

ABSTRACT
Internal-ribosomal entry sites (IRES) are translational elements that allow the initiation machinery to start protein synthesis via internal initiation. IRESs promote tissue-specific translation in stress conditions when conventional cap-dependent translation is inhibited. Since many IRES-containing mRNAs are relevant to diseases, this cellular mechanism is emerging as an attractive therapeutic target for pharmacological and genetic modulations. Indeed, there has been growing interest over the past years in determining the therapeutic potential of IRESs for several disease conditions such as cancer, neurodegeneration and neuromuscular diseases including Duchenne muscular dystrophy (DMD). IRESs relevant for DMD have been identified in several transcripts whose protein product results in functional improvements in dystrophic muscles. Together, these converging lines of evidence indicate that activation of IRES-mediated translation of relevant transcripts in DMD muscle represents a novel and appropriate therapeutic strategy for DMD that warrants further investigation, particularly to identify agents that can modulate their activity.
KEYWORDS: mRNA, IRES, disease, muscle, Duchenne muscular dystrophy, therapy
1. Introduction
The regulation of protein translation is the basis for maintenance of homoeostasis for many cellular mechanisms. In some disease states and under stress conditions, classical cap-dependent protein synthesis can be halted or down-regulated. Thus, alternative translational mechanisms become activated to promote protein production. One such mechanism is the internal ribosome entry site (IRES)-driven initiation. Regulation at the initiation translation step permits a rapid response to changes in cellular physiological conditions. Internal ribosome entry mechanisms were first discovered in encephalomyocarditis virus (EMCV) and Poliovirus [1,2]. Following this discovery, many more viral IRES-harbouring mRNAs were identified along with several cellular mRNAs [3–6]. Previous studies have shown that ~10% of the total mRNA pool within given cells are predicted to contain IRESs. However, many of these remain to be identified [7].
IRES-containing cellular mRNAs are found to be preferentially translated under various conditions that cause inhibition of cap-dependent initiation including for example, cellular stress, episodes of hypoxia, nutrient limitation, pharmaceutical stimulation, cell cycle or differentiation [4,8]. Of particular interest is the fact that IRESs can also be activated under disease conditions thereby revealing their therapeutic potential. Indeed, the development of new methods to target IRES-mediated translation of disease-relevant transcripts via pharmacological interventions, small molecules or genetic manipulation, is gaining attention in order to create novel therapies for a variety of diseases [9–13]. Up until now, the most commonly targeted disorders through IRES regulation are: i) infectious diseases which use small inhibitors to reduce viral activity [12,13]; and ii) tumorigenesis via targeting cellular IRESs within oncogenes, growth factors and programmed cell death regulators [10,11,14]. As the discovery of more cellular IRESs emerged in recent years, IRES-harbouring mRNAs involved in other disorders have also been investigated for their therapeutic potential [15]. Among these, targeting IRES-mediated translation in Alzheimer’s disease [16,17], Parkinson’s disease [18–20], and neuromuscular diseases [21–25] has arose as a novel line of work for therapeutic intervention.
In the context of neuromuscular diseases, many laboratories including our own have been interested in identifying therapeutic strategies for Duchenne muscular dystrophy (DMD) [21–23,26–41]. Recently, a novel IRES was identified in utrophin A mRNAs and considerable efforts were focused in characterizing mechanisms of activation [21–23,27]. Utrophin A is highly relevant for DMD given that it can serve as a surrogate for the missing dystrophin protein in DMD muscles [42,43 and see details below]. Moreover, scanning of the available literature reveals that several other DMD-relevant transcripts also contain IRESs which make them similarly important for developing appropriate DMD therapies based on IRES-mediated translation [24,44–47]. Here, we review our current knowledge of DMD-relevant transcripts containing IRESs and discuss their therapeutic potential for improving sarcolemmal integrity, muscle regeneration and repair as well as angiogenesis in dystrophic muscle. Collectively, this body of work leads us to propose IRES-mediated translation as a key cellular mechanism amenable to appropriate therapeutic interventions for treating DMD.
2. Mechanisms for initiating translation
There are two main mechanisms known to promote translation of mRNAs, namely, cap-dependent and cap-independent translation. Both share similarities in that they each involve four distinct stages: initiation, elongation, termination and ribosomal recycling. Many excellent reviews describe in detail the fundamental regulatory checkpoints of translation (see, for example, Shatsky et al. [48] Yang et al. [49], Shirokikh and Preiss [50]). Therefore, only a summary is provided below.
Cap-dependent translation is the most conventional mechanism to translate mRNAs into functional protein products. The indispensable step for cap-dependent translation initiation of all eukaryotic mRNAs, is the association of the eukaryotic initiation factor (eIF) 4 F protein complex to the 7-methylguanylate cap (m7G), also known as the 5ʹcap. Before this binding, a series of eIFs form a pre-initiation complex. eIF4E is bound by the scaffolding protein eIF4G and the RNA helicase eIF4A. This complex forms eIF4F which is important for 40S recruitment to the 5ʹend of the target mRNA. Additional initiation factors, including eIF3, which recruit the small ribosomal submit to the 5ʹ end of the mRNA as well as eIF2-GTP that delivers the first transfer RNA (tRNA methionine) to the 40S ribosome, are involved in translational initiation. The poly(A) binding protein (PABP) associates with eIF4F and binds to the poly(A) tail of the mRNA to promote its circularization to facilitate translation [51–53]. Cap-dependent translation can be repressed in a variety of ways including for example: i) through the cleavage of eIF4G by viruses and during apoptosis, thereby preventing binding of eIF4E to the m7G cap; ii) phosphorylation of eIF2 that circumscribes recruitment of tRNAs to the ribosome; and iii) increasing the eIF4E-binding protein (4E-BP) which inhibits eIF4E’s ability to bind eIF4G [54–57]. In fact, many physiological, pathological and pharmacological conditions such as during the cell cycle, hypoxia, apoptosis, drug treatments as well as in different disease states, may lead to inhibition of cap-dependent translation and usage of an alternate translation mechanism, i.e., IRES-mediated translation [4,24,27,48,49,58].
