

ABSTRACT
H/ACA ribonucleoproteins catalyse the sequence-dependent pseudouridylation of ribosomal and spliceosomal RNAs. Here, we reconstitute site-specifically fluorophore labelled H/ACA complexes and analyse their structural dynamics using single-molecule FRET spectroscopy. Our results show that the guide RNA is distorted into a substrate-binding competent conformation by specific protein interactions. Analysis of the reaction pathway using atomic mutagenesis establishes a new model how individual protein domains contribute to catalysis. Taken together, these results identify and characterize individual roles for all accessory proteins on the assembly and function of H/ACA RNPs.
KEYWORDS: H/ACA complexes, pseudouridylation, smFRET spectroscopy, RNP assembly, structural dynamics
Introduction
Pseudouridine is the most prevalent modified RNA nucleotide and can be found in all species of cellular RNA [1–4]. Its functions are usually subtle and are associated with processes such as ribosome biogenesis and splicing regulation [5,6]. In prokaryotes, stand-alone pseudouridine synthases [7] recognize structural elements within the target RNA. Archaea and eukaryotes are however equipped with additional capabilities; in these organisms, the H/ACA type ribonucleoprotein complex employs an auxiliary RNA to mediate sequence-specific pseudouridylation. And while individual pseudouridine modifications as well as stand-alone pseudouridine synthases rarely are essential, however, the functionality of the major RNA-guided H/ACA pseudouridine synthase is indispensable [8,9]. The archaeal H/ACA RNP complex consists of a protein tetramer, in which Cbf5, Nop10 and L7Ae bind tightly to a single bulged-hairpin sRNA (‘guide RNA’) (Fig. 1A) that contains the sequence which basepairs with and thus recognizes the substrate RNA [10–12]. The additional protein Gar1 has been shown to increase catalytic efficiency [13–15], even though it does not contact either guide or substrate RNA.
Figure 1.
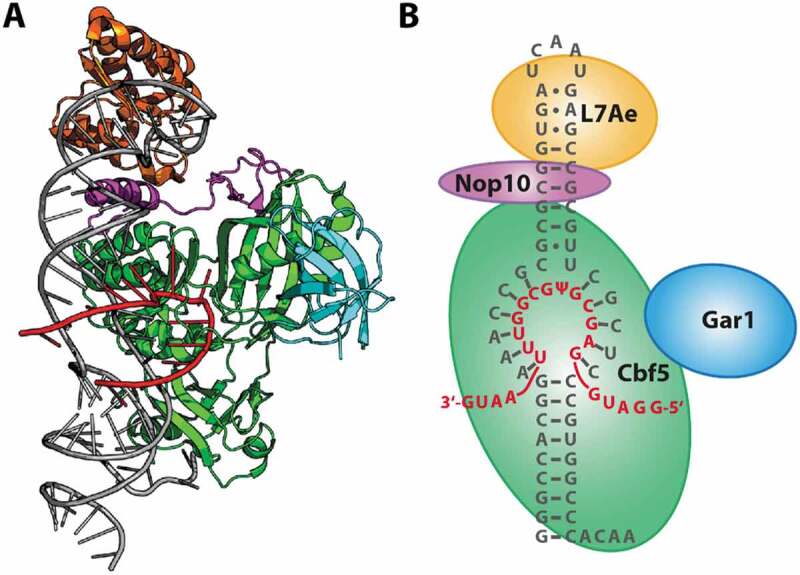
Architecture of a pyrococcal H/ACA complex. (A) Crystal structure (3HAY) [10] showing a representation of a functional RNP. (B) Schematic representation and RNA sequence used in this study
L7Ae tightly binds to the conserved kink-turn or kink-loop motif in the guide RNA and is strictly required for catalysis [16], despite not interacting with the target RNA. The molecular basis for this phenomenon remains unclear, as this requirement is much less prevalent in eukaryotic homologs [17].
Structural and bioinformatic analysis show that two stretches of 4–8 nucleotides in the guide RNA each recognize and recruit the target RNA (Fig. 1B), and place an unpaired uridine into the active site of Cbf5 [18]. Here, the thumb-loop of Cbf5 (Cbf5 thumb loop, CTL) has been shown to play an essential role in modifying the target RNA [10]. Gar1 interacts with the CTL, this however does not sufficiently explain the increase in catalytic efficiency that Gar1 conveys [11].
Binding of substrate analogues has been widely employed in deciphering the mode of binding and catalysis [19,20], and these results show that precise accommodation of the target uridine is required for catalysis. In contrast, binding of 5-fluorouracil (5FU) to the active site results in a trapped intermediate, the nature of which appears to depend on the pseudouridine synthase in question [21].
By stepwise reconstituting catalytically active H/ACA complexes (Fig. 1) and analysing conformations by single-molecule FRET, this work shows how individual proteins interact and distort the guide RNA to adopt a substrate-binding competent conformation.
Within the fully assembled complex, we show how Gar1 mobilizes the CTL, and we furthermore interrogate the conformation of the CTL along the catalytic process.
Together, our results show that assembly of the proteins Cbf5, Nop10 and L7Ae alters the guide RNA conformation to allow for binding of the target RNA. In presence of Gar1, binding of the RNA results in a mobilization of the CTL, which we characterize to be present in three distinct conformational states. The population of these states strongly correlates with the precise accommodation of a uridine into the catalytic pocket. Our results also structurally rationalize how a Gar1-dependent mobilization of the CTL results in an increase of both single- and multiple turnover catalytic efficiency.
Results
Using a combined coexpression/copurification approach, we reconstituted partial or full H/ACA RNA-protein (RNP) complexes from Pyrococcus furiosus [10,20] using a model pyrococcal RNA construct [16]. In order to make these complexes amenable to smFRET spectroscopy, we introduced fluorophores in three different positions into the guide RNA. In order to validate these complexes, we confirmed that the fluorophore labelled complexes retained reduced levels of pseudouridylation activity under multiple turnover conditions (Figure S1).
