

ABSTRACT
Hepatitis C virus (HCV) regulates many cellular genes in modulating the host immune system for benefit of viral replication and long-term persistence in a host for chronic infection. Long noncoding RNAs (lncRNAs) play an important role in the regulation of many important cellular processes, including immune responses. We recently reported that HCV infection downregulates lncRNA Linc-Pint (long intergenic non-protein-coding RNA p53-induced transcript) expression, although the mechanism of repression and functional consequences are not well understood. In this study, we demonstrate that HCV infection of hepatocytes transcriptionally reduces Linc-Pint expression through CCAAT/enhancer binding protein β (C/EBP-β). Subsequently, we observed that the overexpression of Linc-Pint significantly upregulates interferon alpha (IFN-α) and IFN-β expression in HCV-replicating hepatocytes. Using unbiased proteomics, we identified that Linc-Pint associates with DDX24, which enables RIP1 to interact with IFN-regulatory factor 7 (IRF7) of the IFN signaling pathway. We furthermore observed that IFN-α14 promoter activity was enhanced in the presence of Linc-Pint. Together, these results demonstrated that Linc-Pint acts as a positive regulator of host innate immune responses, especially IFN signaling. HCV-mediated downregulation of Linc-Pint expression appears to be one of the mechanisms by which HCV may evade innate immunity for long-term persistence and chronicity.
IMPORTANCE The mechanism by which lncRNA regulates the host immune response during HCV infection is poorly understood. We observed that Linc-Pint was transcriptionally downregulated by HCV. Using a chromatin immunoprecipitation (ChIP) assay, we showed inhibition of transcription factor C/EBP-β binding to the Linc-Pint promoter in the presence of HCV infection. We further identified that Linc-Pint associates with DDX24 for immunomodulatory function. The overexpression of Linc-Pint reduces DDX24 expression, which in turn results in the disruption of DDX24-RIP1 complex formation and the activation of IRF7. The induction of IFN-α14 promoter activity in the presence of Linc-Pint further confirms our observation. Together, our results suggest that Linc-Pint acts as a positive regulator of host innate immune responses. Downregulation of Linc-Pint expression by HCV helps in escaping the innate immune system for the development of chronicity.
KEYWORDS: hepatitis C virus, Linc-Pint, C/EBP-β, DDX24, interferon, IRF7
INTRODUCTION
Hepatitis C virus (HCV) chronically infects a large number of people worldwide and is a leading cause of advanced liver pathogenesis, including cirrhosis and hepatocellular carcinoma (HCC) (1, 2). Available direct-acting antiviral (DAA) agents are effective in reducing viremia to an undetectable level; however, they are unable to prevent reinfection. Immediately after HCV infection, the host innate immune response is triggered by the recognition of pathogen-associated molecular patterns (PAMPs) presented by the infecting virus by specific PAMP receptor factors expressed in host cells. Type I interferons (IFNs) such as IFN-α and IFN-β are rapidly synthesized after virus infection and trigger intracellular signaling events leading to the expression of IFN-stimulated genes (ISGs) that exert inhibitory effects on viral replication (3–7). The induction of type I IFNs is a critical part of the innate immune response following virus attack; however, HCV evades this effect by multiple mechanisms and develops chronic infection in the host (7–12). Furthermore, HCV infection is asymptomatic, infected individuals are diagnosed much later, and most cases develop chronic infection leading to advanced liver diseases. So it is important to better understand the molecular mechanisms underlying the interaction between HCV and the host immune system to effectively eliminate virus from the host.
Long noncoding RNAs (lncRNAs) are >200 nucleotides in length and lack protein-coding potential. lncRNAs can bind to DNA, RNA, and proteins to exert functions in regulating diverse biological processes in cells, such as transcription, mRNA stabilization, and translation (13). Aberrant expression of lncRNAs occurs in various cancers as well as in different viral infections (14–19). Emerging evidence suggests that lncRNAs play important roles during viral infection and the antiviral immune response. For example, the lncRNA nuclear enriched abundant transcript 1 (NEAT1) can enhance the transcription of interleukin-8 and modulate HIV-1 posttranscriptional expression (20). The innate immune response-related lncRNA negative regulator of antiviral response (NRAV) serves as antiviral innate immunity by suppressing the expression of multiple ISGs through interference with histone modification (21). The influenza A virus (IAV)-induced lncRNA VIN was observed to regulate viral replication and protein synthesis of IAV (22), although the mechanisms by which lncRNAs regulate innate immune responses and the consequences of virus-host interactions during HCV infection are still lacking.
We have recently shown that the expression of Linc-Pint (long intergenic non-protein-coding RNA p53-induced transcript) is significantly reduced in chronic HCV-infected human liver biopsy specimens as well as in cell lines infected in vitro with HCV (18). The exogenous expression of Linc-Pint inhibits HCV replication and virus-induced lipogenesis. In this study, a possible mechanism by which HCV downregulates Linc-Pint expression has been investigated. We demonstrate that HCV transcriptionally represses Linc-Pint expression by inhibiting CCAAT/enhancer binding protein β (C/EBP-β) binding to its promoter. On the other hand, the overexpression of Linc-Pint regulates DDX24 (DEAD-box helicase 24)-RIP1 (receptor interacting protein 1)-IRF7 (IFN-regulatory factor 7) signaling to upregulate interferon production by inducing IFN-α14 promoter activity. Therefore, this study provides important information on virus-host associations, which may help in the generation of novel therapeutic modalities against HCV-associated liver pathogenesis.
RESULTS
HCV suppresses Linc-Pint promoter activity.
We recently observed that Linc-Pint RNA expression is significantly repressed in chronic HCV-infected human liver biopsy specimens and in cell lines infected in vitro with HCV (18). To investigate the underlying mechanism for repression, a 2.5-kb Linc-Pint promoter region (bp −2063 to +448 [chromosome 7 {chr7}]) containing the binding sites of the transcriptional factors and cofactors was cloned into a pGL3 Basic vector (Fig. 1A). The promoter construct was separately transfected into Huh7.5 or Rep2a cells harboring the full-length HCV genome to examine its activity. Linc-Pint promoter activity was significantly downregulated as evident from luciferase assays in HCV replicon-expressing Rep2a cells compared to control parental Huh7.5 cells (Fig. 1A). These data suggest that HCV transcriptionally downregulates Linc-Pint expression.
