

ABSTRACT
Uncharacterized viral genomes that encode circular replication-associated proteins of single-stranded DNA viruses have been discovered by metagenomics/metatranscriptomics approaches. Some of these novel viruses are classified in the newly formed family Genomoviridae. Here, we determined the host range of a novel genomovirus, SlaGemV-1, through the transfection of Sclerotinia sclerotiorum with infectious clones. Inoculating with the rescued virions, we further transfected Botrytis cinerea and Monilinia fructicola, two economically important members of the family Sclerotiniaceae, and Fusarium oxysporum. SlaGemV-1 causes hypovirulence in S. sclerotiorum, B. cinerea, and M. fructicola. SlaGemV-1 also replicates in Spodoptera frugiperda insect cells but not in Caenorhabditis elegans or plants. By expressing viral genes separately through site-specific integration, the replication protein alone was sufficient to cause debilitation. Our study is the first to demonstrate the reconstruction of a metagenomically discovered genomovirus without known hosts with the potential of inducing hypovirulence, and the infectious clone allows for studying mechanisms of genomovirus-host interactions that are conserved across genera.
IMPORTANCE Little is known about the exact host range of widespread genomoviruses. The genome of soybean leaf-associated gemygorvirus-1 (SlaGemV-1) was originally assembled from a metagenomic/metatranscriptomic study without known hosts. Here, we rescued SlaGemV-1 and found that it could infect three important plant-pathogenic fungi and fall armyworm (S. frugiperda Sf9) insect cells but not a model nematode, C. elegans, or model plant species. Most importantly, SlaGemV-1 shows promise for inducing hypovirulence of the tested fungal species in the family Sclerotiniaceae, including Sclerotinia sclerotiorum, Botrytis cinerea, and Monilinia fructicola. The viral determinant of hypovirulence was further identified as replication initiation protein. As a proof of concept, we demonstrate that viromes discovered in plant metagenomes can be a valuable genetic resource when novel viruses are rescued and characterized for their host range.
KEYWORDS: Botrytis cinerea, genomovirus, mycovirus, Sclerotinia sclerotiorum, biocontrol, gemygorvirus, virulence determinants
INTRODUCTION
In most high-throughput metagenomic/metatranscriptomic virome sequencing studies, the hosts in which the novel viruses replicate have not been established, and therefore, the etiology, symptoms, and prognosis remain unclear. These new viruses could represent either potential animal and plant health threats or untapped genetic resources (1, 2) for alleviating biotic/abiotic stress (3, 4). Critical efforts are needed to characterize these novel viruses to determine if they are “friends or foes” for animal and plant health. In addition, viruses discovered through high-throughput sequencing (HTS) methods could be reconstructed and engineered as vectors to transfect specific hosts, to develop gene editing for therapeutic purposes or gene-driven strategies to eliminate pests (5).
Members of the recently established family Genomoviridae possess single circular replication-associated protein (Rep) encoded single-stranded DNA (ssDNA) (CRESS DNA) genomes (6, 7). These ssDNA viruses have been discovered in remarkably diverse environments with the use of rolling circle amplification (RCA) and cost-effective metagenomic HTS (8). Most have been detected from complex sources, including plants, cattle, insects, birds, predatory and prey mammals, and even humans, where the hosts have not been confirmed (1, 9–20). A recent review concluded that the detection of ssDNA viruses in clinical cases was incidental and that no evidence of causality exists (21). Genomoviruses have been detected in arthropods, including dragonflies (22), mosquitoes (23), pine beetles (19), thrips (24), ticks (25), and whiteflies (26). So far, only two genomoviruses have been detected in isolates of the fungi Sclerotinia sclerotiorum (27, 28) and Fusarium graminearum (29).
The capsid proteins of the genomoviruses show only low levels of amino acid sequence similarity to CP proteins from animal circoviruses, plant nanoviruses, and geminiviruses (30). Like circoviruses, nanoviruses, and geminiviruses, genomoviruses replicate via an RCA mechanism similar to replication of bacterial plasmids. In fact, it has been hypothesized that CRESS DNA viruses evolved from a fusion of bacterial plasmids and structural proteins from positive-strand RNA viruses, via the rolling circle mode of replication (31). Moreover, the type species of Genomoviridae, Sclerotinia sclerotiorum hypovirulence-associated DNA virus 1 (SsHADV-1), which was the first genomovirus infectious clone constructed (32), replicates in both fungi and insects (27), suggesting that insects may be the main reservoirs of genomoviruses, which could explain why fungi infected by genomoviruses are rare in nature.
Soybean-leaf associated gemycircularvirus 1 (SlaGemV-1) (NCBI accession no. NC_028460) has a 2,234-nucleotide (nt) circular ssDNA genome that encodes two proteins, the 298-amino-acid (aa) capsid protein (CP) and the 332-aa replication-associated protein (Rep) in reverse orientation (1). The SlaGemV-1 genome contains a stem-loop nonanucleotide structure thought to promote the replicational release of virus molecules through RCA, which is similar to other CRESS DNA viruses. The SlaGemV-1 genome contains an intron of 156 nt within the Rep coding region and is classified in the genus Gemygorvirus (1). In contrast, SsHADV-1 is classified in the genus Gemycircularvirus, and its Rep gene sequences does not contain an intron. The CP and Rep amino acid sequences in the two viruses share only 45% and 44% identity, respectively.
The effect of CRESS DNA viral proteins on the host is of great interest because of several evolutionary and ecological advantages that favor emergence and reemergence of this group of viruses (33). In this study, we developed a reverse genetic system for SlaGemV-1, which was discovered from the metatranscriptome of plant leaves without known hosts (11). The complete viral genome was amplified from the soybean DNA extracts and used to construct multimeric infectious clones. Based on its similarity to SsHDAV-1, we hypothesized that SlaGemV-1 could replicate in and debilitate the soybean fungal pathogen S. sclerotiorum. Indeed, the reconstructed virus genome replicated in S. sclerotiorum had produced viral particles. Purified virions were subsequently used to examine SlaGemV-1’s ability to infect other fungi in the family Sclerotiniaceae, including Botrytis cinerea and Monilinia fructicola. Moreover, we identified the viral determinant of hypovirulence by site-specific expression of each SlaGemV-1 protein in one of the hosts we identified, i.e., B. cinerea.
RESULTS
Reconstruction of SlaGemV-1.
The novel virus, SlaGemV-1, was discovered in a metatranscriptomics analysis of field-grown soybean leaves by HTS (1). Two Escherichia coli clones were constructed with one copy of the viral genome (1.1-mer) and 2 copies of the viral genome (2-mer); these copies of the SlaGemV-1 genome were constructed to contain two copies of the nonanucleotide structure. To determine the infectivity of viral genome DNA constructs, the plasmids carrying the viral genomic DNA were transfected to protoplasts of S. sclerotiorum strain DK3 (SsDK3), which is free of virus infection. After at least six transfers to remove residual DNA contamination from transfection, fungal tissue was subjected to extraction to obtain genomic DNA for PCR amplification using primers pjetR and 3′R, resulting in a 2.2-kb amplicon (Fig. 1A). Sanger sequencing of the resulting PCR product confirmed the presence of intact SlaGemV-1 genomes (Fig. 1B). Next, the RCA product was used as the template for PCR using primers 5′F and 3′R, which also resulted in 2.2-kb bands from both 2-mer-transfected and 1.1-mer-transfected cultures (Fig. 1C). Initially, S. sclerotiorum cultures transfected with the 2-mer clone were more debilitated than cultures transfected with the 1.1-mer, suggesting more efficient initiation of replication in the host cells from the 2-mer clone than from the 1.1-mer. However, cultures transfected with the 1.1-mer stably maintained the viral infection and generated a very different morphology, with white aerial mycelia without sectoring (Fig. 1D). Because the 1.1-mer-transfected culture resulted in stable infection, it was used for subsequent studies, including the purification of virions for transmission electron microscopy. As shown in Fig. 1E, icosahedral virus-like particles were observed in SlaGemV-1-transfected S. sclerotium with diameters around 20 to 25 nm. A 2.2-kb band was directly visualized on an agarose gel from the total DNA extracted from sucrose gradient-purified viral particles (Fig. 1F). At the time when the virus was rescued successfully, we observed mutations from 26G to A (R9H) in CP and 873A to G (L277P) in Rep. However, in the subsequent weekly serial transfers for 4 years, we did not see further mutations, suggesting that the initial nucleotide changes were due to either self-correction, sometimes found in the process of rescuing a virus, or adaptation to new hosts. The absence of further mutations differs from the reported equal mutation rates for ssDNA viruses and RNA viruses (34).