A rising number of mRNAs are believed to possess IRESs which permits translation of a transcript independently of its 5ʹ end cap. Conventional scanning from the 5′ end is likely not efficient for most IRES-containing cellular mRNAs because their 5′UTRs are typically long, GC-rich and highly structured [48,49,59]. In cap-independent translation, the ribosome interacts with the IRES with the help of some but not all canonical initiation factors, and allows for continued and/or enhanced expression of proteins when cap-dependent translation is suppressed [4,48,59]. In fact, IRES elements are believed to participate in various interactions with components of the translational machinery including the canonical initiation factors, IRES trans-acting factors (ITAFs) and the 40S ribosomal subunit [4]. ITAFs are suggested to increase the binding affinity between IRESs and canonical initiation factors as well as with the ribosome. ITAFs have been hypothesized to act as RNA chaperones to help the IRES primary sequence attain the appropriate conformational state to promote ribosome binding [60]. Interestingly, it has been reported that some IRESs require a combination of two or three ITAFs to reach efficient translational activity [61–64]. Examples of some of the most studied ITAFs include the human La autoantigen (LA), upstream of N-ras (Unr), poly(rC) binding protein-2 (PCBP2) and polypyrimidine tract-binding protein (PTB) [62–73]. However, many more ITAFs have so far been identified with one or multiple targets. IRES activity is regulated positively or negatively by ITAFs and their relative cellular abundance, as well as, according to tissue specificity and exogenous stimuli [74–76]. Previous reports also demonstrate that the proteins controlling IRES-dependent translation initiation are modulated by their subcellular localization [76]. However, the precise mechanisms regulating protein translation by IRES are still largely unknown and requires additional studies.
3. Therapeutic potential of IRES-mediated translation in Duchenne Muscular Dystrophy
Duchenne muscular dystrophy (DMD) is caused by mutations in the gene that encodes dystrophin, a key protein to maintain the structural integrity of muscle fibres. DMD is one of the most common inherited paediatric neuromuscular diseases and accounts for over 80% of cases of muscular dystrophies around the world [77]. DMD patients suffer from severe muscle degeneration which results in muscle weakness, respiratory impairment, and cardiomyopathy [78–80]. Skeletal muscle fibres of these patients undergo cycles of degeneration and regeneration provoking a devastating domino effect of secondary symptoms such as surges in calcium uptake, critical inflammation and functional ischaemia [78,81,82]. Eventually, the ability of muscle fibres to regenerate fibres runs out leading to the replacement of muscle by adipose and connective tissues and, consequently, loss of muscle strength and function. As the disease progresses, DMD patients suffer from ambulation impairments at approximately 12 years of age and, typically, they will completely lose ambulation in their late teens [77,83]. Assisted ventilation becomes necessary usually around the age of 20 [80,83,84] and death occurs due to respiratory and/or cardiac complications when patients approach their thirties [78,80,85].
The dystrophin gene located on chromosome Xp21.1 contains 79 exons and encodes the 427-kDa dystrophin protein. The DMD gene produces different transcripts encoding various dystrophin isoforms (Fig. 1) that are generated by tissue-specific promoters (brain, muscle and Purkinje cells), alternative splicing and polyA-additional sites [86–92]. Mutations in the DMD gene include deletions (65%-70%) around the 2 hot areas near the N-terminus (exons 3–7) and within exon 45–55 [93–95], duplications (6–10%), small mutations (10%), or point mutations (nonsense or missense) [96–98]. These mutations disrupt the translational reading frame of dystrophin, which may lead to severe Duchenne muscular dystrophy (DMD) with an unfunctional protein or result in a short functional form of the dystrophin protein resulting in the mild disorder Becker muscular dystrophy (BMD) [77].
Figure 1.
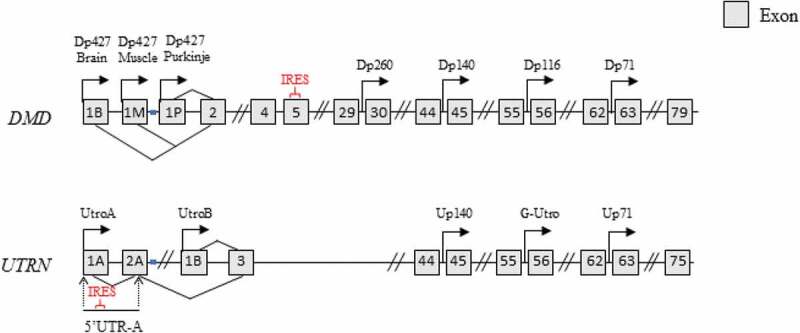
Utrophin and dystrophin genes. Dystrophin (DMD) and utrophin (UTRN) genes with associated promoters and exons. Full-length dystrophin transcription is controlled by 3 promoters (brain, muscle, Purkinje) [91,127,128]. Four alternate promoters result in short isoforms dp260, dp140, dp116, and dp71 [86,129,130]. An IRES is found in exon 5. Dp427 is the main variant produced in muscle [127]. The two isoforms of utrophin (A and B) have distinct promoters and 5ʹUTR regions [131,132]. Utrophin A is expressed primarily at the myotendinous and neuromuscular junction in mature skeletal muscle, in neurons, astrocytes as well as in the brain (choroid plexus and pia mater), whereas utrophin B is primarily localized in vasculature [131–135] The utrophin A transcript contains an IRES between nucleotides 71 and 152 in its 5ʹUTR [22]. The utrophin B promoter is located within the second intron of the utrophin gene resulting in a unique 5ʹ end [131,132]. Three short utrophin isoforms Up140, G-Utrophin and Up71 were reported similar to the dystrophin gene [129,130,136]
The dystrophin protein plays a critical role in supporting muscle fibre integrity by linking the intracellular actin network to the extracellular matrix [99,100] via binding through the dystrophin-associated protein complex (DAPC) at the muscle fibre membrane (also known as the sarcolemma) [101,102]. Such structural organization provides stability to the sarcolemma during repeated cycles of muscle contraction. In the absence of dystrophin, this physical link between the intra- and extracellular compartments of muscle fibres is lost, making the fibres highly susceptible to damage and hence, inducing degeneration. Many potential therapies for DMD have been investigated over the last years including, for example, cell therapy [103–107], gene editing [108–115], gene therapy [116–118], exon skipping [119–123] and suppression of premature stop codons [124–126]. An alternative strategy consists in utilizing a protein highly homologous to dystrophin and to induce its upregulation pharmacologically to compensate for the absence of dystrophin in muscle fibres. An ideal candidate for this role is utrophin A.
3.1. Targeting the utrophin A IRES for treating DMD
Utrophin A is an attractive candidate for treating DMD since it can functionally compensate for the absence of the dystrophin protein in dystrophic muscle. In normal mature skeletal muscle, utrophin A expression is restricted to the neuromuscular and myotendinous junctions. The upregulation of the utrophin protein from these sites to the entire muscle fibre membrane has been shown, to have beneficial effects on the performance, integrity and morphology of dystrophic muscle fibres. Thus, many laboratories have focused their efforts in increasing utrophin A expression through genetic or pharmacological stimulations as a therapeutic approach for the treatment of DMD [26,30–32,137–141].