FIG 1.

HCV inhibits C/EBP-β transcription factor binding to the Linc-Pint promoter region. (A) Schematic diagram of the Linc-Pint full-length promoter region cloned into the pGL3 Basic plasmid (P0). Huh7.5 and Rep2a cells were transfected with the Linc-Pint promoter construct (P0). Promoter activity was measured by a luciferase assay after 48 h of transfection. (B) In silico analysis using the NIH ENCODE data set of C/EBP-β ChIP-Seq signals from HepG2 cells. The result was analyzed from the data set under GEO accession number GSM935622. (C) Schematic diagram of the two deletion mutant constructs of the Linc-Pint promoter. Red boxes show the C/EBP-β binding site (bp −95 to −85). Huh7.5 cells were transfected with Linc-Pint promoter constructs (P0, P1, and P2), and promoter activity was measured by a luciferase assay after 48 h of transfection. (D) Nucleotide sequence of the wild-type (Wt) and mutant C/EBP-β binding sites (top). Changed nucleotides are shown in red. Huh7.5 cells were transfected with the P2 wild-type or mutant promoter construct, and luciferase activity was measured after 48 h of transfection. (E) Linc-Pint P0 promoter construct-transfected Huh7.5 cells were depleted of C/EBP-β using a specific siRNA, and promoter activity was measured by a relative luciferase assay after 48 h of transfection. (F) ChIP analysis of C/EBP-β binding to the Linc-Pint promoter. Huh7.5 cells were mock treated or infected with HCV JFH1 (MOI = 1.0) for 24 h. ChIP analysis was then performed using IgG (negative control) and C/EBP-β antibody. The relative enrichment of Linc-Pint promoter DNA was normalized to the input DNA (5%) for each experiment. Data are presented as means ± SD from three independent experiments. Statistical significance was analyzed using two-tailed Student’s t test. *, P < 0.05; **, P < 0.01.
HCV inhibits C/EBP-β transcription factor binding to the Linc-Pint promoter region.
To determine the cis-regulatory elements present in the Linc-Pint promoter region, we searched databases using TFBIND and Alibaba 2.1 software and found a putative binding site of C/EBP-β at bp −96 to −85 from the transcription start site. This site was confirmed by analyzing the ENCODE C/EBP-β chromatin immunoprecipitation assay sequencing (ChIP-Seq) data set from HepG2 cells (GEO accession no. GSM935622) (Fig. 1B). In our previous studies, we observed that HCV downregulates the promoter activity of C3 complement and microRNA miR-181c by C/EBP-β (23, 24). We therefore postulated that HCV-mediated downregulation of the Linc-Pint promoter may occur by a similar mechanism. To verify this, we constructed two deletion mutant constructs of the Linc-Pint promoter region (P1 and P2) and cloned them into the pGL3 Basic vector (Fig. 1C). The P1 construct does not contain the C/EBP-β binding site, whereas the P2 construct has the C/EBP-β binding site. Our luciferase assay using the Linc-Pint full-length (P0), P1, and P2 promoter constructs transfected into hepatocytes displayed a significant loss of luciferase activity from P1, suggesting that C/EBP-β is necessary for transcription (Fig. 1C). We also mutated the C/EBP-β binding site in the P2 construct of the Linc-Pint promoter, and the changed nucleotides are shown in red in Fig. 1D. Linc-Pint P2 promoter activity was partially reduced in the mutant construct. We next transfected the Linc-Pint P0 promoter construct into C/EBP-β-depleted Huh7.5 cells for luciferase assays and observed a significant downregulation of Linc-Pint promoter activity in the C/EBP-β knockdown cells (Fig. 1E). To further verify that C/EBP-β binding to the Linc-Pint promoter region may be affected in the presence of HCV, we performed a chromatin immunoprecipitation (ChIP) assay in Huh7.5 cells with or without HCV infection using IgG control and C/EBP-β antibodies. The results indicated an enrichment of Linc-Pint promoter DNA in C/EBP-β antibody ChIP samples compared to the IgG control. However, this enrichment was significantly reduced following HCV infection (Fig. 1F). Together, these results indicated that HCV-mediated downregulation of the Linc-Pint promoter occurs involving C/EBP-β.
Linc-Pint induces interferon gene expression.
lncRNAs are reported to induce type I interferon production and play important roles in the regulation of innate immunity during viral infection, especially in HCV infection (19, 25). To investigate whether Linc-Pint exerts a role in IFN signaling, we examined IFN-α and IFN-β expression in pcDNA3 Linc-Pint-overexpressing Huh7.5 cells. Our results indicated a significant upregulation of IFN expression in the presence of Linc-Pint (Fig. 2A). To examine the modulation of IFN systems by Linc-Pint, we examined the expression of ISGs by quantitative real-time PCR (qRT-PCR). Significant upregulation of IFIT1, IFIT2, IFIM1, OAS1, and IFI27 was noted (Fig. 2B). We also examined the status of phospho-STAT1 (p-STAT1) (Tyr701). Exogenous expression of Linc-Pint enhances phospho-STAT1 in Huh7.5 cells (Fig. 2C). Next, we examined IFN-α and IFN-β expression in the presence of Linc-Pint in HCV-infected or -replicating Huh7.5 cells. For this, HCV-infected Huh7.5 or Huh7.5 cells harboring the full-length HCV genome (Rep2a cells) were transfected with pcDNA3 Linc-Pint plasmid DNA and evaluated for IFN gene expression. We observed a significant upregulation of both cytokines in the presence of Linc-Pint (Fig. 2D). We evaluated IFN-α and IFN-β expression in two lncRNAs, ELDR and NORAD, and did not observe a significant difference in IFN gene expression (data not shown). As reported previously, Linc-Pint expression was significantly reduced in HCV replication in infected Huh7.5 cells (Fig. 2E). Together, these data suggested that Linc-Pint induces IFN signaling, and HCV-mediated Linc-Pint suppression may help in evasion of the host immune system.
FIG 2.

(Continued).
Linc-Pint physically interacts with DDX24.