FIG 1.
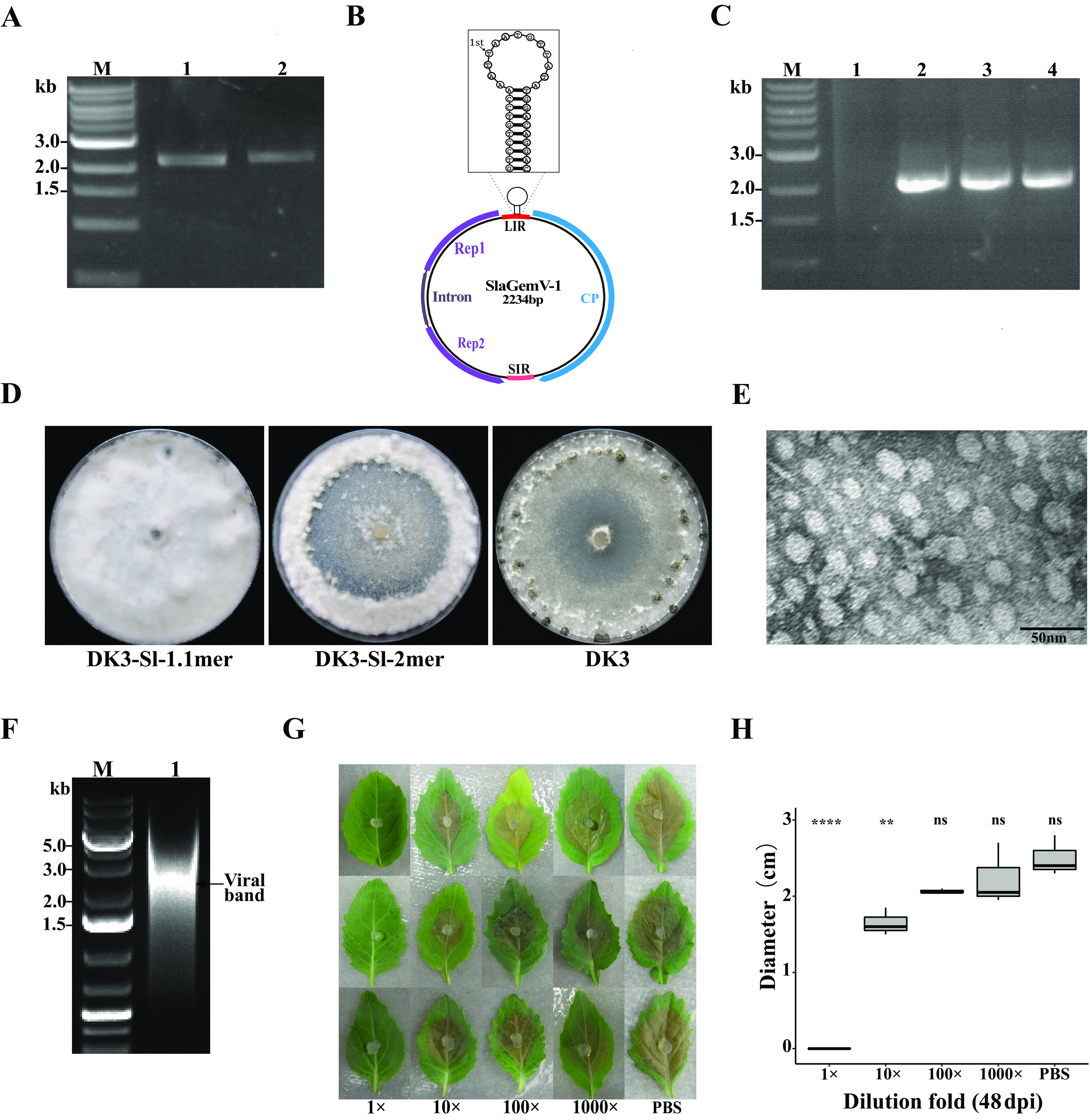
SlaGemV-1 replication in Sclerotinia sclerotiorum after transfection with infectious clones was confirmed. (A) PCR amplification from extracted mycelial DNA of SlaGemV-1-transfected S. sclerotiorum using primers pJetR and 3′R. Lanes: M, 1-kb ladder; 1, 1.1-mer transfection; 2, 2-mer transfection. (B) SlaGemV-1 genome organization with the nonanucleotide structure specified. (C) PCR amplification with primers pJetR and 3′R and with the rolling circle amplification product from the transfected S. sclerotiorum mycelial DNA as the template. The resulting PCR products were subjected to Sanger sequencing to confirm their identities. Lanes: M, 1-kb ladder; 1, RCA product with a smear; 2, PCR product using the RCA product from the 1.1-mer-transfected culture as the template; 3, PCR product of RCA product from 2-mer-transfected culture; 4, positive control from PCR product without RCA from the transfected culture. (D) Comparison of virus-transfected S. sclerotiorum (strain DK3) grown on PDA for 21 days after transfection with pJet-SlaGemV-1 1.1-mer and 2-mer constructs. (Left) pJet-SlaGemV-1 1.1-mer transfection; (middle) pJet-SlaGemV-1-2-mer transfection; (right) original DK3 strain, used for comparison. (E) Transmission electron microscopy image of purified SlaGemV-1 particles. (F) Agarose gel (1%) electrophoresis of DNA extracted from 25% sucrose gradient fractions containing virus particles. (G) Viral particles (1 mg/ml) extracted from infected S. sclerotiorum mycelia were diluted to different concentrations (1 mg/ml, 0.1 mg/ml, 0.01 mg/ml, and 0.001 mg/ml; PBS, phosphate-buffered saline control) used as inoculants (10 µl) on virus free agar plug to inhibit fungal growth. As the viral particles were resuspended in PBS, all dilution used PBS. Each row represents one replication for three replications shown here. (H) Dose-response relationship of undiluted and diluted treatments of viral particles compared to the PBS control. Two-way analysis of variance and Bonferroni post hoc test. **, P < 0.01; ****, P <0.0001.
Effects on Sclerotinia sclerotiorum growth and virulence on detached leaves.
Mycelial growth rates of wild-type and SlaGemV-1-transfected S. sclerotiorum were determined on potato dextrose agar (PDA). The morphology of transfected fungi started to show noticeable differences after ∼15 serial transfers. SlaGemV-1-transfected cultures produced delayed, small, or no sclerotia around the edges of PDA plates, compared to the sclerotia from the virus-free cultures, which are larger, irregular in shape, and randomly distributed on the plates. After months of stable growth on PDA, the SlaGemV-1-transfected culture rarely produced sclerotia, lost pigmentation, and did not sector.
The S. sclerotiorum culture stably transfected with the 1.1-mer clone (SsDK3-V1) had a significantly lower growth rate on PDA and less virulence on spinach leaves than the virus-free SsDK3 culture. The virus-free culture grew significantly faster on PDA (P < 0.05) (Fig. 2A) and caused larger lesions (Fig. 2B) at an average of 2.9 cm in diameter 3 days postinoculation (dpi), compared to an average of 0.4 cm when inoculated by SlaGemV-1-infected culture (P < 0.01). To assess the infectivity of SlaGemV-1 particles purified by ultracentrifugation, the viral particles were diluted for inoculation. A gradient of decreasing inhibition of fungal growth was observed with increasing serial dilutions (Fig. 1G). A significantly reduced virulence of the fungal inoculant was observed for 1 mg/ml viral protein concentration up to 100-fold dilution (Fig. 1H). Thus, the result indicates that an increased dose of virus inoculum leads to a greater inhibition of S. sclerotiorum pathogenicity.
FIG 2.

