

Abstract
Granulomatous dermatitis (GD) is one of the commonest tissue reaction patterns encountered in dermatopathology practice. Granulomatous inflammation in the skin can be seen in a wide range of conditions, thus, granulomatous dermatitis always poses significant challenge even to the astute dermatologists and dermatopathologists. Broadly, granulomatous dermatitis is divided into two groups—infectious and non-infectious, the prevalence of both highly variable and show overlapping pathology. However, there are subtle histological clues, which when combined with clinical features, help to narrow down the differential diagnosis. Thus, a good Clinicopathological correlation (CPC) along with histochemical stains, culture and ancillary techniques including molecular studies are required for arriving at a definite diagnosis. In this review, we shall discuss the histological clues to diagnose non-infectious granulomatous dermatitis (NIGD) and their differential diagnoses.
Keywords: Granulomatous dermatitis, histochemistry, interstitial granuloma, necrobiotic granuloma, sarcoidosis
Introduction
Granulomatous dermatitis (GD) is one of the commonest tissue reaction patterns encountered in dermatopathology practice. Granulomatous inflammation in the skin can be seen in a wide range of conditions, some of which are confined to the skin, while some involve skin as a part of systemic disease. Thus, granulomatous dermatitis always poses significant challenge even to the astute dermatologists and dermatopathologists. Broadly, granulomatous dermatitis is divided into two groups—infectious and non-infectious, the prevalence of both highly variable and show overlapping pathology. In developing countries like India, infectious granulomas clearly outnumber their non-infectious counterparts, whereas in western counties, non-infectious granulomas are more commonly encountered.[1,2] Thus, a good Clinicopathological correlation (CPC) along with histochemical stains, culture and ancillary techniques including molecular studies are required for arriving at a definite diagnosis. In this review, we shall discuss the histological features of non-infectious granulomatous dermatitis (NIGD) and their differential diagnoses.
Approach to a case of granulomatous dermatitis
Careful clinical and histological evaluation.
CPC.
Histochemical stains- connective tissue stains such as alcian blue, mucicarmine, colloidal iron, masson's trichrome, elastic Van Gieson, etc.
To exclude infection: As non-infectious granulomatous dermatoses closely resemble infectious dermatoses, both clinically and histologically, infectious aetiology should always be excluded in a case of GD, especially in developing countries. Special stains for this purpose include Ziehl- Neelsen (ZN), modified ZN, Fites for acid fast bacilli (AFB), periodic acid Schiff (PAS) and Grocott for fungi, Giemsa for leishmaniasis, etc. Culture is considered to be the gold standard for isolating any organism however useful in limited cases only. Molecular techniques, such as polymerase chain reaction (PCR) is sensitive and rapid test which can be used as an adjunct to pin point the cause of GD especially where histochemical stains and culture is not helpful.
Types of granulomatous reaction
Granuloma is composed of epithelioid cells, with or without associated giant cells and varying amount of lymphocytic inflammatory cells. Depending up on the structure of the granuloma and nature of associated inflammatory infiltrates and their extent, granulomas in dermatopathology are classified into the following types-
Sarcoidal granuloma: These are well-formed granulomas, without significant surrounding lymphoid infiltrate (naked granuloma). The granulomas are discrete and compact. The prototype of this type of granuloma is sarcoidosis, although other conditions may also produce this kind of granuloma [see Table 1].
Tuberculoid granuloma: Composed of epithelioid histiocytes, Langhan's type and foreign body type giant cells, with or without central caseation necrosis, these granulomas tend to show fusion and are surrounded by lymphoid cuffing.
Necrobiotic granuloma: Necrobiosis indicates alteration of dermal connective tissue, namely collagen or elastic fiber. These foci of necrobiosis are surrounded by granulomatous reaction. The granulomas are often poorly formed, either diffuse type or palisaded in nature, and are associated with variable inflammatory infiltrate.
Suppurative granuloma: These granulomas contain neutrophils and neutrophilic debris in the center, while the peripheral portion may contain chronic inflammatory infiltrate comprising of lymphocytes, plasma cells, and histiocytes. The prototype of this granuloma is deep cutaneous fungal infection, although this can be seen in other conditions.
Foreign body granuloma: These granulomas contain multinucleated foreign body giant cells, with nuclei arranged haphazardly. Additionally, they may also contain endogenous or exogenous foreign material, although not be demonstrable in some cases.
Xanthogranuloma: These are composed of numerous histiocytes with voluminous pale or foamy cytoplasm, along with variable lymphoplasmacytic infiltrate. Tuton giant cells can be identified in some cases, which are diagnostic of these lesions.
Interstitial granuloma: These are ill defined granulomas, composed of loose collections of histiocytes. They may be associated with scattered giant cells. The histiocytes are often small, present in between collagen bundles.
Table 1.
Different types of granulomas and their aetiology
| Type of granuloma | Associated conditions |
|---|---|
| Sarcoidal | Sarcoidosis, Blau’s syndrome, foreign material, drugs, secondary syphilis, metastatic Crohn’s disease, orofacial granulomatosis, breast cancer, common variable immune deficiency, chronic granulomatous disease |
| Tuberculoid | Tuberculosis, tuberculid, leprosy, late syphilis, leishmaniasis, granulomatous rosacea, perioral dermatitis, lupus miliaris disseminatus faciei, BCG granuloma |
| Necrobiotic | Granuloma annulare, necrobiosis lipoidica, necrobiotic xanthogranuloma, rheumatoid nodule, rheumatic fever nodule, reaction to foreign body |
| Suppurative | Fungal infection, non-tuberculous mycobacterial infection, mycetoma, nocardiosis, actinomycosis, cat scratch disease, lymphogranuloma venereum, pyoderma gangrenosum, ruptured cysts and follicles |
| Foreign body | Endogenous and exogenous foreign material, ruptured cysts and follicles |
| Xanthogranuloma | Juvenile xanthogranuloma, reticulohistiocytoma |
| Interstitial | Giant cell elastolytic granuloma, interstitial granulomatous dermatitis, interstitial granulomatous drug reaction, palisaded neutrophilic and granulomatous dermatitis |
Identifying the type of granuloma helps in limiting the differential diagnoses. However, it should be remembered that there are overlapping histological features among different types of granulomas and sometimes a granuloma may not be accurately placed in one of the above categories. On the other hand, one condition may produce different patterns of granuloma. Thus, a good clinico-pathological correlation is often required. Various conditions commonly producing different types of granulomas have been shown in Table 1.
Red and blue granulomas
A wide variety of non-infectious conditions produces granulomatous dermatitis. Among these, necrobiotic granuloma is the major subtype and constitutes a major bulk of cases. Necrobiotic granulomas are characterized by granulomatous reaction surrounding central altered connective tissue, namely collagen and elastic fibers. A wide range of conditions can bring about this connective tissue alteration. Altered collagen loses its normal staining character and becomes either more eosinophilic (red) or basophilic (blue).[3,4] The increased basophilia can be attributed to deposition of mucin or presence of neutrophils and/or nuclear dust. Conditions producing blue granuloma include granuloma annulare (GA), palisaded neutrophilic and granulomatous dermatitis (PNGD), Wegener's granulomatosis (granulomatosis with polyangiitis, GPA) and rheumatoid vasculitis. The basophilia can be appreciated on routine hematoxylin and eosin (HE) stain. However, the interstitial mucin can be better brought out/highlighted by alcian blue or colloidal iron stain. If the basophilia is due to deposition of mucin, the main consideration is GA, whereas the diagnostic considerations are PNGD, GPA and rheumatoid vasculitis if this basophilia is due to neutrophils or nuclear dust.[3]
On the other hand, red or hypereosinophilic staining character of necrobiotic granuloma is due to hyalinized collagen, fibrin, or degenerated eosinophils. The differential diagnoses of red granulomas include necrobiosis lipoidica (NL), necrobiosis xanthogranuloma (NXG), and rheumatoid nodule (RN). Usually in NL and NXG, the hypereosinophilia is due to hyalinized collagen, whereas in RN, it is due to fibrin deposition.[4] These tinctorial changes provide important diagnostic clues in the diagnosis of non-infectious granulomatous dermatitis, although they are not specific.
Individual conditions
Among the different entities included in the non-infectious granulomatous dermatoses, different studies have found granuloma annulare and sarcoidosis to be most common, whereas some of these conditions are relatively uncommon or rare.[2,5]
Here we discuss the histology of some of the commonly encountered non-infectious granulomatous dermatoses.
Sarcoidosis
Sarcoidosis is an idiopathic, multisystem granulomatous disease, which often involves the skin. Skin involvement is seen in 9-37% of systemic sarcoidosis patients.[6] Cutaneous sarcoidosis presents with myriad of clinical presentation such as papule, nodule, plaque, atrophic, etc., which may often mimic other conditions [Figure 1]. It is a diagnosis of exclusion and is usually based on clinico-pathological features. Although majority of the cases involve skin as a part of systemic sarcoidosis, the disease may be restricted to skin in a substantial proportion of cases, even on long follow up. Wu et al. reported systemic involvement in 27 of 37 patients (73%) of cutaneous sarcoidosis in their series, mostly after the diagnosis of cutaneous disease.[7] Thus, histopathology is mandatory to confirm the diagnosis.
Figure 1.