To understand the mechanism of Linc-Pint-induced interferon signaling, we searched for the interacting partners of Linc-Pint in our proteomics data and identified DEAD-box helicase 24 (DDX24) as a potential candidate. A volcano plot of log10(P values) versus log2(fold changes) of sense/antisense spectrum counts was constructed to display the proteomics data from quantitative analyses, including DDX24 (Fig. 3A). DDX24 has a high spectral count (32.8-fold) in the mass spectrometric analysis. We next verified the RNA-protein interaction using the biotinylated sense or antisense Linc-Pint RNA pulled down from Huh7.5 lysates, followed by Western blotting using DDX24 antibody. An association of DDX24 protein with only the sense strand of Linc-Pint in hepatocytes was observed (Fig. 3B). To further verify the interaction between Linc-Pint and DDX24, we performed a reciprocal immunoprecipitation (IP) assay using DDX24 antibody or the isotype control in Huh7.5 cell lysates followed by qRT-PCR using Linc-Pint-specific primers. A significant enrichment of Linc-Pint RNA was observed in the immunoprecipitated sample using DDX24 antibody compared to that of the isotype control antibody (Fig. 3C). Together, these data suggested that Linc-Pint interacts with DDX24 protein.
FIG 3.

Linc-Pint interacts with DDX24. (A, left) Volcano plot from the mass spectrometry data demonstrating the magnitude and significance of the proteins interacting with the sense strand of Linc-Pint compared with the antisense strand of Linc-Pint. The x axis is the log2(fold change) value, and the y axis is the −log10(P value) showing statistical significance. The horizontal dashed red line shows a P value of 0.05 [−log10(0.05) = 1.3], and the vertical dashed red line shows a fold change of 2 [log2(2) = 1]. An absolute 2-fold change and a P value of 0.05 are used as the threshold cutoffs. (Right) Spectrum counts of antisense or sense Linc-Pint RNA for DDX24 from mass spectrometry data. (B) Western blot analysis was performed from sense or antisense Linc-Pint RNA pulled down from Huh7.5 lysates using a specific antibody, and the presence of DDX24 is shown. (C) Lysates from Huh7.5 cells were immunoprecipitated using isotype control or DDX24 antibody. RNA was isolated from the immunoprecipitated samples, and Linc-Pint RNA enrichment was evaluated by qRT-PCR. Data are presented as means ± SD from three independent experiments. Statistical significance was analyzed using two-tailed Student’s t test. ***, P < 0.001.
Linc-Pint reduces DDX24 protein expression in hepatocytes.
Next, we examined the status of DDX24 in Linc-Pint-overexpressing hepatocytes. For this, vector control or pcDNA3 Linc-Pint plasmid DNA was transfected into Huh7.5 or Rep2a cells. Cell lysates were analyzed by Western blotting using DDX24-specific antibody. A significant downregulation of the DDX24 protein level was observed in Linc-Pint-overexpressing cells compared to the control (Fig. 4A). On the other hand, a significant upregulation of DDX24 expression was noted in Rep2a cells compared to Huh7.5 control cells (Fig. 4B). We further observed that HCV-mediated enhancement of DDX24 was reduced upon exogenous Linc-Pint expression (Fig. 4C). Together, these data suggested that Linc-Pint can downregulate DDX24 expression in hepatocytes.
FIG 4.

Linc-Pint reduces DDX24 protein levels in the presence or absence of HCV. (A) Huh7.5 and Rep2a cells were transfected with control (empty vector) or pcDNA3 Linc-Pint plasmid DNA. Cell lysates were analyzed for DDX24 expression by Western blotting using a specific antibody. The blot was reprobed with antibody to actin for comparison of protein loads. Densitometric scanning results are presented as fold changes in a bar diagram at the bottom. (B) Lysates from Huh7.5 and Rep2a (Huh7.5 cells harboring a genome-length HCV replicon) cells were analyzed for DDX24 expression by Western blotting. The blot was reprobed with antibody to actin for comparison of protein loads. Densitometric scanning results are presented as fold changes in a bar diagram at the bottom. (C) Huh7.5 cells were transfected with the empty vector control (middle lane) or the pcDNA3 Linc-Pint plasmid (right lane) and then infected with HCV JFH1 (MOI = 1.0). Mock-infected cell lysates were used as the control. Cell lysates were analyzed for DDX24 expression by Western blotting using a specific antibody. The blot was reprobed with antibody to actin for comparison of protein loads. Densitometric scanning results are presented as fold changes in a bar diagram at the bottom. Data are presented as means ± SD from three independent experiments. Statistical significance was analyzed using two-tailed Student’s t test. *, P < 0.05; **, P < 0.01.
Linc-Pint-induced IFN expression by RIP1-IRF7 signaling.
DDX24 can reduce interferon production by inhibiting IFN-regulatory factor 7 (IRF7) activation by physical interaction with RIP1 (26). RIP1 positively regulates IRF7 activation (27). We hypothesized that the DDX24-RIP1 interaction will be disrupted in the presence of Linc-Pint, resulting in the activation of IRF7 and the upregulation of the interferon signaling pathway (Fig. 5A). To examine this, we performed a coimmunoprecipitation assay with DDX24 antibody using lysates of Huh7.5 cells transfected with the vector control or pcDNA3 Linc-Pint plasmid DNA, followed by Western blotting with a RIP1 or DDX24 antibody. Our results demonstrated that RIP1 was coprecipitated by DDX24 in control cell lysates, and the interaction was impeded for the presence of Linc-Pint (Fig. 5B). We also observed less interaction with RIP1, resulting in a lower abundance of IRF7, although inputs of RIP1 and IRF7 were similar in both lanes. We further performed a reciprocal experiment by precipitating the protein complex with RIP1 antibody. The DDX24 level was lower in Linc-Pint-transfected samples; however, RIP1 expression levels remained similar in immunoprecipitate and input samples. We also observed higher IRF7 association when precipitated with RIP1 antibody than when precipitated with DDX24 antibody (Fig. 5B). We performed coimmunoprecipitation analysis using RIP1 antibody from lysates of mock- or HCV (JFH1)-infected Huh7.5 cells. Our results showed a reduced level of IRF7 in virus-infected cells, which could be rescued by Linc-Pint overexpression (Fig. 5B, right). We have previously shown that IRF7 directly induces IFN-α production, and IFN-α14 is one of the IFN-α subtypes highly present in hepatocytes (11). We therefore evaluated IFN-α14 promoter activity by overexpressing vector control or pcDNA3 Linc-Pint plasmid DNA. We observed a significant downregulation of IFN-α14 promoter activity in HCV-infected cells and partial restoration of promoter activity upon Linc-Pint overexpression (Fig. 5C). Together, our results demonstrated that Linc-Pint induces IRF7-mediated IFN expression by disrupting the DDX24-RIP1 interaction. Thus, Linc-Pint suppression will have a negative effect on IFN expression.