The extracellular transmission of SlaGemV-1 and its induced hypovirulence in fungi in the family Sclerotiniaceae, including Sclerotinia sclerotiorum, Botrytis cinerea, and Monilinia fructicola, were confirmed. Effects of SlaGemV-1 on (A) fungal growth on PDA at 3 days old and (B) virulence at 4 days postinoculation (dpi) of S. sclerotiorum. Effects of SlaGemV-1 on (C) fungal growth on PDA at 4 days old and (D) virulence at 4 dpi of B. cinerea. Effects of SlaGemV-1 on (E) fungal growth on PDA at 3 days old and (F) virulence at 4 dpi of M. fructicola. All resulted in significantly reduced virulence and growth (*, P < 0.05; **, P < 0.01; ns, not significant).
To compare the infectivity of the purified virions on different strains, we also confirmed that SlaGemV-1 particles are capable of infecting three vegetatively incompatible S. sclerotiorum strains, SsDK3, 1980, and 274. To obtain single-cell lineages, protoplasts were made from Sclerotinia mycelia for the culturing of individual cells. When protoplasts from the three strains were transfected with purified virions without polyethylene glycol (PEG), more than 80% of colonies regenerated from single cells were shown to contain the SlaGemV-1 genome, as determined by PCR amplification of the complete virus genome (Fig. 3A). The transfected fungal colonies showed slow growth, exhibited loss of pigmentation, and produced smaller sclerotia compared to the virus-free S. sclerotiorum strains (Fig. 3B). Hence, viral particles of SlaGemV-1 are highly infectious for S. sclerotiorum regardless of the fungal strain.
FIG 3.

The high infectivity of SlaGemV-1 virions was confirmed. (A) Regardless of Sclerotinia sclerotiorum strain, individual protoplasts as single cells were largely infected by the virion inoculation. Three strains were tested: DK3, 1980, and 274. The whole viral genome was amplified by PCR after the regenerated virus-treated fungal cultures were transferred several times. Protoplast suspensions (100 μl, 107 protoplasts/ml) from different strains were mixed gently with viral particles (20 μl, 1 mg/ml). At the same time, the viral genomic DNA was mixed with protoplasts as a control. The mixture was incubated on ice for 40 min and held at room temperature for an additional 30 min; then, 2 ml of regeneration medium was added and shaken gently for 2 h. The virus-treated protoplasts were regenerated at room temperature for 2 to 3 days. The whole SlaGemV-1 genome was amplified and run on a gel for >10 cells for each strain. (Top) Lanes 1 to 10, virus-treated DK3; 11, viral-DNA-treated DK3. (Middle) Lanes 1 to 10, virus-treated strain 274; 11, viral-DNA-treated strain 274. (Bottom) Lanes 1 to 12, virus-treated strain 1980; 13, viral-DNA-treated strain 1980. (B) Phenotypic changes in the virus-infected (left) cultures compared to virus-free (right) cultures of S. sclerotiorum strains on PDA plates. (C) The infectivity of SlaGemV-1 virions was confirmed by PCR on alternative fungal hosts. The whole viral genome (2.2 kb) was amplified from inoculated B. cinerea and M. fructicola. Lanes: M, ladder; 1, virus-infected B. cinerea; 2, virus-infected M. fructicola; 3, virus-free strain of B. cinerea as a negative control; 4, virus-free strain of M. fructicola as a negative control.
Effects of extracellular transmission of SlaGemV-1 on Botrytis cinerea, Monilinia fructicola, and Fusarium oxysporum.
To determine whether SlaGemV-1 infects, stably replicates, and causes hypovirulence in other fungi, Botrytis cinerea and Monilinia fructicola, relatives within the family Sclerotiniaceae, were inoculated extracellularly with purified virions. After serial transfers of inoculated cultures, SlaGemV-1 gene sequences were detected by PCR in DNA extract of B. cinerea, confirming the infectivity of the purified virion (Fig. 3C). SlaGemV-1 also significantly reduced the vegetative growth on PDA and virulence of B. cinerea cultures inoculated on host plants (Fig. 2C and D). Inoculations of M. fructicola with SlaGemV-1 particles also produced significant reductions in growth and virulence compared to virus-free cultures (P < 0.05) (Fig. 2E and F). SlaGemV-1 was detected in M. fructicola after serial transfers of inoculated cultures and PCR amplification of the SlaGemV-1 genome (Fig. 3C). However, transfection of Fusarium oxysporum, which is outside the family Sclerotiniaceae, did not result in infection (data not shown).
Biocontrol potential of SlaGemV-1 against S. sclerotiorum.
To examine whether crude extracts of SlaGemV-1 viral particles used as foliar sprays are protective against future infection by S. sclerotiorum on whole plants, supernatants of centrifuged fungal homogenate of different optical densities (optical density at 600 nm [OD600] = 4.0, 3.0, and 2.0) were sprayed on intact tomato plants. When the plants were inoculated with virulent S. sclerotiorum, we found that smaller lesions were observed on the leaves of tomato plants sprayed with the viral particles at 6 days postinoculation, while the lesions of controls treated with distilled water expanded to the whole leaves and even stems (Fig. 4A). At 16 days postinoculation, all the control tomato plants wilted due to S. sclerotiorum infection, with most of the plants being defoliated (Fig. 4B). However, all the plants sprayed with viral particles were still alive, with the disease severities in the order OD 4 < OD 3 < OD 2. When the OD of the fungal homogenate before centrifugation was greater than or equal to 2, the average lesion size of white mold was significantly smaller (Fig. 4C). Consistently, SlaGemV-1 viral particles purified by the sucrose gradient and ultracentrifugation also protected the detached leaves from infection by S. sclerotiorum (Fig. 1G and H), which suggested that both crude extract and purified SlaGemV-1 viral particles protect the plants.
FIG 4.

The crude extracts of SlaGemV-1 virions protected the tomato plants from S. sclerotiorum infection. (A) Fungal homogenate was prepared by blending fungal hyphae in PDB and using low-speed centrifugation to separate the hyphal fragments as a method of extracting viral particles, diluted to OD600 values of 4.0, 3.0, and 2.0, and sprayed on plants which were inoculated with hyphal agar plugs applied to two leaves per plant. The control was sprayed with double-distilled water. The fungal-pathogen-infected plants were kept in an incubator with 100% humidity at 22°C, and images of the lesions were taken at 6 dpi. (B) Protective activity of SlaGemV-1 against white mold disease. The crude preparation efficiently protected intact plants from infection with S. sclerotiorum when the OD600 of the viral particles was greater than or equal to 2.0. The images of whole plants were taken at 16 dpi. (C) The disease severity was rated at 16 dpi (*, P < 0.05; ****, P < 0.0001). The rating was performed based on a scale of 0 to 5: 0, no disease; 1, restricted lesion on leaf; 2, expanded lesion on leaf; 3, leaf with expanded lesion and start of stem colonization; 4, severe stem colonization; 5, plant completely dead.
Replication in bacterial cells.
To investigate whether SlaGemV-1 replicates in bacteria, and in which replicated form it exists, Southern hybridization was performed to profile the various conformations of the viral replicons in bacteria harboring the infectious-clone plasmid. The open circular double-stranded DNA (dsDNA), linear dsDNA, supercoiled dsDNA, and ssDNA forms were detected in total DNA from E. coli containing the pJet-1.1-mer construct, which represents the various intermediate forms of the rolling circle replication (Fig. 5A). The total DNA extracted from virus-infected S. sclerotiorum strain DK3 was used as a positive control which exhibited the only SlaGemV-1 DNA form that hybridized to the virus-specific probe, and the E. coli strain carrying the pJet-1.2 vector was the negative control. The results showed that E. coli was able to support the DNA replication of the SlaGemV-1 1.1-mer construct as the form of viral rolling circle replication. In addition, the bacterial culture harboring the 2-mer segment in pJet1.2 and POET-1 vectors had an upward shift in the conformations. In comparison to viral replication in E. coli carrying the pJet-1.1-mer construct, the viral ssDNA form was not observed clearly in E. coli containing the SlaGemV-1 DNA forms, generated from the 2-mer constructs. We found that SlaGemV-1 2-mer construct could not generate the replication of viral DNA species in E. coli, suggesting a lack of dissolution mechanism of the multimers in bacteria tested. Moreover, no viral particles were produced, as evidenced by transmission electron microscopy (TEM) images. Hence, we concluded that SlaGemV-1 replicates in bacteria in the form of a plasmid via rolling circle amplification, without the production of virions.
FIG 5.

