

Abstract
Fibroblast activation protein (FAP) is a membrane protease that is highly expressed by cancer-associated fibroblasts (CAFs). FAP can modulate the tumor microenvironment (TME) by remodeling the extracellular matrix (ECM), and its overexpression on CAFs is associated with poor prognosis in various cancers. The TME is in part accountable for the limited efficacy of chimeric antigen receptor (CAR)-T cell therapy in treatment of solid tumors. Targeting FAP with CAR-T cells is one of the strategies being researched to overcome the challenges in the TME. This review describes the role of FAP in the TME and its potential as a target in CAR-T cell immunotherapy, summarizes the preclinical studies and clinical trials of anti-FAP-CAR-T cells to date, and reviews possible optimizations to augment their cytotoxic efficiency in solid tumors.
Keywords: CAR T-cells, immunotherapy, tumor microenvironment, fibroblasts, fibroblast-activating-protein, solid tumors, stroma
Introduction
Cancer immunotherapy is increasingly recognized as the fifth pillar of cancer treatment. As cancer is seen as a distorted version of our normal selves, the use of our own immune system to recognize and target cancer may render it as the closest solution to combatting cancer. Chimeric antigen receptor (CAR)-T cell therapy has stood out as one of the most promising immunotherapies for cancer, following dramatic results in treatment of hematological malignancies.1 However, research in treatment of solid tumors using CAR-T cell immunotherapies still faces many challenges, the most prominent one being overcoming the hostile tumor microenvironment (TME).
The success of CAR-T cells relies on specific tumor antigen recognition within the TME, but given the heterogeneity of mutations that occur within cancer cells, the target antigens can be masked by the TME and reduce the efficacy of CAR-T cell therapy.2 Targeting non-cancerous cells that modulate the TME is one of the strategies being explored in treatment of solid tumors. Cancer-associated fibroblasts (CAFs) are ideal candidates for this purpose. They are considered to be more genetically stable in the TME.3–6 Fibroblast activation protein (FAP) is preferentially expressed on CAFs and it is one of the biomarkers used to identify CAFs.7 This review summarizes the current literature on the role of CAFs in the TME, as well as pre-clinical and clinical studies of FAP targeting CAR-T cells as a solution to overcome the TME restrictions.
CAR-T Cell Therapies
In 1989, Gross et al. first described a CAR, that enabled T cells to recognize antigens independent of class II major histocompatibility complex (MHC) molecules. They constructed the receptor by replacing the variable (V) antigen recognition domain of T cell receptor with an antibody V domain, as the two shared a high degree of homology. As a result, the re-engineered T cell receptor consisted of antibody-like recognition domain, T cell receptor extracellular constant C domain, transmembrane domain and cytoplasmic domain. It was successfully expressed on T cell surface, and mediated antigen specific target cell killing, having recognized target cells in a non-MHC dependent manner. T cell activation was measured by the release of proinflammatory cytokine interleukin-2 (IL2).8 The construction of a functional CAR paved the way for ex vivo engineering of cytotoxic T lymphocytes for recognition of tumor or virally infected cells.
Three decades later, CAR-T cell therapy has shown remarkable efficacy in targeting of CD19-expressing B cells in hematological malignancies. The first FDA approved CAR-T cell therapy, Kymriah (tisagenlecleucel, CTL019), has achieved 12-month relapse-free survival rates of 55–59% in the treatment of acute lymphoblastic leukemia (ALL).9 Yescarta (KTE-C19 axicabtagene ciloleucel) was approved shortly after Kymriah to treat diffuse large B-cell lymphoma (DLBCL) (NCT02529813). It achieved overall response rate of 83% and complete response rate of 58% in clinical trial.10 Tecartus (brexucabtagene autoleucel, KTE-X19) is the latest FDA approved CAR-T cell therapy for the treatment of relapsed or refractory mantle cell lymphoma (MCL) (NCT03761056). Clinical trial showed 87% of patients responded to the therapy, with 62% achieving complete response (NCT02601313).11
Given the unprecedented success of CAR-T cell therapies in hematological malignancies, there have been worldwide scientific efforts to develop CAR-T cell therapies against solid tumors,12 however demonstration of their efficiency that may result in clinically approved therapies is yet to be achieved.
CAR Construct Evolution
Since their development, CARs have evolved several generations according to the structure of their endo-domains. First-generation CARs consist of a single-chain fragment variable (scFv) for antigen recognition, joined to a transmembrane domain via a spacer taken from a hinge region of IgG1; a hydrophobic alpha helix spans the membrane making up the transmembrane domain; CD3ζ-chain or FcεRIγ form the intracellular domain.13 This form of CAR could redirect T cells and initiate proliferation in pre-activated T cells but not in resting T cells, limiting the anti-tumor efficacy.14 T cell signaling in resting T cells is amplified by co-stimulatory receptor, without which the T cell becomes susceptible to a state of anergy.15 To overcome this, second-generation CARs have incorporated multiple co-stimulatory domains along with CD3ζ, with the most commonly used being 41BB or CD28/B7-1,16,17 so as to achieve full CAR-T cell activation that requires both the stimulatory and co-stimulatory signals. Once TCR binds to the antigen peptide-MHC complex, CD28 is recruited to amplify TCR signaling, which then promotes secretion of IL2, leading to enhanced proliferation, differentiation and survival.18 4-1BB is part of a tumor necrosis factor receptor superfamily (TNFRSF) that also includes OX40, CD27, CD30 and Herpesvirus entry mediator (HVEM). TCR and CD28 signaling leads to transient induction of 4-1BB via ERK and JNK signaling pathways, further enhancing T-cell proliferation, increasing cytokine secretion and preventing T-cell anergy.19 Second-generation CARs with either 41BB or CD28 co-stimulatory domain targeting CD19 has been reported to have complete response rate of up to 90% in a phase I trial for treatment of B-ALL.20,21
Third-generation CAR-T cells started to emerge when Pule et al. developed a diasialoganflisodie (GD2)-targeting chimeric receptor (14g2a) coupled to CD28, OX40 and CD3ξ. This combination showed enhanced proliferation of T cells and increased cytokine release such as IFN-γ and IL2 compared with 14g2a-CD28-ζ or 14g2a-OX40-ζ constructs.22 Efficacy of CAR-T cells was also enhanced when a prostate-specific membrane antigen (PSMA)-targeting CAR had combined CD28 and 41BB co-stimulatory domains; its superior killing capacity was suggested to have been the result of induction of Granzyme-B, GM-CSF and IFN-γ release, resembling the outcomes of physiological T cell activation.23 The majority of the ongoing clinical trials of CAR-T cell therapy use either second- or third-generation CARs.
