the respiratory strategy of its host. eLife 8, e49081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver T, Harris SR, Berriman M, Parkhill J, and McQuillan JA (2012). Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 28, 464–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casjens S. (2003). Prophages and bacterial genomics: what have we learned so far? Mol. Microbiol 49, 277–300. [DOI] [PubMed] [Google Scholar]
- Chen YY, Wang JT, Lin TL, Gong YN, Li TH, Huang YY, and Hsieh YC (2019). Prophage excision in Streptococcus pneumoniae serotype 19A ST320 promote colonization: Insight into its evolution from the ancestral clone Taiwan 19F-14 (ST236). Front. Microbiol 10, 205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citron DM, Baron EJ, Finegold SM, and Goldstein EJ (1990). Short prereduced anaerobically sterilized (PRAS) biochemical scheme for identification of clinical isolates of bile-resistant Bacteroides species. J. Clin. Microbiol 28, 2220–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne MJ, Zitomersky NL, McGuire AM, Earl AM, and Comstock LE (2014). Evidence of extensive DNA transfer between Bacteroidales species within the human gut. MBio 5, e01305–e01314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen TW, Schofield WB, Barry NA, Putnam EE, Rundell EA, Trent MS, Degnan PH, Booth CJ, Yu H, and Goodman AL (2015). Antimicrobial peptide resistance mediates resilience of prominent gut commensals during inflammation. Science 347, 170–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey ME, and de Bruijn FJ (2000). A homologue of the tryptophan-rich sensory protein TspO and FixL regulate a novel nutrient deprivation-induced Sinorhizobium meliloti locus. Appl. Environ. Microbiol 66, 5353–5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deatherage DE, and Barrick JE (2014). Identification of mutations in laboratory-evolved microbes from next-generation sequencing data using breseq. Methods Mol. Biol 1151, 165–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degnan PH, Taga ME, and Goodman AL (2014). Vitamin B12 as a modulator of gut microbial ecology. Cell Metab. 20, 769–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devendran S, Shrestha R, Alves JMP, Wolf PG, Ly L, Hernandez AG, Méndez-García C, Inboden A, Wiley J, Paul O, et al. (2019). Clostridium scindens ATCC 35704: Integration of nutritional requirements, the complete genome sequence, and global transcriptional responses to bile acids. Appl. Environ. Microbiol 85, e00052–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutilh BE, Cassman N, McNair K, Sanchez SE, Silva GGZ, Boling L, Barr JJ, Speth DR, Seguritan V, Aziz RK, et al. (2014). A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat. Commun 5, 4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggerth AH, and Gagnon BH (1933). The Bacteroides of human feces. J. Bacteriol 25, 389–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Kaoutari A, Armougom F, Gordon JI, Raoult D, and Henrissat B. (2013). The abundance and variety of carbohydrate-active enzymes in the human gut microbiota. Nat. Rev. Microbiol 11, 497–504. [DOI] [PubMed] [Google Scholar]
- Feiner R, Argov T, Rabinovich L, Sigal N, Borovok I, and Herskovits AA (2015). A new perspective on lysogeny: prophages as active regulatory switches of bacteria. Nat. Rev. Microbiol 13, 641–650. [DOI] [PubMed] [Google Scholar]
- Göker M, García-Blázquez G, Voglmayr H, Tellería MT, and Martín MP (2009). Molecular taxonomy of phytopathogenic fungi: a case study in Peronospora. PLoS ONE 4, e6319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman AL, McNulty NP, Zhao Y, Leip D, Mitra RD, Lozupone CA, Knight R, and Gordon JI (2009). Identifying genetic determinants needed to establish a human gut symbiont in its habitat. Cell Host Microbe 6, 279–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grau KR, Zhu S, Peterson ST, Helm EW, Philip D, Phillips M, Hernandez A, Turula H, Frasse P, Graziano VR, et al. (2020). The intestinal regionalization of acute norovirus infection is regulated by the microbiota via bile acid-mediated priming of type III interferon. Nat. Microbiol 5, 84–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Kalathur RC, Liu Q, Kloss B, Bruni R, Ginter C, Kloppmann E, Rost B, and Hendrickson WA (2015). Structure and activity of tryptophan-rich TSPO proteins. Science 347, 551–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haft DH, Selengut JD, and White O. (2003). The TIGRFAMs database of protein families. Nucleic Acids Res. 31, 371–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton JP, Xie G, Raufman J-P, Hogan S, Griffin TL, Packard CA, Chatfield DA, Hagey LR, Steinbach JH, and Hofmann AF (2007). Human cecal bile acids: concentration and spectrum. Am. J. Physiol. Gastrointest. Liver Physiol 293, G256–G263. [DOI] [PubMed] [Google Scholar]
