

ABSTRACT
Increased mortality in COVID-19 cases is often associated with microvascular complications. We have recently shown that severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) spike protein promotes an inflammatory cytokine interleukin 6 (IL-6)/IL-6R-induced trans signaling response and alarmin secretion. Virus-infected or spike-transfected human epithelial cells exhibited an increase in senescence, with a release of senescence-associated secretory phenotype (SASP)-related inflammatory molecules. Introduction of the bromodomain-containing protein 4 (BRD4) inhibitor AZD5153 to senescent epithelial cells reversed this effect and reduced SASP-related inflammatory molecule release in TMNK-1 or EAhy926 (representative human endothelial cell lines), when cells were exposed to cell culture medium (CM) derived from A549 cells expressing SARS-CoV-2 spike protein. Cells also exhibited a senescence phenotype with enhanced p16, p21, and senescence-associated β-galactosidase (SA-β-Gal) expression and triggered SASP pathways. Inhibition of IL-6 trans signaling by tocilizumab and inhibition of inflammatory receptor signaling by the Bruton’s tyrosine kinase (BTK) inhibitor zanubrutinib, prior to exposure of CM to endothelial cells, inhibited p21 and p16 induction. We also observed an increase in reactive oxygen species (ROS) in A549 spike-transfected and endothelial cells exposed to spike-transfected CM. ROS generation in endothelial cell lines was reduced after treatment with tocilizumab and zanubrutinib. Cellular senescence was associated with an increased level of the endothelial adhesion molecules vascular cell adhesion molecule 1 (VCAM-1) and intercellular adhesion molecule 1 (ICAM-1), which have in vitro leukocyte attachment potential. Inhibition of senescence or SASP function prevented VCAM-1/ICAM-1 expression and leukocyte attachment. Taken together, we identified that human endothelial cells exposed to cell culture supernatant derived from SARS-CoV-2 spike protein expression displayed cellular senescence markers, leading to enhanced leukocyte adhesion.
IMPORTANCE The present study was aimed at examining the underlying mechanism of extrapulmonary manifestations of SARS-CoV-2 spike protein-associated pathogenesis, with the notion that infection of the pulmonary epithelium can lead to mediators that drive endothelial dysfunction. We utilized SARS-CoV-2 spike protein expression in cultured human hepatocytes (Huh7.5) and pneumocytes (A549) to generate conditioned culture medium (CM). Endothelial cell lines (TMNK-1 or EAhy926) treated with CM exhibited an increase in cellular senescence markers by a paracrine mode and led to leukocyte adhesion. Overall, the link between these responses in endothelial cell senescence and a potential contribution to microvascular complication in productively SARS-CoV-2-infected humans is implicated. Furthermore, the use of inhibitors (BTK, IL-6, and BRD4) showed a reverse effect in the senescent cells. These results may support the selection of potential adjunct therapeutic modalities to impede SARS-CoV-2-associated pathogenesis.
KEYWORDS: SARS-CoV-2, spike protein
INTRODUCTION
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the seventh coronavirus known to infect humans. Among these coronaviruses, severe acute respiratory syndrome coronavirus (SARS-CoV), Middle East respiratory syndrome coronavirus (MERS-CoV), and SARS-CoV-2 can cause severe disease (1). SARS-CoV and MERS-CoV induce a substantial cytopathic effect and dysregulation of host immune responses. Immune-mediated pathogenesis is likely to be a potential factor for severe outcome in MERS-CoV-infected patients. Infection of mononuclear phagocytes (MNPs) is abortive in SARS-CoV; however, MERS-CoV can replicate in monocytes, macrophages, and dendritic cells (2). Evidence of productive SARS-CoV-2 infection in immune cells remains to be determined. Potential mechanisms for disease progression include high rates of viral replication, which could be responsible for enhanced host cell cytolysis, and the strong production of inflammatory cytokines and chemokines by infected epithelial cells (3), which perpetuates virtual damage and excessive accumulation of monocytes, macrophages, and neutrophils. Disease severity correlates with inflammatory cytokines present in the serum. The role of SARS-CoV-2-induced excessive inflammatory responses as a factor contributing to disease severity needs to be critically examined. Acute kidney injury (AKI), cardiac damage, and abdominal pain are the most commonly reported comorbidities of COVID-19 (4, 5). SARS-CoV-2 infection may be associated with damage occurring due to specific pathogenic conditions, including cytokine release syndrome (6).
Cellular senescence is triggered by stressful insults and certain physiological processes, characterized by a prolonged and generally irreversible cell cycle arrest with secretory features, macromolecular damage, and altered metabolism (7). Primary cells normally reach replicative senescence after a limited number of population doublings (8). Senescent cells secrete an excess of soluble factors, including proinflammatory cytokines and chemokines, growth modulators, angiogenic factors, and matrix metalloproteinases (MMPs), which are collectively termed the senescence-associated secretory phenotype (SASP) (9, 10). SASP function constitutes a hallmark of senescent cells and mediates many of their pathophysiological effects.
The present study addresses effects associated with epithelial cells expressing SARS-CoV-2 spike protein and the paracrine-associated response generated in endothelial cells that leads to senescence. Results from this study identified a mechanism by which SARS-CoV-2 infection propagates the paracrine effect, suggesting potential therapeutic means to interdict relevant mechanistic steps impeding deleterious SASP function.
RESULTS
SARS-CoV-2 spike protein expression in A549 cells induces senescence markers.