Replication of the SlaGemV-1 genome in bacteria and insect cells. (A) Southern hybridization to confirm different conformations of the replicative forms of SlaGemV-1 in bacteria. Lanes were loaded with total DNA: 1, virus-infected fungal mycelia; 2, E. coli carrying SlaGemV-1 1.1-mer clone in pJet1.2; 3, E. coli carrying SlaGemV-1 2-mer clone in pJet1.2; 4, E. coli carrying 2-mer clone in pOET-1; 5, E. coli culture without any plasmids. OC, open circular double stranded; Lin, linear; SC, supercoiled double stranded; SS, single-stranded forms. (B) PCR detection of SlaGemV-1 in total DNA extracted from transfected Sf9 insect cells. Lanes: M, ladder, 1, Rep/CP genes amplified from transfected S. sclerotiorum as the positive control for PCR; 2, Rep/CP genes amplified from transfected Sf9 cells; 3, virus-free Sf9 cells as the negative control for PCR. (C) RT-PCR detection of SlaGemV-1 CP/Rep gene expression in total RNA extracted from the supernatant of transfected Sf9 insect cells. Lanes: M, ladder; 1, CP transcript amplified from the supernatant of infected cells; 2, negative amplification of CP transcript from virus-free Sf9 cells; 3, Rep transcript amplified without the intron (650 bp) from the supernatant of transfected Sf9 cells; 4, negative amplification of Rep transcript from the virus-free Sf9 cells. (D) Viral replication kinetics measured at different time points by qPCR in transfected Sf9 cells. (E) Confirmation of positive SlaGemV-1 replication in Sf9 cells. Viral amplification of the cellular and supernatant DNA extracted from the second passage of the infected insect cells. Lanes: 1 and 2, viral CP gene amplification in cellular DNA; 3, viral CP gene amplification in supernatant DNA of virus-infected Sf9 cells; 4, virus-free Sf9 cells; 5 and 6, viral Rep gene amplification in cellular DNA; 7, viral Rep gene amplification in supernatant DNA of virus-infected Sf9 cells; 8, virus-free Sf9 cells. (F) Confirmation by RT-qPCR of the negative replication of SlaGemV-1 in Arabidopsis thaliana mesophyll protoplasts.
Effects of virion inoculation on insect Sf9 cells, Caenorhabditis elegans, and plants.
To test whether SlaGemV-1 replicates in fall armyworm (Spodoptera frugiperda) cells, Sf9 cells were transfected by purified virions and incubated at 27°C for 72 h without passage. Subsequently, cellular DNA was extracted, and both the complete sequences containing CP and Rep genes were amplified by PCR and verified by Sanger sequencing (Fig. 5B). Transcripts of CP and Rep genes were both detected by reverse transcription-PCR (RT-PCR) in total RNA extracted from transfected-cell supernatant, indicating the release of the viral transcripts from the infected and dying cells (Fig. 5C). Furthermore, a time course of the infection by qPCR showed that the titer of the virus increased over time and peaked at 24 h postinoculation (hpi) with direct inoculation of the purified virions. From 48 to 96 hpi, the levels of SlaGemV-1 were relatively stable (Fig. 5D). Moreover, the virus infectivity resulting from the passage of the virus showed that SlaGemV-1 replicates in the second passage of the Sf9 cells, since viral DNA was detected with PCR (Fig. 5E). Taken together, these results indicated that the Sf9 supports SlaGemV-1 replication and serves as a potential host. For Caenorhabditis elegans transfection, we found that SlaGemV-1 was not able to replicate in C. elegans after multiple attempts by both virion inoculation and feeding bacteria harboring the infectious-clone plasmid.
In plants, no replication of SlaGemV-1 was detected in virus-transfected Arabidopsis thaliana protoplasts (Fig. 5F) or in upper noninfiltrated leaves and infiltrated leaves of agroinfiltrated Nicotiana benthamiana based on RT-PCR detection after multiple attempts. RT-PCR detection was the method of choice because the intron would be deleted upon transcription. Additionally, no replication was detected on soybean leaves mechanically inoculated with virions by dusting with carborundum, based on PCR detection.
Identification of viral determinant of hypovirulence.
To identify the direct roles of the two SlaGemV-1 proteins in attenuating fungal pathogenicity, the CP and Rep genes replaced a nonessential bcniaD gene of B. cinerea via homologous recombination and were successfully expressed in B. cinerea individually (Fig. 6A and 7A) as green fluorescent protein (GFP) fusion proteins and were designated BcVB-Rep-S1 and BcVB-CP-S1, respectively. The transcripts of viral CP and Rep without introns were separately detected from transformants of CP- and Rep-expressing strains by RT-PCR to confirm the expression (Fig. 6C and 7B). As shown in Fig. 6B, the phenotypes of severely reduced growth and sparsely distributed hyphae were observed for the transformant (BcVB-Rep-Sl) expressing the Rep protein. The transformant also exhibited impaired pathogenicity, being incapable of causing disease on tomatoes and grapes. The statistical analysis showed that BcVB-Rep-Sl transformant had a significantly reduced growth and lesion length compared to the wild type (P < 0.05). In contrast, the morphology and virulence of the transformant expressing the CP protein, BcVB-CP-Sl, were not significantly different from those of either the wild-type B. cinerea or the control, which is a transformant of the empty OGG vector (Fig. 6B and D to F and Fig. 7C and D). The result suggests that the viral Rep protein was the main determinant of hypovirulence of the fungal host. We also obtained nonfusion mutants of SlaGemV-1 Rep but did not characterize those further. As the existing expression vectors confirmed that fusions occurring at the specific site do not have any effect as long as the nutrient demands are met (35), we utilized the system to answer our inquiries after screening for fusion mutants.
FIG 6.

Site-specific homologous expression of SlaGemV-1 CP or REP in Botrytis cinerea. (A) Construction diagram of displacement of bcniaD by the SlaGemV-1 CP and Rep gene fragments. (B) Comparison of growth on PDA and lesion diameters on tomatoes and grapes. BcVB, wild-type B. cinerea; BcVB-OGG, BcVB transformed by the empty OGG vector; BcVB-CP-Sl, CP transformant; BcBV-V1, SlaGemV-1-infected BcBV; BcVB-Rep-Sl, Rep transformant. (C) Viral Rep and CP gene expression was confirmed from site-directed expression transformants using RT-PCR with specific primers (CP-F/CP-R, Rep-242F/Rep-1072R). Lanes: M, ladder; 1, BcVB-Rep-Sl without intron; 2, BcVB-CP-Sl; 3, BcVB-OGG as the negative control. (D) Mycelial growth on PDA measured by diameters. (E) Lesion diameters on tomatoes. (F) Lesion diameters on grapes.
FIG 7.

Confirmation of site-directed expression of Rep and CP in B. cinerea. (A) Homologous recombination at the 5′- and 3′-flanking regions was validated for transformants BcVB-CP-Sl and BcVB-Rep-Sl using the diagnostic PCR primers (bcniaD-hi5F/TgluchiF, hph-hiF/bcniaD-hi3R) (35). (Left) Confirmation of the size of the 5′ region of homologous integration transformants; (right) confirmation of the 3′ region. Lanes: M, ladder; 1, BcVB-CP-Sl transformant; 2, BcVB-Rep-Sl transformant; 3, BcVB-OGG transformant. (B) Sequencing was conducted to confirm the intron (red bases) removal of the BcVB-Rep-Sl transformant in B. cinerea. (C) Confirmation of virulence determinant of SlaGemV-1 by BcVB-CP-Sl and BcVB-Rep-Sl transformants on grapes. Lesion diameters were compared on green grapes inoculated with different transformants. BcVB, wild-type B. cinerea; BcVB-OGG, BcVB transformed by empty OGG vector; BcVBCP-Sl, CP transformant; BcBV-VI, SlaGemV-1-infected BcBV; BcVB-Rep-Sl, Rep transformant. (D) Average lesion diameters measured on 8 grapes after 24, 48, 72, and 96 h after inoculation with the strains from one of the three trials. All three trials showed reduced virulence caused by BcVB-Rep-Sl.