- Han M, Yang P, Zhong C, and Ning K. (2018). The human gut virome in hypertension. Front. Microbiol 9, 3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannigan GD, Duhaime MB, Ruffin MT 4th, Koumpouras CC, and Schloss PD (2018). Diagnostic potential and interactive dynamics of the colorectal cancer virome. MBio 9, e02248–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Doria JD, and Sperandio V. (2018). Bacteriophage transcription factor Cro regulates virulence gene expression in enterohemorrhagic Escherichia coli. Cell Host Microbe 23, 607–617.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey CA, Kuhn KA, Donermeyer DL, Porter NT, Jin C, Cameron EA, Jung H, Kaiko GE, Wegorzewska M, Malvin NP, et al. (2015). Colitogenic Bacteroides thetaiotaomicron antigens access host immune cells in a sulfatase-dependent manner via outer membrane vesicles. Cell Host Microbe 17, 672–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hills RD Jr., Pontefract BA, Mishcon HR, Black CA, Sutton SC, and Theberge CR (2019). Gut microbiome: profound implications for diet and disease. Nutrients 11, 1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard-Varona C, Hargreaves KR, Abedon ST, and Sullivan MB (2017). Lysogeny in nature: mechanisms, impact and ecology of temperate phages. ISME J. 11, 1511–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu BB, Gibson TE, Yeliseyev V, Liu Q, Lyon L, Bry L, Silver PA, and Gerber GK (2019). Dynamic modulation of the gut microbiota and metabolome by bacteriophages in a mouse model. Cell Host Microbe 25, 803–814.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt D, Chen G-L, Locascio PF, Land ML, Larimer FW, and Hauser LJ (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jermyn WS, and Boyd EF (2002). Characterization of a novel Vibrio pathogenicity island (VPI-2) encoding neuraminidase (nanH) among toxigenic Vibrio cholerae isolates. Microbiology 148, 3681–3693. [DOI] [PubMed] [Google Scholar]
- Jofre J, Blanch AR, Lucena F, and Muniesa M. (2014). Bacteriophages infecting Bacteroides as a marker for microbial source tracking. Water Res. 55, 1–11. [DOI] [PubMed] [Google Scholar]
- Johnson JL (1978). Taxonomy of the Bacteroides. Int. J. Syst. Evol. Microbiol 28, 245–256. [Google Scholar]
- Joyce SA, MacSharry J, Casey PG, Kinsella M, Murphy EF, Shanahan F, Hill C, and Gahan CGM (2014). Regulation of host weight gain and lipid metabolism by bacterial bile acid modification in the gut. Proc. Natl. Acad. Sci. USA 111, 7421–7426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, and Goto S. (2000). KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, Wass MN, and Sternberg MJE (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc 10, 845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan Mirzaei M, Khan MAA, Ghosh P, Taranu ZE, Taguer M, Ru J, Chowdhury R, Kabir MM, Deng L, Mondal D, and Maurice CF (2020). Bacteriophages isolated from stunted children can regulate gut bacterial communities in an age-specific manner. Cell Host Microbe 27, 199–212.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kongari R, Rajaure M, Cahill J, Rasche E, Mijalis E, Berry J, and Young R. (2018). Phage spanins: diversity, topological dynamics and gene convergence. BMC Bioinformatics 19, 326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV, and Yutin N. (2020). The crAss-like phage group: how metagenomics reshaped the human virome. Trends Microbiol. 28, 349–359. [DOI] [PubMed] [Google Scholar]
- Koropatkin NM, Martens EC, Gordon JI, and Smith TJ (2008). Starch catabolism by a prominent human gut symbiont is directed by the recognition of amylose helices. Structure 16, 1105–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krinos CM, Coyne MJ, Weinacht KG, Tzianabos AO, Kasper DL, and Comstock LE (2001). Extensive surface diversity of a commensal microorganism by multiple DNA inversions. Nature 414, 555–558. [DOI] [PubMed] [Google Scholar]
- Kropinski AM, Mazzocco A, Waddell TE, Lingohr E, and Johnson RP (2009). Enumeration of bacteriophages by double agar overlay plaque assay. Methods Mol. Biol 501, 69–76. [DOI] [PubMed] [Google Scholar]