Cellular senescence may play a role in poor clinical outcomes in COVID-19 patients (11). We examined whether A549 cells transfected with the full-length spike plasmid exhibit senescence marker expression. Examination by immunofluorescence of spike-transfected A549 cells showing spike protein expression showed enhanced expression of the senescence marker senescence-associated β-galactosidase (SA-β-Gal). On the other hand, a similar or reduced expression of SA-β-Gal was observed in the apparent absence of spike protein expression in cells, which may be due to a paracrine effect from spike-expressing neighboring cells. The increased expression was seen most prominently in single cells, with lesser fluorescence visible in surrounding cells (Fig. 1A). A549 cells exhibited an extremely low level of infection with SARS-CoV-2. However, lysates from virus-infected cells exhibited clearly enhanced p21 and p16 expression. Likewise, A549 cells expressing spike protein also displayed enhanced p21 and p16 markers (Fig. 1B). In a similar manner, virus-infected or spike-transfected Huh7.5 cells also displayed elevated levels of p21 and p16 (Fig. 1C).
FIG 1.

SARS-CoV-2 spike protein expression induces senescence markers in cells. Spike-transfected A549 cells exhibited rounding and enhanced senescence-associated β-galactosidase (SA-β-Gal) expression in the presence of SARS-CoV-2 spike protein (A). Virus-infected or spike protein-expressing A549 (B) or Huh7.5 (C) cells also exhibited induction of senescence markers p21 and p16, as well as SARS-CoV-2 spike protein expression. Expression level of actin in each lane from the same gel is shown as a total protein load for comparison. Experiments were performed in triplicate.
Oxidative stress is associated with the pathology of SARS-CoV-2 infection, including its amplification of cytokines (12), and reactive oxygen species (ROS) play a significant role in oxidative stress. Senescent cells are characterized by increased ROS levels. ROS generation is associated with the induction of senescence and maintenance of a viable senescent state in A549 cells (13, 14). Here, we measured intracellular ROS and observed an approximately 3-fold increase in ROS in A549 spike-transfected cells compared to levels in empty plasmid-transfected control cells (Fig. 2A). Increased production of intracellular ROS contributes to DNA damage, leading to cellular senescence. We observed an increase in the DNA damage response marker γ-H2AX in cells transfected with the viral spike gene (Fig. 2B). γ-H2AX was found to be localized to the nucleus in viral spike-transfected A549 cells (Fig. 2C). Our results indicated that SARS-CoV-2 spike protein expression induces a senescent state in spike-transfected A549 cells that is associated with the DNA damage response and increased ROS generation.
FIG 2.

SARS-CoV-2 spike protein expression generates oxidative stress in A549 cells. SARS-CoV-2 spike transfection induced reactive oxygen species (ROS) release (A), γ-H2AX expression (B), and nuclear localization of γ-H2AX (C). Experiments were performed in triplicate and results are presented as mean ± standard deviation. **, P < 0.005. DAPI, 4′,6-diamidino-2-phenylindole.
Bromodomain-containing protein 4 inhibitor represses the senescence-associated secretory phenotype.
Bromodomain-containing protein 4 (BRD4) is a novel regulator of the senescence mechanism. BRD4 expression is required in senescence immune surveillance and SASP-associated signaling (15). Spike gene transfection of A549 cells led to an enhanced expression of BRD4 (Fig. 3A). A549 cells were transfected with the SARS-CoV-2 spike protein and incubated for 48 h prior to exposure for 16 h to the BRD4 inhibitor AZD5153. Spike protein expression induced p21 and p16 senescence markers, the levels of which were reduced by the addition of AZD5153 (Fig. 3B). Spike-transfected A549 cells exhibited enhanced secretion of SASP-related inflammatory molecules like the alarmin HMGB1 and cytokines interleukin 1α (IL-1α) and IL-6. Treatment with the BRD4 inhibitor AZD5153 in spike-transfected A549 cells (as described above) reduced secretory levels of these inflammation molecules (Fig. 3C). Our results suggest that the presence of SARS-CoV-2 spike protein in A549 cells induces release of SASP-related inflammatory molecules that could be impeded using an inhibitor of BRD4.
FIG 3.

SARS-CoV-2 spike-transfected A549 cells display BRD4 induction, senescence markers, and enhanced HMGB1 and cytokine release. Spike transfection enhanced BRD4 expression (A) and induced both p21 and p16 expression, which were inhibited by the addition of AZD5153 inhibitor (B). SARS-CoV-2 spike protein expression is also shown in the corresponding panels. Transfected cells exhibited enhanced secretion of HMGB1, IL-1α, and IL-6 (C). Cytokine expression was inhibited by AZD5153. Experiments were performed in triplicate. The results are presented as mean ± standard deviation. *, P < 0.05; **, P < 0.005.
Conditioned culture medium from spike-transfected epithelial cells induces a paracrine mode of senescence in endothelial cells.
Senescent cells secrete inflammatory molecules that may induce paracrine senescence in their surrounding cells. We examined whether senescent epithelial cells generated by spike transfection stimulate paracrine senescence in nearby endothelial cells where SARS-CoV-2 spike protein expression is absent. For this, we incubated endothelial TMNK-1 cells with conditioned culture medium (CM) collected from SARS-CoV-2 spike-expressing A549 cells. We observed green fluorescence associated with SA-β-Gal expression in spike CM-treated cells (Fig. 4A). We also observed a strong expression of the senescence markers p21 and p16 from spike CM-treated TMNK-1 cells. However, incubation with CM from spike-expressing A549 cells previously treated with the BRD inhibitor AZD5153 led to a loss of enhancement of both p21 and p16 in TMNK-1 cells (Fig. 4B). We previously observed that SARS-CoV-2 spike induces senescence in Huh7.5 cells. To verify whether the inflammatory SASP molecules remaining in the CM induced a paracrine effect on endothelial cells, we performed an SA-β-Gal assay in TMNK-1 cells incubated with CM from spike-expressing Huh7.5 cells. A similar outcome was noted with SA-β-Gal expression from TMNK-1 cells upon incubation with CM collected from SARS-CoV-2 spike-expressing Huh7.5 cells treated with AZD5153 (Fig. 4C). Release of SASP molecules is a signature of the senescence mechanism. An increase in the levels of SASP release-related inflammatory molecules HMGB1, IL-1α, IL-6, and monocyte chemoattractant protein 1 (MCP-1) was also observed in spike CM-treated TMNK-1 cells (Fig. 4D). Thus, our results indicated an induction of paracrine endothelial cell senescence as a bystander effect from SARS-CoV-2 spike-expressing epithelial cells.