As catalytic activity in archaeal H/ACA complexes most significantly depends on the presence of the two auxiliary proteins Nop10 and L7Ae [22], we first tested the effect of binding of these proteins to the guide RNA. To monitor conformational changes in the guide RNA, we devised two different constructs which allow monitoring distance changes either within the apical, kink-turn containing motif or across the pseudouridylation pocket (Fig. 2). We determined FRET efficiencies of the different reconstituted RNP complexes, containing either the RNA alone or with L7Ae, with the Nop10-Cbf5- Gar1 trimer (NCG), or with all four proteins (LNCG). For most of the assemblies tested in these experiments, structures of the complexes have not been determined to date, with the exception of the substrate-free sRNA-LNCG complex [11]. Based on the high affinity of L7Ae to the RNA [23–26], previous work on the same protein [27], the conditions used for reconstitution and immobilization, and the results obtained (see below), complex formation in this experiment is near-quantitative [19,27]. For the RNA alone, smFRET analysis yields one predominant population at an EFRET = 0.68, indicating a homogeneous fold of the RNA. Within the apical hairpin (Fig. 2A), L7Ae binding to the kink-turn motif does not result in a change in the apparent FRET efficiency. The addition of the Nop10-Cbf5- Gar1 protein complex also does not result in a significant shift in FRET efficiency. Incubation with all four LNCG proteins however significantly increases the FRET efficiency of a large number (45%) of molecules, indicative of a compaction of the apical loop structure.
Figure 2.
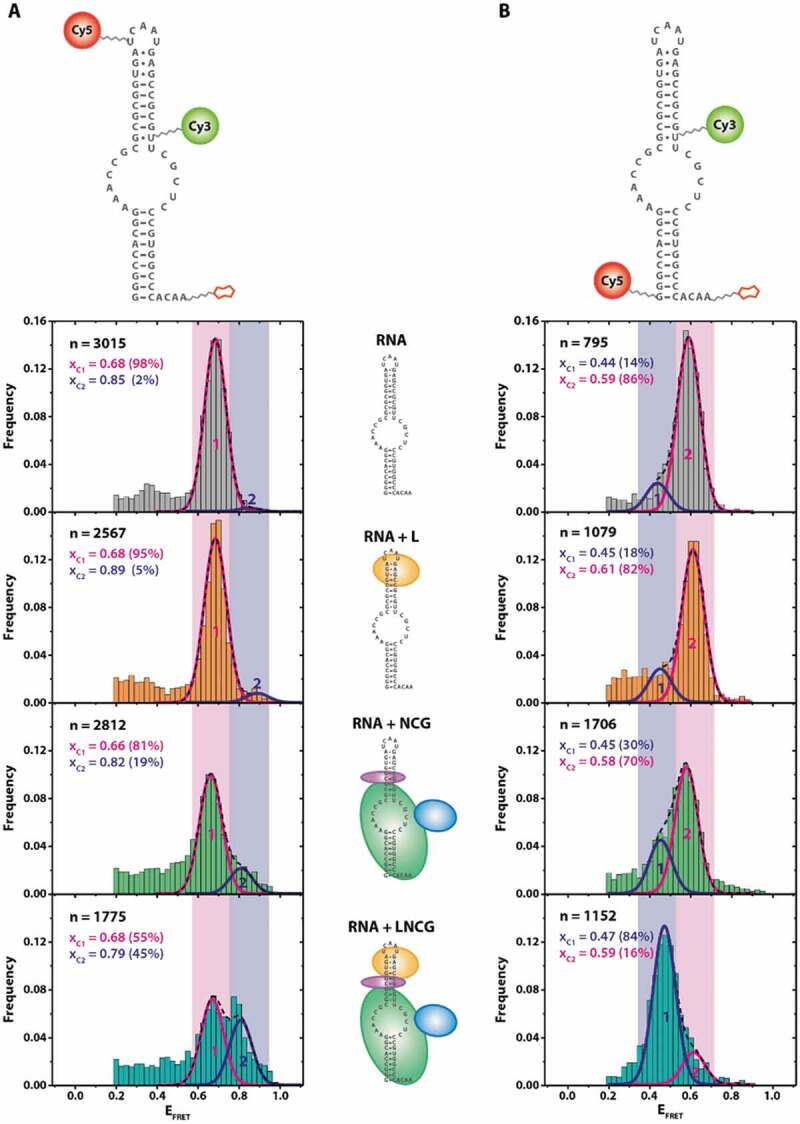
FRET populations in RNP reconstitution experiments show RNA distortion upon protein binding. (A) FRET construct used for assessing the conformation of the upper stem-loop structure of the sRNA. Stepwise assembly is indicated for each FRET histogram. (B) FRET construct used for assessing conformation of the pseudouridylation pocket. Complex composition: RNA + proteins L7Ae (“L), Nop10 (“N), Cbf5 (“C), Gar1 (‘G’). n = number of molecules analysed. Xci = centre of FRET distributions. The histograms were cropped for donor-only molecules at EFRET<0.2 (see Material and Methods)
The FRET efficiency across the pseudouridylation pocket shows a slightly different behaviour upon stepwise complex reconstitution (Fig. 2B). While the RNA alone yields an EFRET = 0.59 for 86% of the molecules, binding of L7Ae alone does not change the apparent FRET efficiency. Binding of NCG however decreases FRET efficiency for a smaller amount (30% versus 14%) of molecules, which suggests an opening or stretching of the RNA upon binding to the protein trimer. This effect becomes significantly more pronounced when all four proteins are assembled on the RNA; here, a near-quantitative (84% versus 14%) shift towards a state with a lower FRET efficiency can be observed. These states show no interconversion in time-dependent FRET analysis (Figure S2), which was the case for all experiments described in Fig. 2, as judged by analysis of >100 individual fluorescence traces up to 2 minutes in length.
As L7Ae, Cbf5 and Nop10 are all strictly required for catalysis ([19], and Figure S3), the question arises whether the conformational change of the guide RNA upon RNP reconstitution is of functional relevance for pseudouridylation activity of the complex. We, therefore, tested whether the (sub-) complexes generated in our experiments (Fig. 2) were substrate-binding competent. To this end, we designed a single-molecule experiment where we decorated a target RNA containing 5-fluorouracil (5FU) in the site of modification with the FRET acceptor dye (Fig. 3). This results in complexes without target RNA (‘apo’ form) showing only donor fluorescence, and complexes that recruited the target RNA (‘holo’ form) exhibiting additional FRET fluorescence. 5FU can act as an inhibitor and results in a high-affinity complex between the H/ACA RNP and the target RNA, as evidenced by a number of structural and biochemical studies [19–21].