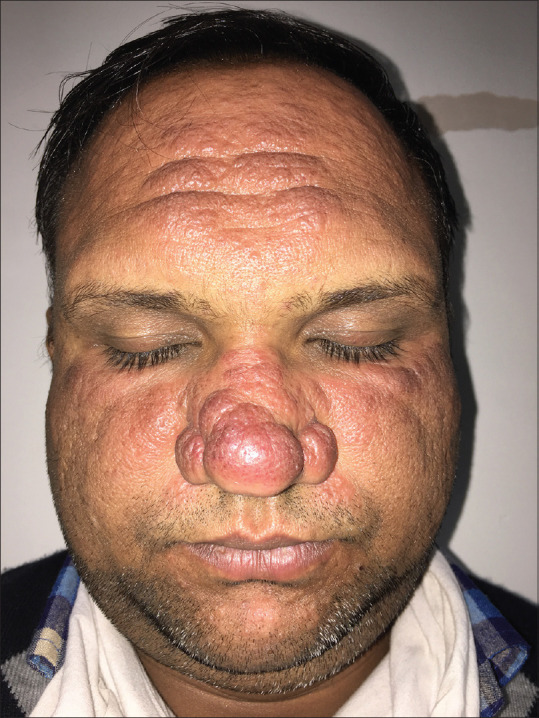
Case of sarcoidosis showing erythematous papules coalescing to form plaques on the forehead, nose (bridge and alae), nasolabial fold and malar areas of both cheeks
Histopathology
Sarcoidosis is characterized by presence of sarcoidal granulomas. Overlying epidermis is usually unremarkable. Sarcoidosis may rarely present as verrucous or ichthiosiform lesion, in which epidermis shows acanthosis and hyperkeratosis, respectively. Sarcoidal granuloma is well-formed epithelioid cell granuloma without lymphoid cuffing [Figure 2]. These granulomas are discrete and devoid of central caseation necrosis. The granulomas are distributed throughout the dermis or remain confined to upper dermis. The different patterns of dermal involvement include extensive nodular, sausage-like pattern along the neurovascular plexus, dense diffuse, and dispersed nodular pattern. Among these, extensive nodular pattern is most commonly encountered.[7] Sarcoidal granulomas are commonly found adjacent to arterioles, and also in periadnexal and perineural location. Because of perineural location of granulomas, sarcoid may resemble leprosy. Ball et al. reported linear perineural granulomas in 25% of their cases.[8] Although the granulomas are seen in perivascular location, granulomatous vasculitis is extremely rare.[6] Although sarcoidal granulomas are characteristic of sarcoidosis, other types of granulomas are also rarely encountered in sarcoidosis. Ball et al. reviewed 28 cases of cutaneous sarcoidosis and found 4 (14.3%) cases showed tuberculoid granuloma with perigranulomatous lymphocytic cuffing. They also fund interstitial granulomas in 5 (18%) cases.[8] Wu et al. reported most of the cases in their series demonstrated variable combination of sarcoidal and tuberculoid granulomas and 4% of their cases morphologically resembled necrobiotic granuloma.[7] Granuloma can focally extend into superficial subcutis but that doesn’t qualify for subcutaneous sarcoidosis (discussed latter). Focal fibrinoid necrosis may be encountered, and their presence is variably reported in literature. Ball et al. reported focal necrosis in as many as 43% of their cases.[8] This should not be misdiagnosed as caseation necrosis, which is a feature of tuberculous granuloma. Caseation necrosis has a granular appearance and contains nuclear debris, which is absent in fibrinoid necrosis. Transepidermal elimination has been reported in sarcoidosis. The histopathology shows epidermal necrosis with elimination track. Variable plasma cell infiltrate can be seen in sarcoidosis.[6] Sarcoidosis can produce mild to moderate dermal fibrosis, but marked dermal fibrosis is usually not seen.
Figure 2.
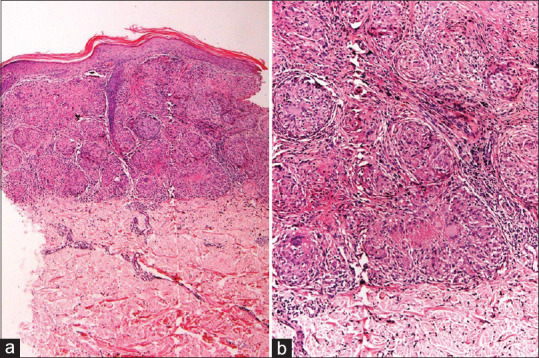
Sarcoidosis: (a) Skin biopsy showing multiple epithelioid cell granulomas in upper and mid dermis (hematoxylin and eosin, x100). (b) The granulomas are well-formed, compact, without lymphoid cuffing. Occasional granulomas show central fibrinoid necrosis (hematoxylin and eosin, x200)
Both Langhan's type and foreign body type giant cells are seen in sarcoidosis. The giant cells may contain different types of structures, such as asteroid body, Schaumann body, Hamazaki Wezenberg body. Asteroid bodies are star shaped inclusions, probably derived from cytoskeletal proteins. Schaumann bodies are crystalline inclusions composed of calcium and proteins. These structures are found in sarcoidosis with variable frequency and found more commonly lymph node compared to skin. These are neither universally present nor specific for diagnosis of sarcoidosis and can also be found in other granulomatous inflammation. Minor percentage of cases can show presence of osteoclast like giant cells.[7]
Foreign material is frequently found in sarcoidosis. Various studies have reported polarizable foreign bodies in giant cells in 22-50% of sarcoidosis.[6,8,9,10,11] Presence of foreign material doesn’t exclude a diagnosis of sarcoidosis.
Subcutaneous sarcoidosis is a rare manifestation of sarcoidosis, and their reported frequency ranges from 1.4 to 6% in patients of systemic sarcoidosis.[12,13,14] In a large series on subcutaneous sarcoidosis, Marcoval et al. reported 28 of 640 patients (4.38%) of systemic sarcoidosis had subcutaneous sarcoidosis.[15] Subcutaneous sarcoidosis presents as discrete subcutaneous nodules and most commonly involves forearm. Some authors believe subcutaneous sarcoidosis should be restricted to subcutis and not involve dermis. However, some extent of lower dermal involvement is acceptable. Marcoval et al. reported 10 out of 28 (35.7%) cases in their series had granulomas in deeper dermis.[15] To start with, subcutaneous sarcoidosis shows lobular involvement, and with disease progression can also involve the septa, producing both lobular and septal granulomatous panniculitis. Isolated septal involvement is usually not seen.[16,17] Subcutaneous sarcoidosis can be associated with significant fibrosis.[18,19] Marcovel et al. reported 75% cases of subcutaneous sarcoidosis demonstrated fibrosis, which were of septal, intergranulomatous, or diffuse form. However, fibrosis in subcutaneous sarcoidosis doesn’t correlate with pulmonary fibrosis.[15]
Up to 25% cases of cutaneous sarcoidosis don’t show systemic involvement even on long term follow up.[7] These cases are difficult to diagnose. The diagnosis in these cases typically rely on a good clinico-pathological correlation and exclusion of other granulomatous dermatoses.[20]
Pearls for diagnosis of cutaneous sarcoid:
Sarcoidal granulomas
Langhans and foreign body giant cells
Absence of caseation necrosis
Giant cells containing inclusions such as asteroid body, Schaumann body
Pitfalls:
Minority of cases show tubercular and interstitial granuloma
Fibrinoid necrosis is common, not to confuse with caseous necrosis.
Inclusion bodies are neither sensitive nor a specific feature of sarcoidosis
Exclusion of an infective aetiology is essential
Necrobiotic granulomas
We shall discuss granuloma annulare, necrobiosis lipoidica and rheumatoid nodule in this section.
Granuloma annulare
Granuloma annulare (GA) is a prototype of necrobiotic granuloma. It is a self-limiting non-infectious granulomatous dermatitis, described more than 100 years ago.[21] Commonest clinical presentation is skin-colored to erythematous papular lesions in an annular configuration, predominantly involving the extremities [Figure 3].
Figure 3.
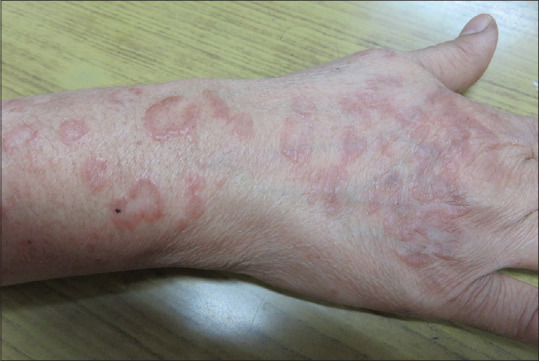
Case of granuloma annulare showing erythematous papules coalescing in an annular fashion on the dorsum of hand and forearm
Histopathology
Histologically, GA is characterized by the presence of necrobiotic granulomas involving predominantly upper and mid dermis [Figure 4]. Palisading and interstitial patterns of granuloma are commonly encountered. The palisading pattern is characterized by central zone of necrobiotic collagen walled off by palisaded histiocytes admixed with other inflammatory cells [Figure 5a]. The interstitial granulomatous pattern is characterized by collection of histiocytes in between the collagen bundles without forming well-defined aggregate.[22] Well formed sarcoidal or tuberculoid granulomas are rare in GA. There is disagreement among different studies regarding the commonest pattern of granuloma in GA. Umbert et al. examined 207 patients of GA and found interstitial pattern in as many as 71% of the cases.[23] Similarly, Chatterjee et al. in a previous study from India found interstitial granuloma to be the commonest pattern (44%).[24] Cheng et al., however, found palisading histiocytes to be the predominant pattern of infiltration.[25] Presence of degenerated collagen and is almost universally observed, while elastic fibers may be normal, reduced, or absent.[25,26] Dermal mucin is observed frequently [Figure 5b], and with aid of special stain such as Alcian blue or colloidal iron, it can be demonstrated in as many as 93% of the cases.[25] The granulomatous inflammation in GA usually involves upper and mid dermis. Lower dermal and subcutaneous involvement is less frequent. Subcutaneous GA is seen exclusively in pediatric age group and is characterized by presence of firm, painless subcutaneous nodules predominantly on the extremities, which may either occur alone or in association with lesion in the dermis.[27] To qualify for subcutaneous GA, the brunt of the disease should be present in the subcutaneous fat with variable dermal involvement. Dominant dermal involvement with focal subcutaneous extension of granulomas does not qualify for subcutaneous GA. Stefenaki et al. studied the histologic pattern of GA in children and found subcutaneous extension of inflammation in 6 of 13 cases (46%).[28]
Figure 4.