More recent developments focused on improving CAR-T cell persistence and efficacy in the challenging solid tumor microenvironment. T cells redirected for universal cytokine-mediated killing (TRUCK) utilize therapeutic proteins (“payload”) released upon CAR-T cell target engagement. These CAR-T cells can be modified to release various cytokines such as IL12, IL15 and IL18. The inducible release of cytokines can attract and activate innate immune cells, and eliminate target cells that have escaped CAR-T cell recognition, thus reducing the chances of tumor relapse.24 Experiments on CAR-T cells releasing inducible IL18 reported superior killing activity against carcinoembryonic antigen (CEA) in a pancreatic tumor model.25 As well as releasing cytokines upon recognition of target antigen, CAR-T cells can also be engineered to modulate cytokine functions in the TME. A PSMA targeting second-generation CAR-T cell designed to co-express dominant negative TGF-β receptor (dnTGF-β RII), of which the signaling domain of the receptor was removed to reduce TGF-β mediated immunosuppressive effects in the TME, has shown improved proliferation, persistence, and enhanced anti-tumor efficacy of the CAR-T cells in vivo.26 This finding has led to an ongoing phase I clinical trial using the PSMA targeting fourth-generation CAR-T cells for the treatment of castrate-resistant prostate cancer (NCT03089203).
Target Selection for CAR-T Cell Therapy
CAR-T cells have been designed to recognize antigens independent of MHC expression, which enables recognition of a broad spectrum of antigens expressed on the cell surface, including proteins, carbohydrates and glycolipids.27 However, there are many challenges in identifying an ideal target for treatment of solid tumors.
As cancer forms following malignant transformation, it continuously evolves and proliferates. This continuous evolution results in cancer cells constantly undergoing genetic, epigenetic, transcriptomic and phenotypic changes, ultimately resulting in highly heterogeneous populations within a tumor site.28 This heterogeneity can be distinguished into inter-tumoral and intra-tumoral heterogeneity. Inter-tumoral heterogeneity exists between different patients with the same types of cancer. Individual patient genetic profile and environmental factors are the causes of inter-tumoral heterogeneity. Intra-tumoral heterogeneity refers to the difference among the tumor cells in one patient.28 Antigens selected for CAR-T cell targeting should be overexpressed on tumor cell surfaces, while exhibiting low or no expression in healthy cells. Tumor-specific antigens (TSAs) are specifically expressed on tumor cells but not on healthy tissues, however, due to the heterogeneity, it is difficult to find an antigen uniformly expressed across different patients with the same cancer or even across different tumor sites of the same patient.27 One example of TSA is epidermal growth factor receptor variant III (EGFRvIII). It is specifically expressed in the majority of patients with glioblastoma, as well as breast carcinomas, non-small cell lung carcinomas and gliomas.27,29 One example of TSA is epidermal growth factor receptor variant III (EGFRvIII), which is specifically expressed in the majority of glioblastoma, breast carcinoma, non-small cell lung carcinoma and glioma tumors.27,29 A number of glioblastoma trials have used EGFRvIII targeting CAR-T cells (NCT01454596; NCT02209376). In these, CAR-T cell treatment resulted in recurrence of tumors, due to heterogeneous expression levels of EGFRvII on tumor cells. To overcome this issue, synNotch-CAR-T cells for the treatment of glioblastoma were developed. The synNotch-CAR targets multiple antigens in the TME and utilizes prime-and-kill circuits to achieve specific and controlled killing of tumor cells.30
Tumor-associated antigens (TAAs) are overexpressed by tumor cells and, at low levels, by healthy cells. The three currently approved CAR-T cell therapies, Yescarta, Kymriah and Tecartus, target CD19 on B lymphocytes. CD19 is a biomarker for normal and neoplastic B cells, expressed in hematological malignancies such as acute lymphoblastic leukemia (ALL), chronic lymphocytic leukemia (CLL), and B cell lymphoma. Its expression is also seen in normal plasma cells,31 targeting CD19 can therefore eliminate the B-cell compartment in the immune system. However, this is tolerated by the immune system because the T cell compartment is spared,32 which is one of the reasons for the success of CAR-T cell therapies in hematological malignancies. In the case of solid tumors, the expression of TAAs on vital organs can lead to adverse effects such as on-target off-tumor toxicity or off-target off-tumor cross reactivity. A clinical trial on the safety and activity of affinity enhanced autologous T cells targeting MAGE-A3, a TAA expressed on melanoma and myeloma, resulted in two patient deaths due to cross reactivity of antigens expressed on normal cardiac tissue (NCT01350401; NCT01352286).33 Therefore, finding a suitable target for CAR-T cell therapy in the treatment of solid tumors is paramount.
Another option in choosing target antigens for CAR-T cell therapies is targeting stromal cells that support the TME rather than the tumor cells themselves. As stromal cells are genetically stable compared with tumor cells, CAR-T cells can maintain recognition, avoid tumor escape and break down the support for the tumor cells, ultimately reducing tumor growth. The role of stromal cells is further discussed in the following sections.