Figure 3.
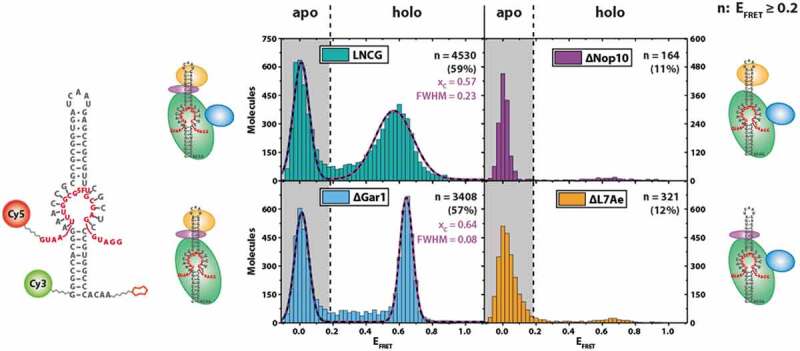
Single-molecule target RNA binding for different RNP compositions. Left: construct used for pulldown experiment of substrate-binding competent RNP complexes, with the biotin moiety attached to the 3ʹ end and Cy3 attached to the 5ʹ end of the guide RNA. Cy5 is attached to the 3ʹ end of the target RNA. Right: FRET histograms showing EFRET of immobilized molecules. n indicates the number of molecules exhibiting EFRET > 0.2.xc: centre of FRET efficiency population. FWHM: full width at half maximum of FRET efficiency population
When examining the LNCG complex in these experiments, two populations were observed: as expected, a significant fraction of complexes (41%) showed only donor fluorescence (EFRET < 0.2) (Fig. 3). Complexes that were either devoid of Nop10 or L7Ae showed only minute amounts of molecules at EFRET = 0.6 (11% and 12%, respectively), indicating that most of the complexes did not bind the target RNA. Complexes with either LNC or LNCG protein composition, however, showed a significant proportion of molecules at EFRET = 0.6 (57% and 59%, respectively), indicating stable binding of the target RNA. In absence of all proteins, no discernible binding events were observed (data not shown).
When reconstituting the target RNA-bound LNC complex, the resulting FRET distribution is quite sharp (Full width at half maximum (FWHM) = 0.08), suggesting that the resulting conformation is very homogeneous (Fig. 3). Interestingly, the presence of Gar1 results in a slightly shifted (xc = 0.57 for LNCG versus xc = 0.64 for LNC) and at the same time more broadly distributed overall population (FWHM = 0.23), as compared to the LNC complex. The corresponding FRET traces did not show dynamic behaviour (Figure S4).
Conformation of the guide RNA upon binding of target RNA
To identify whether the additional RNA conformation observed in the reconstitution experiments (Fig. 2A) is functionally relevant, we devised a strategy that allowed us to specifically observe fully reconstituted complexes bound to a target RNA. Using a biotinylated target RNA carrying different nucleotides in the modification site, we pulled down complexes on these target RNAs (Fig. 4). As established before (Fig. 3), these complexes carry at least Cbf5, Nop10 and L7Ae; based on these reconstitution conditions, we can also assume the presence of Gar1 in the majority of complexes.
Figure 4.
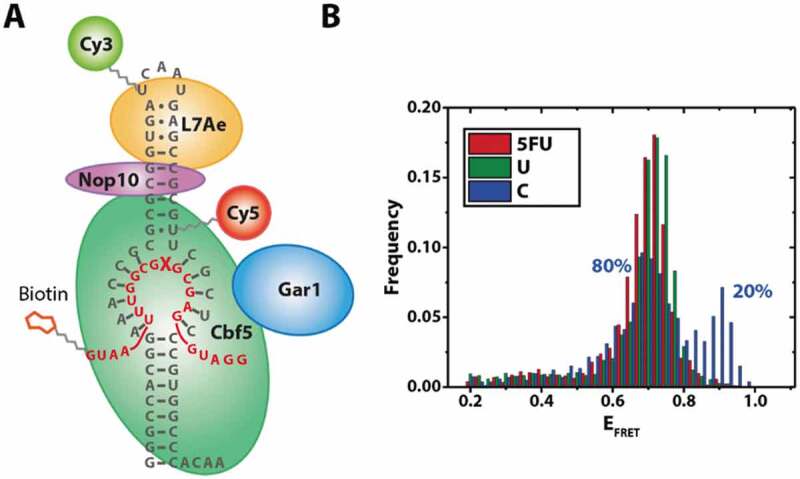
Pulldown experiment of fully assembled RNP complexes via the target RNA. Left: Construct used for pulldown experiments using a biotinylated target RNA. X: Position of target nucleotide. Right: FRET histograms obtained with different target nucleotides
Molecules immobilized via the target RNAs exhibit one predominant population (Fig. 4). With the exception of cytosine, this was largely independent of the target nucleotide (Fig.s 4, S5). The only significant population observed for these fully assembled complexes with 5FU or U as the target nucleotide is centred at an EFRET = 0.71, in between the two populations observed in experiments without target RNA (EFRET = 0.68 and 0.79) (Fig. 2A). For C as the target nucleotide, we see an additional population (20%) centred at EFRET = 0.9,
The most crucial domain for catalytic activity is the CTL, which is usually mobile and therefore not fully resolved in some of the available crystal structures [10,11,16,20,28]. The CTL also interacts with Gar1 in some of these structures, and the current hypothesis is that a conformational change of the CTL is required during catalysis [10,29]. In order to experimentally test this hypothesis, we devised a FRET construct in which one of the fluorophores is placed within the CTL. Based on available sequences and data on the importance of single CTL residues [10] on catalytic activity, we chose L145 of Cbf5 as the labelling site. To obtain site-specifically labelled protein, we employed the pEVOL system [27,30] to recombinantly express protein that carries a propargyllysine (PrK) residue at position 145 of Cbf5. As this modification may affect the functionality of Cbf5, we verified the catalytic activity of the PrK-mutated protein Cbf5 after fluorophore attachment (Figure S6). These constructs displayed an only slightly reduced catalytic activity (38% versus 50% in absence of Gar1, and 60% versus 56% in presence of Gar1). In order to selectively observe complexes that are substrate-binding competent, we placed the other FRET partner into the 3ʹ end of the target RNA. Here, a FRET signal can only be obtained when both Cbf5 and the target RNA are present. As shown in experiments above (Fig. 3), target RNA can only be efficiently recruited in presence of at least the LNC complex; therefore, the presence of a FRET signal is a very strong indication of a reconstituted complex (Fig. 5).