FIG 4.

Senescence markers in TMNK-1 following incubation with CM from SARS-CoV-2 spike protein-expressing epithelial cells. CM from spike-transfected A549 cells induced SA-β-Gal expression, shown by immunofluorescence (A), and p21/p16 protein expression, shown by Western blot (B). CM from spike-transfected Huh7.5 cells also induced SA-β-Gal expression, shown by immunofluorescence (C). The use of AZD5153 inhibitor inhibited senescence marker expression from both A549 (B) and Huh-7.5 (C) spike-transfected CM. The release of SASP-related inflammatory molecules, namely, HMGB1, IL-1α, IL-6, and MCP-1, from spike-expressing A549 CM was measured by enzyme-limited immunosorbent assay (ELISA) (D). Experiments were performed in triplicate, and results are presented as mean ± standard deviation. **, P < 0.005.
Inhibition of inflammatory receptor-mediated signaling reverses the paracrine mode of endothelial cell senescence.
We have shown here that exposure of CM from SARS-CoV-2 spike-expressing epithelial cells stimulates paracrine endothelial cell senescence due to the presence of higher levels of SASP-related inflammatory molecules. It is anticipated that the inflammatory molecules present in CM from spike-expressing cells, such as HMGB1, IL-1α, and IL-6, may bind to different receptors on the endothelial cell surface and stimulate Toll-like receptor (TLR)-mediated signaling. Stimulation of TLR signaling leads to activation of the downstream molecules Akt and p38-MAPK, and these active molecules are crucial regulators of the senescence phenomenon. We observed that exposure of TMNK-1 and EAhy926 endothelial-like cell lines to spike CM resulted in an enhancement in phosphorylation of Akt and p38-MAPK molecules (Fig. 5A and B). The Bruton’s tyrosine kinase (BTK) inhibitor zanubrutinib is used to treat B-cell malignancies and prevents a vast range of inflammatory receptor signaling, including that by the tumor necrosis factor receptor (TNFR), IRAK, and all TLRs except TLR3 (16). Treatment of TMNK-1 and EAhy926 endothelial cells with zanubrutinib prior to exposure to spike CM caused significant attenuation in Akt and p38-MAPK phosphorylation (Fig. 5A and B).
FIG 5.

Modulation of Akt and p38 activation in endothelial cells and induction of cell senescence markers by CM from viral spike-expressing A549 cells as a paracrine response. TMNK-1 or EAhy926 cells were exposed to A549 spike CM for 24 h and measured by Western blot analysis for phosphorylation of Akt, p38-MAPK (A and B), p21, and p16 (C and D). At exposure, individual wells were treated with zanubrutinib or tocilizumab, as indicated.
Our previous study revealed that exposure of CM from SARS-CoV-2 spike-expressing epithelial cells triggers STAT3 activation in TMNK-1 endothelial cells, which is blocked by treatment with tocilizumab (a monoclonal antibody that inhibits IL-6 signaling) (17). STAT3 is also one of the key regulators of the senescence mechanism. As the spike-expressing A549 CM contained HMGB1-, IL-1α-, and IL-6-like inflammatory molecules, we examined whether introduction of tocilizumab and zanubrutinib can prevent paracrine senescence generated in this manner. We observed that the elevated expression of senescence markers p21 and p16 was hindered individually by either tocilizumab or zanubrutinib treatment in TMNK-1 and EAhy926 cells incubated with spike CM (Fig. 5C and D). These results further verified that endothelial cell senescence occurs due to a paracrine effect related to SARS-CoV-2 spike senescence established in the transfected pneumocyte cell line A549.
Spike-transfected A549 culture medium induces reactive oxygen species generation in endothelial cells.
Elevated levels of ROS are commonly observed in senescent cells (13). Both intracellular and extracellular ROS have been shown to contribute to senescence induction. Here, we examined ROS production in endothelial cells exposed to CM from A549 cells expressing spike in the presence of specific inhibitors. ROS levels were increased approximately 2-fold in endothelial cells (TMNK-1 and EAhy926) exposed to A549 spike-expressing CM, in comparison to those exposed to control medium. The use of tocilizumab and zanubrutinib inhibited ROS production in endothelial cells cultured in the presence of A549 spike CM (Fig. 6A and B).
FIG 6.

A549 spike-transfected culture medium induces ROS generation in endothelial cells. ROS production in two endothelial cells (A and B) exposed to A549 spike CM was measured in the presence or absence of inhibitors. CM decreased the proliferative ability of the endothelial cells, as displayed by progressively reduced MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide] activity compared to that of control CM. Experiments were performed in triplicate. ROS levels were increased approximately 2-fold in experimental cells compared to those in mock-treated control cells. Experiments were performed in triplicate. The percentage of adhesion was determined by a release of fluorescence from human monocytes (THP-1) after 6 h of adhesion on treated TMNK-1 (C) and EAhy926 (D) cells. Experiments were performed in duplicate. The results are presented as mean ± standard deviation. *, P < 0.05.