DISCUSSION
The current study establishes for the first time that metagenomic/metatranscriptomic-discovered genomoviruses from environmental or plant samples can be rescued by introducing multimer clones into potential hosts. Recent high-throughput sequencing studies have discovered hundreds of novel genomoviruses from the environment, sewage, river basins, plants, animals, and animal feces (reviewed in reference 8), all without known hosts. This study showed that the new single-stranded DNA virus, SlaGemV-1, can cause reduction of virulence in S. sclerotiorum, the white mold fungus, that infects >400 plant species, many of which are economically important crops (31, 32). As the virus represents a member of the genus Gemygorvirus, rather than Gemycircularvirus, we propose to change the virus name from “soybean-leaf associated gemycircularvirus 1” to “soybean-leaf associated gemygorvirus 1” and will present this to the ICTV after publication. Most importantly, replication of the virus and reductions of virulence were observed in SlaGemV-1-infected members of the family Sclerotiniaceae, such as B. cinerea, the gray mold fungus, which infects more than 800 plant species and M. fructicola, the causal agent of brown rot of stone fruits. SlaGemV-1 virions, however, cannot directly transfect F. oxysporum with or without protoplasting. It is likely that a conserved mechanism was involved in ssDNA virus uptake and there is a lack of host factors that exist for SlaGemV-1 intracellular replication. Interestingly, viral genomic DNA cannot be readily seen on agarose gels upon the extraction of total DNA from infected mycelia, suggesting a lower titer compared to the other two genomoviruses, SsHADV-1 and FgGMTV1. However, Southern hybridization could detect the viral DNA using a chemifluorescent method which is more sensitive than visualizing on agarose gels.
To understand the genetic determinant of hypovirulence, we expressed the two SlaGemV-1 proteins separately and established that the main virulence determinant is Rep. In 2009, Kittelmann et al. showed that the Rep protein of a plant geminivirus induced DNA replication in Schizosaccharomyces pombe that disrupted the morphology of cells (36). Since CRESS DNA viruses do not encode their own polymerases, to activate the host DNA synthesis machinery, Rep induces differentiated cells to reenter the cell cycle. It is possible that the Rep proteins from different genomoviruses could have different specificities or strengths in inducing replication. In contrast, the CP protein of the same plant geminiviruses did not change the cell morphology when expressed in S. pombe (37), similar to the CP from SlaGemV-1 when expressed in B. cinerea which has all the site-specific gene displacement vectors designed (35). Therefore, we took advantage of the developed system for pinpointing the effects on B. cinerea, which will also allow us to confirm the protein localizations in the future.
A previous study showed that in the case of a tripartite genomovirus, FgGMTV1, DNA-C serves as a symptom alleviation determinant and a replication enhancer (29). The spontaneous uptake of purified SlaGemV-1 virions by more than one fungal species reported in this study is the first example for gemygorvirus and is of great interest. While a similar phenomenon was observed for two genomoviruses in a different genus, this is the first gemygorvirus reported to have such an extracellular transmission property. While the molecular mechanisms involved in the uptake of virions is still unclear, such a mechanism is likely conserved across Genomoviridae.
Like SsHADV-1 (28), SlaGemV-1 does not readily replicate in N. benthamiana and causes asymptomatic infection in insect cells, which are two important considerations when biopesticides are being developed. SlaGemV-1 is classified among feces-associated gemycircularviruses, closely related to feces-associated gemycircularvirus 10. Unlike the ancient caribou feces-associated virus (aCFV) that was reported to replicate in N. benthamiana (8), SlaGemV-1 did not replicate in plants we tested using either the approach of agroinfiltration in N. benthamiana for detection from the local and upper leaves or the A. thaliana protoplast PEG-mediated transformation based on RT-qPCR. The use of carborundum dusting for mechanically inoculated virions did not result in replication or symptoms. An interesting question closely relevant to this study is whether SlaGemV-1-infected fungi (S. sclerotiorum, B. cinerea, and M. fructicola) undergo endophytic life on their host plants, as shown in a previous study (38). The overall finding describes a unique approach to harness the many HTS-discovered genomoviruses by specifically transfecting the plant-pathogenic fungi, followed by site-specific gene displacement to dissect the role of viral determinant of hypovirulence. Besides the biocontrol potential, the available infectious clone and the three related fungal hosts allow further exploration of genomovirus-host interactions that are phylogenetically conserved.
The finding that SlaGemV-1 does not replicate in C. elegans and therefore cannot be used as a model system to study virus-host interactions also suggests that SlaGemV-1 may not be capable of infecting humans. It would be of further agricultural interest to compare the stable virus titers SlaGemV-1 reaches in different plant-pathogenic fungi and to determine whether certain mutations are beneficial for the virus to adapt to different hosts. Frequent reversion of replication protein mutants was observed in other ssDNA viruses infecting plants, depending on the viral background and host components (39). It would still require more in-depth studies to determine whether the mutations also revert in SlaGemV-1. To elucidate the effect of the mutations within the SlaGemV-1 genome discovered due to adaptations to different Sclerotiniaceae hosts leading to increased viral fitness, future studies related to the evolution of the viral protein structures and their interactions are needed.
MATERIALS AND METHODS
Fungal strains and culture maintenance.
Sclerotinia sclerotiorum strains DK3, 1980, and 274, B. cinerea, and M. fructicola were used in this study; strain 274 was a gift from W. Underwood at USDA-ARS in Fargo, ND, and strain 1980 was a gift from J. Rollins at University of Florida. The three S. sclerotiorum strains were maintained in the laboratory. The B. cinerea and M. fructicola strains were isolated from store-bought rotten blueberries and peaches. Their identities were confirmed by Sanger sequencing of amplicons generated using fungal universal internal transcribed spacer PCR primers. All the fungal cultures were maintained on PDA (Difco) at 22 to 24°C. The virus-infected and virus-free fungal cultures used for nucleic acid extraction were grown for 3 to 4 days on the PDA plates overlaid with cellophane (Promega).
Construction of the infectious clones.
The originally published sequence of SlaGemV-1, cloned in the pJet1.2 vector, inadvertently lacked the nonanucleotide motif which was necessary for the initiation of rolling circle DNA replication. As the sequence was present in the original Sanger sequencing file from a PCR amplicon of the soybean DNA extract, a DNA fragment (294 bp) including the stem-loop motif was chemically synthesized (5′-AGCCCCGCGGACTGACTGTAAGTGATGAGAAAGTATTTAGCATTGCAATGGAAATCGGGCATGTGTCCCAGAGTCCTGTCCAAATTAATGTTATATGGACAGGACACAGGACACAGGACACAGGACCCCTATAAATAGGCGGCCTCCCCACCTGAAGTGGGCAATGTTTTGCGATCTCGACACACCGCACGTTTGTCACCCCGCGCGCTCTTGCGTGGCCACCGACCCCTGTGAGGCCCCCCACTATTGTTTTATTTGTCACCGCAACTATGGCATCACGCAGGAGCTCCAAGC-3′) and cloned in a pUC57 vector. Both plasmids were double digested with ScaI and SacI. The 2,068-bp fragment derived from pUC57 with the correct motif sequence was used to replace the original mutant site in the pJet1.2_GemV1 plasmid via ligation to reconstruct the functional SlaGemV-1. As the synthetic DNA fragment was designed to include an 86-nt duplication of the large intergenic region (LIR) added to the original SlaGemV-1 sequence, the resulting SlaGemV-1 was considered a 1.1-mer clone. Due to the duplication of the synthetic fragment, one more SacII site was introduced. By using SacII enzyme digestion, a 2,234-bp full genome was gel extracted and then ligated with dephosphorylated pJet1.2 vector digested with SacII. Specifically, a 1.1-mer of the viral genome (GenBank accession no. KT598248.2) of SlaGemV-1 was cloned in pJet1.2 vector (CloneJET PCR cloning kit; Thermo Fisher), including two copies of the nonanucleotide motif. The two copies of the SlaGemV-1 genome were cloned simultaneously in the pJet1.2 vector, and the resulting construct was named 2-mer. All primer sequences are available upon request.
Fungal protoplast preparation, transformation, and confirmation of infectivity of the constructs.
Protoplasts of S. sclerotiorum were produced by following the procedures described by Ge et al. (40). Both the SlaGemV-1 1.1-mer and 2-mer DNA were transfected into virus-free S. sclerotiorum protoplasts (strain DK3) by electroporation at 1,500 kV (41). Mycelial plugs were transferred to PDA from the recovered cultures. After several serial transfers to fresh PDA plates at 22°C, infection was confirmed by PCR amplification using virus-specific primers, with the product subjected to Sanger sequencing.