FIG 5.

Linc-Pint induces IFN production by the RIP1-IRF7 pathway. (A) Schematic diagram showing the mechanism of Linc-Pint-mediated IFN signaling. (B) Huh7.5 cells were transfected with the vector control and the pcDNA3 Linc-Pint plasmid. Cell lysates were immunoprecipitated using DDX24 or RIP1 antibody. The association of DDX24, RIP1, and IRF7 was detected by Western blotting (WB) using specific antibodies. Huh7.5 cells were infected with HCV and transfected with the vector control or the pcDNA3 Linc-Pint plasmid. Cell lysates were immunoprecipitated using RIP1 antibody. The association of RIP1 and IRF7 was detected by Western blotting using specific antibodies. The cell lysates were also used for Western blot analysis using specific antibodies, and the blot was reprobed with antibody to actin for comparison of protein loads. (C) Huh7.5 cells were mock treated or infected with HCV (JFH1) and then cotransfected with the IFN-α14 luciferase promoter plasmid and vector control or pcDNA3 Linc-Pint plasmid DNA. IFN-α14 promoter activity was measured by a luciferase assay after 48 h of transfection. Data are presented as means ± SD from three independent experiments. Statistical significance was analyzed using two-tailed Student’s t test. *, P < 0.05; **, P < 0.01.
DISCUSSION
Emerging evidence suggests that lncRNAs play important roles during virus infection and antiviral immune responses (28–30). RNA viruses have evolved mechanisms to manipulate and hijack host lncRNAs to promote viral replication (31). Specific lncRNA expression signatures can induce part of the innate immune response to viral infection. We have recently shown that Linc-Pint is the only lncRNA reduced in HCV-replicating hepatocytes (18). We demonstrated two major observations from this study: (i) HCV transcriptionally downregulates Linc-Pint via the C/EBP-β transcription factor, and (ii) Linc-Pint expression enhances IFN signaling by interacting with and inhibiting DDX24. Thus, downregulation of Linc-Pint may impair IFN expression in HCV-infected cells.
Linc-Pint is positively regulated by p53, and there are three p53 response elements in Linc-Pint genomic sequences (32). Although HCV infection may disrupt p53 function by diverse mechanisms (33–36), we observed that Linc-Pint expression is reduced in HCV-replicating or -infected Huh7.5 cells, where p53 is mutated. We hypothesized that other factors along with p53 are involved in HCV-mediated inhibition of Linc-Pint. We found that C/EBP-β binds to regions near the Linc-Pint promoter by analyzing the NIH ENCODE data sets for C/EBP-β (GEO accession no. GSM935622) and visualizing the results at the Linc-Pint locus using Integrated Genome Viewer (version 2.5.1). Furthermore, by examining histone marks using the same ENCODE data set, we have obtained evidence that Linc-Pint transcription is primarily regulated at the basal transcription step but not at the chromatin remodeling step. We show here that C/EBP-β plays an important role in regulating Linc-Pint promoter activity. A subsequent study using ChIP assays suggested that the enrichment of Linc-Pint promoter DNA by C/EBP-β antibody pulldown was much higher in mock-treated cells than in HCV-infected cells.
The host innate immune system is triggered by viral PAMPs during HCV infection, although activation was not sufficient to trigger high-enough antiviral responses (5, 7, 10, 37, 38). HCV has evolved multiple strategies to evade the host innate immune response for its long-term persistence. We have demonstrated here that Linc-Pint overexpression significantly induced IFN gene expression in HCV-replicating Huh7.5 cells. To investigate the mechanism, we identified that Linc-Pint interacts with DDX24 protein; downregulates its expression, resulting in a disruption of the DDX24-RIP1 axis; and activates IRF7-mediated interferon signaling.
Type I interferons are a family of cytokines that function primarily to elicit immune responses and serve as important components of protective immunity against viral infection. lncRNAs have been reported to play important roles in the regulation of the IFN response and ISG induction during HCV infection (25, 39). HCV may be involved in the deregulation of the expression of several lncRNAs, which favors viral replication and persistence. Overexpression of lncRNA-IFI6 has been implicated in the control of antiviral gene IFI6 expression by histone modification and the induction of HCV growth (40). Very few lncRNAs have been described that increase after HCV infection and reduce viral replication. One appropriate example is lncRNA GAS5 (growth arrest-specific transcript 5), which is upregulated after HCV infection, and the GAS5 5′ end binds viral NS3 protein and blocks its function (41). NS3 is a protease involved in cleaving cellular factors that activate the immune response, such as MAVS and TRIF (42). Thus, GAS5 induction and NS3 blocking lead to increased levels of MAVS and TRIF and increased ISG expression for an antiviral role in HCV infection. lncRNAs were also found to interact with different cellular proteins to modulate IFN signaling. lncRNA lung cancer-associated transcript 1 (LUCAT1), which acts as a negative regulator that limits the type I IFN response in human myeloid cells, was identified to interact with STAT1 in the nucleus, resulting in the alteration of STAT1 function (38).