Exposure of endothelial cells to A549 spike-transfected culture medium exhibits decreased proliferation.
Senescence is an irreversible arrest of cell proliferation while the cell maintains metabolic function. Here, we compared the proliferative capacity of endothelial cells treated with control or A549 spike CM and the effect of inhibitor treatment. A549 spike CM decreased the proliferative ability of the endothelial cell line, as displayed by progressively reduced MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide] conversion (44% on TMNK-1 and 48% on EAhy926 cells) on day 3 postexposure, compared to control CM. The use of tocilizumab and zanubrutinib restored the proliferative capacity of CM-treated cells (Fig. 6C and D).
Senescent endothelial cells express adhesion molecules for leukocyte attachment.
The adhesion molecules vascular cell adhesion molecule 1 (VCAM-1) and intercellular adhesion molecule 1 (ICAM-1) are expressed on the endothelial cell surface in response to cytokines, contributing to leukocyte attachment for initiating vasculopathy. We observed that the endothelial cell lines TMNK-1 and EAhy926 exhibit a paracrine mode of senescence state after introduction of CM from SARS-CoV-2 spike-expressing epithelial cells. We examined whether senescent endothelial cells express adhesion molecules. Our Western blot analysis showed that both TMNK-1 and EAhy926 cells express increased levels of VCAM-1 and ICAM-1 molecules after exposure to CM from SARS-CoV-2 spike-expressing A549 cells (Fig. 7A and B). These enhanced levels of VCAM-1 and ICAM-1 were reduced after rescue from senescence following treatment with tocilizumab or zanubrutinib. To further determine whether these contrasting phenotypes are seen under physiological conditions, we assessed leukocyte adhesion using a fluorometric assay on the human monocyte cell line THP-1. A significant cellular adhesion of THP-1 cells on the surface of TMNK-1 and EAhy926 cells was detected in the presence of CM from SARS-CoV-2 spike-expressing A549 cells, in association with elevated expression of the adhesion molecules (Fig. 7C and D). The loss of leukocyte adhesion is also corroborated by the reduction of VCAM-1 and ICAM-1 after treatment with tocilizumab or zanubrutinib. These results indicate that leukocyte trafficking occurs to endothelial cells in an in vitro cellular environment, which is influenced by SARS-CoV-2 spike expression and paracrine senescence in surrounding epithelial cells.
FIG 7.

Bystander senescence induces endothelial adhesiveness. Expression of adhesion molecules, VCAM-1 and ICAM-1, was analyzed by Western blot from TMNK-1 and EAhy926 cell lysates prepared after 24 h of exposure of CM from spike-expressing A549 cells with or without tocilizumab or zanubrutinib treatment (A and B). The blot in panel A was spliced to remove an extra lane, indicated by the black tooling line. The expression level of actin in each lane is shown as a total protein load control for comparison, and the background in the blot is decreased for clarity of the image. A comparative analysis of leukocyte (THP-1 cells were used as a model cell line for leukocytes) adhesion on endothelial cells (TMNK-1 and EAhy926) after 24-h exposure of CM in the presence or absence of tocilizumab or zanubrutinib is shown (C and D). Experiments were performed in triplicate, and results are presented as mean ± standard deviation. **, P < 0.005.
DISCUSSION
We previously reported a marked elevation of SASP-related inflammatory molecules IL-6, IL-1α, and HMGB-1 in the culture supernatant of SARS-CoV-2 spike-expressing epithelial cells (17). The present study revealed that SARS-CoV-2 infection or viral spike protein expression induces epithelial cell senescence and increases the levels of SA-β-Gal and p16 and p21 marker proteins. A recent study suggested that alveolar type II lung cells harboring SARS-CoV-2 exhibit senescence with a proinflammatory phenotype, in association with the presence of spike protein in COVID-19 patients (11). BRD4, one of the bromodomain and extraterminal (BET) family proteins, plays a vital role in cellular senescence, including stimulation of SASP (18). We observed that inhibition of BRD4 by AZD5153 reduces the release of SASP-related inflammatory molecules and expression of senescence associated marker proteins. A recent study also suggested that BET inhibition blocks inflammation-induced cardiac dysfunction in COVID-19 patients (19).
ROS are generated as a by-product of cellular metabolism. Accumulation of ROS causes cytostatic effects due to their ability to damage DNA, protein, and lipid molecules in cells. ROS cause a variety of lesions in DNA that lead to DNA double-strand breaks (20). Systemic oxidative stress status is raised in critically ill COVID-19 patients (21). Oxidative stress-mediated DNA damage accelerates cellular senescence (22). Our data suggest that the presence of SARS-CoV-2 spike protein generates ROS in epithelial cells. A clinical trial (ClinicalTrials registration no. NCT04377789) is continuing to assess the effect of the senolytic drug quercetin for prevention and treatment of COVID-19, indicating the perceived importance of senescence in COVID-19 pathogenesis.