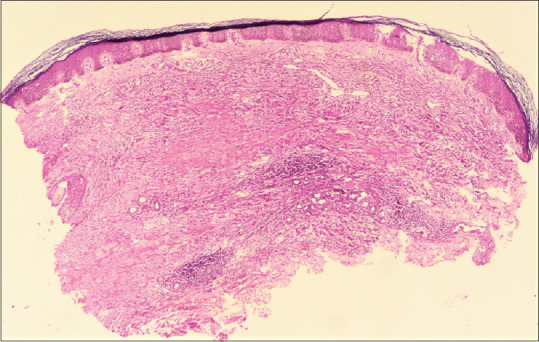
Granuloma annulare: biopsy shows areas of necrobiosis involving upper and mid dermis (hematoxylin and eosin, x40)
Figure 5.
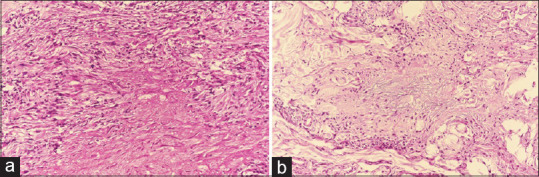
Granuloma annulare: (a) Areas of altered collagen and fragmented elastic fibers, surrounded by palisading histiocytes (hematoxylin and eosin, x200), (b) Mucin production and interstitial histiocytic infiltrate, so called blue granuloma (hematoxylin and eosin, x200)
A number of studies in the literature have found eosinophils infiltrate in GA, the frequency (18–66%) and the extent of which is highly variable in different studies.[29,30,31,32] Romero et al. in a retrospective study found eosinophils in 66% (51/77) of the biopsies, more commonly associated with a palisaded pattern of infiltrate.[31] Chatterjee et al. found eosinophils in 20% of their GA cases, but it didn’t show any correlation with the pattern of granuloma.[24] Cheng et al. reported a positive correlation between eosinophilic infiltrate and subcutaneous GA.[25] Although the presence of eosinophils in GA is known since long, their role in the pathogenesis of the disease remains unexplained.[30,32,33] Small-vessel vasculitis has been variably reported in GA. Dahl et al. found vascular changes such as vessel wall necrosis, fibrinoid change, thickening, and occlusion in 33 of 38 GA patients (86%).[34] This, however, is an exceptional finding as no other study has reported such high frequency of vasculitis in GA. Chatterjee et al. reported small vessel vasculitis in 20% of their GA biopsies.[24] Chaitra et al. reported vasculitis in 10% of the cases and postulated that vasculitis causes microinfarct in the dermis, which consequently leads to collagen alteration and histocytic infiltration in GA.[29] However, leukocytoclastic vasculitis was not detected in any of the cases by Gunes et al., who, therefore suggested that pathogenesis of GA does not involve vascular changes.[26] Chatterjee et al. reported plasma cell infiltrate in 33% of their GA cases.[24]
The differential diagnosis of GA includes other granulomatous inflammation, such as NBL, sarcoidosis, rheumatoid nodule, palisaded neutrophilic granulomatous dermatitis, and various infectious dermatosis. In most of the cases, the diagnosis is straight forward, presence of collagen degeneration and stromal mucin deposition confirm the diagnosis.[29] However, diagnosis may be challenging in cases lacking mucin deposition. GA can closely resemble NBL histologically, however these two diseases differ in clinical presentation. Histologically, NBL shows predominantly lower dermal involvement as compared to upper dermal involvement in GA, and it lacks abundant mucin deposition. Sometimes GA and NBL can be present simultaneously. Although GA can be distinguished from NBL in most of the cases, sometimes it may be practically impossible to distinguish these two entities, both clinically and histologically. Rheumatoid nodule usually involves lower dermis and subcutaneous tissue. Histologically, it is characterized by central area of fibrinoid necrosis, surrounded by palisading histiocytes. Mucin deposition is extremely rare. It should be differentiated from subcutaneous GA. Sarcoidosis shows compact, well-formed granulomas, with minimal lymphoid infiltrate. A possibility of infectious granuloma should be carefully excluded in all cases using special stains such as ZN, PAS, etc.
Diagnostic pearls
Predominantly upper dermal involvement.
Necrobiosis shows areas of altered collagen and elastic tissue with palisaded or interstitial histiocytes.
Mucin deposition in areas of necrobiosis.
Pitfalls
Sometimes difficult to distinguish from necrobiosis lipoidica, especially in cases with inconspicuous mucin deposition.
Scattered interstitial histiocytic infiltrate can be missed in inexperienced eyes.
Necrobiosis lipoidica
Necrobiosis lipoidica (NBL) is a chronic non-infectious granulomatous dermatitis. It was previously known as necrobiosis lipoidica diabeticorum, as it was mostly seen in patients with diabetes. However, <1% of the diabetics develop NBL and it is now recognized that NBL can be seen in many patients without diabetes.[35] NBL commonly affects pretibial skin and presents as well demarcated areas of shiny and thinned skin.
Histopathology
NBL usually affects the entire or lower part of dermis [Figure 6a]. Isolated upper dermal involvement is rare. The overlying epidermis can be normal, atrophic or hypertrophic. Sometimes, NBL can show ulceration of the epidermis. The necrobiosis in NBL is usually not as prominent as GA. It can show small areas of necrobiosis as seen as areas of altered collagen, with interstitial excess of epithelioid histiocytes. Some cases show well demarcated areas of necrobiosis, with palisaded histiocytes and well-formed epithelioid cell granulomas, with or without giant cells. The areas of necrobiosis sometimes alter with areas of normal collagen in a layered manner with inflammatory cells present in between the layers of necrobiosis (”wedding cake” or “sandwich” appearance).[4] The necrobiosis appears red on hematoxylin and eosin (red granuloma) because of hyalinization [Figure 6b].[36,37] Mucin deposition is usually absent, however, some amount of mucin deposition has been reported in rare cases of NBL, making it difficult to differentiate from GA.[38] Nuclear dust may be present inside the area of necrobiosis and elastic fibers are usually diminished in the center. The necrobiosis often extends into the superficial subcutis and entraps degenerated fat. Presence of perivascular lymphoplasmacytic infiltrate in the deeper dermis can be observed. There may be lymphoid aggregates with or without germinal center in the dermis-hypodermis junction, with extension of the infiltrate along the subcutaneous fat septa.[36] Long-standing cases may show significant fibrosis of the stroma.
Figure 6.
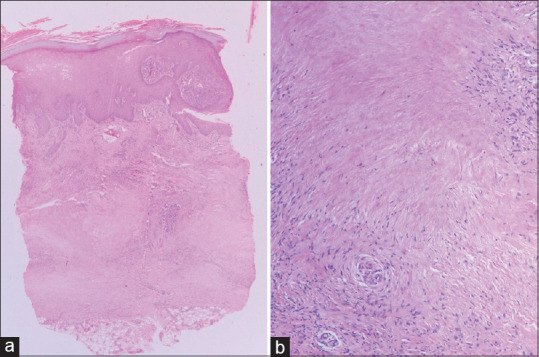
Necrobiosis lipoidica: (a) showing necrobiosis mainly involving middle and lower dermis (hematoxylin and eosin, x40), (b) Areas of necrobiosis without mucin, surrounded by palisaded histiocytes (hematoxylin and eosin, x200)
NBL can be present along with other non-infectious granulomatous dermatoses, such as GA and sarcoidosis.[39,40,41] Squamous cell carcinoma is a rare but well recognized complication.[42] The differential diagnoses of NBL include GA and other necrobiotic NIGD [Table 2].
Table 2.
Differential diagnoses of necrobiotic granuloma
| Features | Granuloma annulare | Necrobiosis lipoidica | Rheumatoid nodule |
|---|---|---|---|
| Clinical | |||
| Age of presentation, gender | Children to young adults, equally affects male and female | Middle age, more common in female | Adults with rheumatoid factor positive rheumatoid arthritis, more common in females |
| Site involved | Arm, hand, leg, feet | Shin, ankle, calves, thighs | Extensor prominences, proximal ulna, olecranon process, metacarpophalangeal joint, proximal interphalangeal joint |
| Type of lesion | Small, firm, asymptomatic papules, pale red in color | Single or multiple, irregular, sharply demarcated patches or plaques | Deep seated form nodules ranging in size from few mm to 5 cm |
| Histopathology | |||
| Area involved | Upper and mid dermis | Entire dermis or lower two thirds | Subcutaneous fat, can also involve dermis |
| Extent of necrobiosis | Usually large areas of necrobiosis | Early lesions show small areas of necrobiosis separated by normal collagen (sandwich sign), late lesions show larger areas of necrobiosis | Small nodular areas of necrobiosis in the subcutaneous tissue |
| Nature of granuloma | Areas of necrobiosis with palisaded or interstitial histiocytic infiltrate, well-formed granuloma is rare | Areas of necrobiosis surrounded by interstitial infiltrate by histiocytes, well-formed granuloma is rare | Nodular granulomatous inflammation, with central fibrinoid necrosis and peripheral palisading of histiocytes |
| Mucin/fibrin deposition | Mucin deposition common (blue granuloma) | Can show fibrin deposition (red granuloma), mucin deposition rare | Fibrinoid necrosis common (red granuloma) |
| Associated inflammation | Lymphoid infiltrate in the adjacent dermis | Lymphocyte and plasma cell infiltrate common in the junction of dermis and subcutaneous fat | Lymphocyte, plasma cell and neutrophils |
| Vasculitis | Can be seen (variable) | Rare | Rare |
| Panniculitis | Rare | Can be present | Commonly seen |
| Fibrosis | Rare | Can be seen in late stage | Commonly seen |
Diagnostic pearls
Involvement of entire or lower two thirds of dermis by necrobiosis, with extension into subcutaneous fat.
Areas of necrobiosis with hyalinization of collagen (red granuloma)
Palisaded or interstitial histiocytes, and absence of mucin.
Lymphoplasmacytic infiltrate in deeper dermis.