Figure 5.
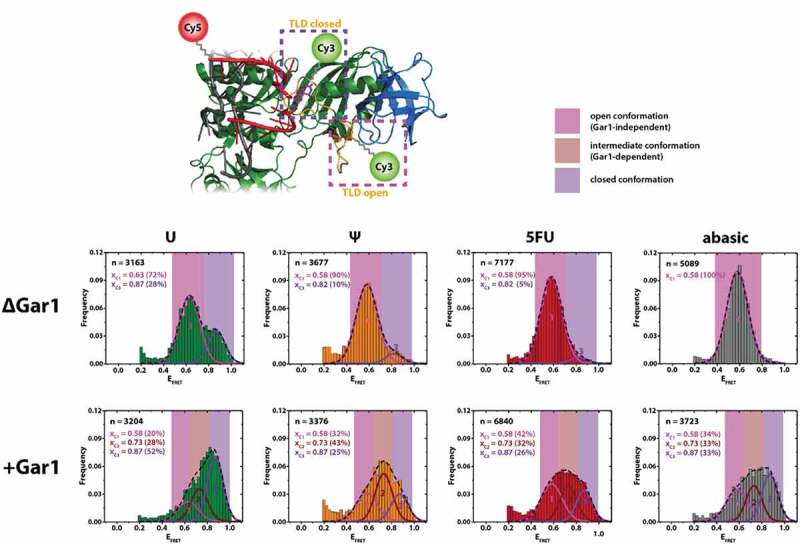
Construct used for monitoring CTL dynamics, and smFRET analysis. Top: CTL-labelled region of the H/ACA RNP construct used for monitoring thumb-loop dynamics (overlay of structures [10]), derived from models showing two distinct proposed CTL conformations (open and closed, respectively). Bottom: smFRET histogram analysis using different target nucleotides in presence or absence of Gar1. n = number of molecules analysed. Xci = centre of FRET distributions. The histograms were cropped for donor-only molecules at EFRET<0.2 (see Material and Methods)
This approach also allows placing different target nucleotides into the substrate-binding pocket of Cbf5 [19,20]. We chose to investigate the effect of an abasic site (corresponding to a pre-catalytic state, with a catalytic pocket that is devoid of the free nucleobase), cytosine (mismatch), 3-methyl-uridine (near-substrate inhibitor), 5-fluorouracil (transition state mimic), pseudouridine (product) and uridine (bona fide substrate) on the behaviour of the complex, resembling all feasibly attainable states within the reaction process of pseudouridylation [20].
In absence of Gar1, for each of the target nucleotides, a similar behaviour can be observed, in that most of the complexes assume a conformation that yields an EFRET = 0.58, which we assign to be a conformation corresponding to an open CTL. In case of a target uracil, a slight deviation can be seen with EFRET = 0.63. In each case except for uracil, this conformation accounts for more than 90% of the molecules observed. With uracil as the target nucleobase, about 28% of molecules adopt a higher EFRET = 0.87.
In presence of Gar1 however, the complex assumes three different CTL conformations irrespective of the target nucleotide. These were distinguished by fitting of the overlaying populations in the histograms in Fig. 5 and were verified by analysis of individual traces (Figure S7). For each of the nucleotides tested, the population distribution followed a similar trend: In addition to the conformation at EFRET = 0.58, we find two additional populations at EFRET = 0.73 and EFRET = 0.87. We term these conformations the open, intermediate and closed conformation, respectively.
For all target nucleotides except for uracil, these populations are similarly populated (25–43%). For uracil, we again found the closed conformation at EFRET = 0.87 populated to a higher extent (50%) on the expense of the open conformation (EFRET = 0.58, 20%).
Discussion
Our results for the first time allow the direct observation of heterogeneous populations covering different conformational states within H/ACA RNPs on the level of individual molecules. We have validated all constructs used for single-molecule analysis by verifying catalytic activity. While some of the constructs with labelled RNA showed a slightly reduced level of pseudouridylation under multiple turnover conditions, the labels introduced into any of the H/ACA RNPs did neither lead to misfolding nor cause assembly perturbation.
The smFRET experiments in this study were performed at ambient temperature, and while activity has been verified at 70°C, the usual temperature for these complexes, several other studies have shown that valid functional and structural conclusions from such comparisons at different temperatures can be made [16,19,31]. As we do not observe structural dynamics within the timescales amenable to our analysis, we conclude that the conformationally heterogeneous populations observed in our experiments are indicative of structural dynamics.
We have reconstituted complexes using a model Pyrococcus furiosus sRNA that has been used before [16], from the high degree of secondary structure conservation in these RNAs [12,24] we expect that our results are valid for other sRNAs as well.
The data obtained on partially reconstituted complexes observing RNA conformations (Fig. 2) suggest that during assembly of the RNA-protein complex, binding of L7Ae to the kink-turn motif (which is strictly required to be folded for L7Ae binding [23,24]) only has a minor impact on the overall guide RNA conformation. A similar finding can be assigned to the binding of the Cbf5-Nop10-Gar1 trimer. The combination of all four proteins however leads to a marked conformational change in the apical region of the guide RNA. The only reported interaction between L7Ae and the NCG trimer not involving the guide RNA is between L7Ae and Nop10 [10]. It can therefore be concluded that the interaction between L7Ae and Nop10 is required for the observed alteration in the RNA conformation. This is also in line with findings from evolutionarily more divergent organisms (yeast [29] or human [32]), where the interaction between Nop10 and L7Ae homologs (i.e. Nhp2) is highly conserved, which also holds true for the ‘backbone’ described for the Cbf5-Nop10-L7Ae trimer [12,33].