Induction of inflammatory molecules is attributed to SASP function (23). Cellular senescence is a stress inducer in tissues, leading to the expansion of senescence to normal bystander cells through SASP-related inflammatory molecules (24). The above-described observations lead to further examination of whether senescent epithelial cells may generate a bystander senescence response in nearby endothelial cells. We observed that endothelial cells exposed to CM from SARS-CoV-2 spike-expressing epithelial cells exhibited sign of senescence that induced senescence markers, including SASP molecules and ROS generation. Our previous study also demonstrated that treatment of endothelial cells with CM from SARS-CoV-2 spike-expressing epithelial cells containing elevated IL-6 activates STAT3 tyrosine phosphorylation, resulting in induction of MCP-1 expression; addition of tocilizumab inhibits MCP-1 expression (17). A recent study of postmortem liver specimens of COVID-19 patients suggested the involvement of IL-6 trans signaling in endotheliopathy, increased expression of adhesion molecules, and leukocyte extravasation (25). MCP-1 is an SASP-related chemokine, and its secretion was inhibited by using tocilizumab in endothelial cells. We suggest that endothelial senescence may occur as a bystander effect of senescent epithelial cells in a paracrine manner by inflammatory receptor-based signaling. To verify, we included treatment with zanubrutinib and tocilizumab to block inflammatory receptor-based signaling. The use of zanubrutinib inhibited Akt and p38-MAPK phosphorylation, which is important in triggering the senescence mechanism. Elevated senescence markers were lowered by the treatment of these inhibitors. Thus, our results indicated that the SASP-related inflammatory molecules present in the CM from SARS-CoV-2 spike-expressing epithelial cells lead to senescence in endothelial cells and support a recent observation from another group of investigators (26). Blocking inflammatory receptor-mediated signaling prevents the paracrine effect in endothelial cells (Fig. 8). Application of tocilizumab or BTK inhibitors in severe COVID-19 cases may improve treatment strategy (27–29).
FIG 8.
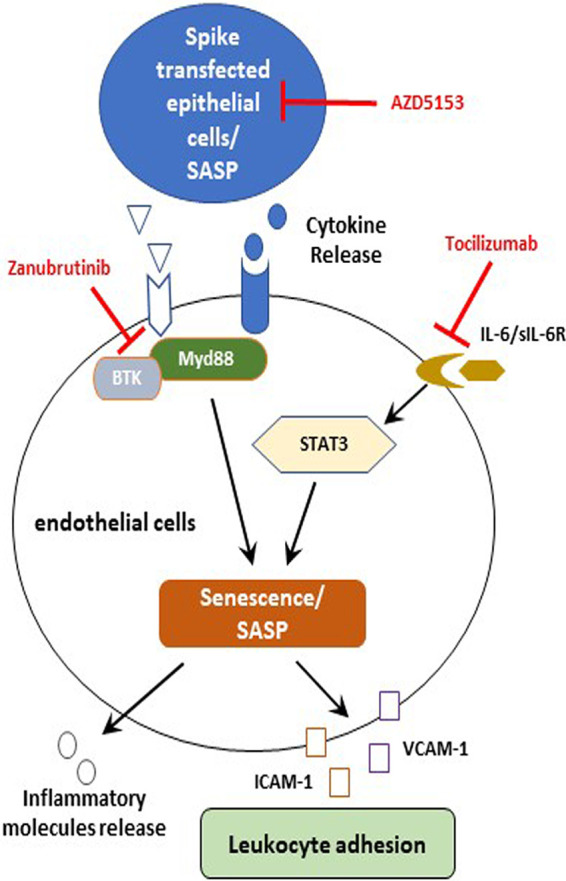
Schematic presentation of viral spike-expressing epithelial cells leading to downstream signaling for functional consequences. The presentation reflects the potential mechanisms of SARS-CoV-2 spike to promote paracrine endothelial senescence favoring leukocyte attachment. Potential inhibitory steps of these signaling events are shown by blunt arrows. sIL-6R, soluble receptor of interleukin-6.
Endothelial senescence may lead to microvascular complications by secretion of the cellular adhesion molecules VCAM-1/ICAM-1, which may cause leukocyte adhesion on the surface of the endothelium and may increase coronary blockade (30). The increased expression of endothelial cell adhesion molecules has been noted in COVID-19 patients (31). Our study suggested that the use of tocilizumab and zanubrutinib reversed the senescence effect in endothelial cells, prevented VCAM-1/ICAM-1 expression, and reduced leukocyte attachment.
A nonreplicable form of SARS-CoV-2 spike protein is used in the vaccines for immunization. Therefore, viral spike protein expression and duration of antigenic stimulation to the immune system in the injected tissue (although expected for a brief period) may not be sufficient to exhibit significant senescence or deleterious effect to adjacent endothelial cells. In this scenario, senior or elderly people who already have an accumulation of senescent cells may have a higher possibility of paracrine senescence after vaccination (32).
COVID-19 epidemiological data show that the SARS-CoV-2 infection mortality rate rises with age, particularly in individuals of advanced age. Aging is a physiological decline of organismal functions involving accumulation of cells that undergo a senescence state. Therefore, cellular senescence could hypothetically be a contributor to COVID-19 pathogenesis. Our present study revealed that SARS-CoV-2-infected epithelial cells undergo senescence, which promotes paracrine signaling to induce senescence in other tissues or cells, leading to endothelial dysfunction or microvascular complication. The reversal of senescence demonstrated in this study may be extended to clinical applications to alleviate disease severity and act as a potential adjunct therapy to reduce COVID-19 mortality.
MATERIALS AND METHODS
Cell culture and transient transfection.
Transformed human lung epithelial cells (A549), liver epithelial cells (Huh7.5), liver sinusoidal endothelial cells (TMNK-1) (kindly provided by A. Soto-Gutierrez, University of Pittsburg, Pittsburg, PA), and EAhy926 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; HyClone) containing 10% fetal bovine serum (FBS; Sigma), 100 U of penicillin/ml, and 100 μg of streptomycin/ml (Sigma). The cells were maintained in a humidified atmosphere at 37°C with 5% CO2.
Cells were subcultured in a 6-well plate to ∼60% confluence overnight and transfected with plasmid DNA (pcDNA3.1-SARS-Cov-2-Spike MC-0101087-5834, kindly provided from BEI Resources) or empty vector construct (2.5 μg/well) using Lipofectamine 3000 (Life Technologies) following the manufacturer’s instructions. Cell lysates were prepared for analyses after 72 h of transfection. Culture supernatant was collected after 72 h (A549 spike CM), with or without 16 h of incubation with inhibitor.