We show that the overexpression of Linc-Pint significantly increases IFN-α/β expression in hepatocytes by interacting with DDX24 protein. DDX24 belongs to the DExH/D family, which contains at least 59 proteins conserved from bacteria to humans. DExH/D helicases are broadly involved in many RNA-related processes such as transcription, translation, ribosome biogenesis, and RNA transportation (43). In addition to RIG-I, MDA5, and LGP2, DExH/D helicases have also been reported to be key sensors in RNA virus-mediated innate immune signaling processes (44). DDX24 interacts with RIP1 and acts as a competitive protein to disrupt the association between RIP1 and IRF7, thereby negatively regulating the IFN pathway (26). The loss of RIP1 leads to defective type I IFN production and significantly increased susceptibility to RNA virus infection (45). IRF7 is a master regular of IFN-α production. IRF7 undergoes phosphorylation when activated and translocates into the nucleus. It amplifies the type I IFN response by inducing IFN-α expression, which also acts in both autocrine and paracrine manners through the IFN-α/β receptor (9). Linc-Pint-mediated enhancement of IFN-α14 activity is implicated in the disruption of the DDX24-RIP1 axis. Together, our results suggest that Linc-Pint acts as a positive regulator of innate immune pathways, especially interferon signaling, and that HCV inhibits Linc-Pint expression for the establishment of chronic infection. Our observations contribute to the understanding of the mechanistic role of Linc-Pint in HCV-host interactions toward pathogenesis (Fig. 6). These results will help in understanding the molecular basis of lncRNA-mediated regulation of host innate immune responses and identifying a novel molecular mechanism of innate immune regulation by HCV.
FIG 6.
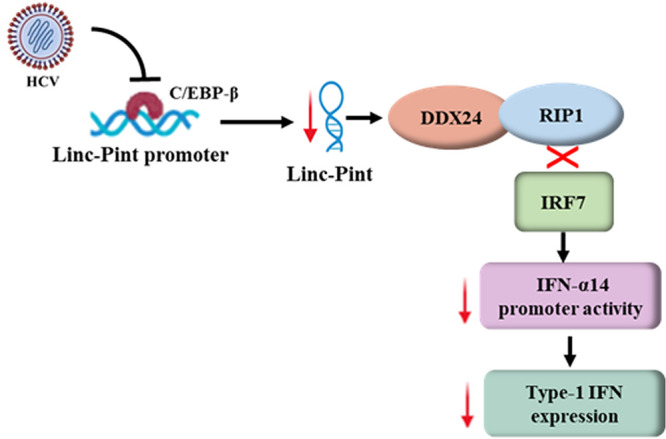
Schematic diagram showing the mechanism of HCV-mediated suppression of IFN signaling by downregulating Linc-Pint expression. Down arrows (red color) denote downregulation.
MATERIALS AND METHODS
Cloning of Linc-Pint promoter constructs.
A 2.5-kb Linc-Pint promoter region (bp −2063 to +448 [chr7]) containing the binding sites of transcriptional factors and cofactors was PCR amplified from Huh7.5 genomic DNA using specific primers (Table 1). The fragment was digested with KpnI and HindIII restriction enzymes and cloned into the pGL3 Basic vector (P0). Two deletion mutant constructs of the Linc-Pint promoter region (P1 and P2) were PCR amplified from the full-length Linc-Pint promoter plasmid (P0) and cloned into the pGL3 Basic vector similarly. Full-length and deletion mutant constructs of the Linc-Pint promoter were used for luciferase assays.
TABLE 1.
List of primer pairs
| Primer target | Sequence |
|---|---|
| Cloning | |
| Linc-Pint full-length promoter (P0) | Forward, 5′-AAAGACAATTGGTACCCAAAGTGATG-3′ |
| Reverse, 5′-ATCTCTCCCAAGCTTTACCTTCCTATC-3′ | |
| Linc-Pint P1 | Forward, 5′-AAAGACAATTGGTACCCAAAGTGATG-3′ |
| Reverse, 5′-GTTGACCCCAAGCTTCACAGAGGG-3′ | |
| Linc-Pint P2 | Forward, 5′-TACGGCCTGGGTACCTCTCAGCTG-3′ |
| Reverse, 5′-ATCTCTCCCAAGCTTTACCTTCCTATC-3′ | |
| ChIP assay | |
| Linc-Pint promoter | Forward, 5′-CGCCTCCTTCTTGCATGACAC-3′ |
| Reverse, 5′-CACAGTGGGGCCAAGGATC-3′ | |
| PCR | |
| Linc-Pint | Forward, 5′-CGTGGGAGCCCCTTTAAGTT-3′ |
| Reverse, 5′-GGGAGGTGGCGTAGTTTCTC-3′ | |
Cell culture, transfection, and infection.
Huh7.5 cells and Huh7.5 cells harboring the genome-length HCV replicon (Rep2a cells) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin at 37°C in a 5% CO2 atmosphere. Cells were seeded into a 6-well plate at a density of 3 × 105 cells/well and transfected with 1 μg/well of Linc-Pint promoter constructs (P0, P1, and P2) and pcDNA3 Linc-Pint plasmid DNA (GenBank accession no. BC130416) using Lipofectamine (Invitrogen). Cells were harvested for luciferase assays and RNA/protein analyses.
Transfection of C/EBP-β small interfering RNA (siRNA) into Huh7.5 cells was performed using Lipofectamine RNAiMAX (Invitrogen). Briefly, Huh7.5 cells were transfected with 25 nM C/EBP-β siRNA or control siRNA (Santa Cruz Biotechnology). After 48 h of transfection, cells were harvested for luciferase assays as described previously (24).
HCV genotype 2a (clone JFH1) was grown in Huh7.5 cells as described previously (18). For infection, Huh7.5 cells were incubated with HCV JFH1 (multiplicity of infection [MOI] = 1.0), and cells were harvested for RNA/protein analyses and ChIP assays.
RNA isolation and quantitative real-time PCR.
Total RNA was isolated using TRIzol reagent (Invitrogen), and cDNA was generated by reverse transcription using random hexamers and a Superscript III reverse transcriptase kit (Invitrogen). For gene expression, quantitative real-time PCR (qRT-PCR) was performed with a 7500 real-time PCR system using SYBR green PCR master mix and specific primer pairs (Table 1). 18S rRNA was used as an internal control. IFN-α, IFN-β, IFIT1, IFIT2, IFIM1, OAS1, and IFI27 genes were quantified using a TaqMan assay (IFN-α, assay identifier Hs00353738_s1; IFN-β, Hs02621180_s1; IFIT1, Hs00356631_91; IFIT2, Hs00533665_M1; IFITM1, Hs00705137-S1; OAS1, Hs00271467_M1; IFI27, Hs0027146_M1; HCV RNA, AI6Q1GI). Relative gene expression was analyzed by the 2−ΔΔCT formula (ΔΔCT = ΔCT of the sample − ΔCT of the untreated control).