- Krupovic M, and Forterre P. (2011). Microviridae goes temperate: microvirus-related proviruses reside in the genomes of Bacteroidetes. PLoS ONE 6, e19893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagesen K, Hallin P, Rødland EA, Staerfeldt H-H, Rognes T, and Ussery DW (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange A, Beier S, Steimle A, Autenrieth IB, Huson DH, and Frick J-S (2016). Extensive mobilome-driven genome diversification in mouse gut-associated Bacteroides vulgatus mpk. Genome Biol. Evol 8, 1197–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leplae R, Lima-Mendez G, and Toussaint A. (2010). ACLAME: a CLAssification of Mobile genetic Elements, update 2010. Nucleic Acids Res. 38, D57–D61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Liu J, Zheng Y, Garavito RM, and Ferguson-Miller S. (2015a). Protein structure. Crystal structures of translocator protein (TSPO) and mutant mimic of a human polymorphism. Science 347, 555–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Liu J, Garavito RM, and Ferguson-Miller S. (2015b). Evolving understanding of translocator protein 18 kDa (TSPO). Pharmacol. Res 99, 404–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima-Mendez G, Van Helden J, Toussaint A, and Leplae R. (2008). Prophinder: a computational tool for prophage prediction in prokaryotic genomes. Bioinformatics 24, 863–865. [DOI] [PubMed] [Google Scholar]
- Liu J, Rone MB, and Papadopoulos V. (2006). Protein-protein interactions mediate mitochondrial cholesterol transport and steroid biosynthesis. J. Biol. Chem 281, 38879–38893. [DOI] [PubMed] [Google Scholar]
- Lowe TM, and Eddy SR (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClure R, Balasubramanian D, Sun Y, Bobrovskyy M, Sumby P, Genco CA, Vanderpool CK, and Tjaden B. (2013). Computational analysis of bacterial RNA-Seq data. Nucleic Acids Res. 41, e140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier-Kolthoff JP, and Göker M. (2017). VICTOR: genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 33, 3396–3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minot S, Sinha R, Chen J, Li H, Keilbaugh SA, Wu GD, Lewis JD, and Bushman FD (2011). The human gut virome: inter-individual variation and dynamic response to diet. Genome Res. 21, 1616–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzaei MK, and Maurice CF (2017). Ménage àtrois in the human gut: interactions between host, bacteria and phages. Nat. Rev. Microbiol 15, 397–408. [DOI] [PubMed] [Google Scholar]
- Monaco CL, Gootenberg DB, Zhao G, Handley SA, Ghebremichael MS, Lim ES, Lankowski A, Baldridge MT, Wilen CB, Flagg M, et al. (2016). Altered virome and bacterial microbiome in Human Immunodeficiency Virus-associated Acquired Immunodeficiency Syndrome. Cell Host Microbe 19, 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon K, Shoemaker NB, Gardner JF, and Salyers AA (2005). Regulation of excision genes of the Bacteroides conjugative transposon CTnDOT. J. Bacteriol 187, 5732–5741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Gallego JL, Chou S-P, Di Rienzi SC, Goodrich JK, Spector TD, Bell JT, Youngblut ND, Hewson I, Reyes A, and Ley RE (2019). Virome diversity correlates with intestinal microbiome diversity in adult monozygotic twins. Cell Host Microbe 25, 261–272.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mount DW (2007). Using the Basic Local Alignment Search Tool (BLAST). CSH Protoc. 2007, pdb.top17. [DOI] [PubMed] [Google Scholar]
- Nakayama K, Kanaya S, Ohnishi M, Terawaki Y, and Hayashi T. (1999). The complete nucleotide sequence of 4 CTX, a cytotoxin-converting phage of Pseudomonas aeruginosa: implications for phage evolution and horizontal gene transfer via bacteriophages. Mol. Microbiol 31, 399–419. [DOI] [PubMed] [Google Scholar]
- Nawrocki EP, and Eddy SR (2013). Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi H, Taniguchi T, and Itoh T. (2008). MetaGeneAnnotator: detecting species-specific patterns of ribosomal binding site for precise gene prediction in anonymous prokaryotic and phage genomes. DNA Res. 15, 387–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman JM, Handley SA, Baldridge MT, Droit L, Liu CY, Keller BC, Kambal A, Monaco CL, Zhao G, Fleshner P, et al. (2015). Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell 160, 447–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northfield TC, and McColl I. (1973). Postprandial concentrations of free and conjugated bile acids down the length of the normal human small intestine. Gut 14, 513–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurk S, Meleshko D, Korobeynikov A, and Pevzner PA (2017). meta-SPAdes: a new versatile metagenomic assembler. Genome Res. 27, 824–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogilvie LA, Bowler LD, Caplin J, Dedi C, Diston D, Cheek E, Taylor H, Ebdon JE, and Jones BV (2013). Genome signature-based dissection of human gut metagenomes to extract subliminal viral sequences. Nat. Commun 4, 