For infection with SARS-CoV-2, A549 cells were maintained in DMEM containing 2% FBS prior to collection. A SARS-CoV-2 isolate (USA-WA1/2020, BEI Resources) derived from an isolate sourced to Wuhan, China, was used to infect cells at a multiplicity of infection of 0.5. All live virus experiments were performed in a P3 facility approved by the Institutional Biosafety Committee. Cell lysates were collected 48 h after infection.
Senescence-associated β-galactosidase expression.
Cells were examined using the SPiDER-β-Gal cellular senescence detection kit (Dojindo). Spike-transfected A549 cells were measured for SA-β-Gal expression at 72 h posttransfection. TMNK-1 cells were measured at 24 h postexposure to A549 spike-conditioned culture medium (CM). Cells were initially exposed to bafilomycin A1 for 1 h to inhibit endogenous β-Gal activity. Cells were treated with SPiDER-β-Gal reagent and incubated for an additional 30 min prior to visualization at ×40 magnification by immunofluorescence microscope following the supplier’s protocol.
Western blot analysis.
Cell lysates were electrophoresed to resolve proteins by SDS-PAGE, transferred onto a nitrocellulose membrane, and blocked with 4% nonfat dry milk. The membrane was incubated at 4°C overnight with specific primary antibody, followed by a secondary antibody conjugated with horseradish peroxidase. The blot from the same run was reprobed with β-actin (Sigma) horseradish peroxidase (HRP)-conjugated antibody to compare protein load in each lane. Commercially available antibodies for phospho-p38 MAPK (Thr180/Tyr182), phospho-Akt (S473), p21, γ-H2AX (Cell Signaling), VCAM-1, ICAM-1, and p16 (Santa Cruz Biotechnologies) were used. Western blots were developed with the SuperSignal West Pico chemiluminescence kit (Thermo Scientific) using the manufacturer’s protocol. Densitometric scanning results are exhibited as an average of three separate experiments.
Immunofluorescence.
A549 cells were transfected by Lipofectamine 3000 with SARS-CoV-2 spike protein using an enhanced green fluorescent protein (EGFP) linker construct. After 48 h, cells were fixed, and γ-H2AX antibody (Cell Signaling) was used for immunofluorescence (IF). γ-H2AX was visualized using anti-rabbit Alexa Fluor 594 (Invitrogen). Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). Fluorescence was visualized at ×40 magnification with an immunofluorescence microscope (Leica).
Inhibitor treatment.
Cells were incubated in the presence of tocilizumab (5 mg/ml; Absolute Antibody) for 24 h (33), AZD5153 (500 ng/ml; Cayman Chemical) for 24 h (34), or zanubrutinib (500 ng/ml; SelleckChem) for 24 h (35). AZD5153 and zanubrutinib were dissolved in DMSO.
ELISA.
Cell culture medium (CM) from SARS-CoV-2 spike-transfected cells were analyzed for the presence of secreted IL-6, MCP-1, IL-1α (Sigma), and HMGB-1 (Novus Biologicals) using enzyme-limited immunosorbent assay (ELISA) kits following the manufacturer’s instructions. TMNK-1 cell culture medium exposed to A549 spike CM and CM from spike-expressing A549 cells in the presence or absence of inhibitors were also used.
ROS assay.
A549 cells were cultured after transfection of SARS-CoV-2 spike protein for 48 h and were treated with inhibitors for 24 h in a 37°C CO2 incubator. TMNK-1 cells were exposed to A549 spike CM and incubated as described above 24 h prior to measurement. Intracellular ROS were measured using a commercially available ROS detection cell-based assay kit (Cayman Chemicals).
Leukocyte adhesion assay.
Leukocyte attachment on an endothelial cell surface was performed using monocyte-derived cells (THP-1) with TMNK-1 and EAhy926 endothelial cells. Adhesion of THP-1 cells was measured using the CytoSelect leukocyte-endothelium adhesion assay kit (Cell Biolabs, Inc.) following the supplier’s protocol.
Statistical analysis.
Graph Pad Prism 7 was used to analyze the experimental data. All experiments were performed at least three times for reproducibility. The results are presented as mean ± standard deviation. Paired two-tailed t test analyses were performed to compare the mean values between the two groups. A P value of <0.05 was considered statistical significance.
ACKNOWLEDGMENTS
We thank Daniel Hoft from Infectious Diseases, James Brien from Molecular Microbiology & Immunology, and Ratna B. Ray from Pathology for suggestions on our study.
The study was supported by seed grant funding from Saint Louis University.
The funders had no role in study design, data collection and analysis, the decision to publish, or preparation of the manuscript.
We declare that we do not have conflicts of interest.
T. Patra and R. Ray designed the experiments. K. Meyer, T. Patra, and Vijayamahantesh conducted the experiments. R. Ray analyzed the results. All authors drafted the manuscript. All authors read and approved the final manuscript.
Contributor Information
Ranjit Ray, Email: rayr@slu.edu.
Tom Gallagher, Loyola University Chicago.