The Challenges of the Solid Tumor Microenvironment (TME)
The success of CAR-T cell therapy in hematological malignancies has not been replicated in the treatment of solid tumors due to a number of factors. Firstly, there is a lack of exclusive tumor-specific antigens; although most are highly expressed on tumors, they are also expressed at low levels on some healthy tissues, thus compromising target recognition specificity and increasing the likelihood of on-target off-tumor toxicity. Secondly, solid tumors display intra-tumor antigen heterogeneity, with tumor cells showing various levels of antigen expression as well as lack of expression resulting in the reduction of antigen recognition by CAR-T cells. Finally, tumors develop a highly suppressive TME that is hostile towards CAR-T cells.2
The TME is a microcosmos of physical, cellular, and chemical factors, aiming to counteract the body’s immune defenses and aid in the growth and progression of the tumor. The immunosuppressive nature of the TME is such that any immune response from infiltrated T cells is quickly dampened. The uncontrollable proliferation of tumors limits oxygen availability (hypoxia) and reduces blood supply in the microenvironment,34 which drives abnormal angiogenesis to supply nutrients and oxygen to the tumor. The newly developed blood vessels have high vascular permeability and enhanced permeation resulting in abnormal vasculature, desmoplasia, and inflammation, leading to tumor progression and therapeutic resistance. The hypoxic environment is ideal for glycolysis and lactic acid production, which results in low pH in the environment.35 Studies have shown that acidic intracellular pH in tumor cells can aid in metastasis by promoting proteolytic reactions.36
Cellular and Soluble Factors of the TME
Immune suppressive cells inhibit anti-tumor responses in the TME. These cells include Tregs, myeloid-derived suppressor cells (MDSCs) and tumor-associated macrophages (TAMs). TAMs are derived from mononuclear cells and they are the most common tumor infiltrating immune cells in the TME. They can make tumor cells resistant to therapies, promote tumor growth by promoting angiogenesis and increase tumorigenesis.37 Most importantly, various chemokines, cytokines and enzymes secreted by TAMs can directly inhibit CD4+ and CD8+ T cells function, reducing the efficacy of immunotherapy.37 MDSCs can induce cytokine release from macrophages, they are differentiated from TAMs and are known as potent suppressors of T lymphocytes.38 MDSCs and TAMs secrete suppressive factors and cytokines such as TGF-β and IL10, all of which dampen the anti-tumor response.39 TGF-β has been shown to hamper T cell proliferation through multiple pathways,40 such as blocking IL2-dependent proliferation of activated T cells, and promoting SMAD3-dependent inhibition of IL-2 promoter activity.41 TGF-β along with other tumor-derived factors such as vascular endothelial growth factor (VEGF), stromal cell-derived factor 1 and IL-10 have been known to induce Tregs in the TME.42 The Tregs, an immunosuppressive subtype of CD4 T cells, can then produce adenosine and prostaglandin E2 (PGE2), which collaborate in suppressing functions of immune cells within the TME.43
The solid TME can also downregulate T cell activation by upregulating protein tyrosine phosphatase SHP-1 which in turn inhibits activation of P56lck, P59fyn, and Zap-70, molecules downstream of TCR signalling.44 TGF-β-mediated mechanisms can severely suppress T cell effector functions that include production of perforin, granzyme B and IFN-γ, and expression of Fas ligand, which is important in T cell-mediated apoptosis.40
Chronic antigen stimulation and cytokine secretion upregulate inhibitory receptors, which are another barrier to the function of T cells within TME. The most prominent examples of inhibitory receptors are cytotoxic T lymphocyte antigen (CTLA4) and programmed cell death (PD1). CTLA4 is expressed at low basal levels, T cell activation increases CTLA4 expression which then competes with CD28 for binding to B7-1. As CD28 has a lower binding affinity, CTLA4 can therefore interfere with CD28-B7-1 interaction, inhibiting T cell activation and proliferation.45 Furthermore, PDL1 expressed on antigen-presenting cells bind to PD-1 on activated T cells, leading to inhibition of T cell proliferation and cytokine secretion.46
Cancer-Associated Fibroblasts (CAFs) as Physical Barrier of the TME
The extracellular matrix (ECM) of tumor stroma creates a physical barrier to cancer therapies by preventing infiltration of therapeutic agents into tumors. ECM is made up of various structural molecules such as fibrous proteins, glycosaminoglycans, and proteoglycans. These are produced by tumors and cancer-associated fibroblasts (CAFs) that contribute to tumorigenesis.47 In the TME, the CAFs can build up and remodel the structure of ECM. Extensive in vitro studies have been performed on CAFs and showed that they are easy to isolate and have inherent phenotypic plasticity, however the exact definition of CAFs is still unclear due to their heterogeneous population and lack of specific marker expression.48–50
Fibroblasts in normal tissue have a thin, elongated spindle shape extending from front to back, and they are generally considered to be in a resting state. Fibroblasts are found in the interstitial stroma in-between the layers of functional parenchyma adult tissue,51 normally activated in response to tissue injury and stimuli such as stress, hypoxia and cytokines. The activated fibroblasts secrete higher levels of collagens, laminins and fibronectin, factors that constitute the ECM,52 therefore they can help with wound repairs by remodeling the ECM. As the wound is repaired, the activated fibroblasts decrease in number and most likely reverse back to the resting state.53 However in tumor fibrosis it is considered that tumors are wounds that are in a chronic state of healing, and fibroblasts are hyper-activated through mediating factors such as TGF-β, platelet-derived growth factor (PDGF) and fibroblast growth factor 2 (FGF2). CAFs are therefore considered as an irreversibly activated heterogeneous population of fibroblasts with distinct functions.51 They greatly contribute to the TME’s immunosuppression by secreting various chemokines and cytokines, including TGF-β, IL6, IL8, IL13 and CX-chemokine ligand 12 (CXCL12), and vascular endothelial growth factor A (VEGF).54
The Role of FAP in the TME
Expression of fibroblast activation protein α (FAP) is an important characteristic of CAFs.55 Through quantitative secretome profiling analysis, Koczorowska et al. found that FAP activity impacts the secreted CAF proteome, leading to reduced levels of anti-angiogenic factors (PEDF, angiopoietin-1, VEGFC), increased levels of TGF-β, and regulates matrix processing enzymes.56
FAP is a type II membrane protease that belongs to the prolyl oligopeptidase family. It can be further classified into the dipeptidyl peptidase (DPP) subfamily; enzymes in this family have been shown to hydrolyze a prolyl bond that is two amino acids from the N-terminus of a protein, which is termed as dipeptidyl peptidase activity. In addition to this activity, FAP has also been shown to have an endopeptidase activity, meaning that it can cleave after a glycine-proline (Gly-Pro) bond that is more than two amino acids away from the N-terminus of a protein.57 The Gly-Pro bond is commonly found in collagens that populate the ECM.58 FAP can therefore remodel the ECM through the endopeptidase activity by cleaving the collagen and modifying bioactive signaling peptides in cancer.59
During tissue remodeling, FAP is detected in activated fibroblasts that are involved in wound healing, but not in other healthy adult tissues.60,61 However in the disease state, FAP expression was shown to have moderate to strong expression on the surface of breast, lung, colorectal, prostate, stomach, pancreatic, prostate, thyroid, cervical and urothelial cancers.62 A representative image of FAP-positive CAFs in a human lung tumor is shown in Figure 1.
Figure 1.
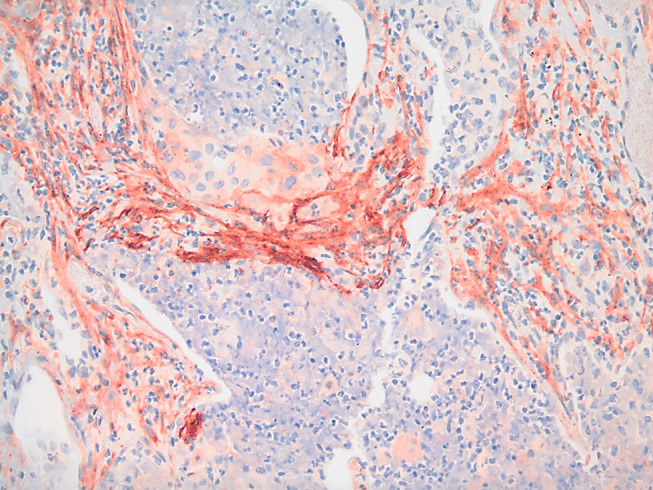
FAP expression in cancer tissue. Strong FAP expression (red) is observed in the stromal cells (CAFs) of a human squamous cell carcinoma stained with anti-FAP rabbit polyclonal antibody (dilution 1:100) IHC (20X image).