Purification of virions and TEM.
SlaGemV-1-infected S. sclerotiorum was grown at 22°C for 7 days in PDB medium. The mycelia were harvested and homogenized with 0.1 M sodium phosphate buffer with 0.2 M KCl (pH 7.4) using a blender for 5 min, and the homogenate was incubated at 4 to 10°C for 30 min with gentle shaking. The homogenate was extracted with chloroform and centrifuged at 8,000 × g at 4°C for 10 min to remove the fungal tissue. The supernatant was transferred to a new tube for ultracentrifugation at 30,000 × g for 2.5 h to produce a pellet that was resuspended in 0.01 M sodium phosphate buffer overnight on ice. The next day, a low-speed centrifugation was performed to remove the fungal debris. The supernatant was subjected to sucrose gradient ultracentrifugation at 35,000 rpm for 3 h, and the fractions collected from the 25% sucrose layer went through an additional ultracentrifugation for 3 h. The enrichment of virions originating from the 25% layer was confirmed by PCR amplification using virus-specific primers. The samples were submitted to the Materials Research Lab at University of Illinois in Urbana-Champaign.
Morphology on PDA, virulence assay, and extracellular transmission test.
A fungal growth assay was conducted on PDA plates with at least 5 replicates each of virus-free strain DK3 and 1.1-mer and 2-mer SlaGemV-1-transfected cultures. PDA discs (5 mm) were taken from the edges of actively growing 2-day-old cultures and inoculated onto fresh PDA plates. Hyphal diameter was measured daily from 1 to 3 dpi. For virulence assay of SlaGemV-1-infected fungi, canola leaves were inoculated with a single 5-mm PDA plug from the actively growing edge of virus-infected S. sclerotiorum DK3. The lesion size was measured daily from 1 to 4 dpi. Similarly, to test the effect of SlaGemV-1 on the virulence of other fungi, tomatoes were inoculated with a virus-infected B. cinerea agar plug and cherries with a virus-infected M. fructicola agar plug. Lesion size was measured daily from 1 to 4 dpi. At least five replicates of the samples were incubated at room temperature on a lab bench in petri dishes, and all the virulence assays were repeated at least three times.
For the extracellular transmission test, the purified SlaGemV-1 particles from the infected host were used as inoculants onto growing fungal hyphae of B. cinerea, M. fructicola, and F. oxysporum three times with 20 μl (1 mg/ml) in each inoculation. After more than 6 serial transfers to fresh PDA, genomic DNAs of virus-treated cultures were extracted using a PureLink Microbiome DNA purification kit (Invitrogen) to confirm the replication of SlaGemV-1, following the manufacturer’s instructions, to be detected by PCR. Sanger sequencing confirmed the identity of the PCR amplicons of expected sizes.
Testing the biocontrol potential of SlaGemV-1.
To investigate the protective effect of crude viral particles against infection by S. sclerotiorum. The crude viral particles were extracted following a preparation method described previously (33) with minor modifications. SlaGemV-1-infected S. sclerotiorum strain DK3 was cultured in PDB medium in a 1,000-ml flask at 22°C for 2 weeks, and mycelium was collected and homogenized in a prechilled Waring blender for 30 s. The fungal homogenate was diluted to 4.0, 3.0, and 2.0 OD600 units, respectively, with distilled water and sprayed on the tomato plants (15 ml crude preparation/plant) after centrifugation at 12,000 rpm for 10 min, and the supernatant was passed through a 0.22-μm syringe filter. Eight replicate plants were treated with preparations at each optical density and a control with water only. The agar plugs from the edges of actively growing colonies of S. sclerotiorum strains were cultured on minimal medium and were randomly inoculated on leaves of virus-treated plants after 24 h on two leaves each plant. The treated plants were kept in an incubator at 22°C with 100% humidity. Pictures of intact plants were taken at 6 and 16 dpi.
Confirmation of SlaGemV-1 replication in bacteria by Southern blots.
Total DNA was extracted from E. coli harboring pJet-1.2 carrying the 1.1-mer and 2-mer constructs using a phenol-chloroform method (39). Total DNA (1 mg) was separated using 0.8% agarose gels in Tris-borate-EDTA. The gel was submerged in 0.25 M HCl, followed by denaturation and neutralization steps, and capillary blotted onto the nylon membrane (Amersham Hybond N+; GE Healthcare). Biotin-labeled DNA probes were prepared by a random primer labeling kit manufactured by North2South (Thermo Scientific) using dimeric viral DNA fragments (4.2 kb) as the template. After prehybridization and hybridization, the nylon membrane was exposed and detected by a chemiluminescent nucleic acid detection module (Thermo Scientific). All primer sequences are available upon request.
Confirmation of SlaGemV-1 replication in Sf9 insect cells and C. elegans.
Immediately after retrieving the stock Sf9 cells from a liquid nitrogen storage dewar, 1 ml of Sf9 cells were thawed and inoculated in 10 ml of Sf-900 II SFM medium (Gibco, Invitrogen) with 10 μl of 100 μl/ml of penicillin-streptomycin in 75-ml cell culture flasks at 27°C without shaking. To determine whether SlaGemV-1 replicates in insect cells, purified virions (200 μl, 20 ng/μl) were passed through a 0.22-μm biofilter and inoculated on S. frugiperda (Sf9) cells in a 6-well plate (2 ml at 2 × 105 cells per well), then incubated at 27°C, and harvested after 0 h, 12 h, 24 h, 72 h, and 96 h. The cells were washed twice with Sf-900 II SFM medium. DNA was extracted from the inoculated cell pellets with a GeneJET Viral DNA/RNA purification kit (Thermo Scientific) and used for PCR and qPCR analysis. The primer pairs used for this analysis are available upon request. Cells were passaged as described by Liu et al. (27). Briefly, Sf9 cells (P0) were inoculated with virions and incubated for 24 h, at which time the old medium was discarded and attached cells were washed with fresh medium twice. After the cells had been incubated in fresh medium for 72 h, the supernatant was collected by low-speed spinning and used for inoculation of new cells (P1), which were incubated for 72 h. Then, the supernatant was collected from P1 cells and used to inoculate new cells (P2). At 24 hpi, P2 cells were subjected to extraction to obtain DNA/RNA for virus detection.
The C. elegans strains WM27 rde-1(ne219) and CB1467 him-5(e1467) were obtained from the Caenorhabditis Genetics Center (CGC), and cultured on nematode growth medium (NGM) plates that had been spread with Escherichia coli strain K-12 as a source of feeding in a 20°C incubator for 3 to 4 days. Fifty to 100 eggs or L1 larvae were transferred to a new NGM plate seeded with E. coli K-12 and inoculated with 20 μl viral particles (1 mg/ml). The virus-inoculated cultures were maintained at 20°C for 3 to 4 days, and then the new generation of eggs or L1 larvae were serially transferred to new NGM plates with E. coli K-12 6 times. Furthermore, 50 to 100 eggs or larvae were transferred to the NGM plates seeded with E. coli harboring the SlaGemV-1 1.1-mer infectious clone. Nematodes were collected and washed with M9 buffer twice at 3 and 4 dpi, and larvae were continuously transferred 6 times to new NGM plates with E. coli K-12. After serial transfers, the nematodes were collected to extract the DNA and RNA.
Agroinfiltration of infectious clones into Nicotiana benthamiana.
The same 1.1-mer and 2-mer segments were cloned into pCambia1302 using XbaI and HindIII sites, and the resulting constructs were named pCambia-1.1-mer and pCambia-2-mer. The cultures of A. tumefaciens containing pCambia-1.1-mer and pCambia-2-mer were streaked on LB plates with the appropriate antibiotic and allowed to grow for 1 to 2 days at 30°C. The cultures were collected from the plates with a sterile loop and diluted in buffer (10 mM MES [morpholineethanesulfonic acid] [pH 5.6] and 10 mM MgCl2) to an OD600 of 0.6. For each 1 ml of culture volume, 1.5 μl of 0.1 M acetosyringone (in dimethyl sulfoxide [DMSO]) was added (final concentration = 150 μM). The cultures were left without shaking at room temperature for 2 to 3 h.