Western blot analysis.
Cell lysates from the vector control or Linc-Pint-overexpressing and HCV-infected cells were subjected to SDS-PAGE and transferred onto a nitrocellulose membrane. The membrane was blocked in 5% nonfat dried milk and incubated with specific primary antibody overnight at 4°C, followed by incubation with a secondary antibody conjugated to horseradish peroxidase (HRP) for 1 h. Proteins were detected by an enhanced chemiluminescence for Western blotting substrate (Thermo Fisher Scientific). Membranes were reprobed with HRP-conjugated β-actin antibody (Santa Cruz Biotechnology) to determine the protein load. Densitometric analyses of protein band images were performed using ImageJ software. Commercially available antibodies to DDX24 (catalog no. NB100-2226; Novus Biologicals), p-STAT1 (Y701) and STAT1 (Cell Signaling), IRF7 (catalog no. 9038; Santa Cruz Biotechnology), and RIP1 (catalog no. 610459; BD Biosciences) were used for Western blot analysis.
RNA pulldown and mass spectrometry.
To identify the binding partner of Linc-Pint, pulldown and mass spectrometry were performed as described previously (18). Briefly, Linc-Pint sense or antisense RNA was in vitro transcribed from the pcDNA3 Linc-Pint plasmid (500 ng) using biotin RNA labeling mix and T7 or SP6 RNA polymerase (AmpliScribe T7-Flash transcription kit; Lucigen) as described previously (18). Purified biotinylated sense or antisense Linc-Pint RNA (20 pmol) was labeled with streptavidin magnetic beads (Pierce, Thermo Fisher Scientific) for 1 h at room temperature. Huh7.5 cells harboring the genome-length HCV replicon (Rep2a cells) were lysed in IP buffer (25 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM EDTA, 5% glycerol, 1% NP-40, 1× protease inhibitor, and 100 U/ml RNase inhibitor), and an RNA pulldown assay was performed by using a Pierce magnetic RNA-protein pulldown kit (Thermo Fisher Scientific). Cell lysates (5 mg) were incubated with beads containing sense or antisense RNA for 3 h at 4°C. After washing the beads, the bound proteins were eluted with elution buffer at 37°C for 1 h with agitation. The eluted proteins were used for mass spectrometric analysis. The mass spectrometer used for data acquisition was a Thermo Q-Exactive system. Peptides were separated on an EASYnLC system with a Thermo ES803 PepMap C18 column; data were acquired in DDA (data-dependent acquisition) mode (top 10 m/z values for MS2 per cycle). Candidate proteins were defined as those having at least 2-fold enrichment for the sense strand compared to antisense RNA pulldown. In another set of experiments, the beads containing RNA were incubated with extracts of Huh7.5 cells, followed by Western blot analysis.
RNA immunoprecipitation.
Huh7.5 cells were lysed with IP lysis buffer (25 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM EDTA, 5% glycerol, 1% NP-40, 1× protease inhibitor, and 100 U/ml RNase inhibitor). Cell lysates were centrifuged and incubated overnight with antibody against DDX24 (catalog no. NB100-2226; Novus Biologicals) or an isotype control antibody at 4°C. Cell lysates were incubated with protein G-Sepharose beads (Amersham Biosciences) for 2 h. After washing, RNA was isolated from beads using TRIzol reagent, cDNA was synthesized, and the relative enrichment of Linc-Pint RNA was examined by qRT-PCR as described above.
Coimmunoprecipitation.
Huh7.5 cells were transfected with the vector control or the pcDNA3 Linc-pint plasmid. After 48 h of transfection, cells were lysed using IP lysis buffer (25 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM EDTA, 5% glycerol, 1% NP-40, 1× protease inhibitor, and 100 U/ml RNase inhibitor). Cell lysates were clarified by centrifugation at 14,000 rpm for 20 min, and small portions of the cell lysates were separated as the input samples for the detection of specific proteins by Western blotting. The remaining lysates were incubated with antibody against DDX24 (catalog no. NB100-2226; Novus Biologicals) or RIP1 overnight at 4°C. Cell lysates were incubated with protein G-Sepharose beads (Amersham Biosciences) for 5 h at 4°C. After washing the beads three times with lysis buffer, immunoprecipitated proteins were boiled for 10 min in sample buffer and subjected to Western blot analysis using specific antibodies.
ChIP assay.
Mock-treated or HCV-infected Huh7.5 cells were selected for ChIP assays using the SimpleCHIP enzymatic chromatin IP kit (Cell Signaling Technologies). Briefly, 1 × 107 cells were fixed with 1% formaldehyde for 10 min at room temperature, followed by quenching of the formaldehyde with 10× glycine for 5 min at room temperature. Cells were lysed, and the nuclear fraction was pelleted by centrifugation. The chromatin was digested to a length of approximately 150 to 900 bp by micrococcal nuclease digestion for 20 min at 37°C. After brief sonication to rupture the nuclear membrane, the lysate was immunoprecipitated with 5 μg of anti-C/EBP-β antibody (clone C-19, catalog no. SC-150). Antibody against normal rabbit IgG (catalog no. 2729) was used as a negative control. Elution of protein-DNA complexes was performed using ChIP-grade protein G magnetic beads (catalog no. 9006). Next, reverse cross-linking of protein-DNA complexes to free the DNA was performed using ChIP elution buffer. The DNA was purified, and enrichment of Linc-Pint promoter DNA in immunoprecipitated samples was evaluated by qRT-PCR using ChIP primers for the site of C/EBP-β binding to the Linc-Pint promoter (Table 1).
Statistical analysis.
All the experiments were performed at least in triplicates, and data are presented as means ± standard deviations (SD). Statistical analysis was performed by Student’s t test with a two-tailed distribution. A P value of <0.05 was considered statistically significant.
ACKNOWLEDGMENTS
We thank Oskar Marín-Béjar for providing Linc-Pint plasmid DNA.