FAP expression in cancer is thought to be associated with poor prognosis.63 Lo et al. demonstrated that overexpression of FAP in the TME negatively impacts both the overall and disease-free survival of pancreatic ductal adenocarcinomas (PDAs) patients.64 Cohen et al. showed that higher expression of FAP in tumor-associated myofibroblasts surrounding tumors, correlated with a higher chance of lymph nodes positive for patients with PDA.65 In FAP-depleted transgenic mice injected with Lewis lung carcinomas, rapid hypoxic necrosis was observed in the cancer and stromal cells that was mediated via IFN-γ and tumor necrosis factor-α, showing that FAP-expressing cells have an immunosuppressive role in the TME.66 The effect of FAP depletion was tested in a PDA mouse model, and it was found that FAP deficiency delayed the onset of PDA in mice, and that the deletion of FAP increased the survival of mice, highlighting that FAP plays an important role in promoting disease progression. In FAP knock-out mice, PDA metastasis was reduced in liver, lung, mesentery and diaphragm.64–66 These studies highlighted the potential of using FAP as a therapeutic target in PDA.
Mechanism of FAP Mediated Immunosuppression
In a study that examined the function of FAP in vitro, it was shown that expression of FAP enhanced tumorigenesis of cells in ovarian cancer cell line in a dose-dependent manner.67 To investigate the mechanism with which FAP promotes immunosuppression, Yang et al. reported that FAP induces inflammatory CAFs by activating the transcription factor STAT2 via the Urokinase-type plasminogen activator receptor (uPAR)-FAK-c-Src-JAK2 pathway in a uPAR-dependent manner. The FAP-STAT3 axis was found to drive the CAFs towards mediating tumor immunosuppression by recruiting myeloid-derived suppressor cells (MDSCs) in the TME and furthermore, STAT3 was found to control the fibroblastic expression of CCL2, allowing for the CAFs to further promote tumor growth.68
FAP has also been reported to have a pro-angiogenic function. This was demonstrated in vitro using a human breast cancer cell line that expressed FAP; the cell line displayed a much higher growth rate compared with control cell line that did not express FAP.69 Sites of tumors are rich in blood supply to facilitate tumor growth and its integration into the surrounding tissue; the study also measured micro-vessel density of a tumor from the animals by immunohistochemistry. It was found that the average micro-vessel density for FAP-expressing cells is three times that of the control cells, indicating a strong role of FAP in angiogenesis.69 Researchers at Johns Hopkins University observed that increasing the amount of FAP in vitro increases the concentration of VEGF detected in human prostate cancer cells (PC3), signifying the immunosuppressive role of FAP in the TME.70 However, there are also studies suggesting that FAP can act as a tumor suppressor. Expression of FAP at physiological levels reduces the proliferation and tumorigenicity of transformed melanocytic cells.71 A similar observation was made when the expression of FAP was found to be inversely correlated with tumorigenicity in HeLa carcinoma cells.72 The discrepancy between the function of FAP in tumor promotion and suppression suggests that the exact function of FAP may depend on the exact context of the different TME.
FAP-CAR-T Cell Therapies
FAP-CAR-T Cell Preclinical Studies
FAP-targeting CAR-T cells have been engineered to target CAFs in various solid cancers, such as mesothelioma, lung and pancreatic cancers (Figure 2).47 A number of studies have showed anti-tumor activity in preclinical models.73–75
Figure 2.

Anti-FAP CAR-T cells in the solid tumor TME. (A) The anti-FAP CAR constructs consist of an anti-FAP targeting moiety (scFv), a spacer, a transmembrane domain (TM), and signaling domains (CD28/41BB and CD3ζ). These are transduced and expressed on T cells. The developed anti-FAP CAR-T cells can then bind to FAP expressed on CAFs. (B) Binding of anti-FAP CAR-T cells to FAP on CAFs within the hostile, multicellular TME.
One of the important measures in these studies is on-target off-tumor toxicity, because while FAP is highly expressed in these cancers, it is also found to express at low level in skeletal muscle, adipose tissue, and pancreas in mice.76 When Roberts et al. depleted FAP+ stromal cells in adult mice using genetic approaches, they found that the mice had rapid weight loss, bone marrow hypoplasia and pancreatic activity and anemia.76 The causes of the toxicity were explored in a study targeting FAP with a second-generation FAP-5-scFv-CAR in various mouse models.77 FAP5 is a mouse antibody that has high affinity for both mouse and human FAP. The study showed that FAP-CAR-T cells had weak anti-tumor effect in mice bearing either melanoma or colon cancer, and minimal to modest antitumor effect in mice with fibrosarcoma, breast or Renca kidney tumors. The latter were reported to have experienced severe bone marrow hypocellularity and cachexia.77
To explain the toxicity the study observed that FAP is expressed by both tumor stromal fibroblasts and multipotent bone marrow stromal cells (BMSCs), suggesting that BMSCs may be a source of tumor stromal cells, and that anti-FAP-CAR-T cells can target both BMSCs and tumor stromal cells.77 However such toxicity was not observed when FAP was targeted by a FAP-targeting vaccine.73–75 This could be explained by the fact that T cell-inducing vaccines may be able to target FAP expressed by tumor stromal cells due to the way FAP epitopes are processed and presented by MHC molecules, whereas CAR-T cells mediate a robust anti-tumor response and can directly recognize cell surface FAP without MHC molecule presentation.77
Kakarla et al. had also developed a second-generation FAP-CAR (CD28/CD3ζ) which targeted both murine and human FAP using scFv MO36 as the antigen recognition domain.78 These CAR-T cells significantly decreased the growth of tumors in a A549 lung cancer model and improved the survival of these mice compared with control mouse models. In addition to targeting the tumor stromal cells using the MO36-FAP-CAR, they also targeted A549 tumor-associated antigen erythropoietin-producing hepatocellular carcinoma A2 (EphA2), using EphA2-CAR. EphA2 is overexpressed in A549 lung cancer cells; by targeting both the tumor stromal cells and the cancer cells in mice, the group observed a superior anti-tumor activity. Nevertheless, all mice had eventually relapsed, however the reasons remained unclear. No significant toxicity was observed for at least two days after the treatment with CAR-T cells.78
Similarly, Wang et al. at the University of Pennsylvania, did not observe significant toxicity when they treated mice with FAP-CAR-T cells. The researchers developed a second-generation retroviral CAR targeting mouse FAP, consisting of scFv FAP (mab 73.3) with CD8 stalk, human CD3ζ and 4–1BB domains. The FAP-CAR-T cells were tested in vivo in three different established models, with the growth of tumors significantly reduced by 35–50% following treatment.79 The anti-tumor effect was seen in fully immune-competent mice, but not in immune-deficient mice, suggesting the involvement of an acquired immune system in the anti-tumor effect.79 The effect had also diminished in FAP-null mice, proving that the anti-tumor activity was dependent on expression of FAP in the mice.79 In a quest to increase the anti-tumor activity, they successfully enhanced the efficacy of the CAR-T cell therapy by either injecting a second dose or combining the CAR-T cell therapy with a vaccine HPV-E7 (Ad.E7). The persistence of transduced T cells was also increased when they transduced mouse T cells that had depleted inhibitory enzyme diacylglycerol kinase zeta (DGKζ).79
The studies mentioned so far have demonstrated that depletion of FAP+ cells decreases tumor growth in an immune-dependent manner, which provides support for combining FAP-CAR-T cell therapy with cancer vaccines or immune check point inhibitors such as anti-CTLA5 and anti-PD-1, depleting or inhibiting immunosuppressive factors such as DGKξ and TGF-β.