REFERENCES
- 1.Corman VM, Lienau J, Witzenrath M. 2019. [Coronaviruses as the cause of respiratory infections]. Internist (Berl) 60:1136–1145. (In German.) 10.1007/s00108-019-00671-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Merad M, Martin JC. 2020. Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat Rev Immunol 20:355–362. 10.1038/s41577-020-0331-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sur S, Khatun M, Steele R, Isbell TS, Ray R, Ray RB. 2021. Exosomes from COVID-19 patients carry tenascin-C and fibrinogen-beta in triggering inflammatory signals in cells of distant organ. Int J Mol Sci 22:3184. 10.3390/ijms22063184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chu KH, Tsang WK, Tang CS, Lam MF, Lai FM, To KF, Fung KS, Tang HL, Yan WW, Chan HW, Lai TS, Tong KL, Lai KN. 2005. Acute renal impairment in coronavirus-associated severe acute respiratory syndrome. Kidney Int 67:698–705. 10.1111/j.1523-1755.2005.67130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zheng X, Zhao Y, Yang L. 2020. Acute kidney injury in COVID-19: the Chinese experience. Semin Nephrol 40:430–442. 10.1016/j.semnephrol.2020.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perico L, Benigni A, Remuzzi G. 2020. Should COVID-19 concern nephrologists? Why and to what extent? The emerging impasse of angiotensin blockade. Nephron 144:213–221. 10.1159/000507305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, Campisi J, Collado M, Evangelou K, Ferbeyre G, Gil J, Hara E, Krizhanovsky V, Jurk D, Maier AB, Narita M, Niedernhofer L, Passos JF, Robbins PD, Schmitt CA, Sedivy J, Vougas K, von Zglinicki T, Zhou D, Serrano M, Demaria M. 2019. Cellular senescence: defining a path forward. Cell 179:813–827. 10.1016/j.cell.2019.10.005. [DOI] [PubMed] [Google Scholar]
- 8.Stewart SA, Weinberg RA. 2002. Senescence: does it all happen at the ends? Oncogene 21:627–630. 10.1038/sj.onc.1205062. [DOI] [PubMed] [Google Scholar]
- 9.Coppe JP, Desprez PY, Krtolica A, Campisi J. 2010. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 5:99–118. 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuilman T, Peeper DS. 2009. Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev Cancer 9:81–94. 10.1038/nrc2560. [DOI] [PubMed] [Google Scholar]
- 11.Evangelou K, Veroutis D, Foukas PG, Paschalaki K, Kittas C, Tzioufas AG, de Leval L, Vassilakos D, Barnes PJ, Gorgoulis VG. 2021. Alveolar type II cells harbouring SARS-CoV-2 show senescence with a proinflammatory phenotype. bioRxiv 10.1101/2021.01.02.424917. [DOI]
- 12.Cecchini R, Cecchini AL. 2020. SARS-CoV-2 infection pathogenesis is related to oxidative stress as a response to aggression. Med Hypotheses 143:110102. 10.1016/j.mehy.2020.110102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nair RR, Bagheri M, Saini DK. 2015. Temporally distinct roles of ATM and ROS in genotoxic-stress-dependent induction and maintenance of cellular senescence. J Cell Sci 128:342–353. 10.1242/jcs.159517. [DOI] [PubMed] [Google Scholar]
- 14.Zheng H, Huang Q, Huang S, Yang X, Zhu T, Wang W, Wang H, He S, Ji L, Wang Y, Qi X, Liu Z, Lu L. 2018. Senescence inducer shikonin ROS-dependently suppressed lung cancer progression. Front Pharmacol 9:519. 10.3389/fphar.2018.00519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tasdemir N, Banito A, Roe JS, Alonso-Curbelo D, Camiolo M, Tschaharganeh DF, Huang CH, Aksoy O, Bolden JE, Chen CC, Fennell M, Thapar V, Chicas A, Vakoc CR, Lowe SW. 2016. BRD4 connects enhancer remodeling to senescence immune surveillance. Cancer Discov 6:612–629. 10.1158/2159-8290.CD-16-0217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spaargaren M, de Rooij MF, Kater AP, Eldering E. 2015. BTK inhibitors in chronic lymphocytic leukemia: a glimpse to the future. Oncogene 34:2426–2436. 10.1038/onc.2014.181. [DOI] [PubMed] [Google Scholar]
- 17.Patra T, Meyer K, Geerling L, Isbell TS, Hoft DF, Brien J, Pinto AK, Ray RB, Ray R. 2020. SARS-CoV-2 spike protein promotes IL-6 trans-signaling by activation of angiotensin II receptor signaling in epithelial cells. PLoS Pathog 16:e1009128. 10.1371/journal.ppat.1009128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi J, Vakoc CR. 2014. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol Cell 54:728–736. 10.1016/j.molcel.2014.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mills RJ, Humphrey SJ, Fortuna PRJ, Lor M, Foster SR, Quaife-Ryan GA, Johnston RL, Dumenil T, Bishop C, Rudraraju R, Rawle DJ, Le T, Zhao W, Lee L, Mackenzie-Kludas C, Mehdiabadi NR, Halliday C, Gilham D, Fu L, Nicholls SJ, Johansson J, Sweeney M, Wong NCW, Kulikowski E, Sokolowski KA, Tse BWC, Devilee L, Voges HK, Reynolds LT, Krumeich S, Mathieson E, Abu-Bonsrah D, Karavendzas K, Griffen B, Titmarsh D, Elliott DA, McMahon J, Suhrbier A, Subbarao K, Porrello ER, Smyth MJ, Engwerda CR, MacDonald KPA, Bald T, James DE, Hudson JE. 2021. BET inhibition blocks inflammation-induced cardiac dysfunction and SARS-CoV-2 infection. Cell 184:2167–2182.e22. 10.1016/j.cell.2021.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gonzalez-Hunt CP, Wadhwa M, Sanders LH. 2018. DNA damage by oxidative stress: measurement strategies for two genomes. Current Opinion in Toxicology 7:87–94. 10.1016/j.cotox.2017.11.001. [DOI] [Google Scholar]