3.1.1. Utrophin A as a surrogate for dystrophin
Similarities between utrophin A and dystrophin clearly highlight the potential of utrophin A to functionally compensate for dystrophin [142,143]. Utrophin A is expressed primarily at the myotendinous and neuromuscular junctions (NMJ), in neurons, astrocytes as well as in the brain (choroid plexus and pia mater) [131–135]. Utrophin A displays varying localization patterns in skeletal muscle through embryonic to adult development. In fact, utrophin A is expressed throughout the sarcolemma during embryogenesis, reaching maximal expression levels at approximately 17 weeks’ gestation, and gradually decreasing to negligible levels at week 26, which coincides with increasing levels of dystrophin [144,145]. In adult tissues, utrophin A is mostly restricted to the NMJ but in regenerating muscles, utrophin A is expressed along the entire sarcolemma as a part of the repair process [146–148]. More interestingly, utrophin A is up-regulated in dystrophin-deficient muscle fibres and, in part, localizes to the sarcolemma [132]. Thus, these studies suggest that utrophin A plays a role similar to that of dystrophin during certain developmental stages of muscle fibres and could compensate for the loss of dystrophin in DMD.
Utrophin A has similar protein binding functions to dystrophin and has approximately 80% sequence homology in the actin-binding domain [43,142,149–152] and the carboxy terminus, including the cysteine-rich domain [142,149–151] (Fig. 2). In addition, utrophin A has been shown to associate to F-actin filaments with its N-terminal actin-binding domain and interacts with members of DAPC, notably α-syntrophin, β-dystroglycan and α-dystrobrevin-1 at its C-terminal area [153–157]. The major structural difference between dystrophin and utrophin A is in the rod domain at an identity less than 30%, where utrophin is missing repeats 15 and 19 as well as two hinge regions [142,157]. These differences could explain why utrophin is unable to perform all of dystrophin’s functions. For instance, transgenic utrophin overexpression in mdx mouse models (an x-linked muscular dystrophy mouse mutant model widely used in DMD research) does not restore the localization of neural nitric oxide synthase (nNOS) to the sarcolemma of muscle fibres, which is the main source of nitric oxide in skeletal muscles and is indispensable for skeletal muscle integrity as well as its contractile performance [158–160]. Whereas dystrophin can bind nNOS to promote angiogenesis [161,162]. Additionally, unlike dystrophin, utrophin A is unable to bind microtubules to form an organized lattice at the sarcolemma which also protects against contraction-induced injury [163–165].
Figure 2.
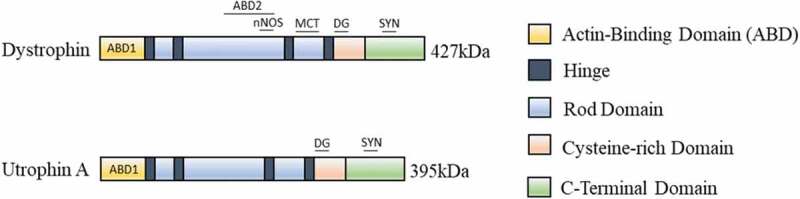
Utrophin A and dystrophin protein domains. Similar to full-length dystrophin, utrophin A can bind DAPC members at the sarcolemma of muscle fibres to provide a link between the ECM and the intracellular cytoskeleton. Utrophin A has a rod domain and four hinge regions (H1, H2, H3, and H4) and a cysteine-rich region (CR) that binds β-dystroglycan. Only the N-terminal domain (N) of utrophin A can interact with F-actin filaments [171]. The main difference between utrophin and dystrophin is the lack of the spectrin-like repeats 15 and 19, and 2 hinge regions [142,157]. Utrophin A and dystrophin associate with α-dystrobrevin, syntrophins α and β at their C-terminal domains [153–157]. Via their PDZ domain, the syntrophins recruit nNOS at the muscle fibre membrane to mediate blood flow to the muscle and this is also supported by dystrophin which binds to nNOS via its spectrin repeats 16 and 17 for proper localization to the muscle membrane [172]. However, utrophin A cannot recruit nNOS at the sarcolemma via direct binding like to dystrophin [161,162]. Additionally, utrophin A is unable to bind microtubules (MCT) to form an organized lattice at the sarcolemma [163–165]
Despite these differences, increasing utrophin A expression throughout the sarcolemma of dystrophic muscle fibres has proven to be an efficient approach to compensate for the lack of dystrophin [139]. In dystrophic muscles, there is a slight increase in utrophin A at the sarcolemma, that occurs naturally as part of the repair process [146,166,167]. Transgenic overexpression of a truncated utrophin using the human skeletal α-actin (HSA) promoter improved the dystrophic muscle phenotype of mdx mice significantly. This includes a decrease in the serum levels of the enzyme creatine kinase (CK) that leaks out of damaged muscle, a reduction of centrally nucleated myofibers (indicative of fewer regenerating fibres), as well as a rescue of utrophin A and DAPC members at the sarcolemma [140]. Transgenic mouse models expressing full-length utrophin A elicited full recovery of mechanical functions of the diaphragm with only a 2-Fold induction of utrophin A as compared to the mdx control and with no adverse effects on non-muscle cells [139,168]. As little as a 1.5-fold increase of utrophin A can be beneficial as long as the localization of utrophin A is at the level of the sarcolemma in dystrophic muscle fibres [139]. Furthermore, a recent study showed that AAV9-mediated micro-utrophin expression can also strongly alleviate the cardiac defects of a more severe mdx mouse model [169]. Similar beneficial effects were also observed in the golden retriever muscular dystrophy (GRMD) dog model of DMD with AAV-mediated mini-utrophin gene transfer [170]. Thus, it appears that utrophin A is a suitable replacement for dystrophin and, accordingly, it represents an attractive therapeutic target for DMD.