Aiming to improve FAP-CAR-T cell efficacy, a new FAP-targeting construct was developed that consisted of transmembrane and cytoplasmic domains of killer cell immunoglobulin-like receptor, two Ig domains and short cytoplasmic tail 2 (KIR2DS2) – a stimulatory killer immunoglobulin-like receptor (KIR), derived from natural killer cells – instead of the conventional cytoplasmic domains that are commonly used (CD28 and 4–1BB).80 The anti-FAP-KIR-based CAR (FAP-KIRS2) was able to activate T cells in a manner comparable to CAR-T cells that utilize intracellular signaling domains from T cell co-stimulatory receptors. An enhanced anti-tumor effect was observed when established human mesothelioma (EMMESO) tumors in NSG mice were treated with FAP-KIRS2-CAR-T cells compared with CD3ζ based CAR-T cells.80 However toxicity that was not observed in CD3ζ CAR-T cells was reported with the KIR-based CAR-T cells, which could potentially be attributed to the more robust depletion of FAP+ cells.80
Researchers at the University of Zurich developed another FAP targeting CAR, anti-FAP-F19-ΔCD28/CD3ζ.81 They removed the binding site of lck from the CD28 signaling domain; this modification has been shown to abolish IL2 release upon CAR-T cell engagement with its target, and subsequently reduces Treg cell persistence.82 The CH2/CH3 domain of the CAR was also modified to reduce binding of FcγR+ cells, reducing off-target toxicities of CAR-T cells. These anti-FAP-CAR-T cells showed efficacy both in vitro in FAP-positive mesothelioma cells, and in vivo in an established FAP-expressing tumor model,81 leading to initiation of a phase I clinical trial (discussed in the clinical trials section). The same group had explored a combination therapy of anti-FAP-CAR-T cells and PD-1 blockade. They inoculated humanized NSG mice with human fibrosarcoma cells expressing FAP, and administered PD-1 blocking antibody alongside CAR-T cells. Only the anti-FAP-F19-ΔCD28/CD3ζ-CAR-T cells demonstrated significant anti-tumor activity, proving the advantage of combining checkpoint inhibitors with CAR-T cell therapy.83
FAP CAR-T Cell Clinical Trials
Currently, only two clinical trials using anti-FAP CAR-T cell therapies have been conducted. A phase I clinical trial using F19 FAP-CAR-T-cells (CD8 positive CD3ζ/CD28 T cells) was initiated in 2012 (NCT01722149).84 A single dose of 1×106 transduced T cells was injected in patients with malignant pleural mesothelioma, with pleural effusion,84 a condition that describes a build-up of fluid in the pleural space of the lungs.85 The local injection of CAR-T cells within the pleural effusion was expected to help reduce systemic side effects and prevent on-target off-tumor toxicity. A previous study had shown that localized injection of a mesothelin targeted CAR-T cell therapy generated longer tumor immunity compared with systemically delivered CAR-T cells in mouse models.85 The first patient report following localized anti-FAP CAR-T cell treatment was recorded in 2015 and no cytokine storm or inflammation was observed.86 In total three patients with malignant pleural mesothelioma were treated with the FAP-CAR-T cells and no significant toxicity was observed. One patient received a PD-1 blocking antibody 8 months after the treatment with anti-FAP CAR-T therapy cell and no toxicity was reported.87
The most recent clinical trial aims to use a fourth-generation CAR-T targeting Nectin4/FAP in Nectin4-positive advanced malignant solid tumors (NCT03932565). Nectin4 is a triple-negative breast cancer (TNBC) specific cell surface molecule, that is overexpressed in cancer tissues such as pancreatic, ovarian, non-small cell lung and breast cancer.88 The CAR-T cell has been designed to inductively release IL7 and CCL19, or IL12 upon engagement with target antigen. This is the first FAP-targeting fourth-generation CAR-T cell trial.
Conclusion
CAR-T cell therapy has the potential to be a versatile treatment for various cancers and diseases. Engineering and modifying different components of CAR can lead to development of diverse CAR-T cell modalities, targeting different tumor types with improved CAR-T cell efficacy. These include fourth-generation (armored/TRUCK) CAR T cells, which are designed to increase infiltration and trafficking into solid tumors, prolong persistence of CAR-T cells in the TME and enhance their killing efficiency; and synNotch-CAR-T cells that can target multiple antigens and induce precise killing of target cells via dual receptor circuit. FAP is an ideal target for treatment of solid tumors, as the preclinical studies presented in this review have shown, targeting FAP using CAR-T cells can effectively increase killing of tumor cells in vitro. More clinical trials are necessary to ensure the safety and efficacy of the therapy. Targeting FAP alone in the TME may not provide complete solid tumor clearance; therefore, combined therapies or targeting cancer cells as well as fibroblasts should also be investigated.
Acknowledgments
We thank Prof Steven M Albelda (University of Pennsylvania) for supplying the microscopy figure included in the manuscript.
Funding Statement
AK is funded by the Cris Cancer Foundation.