- 21.Pincemail J, Cavalier E, Charlier C, Cheramy-Bien JP, Brevers E, Courtois A, Fadeur M, Meziane S, Goff CL, Misset B, Albert A, Defraigne JO, Rousseau AF. 2021. Oxidative stress status in COVID-19 patients hospitalized in intensive care unit for severe pneumonia: pilot study. Antioxidants 10:257. 10.3390/antiox10020257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kurz DJ, Decary S, Hong Y, Trivier E, Akhmedov A, Erusalimsky JD. 2004. Chronic oxidative stress compromises telomere integrity and accelerates the onset of senescence in human endothelial cells. J Cell Sci 117:2417–2426. 10.1242/jcs.01097. [DOI] [PubMed] [Google Scholar]
- 23.Nelson G, Kucheryavenko O, Wordsworth J, von Zglinicki T. 2018. The senescent bystander effect is caused by ROS-activated NF-κB signalling. Mech Ageing Dev 170:30–36. 10.1016/j.mad.2017.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nelson G, Wordsworth J, Wang C, Jurk D, Lawless C, Martin-Ruiz C, von Zglinicki T. 2012. A senescent cell bystander effect: senescence-induced senescence. Aging Cell 11:345–349. 10.1111/j.1474-9726.2012.00795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McConnell MJ, Kawaguchi N, Kondo R, Sonzogni A, Licini L, Valle C, Bonaffini PA, Sironi S, Alessio MG, Previtali G, Seghezzi M, Zhang X, Lee A, Pine AB, Chun HJ, Zhang X, Fernandez-Hernando C, Qing H, Wang A, Price C, Sun Z, Utsumi T, Hwa J, Strazzabosco M, Iwakiri Y. 2021. Liver injury in COVID-19 and IL-6 trans-signaling-induced endotheliopathy. J Hepatol 10.1016/j.jhep.2021.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hui KP, Cheung MC, Lai KL, Ng KC, Ho JC, Peiris M, Nicholls JM, Chan MC. 2021. Role of epithelial-endothelial cell interaction in the pathogenesis of SARS-CoV-2 infection. Clin Infect Dis 10.1093/cid/ciab406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patra T, Ray R. 2021. IL-6 induction and signaling: horizons of COVID-19-related pathogenesis. DNA Cell Biol 40:639–642. 10.1089/dna.2021.0152. [DOI] [PubMed] [Google Scholar]
- 28.Roschewski M, Lionakis MS, Sharman JP, Roswarski J, Goy A, Monticelli MA, Roshon M, Wrzesinski SH, Desai JV, Zarakas MA, Collen J, Rose K, Hamdy A, Izumi R, Wright GW, Chung KK, Baselga J, Staudt LM, Wilson WH. 2020. Inhibition of Bruton tyrosine kinase in patients with severe COVID-19. Sci Immunol 5:eabd0110. 10.1126/sciimmunol.abd0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Treon SP, Castillo JJ, Skarbnik AP, Soumerai JD, Ghobrial IM, Guerrera ML, Meid K, Yang G. 2020. The BTK inhibitor ibrutinib may protect against pulmonary injury in COVID-19-infected patients. Blood 135:1912–1915. 10.1182/blood.2020006288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Minamino T, Miyauchi H, Yoshida T, Ishida Y, Yoshida H, Komuro I. 2002. Endothelial cell senescence in human atherosclerosis: role of telomere in endothelial dysfunction. Circulation 105:1541–1544. 10.1161/01.cir.0000013836.85741.17. [DOI] [PubMed] [Google Scholar]
- 31.Tong M, Jiang Y, Xia D, Xiong Y, Zheng Q, Chen F, Zou L, Xiao W, Zhu Y. 2020. Elevated expression of serum endothelial cell adhesion molecules in COVID-19 patients. J Infect Dis 222:894–898. 10.1093/infdis/jiaa349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kowarz E, Krutzke L, Reis J, Bracharz S, Kochanek S, Marschalek R. 2021. “Vaccine-induced Covid-19 mimicry” syndrome: splice reactions within the SARS-CoV-2 spike open reading frame result in spike protein variants that may cause thromboembolic events in patients immunized with vector-based vaccines. Res Square 10.21203/rs.3.rs-558954/v1. [DOI] [Google Scholar]
- 33.Wan S, Zhao E, Kryczek I, Vatan L, Sadovskaya A, Ludema G, Simeone DM, Zou W, Welling TH. 2014. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology 147:1393–1404. 10.1053/j.gastro.2014.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rhyasen GW, Hattersley MM, Yao Y, Dulak A, Wang W, Petteruti P, Dale IL, Boiko S, Cheung T, Zhang J, Wen S, Castriotta L, Lawson D, Collins M, Bao L, Ahdesmaki MJ, Walker G, O’Connor G, Yeh TC, Rabow AA, Dry JR, Reimer C, Lyne P, Mills GB, Fawell SE, Waring MJ, Zinda M, Clark E, Chen H. 2016. AZD5153: a novel bivalent BET bromodomain inhibitor highly active against hematologic malignancies. Mol Cancer Ther 15:2563–2574. 10.1158/1535-7163.MCT-16-0141. [DOI] [PubMed] [Google Scholar]
- 35.Tarantelli C, Zhang L, Curti E, Gaudio E, Spriano F, Priebe V, Cascione L, Arribas AJ, Zucca E, Rossi D, Stathis A, Bertoni F. 2019. The Bruton tyrosine kinase inhibitor zanubrutinib (BGB-3111) demonstrated synergies with other anti-lymphoma targeted agents. Haematologica 104:e307–e309. 10.3324/haematol.2018.214759. [DOI] [PMC free article] [PubMed] [Google Scholar]