For the construct spanning the pseudouridylation pocket, we observe a gradual shift towards an open conformation upon stepwise assembly of the complex (Fig. 2B). This shift is near quantitative for the LNCG reconstitution, in turn demonstrating that our experimental approach yields a large fraction fully assembled H/ACA RNPs.
Taking together, the conformational changes in the apical part of the guide RNA and across the pseudouridylation pocket, the observed changes suggest a concerted distortion of the target RNA upon assembly of the full complex, mediated mainly by binding of Cbf5 and L7Ae to the H/ACA RNA, and the interaction between L7Ae, Nop10 and Cbf5. The notion that in the available crystal structures [10,16,20,28,29] the conformation of the guide RNA varies between complexes with different lengths of the RNA supports this hypothesis.
The experimental approach used for determining substrate-binding capabilities showed significantly different results for various complexes (Fig. 3). The differences between complexes with both L7Ae and Nop10 and those without either protein show that the former are largely substrate-binding incompetent when compared to the LNC complex (164 and 321 molecules, respectively, versus 3408 molecules) assembled under identical conditions. This again indicates that the protein backbone formed between Cbf5, Nop10 and L7Ae, which was shown to distort the guide RNA in experiments shown above, allows the RNA to adopt a conformation which enables the facial interaction between guide and target RNA [17]. This interaction is sufficiently stable to be observed in our smFRET experiments and immediately suggests that both Nop10 and L7Ae are required to transform the complex into a target RNA binding competent conformational state. This also offers a novel explanation for the impact of both Nop10 and L7Ae on catalytic activity, as their absence already prevents formation of the enzyme-substrate complex.
By dissecting the individual contribution of each protein as well as the target RNA on guide RNA conformation, our results complement and provide a more detailed mechanistic view on the assembly of H/ACA RNP complexes on the level of individual molecules [ref Liang 2008].
When observing only substrate-binding competent complexes using the single-molecule target RNA pulldown (Fig. 4), we find that in comparison with the results in Fig. 2B, less heterogeneous populations and thus likely less dynamics of the sRNA can be observed. The results for complexes containing either uracil or 5FU as target nucleotide are virtually identical, indicating that this part of the RNA may be conformationally stable during catalysis. For cytosine as the target nucleotide, the additional conformation at a higher EFRET likely resembles a non-productive state of assembly and may hint at a possible function that helps discriminating against near-cognate target sequences. Another possibility is that this population arises from complexes that are trapped in a pre-product-release state. This would be in line with previous findings [19] and is supported by the small percentage of Ψ-containing (11%) complexes in the same experiment (Figure S5).
Together, these experiments demonstrate that all proteins comprising a functional H/ACA RNP complex work together in shaping the RNA conformation, and thus alter the architecture of the RNP to render the complex catalytically active.
While the RNA conformation is determined by the assembly state, protein conformations are considered to be required for pseudouridylation catalysis. We, therefore, assessed CTL dynamics in the reconstituted RNP complexes (Fig. 5). The major population at EFRET = 0.58 in absence of Gar1 shows that the CTL is in one predominant state, in line with an open conformation.
The presence of the high-FRET state exclusively in presence of a target uracil may likely be due to partial turnover into pseudouridine (Figure S6), and we find about 30% of the molecules in a closed conformation. This is in line with the fact that catalytic turnover can be observed in complexes devoid of Gar1 (Figure S3). The closed conformation can however not be observed for 5FU. This points to differences in the enzyme’s handling of 5FU and a partial catalytic turnover that progresses further along the reaction trajectory for uracil.
Comparing the effects with complexes containing Gar1, the populations at EFRET = 0.73 and EFRET = 0.87 are strictly dependent on the presence of Gar1, and we describe them as an intermediate conformation and closed conformation, respectively. Our results of both smFRET and activity analysis show that there is a significant effect arising from Gar1 binding, but leave open the possibility that we do not quantitatively reconstitute all RNAs into functional RNPs containing Gar1. As a result, the peak at EFRET ~ 0.6 in Fig. 5 for all nucleotides may also arise from complexes that do not contain Gar1 (but contain Cbf5, Nop10 and L7Ae, as discussed above). We, therefore, designate the population at EFRET ~ 0.6 the Gar1-independent open conformation.
The presence of both these conformations shows that Gar1 mobilizes the CTL. As both the intermediate and closed conformations can also be observed for the target RNA containing an abasic site, this effect does not require a nucleobase to be present within the active site. The closed conformation can otherwise only be observed for uracil in absence of Gar1. For pseudouridine, the most populated state in presence of Gar1 is the intermediate conformation. Compared to uracil, the population of the intermediate and closed states, respectively, is almost inverted (25% versus 52% for the closed, and 43% versus 28% for the intermediate state) (Fig. 5). This suggests that the complex with pseudouridine as target nucleotide indeed represents a postcatalytic state.
From these findings, we attribute the conformations to different steps in catalysis: in absence of Gar1, the CTL is in an open conformation. In presence of Gar1 and upon substrate binding, the CTL undergoes conformational changes via the intermediate into the closed conformation, where catalysis occurs. Following pseudouridylation, the CTL undergoes an opening movement into the intermediate state to facilitate product release.
In this model, the higher mobility and thus an energetically favoured closing of the CTL in presence of Gar1 also explain a higher catalytic turnover under single-turnover conditions [10] (Figure S3). In turn, the presence of all conformations upon pseudouridine product binding likely reflects that opening of the CTL postcatalysis is also facilitated by Gar1.
Combining these two findings, it can be inferred that both closing as well as opening of the CTL are accelerated by Gar1. This is excellently reflected in the fact that both these effects are discernible in activity assays: an accelerated closing of the CTL results in a higher single-turnover activity, as the catalytic conformation can be reached quicker. In turn, opening of the CTL postcatalysis facilitates product release and thus results in an even more accelerated multiple turnover activity. These findings are also in excellent agreement with published activity assays [10].