Author Contributions
RB and AK conceptualized, wrote and edited the manuscript. PD and RRD wrote and edited the manuscript. RB and AK designed and edited the figures. All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Park JH, Geyer MB, Brentjens RJ. CD19-targeted CAR T-cell therapeutics for hematologic malignancies: interpreting clinical outcomes to date. Blood. 2016;127(26):3312–3320. doi: 10.1182/blood-2016-02-629063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen N, Li X, Chintala NK, Tano ZE, Adusumilli PS. Driving CARs on the uneven road of antigen heterogeneity in solid tumors. Curr Opin Immunol. 2018;51:103–110. doi: 10.1016/j.coi.2018.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hosein AN, Wu M, Arcand SL, et al. Breast carcinoma-associated fibroblasts rarely contain p53 mutations or chromosomal aberrations. Cancer Res. 2010;70(14):5770–5777. doi: 10.1158/0008-5472.CAN-10-0673 [DOI] [PubMed] [Google Scholar]
- 4.Arandkar S, Furth N, Elisha Y, et al. Altered p53 functionality in cancer-associated fibroblasts contributes to their cancer-supporting features. Proc Natl Acad Sci USA. 2018;115(25):6410–6415. doi: 10.1073/pnas.1719076115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bianchi-Frias D, Basom R, Delrow JJ, et al. Cells comprising the prostate cancer microenvironment lack recurrent clonal somatic genomic aberrations. Mol Cancer Res. 2016;14(4):374–384. doi: 10.1158/1541-7786.MCR-15-0330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Campbell I, Polyak K, Haviv I. Clonal mutations in the cancer-associated fibroblasts: the case against genetic coevolution. Cancer Res. 2009;69(17):6765–6768; discussion 6769. doi: 10.1158/0008-5472.CAN-08-4253 [DOI] [PubMed] [Google Scholar]
- 7.Han C, Liu T, Yin R. Biomarkers for cancer-associated fibroblasts. Biomark Res. 2020;8(1):64. doi: 10.1186/s40364-020-00245-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci USA. 1989;86(24):10024–10028. doi: 10.1073/pnas.86.24.10024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Halford Z, Anderson MK, Bennett LL, Moody J. Tisagenlecleucel in acute lymphoblastic leukemia: a review of the literature and practical considerations. Ann Pharmacother. 2021;55(4):466–479. doi: 10.1177/1060028020948165 [DOI] [PubMed] [Google Scholar]
- 10.Halford Z, Anderson MK, Bennett LL. Axicabtagene ciloleucel: clinical data for the use of CAR T-cell therapy in relapsed and refractory large B-cell lymphoma. Ann Pharmacother. 2021;55(3):390–405. doi: 10.1177/1060028020944233 [DOI] [PubMed] [Google Scholar]
- 11.Mian A, Hill BT. Brexucabtagene autoleucel for the treatment of relapsed/refractory mantle cell lymphoma. Expert Opin Biol Ther. 2021;21:435–441. doi: 10.1080/14712598.2021.1889510 [DOI] [PubMed] [Google Scholar]
- 12.Ma S, Li X, Wang X, et al. Current progress in CAR-T cell therapy for solid tumors. Int J Biol Sci. 2019;15(12):2548–2560. doi: 10.7150/ijbs.34213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci USA. 1993;90(2):720–724. doi: 10.1073/pnas.90.2.720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brocker T, Karjalainen K. Signals through T cell receptor-zeta chain alone are insufficient to prime resting T lymphocytes. J Exp Med. 1995;181(5):1653–1659. doi: 10.1084/jem.181.5.1653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jenkins MK, Chen CA, Jung G, Mueller DL, Schwartz RH. Inhibition of antigen-specific proliferation of type 1 murine T cell clones after stimulation with immobilized anti-CD3 monoclonal antibody. J Immunol. 1990;144(1):16–22. [PubMed] [Google Scholar]
- 16.Maher J, Brentjens RJ, Gunset G, Rivière I, Sadelain M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRζ /CD28 receptor. Nat Biotechnol. 2002;20(1):70–75. doi: 10.1038/nbt0102-70 [DOI] [PubMed] [Google Scholar]
- 17.Imai C, Mihara K, Andreansky M, et al. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. 2004;18(4):676–684. doi: 10.1038/sj.leu.2403302 [DOI] [PubMed] [Google Scholar]
- 18.Viola A, Lanzavecchia A. T cell activation determined by T cell receptor number and tunable thresholds. Science. 1996;273(5271):104–106. doi: 10.1126/science.273.5271.104 [DOI] [PubMed] [Google Scholar]
- 19.Van Der Stegen SJC, Hamieh M, Sadelain M. The pharmacology of second-generation chimeric antigen receptors. Nat Rev Drug Discov. 2015;14(7):499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davila ML, Riviere I, Wang X, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6(224):224ra225. doi: 10.1126/scitranslmed.3008226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385(9967):517–528. doi: 10.1016/S0140-6736(14)61403-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pule M, Straathof K, Dotti G, Heslop H, Rooney C, Brenner M. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol Ther. 2005;12(5):933–941. [DOI] [PubMed] [Google Scholar]
- 23.Zhong XS, Matsushita M, Plotkin J, Riviere I, Sadelain M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol Ther. 2010;18(2):413–420. doi: 10.1038/mt.2009.210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hawkins ER, D’Souza RR, Klampatsa A. Armored CAR T-cells: the next chapter in T-cell cancer immunotherapy. Biologics. 2021;15:95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chmielewski M, Abken H. CAR T cells releasing IL-18 convert to T-Bethigh FoxO1low effectors that exhibit augmented activity against advanced solid tumors. Cell Rep. 2017;21(11):3205–3219. doi: 10.1016/j.celrep.2017.11.063 [DOI] [PubMed] [Google Scholar]
- 26.Kloss CC, Lee J, Zhang A, et al. Dominant-negative TGF-beta receptor enhances PSMA-targeted human CAR T cell proliferation and augments prostate cancer eradication. Mol Ther. 2018;26(7):1855–1866. doi: 10.1016/j.ymthe.2018.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu B, Yan L, Zhou M. Target selection of CAR T cell therapy in accordance with the TME for solid tumors. Am J Cancer Res. 2019;9(2):228–241. [PMC free article] [PubMed] [Google Scholar]
- 28.Dagogo-Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol. 2018;15(2):81–94. doi: 10.1038/nrclinonc.2017.166 [DOI] [PubMed] [Google Scholar]
- 29.Wikstrand CJ, Hale LP, Batra SK, et al. Monoclonal antibodies against EGFRvIII are tumor specific and react with breast and lung carcinomas and malignant gliomas. Cancer Res. 1995;55(14):3140–3148. [PubMed] [Google Scholar]