Pitfalls
Inconspicuous granulomatous inflammation
Inconspicuous focus of necrobiosis.
Rheumatoid nodule (RN)
RN is the most common extra-articular manifestation of rheumatoid arthritis (RA). It is seen in up to 25% patients of seropositive RA. However, RN can be seen in association with other autoimmune disease, in absence of RA. It presents as deeply seated firm, nodular mass on the extensor surfaces, such as proximal ulna, olecranon process, metacarpophalangeal joint and proximal interphalangeal joint.[43,44]
Histopathology
The pathology shows nodular lesion involving subcutaneous fat with or without lower dermis. It shows areas of fibrinoid necrosis of collagen. The necrosis can be inconspicuous in early cases, which are difficult to diagnose. Established cases show well demarcated areas of fibrinoid necrosis, which appears bright red on routine hematoxylin and eosin (H and E) stain, surrounded by palisaded histiocytes [Figure 7]. These macrophages are mostly HLA-DR+ and admixed with foreign body giant cells in majority of cases. This area of necrosis usually contains nuclear debris, but mucin is not present. In between the areas of necrobiosis, the stroma shows fibrosis, vascular proliferation and variable degree of inflammation. The inflammatory infiltrate is composed of combination of neutrophils, lymphocytes and plasma cells, admixed with histiocytes.[45,46] In perforating RN, the necrobiosis process extends through epidermis, producing transepidermal elimination. Vasculitis is rarely described in RN.[47] The main differential diagnosis is subcutaneous GA, which can be differentiated by presence of mucin deposition.
Figure 7.
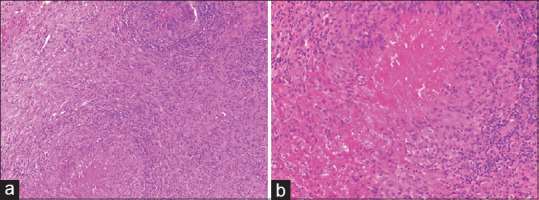
Rheumatoid nodule: (a) Multiple epithelioid cell granulomas with intervening areas of fibrosis and inflammation (hematoxylin and eosin, x100), (b) The granulomas show central fibrinoid necrosis with peripheral palisading histiocytes, so called red granuloma (hematoxylin and eosin, x200)
Metastatic Crohn's disease (MCD)
Crohn's disease (CD) is a chronic, inflammatory granulomatous disorder, which predominantly involves the gastrointestinal (GI) tract, usually affecting young adults.[48] Extra-intestinal manifestations of CD include cutaneous, ocular, articular and oral involvement.[49] Metastatic CD (MCD) is the rarest form of cutaneous involvement by CD, and has been defined as sterile granulomatous inflammation of the skin, occurring at sites non-contiguous from the GI tract in patients of CD.[50,51] MCD usually affects young adults. Vulval involvement in commonest in MCD and presents as vulval edema, swelling, ulceration, and fissure [Figure 8].[52,53,54] Up to 80% of vulval MCD is associated with intestinal CD. MCD can be the presenting feature of CD, and precede the diagnosis of intestinal CD in 25-30% cases.[26,55]
Figure 8.

A case of metastatic Crohn's disease showing well defined plaque with surrounding diffuse vulval edema
Histopathology
Microscopically, MCD is characterized by presence of non-caseating epithelioid cell granulomas with Langhans and foreign body type giant cells [Figure 9a]. However, the incidence of granulomatous inflammation can be quite variable. In a study by Bhoyrul et al., only 46% cases of vulval MCD demonstrated granulomas.[52] The authors suggested that dependence on non-caseating granuloma as an absolute criterion for diagnosis of MCD is unreliable. However, in a subsequent study of 12 cases of MCD, Patrick et al. found epithelioid cell granulomas in all cases.[56] Chatterjee et al. in a previous study from India reported granulomatous inflammation in 11 out of 12 (92%) cases, with the remaining case showing scattered giant cells.[55] They have reported perifollicular distribution of granulomas with follicular destruction as a histological clue for MCD diagnosis. Granuloma in MCD is mostly sarcoidal type, without significant lymphoid cuffing or necrosis. Other histological features include panniculitis, vasculitis, eosinophil, and plasma cell infiltrate [Figure 9b], which are present to a variable extent.[55,57] The main histological differential diagnoses are sarcoidosis and tuberculosis (TB). Both tuberculosis and CD can involve GI track and skin. Sometimes these are indistinguishable without polymerase chain reaction (PCR) for confirming the diagnosis of TB. Some cases are mislabeled as sarcoidosis or TB, and the final diagnosis of MCD is established after repeated clinicopathological correlation.[55]
Figure 9.
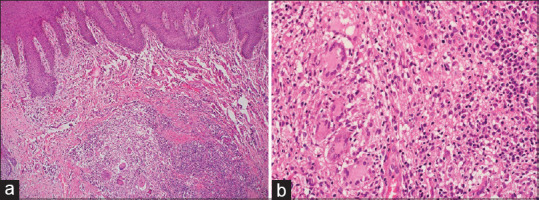
Metastatic Crohn's disease: (a) Skin biopsy shows granulomatous inflammation in the dermis with surrounding inflammation (hematoxylin and eosin, x40), (b) Granulomas shows foreign body and Langhan's type giant cells with adjacent lymphoplasmacytic infiltrate (hematoxylin and eosin, x400)
Granulomatous rosacea
Granulomatous rosacea (GR) is a variant of rosacea, characterized by periorifacial yellow, brown, or red monomorphic papules or nodules.[58] Though considered a variant, the patients may not have all the classical symptoms of rosacea like flushing and erythema.[59]
Histopathology
On histological examination, GR shows variable pattern of inflammation but the diagnostic hallmark is presence of tuberculoid granulomas in the dermis, admixed with lymphoid infiltrate and dilated capillaries in the upper dermis. Usually there is no necrosis or nuclear debris.[60] The granulomas are usually small and located in the perifollicular location. Upper dermal inflammation is more common and upper dermis looks busy under lower magnification [Figure 10]. The demonstration of granulomas in GR cases also depends on the age of the lesion at the time of biopsy. Histologically, GR was classified by Sanchez et al. based on the location of the inflammatory infiltrate into perifollicular, nodular, diffuse, and combined patterns.[61] Demodex folliculorum infestation is commonly found.
Figure 10.
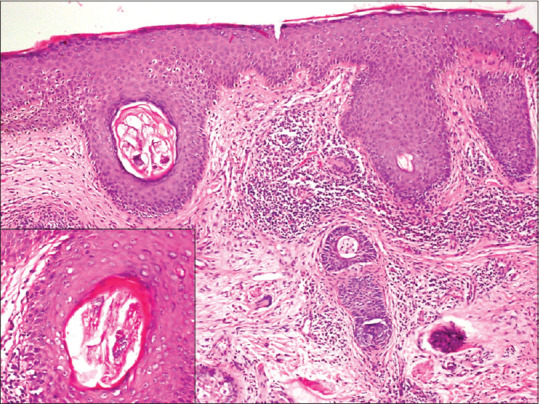
Granulomatous rosacea: nodular pattern of inflammation, with presence of tiny epithelioid cell granulomas with occasional giant cells. Inset showing degenerated fragments of Demodex folliculorum (hematoxylin and eosin, x100)
Lupus miliaris disseminatus faciei (LMDF)
LMDF is an idiopathic granulomatous inflammatory condition characterized by multiple discrete red-brown, dome-shaped papules on the medial and lateral areas of the face, with frequent involvement of the neck and chin [Figure 11].[62] The exact etiology of LMDF is not known, but previously it was thought to be associated with tuberculosis, sarcoidosis, and rosacea.[63] Some authors have suggested the terminology L. F.I.GU.R.E.: facial idiopathic granulomas with regressive evolution instead of LMDF, however, this is still not a well-accepted terminology.
Figure 11.
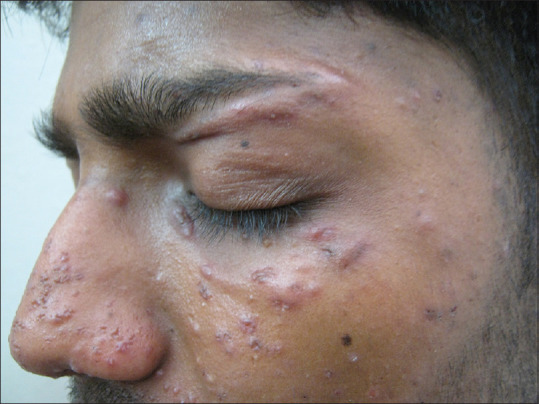
Case of lupus miliaris disseminatus faciei showing erythematous papules with central necrosis on the nose, eyelids, and malar area, with areas of scarring
Histopathology
Histologically, LMDF is characterized by presence of large granulomas in the dermis, mostly in relationship to pilosebaceous units [Figure 12a]. The center of the granulomas contain necrosis and nuclear debris, resembling tuberculosis closely [Figure 12b]. The necrosis can be both caseous and fibrinoid type.[60] The adjacent dermis shows mild lymphomononuclear infiltrate. Usually there is no capillary dilatation or Demodex folliculorum infestation. The main differential diagnosis is tuberculosis, from which it can be distinguished based on clinical and PCR findings.[60]
Figure 12.
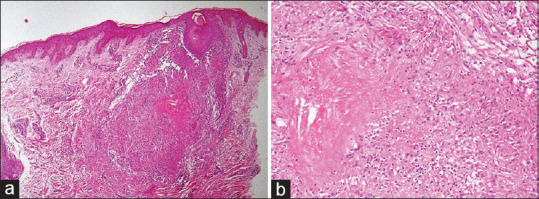
Lupus miliaris disseminatus faciei: (a) Dermis showing a well-formed granuloma in perifollicular location (hematoxylin and eosin, x40), (b) The granuloma shows central areas of caseation necrosis and nuclear debris (hematoxylin and eosin, x200)
Interstitial granuloma patterns
We shall discuss interstitial granulomatous dermatitis, interstitial granulomatous drug reaction and palisaded neutrophilic and granulomatous dermatitis in this section.