Summary
Our results for the first time show that a pronounced distortion of the guide RNA is required for efficient substrate binding. This RNA distortion is facilitated by tight binding of the RNA to both Cbf5 and L7Ae, and by a concomitant interaction between Cbf5, Nop10 and L7Ae. This three-protein backbone of the RNP keeps the guide RNA in a substrate-binding competent conformation. Gar1, on the other hand, is required for a newly described mobilization of the full RNP, as evidenced by structural dynamics upon substrate binding, and by the Gar1-dependent mobilization of the Cbf5 thumb loop. This mobilization offers a novel, direct demonstration of structural dynamics, which are in excellent agreement with existing structural and biochemical data.
Supplementary Material
Acknowledgments
This project was supported by DFG, SFB902 and EXC115. L7Ae plasmid was a gift from Hong Li. pEVOL plasmids were a gift from Edward Lemke. The authors would like to thank Prof. Harald Schwalbe for generous support, and Prof. Mike Heilemann for access to instrumentation.
Funding Statement
This work was supported by the Deutsche Forschungsgemeinschaft (DE) [SFB902].
Disclosure Statement
No potential conflicts of interest were disclosed.
Supplementary material
Supplemental data for this article can be accessed here.
Materials and methods
Oligonucleotide sequences
The modified RNA oligonucleotides were purchased from GE Dharmacon.
Modification abbreviations: p = Phosphate, bio = Biotin, U = 5-Aminoallyl-uridine, G = 5ʹ-Amino modified C6-linker, C = 2ʹ-Amino-cytidine, Ψ = Pseudouridine, 5FU = 5-Fluoro-uridine
Oligonucleotides used to generate labelled H/ACA-constructs (amino-modified nucleotides are marked in underlined letters and numbers represent nucleotide numbering in the native full-length H/ACA-RNA):
Oligo-G1 (1-30):GGGCCACGGAAACCGCGCGCGGUGAUCAAU
Oligo-U26 (1-30):GGGCCACGGAAACCGCGCGCGGUGAUCAAU
Oligo-C37 (31-58):pGAGCCGCGUUCGCUCCCGUGGCCCACAAbio
Oligo-U39 (31-58):pGAGCCGCGUUCGCUCCCGUGGCCCACAAbio
Modified target RNA and its analogues (the target nucleotides for pseudouridylation are highlighted in bold letters. At the terminal uridine, the RNAs carry a 5ʹ-Amino modified C6-linker ‘-N6’):
U (substrate):GAUGGAGCGUGCGGUUUAAU-N6
Ψ (product):GAUGGAGCGΨGCGGUUUAAU-N6
C (mismatch):GAUGGAGCGCGCGGUUUAAU-N6
5FU (inhibitor):GAUGGAGCG5FUGCGGUUUAAU-N6
Dye coupling and purification
The RNA oligonucleotides were purchased in the 2ʹ-bis(2-acetoxyethoxy)-methyl(ACE)-protected form and dissolved in ddH2O. Following ethanol precipitation, 10 nmol (5FU-Sub) or 30 nmol (H/ACA-oligos) of RNA were used for coupling with the amine-reactive dyes Cy3 or Cy5 (Amersham CyDye Mono-Reactive Dye Packs, GE Healthcare). The RNA pellet was dissolved in freshly prepared 20 µL 0.1 M NaHCO3 and the dye was dissolved in 20 µL DMSO. Both mixtures were combined and incubated for 90 min at room temperature in the dark. Following ethanol precipitation, the deprotection was done in 100-300 µL deprotection buffer (provided by Dharmacon) for 30 min at 60°C (90 min for biotinylated oligos). After another ethanol precipitation, the RNAs were dissolved in HPLC buffer A (0.1 M TEAA, pH 7.0) for HPLC purification. The chromatography was done in a gradient from 100% HPLC buffer A to 100% acetonitrile on an Äkta Purifier 10 system, using a C8 column (Kromasil 100 C8 7 µm 250 × 4.6 mm). The peaks showing RNA and dye absorption were pooled, the acetonitrile was evaporated in a Speedvac, the RNA was precipitated by ethanol, and then redissolved in ddH2O. Concentrations were determined using a Nanodrop ND-1000 UV spectrophotometer.
DNA-splinted RNA ligations
For mono-labelled (Cy3 or Cy5 only) H/ACA-RNAs, one labelled and one unlabelled modified oligonucleotide from Dharmacon were used. Therefore, 10 nmol of unlabelled oligo was deprotected in provided deprotection buffer, ethanol precipitated and resuspended in ddH2O. The concentrations were determined using a Nanodrop ND-1000 UV spectrophotometer.
The ligation of the fluorophore-labelled H/ACA-constructs was done using DNA splints. Both RNA oligonucleotides and the DNA splint were used in equimolar amounts (2-10 µM). The oligonucleotides were annealed by heating for 5 min at 80°C and slow cooling to room temperature in 0.5x T4 DNA Ligase buffer. Samples were adjusted to 1x T4 DNA Ligase buffer, 40 U/µL T4 DNA Ligase (NEB) and 1 U/µL Recombinant RNasin (Promega) were added. The reaction mixture was incubated at 16°C overnight. Subsequently, the DNA splint was hydrolyzed by addition of 0.06 U/µL DNase (TURBO DNase, Ambion) and incubation at 37°C for 30 min. The samples were phenol/diethyl ether extracted, ethanol precipitated and the ligation products were separated by denaturing urea PAGE (8-10%). The ligation products were identified by eye (coloured bands of dual Cy3-Cy5-labelled RNA). Target bands were excised and eluted by shaking in 0.5 M NH4OAc at 25°C overnight. The RNA was precipitated by ethanol and resuspended in ddH2O. The concentrations were determined using a Nanodrop ND-1000 UV spectrophotometer.