- 30.Choe JH, Watchmaker PB, Simic MS, et al. SynNotch-CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci Transl Med. 2021;13:591. doi: 10.1126/scitranslmed.abe7378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang K, Wei G, Liu D. CD19: a biomarker for B cell development, lymphoma diagnosis and therapy. Exp Hematol Oncol. 2012;1(1):36. doi: 10.1186/2162-3619-1-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Posey AD, Schwab RD, Boesteanu AC, et al. Engineered CAR T cells targeting the cancer-associated Tn-glycoform of the membrane mucin MUC1 control adenocarcinoma. Immunity. 2016;44(6):1444–1454. doi: 10.1016/j.immuni.2016.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Linette GP, Stadtmauer EA, Maus MV, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. 2013;122(6):863–871. doi: 10.1182/blood-2013-03-490565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jing X, Yang F, Shao C, et al. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol Cancer. 2019;18(1):157. doi: 10.1186/s12943-019-1089-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jain RK. Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell. 2014;26(5):605–622. doi: 10.1016/j.ccell.2014.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pilon-Thomas S, Kodumudi KN, El-Kenawi AE, et al. Neutralization of tumor acidity improves antitumor responses to immunotherapy. Cancer Res. 2016;76(6):1381–1390. doi: 10.1158/0008-5472.CAN-15-1743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen Y, Song Y, Du W, Gong L, Chang H, Zou Z. Tumor-associated macrophages: an accomplice in solid tumor progression. J Biomed Sci. 2019;26(1):78. doi: 10.1186/s12929-019-0568-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9(3):162–174. doi: 10.1038/nri2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gowrishankar K, Birtwistle L, Micklethwaite K. Manipulating the tumor microenvironment by adoptive cell transfer of CAR T-cells. Mamm Genome. 2018;29(11–12):739–756. doi: 10.1007/s00335-018-9756-5 [DOI] [PubMed] [Google Scholar]
- 40.Dahmani A, Delisle J-S. TGF-β in T cell biology: implications for cancer immunotherapy. Cancers. 2018;10(6):194. doi: 10.3390/cancers10060194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McKarns SC, Schwartz RH, Kaminski NE. Smad3 is essential for TGF- 1 to suppress IL-2 production and TCR-induced proliferation, but not IL-2-induced proliferation. J Immunol. 2004;172(7):4275–4284. [DOI] [PubMed] [Google Scholar]
- 42.Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer. 2005;5(4):263–274. doi: 10.1038/nrc1586 [DOI] [PubMed] [Google Scholar]
- 43.Mandapathil M, Szczepanski MJ, Szajnik M, et al. Adenosine and prostaglandin E2 cooperate in the suppression of immune responses mediated by adaptive regulatory T cells. J Biol Chem. 2010;285(36):27571–27580. doi: 10.1074/jbc.M110.127100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Choudhry MA, Sir O, Sayeed MM. TGF-beta abrogates TCR-mediated signaling by upregulating tyrosine phosphatases in T cells. Shock. 2001;15(3):193–199. doi: 10.1097/00024382-200115030-00006 [DOI] [PubMed] [Google Scholar]
- 45.Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. 2020;20(11):651–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the Pd-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192(7):1027–1034. doi: 10.1084/jem.192.7.1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rodriguez-Garcia A, Palazon A, Noguera-Ortega E, Powell DJ, Guedan S. CAR-T cells hit the tumor microenvironment: strategies to overcome tumor escape. Front Immunol. 2020;11. doi: 10.3389/fimmu.2020.01109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Costa A, Kieffer Y, Scholer-Dahirel A, et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell. 2018;33(3):463–479 e410. doi: 10.1016/j.ccell.2018.01.011 [DOI] [PubMed] [Google Scholar]
- 49.Bartoschek M, Oskolkov N, Bocci M, et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat Commun. 2018;9(1):5150. doi: 10.1038/s41467-018-07582-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ohlund D, Handly-Santana A, Biffi G, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med. 2017;214(3):579–596. doi: 10.1084/jem.20162024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16(9):582–598. doi: 10.1038/nrc.2016.73 [DOI] [PubMed] [Google Scholar]
- 52.Castor CW, Wilson SM, Heiss PR, Seidman JC. Activation of lung connective tissue cells in vitro. Am Rev Respir Dis. 1979;120(1):101–106. [DOI] [PubMed] [Google Scholar]
- 53.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3(5):349–363. doi: 10.1038/nrm809 [DOI] [PubMed] [Google Scholar]
- 54.Liu T, Han C, Wang S, et al. Cancer-associated fibroblasts: an emerging target of anti-cancer immunotherapy. J Hematol Oncol. 2019;12(1):86. doi: 10.1186/s13045-019-0770-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Garin-Chesa P, Old LJ, Rettig WJ. Cell surface glycoprotein of reactive stromal fibroblasts as a potential antibody target in human epithelial cancers. Proc Natl Acad Sci USA. 1990;87(18):7235–7239. doi: 10.1073/pnas.87.18.7235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Koczorowska MM, Tholen S, Bucher F, et al. Fibroblast activation protein-alpha, a stromal cell surface protease, shapes key features of cancer associated fibroblasts through proteome and degradome alterations. Mol Oncol. 2016;10(1):40–58. doi: 10.1016/j.molonc.2015.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hamson EJ, Keane FM, Tholen S, Schilling O, Gorrell MD. Understanding fibroblast activation protein (FAP): substrates, activities, expression and targeting for cancer therapy. Proteomics Clin Appl. 2014;8(5–6):454–463. doi: 10.1002/prca.201300095 [DOI] [PubMed] [Google Scholar]
- 58.Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. J Cell Sci. 2010;123(24):4195–4200. doi: 10.1242/jcs.023820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kelly T. Fibroblast activation protein-alpha and dipeptidyl peptidase IV (CD26): cell-surface proteases that activate cell signaling and are potential targets for cancer therapy. Drug Resist Updat. 2005;8(1–2):51–58. doi: 10.1016/j.drup.2005.03.002 [DOI] [PubMed] [Google Scholar]
- 60.Jacob M, Chang L, Pure E. Fibroblast activation protein in remodeling tissues. Curr Mol Med. 2012;12(10):1220–1243. doi: 10.2174/156652412803833607 [DOI] [PubMed] [Google Scholar]
- 61.Levy MT, McCaughan GW, Abbott CA, et al. Fibroblast activation protein: a cell surface dipeptidyl peptidase and gelatinase expressed by stellate cells at the tissue remodelling interface in human cirrhosis. Hepatology. 1999;29(6):1768–1778. doi: 10.1002/hep.510290631 [DOI] [PubMed] [Google Scholar]
- 62.Atlas THP. FAP; 2021. Available from: https://www.proteinatlas.org/ENSG00000078098-FAP/pathology. Accessed July20, 2021.