Interstitial granulomatous dermatitis (IGD)
IGD is a rare form of non-infectious granulomatous dermatosis, which is often associated with autoimmune disorders. It is seen in adults with female preponderance. It is usually seen in the setting of underlying systemic autoimmune diseases, most commonly rheumatoid arthritis and systemic lupus erythematosus. Association with systemic malignancy, some drugs have also been reported. Clinically it presents as erythematous to violaceous patches and plaques on the upper trunk and proximal limbs. Some cases (approximately 10%) show presence of linear subcutaneous cords or ropes (known as rope sign), which is considered very characteristic.[64,65]
Histologically, IGD shows small foci of degenerated collagen, with interstitial epithelioid histiocytes [Figure 13]. The density of histiocytes can be variable and the infiltrate usually involves the entire dermis including deeper dermis. Small granuloma formation may be seen. The histiocytic rimming of abnormal collagen may form cleft from the adjacent normal dermis, also known as “floating sign”.[65,66] Epidermis usually appears normal, and the dermis shows variable extent of inflammatory infiltrate composed of lymphocytes, neutrophils, and eosinophils. Mucin deposition and vasculitis are not encountered.
Figure 13.
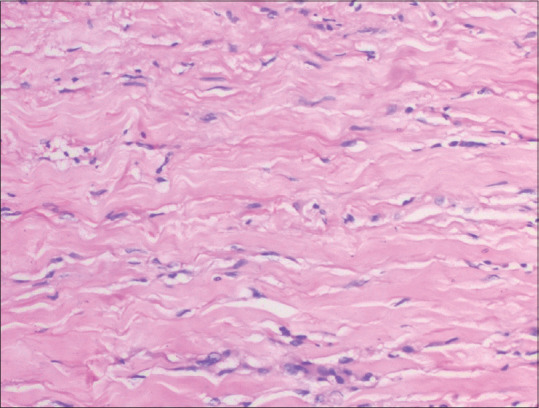
Interstitial granulomatous dermatitis: dermis shows interstitial infiltration of histiocytes with occasional giant cells and small areas of altered collagen (hematoxylin and eosin, x200)
Interstitial granulomatous drug reaction (IGDR)
Although various drugs may produce IGD, IGDR deserves separate discussion. Many authors have used the terminologies drug-induced IGD and IGDR interchangeably, but they constitute different entity. The list of drugs that can produce IGDR is long, the common ones include calcium channel inhibitors, beta blockers, ACE inhibitors, statins etc.[67,68] IGDR presents as erythematous to violaceous plaques on the proximal arms, medial thighs and proximal trunk.
Histologically, IGDR shows features similar to IGD. It is characterized by small areas of degenerated collagen, with interstitial infiltration by epithelioid histiocytes, with formation of ill-formed granulomas. Giant cells can be rarely seen. Mucin deposition and vasculitis are rare.[67] However, in contrast to IGD, IGDR shows interface dermatitis with vacuolar generation of basal keratinocytes, apoptotic keratinocytes, and eosinophilic infiltrate in the dermis. These three features help to distinguish IGDR from IGD.[69] Sometimes, the lymphoid cells may show nuclear indentation and epidermotropism.[67]
Palisaded neutrophilic and granulomatous dermatitis (PNGD)
PNGD is another rare type of NIGD with significant systemic association. Most of the PNGD cases are associated with systemic autoimmune diseases, particularly rheumatoid arthritis and systemic lupus erythematosus. It classically presents as flesh colored erythematous papule with central umbilication, present on the extremities, particularly elbow.[70,71,72,73] Rarely PNGD may be associated with sarcoidosis.[74]
Histological features of PNGD depends on the stage of the disease. At initial stage, it usually shows leukocytoclastic vasculitis, stromal neutrophilic infiltrate and karyhorrectic debris.[71,75,76] However, the picture is never dominated by pure vasculitis, usually there is more intense neutrophilic infiltrate and debris. Scattered interstitial epithelioid histiocytes may be encountered. In the more advanced stage of the disease, there is central collection of neutrophils and nuclear debris along with degenerated collagen, surrounded by palisaded histiocytes [Figure 14]. Well-formed granuloma is rarely observed. Mucin deposition is rare. Leukocytoclastic vasculitis may be present in the advanced stage in a minority of patients, which if present, helps to distinguish PNGD from IGD. Eventually, the inflammation is replaced by fibrosis.[70,76] Many authors believe that IGD and PNGD are not separate diseases, they represent different spectrum of the same disease. However, this is a still unresolved question.
Figure 14.
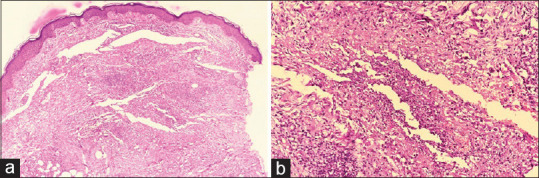
Palisaded neutrophilic granulomatous dermatitis: (a) Dermis shows areas of neutrophilic collection (hematoxylin and eosin, x40), (b) Dermis shows areas of altered collagen, collection of neutrophils, with palisaded histiocytes at the periphery (hematoxylin and eosin, x200)
Annular elastolytic giant cell granuloma (AEGCG)
AEGCG is a rare form of granulomatous dermatitis, characterized by presence of elastolysis and engulfed elastic fibers within giant cells. It is essentially a histological diagnosis. The clinical spectrum ranges from large annular plaques to papules and reticular form. Although it commonly involves sun-exposed areas, it can also exam non-sun-exposed areas as well. Since all cases do not present as annular plaques, some authors prefer the term giant cell elastolytic granuloma (GCEG).[77]
Histological examination reveals granulomatous inflammation commonly involving upper dermis, but it can also involve middle and deeper dermis [Figure 15a]. There is elastic tissue degeneration and evidence of elastophagocytosis by the giant cells [Figure 15b]. This phenomenon can be appreciated on hematoxylin and eosin stain, but can be better highlighted on elastic tissue stain such as elastic Van Gieson (EVG) or Verhoeff- Van Gieson (VVG) stain.[77] The granulomas are usually diffuse and interstitial type, rather than well-formed compact granulomas.[78] Areas of altered collagen (necrobiosis), mucin deposition, vasculitis is not encountered.
Figure 15.
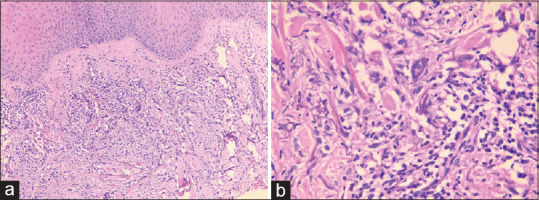
Elastolytic giant cell granuloma: (a) Dermis shows areas of elastolysis with formation of ill-formed granuloma (hematoxylin and eosin, x100), (b) Dermis shows fragmented elastic fibers and elastophagocytosis by the giant cells (hematoxylin and eosin, x400)
Granulomatous vasculitis
Granulomatosis with polyangiitis (GPA): GPA, previously known as Wegener's granulomatosis, is a systemic small vessel vasculitis, which commonly affects respiratory track and kidney. However, it can variably involve skin (10-50%). It commonly presents as cutaneous ulcers.[79] In minority of cases, the cutaneous manifestations may precede the systemic manifestations.[80] Histological examination commonly shows necrotizing small vessel vasculitis or leukocytoclastic vasculitis. Granulomatous vasculitis is rarely encountered in cutaneous GPA.[81] Some cases show transmural inflammation of the vessel wall with presence of giant cells, which provides important diagnostic clue. Small foci of necrosis and palisaded histiocytes may be seen [Figure 16]. The histology in some cases may resemble PNGD.[80] Rarely GPA can present as pure granulomatous vasculitis involving small arteries. There is limited differential diagnosis of granulomatous vasculitis of skin. Thus, a diagnosis of GPA should be suspected and the same should be confirmed by demonstrating ANCA (anti neutrophilic cytoplasmic antibody). Other ANCA associated vasculitis (AAV) can also variably involve the skin with presence of granulomatous inflammation, but presentation as granulomatous vasculitis is extremely rare.
Figure 16.
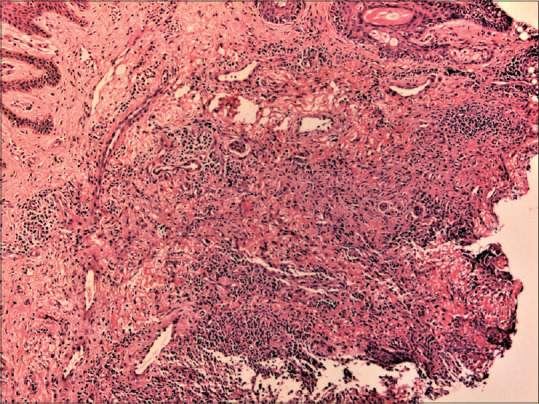
A case of granulomatosis with polyangiitis: dermis shows granulomatous inflammation, small areas of necrosis and small vessel vasculitis (hematoxylin and eosin, x100)
Granulomatous lymphoproliferative disorders
Granulomatous reaction is a rare manifestation of cutaneous lymphoproliferative disorders. Two lymphomas that produces granulomatous reaction pattern include cutaneous T cell lymphoma (CTCL) and lymphomatoid granulomatosis. However, it should be remembered that any lymphoma can produce secondary granulomatous inflammation as a reaction to tumor antigen.