The cultures were mixed and infiltrated on the underside of the lower leaves of N. benthamiana seedlings at the 4-leaf stage (3 weeks) using a 1-ml syringe. The infection was confirmed by extracting RNA from leaves to be used as the RT-PCR template. The primer pair Rep-F and Rep-R was used to amplify the replication protein coding region, and the primer pair CP-F and CP-R was used for the CP.
Plant protoplast preparation, virus transfection, and inoculation.
The protocol of Yoo et al. (42) for preparation and transfection of Arabidopsis thaliana Col-0 protoplasts with virus, with modification of pectinase (TCI America, CAS registry no. 9032-75-1) instead of Macro-Zyme, was used. Because of differences in enzymatic activities from different manufacturers, 3.2% (wt/vol) of pectinase was determined to be optimal. The concentration of cellulase (Worthington Biochemical, catalog no. LS002601) and other reagents remained the same. We mixed 150 μl of the virions with 300 μl protoplasts (6 × 104 protoplasts) and an equal volume (450 μl) of freshly prepared PEG solution and then diluted the mixture with an equal volume of W5 solution (900 μl). Protoplasts were harvested at 0 and 48 hpi with three biological replicates at each time point for RNA extraction using the RNeasy plant minikit (Qiagen), followed by DNA removal using Turbo DNA-free kit (Thermo Fisher Scientific). The concentration of RNA was quantified with a NanoDrop spectrophotometer (Thermo Scientific). qRT-PCR was performed using Verso SYBR green 1-step qRT-PCR ROX mix (Thermo Scientific) with EF1α (AT5G60390) as the internal control for normalization, and the primer set S1-CP-qPCR-F and -R was used to quantify viral replication. Additionally, 10 μl of purified viral particles was mechanically inoculated by rubbing on soybean leaves with carborundum following a standard protocol (43).
Site-specific integration of viral proteins expressed in Botrytis cinerea.
To determine the role of each SlaGemV-1 protein in hypovirulence, the coding sequences of Rep or CP genes were individually cloned into pNDH-OGG (35), which contains a hygromycin resistance gene cassette, using the NotI site. The advantage of the system is that the plasmids were designed to replace the endogenous bcniaD gene, which was previously determined to be nonessential in B. cinerea under normal growth conditions. PEG transformation of the protoplasts with the constructs was performed as described above, and transformants were selected on PDA plates containing hygromycin (29). DNA and RNA extracted from the transformants were screened for homologous recombination by diagnostic PCR and RT-PCRs described elsewhere (35). Only transformants confirmed to result from site-specific gene displacement were further compared for morphology and virulence effects on tomatoes and grapes.
Data analysis.
Experimental data of fungal growth rate and virulence assays of virus-infected and virus-free fungi were analyzed using a two-sample Wilcoxon-test in R/Bioconductor to calculate the P values (36). Statistical analysis of values from site-specific integration transformants was performed using Prism version 5.0 (GraphPad, USA). Significance of differences in pathogenicity among different treatments were evaluated using a two-way analysis of variance followed by the Bonferroni posttest, and the treatment means were calculated using the least significant difference (LSD) at a P value of 0.05.
Contributor Information
Shin-Yi Lee Marzano, Email: shinyi.marzano@usda.gov.
Anne E. Simon, University of Maryland, College Park
REFERENCES
- 1.Marzano SYL, and, Domier LL. 2016. Novel mycoviruses discovered from metatranscriptomics survey of soybean phyllosphere phytobiomes. Virus Res 213:332–342. doi: 10.1016/j.virusres.2015.11.002. [DOI] [PubMed] [Google Scholar]
- 2.Neupane A, Feng C, Feng J, Kafle A, Bücking H, Lee Marzano S-Y. 2018. Metatranscriptomic analysis and in silico approach identified mycoviruses in the arbuscular mycorrhizal fungus Rhizophagus spp. Viruses 10:707. doi: 10.3390/v10120707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Márquez LM, Redman RS, Rodriguez RJ, Roossinck MJ. 2007. A virus in a fungus in a plant: three-way symbiosis required for thermal tolerance. Science 315:513–515. doi: 10.1126/science.1136237. [DOI] [PubMed] [Google Scholar]
- 4.Bao X, Roossinck MJ. 2013. Multiplexed interactions: viruses of endophytic fungi. Adv Virus Res 86:37–58. doi: 10.1016/B978-0-12-394315-6.00002-7. [DOI] [PubMed] [Google Scholar]
- 5.Li C, and, Samulski RJ. 2020. Engineering adeno-associated virus vectors for gene therapy. Nat Rev Genet 21:255–272. doi: 10.1038/s41576-019-0205-4. [DOI] [PubMed] [Google Scholar]
- 6.Krupovic M, Ghabrial SA, Jiang D, Varsani A. 2016. Genomoviridae: a new family of widespread single-stranded DNA viruses. Arch Virol 161:2633–2643. doi: 10.1007/s00705-016-2943-3. [DOI] [PubMed] [Google Scholar]
- 7.Varsani A, and, Krupovic M. 2017. Sequence-based taxonomic framework for the classification of uncultured single-stranded DNA viruses of the family Genomoviridae. Virus Evol 3:vew037. doi: 10.1093/ve/vew037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ng TFF, Chen L-F, Zhou Y, Shapiro B, Stiller M, Heintzman PD, Varsani A, Kondov NO, Wong W, Deng X, Andrews TD, Moorman BJ, Meulendyk T, MacKay G, Gilbertson RL, Delwart E. 2014. Preservation of viral genomes in 700-y-old caribou feces from a subarctic ice patch. Proc Natl Acad Sci U S A 111:16842–16847. doi: 10.1073/pnas.1410429111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fontenele RS, Roumagnac P, Richet C, Kraberger S, Stainton D, Aleamotu‘A M, Filloux D, Bernardo P, Harkins GW, McCarthy J, Charles LS, Lamas NS, Abreu EFM, Abreu RA, Batista GB, Lacerda ALM, Salywon A, Wojciechowski MF, Majure LC, Martin DP, Ribeiro SG, Lefeuvre P, Varsani A. 2020. Diverse genomoviruses representing twenty-nine species identified associated with plants. Arch Virol 165:2891–2901. doi: 10.1007/s00705-020-04801-5. [DOI] [PubMed] [Google Scholar]
- 10.Chabi-Jesus C, Najar A, Fontenele RS, Kumari SG, Ramos-González PL, Freitas-Astúa J, Kraberger S, Varsani A. 2020. Viruses representing two new genomovirus species identified in citrus from Tunisia. Arch Virol 165:1225–1229. doi: 10.1007/s00705-020-04569-8. [DOI] [PubMed] [Google Scholar]
- 11.Dayaram A, Opong A, Jäschke A, Hadfield J, Baschiera M, Dobson RCJ, Offei SK, Shepherd DN, Martin DP, Varsani A. 2012. Molecular characterisation of a novel cassava associated circular ssDNA virus. Virus Res 166:130–135. doi: 10.1016/j.virusres.2012.03.009. [DOI] [PubMed] [Google Scholar]
- 12.de Rezende RR, Mar TB, Páez LMC, Xavier ADS, Xavier CAD, Navas-Castillo J, Zerbini FM, Alfenas-Zerbini P. 2018. Complete genome sequences of two gemycircularviruses associated with non-cultivated plants in Brazil. Arch Virol 163:3163–3166. doi: 10.1007/s00705-018-3924-5. [DOI] [PubMed] [Google Scholar]
- 13.Kraberger S, Farkas K, Bernardo P, Booker C, Argüello-Astorga GR, Mesléard F, Martin DP, Roumagnac P, Varsani A. 2015. Identification of novel Bromus- and Trifolium-associated circular DNA viruses. Arch Virol 160:1303–1311. doi: 10.1007/s00705-015-2358-6. [DOI] [PubMed] [Google Scholar]