- 63.Busek P, Mateu R, Zubal M, Kotackova L, Sedo A. Targeting fibroblast activation protein in cancer - prospects and caveats. Front Biosci (Landmark Ed). 2018;23:1933–1968. doi: 10.2741/4682 [DOI] [PubMed] [Google Scholar]
- 64.Lo A, Li CP, Buza EL, et al. Fibroblast activation protein augments progression and metastasis of pancreatic ductal adenocarcinoma. JCI Insight. 2017;2(19):e92232. doi: 10.1172/jci.insight.92232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cohen SJ, Alpaugh RK, Palazzo I, et al.Fibroblast activation protein and its relationship to clinical outcome in pancreatic adenocarcinoma. Pancreas. 2008;37(2):154–158. doi: 10.1097/MPA.0b013e31816618ce [DOI] [PubMed] [Google Scholar]
- 66.Kraman M, Bambrough PJ, Arnold JN, et al.Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science. 2010;330(6005):827–830. doi: 10.1126/science.1195300 [DOI] [PubMed] [Google Scholar]
- 67.Chen H, Yang WW, Wen QT, Xu L, Chen M. TGF-beta induces fibroblast activation protein expression; fibroblast activation protein expression increases the proliferation, adhesion, and migration of HO-8910PM [corrected]. Exp Mol Pathol. 2009;87(3):189–194. [DOI] [PubMed] [Google Scholar]
- 68.Yang X, Lin Y, Shi Y, et al. FAP promotes immunosuppression by cancer-associated fibroblasts in the tumor microenvironment via STAT3-CCL2 signaling. Cancer Res. 2016;76(14):4124–4135. doi: 10.1158/0008-5472.CAN-15-2973 [DOI] [PubMed] [Google Scholar]
- 69.Huang Y, Wang S, Kelly T. Seprase promotes rapid tumor growth and increased microvessel density in a mouse model of human breast cancer. Cancer Res. 2004;64(8):2712–2716. doi: 10.1158/0008-5472.CAN-03-3184 [DOI] [PubMed] [Google Scholar]
- 70.Brennen W, J Thorek DL, Jiang W. Overcoming stromal barriers to immuno-oncological responses via fibroblast activation protein-targeted therapy. Immunotherapy. 2021;13(2):155–175. doi: 10.2217/imt-2020-0066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ramirez-Montagut T, Blachere NE, Sviderskaya EV, et al. FAPalpha, a surface peptidase expressed during wound healing, is a tumor suppressor. Oncogene. 2004;23(32):5435–5446. doi: 10.1038/sj.onc.1207730 [DOI] [PubMed] [Google Scholar]
- 72.Tsujimoto H, Nishizuka S, Redpath JL, Stanbridge EJ. Differential gene expression in tumorigenic and nontumorigenic HeLa x normal human fibroblast hybrid cells. Mol Carcinog. 1999;26(4):298–304. doi: [DOI] [PubMed] [Google Scholar]
- 73.Loeffler M, Kruger JA, Niethammer AG, Reisfeld RA. Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J Clin Invest. 2006;116(7):1955–1962. doi: 10.1172/JCI26532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lee J, Fassnacht M, Nair S, Boczkowski D, Gilboa E. Tumor immunotherapy targeting fibroblast activation protein, a product expressed in tumor-associated fibroblasts. Cancer Res. 2005;65(23):11156–11163. doi: 10.1158/0008-5472.CAN-05-2805 [DOI] [PubMed] [Google Scholar]
- 75.Wen Y, Wang CT, Ma TT, et al. Immunotherapy targeting fibroblast activation protein inhibits tumor growth and increases survival in a murine colon cancer model. Cancer Sci. 2010;101(11):2325–2332. doi: 10.1111/j.1349-7006.2010.01695.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Roberts EW, Deonarine A, Jones JO, et al. Depletion of stromal cells expressing fibroblast activation protein-alpha from skeletal muscle and bone marrow results in cachexia and anemia. J Exp Med. 2013;210(6):1137–1151. doi: 10.1084/jem.20122344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tran E, Chinnasamy D, Yu Z, et al. Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J Exp Med. 2013;210(6):1125–1135. doi: 10.1084/jem.20130110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kakarla S, Chow KK, Mata M, et al. Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol Ther. 2013;21(8):1611–1620. doi: 10.1038/mt.2013.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang LC, Lo A, Scholler J, et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res. 2014;2(2):154–166. doi: 10.1158/2326-6066.CIR-13-0027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang E, Wang LC, Tsai CY, et al. Generation of potent T-cell immunotherapy for cancer using DAP12-based, multichain, chimeric immunoreceptors. Cancer Immunol Res. 2015;3(7):815–826. doi: 10.1158/2326-6066.CIR-15-0054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schuberth PC, Hagedorn C, Jensen SM, et al. Treatment of malignant pleural mesothelioma by fibroblast activation protein-specific re-directed T cells. J Transl Med. 2013;11:187. doi: 10.1186/1479-5876-11-187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kofler DM, Chmielewski M, Rappl G, et al. CD28 costimulation impairs the efficacy of a redirected t-cell antitumor attack in the presence of regulatory t cells which can be overcome by preventing Lck activation. Mol Ther. 2011;19(4):760–767. doi: 10.1038/mt.2011.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gulati P, Ruhl J, Kannan A, et al. Aberrant Lck signal via CD28 costimulation augments antigen-specific functionality and tumor control by redirected T cells with PD-1 blockade in humanized mice. Clin Cancer Res. 2018;24(16):3981–3993. doi: 10.1158/1078-0432.CCR-17-1788 [DOI] [PubMed] [Google Scholar]
- 84.Petrausch U, Schuberth PC, Hagedorn C, et al. Re-directed T cells for the treatment of fibroblast activation protein (FAP)-positive malignant pleural mesothelioma (FAPME-1). BMC Cancer. 2012;12:615. doi: 10.1186/1471-2407-12-615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Karkhanis VS, Joshi JM. Pleural effusion: diagnosis, treatment, and management. Open Access Emerg Med. 2012;4:31–52. doi: 10.2147/OAEM.S29942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pircher M, Schuberth P, Gulati P, et al. FAP-specific re-directed T cells first in-man study in malignant pleural mesothelioma: experience of the first patient treated. BMC Cancer. 2015;3:1. [Google Scholar]
- 87.Curioni A, Britschgi C, Hiltbrunner S, et al. A phase I clinical trial of malignant pleural mesothelioma treated withlocally delivered autologous anti-FAP-targeted CAR T-cells. Ann Oncol. 2019;30:v501. [Google Scholar]
- 88.Zeindler J, Soysal SD, Piscuoglio S, et al. Nectin-4 expression is an independent prognostic biomarker and associated with better survival in triple-negative breast cancer. Front Med (Lausanne). 2019;6:200. doi: 10.3389/fmed.2019.00200 [DOI] [PMC free article] [PubMed] [Google Scholar]