Granulomatous cutaneous T cell lymphoma
Granulomatous inflammation is a rare manifestation of CTCL (<2%).[82] Two forms of CTCL are associated with granulomatous inflammation, namely granulomatous mycosis fungoides (GMF) and granulomatous slack skin (GSS). The clinical manifestation of GMF is similar to classical MF, and may present with patch, plaque, tumor, erythroderma, poikilodermatous patches or annular lesions. GSS is a very rare form of CTCL which presents as bulky loose skin folds with predilection for axilla and groin.[83]
On histological examination, GMF and GSS shows overlapping features and they can’t be distinguished by histology alone. GMF shows lichenoid infiltrate by atypical lymphoid cells, which show CD4+/CD8- phenotype. There is interstitial and perivascular granulomas, resembling sarcoidosis. In GSS, histology classically shows diffuse dermal granulomatous inflammation with presence of giant cells exhibiting elastophagocytosis. There is loss of elastic fibers in the dermis.[84] There is atypical lymphoid cell infiltrate in the dermis, which shows convoluted nuclei. Sometimes, the lymphoid cells may be scanty and get obscured by the granulomas, delaying the diagnosis and treatment.[83]
Orofacial granulomatosis (OFG)
A number of conditions present as granulomatous inflammation involving orofacial area, which include both infectious and non-infectious etiology. Clinically, OFG can include any part of oral cavity or face.[85,86] When the disease it restricted to the lip, it is also known as granulomatous cheilitis or cheilitis granulomatosa.[87] Among the non-infectious causes of orofacial granulomatosis, it may be primary or idiopathic (idiopathic OFG) or it may be secondary to various systemic diseases (such as sarcoidosis and Crohn's disease). The clinical presentation of OFG may involve entire facial tissue, but most commonly involves lip. It presents as painless swelling, which may involve one or both lips. Idiopathic OFG, also known as Melkersson Rosenthal syndrome, is usually associated with facial nerve palsy.[85,86]
Histology of OFG shows usually unremarkable epidermis/epithelium. Superficial lamina propria shows edema and ectatic vascular channels. There is interstitial and perivascular histiocytic collections forming loose granulomas, which may reach deeper up to minor salivary glands and skeletal muscles [Figure 17]. Some cases may show presence of well-formed compact “sarcoid like” granulomas.[86,87,88] These granulomas are accompanied by mild lymphoplasmacytic infiltrate. It is important to exclude other secondary causes such as infections, sarcoidosis or Crohn's disease before a diagnosis of idiopathic OFG/idiopathic granulomatous cheilitis is established.[86,87]
Figure 17.
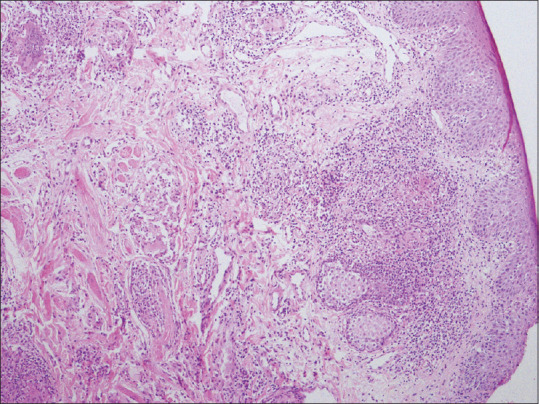
Granulomatous cheilitis showing lamina propria edema, dilated lymphatic channels, epithelioid cell granulomas accompanied by mild lymphoid infiltrate extending into skeletal muscle (hematoxylin and eosin, x100)
Foreign body granulomas
Granulomatous dermatitis is a common pattern of tissue reaction to various foreign bodies that enters the skin. Various agents, such as tattoo, splinter, suture material can produce foreign body type granulomas. Sometimes endogenous substances can also produce foreign body granulomas, such as ruptured hair follicle, ruptured epidermal inclusion cyst; where keratin incites granulomatous reaction. Histologically, these granulomas show presence of foreign body giant cells. These are large cells with numerous nuclei arranged haphazardly. These giant cells may contain ingested foreign body.
Granulomatous dermatitis to cosmetic agents
Soft tissue augmentation is a common procedure in cosmetology practice. Injectable hyaluronic acid (HA) derivatives are most commonly used reabsorbable dermal fillers. Although these agents are considered safe, there are few reports of local adverse reaction to these agents.[89,90] They produce foreign body granulomatous response. However, these conditions are self-resolving.
Thus, a wide range of non-infectious conditions can produce granulomatous dermatitis. Recognition of the pattern of granuloma and looking for subtle histological clue are extremely important for making a correct diagnosis. It should be worth mentioning that many conditions can show overlapping histological features; thus, a good clinical correlation is often required.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Bal A, Mohan H, Dhami GP. Infectious granulomatous dermatitis: A clinico pathological study. Indian J Dermatol. 2006;51:217–20. [Google Scholar]
- 2.Queiros CS, Uva L, Soares de Almeida L, Filipe P. Granulomatous skin diseases in a tertiary care Portuguese hospital: A 10-year retrospective study. Am J Dermatopathol. 2020;42:157–64. doi: 10.1097/DAD.0000000000001441. [DOI] [PubMed] [Google Scholar]
- 3.Lynch JM, Barrett TL. Collagenolytic (necrobiotic) granulomas: Part 1--The “blue” granulomas. J Cutan Pathol. 2004;31:353–61. doi: 10.1111/j.0303-6987.2004.00194.x. [DOI] [PubMed] [Google Scholar]
- 4.Lynch JM, Barrett TL. Collagenolytic (necrobiotic) granulomas: Part II--The ‘red’ granulomas. J Cutan Pathol. 2004;31:409–18. doi: 10.1111/j.0303-6987.2004.00208.x. [DOI] [PubMed] [Google Scholar]
- 5.Mohan H, Bal A, Dhami GP. Non-infectious granulomatous dermatitis: A clinicopathological study. J Cutan Pathol. 2006;33:767–71. doi: 10.1111/j.1600-0560.2006.00566.x. [DOI] [PubMed] [Google Scholar]
- 6.Mangas C, Fernandez-Figueras MT, Fite E, Fernandez-Chico N, Sabat M, Ferrandiz C. Clinical spectrum and histological analysis of 32 cases of specific cutaneous sarcoidosis. J Cutan Pathol. 2006;33:772–7. doi: 10.1111/j.1600-0560.2006.00563.x. [DOI] [PubMed] [Google Scholar]
- 7.Wu MC, Lee JY. Cutaneous sarcoidosis in southern Taiwan: Clinicopathologic study of a series with high proportions of lesions confined to the face and angiolupoid variant. J Eur Acad Dermatol Venereol. 2013;27:499–505. doi: 10.1111/j.1468-3083.2012.04473.x. [DOI] [PubMed] [Google Scholar]
- 8.Ball NJ, Kho GT, Martinka M. The histologic spectrum of cutaneous sarcoidosis: A study of twenty-eight cases. J Cutan Pathol. 2004;31:160–8. doi: 10.1111/j.0303-6987.2004.00157.x. [DOI] [PubMed] [Google Scholar]
- 9.Marcoval J, Mana J, Moreno A, Gallego I, Fortuno Y, Peyri J. Foreign bodies in granulomatous cutaneous lesions of patients with systemic sarcoidosis. Arch Dermatol. 2001;137:427–30. [PubMed] [Google Scholar]
- 10.Kim YC, Triffet MK, Gibson LE. Foreign bodies in sarcoidosis. Am J Dermatopathol. 2000;22:408–12. doi: 10.1097/00000372-200010000-00004. [DOI] [PubMed] [Google Scholar]
- 11.Callen JP. The presence of foreign bodies does not exclude the diagnosis of sarcoidosis. Arch Dermatol. 2001;137:485–6. [PubMed] [Google Scholar]
- 12.Marcoval J, Mana J, Moreno A, Peyri J. Subcutaneous sarcoidosis--Clinicopathological study of 10 cases. Br J Dermatol. 2005;153:790–4. doi: 10.1111/j.1365-2133.2005.06815.x. [DOI] [PubMed] [Google Scholar]
- 13.Higgins EM, Salisbury JR, Du Vivier AW. Subcutaneous sarcoidosis. Clin Exp Dermatol. 1993;18:65–6. doi: 10.1111/j.1365-2230.1993.tb00972.x. [DOI] [PubMed] [Google Scholar]
- 14.Mayock RL, Bertrand P, Morrison CE, Scott JH. Manifestations of sarcoidosis. Analysis of 145 Patients, with a review of nine series selected from the literature. Am J Med. 1963;35:67–89. doi: 10.1016/0002-9343(63)90165-7. [DOI] [PubMed] [Google Scholar]
- 15.Marcoval J, Penin RM, Mana J. Histopathological features of subcutaneous sarcoidosis. Am J Dermatopathol. 2020;42:233–43. doi: 10.1097/DAD.0000000000001509. [DOI] [PubMed] [Google Scholar]
- 16.Requena L, Sanchez Yus E. Panniculitis. Part II. Mostly lobular panniculitis. J Am Acad Dermatol. 2001;45:325–61. doi: 10.1067/mjd.2001.114735. quiz 62-4. [DOI] [PubMed] [Google Scholar]
- 17.Vainsencher D, Winkelmann RK. Subcutaneous sarcoidosis. Arch Dermatol. 1984;120:1028–31. [PubMed] [Google Scholar]
- 18.Resnik KS. Subcutaneous sarcoidosis histopathologically manifested as fibrosing granulomatous panniculitis. J Am Acad Dermatol. 2006;55:918–9. doi: 10.1016/j.jaad.2006.04.080. [DOI] [PubMed] [Google Scholar]