- 14.Lamas NS, Fontenele RS, Melo FL, Costa AF, Varsani A, Ribeiro SG. 2016. Complete genome sequence of a genomovirus associated with common bean plant leaves in Brazil. Genome Announc 4:e01247-16. doi: 10.1128/genomeA.01247-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Male MF, Kami V, Kraberger S, Varsani A. 2015. Genome sequences of Poaceae-associated gemycircularviruses from the Pacific Ocean island of Tonga. Genome Announc 3:e01144-15. doi: 10.1128/genomeA.01144-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sikorski A, Massaro M, Kraberger S, Young LM, Smalley D, Martin DP, Varsani A. 2013. Novel myco-like DNA viruses discovered in the faecal matter of various animals. Virus Res 177:209–216. doi: 10.1016/j.virusres.2013.08.008. [DOI] [PubMed] [Google Scholar]
- 17.Uch R, Fournier P-E, Robert C, Blanc-Tailleur C, Galicher V, Barre R, Jordier F, de Micco P, Raoult D, Biagini P. 2015. Divergent gemycircularvirus in HIV-positive blood, France. Emerg Infect Dis 21:2096–2098. doi: 10.3201/eid2111.150486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou C, Zhang S, Gong Q, Hao A. 2015. A novel gemycircularvirus in an unexplained case of child encephalitis. Virol J 12:197. doi: 10.1186/s12985-015-0431-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kraberger S, Hofstetter RW, Potter KA, Farkas K, Varsani A. 2018. Genomoviruses associated with mountain and western pine beetles. Virus Res 256:17–20. doi: 10.1016/j.virusres.2018.07.019. [DOI] [PubMed] [Google Scholar]
- 20.Wang H, Li S, Mahmood A, Yang S, Wang X, Shen Q, Shan T, Deng X, Li J, Hua X, Cui L, Delwart E, Zhang W. 2018. Plasma virome of cattle from forest region revealed diverse small circular ssDNA viral genomes. Virol J 15:11. doi: 10.1186/s12985-018-0923-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown JR, Bharucha T, and, Breuer J. 2018. Encephalitis diagnosis using metagenomics: application of next generation sequencing for undiagnosed cases. J Infect 76:225–240. doi: 10.1016/j.jinf.2017.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dayaram A, Potter KA, Pailes R, Marinov M, Rosenstein DD, Varsani A. 2015. Identification of diverse circular single-stranded DNA viruses in adult dragonflies and damselflies (Insecta: Odonata) of Arizona and Oklahoma, USA. Infect Genet Evol 30:278–287. doi: 10.1016/j.meegid.2014.12.037. [DOI] [PubMed] [Google Scholar]
- 23.Xia H, Wang Y, Shi C, Atoni E, Zhao L, Yuan Z. 2018. Comparative metagenomic profiling of viromes associated with four common mosquito species in China. Virol Sin 33:59–66. doi: 10.1007/s12250-018-0015-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kraberger S, Polston JE, Capobianco HM, Alcalá-Briseño RI, Fontenele RS, Varsani A. 2017. Genomovirus genomes recovered from Echinothrips americanus sampled in Florida, USA. Genome Announc 5:e00445-17. doi: 10.1128/genomeA.00445-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Waits K, Edwards MJ, Cobb IN, Fontenele RS, Varsani A. 2018. Identification of an anellovirus and genomoviruses in ixodid ticks. Virus Genes 54:155–159. doi: 10.1007/s11262-017-1520-5. [DOI] [PubMed] [Google Scholar]
- 26.Nakasu EYT, Melo FL, Michereff-Filho M, Nagata T, Ribeiro BM, Ribeiro SG, Lacorte C, Inoue-Nagata AK. 2017. Discovery of two small circular ssDNA viruses associated with the whitefly Bemisia tabaci. Arch Virol 162:2835–2838. doi: 10.1007/s00705-017-3425-y. [DOI] [PubMed] [Google Scholar]
- 27.Liu S, Xie J, Cheng J, Li B, Chen T, Fu Y, Li G, Wang M, Jin H, Wan H, Jiang D. 2016. Fungal DNA virus infects a mycophagous insect and utilizes it as a transmission vector. Proc Natl Acad Sci U S A 113:12803–12808. doi: 10.1073/pnas.1608013113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu X, Li B, Fu Y, Xie J, Cheng J, Ghabrial SA, Li G, Yi X, Jiang D. 2013. Extracellular transmission of a DNA mycovirus and its use as a natural fungicide. Proc Natl Acad Sci U S A 110:1452–1457. doi: 10.1073/pnas.1213755110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li P, Wang S, Zhang L, Qiu D, Zhou X, Guo L. 2020. A tripartite ssDNA mycovirus from a plant pathogenic fungus is infectious as cloned DNA and purified virions. Sci Adv 6:eaay9634. doi: 10.1126/sciadv.aay9634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosario K, Duffy S, Breitbart M. 2012. A field guide to eukaryotic circular single-stranded DNA viruses: insights gained from metagenomics. Arch Virol 157:1851–1871. doi: 10.1007/s00705-012-1391-y. [DOI] [PubMed] [Google Scholar]
- 31.Koonin EV, Dolja VV, and, Krupovic M. 2015. Origins and evolution of viruses of eukaryotes: the ultimate modularity. Virology 479–480:2–25. doi: 10.1016/j.virol.2015.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mochama P, Jadhav P, Neupane A, Lee Marzano S-Y. 2018. Mycoviruses as triggers and targets of RNA silencing in white mold fungus Sclerotinia sclerotiorum. Viruses 10:214. doi: 10.3390/v10040214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao L, Rosario K, Breitbart M, Duffy S. 2019. Eukaryotic circular rep-encoding single-stranded DNA (CRESS DNA) viruses: ubiquitous viruses with small genomes and a diverse host range. Adv Virus Res 103:71–133. doi: 10.1016/bs.aivir.2018.10.001. [DOI] [PubMed] [Google Scholar]
- 34.van der Walt E, Martin DP, Varsani A, Polston JE, Rybicki EP. 2008. Experimental observations of rapid Maize streak virus evolution reveal a strand-specific nucleotide substitution bias. Virol J 5:104–111. doi: 10.1186/1743-422X-5-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schumacher J. 2012. Tools for Botrytis cinerea: new expression vectors make the gray mold fungus more accessible to cell biology approaches. Fungal Genet Biol 49:483–497. doi: 10.1016/j.fgb.2012.03.005. [DOI] [PubMed] [Google Scholar]
- 36.Kittelmann K, Rau P, Gronenborn B, Jeske H. 2009. Plant geminivirus Rep protein induces rereplication in fission yeast. J Virol 83:6769–6778. doi: 10.1128/JVI.02491-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hipp K, Schäfer B, Kepp G, Jeske H. 2016. Properties of African cassava mosaic virus capsid protein expressed in fission yeast. Viruses 8:190. doi: 10.3390/v8070190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang H, Xie J, Fu Y, Cheng J, Qu Z, Zhao Z, Cheng S, Chen T, Li B, Wang Q, Liu X, Tian B, Collinge DB, Jiang D. 2020. A 2-kb mycovirus converts a pathogenic fungus into a beneficial endophyte for Brassica protection and yield enhancement. Mol Plant 13:1420–1433. doi: 10.1016/j.molp.2020.08.016. [DOI] [PubMed] [Google Scholar]
- 39.Arguello-Astorga G, Ascencio-Ibáñez JT, Dallas MB, Orozco BM, Hanley-Bowdoin L. 2007. High-frequency reversion of geminivirus replication protein mutants during infection. J Virol 81:11005–11015. doi: 10.1128/JVI.00925-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ge C-Y, Duan Y-B, Zhou M-G, Chen C-J. 2013. A protoplast transformation system for gene deletion and complementation in Sclerotinia sclerotiorum. J Phytopathol 161:800–806. doi: 10.1111/jph.12137. [DOI] [Google Scholar]
- 41.Marzano S-YL, Hobbs HA, Nelson BD, Hartman GL, Eastburn DM, McCoppin NK, Domier LL. 2015. Transfection of Sclerotinia sclerotiorum with in vitro transcripts of a naturally occurring interspecific recombinant of Sclerotinia sclerotiorum hypovirus 2 significantly reduces virulence of the fungus. J Virol 89:5060–5071. doi: 10.1128/JVI.03199-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yoo S-D, Cho Y-H, and, Sheen J. 2007. Arabidopsis mesophyll protoplasts: a versatile cell system for transient gene expression analysis. Nat Protoc 2:1565–1572. doi: 10.1038/nprot.2007.199. [DOI] [PubMed] [Google Scholar]
- 43.Hull R. 2009. Mechanical inoculation of plant viruses. Curr Protoc Microbiol Chapter 16:Unit 16B.6. doi: 10.1002/9780471729259.mc16b06s13. [DOI] [PubMed] [Google Scholar]