3.1.2. Utrophin A 5ʹUTR contains an IRES
To date, transcriptional regulation of utrophin A has been the focus of most studies interested in upregulating its expression levels in dystrophic muscle [36,38,39,43,134,173–178]. However, over the past 15 years, a growing number of reports have highlighted the fact that regulation of utrophin A expression is also under the tight control of post-transcriptional events [33,34,179–182]. In this context, the notion of potential alternative translational regulation of utrophin A arose several years ago from studies conducted in our lab and that of others, showing elevated utrophin A protein levels in muscle, with little to no increase in mRNA expression [36,37,132,183,184]. The utrophin A protein of 395 kDa is encoded by a 13kb transcript which consists of 74 exons and is located on chromosome 6q24 [143,150]. Moreover, utrophin A has a long 5ʹUTR consisting of 560 nucleotides for humans (508 for murine), that expands from the transcriptional start site through exons 1A and 2A to the translation initiation site for utrophin A [131,134]. Similar to other IRES-containing mRNAs, the long utrophin A 5ʹUTR is predicted to have a high degree of secondary structure and is expected to be translationally repressed through both its 5ʹ and 3ʹUTR’s [21,181,185]. The repression via the 3ʹUTR has been attributed to miRNAs and ARE-mediated degradation, but the exact inhibitory mechanisms that operate through its 5ʹ-UTR remain nebulous [181].
Studies have shown that utrophin A can be translated through both cap-dependent and IRES-dependent translation [21–23,186]. With the use of a bicistronic reporter construct containing the utrophin A 5ʹUTR, Miura et al. demonstrated that in intact muscles, little utrophin A IRES activity is detected. However, a ~ 9-fold increase in reporter activity was observed in muscles subjected to degeneration and regeneration cycles by cardiotoxin injections, thus first revealing the possible cap-independent translational regulation of utrophin A in regenerating muscles [21]. Control experiments were performed in parallel to ensure that the bicistronic mRNA did not undergo aberrant splicing, and that the utrophin A 5ʹUTR did not contain cryptic promoter activity [21]. In addition, a study shows that glucocorticoids, the main intervention used for treating DMD patients [77,187,188], can drive IRES-mediated translation of utrophin A [22]. In fact, a 2–3 day treatment of C2C12 myotubes, an immortalized skeletal muscle cell line, with 6α-methylprednisolone-21 sodium succinate, promotes utrophin A IRES activation as detected using a bicistronic reporter assay [22]. Additionally, a polysome profiling assay demonstrates that the treatment with 6α-methylprednisolone-21 sodium succinate reduced global protein synthesis, while increasing the polysome association of the reporter mRNA harbouring the utrophin A 5ʹUTR IRES [22]. This indicates that the activity of this IRES can indeed be modulated pharmacologically. This work also suggests that the glucocorticoid corticosteroid, prednisolone, known for its anti-inflammatory actions [189,190], may promote combinatorial benefits in dystrophic muscle by reducing inflammation an increasing utrophin A expression in parallel.
To determine whether the utrophin A IRES displayed tissue-specific activity in vivo, another study aimed to generate transgenic mice harbouring control (CMV/betaGAL/CAT) or utrophin A 5ʹUTR (CMV/betaGAL/UtrA/CAT) bicistronic reporter transgenes. Examination of multiple tissues from two CMV/betaGAL/UtrA/CAT lines revealed that the utrophin A 5ʹUTR preferentially drives cap-independent translation of the reporter gene in skeletal muscles [23].
Since DNA-based bicistronic constructs used for IRES identification can sometimes give false positives, a study from Ghosh et al. took a different approach to confirm the presence of an IRES in the mouse utrophin-A 5ʹUTR with an alternate strategy [186]. In this study, the relative contribution of cap-independent and cap-dependent translation with the mouse utrophin A 5ʹUTR was compared through an m7G-capped and A-capped mRNA transfection-based reporter assay. As A-capped mRNA is poorly translated through cap-dependent events, its expression provides reliable estimation of cap-independent translation. The results confirm that indeed utrophin A 5ʹUTR can be translated through a cap-independent manner but also demonstrate that the 5ʹUTR plays a key role in the inhibition of cap-dependent translation of utrophin A [186].
3.1.3. eEF1A2 regulates utrophin A-IRES activity
Recent work using RNA-affinity chromatography, identified the elongation factor 1A2 (eEF1A2) as a putative ITAF. The initial results showed that eEF1A2 associates to the 5ʹUTR of utrophin A and mediates utrophin A’s IRES-dependent translation in skeletal muscles [23]. In addition, ectopic expression of eEF1A2 in tibialis anterior muscle of mdx and wild-type mice results in upregulation of utrophin A protein levels and utrophin A IRES-activation in CMV/betaGAL/UtrA/CAT bicistronic reporter harbouring transgenic mice [27]. These data are in agreement with a recent study reporting that another elongation factor, eukaryotic elongation factor 2 (eEF2), acts as an ITAF to regulate IRES-mediated translation of XIAP and FGF-2 mRNAs [191].
There are two eEF1A isoforms: eEFlAl and eEFlA2 which share ~92% sequence identity [192]. Both eEF1A isoforms function in regulating transport of aminoacyl-tRNA throughout translation elongation, however, eEF1A2 also plays a role in skeletal muscle, where it prevents apoptosis [193]. Several other functions have also been attributed to eEF1A including interaction with the cytoskeleton, binding and bundling of actin filaments, as well as inducing a severing effect on microtubules [194,195]. In contrast to eEF1A1 that is generally expressed ubiquitously, eEF1A2 is preferentially expressed in skeletal muscle, heart and brain [196,197], thereby suggesting that eEF1A2 could indeed play a distinct role in skeletal muscle. Given these observations, it seemed important to identify pharmacological compounds that target eEF1A2 in order to increase endogenous levels of utrophin A expression, via IRES-mediated translation. Such an approach could serve as a drug-based therapy to treat DMD, which offers several advantages over other methods including, ease of administration, positive impact on all skeletal muscles, and applicability to all DMD patients regardless of their dystrophin mutation.
With this in mind, a recent study set-out to identify and characterize FDA-approved drugs that target eEF1A2 and cause upregulation of utrophin A in skeletal muscle. Using FDA-approved drugs has the distinct benefit of potentially greatly accelerating translation of the findings into the clinical setting. Using an in-cell ELISA-based high-throughput screening approach, 11 FDA-approved drugs were identified that significantly and concomitantly raised protein expression levels of eEF1A2 and utrophin A in skeletal muscle cells. Among these drugs, many had common therapeutic roles including anti-diabetic, anti-peptic ulcer, cholesterol-lowering and beta-adrenergic blocking agents [27]. Five lead drugs (Acarbose, Betaxolol, Labetalol, Pravastatin, and Telbivudine) were able to stimulate reporter activation from a bicistronic reporter construct harbouring the utrophin A 5ʹUTR IRES, showing that, indeed, these two drugs act via IRES-mediated regulation of utrophin A translation.