This work was supported by the National Institutes of Health (DK081817 and DK113645).
We have no potential conflict of interest to report.
Contributor Information
Ratna B. Ray, Email: ratna.ray@health.slu.edu.
J.-H. James Ou, University of Southern California.
REFERENCES
- 1.Roger S, Ducancelle A, Le Guillou-Guillemette H, Gaudy C, Lunel F. 2021. HCV virology and diagnosis. Clin Res Hepatol Gastroenterol 45:101626. 10.1016/j.clinre.2021.101626. [DOI] [PubMed] [Google Scholar]
- 2.Khatun M, Ray RB. 2019. Mechanisms underlying hepatitis C virus-associated hepatic fibrosis. Cells 8:1249. 10.3390/cells8101249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schoggins JW, Rice CM. 2011. Interferon-stimulated genes and their antiviral effector functions. Curr Opin Virol 1:519–525. 10.1016/j.coviro.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Metz P, Dazert E, Ruggieri A, Mazur J, Kaderali L, Kaul A, Zeuge U, Windisch MP, Trippler M, Lohmann V, Binder M, Frese M, Bartenschlager R. 2012. Identification of type I and type II interferon-induced effectors controlling hepatitis C virus replication. Hepatology 56:2082–2093. 10.1002/hep.25908. [DOI] [PubMed] [Google Scholar]
- 5.Rosen HR. 2013. Emerging concepts in immunity to hepatitis C virus infection. J Clin Invest 123:4121–4130. 10.1172/JCI67714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Horner SM. 2014. Activation and evasion of antiviral innate immunity by hepatitis C virus. J Mol Biol 426:1198–1209. 10.1016/j.jmb.2013.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patra T, Ray RB, Ray R. 2019. Strategies to circumvent host innate immune response by hepatitis C virus. Cells 8:274. 10.3390/cells8030274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bowie AG, Unterholzner L. 2008. Viral evasion and subversion of pattern-recognition receptor signalling. Nat Rev Immunol 8:911–922. 10.1038/nri2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raychoudhuri A, Shrivastava S, Steele R, Dash S, Kanda T, Ray R, Ray RB. 2010. Hepatitis C virus infection impairs IRF-7 translocation and alpha interferon synthesis in immortalized human hepatocytes. J Virol 84:10991–10998. 10.1128/JVI.00900-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heim MH. 2013. Innate immunity and HCV. J Hepatol 58:564–574. 10.1016/j.jhep.2012.10.005. [DOI] [PubMed] [Google Scholar]
- 11.Chowdhury JB, Kim H, Ray R, Ray RB. 2014. Hepatitis C virus NS5A protein modulates IRF-7-mediated interferon-alpha signaling. J Interferon Cytokine Res 34:16–21. 10.1089/jir.2013.0038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ray RB, Ray R. 2019. Hepatitis C virus manipulates humans as its favorite host for a long-term relationship. Hepatology 69:889–900. 10.1002/hep.30214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iyer MK, Niknafs YS, Malik R, Singhal U, Sahu A, Hosono Y, Barrette TR, Prensner JR, Evans JR, Zhao S, Poliakov A, Cao X, Dhanasekaran SM, Wu YM, Robinson DR, Beer DG, Feng FY, Iyer HK, Chinnaiyan AM. 2015. The landscape of long noncoding RNAs in the human transcriptome. Nat Genet 47:199–208. 10.1038/ng.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Balas MM, Johnson AM. 2018. Exploring the mechanisms behind long noncoding RNAs and cancer. Noncoding RNA Res 3:108–117. 10.1016/j.ncrna.2018.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cipolla GA, de Oliveira JC, Salviano-Silva A, Lobo-Alves SC, Lemos DS, Oliveira LC, Jucoski TS, Mathias C, Pedroso GA, Zambalde EP, Gradia DF. 2018. Long non-coding RNAs in multifactorial diseases: another layer of complexity. Noncoding RNA 4:13. 10.3390/ncrna4020013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin C, Yang L. 2018. Long noncoding RNA in cancer: wiring signaling circuitry. Trends Cell Biol 28:287–301. 10.1016/j.tcb.2017.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rajput R, Sharma J, Nair MT, Khanna M, Arora P, Sood V. 2020. Regulation of host innate immunity by non-coding RNAs during dengue virus infection. Front Cell Infect Microbiol 10:588168. 10.3389/fcimb.2020.588168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khatun M, Sur S, Steele R, Ray R, Ray RB. 24November2020. Inhibition of long noncoding RNA Linc-Pint by hepatitis C virus in infected hepatocytes enhances lipogenesis. Hepatology 10.1002/hep.31656. [DOI] [PMC free article] [PubMed]
- 19.Wang Y, Wang P, Zhang Y, Xu J, Li Z, Li Z, Zhou Z, Liu L, Cao X. 2020. Decreased expression of the host long-noncoding RNA-GM facilitates viral escape by inhibiting the kinase activity TBK1 via S-glutathionylation. Immunity 53:1168–1181.e7. 10.1016/j.immuni.2020.11.010. [DOI] [PubMed] [Google Scholar]
- 20.Imamura K, Imamachi N, Akizuki G, Kumakura M, Kawaguchi A, Nagata K, Kato A, Kawaguchi Y, Sato H, Yoneda M, Kai C, Yada T, Suzuki Y, Yamada T, Ozawa T, Kaneki K, Inoue T, Kobayashi M, Kodama T, Wada Y, Sekimizu K, Akimitsu N. 2014. Long noncoding RNA NEAT1-dependent SFPQ relocation from promoter region to paraspeckle mediates IL8 expression upon immune stimuli. Mol Cell 53:393–406. 10.1016/j.molcel.2014.01.009. [DOI] [PubMed] [Google Scholar]