Protein expression and purification
E.coli codon-optimized genes of P. furiosus Cbf5, Gar1 and Nop10 were purchased from Eurofins Genomics in pEX-K4 (Cbf5) or pEX-A2 (Nop10 and Gar1) vectors. The genes of Cbf5 and Nop10 were cloned into pET-Duet-1, and Gar1 was cloned into pET-28b(+). L7ae was expressed from a pET15 plasmid as described [27]. Cbf5 contains an engineered C- or N-terminal 6xHis-Tag. For expression of the ternary NCG-subcomplex, the plasmids pET-Duet-1 (Cbf5 and Nop10) and pET-28b(+) (Gar1) were double transformed into E.coli BL21(DE3) cells. For expression of the secondary NC-subcomplex only pET-Duet-1 (Cbf5 and Nop10) was transformed into E.coli BL21(DE3) cells. For expression of Cbf5 as stand-alone protein, the pET-Duet-1 vector containing only the Cbf5 gene was transformed into E.coli BL21(DE3) cells. The ternary NCG as well as the secondary NC-subcomplexes were coexpressed and copurified in the same manner like the stand-alone protein Cbf5. Cells expressing NCG, NC or Cbf5 were mixed in buffer A (1 M NaCl, 20 mM imidazole, 50 mM phosphate, pH 6.0) and lysed by sonication. The cell lysate was clarified by centrifugation, heated at 70°C for 20 minutes, again clarified by centrifugation and after adding a 5% v/v polyethyleneimine solution for nucleic acid precipitation, the suspension was again centrifuged. The supernatant was filtered (0.22 µm) before loading onto a HisTrap column. Proteins were eluted by gradient elution from buffer A to buffer B (1 M NaCl, 500 mM imidazole, 50 mM phosphate, pH 6.0).
The final purification was either done by size exclusion (Superdex 200 Increase 10/300 GL from GE) for NC and CG in storage buffer (500 mM KCl, 25 mM HEPES pH 7.5, 10% (v/v) glycerol) or with a Heparin column (GE) for NCG. All samples were frozen in liquid nitrogen and stored at −80°C in storage buffer. Expression of modified Cbf5 constructs was performed according to published protocols [27]. Purification was performed as described above.
Activity assays
The activity was determined using a substrate RNA that was site-specifically 32P-labelled at the target nucleotide [29]. The 32P-labelled substrate was prepared by ligation of a 5ʹ-half RNA (5ʹ-GAUGGAGCG-3ʹ) and a 5ʹ-32P-labelled 3ʹ-half RNA (5ʹ-32P-UGCGGUUUAAU-3ʹ). This was done by DNA-splinted ligation using T4-DNA-ligase (NEB). After digestion of the splint using TurboDNase (Ambion), the substrate was gel-purified and extracted.
For pseudouridylation 0.4 µM 32P-labelled substrate RNA with 0.2 µM transcribed or dye-labelled H/ACA RNA, 0.4 µM NCG and 5 µM L7Ae were incubated at 70°C for 90 min in assay-buffer (100 mM NH4OAc, 100 mM Tris pH 8.0, 5 mM MgCl2). The reaction mixtures were phenol extracted, RNase P1 digested, the resulting mononucleotides were separated by thin-layer chromatography and visualized with a storage phosphor screen as described [29].
Single-molecule measurements
Single-molecule FRET experiments were performed on a total internal reflection fluorescence (TIRF) setup as described [27]. For investigation of the RNA conformation, RNP samples, carrying fluorophores only on the RNAs, were prepared in a 5 µL scale containing 50 nM guide RNA and regarding the sample 400 nM NCG/NC/CG, 5 µM L7Ae and 70 nM substrate. Incubation was performed at 70°C in assay-buffer for 5 min (data in Fig. 4) or 15 min (data in figures 2 and 3), and the samples were diluted 1:500 with assay buffer at 4°C. To reconstitute the H/ACA RNP complex for analysis of the CTL conformation, 300 nM Cbf5L145X-Sulfo-Cy3/Nop10 (position L145), 720 nM biotinylated sRNA, 1.25 μM L7Ae, 1 μM Cy5-target RNA and ± 1.25 μM Gar1 were incubated for 5 min at 70°C, then cooled, diluted and immobilized at ~100pM.
Biotinylated constructs were immobilized on homemade surface passivated (PEG MW 5000) glass slides as described [34]. Diluted samples were immobilized, washed with assay-buffer and rebuffered in imaging-buffer (100 mM NH4OAc, 100 mM Tris, 5 mM MgCl2, 0.1 mM dodecylmaltoside, 0.08 mg/mL glucose oxidase, 0.02 mg/mL catalase, Trolox (saturated), 10% w/v glucose, pH 8.0) for recording of single-molecule traces. Excitation of the FRET donor was done at 532 nm. Measurements were performed at ambient temperatures (21°C) on an objective-type total internal reflection microscopy setup with an EMCCD camera (iXon, Andor Technology) at 100 ms integration time.
For data acquisition, typically 20 2-second-videos were recorded for obtaining histogram data. Typically, five 2-minute-videos were recorded for individual traces, resulting in approx. 50–200 distinguishable molecules per video. Raw data were processed with IDL and imported into MATLAB to calculate corresponding FRET efficiencies as described [34]. Donor leakage was adjusted (typically 12%) for every video by calibrating the donor-only peak to EFRET = 0. Donor (ID) and acceptor (IA) intensities were averaged over 20 frames, FRET efficiency was calculated as EFRET = IA/(IA+ID) and binned into intervals of 0.025. Plotting and Gaussian distribution fitting of obtained histogram data points was done in Origin (OriginPro 2017, OriginLab). In cases where donor-only molecules were removed, the histogram data were cropped below EFRET=0.2.