This work focused on the two most promising candidates, the selective beta-1 adrenergic blocker Betaxolol, and the cholesterol-lowering agent Pravastatin. A 7-day treatment of transgenic mice containing the bicistronic utrophin A 5ʹUTR IRES reporter construct with Betaxolol or Pravastatin, demonstrated the potential of both drugs to activate utrophin A IRES-dependent translation in vivo. Chronic treatment of mdx mice with either of these two drugs resulted in increased expression of utrophin A via IRES activation and, localization of utrophin A at the sarcolemma of muscle fibres, while causing significant beneficial effects on the dystrophic phenotype. In fact, a grip strength assessment and ex-vivo force drop analysis demonstrated that the 4-week treatment with Betaxolol and Pravastatin significantly improved muscle strength. In addition, histological and immunofluorescence experiments showed a decrease in central nucleation, a marker of muscle fibre regeneration [198], and an improvement of muscle fibre integrity [27]. Finally, Pravastatin treatment of ‘wasted’ mice, which are genetically deficient in eEF1A2, failed to induce utrophin A expression in skeletal muscle, thus clearly demonstrating the key role of this ITAF in IRES-mediated translation of utrophin A. Altogether, these proof-of-principle studies illustrate the feasibility of specifically targeting the utrophin A IRES with pharmacological agents, as a therapeutically viable approach for treating DMD.
3.2. IRES-mediated translation of dystrophin
Recent studies revealed that a possible approach to recover partial dystrophin expression in DMD muscles is to target an IRES at the 5ʹend of the dystrophin transcript [24]. In fact, this finding aligns well with the original observation on the presence of an IRES in the utrophin A 5ʹUTR since the utrophin and dystrophin genes have common ancestral origins [142,143]. Such findings further suggest that the DMD disease state can suppress cap-dependent translation and that IRES-mediated translation has evolutionarily been conserved for both gene products.
The severity of the symptoms of muscular dystrophy patients is known to be dependent on the nature of the dystrophin gene mutation. More specifically, DMD, the most severe form, occurs when the mutation provokes a complete absence of the dystrophin protein whereas occasionally, the mutation still allows for production of a truncated, yet functional protein. For example, there is evidence of patients affected by a nonsense mutation in exon 1 of the dystrophin gene, who show mild symptoms and are known to be afflicted with the milder form of DMD, BMD. In some cases, ambulation of Becker patients over the age of 60 years has been reported. Although this nonsense mutation at exon 1 is predicted to result in no protein translation, muscle biopsies indicated that these patients produced a short but functional N-terminally truncated isoform of dystrophin, thus suggesting alternate translation initiation [199]. A similar outcome was recently reported in a patient affected by a nonsense mutation in exon 2 of the DMD gene, and who demonstrated mild BMD symptoms [200]. In 2014, Wein et al. reported that the production of a short dystrophin isoform protein in a situation where the mutation is present in the second exon of the DMD gene, is due to translation resulting from an IRES within exon 5 of the DMD gene [24]. Results from this study further demonstrated that IRES-mediated translation of the short dystrophin isoform is achieved in vitro by expressing N-terminally truncated dystrophin reporter constructs or by promoting exon skipping of the upstream reading frame. In addition, exon skipping of exon 2 in a mouse model affected by a duplicated exon 2 mutation in the DMD gene, resulted in sarcolemmal recovery of the dystrophin complex, morphological improvements of the skeletal muscle fibres and amelioration of muscle strength [24].
Similar to utrophin A, IRES activation of dystrophin is stimulated by glucocorticoid treatment [24]. However, IRESs are not defined by a consensus sequence or distinct RNA structure [201] and, consequently, despite the many similarities between utrophin A and dystrophin, the regulation of the dystrophin IRES may not be comparable to utrophin A’s. More specifically, an ITAF that regulate the dystrophin IRES has yet to be identified, but it would seem unlikely, given the current knowledge of ITAFs and their degree of specificity, that eEF1A2 could both regulate the utrophin A and dystrophin IRESs. Nonetheless, targeting IRES-mediated dystrophin protein translation, represents an attractive target for patients containing mutations at the 5ʹend of the DMD gene which consists of 6% of DMD patients [93].
3.3. IRES-mediated translation of growth factors
In disease state such as muscular dystrophies where cap-dependant translation may be compromised, targeting IRES activity of key growth factors may be another attractive strategy to improve the dystrophic muscle phenotype. Over the years, many laboratories worldwide have examined the potential therapeutic impact of various growth factors for dystrophic muscles [202–206]. Interestingly, several of these highly DMD-relevant growth factors, including VEGF, FGF-1, FGF-2, IGF-2 and IGF-1 R have been previously shown to contain IRESs.
3.3.1. Targeting IRES-mediated translation of VEGF
It has been established that there are beneficial effects of rising tissue perfusion by increasing the density of the vasculature in dystrophic muscles as a therapy for DMD. DMD patients are known to also suffer from abnormal blood flow after muscle contraction, thus, reducing the effects of functional ischaemia might decrease muscle damage [207–209] and improve dystrophic muscle function. One potential target in this case is vascular endothelial growth factor (VEGF), a known angiogenesis regulator. Interestingly, VEGF contains a long and structured 5′UTR containing two independent IRESs involved in its translation [210–213]. VEGF-A ligand can associate with the VEGF receptors to promote vascular permeability while also inducing proliferation and survival of newly formed endothelial cells, providing the basic structure of novel vasculatures [214]. Four-week post-intramuscular injection of a rAAV-VEGF vector in the bicep and tibialis anterior (TA) muscles of mdx mice, resulted in increased capillary density, reduced necrotic fibres, and significant improvements in forelimb strength, compared to AAV-LacZ-injected control mdx mouse muscle [215]. Thus, VEGF is an interesting and potentially important therapeutic target for DMD. VEGF-A expression has been shown to be regulated by IRES-mediated translation during ischaemic and hypoxic stress [213,216] via the ITAF heterogeneous nuclear ribonucleoprotein (hnRNP)L [216]. Although the implication of hnRNP L in DMD has yet to be examined, one can certainly envisage nonetheless, designing pharmacological interventions aimed at boosting VEGF expression via this translational mechanism.