- 19.Llamas-Velasco M, Godoy A, Fraga J, Sanchez-Perez J. Fibrosing cutaneous sarcoidosis. Actas Dermosifiliogr. 2012;103:331–3. doi: 10.1016/j.ad.2011.06.009. [DOI] [PubMed] [Google Scholar]
- 20.Bhattacharjee R, Chatterjee D, De D. A man with infiltrated plaques on the pretibial area. JAMA Dermatol. 2018;154:955–6. doi: 10.1001/jamadermatol.2017.6449. [DOI] [PubMed] [Google Scholar]
- 21.Fox TC. Ringed eruption of the fingers. Br J Dermatol. 1895;7:91. [Google Scholar]
- 22.Piette EW, Rosenbach M. Granuloma annulare: Clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457–65. doi: 10.1016/j.jaad.2015.03.054. [DOI] [PubMed] [Google Scholar]
- 23.Umbert P, Winkelmann RK. Histologic, ultrastructural and histochemical studies of granuloma annulare. Arch Dermatol. 1977;113:1681–6. [PubMed] [Google Scholar]
- 24.Chatterjee D, Kaur M, Punia RPS, Bhalla M, Handa U. Evaluating the unusual histological aspects of granuloma annulare: A study of 30 cases. Indian Dermatol Online J. 2018;9:409–13. doi: 10.4103/idoj.IDOJ_75_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheng YW, Tsai WC, Chuang FC, Chern E, Lee CH, Sung CH. A retrospective analysis of 44 patients with granuloma annulare during an 11-year period from a tertiary medical center in south Taiwan. Dermatol Sin. 2016;34:121–5. [Google Scholar]
- 26.Gunes P, Goktay F, Mansur AT, Koker F, Erfan G. Collagen-elastic tissue changes and vascular involvement in granuloma annulare: A review of 35 cases. J Cutan Pathol. 2009;36:838–44. doi: 10.1111/j.1600-0560.2008.01169.x. [DOI] [PubMed] [Google Scholar]
- 27.Ko CJ, Glusac EJ, Shapito PE. Noninfectious granulomas. In: Elder DE, editor. Lever's Histopathology of the Skin. 9th ed. Philadelphia, PA: Lippincott Williams and Wilkins; 2004. pp. 361–87. [Google Scholar]
- 28.Stefanaki K, Tsivitanidou-Kakourou T, Stefanaki C, Valari M, Argyrakos T, Konstantinidou CV, et al. Histological and immunohistochemical study of granuloma annulare and subcutaneous granuloma annulare in children. J Cutan Pathol. 2007;34:392–6. doi: 10.1111/j.1600-0560.2006.00626.x. [DOI] [PubMed] [Google Scholar]
- 29.Chaitra V, Inchara YK, Rajalakshmi T, Antony M. Granuloma annulare-Histology reconsidered. Indian J Dermatol Venereol Leprol. 2010;76:568–9. doi: 10.4103/0378-6323.69050. [DOI] [PubMed] [Google Scholar]
- 30.Yun JH, Lee JY, Kim MK, Seo YJ, Kim MH, Cho KH, et al. Clinical and pathological features of generalised granuloma annulare with their correlation: A retrospective multicentre study in Korea. Ann Dermatol. 2009;21:113–9. doi: 10.5021/ad.2009.21.2.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Romero LS, Kantor GR. Eosinophils are not a clue to the pathogenesis of granuloma annulare. Am J Dermatopathol. 1998;20:29–34. doi: 10.1097/00000372-199802000-00006. [DOI] [PubMed] [Google Scholar]
- 32.Silverman RA, Rabinowitz AD. Eosinophils in the cellular infiltrate of granuloma annulare. J Cutan Pathol. 1985;12:13–7. doi: 10.1111/j.1600-0560.1985.tb00424.x. [DOI] [PubMed] [Google Scholar]
- 33.Piette EW, Rosenbach M. Granuloma annulare: Pathogenesis, disease associations and triggers, and therapeutic options. J Am Acad Dermatol. 2016;75:467–79. doi: 10.1016/j.jaad.2015.03.055. [DOI] [PubMed] [Google Scholar]
- 34.Dahl MV, Ullman S, Goltz RW. Vasculitis in granuloma annulare: Histopathology and direct immunofluorescence. Arch Dermatol. 1977;113:463–7. [PubMed] [Google Scholar]
- 35.Lima AL, Illing T, Schliemann S, Elsner P. Cutaneous manifestations of diabetes mellitus: A Review. Am J Clin Dermatol. 2017;18:541–53. doi: 10.1007/s40257-017-0275-z. [DOI] [PubMed] [Google Scholar]
- 36.Muller SA, Winkelmann RK. Necrobiosis lipoidica diabeticorum histopathologic study of 98 cases. Arch Dermatol. 1966;94:1–10. doi: 10.1001/archderm.94.1.1. [DOI] [PubMed] [Google Scholar]
- 37.Lowitt MH, Dover JS. Necrobiosis lipoidica. J Am Acad Dermatol. 1991;25:735–48. doi: 10.1016/s0190-9622(08)80961-9. [DOI] [PubMed] [Google Scholar]
- 38.Take N, Mitoma C, Furue M. Necrobiosis lipoidica with mucin deposition in a patient with autoimmune thyroiditis. J Dermatol. 2018;45:e193–4. doi: 10.1111/1346-8138.14250. [DOI] [PubMed] [Google Scholar]
- 39.Valecha N, Bennett G, Yip L. A granulomatous conundrum: Concurrent necrobiosis lipoidica, cutaneous sarcoidosis and erythema nodosum in a nondiabetic patient. Australas J Dermatol. 2017;58:e232–5. doi: 10.1111/ajd.12572. [DOI] [PubMed] [Google Scholar]
- 40.Harb JN, George EV, Walker A, Schoch JJ. Concomitant granuloma annulare and necrobiosis lipoidica: Do they have a related pathogenesis? Clin Exp Dermatol. 2019;44:674–6. doi: 10.1111/ced.13844. [DOI] [PubMed] [Google Scholar]
- 41.Lee J, Flowers RH, Cocks MM, Noland MB. Necrobiosis lipoidica associated with sarcoidosis. J Cutan Pathol. 2018;45:944–8. doi: 10.1111/cup.13357. [DOI] [PubMed] [Google Scholar]
- 42.Lim C, Tschuchnigg M, Lim J. Squamous cell carcinoma arising in an area of long-standing necrobiosis lipoidica. J Cutan Pathol. 2006;33:581–3. doi: 10.1111/j.1600-0560.2006.00487.x. [DOI] [PubMed] [Google Scholar]
- 43.Arnold C. The management of rheumatoid nodules. Am J Orthop (Belle Mead NJ) 1996;25:706–8. [PubMed] [Google Scholar]
- 44.Magro CM, Crowson AN. The spectrum of cutaneous lesions in rheumatoid arthritis: A clinical and pathological study of 43 patients. J Cutan Pathol. 2003;30:1–10. doi: 10.1034/j.1600-0560.2003.300101.x. [DOI] [PubMed] [Google Scholar]
- 45.Ziff M. The rheumatoid nodule. Arthritis Rheum. 1990;33:761–7. doi: 10.1002/art.1780330601. [DOI] [PubMed] [Google Scholar]
- 46.Fukase M, Koizumi F, Wakaki K. Histopathological analysis of sixteen subcutaneous rheumatoid nodules. Acta Pathol Jpn. 1980;30:871–82. doi: 10.1111/j.1440-1827.1980.tb03277.x. [DOI] [PubMed] [Google Scholar]
- 47.Rasker JJ, Kuipers FC. Are rheumatoid nodules caused by vasculitis? A study of 13 early cases. Ann Rheum Dis. 1983;42:384–8. doi: 10.1136/ard.42.4.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Crohn BB, Ginzburg L, Oppenheimer GD. Regional ileitis; a pathologic and clinical entity. Am J Med. 1952;13:583–90. doi: 10.1016/0002-9343(52)90025-9. [DOI] [PubMed] [Google Scholar]
- 49.Ephgrave K. Extra-intestinal manifestations of Crohn's disease. Surg Clin North Am. 2007;87:673–80. doi: 10.1016/j.suc.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 50.Parks AG, Morson BC, Pegum JS. Crohn's Disease with cutaneous involvement. Proc R Soc Med. 1965;58:241–2. doi: 10.1177/003591576505800419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mountain JC. Cutaneous ulceration in Crohn's disease. Gut. 1970;11:18–26. doi: 10.1136/gut.11.1.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bhoyrul B, Lyon C. Crohn's disease of the vulva: A prospective study. J Gastroenterol Hepatol. 2018;33:1969–74. doi: 10.1111/jgh.14291. [DOI] [PubMed] [Google Scholar]
- 53.Barret M, de Parades V, Battistella M, Sokol H, Lemarchand N, Marteau P. Crohn's disease of the vulva. J Crohns Colitis. 2014;8:563–70. doi: 10.1016/j.crohns.2013.10.009. [DOI] [PubMed] [Google Scholar]
- 54.Laftah Z, Bailey C, Zaheri S, Setterfield J, Fuller LC, Lewis F. Vulval Crohn's disease: A clinical study of 22 patients. J Crohns Colitis. 2015;9:318–25. doi: 10.1093/ecco-jcc/jjv037. [DOI] [PubMed] [Google Scholar]
- 55.Chatterjee D, Bhattacharjee R, Khullar G, Kumaran S, De D, Saikia UN, et al. Metastatic Crohn disease: A clinicohistological appraisal from a tertiary care center in India. Am J Dermatopathol. 2020;42:506–12. doi: 10.1097/DAD.0000000000001543. [DOI] [PubMed] [Google Scholar]
- 56.Emanuel PO, Phelps RG. Metastatic Crohn's disease: A histopathologic study of 12 cases. J Cutan Pathol. 2008;35:457–61. doi: 10.1111/j.1600-0560.2007.00849.x. [DOI] [PubMed] [Google Scholar]
- 57.Siroy A, Wasman J. Metastatic Crohn disease: A rare cutaneous entity. Arch Pathol Lab Med. 2012;136:329–32. doi: 10.5858/arpa.2010-0666-RS. [DOI] [PubMed] [Google Scholar]
- 58.Wilkin J, Dahl M, Detmar M, Drake L, Feinstein A, Odom R, et al. Standard classification of rosacea: Report of the National Rosacea Society Expert Committee on the Classification and Staging of Rosacea. J Am Acad Dermatol. 2002;46:584–7. doi: 10.1067/mjd.2002.120625. [DOI] [PubMed] [Google Scholar]