References
- [1].Bohnsack, M.T. and Sloan K.E.. Modifications in small nuclear RNAs and their roles in spliceosome assembly and function. Biol Chem. 2018;399(11):1265–1276. [DOI] [PubMed] [Google Scholar]
- [2].Gilbert, W.V., Bell T.A., and Schaening C.. Messenger RNA modifications: form, distribution, and function. Science. 2016;352(6292):1408–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Karijolich, J., Yi C., and Yu Y.T.. Transcriptome-wide dynamics of RNA pseudouridylation. Nat Rev Mol Cell Biol. 2015;16(10):581–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Spenkuch, F., Motorin Y., and Helm M.. Pseudouridine: still mysterious, but never a fake (uridine)! RNA Biol. 2014;11(12):1540–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Wu, G., Xiao, Mu, Yang, C., et al. U2 snRNA is inducibly pseudouridylated at novel sites by Pus7p and snR81 RNP. Embo J. 2011;30(1):79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sloan, K.E., Warda, A S., Sharma, S., et al.. Tuning the ribosome: the influence of rRNA modification on eukaryotic ribosome biogenesis and function. RNA Biol. 2017;14(9):1138–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Rintala-Dempsey, A.C. and Kothe U.. Eukaryotic stand-alone pseudouridine synthases - RNA modifying enzymes and emerging regulators of gene expression? RNA Biol. 2017;14(9):1185–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Giordano, E., Peluso, I., Senger, S., et al.. minifly, a Drosophila gene required for ribosome biogenesis. J Cell Biol. 1999;144(6):1123–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].He, J., Navarrete, S., Jasinski, M., et al.. Targeted disruption of Dkc1, the gene mutated in X-linked dyskeratosis congenita, causes embryonic lethality in mice. Oncogene. 2002;21(50):7740–7744. [DOI] [PubMed] [Google Scholar]
- [10].Duan, J., Li, L., Lu, J., et al.. Structural mechanism of substrate RNA recruitment in H/ACA RNA-guided pseudouridine synthase. Mol Cell. 2009;34(4):427–439. [DOI] [PubMed] [Google Scholar]
- [11].Li, L. and Ye K.. Crystal structure of an H/ACA box ribonucleoprotein particle. Nature. 2006;443(7109):302–307. [DOI] [PubMed] [Google Scholar]
- [12].Baker, D.L., et al.. RNA-guided RNA modification: functional organization of the archaeal H/ACA RN. Genes Dev. 2005;19(10):1238–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Fujikane, R., et al. Contribution of protein Gar1 to the RNA-guided and RNA-independent rRNA:Psi-synthase activities of the archaeal Cbf5 protein. Sci Rep. 2018;8(1):13815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kamalampeta, R. and Kothe U.. Archaeal proteins Nop10 and Gar1 increase the catalytic activity of Cbf5 in pseudouridylating tRNA. Sci Rep. 2012;2:663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wang, P., Yang, L., Gao, Y Q., et al.. Accurate placement of substrate RNA by Gar1 in H/ACA RNA-guided pseudouridylation. Nucleic Acids Res. 2015;43(15):7207–7216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Liang, B., Zhou, J., Kahen, E., et al.. Structure of a functional ribonucleoprotein pseudouridine synthase bound to a substrate RNA. Nat Struct Mol Biol. 2009;16(7):740–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Caton, E.A., Kelly, E K., Kamalampeta, R., et al.. Efficient RNA pseudouridylation by eukaryotic H/ACA ribonucleoproteins requires high affinity binding and correct positioning of guide RNA. Nucleic Acids Res. 2018;46(2):905–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Schattner, P.A computational screen for mammalian pseudouridylation guide H/ACA RNAs. RNA. 2006;12(1):15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Yang, X., et al. Kinetic and thermodynamic characterization of the reaction pathway of box H/ACA RNA-guided pseudouridine formation. Nucleic Acids Res. 2012;40(21):10925–10936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zhou, J., Liang B., and Li H.. Functional and structural impact of target uridine substitutions on the H/ACA ribonucleoprotein particle pseudouridine synthase. Biochemistry. 2010;49(29):6276–6281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Veerareddygari, G.R., Singh S.K., and Mueller E.G.. The pseudouridine synthases proceed through a glycal intermediate. J Am Chem Soc. 2016;138(25):7852–7855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Rashid, R., Liang, Bo, Baker, D L., et al.. Crystal structure of a Cbf5-Nop10-Gar1 complex and implications in RNA-guided pseudouridylation and dyskeratosis congenita. Mol Cell. 2006;21(2):. 249–60. [DOI] [PubMed] [Google Scholar]
- [23].Huang, L. and Lilley D.M.. The molecular recognition of kink-turn structure by the L7Ae class of proteins. RNA. 2013;19(12):1703–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Rozhdestvensky, T.S., et al.. Binding of L7Ae protein to the K-turn of archaeal snoRNAs: a shared RNA binding motif for C/D and H/ACA box snoRNAs in Archaea. Nucleic Acids Res. 2003;31(3):869–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Turner, B., et al.. Induced fit of RNA on binding the L7Ae protein to the kink-turn motif. RNA. 2005;11(8):1192–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Nolivos, S., Carpousis A.J., and Clouet-d’Orval B.. The K-loop, a general feature of the Pyrococcus C/D guide RNAs, is an RNA structural motif related to the K-turn. Nucleic Acids Res. 2005;33(20):6507–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Schmidt, A., Altincekic, N., Gustmann, H., et al.. The protein microenvironment governs the suitability of labeling sites for single-molecule spectroscopy of RNP complexes. ACS Chem Biol. 2018;13(9):2472–2483. [DOI] [PubMed] [Google Scholar]
- [28].Liang, B., Xue, S., Terns, R M., et al.. Substrate RNA positioning in the archaeal H/ACA ribonucleoprotein complex. Nat Struct Mol Biol. 2007;14(12):1189–1195. [DOI] [PubMed] [Google Scholar]
- [29].Li, S., Duan, J., Li, D., et al.. Reconstitution and structural analysis of the yeast box H/ACA RNA-guided pseudouridine synthase. Genes Dev. 2011;25(22):2409–2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Plass, T., Milles, S., Koehler, C., et al.. Genetically encoded copper-free click chemistry. Angew Chem Int Ed Engl. 2011;50(17):3878–3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Liang, B., Kahen, E J., Calvin, K., et al.. Long-distance placement of substrate RNA by H/ACA proteins. RNA. 2008;14(10):2086–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Dragon, F., Pogacic V., and Filipowicz W.. In vitro assembly of human H/ACA small nucleolar RNPs reveals unique features of U17 and telomerase RNAs. Mol Cell Biol. 2000;20(9):3037–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Charpentier, B., Muller S., and Branlant C.. Reconstitution of archaeal H/ACA small ribonucleoprotein complexes active in pseudouridylation. Nucleic Acids Res. 2005;33(10):3133–3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hengesbach, M., Kim, N-K., Feigon, J., et al.. Single-molecule FRET reveals the folding dynamics of the human telomerase RNA pseudoknot domain. Angew Chem Int Ed Engl. 2012;51(24):5876–5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.