2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh JH, Lin XB, Zhang S, Tollenaar SL, Özçam M, Dunphy C, Walter J, and van Pijkeren J-P (2019). Prophages in Lactobacillus reuteri are associated with fitness trade-offs but can increase competitiveness in the gut ecosystem. Appl. Environ. Microbiol 86, e01922–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick S, Blakely GW, Houston S, Moore J, Abratt VR, Bertalan M, Cerdeño-Tárraga AM, Quail MA, Corton N, Corton C, et al. (2010). Twenty-eight divergent polysaccharide loci specifying within- and amongst-strain capsule diversity in three strains of Bacteroides fragilis. Microbiology 156, 3255–3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planer JD, Peng Y, Kau AL, Blanton LV, Ndao IM, Tarr PI, Warner BB, and Gordon JI (2016). Development of the gut microbiota and mucosal IgA responses in twins and gnotobiotic mice. Nature 534, 263–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter NT, Hryckowian AJ, Merrill BD, Fuentes JJ, Gardner JO, Glowacki RWP, Singh S, Crawford RD, Snitkin ES, Sonnenburg JL, and Martens EC (2020). Phase-variable capsular polysaccharides and lipoproteins modify bacteriophage susceptibility in Bacteroides thetaiotaomicron. Nat. Microbiol 5, 1170–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MN, Dehal PS, and Arkin AP (2010). FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE 5, e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig M, and Gironés R. (1999). Genomic structure of phage B40–8 of Bacteroides fragilis. Microbiology 145, 1661–1670. [DOI] [PubMed] [Google Scholar]
- Rabinovich L, Sigal N, Borovok I, Nir-Paz R, and Herskovits AA (2012). Prophage excision activates Listeria competence genes that promote phagosomal escape and virulence. Cell 150, 792–802. [DOI] [PubMed] [Google Scholar]
- Reyes A, Wu M, McNulty NP, Rohwer FL, and Gordon JI (2013). Gnotobiotic mouse model of phage-bacterial host dynamics in the human gut. Proc. Natl. Acad. Sci. USA 110, 20236–20241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A, Blanton LV, Cao S, Zhao G, Manary M, Trehan I, Smith MI, Wang D, Virgin HW, Rohwer F, and Gordon JI (2015). Gut DNA viromes of Malawian twins discordant for severe acute malnutrition. Proc. Natl. Acad. Sci. USA 112, 11941–11946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridlon JM, Harris SC, Bhowmik S, Kang D-J, and Hylemon PB (2016). Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes 7, 22–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley AB, Kim D, and Hansen AK (2017). Genome sequence of ‘‘Candidatus Carsonella ruddii’’ strain BC, a nutritional endosymbiont of Bactericera cockerelli. Genome Announc. 5, e00236–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts JW, Roberts CW, and Craig NL (1978). Escherichia coli recA gene product inactivates phage lambda repressor. Proc. Natl. Acad. Sci. USA 75, 4714–4718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rone MB, Midzak AS, Issop L, Rammouz G, Jagannathan S, Fan J, Ye X, Blonder J, Veenstra T, and Papadopoulos V. (2012). Identification of a dynamic mitochondrial protein complex driving cholesterol import, trafficking, and metabolism to steroid hormones. Mol. Endocrinol 26, 1868–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux S, Enault F, Hurwitz BL, and Sullivan MB (2015). VirSorter: mining viral signal from microbial genomic data. PeerJ 3, e985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salyers AA, Bonheyo G, and Shoemaker NB (2000). Starting a new genetic system: lessons from Bacteroides. Methods 20, 35–46. [DOI] [PubMed] [Google Scholar]
- Salyers AA, Vercellotti JR, West SE, and Wilkins TD (1977). Fermentation of mucin and plant polysaccharides by strains of Bacteroides from the human colon. Appl. Environ. Microbiol 33, 319–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt TSB, Raes J, and Bork P. (2018). The human gut microbiome: from association to modulation. Cell 172, 1198–1215. [DOI] [PubMed] [Google Scholar]