3.3.2. IRES activation of the fibroblast growth factors: FGF-1 and FGF-2
Several other growth factors regulated by IRES translation are members of the fibroblast growth factor (FGF) family including FGF-1 and FGF-2. FGFs have been closely investigated over the years in muscular disorders due to their ability to promote muscle fibre regeneration and tissue damage repair [202,203,217,218]. More specifically, FGF-1 has been shown to be involved in muscle development and regeneration of muscle fibres. The FGF-1 gene is comprised of four tissue-specific promoters able to produce four transcripts that vary at their 5ʹ end. One such transcript, FGF-1A, contains an IRES that is activated during myoblast development and muscle regeneration, and is further stimulated by a cis-acting element in its promoter. This reveals a combined mechanistic process of mRNA transcription and translation coupling, in regenerating muscle fibres [204]. FGF-1A’s IRES is activated in hypoxic cells where expression of its known ITAF, vasohibin1, increases in the nucleus, is translocated to the cytoplasm (may be translocated in part to stress granules) and binds the IRES of FGF-1A mRNA to recruit the transcript to the ribosome [219]. In agreement with these findings, FGF-1 is upregulated in regenerating muscle cells of mdx mice and in skeletal muscles of Facioscapulohumoral muscular dystrophy patients [205,220]. Considering that DMD patients suffer from muscle degeneration, this suggests that targeting the FGF-1 IRES may be of therapeutic benefit to stimulate regeneration of dystrophic muscle fibres. Currently, it is unknown whether vasohibin1 plays a role in DMD muscle but its regulation of FGF-1 suggests that it may be a good target to increase FGF-1 expression levels in dystrophic muscles via cap-independent translation.
Another key FGF that contains an IRES is FGF-2. Translation of FGF-2 results in four isoforms differentially localized in cells, with all isoforms being potentially synthesized through IRES-mediated translation [221,222]. FGF-2 is embedded within the basal lamina and extracellular matrix of uninjured skeletal muscle tissue where it plays a role in skeletal muscle regeneration by maintaining the pool of muscle precursor cells [217,223]. By repressing myogenesis, FGF-2 permits satellite cell self-renewal and is required for maintenance and repair of skeletal muscle [217]. FGF-2 is available to satellite cells following its production by myofibers, fibroblasts and other satellite cells [202,224–227].
Considering the key role of FGF-2 in muscle fibre repair [202,203,217], it may thus represent another attractive target for DMD therapy. In fact, like FGF-1, expression of FGF-2 is increased in regenerating muscle fibres including dystrophic muscles [225,228]. IRES-mediated translation of FGF-2 has been shown to be activated in vivo during limb ischaemia and in hypoxic conditions [229]. FGF-2 IRES activity is also sensitive to oxidative stress. These conditions promote the nuclear-cytoplasmic translocation of eukaryotic eEF2 where it then assumes the role of an ITAF to drive FGF-2 IRES-mediated translation [191]. This suggests that this cellular IRES may be a good target for FGF-2 protein expression in different pathological conditions such as muscle atrophy, ischaemia and apoptosis, conditions often seen in muscular disorders where cap-dependent translation may be repressed. Along those lines, genetic ablation of FGF-2 in mdx mice promotes a more severe dystrophic phenotype than that seen in mdx mice, demonstrating its importance for proper muscle maintenance [206]. In addition, FGF-2 expression has also been shown to have protective effects against glucocorticoid-induced deterioration of bone through inhibition of the Wnt antagonist sclerostin [230]. Keeping in mind that glucocorticoid-treated DMD patients frequently develop osteoporosis, resulting in reduction of bone strength and long-bone fractures, IRES-mediated expression of FGF-2 in these patients might offer therapeutic benefits for improving muscle repair and bone reinforcement [78].
3.3.3. Targeting IRES-mediated Translation of IGF-2
Insulin-like growth factors (IGF) are amongst the growth factors that are the most studied in the DMD context [44–47]. One such IGF, IGF-2 has been studied in muscular dystrophies for its ability to modulate regulatory networks involved in programmed cell death [47]. IGF-2 is a potent growth factor with anti-apoptotic capacity in a variety of cell types [231–233]. IGF-2 can interact with the IGF-1 receptor (IGF-1 R) to activate phosphatidylinositol 3-kinase/Akt signalling, which in turn blocks proteolytic processing of key caspases and prevents initiation of the apoptotic cascade [231,234]. During foetal development, the IGF-2 gene produces three mRNAs containing identical coding regions but varying only at their 5ʹUTR. One isoform, IGF-2 leader 2, has a long 5ʹUTR containing an IRES which is activated in dividing cells [235]. The IGF-2 mRNA-binding protein 2 (IMP2) has been identified as an ITAF, phosphorylated by mTOR to modulate IGF-2 IRES activity in a mouse embryogenesis model [236].
The beneficial impact of IGF-2 has previously been investigated in the mdx mouse. For example, IGF-2 overexpression in mdx mice inhibits the normally seen elevated levels of programmed cell death in DMD muscle stem cells. This ectopic expression of IGF-2 results also in significant improvements in the initial histopathological changes observed in dystrophic skeletal muscles [47]. Thus, this provides evidence that that IGF-2 is a strong candidate for therapeutic intervention in DMD. In addition, its receptor IGF-1 R harbours an IRES at the 5ʹUTR [237] that is activated by thiazolidinediones antidiabetic compounds (rosiglitazone and pioglitazone) in smooth muscle cells and promotes beneficial effects on cell survival potentially via the ITAF HuR [238,239]. As the effects of IGF-2 can be mediated through IGF-1 R, pharmacologically promoting translation of IGF-1 R, through IRES activation in addition to targeting IRES-mediated translation of IGF-2, might represent another therapeutic approach for DMD. This seems particularly attractive given the known roles of HuR in skeletal muscle development and plasticity [240–242].
4. Conclusion and perspectives
Directing IRES-mediated translation of key transcripts involved in various diseases is gaining greater interest as an alternative therapeutic approach. This alternative translation avenue permits a rapid protein response to changes in physiological and pathological cellular conditions, bypassing the steps of transcription and post-transcription. In addition, this targeting approach achieves stimulation of protein expression or in turn, attenuates expression levels of deregulated or damaging signalling molecules in a stress and disease state, when cap-dependent translation might be compromised. In fact, laboratories around the world have been identifying and developing new drugs or small molecules to regulate IRES-mediated translation of target mRNAs in order to create new therapies for an ever-growing array of diseases [9–13,22,24,27]. Many of these molecules target and inhibit stress-induced, IRES-mediated translation such as antisense oligonucleotides, peptide nucleic acids, ribozymes, short hairpin RNAs and small interfering RNAs [243].