- 59.Crawford GH, Pelle MT, James WD. Rosacea: I.Etiology, pathogenesis, and subtype classification. J Am Acad Dermatol. 2004;51:327–41. doi: 10.1016/j.jaad.2004.03.030. quiz 42.4. [DOI] [PubMed] [Google Scholar]
- 60.Chougule A, Chatterjee D, Yadav R, Sethi S, De D, Saikia UN. Granulomatous rosacea versus lupus miliaris disseminatus faciei-2 faces of facial granulomatous disorder: A clinicohistological and molecular study. Am J Dermatopathol. 2018;40:819–23. doi: 10.1097/DAD.0000000000001243. [DOI] [PubMed] [Google Scholar]
- 61.Sanchez JL, Berlingeri-Ramos AC, Dueno DV. Granulomatous rosacea. Am J Dermatopathol. 2008;30:6–9. doi: 10.1097/DAD.0b013e31815bc191. [DOI] [PubMed] [Google Scholar]
- 62.van de Scheur MR, van der Waal RI, Starink TM. Lupus miliaris disseminatus faciei: A distinctive rosacea-like syndrome and not a granulomatous form of rosacea. Dermatology. 2003;206:120–3. doi: 10.1159/000068457. [DOI] [PubMed] [Google Scholar]
- 63.Hodak E, Trattner A, Feuerman H, Frinmesser M, Tsvieli R, Mitrani-Rosenbaum S, et al. Lupus miliaris disseminatus faciei--the DNA of Mycobacterium tuberculosis is not detectable in active lesions by polymerase chain reaction. Br J Dermatol. 1997;137:614–9. doi: 10.1111/j.1365-2133.1997.tb03797.x. [DOI] [PubMed] [Google Scholar]
- 64.Dykman CJ, Galens GJ, Good AE. Linear subcutaneous bands in rheumatoid arthritis. An unusual form of rheumatoid granuloma. Ann Intern Med. 1965;63:134–40. doi: 10.7326/0003-4819-63-1-134. [DOI] [PubMed] [Google Scholar]
- 65.Peroni A, Colato C, Schena D, Gisondi P, Girolomoni G. Interstitial granulomatous dermatitis: A distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775–83. doi: 10.1111/j.1365-2133.2011.10727.x. [DOI] [PubMed] [Google Scholar]
- 66.Altaykan A, Erkin G, Boztepe G, Gokoz A. Interstitial granulomatous dermatitis with arthritis. Hum Pathol. 2004;35:892–4. doi: 10.1016/j.humpath.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 67.Magro CM, Crowson AN, Schapiro BL. The interstitial granulomatous drug reaction: A distinctive clinical and pathological entity. J Cutan Pathol. 1998;25:72–8. doi: 10.1111/j.1600-0560.1998.tb01693.x. [DOI] [PubMed] [Google Scholar]
- 68.Siami K, Wilkerson M, Clark SH, Crowson AN. Pathologic quiz case: An indurated plaque on the ankle of a 74-year-old woman. Interstitial granulomatous drug reaction. Arch Pathol Lab Med. 2004;128:e129–30. doi: 10.5858/2004-128-e129-PQCAIP. [DOI] [PubMed] [Google Scholar]
- 69.Rosenbach M, English JC., 3rd Reactive granulomatous dermatitis: A review of palisaded neutrophilic and granulomatous dermatitis, interstitial granulomatous dermatitis, interstitial granulomatous drug reaction, and a proposed reclassification. Dermatol Clin. 2015;33:373–87. doi: 10.1016/j.det.2015.03.005. [DOI] [PubMed] [Google Scholar]
- 70.Chu P, Connolly MK, LeBoit PE. The histopathologic spectrum of palisaded neutrophilic and granulomatous dermatitis in patients with collagen vascular disease. Arch Dermatol. 1994;130:1278–83. [PubMed] [Google Scholar]
- 71.Finan MC, Winkelmann RK. The cutaneous extravascular necrotizing granuloma (Churg-Strauss granuloma) and systemic disease: A review of 27 cases. Medicine (Baltimore) 1983;62:142–58. doi: 10.1097/00005792-198305000-00002. [DOI] [PubMed] [Google Scholar]
- 72.Hantash BM, Chiang D, Kohler S, Fiorentino D. Palisaded neutrophilic and granulomatous dermatitis associated with limited systemic sclerosis. J Am Acad Dermatol. 2008;58:661–4. doi: 10.1016/j.jaad.2007.09.019. [DOI] [PubMed] [Google Scholar]
- 73.Hunt RD, Hartman RD, Molho-Pessach V, Votava HJ, Schaffer JV. Palisaded neutrophilic and granulomatous dermatitis in an adolescent girl with perinuclear antineutrophil cytoplasmic antibody-positive pauci-immune glomerulonephritis and arthritis. J Am Acad Dermatol. 2012;67:e164–6. doi: 10.1016/j.jaad.2012.01.009. [DOI] [PubMed] [Google Scholar]
- 74.Mahmoodi M, Ahmad A, Bansal C, Cusack CA. Palisaded neutrophilic and granulomatous dermatitis in association with sarcoidosis. J Cutan Pathol. 2011;38:365–8. doi: 10.1111/j.1600-0560.2010.01560.x. [DOI] [PubMed] [Google Scholar]
- 75.Misago N, Shinoda Y, Tago M, Narisawa Y. Palisaded neutrophilic granulomatous dermatitis with leukocytoclastic vasculitis in a patient without any underlying systemic disease detected to date. J Cutan Pathol. 2010;37:1092–7. doi: 10.1111/j.1600-0560.2009.01466.x. [DOI] [PubMed] [Google Scholar]
- 76.Sangueza OP, Caudell MD, Mengesha YM, Davis LS, Barnes CJ, Griffin JE, et al. Palisaded neutrophilic granulomatous dermatitis in rheumatoid arthritis. J Am Acad Dermatol. 2002;47:251–7. doi: 10.1067/mjd.2002.124620. [DOI] [PubMed] [Google Scholar]
- 77.Chen WT, Hsiao PF, Wu YH. Spectrum and clinical variants of giant cell elastolytic granuloma. Int J Dermatol. 2017;56:738–45. doi: 10.1111/ijd.13502. [DOI] [PubMed] [Google Scholar]
- 78.Arora S, Malik A, Patil C, Balki A. Annular elastolytic giant cell granuloma: A report of 10 cases. Indian Dermatol Online J. 2015;6(Suppl 1):S17–20. doi: 10.4103/2229-5178.171055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kaushik A, Bhattacharjee R, Vinay K, Chatterjee D. Recurrent skin ulceration: A recherche manifestation of granulomatosis with polyangiitis. Am J Med. 2020;133:814–6. doi: 10.1016/j.amjmed.2019.11.020. [DOI] [PubMed] [Google Scholar]
- 80.Comfere NI, Macaron NC, Gibson LE. Cutaneous manifestations of Wegener's granulomatosis: A clinicopathologic study of 17 patients and correlation to antineutrophil cytoplasmic antibody status. J Cutan Pathol. 2007;34:739–47. doi: 10.1111/j.1600-0560.2006.00699.x. [DOI] [PubMed] [Google Scholar]
- 81.Barksdale SK, Hallahan CW, Kerr GS, Fauci AS, Stern JB, Travis WD. Cutaneous pathology in Wegener's granulomatosis. A clinicopathologic study of 75 biopsies in 46 patients. Am J Surg Pathol. 1995;19:161–72. [PubMed] [Google Scholar]
- 82.Gangar P, Venkatarajan S. Granulomatous lymphoproliferative disorders: Granulomatous slack skin and lymphomatoid granulomatosis. Dermatol Clin. 2015;33:489–96. doi: 10.1016/j.det.2015.03.013. [DOI] [PubMed] [Google Scholar]
- 83.Li JY, Pulitzer MP, Myskowski PL, Dusza SW, Horwitz S, Moskowitz A, et al. A case-control study of clinicopathologic features, prognosis, and therapeutic responses in patients with granulomatous mycosis fungoides. J Am Acad Dermatol. 2013;69:366–74. doi: 10.1016/j.jaad.2013.03.036. [DOI] [PubMed] [Google Scholar]
- 84.Kempf W, Ostheeren-Michaelis S, Paulli M, Lucioni M, Wechsler J, Audring H, et al. Granulomatous mycosis fungoides and granulomatous slack skin: A multicenter study of the Cutaneous Lymphoma Histopathology Task Force Group of the European Organization For Research and Treatment of Cancer (EORTC) Arch Dermatol. 2008;144:1609–17. doi: 10.1001/archdermatol.2008.46. [DOI] [PubMed] [Google Scholar]
- 85.Troiano G, Dioguardi M, Giannatempo G, Laino L, Testa NF, Cocchi R, et al. Orofacial granulomatosis: Clinical signs of different pathologies. Med Princ Pract. 2015;24:117–22. doi: 10.1159/000369810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Müller S. Non-infectious granulomatous lesions of the orofacial region. Head Neck Pathol. 2019;13:449–56. doi: 10.1007/s12105-018-00997-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Critchlow WA, Chang D. Cheilitis granulomatosa: A review. Head Neck Pathol. 2014;8:209–13. doi: 10.1007/s12105-013-0488-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Marcoval J, Penín RM. Histopathological features of orofacial granulomatosis. Am J Dermatopathol. 2016;38:194–200. doi: 10.1097/DAD.0000000000000343. [DOI] [PubMed] [Google Scholar]
- 89.Ghislanzoni M, Bianchi F, Barbareschi M, Alessi E. Cutaneous granulomatous reaction to injectable hyaluronic acid gel. Br J Dermatol. 2006;154:755–8. doi: 10.1111/j.1365-2133.2005.07074.x. [DOI] [PubMed] [Google Scholar]
- 90.Sidwell RU, Dhillon AP, Butler PE, Rustin MH. Localized granulomatous reaction to a semi-permanent hyaluronic acid and acrylic hydrogel cosmetic filler. Clin Exp Dermatol. 2004;29:630–2. doi: 10.1111/j.1365-2230.2004.01625.x. [DOI] [PubMed] [Google Scholar]