- Seguritan V Jr., Alves N Jr., Arnoult M, Raymond A, Lorimer D, Burgin AB Jr., Salamon P, and Segall AM (2012). Artificial neural networks trained to detect viral and phage structural proteins. PLoS Comput. Biol 8, e1002657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekulovic O, and Fortier L-C (2015). Global transcriptional response of Clostridium difficile carrying the CD38 prophage. Appl. Environ. Microbiol 81, 1364–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Giardino Torchia ML, Lawson GW, Karp CL, Ashwell JD, and Mazmanian SK (2012). Outer membrane vesicles of a human commensal mediate immune regulation and disease protection. Cell Host Microbe 12, 509–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shkoporov AN, Khokhlova EV, Fitzgerald CB, Stockdale SR, Draper LA, Ross RP, and Hill C. (2018). FCrAss001 represents the most abundant bacteriophage family in the human gut and infects Bacteroides intestinalis. Nat. Commun 9, 4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker NB, Vlamakis H, Hayes K, and Salyers AA (2001). Evidence for extensive resistance gene transfer among Bacteroides spp. and among Bacteroides and other genera in the human colon. Appl. Environ. Microbiol 67, 561–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon R, Priefer U, and Pühler A. (1983). A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram negative bacteria. Nat. Biotechnol 1, 784–791. [Google Scholar]
- Sullivan MB, Coleman ML, Weigele P, Rohwer F, and Chisholm SW (2005). Three Prochlorococcus cyanophage genomes: signature features and ecological interpretations. PLoS Biol. 3, e144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartera C, and Jofre J. (1987). Bacteriophages active against Bacteroides fragilis in sewage-polluted waters. Appl. Environ. Microbiol 53, 1632–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumder R, Mekhedov SL, Nikolskaya AN, et al. (2003). The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton RF, Murphy EC, Kagawa TF, O’Toole PW, and Cooney JC (2012). The effect of environmental conditions on expression of Bacteroides fragilis and Bacteroides thetaiotaomicron C10 protease genes. BMC Microbiol. 12, 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuncil YE, Xiao Y, Porter NT, Reuhs BL, Martens EC, and Hamaker BR (2017). Reciprocal prioritization to dietary glycans by gut bacteria in a competitive environment promotes stable coexistence. MBio 8, e01068–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittle G, Hund BD, Shoemaker NB, and Salyers AA (2001). Characterization of the 13-kilobase ermF region of the Bacteroides conjugative transposon CTnDOT. Appl. Environ. Microbiol 67, 3488–3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter SE, Thiennimitr P, Winter MG, Butler BP, Huseby DL, Crawford RW, Russell JM, Bevins CL, Adams LG, Tsolis RM, et al. (2010). Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature 467, 426–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao L, Seaton SC, Ndousse-Fetter S, Adhikari AA, DiBenedetto N, Mina AI, Banks AS, Bry L, and Devlin AS (2018). A selective gut bacterial bile salt hydrolase alters host metabolism. eLife 7, e37182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeliseev AA, and Kaplan S. (1999). A novel mechanism for the regulation of photosynthesis gene expression by the TspO outer membrane protein of Rhodobacter sphaeroides 2.4.1. J. Biol. Chem 274, 21234–21243. [DOI] [PubMed] [Google Scholar]
- Young R. (2014). Phage lysis: three steps, three choices, one outcome. J. Microbiol 52, 243–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu CS, Cheng CW, Su WC, Chang KC, Huang SW, Hwang JK, and Lu CH (2014). CELLO2GO: a web server for protein subCELlular LOcalization prediction with functional gene ontology annotation. PLoS ONE 9, e99368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yutin N, Makarova KS, Gussow AB, Krupovic M, Segall A, Edwards RA, and Koonin EV (2018). Discovery of an expansive bacteriophage family that includes the most abundant viruses from the human gut. Nat. Microbiol 3, 38–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeno S, Veenman L, Katz Y, Bode J, Gavish M, and Zaaroor M. (2012). The 18 kDa mitochondrial translocator protein (TSPO) prevents accumulation of protoporphyrin IX. Involvement of reactive oxygen species (ROS). Curr. Mol. Med 12, 494–501. [DOI] [PubMed] [Google Scholar]
- Zhao S, Lieberman TD, Poyet M, Kauffman KM, Gibbons SM, Groussin M, Xavier RJ, and Alm EJ (2019). Adaptive evolution within gut microbiomes of healthy people. Cell Host Microbe 25, 656–667.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Trimmed DNA sequencing reads from mutant genomes generated in this study are deposited in the NCBI SRA under PRJNA655911. Trimmed RNA sequencing reads from this study are deposited in the NCBI SRA under PRJNA622597. Trimmed wastewater virome DNA sequencing reads are deposited in the NCBI SRA under PRJNA622299. Ten new assembled B. vulgatus genomes are deposited in NCBI GenBank under PRJNA622758. Untrimmed phage DNA sequencing reads from culture supernatants are deposited in the NCBI SRA under PRJNA655697. All NCBI BioSample Accession numbers are listed in Table S3. The code for annotation of bacterial genomes is available at https://github.com/phdegnan/MICROBIALOMICS.