An important aspect to consider with targeting IRES-translation for therapy, is the potential for off-target effects. ITAFs likely have multiple mRNA targets and altering their normal expression patterns in certain tissues might result in detrimental secondary effects. For instance, VEGF has been shown to be involved in tumour survival through inducing tumour angiogenesis [244,245] whereas eEF1A2 has been shown to be upregulated in many types of cancers including breast, ovarian, pancreatic, prostate and lung cancers [246–252]. Similarly, growth factors such as FGF-2 which is particularly widespread, are known to be elevated in many tumours including melanomas as well as in breast, pancreas, bladder, head and neck, prostate, and hepatocellular tumours [253,254,255]. Thus, this type of therapy will need proper safety assessments and may ideally be designed to target-specific tissue(s).
In the context of neuromuscular disorders, a variety of DMD-relevant transcripts have been shown to contain IRESs as described above. Moreover, for some of these mRNAs, ITAFs have also been identified and characterized. All of these thus represent potential targets for pharmacological interventions aimed at modulating their activity leading to protein production (Fig. 3). This therapeutic concept seems well advanced for the utrophin A IRES and its ITAF eEF1A2, with several FDA-approved drugs recently identified and now known to be able to activate utrophin A expression. One could even envisage that, therapeutically, a rationally designed pharmacological intervention could potentially target several DMD-relevant IRES-containing transcripts thereby leading to multiple complementary benefits in dystrophic muscle. The fact that up to 10% of cellular mRNAs are predicted to contain an IRES [254], suggests that the search for additional DMD-relevant, IRES-containing transcripts, and their regulating ITAFs, should continue, perhaps even accelerate given the promising findings obtained to date. In fact, this may well be important and extended to other neuromuscular conditions including for example amyotrophic lateral sclerosis, in which IRES-mediated translation of VEGF may play a modifying role [255,256]. Overall, our knowledge of cellular IRESs, their structure, mechanisms of regulation, and underlying signalling pathways is still in its infancy. Accordingly, increased efforts in this area appear warranted particularly if the goal is to capitalize and exploit the full therapeutic potential of IRESs and ITAFs for multiple diseases including neuromuscular conditions such as DMD which, despite the discovery of the dystrophin gene as causative of the disease more than 30 years ago, remains today without a cure or effective treatment.
Figure 3.
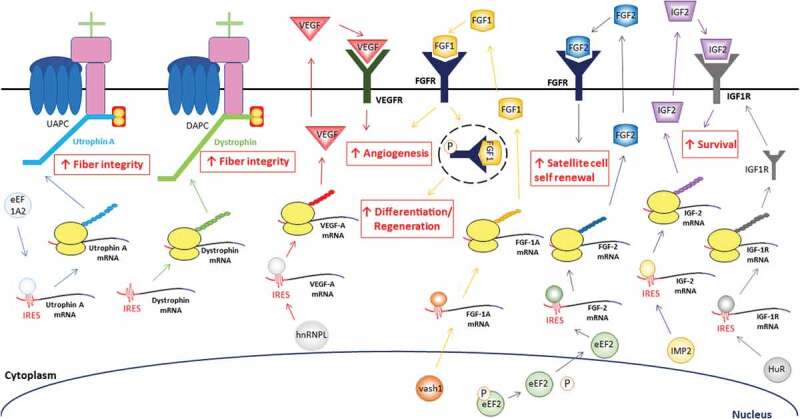
Potential DMD-relevant mRNAs that contain IRESs for the treatment of DMD. eEF1A2 acts as an ITAF, driving utrophin A IRES-mediated translation. This results in increased levels of utrophin A, rescue of the utrophin-glycoprotein complex (UAPC) at the sarcolemma of muscle fibres and improvement of fibre integrity. Dystrophin’s IRES can drive its translation in dystrophic muscles and also provides stability at the sarcolemma with the DAPC. VEGF-A expression has been shown to be regulated by IRES-mediated translation via the ITAF hnRNP L [216]. VEGF-A interacts with the VEGF receptor (VEGFR) and induces angiogenesis, cell proliferation and survival [214]. FGF-1A’s IRES is activated under stress conditions where vasohibin1 (VASH1) expression increase in the nucleus, translocate to the cytoplasm and bind the IRES of FGF-1A mRNA to recruit the transcript to the ribosome [219]. FGF-1A protein can either bind an external FGF receptor (FGFR) to promote angiogenesis or an intracellular FGFR to induce cell differentiation and regeneration [225,228]. FGF-2 IRES activity is sensitive to oxidative stress. This promotes translocation of nuclear eEF2 to the cytoplasm where it acts as an ITAF and drives FGF-2 IRES-mediated translation. FGF-2 protein binds FGFR, induces satellite cell self renewal by maintaining the pool of muscle precursor cells and inhibits myogenesis [191]. The IRES of IGF-2 has been shown to be activated via ITAF IMP2 [236]. The IGF-2 protein weakly binds the IGF-1 receptor (IGF-1 R) and promotes cell survival by modulating the regulatory networks involved in programmed cell death [47]. This is further stimulated by increasing the number of available IGF-1 R which in part is also regulated through IRES-mediated translation via HuR
Acknowledgments
The authors are grateful to many collaborators and previous trainees within the Jasmin lab, the University of Ottawa Eric Poulin Centre for Neuromuscular Disease, and the Faculty of Medicine RNA Research Group. We are especially indebted to Drs. Martin Holcik and Pedro Miura for their important contributions over the years as the work on utrophin A translation was being pursued in the Jasmin lab. Work in the Jasmin lab has been consistently supported over the years by (in alphabetical order), the Association Française contre les Myopathies, the Canadian Institutes of Health Research, the Canadian Space Agency, the Cancer Research Society, Jesse’s Journey, and the Muscular Dystrophy Association (USA).
Funding Statement
This work was supported by the Association Française contre les Myopathies; Canadian Institutes of Health Research; Canadian Space Agency; Muscular Dystrophy Association; the Cancer Research Society; Jesse’s Journey.
Disclosure statement
No potential conflicts of interest were disclosed.