- 21.Ouyang J, Zhu X, Chen Y, Wei H, Chen Q, Chi X, Qi B, Zhang L, Zhao Y, Gao GF, Wang G, Chen JL. 2014. NRAV, a long noncoding RNA, modulates antiviral responses through suppression of interferon-stimulated gene transcription. Cell Host Microbe 16:616–626. 10.1016/j.chom.2014.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Winterling C, Koch M, Koeppel M, Garcia-Alcalde F, Karlas A, Meyer TF. 2014. Evidence for a crucial role of a host non-coding RNA in influenza A virus replication. RNA Biol 11:66–75. 10.4161/rna.27504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mazumdar B, Kim H, Meyer K, Bose SK, Di Bisceglie AM, Ray RB, Ray R. 2012. Hepatitis C virus proteins inhibit C3 complement production. J Virol 86:2221–2228. 10.1128/JVI.06577-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sasaki R, Sur S, Cheng Q, Steele R, Ray RB. 2019. Repression of microRNA-30e by hepatitis C virus enhances fatty acid synthesis. Hepatol Commun 3:943–953. 10.1002/hep4.1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Valadkhan S, Fortes P. 2018. Regulation of the interferon response by lncRNAs in HCV infection. Front Microbiol 9:181. 10.3389/fmicb.2018.00181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma Z, Moore R, Xu X, Barber GN. 2013. DDX24 negatively regulates cytosolic RNA-mediated innate immune signaling. PLoS Pathog 9:e1003721. 10.1371/journal.ppat.1003721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huye LE, Ning S, Kelliher M, Pagano JS. 2007. Interferon regulatory factor 7 is activated by a viral oncoprotein through RIP-dependent ubiquitination. Mol Cell Biol 27:2910–2918. 10.1128/MCB.02256-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Landeras-Bueno S, Ortin J. 2016. Regulation of influenza virus infection by long non-coding RNAs. Virus Res 212:78–84. 10.1016/j.virusres.2015.08.008. [DOI] [PubMed] [Google Scholar]
- 29.Agliano F, Rathinam VA, Medvedev AE, Vanaja SK, Vella AT. 2019. Long noncoding RNAs in host-pathogen interactions. Trends Immunol 40:492–510. 10.1016/j.it.2019.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Damas ND, Fossat N, Scheel TKH. 2019. Functional interplay between RNA viruses and non-coding RNA in mammals. Noncoding RNA 5:7. 10.3390/ncrna5010007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kotzin JJ, Mowel WK, Henao-Mejia J. 2017. Viruses hijack a host lncRNA to replicate. Science 358:993–994. 10.1126/science.aar2300. [DOI] [PubMed] [Google Scholar]
- 32.Marin-Bejar O, Marchese FP, Athie A, Sanchez Y, Gonzalez J, Segura V, Huang L, Moreno I, Navarro A, Monzo M, Garcia-Foncillas J, Rinn JL, Guo S, Huarte M. 2013. Pint lincRNA connects the p53 pathway with epigenetic silencing by the Polycomb repressive complex 2. Genome Biol 14:R104. 10.1186/gb-2013-14-9-r104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ray RB, Steele R, Meyer K, Ray R. 1997. Transcriptional repression of p53 promoter by hepatitis C virus core protein. J Biol Chem 272:10983–10986. 10.1074/jbc.272.17.10983. [DOI] [PubMed] [Google Scholar]
- 34.Majumder M, Ghosh AK, Steele R, Ray R, Ray RB. 2001. Hepatitis C virus NS5A physically associates with p53 and regulates p21/waf1 gene expression in a p53-dependent manner. J Virol 75:1401–1407. 10.1128/JVI.75.3.1401-1407.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bittar C, Shrivastava S, Bhanja Chowdhury J, Rahal P, Ray RB. 2013. Hepatitis C virus NS2 protein inhibits DNA damage pathway by sequestering p53 to the cytoplasm. PLoS One 8:e62581. 10.1371/journal.pone.0062581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mitchell JK, Midkiff BR, Israelow B, Evans MJ, Lanford RE, Walker CM, Lemon SM, McGivern DR. 2017. Hepatitis C virus indirectly disrupts DNA damage-induced p53 responses by activating protein kinase R. mBio 8:e00121-17. 10.1128/mBio.00121-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chigbu DI, Loonawat R, Sehgal M, Patel D, Jain P. 2019. Hepatitis C virus infection: host-virus interaction and mechanisms of viral persistence. Cells 8:376. 10.3390/cells8040376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Agarwal S, Vierbuchen T, Ghosh S, Chan J, Jiang Z, Kandasamy RK, Ricci E, Fitzgerald KA. 2020. The long non-coding RNA LUCAT1 is a negative feedback regulator of interferon responses in humans. Nat Commun 11:6348. 10.1038/s41467-020-20165-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Unfried JP, Fortes P. 2020. LncRNAs in HCV infection and HCV-related liver disease. Int J Mol Sci 21:2255. 10.3390/ijms21062255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu X, Duan X, Holmes JA, Li W, Lee SH, Tu Z, Zhu C, Salloum S, Lidofsky A, Schaefer EA, Cai D, Li S, Wang H, Huang Y, Zhao Y, Yu ML, Xu Z, Chen L, Hong J, Lin W, Chung RT. 2019. A long noncoding RNA regulates hepatitis C virus infection through interferon alpha-inducible protein 6. Hepatology 69:1004–1019. 10.1002/hep.30266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qian X, Xu C, Zhao P, Qi Z. 2016. Long non-coding RNA GAS5 inhibited hepatitis C virus replication by binding viral NS3 protein. Virology 492:155–165. 10.1016/j.virol.2016.02.020. [DOI] [PubMed] [Google Scholar]
- 42.Moradpour D, Penin F. 2013. Hepatitis C virus proteins: from structure to function. Curr Top Microbiol Immunol 369:113–142. 10.1007/978-3-642-27340-7_5. [DOI] [PubMed] [Google Scholar]
- 43.Cordin O, Banroques J, Tanner NK, Linder P. 2006. The DEAD-box protein family of RNA helicases. Gene 367:17–37. 10.1016/j.gene.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 44.Taschuk F, Cherry S. 2020. DEAD-box helicases: sensors, regulators, and effectors for antiviral defense. Viruses 12:181. 10.3390/v12020181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Balachandran S, Venkataraman T, Fisher PB, Barber GN. 2007. Fas-associated death domain-containing protein-mediated antiviral innate immune signaling involves the regulation of Irf7. J Immunol 178:2429–2439. 10.4049/jimmunol.178.4.2429. [DOI] [PubMed] [Google Scholar]