References
- [1].Pelletier J, Sonenberg N.. Internal initiation of translation of eukaryotic mRNA directed by a sequence derived from poliovirus RNA. Nature. 1988;334:320–325. [DOI] [PubMed] [Google Scholar]
- [2].Jang SK, Kräusslich HG, Nicklin MJ, et al. A segment of the 5ʹ nontranslated region of encephalomyocarditis virus RNA directs internal entry of ribosomes during in vitro translation. J Virol. 1988;62:2636–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Fitzgerald KD, Semler BL. Bridging IRES elements in mRNAs to the eukaryotic translation apparatus. Biochim Biophys Acta. 2009;1789:518–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Komar AA, Hatzoglou M. Cellular IRES-mediated translation: the war of ITAFs in pathophysiological states. Cell Cycle. 2011;10:229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Martinez-Salas E, Francisco-Velilla R, Fernandez-Chamorro J, et al. Insights into structural and mechanistic features of viral ires elements. Front Microbiol. 2018;8:2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Godet A-C, David F, Hantelys F, et al. IRES trans-acting factors, key actors of the stress response. Int J Mol Sci. 2019;20:924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Weingarten-Gabbay S, Elias-Kirma S, Nir R, et al. Systematic discovery of cap-independent translation sequences in human and viral genomes. Science. 2016;351:240-240. [DOI] [PubMed] [Google Scholar]
- [8].Komar AA, Hatzoglou M. Internal ribosome entry sites in cellular mRNAs: mystery of their existence. J Biol Chem. 2005;280:23425–23428. [DOI] [PubMed] [Google Scholar]
- [9].Berry KE, Peng B, Koditek D, et al. Optimized high-throughput screen for hepatitis C virus translation inhibitors. J Biomol Screen. 2011;16:211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Didiot M-C, Hewett J, Varin T, et al. Identification of cardiac glycoside molecules as inhibitors of c-Myc IRES-mediated translation. J Biomol Screen. 2013;18:407–419. [DOI] [PubMed] [Google Scholar]
- [11].Vaklavas C, Meng Z, Choi H, et al. Small molecule inhibitors of IRES-mediated translation. Cancer Biol Ther. 2015;16:1471–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Venkatesan A, Dasgupta A. Novel fluorescence-based screen to identify small synthetic internal ribosome entry site elements. Mol Cell Biol. 2001;21:2826–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zhou S, Rynearson KD, Ding K, et al. Screening for inhibitors of the hepatitis C virus internal ribosome entry site RNA. Bioorg Med Chem. 2013;21:6139–6144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Holcik M. Targeting translation for treatment of cancer - a novel role for IRES? Current Cancer Drug Targets. 2004;4:299–311. [DOI] [PubMed] [Google Scholar]
- [15].Holcik M. Internal ribosome entry site-mediated translation in neuronal protein synthesis. In: The oxford handbook of neuronal protein synthesis. Oxford University Press, New York, USA; Edited by Wayne S. Sossin 2018. p. 1–25. [Google Scholar]
- [16].Veo BL, Krushel LA. Secondary RNA structure and nucleotide specificity contribute to internal initiation mediated by the human tau 5ʹ leader. RNA Biol. 2012;9:1344–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Beaudoin ME, Poirel V-J, Krushel LA. Regulating amyloid precursor protein synthesis through an internal ribosomal entry site. Nucleic Acids Res. 2008;36:6835–6847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Koukouraki P, Doxakis E. Constitutive translation of human α-synuclein is mediated by the 5ʹ-untranslated region. Open Biol. 2016;6:160022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Pacheco A, Twiss JL. Localized IRES-dependent translation of ER chaperone protein mRNA in sensory axons. Plos One. 2012;7:e40788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Willis D, Li KW, Zheng J-Q, et al. Differential transport and local translation of cytoskeletal, injury-response, and neurodegeneration protein mRNAs in axons. J Neurosci. 2005;25:778–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Miura P, Thompson J, Chakkalakal JV, et al. The utrophin A 5ʹ-untranslated region confers internal ribosome entry site-mediated translational control during regeneration of skeletal muscle fibers. J Biol Chem. 2005;280:32997–33005. [DOI] [PubMed] [Google Scholar]
- [22].Miura P, Andrews M, Holcik M, et al. IRES-mediated translation of utrophin a is enhanced by glucocorticoid treatment in skeletal muscle cells. Plos One. 2008;3:e2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Miura P, Coriati A, Bélanger G, et al. The utrophin A 5′-UTR drives cap-independent translation exclusively in skeletal muscles of transgenic mice and interacts with eEF1A2. Hum Mol Genet. 2010;19:1211–1220. [DOI] [PubMed] [Google Scholar]
- [24].Wein N, Vulin A, Falzarano MS, et al. Translation from a DMD exon 5 IRES results in a functional dystrophin isoform that attenuates dystrophinopathy in humans and mice. Nat Med. 2014;20:992–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lehmkuhl EM, Zarnescu DC. Lost in translation: evidence for protein synthesis deficits in ALS/FTD and related neurodegenerative diseases. Adv Neurobiol. 2018;20:283–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Miura P, Chakkalakal JV, Boudreault L, et al. Pharmacological activation of PPARβ/δ stimulates utrophin A expression in skeletal muscle fibers and restores sarcolemmal integrity in mature mdx mice. Hum Mol Genet. 2009;18:4640–4649. [DOI] [PubMed] [Google Scholar]
- [27].Péladeau C, Adam N, Bronicki LM, et al. Identification of therapeutics that target eEF1A2 and upregulate utrophin A translation in dystrophic muscles. Nat Commun. 2020;11:1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Péladeau C, Ahmed A, Amirouche A, et al. Combinatorial therapeutic activation with heparin and AICAR stimulates additive effects on utrophin A expression in dystrophic muscles. Hum Mol Genet. 2016;25:24–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Péladeau C, Adam NJ, Jasmin BJ. Celecoxib treatment improves muscle function in mdx mice and increases utrophin A expression. Faseb J. 2018;32:5090–5103. [DOI] [PubMed] [Google Scholar]
- [30].Ljubicic V, Jasmin BJ. Metformin increases peroxisome proliferator-activated receptor γ Co-activator-1α and utrophin a expression in dystrophic skeletal muscle. Muscle Nerve. 2015;52:139–142. [DOI] [PubMed] [Google Scholar]
- [31].Ljubicic V, Burt M, Lunde JA, et al. Resveratrol induces expression of the slow, oxidative phenotype in mdx mouse muscle together with enhanced activity of the SIRT1-PGC-1α axis. Am J Physiol Cell Physiol. 2014;307:C66–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ljubicic V, Miura P, Burt M, et al. Chronic AMPK activation evokes the slow, oxidative myogenic program and triggers beneficial adaptations in mdx mouse skeletal muscle. Hum Mol Genet. 2011;20:3478–3493. [DOI] [PubMed] [Google Scholar]
- [33].Amirouche A, Tadesse H, Lunde JA, et al. Activation of p38 signaling increases utrophin A expression in skeletal muscle via the RNA-binding protein KSRP and inhibition of AU-rich element-mediated mRNA decay: implications for novel DMD therapeutics. Hum Mol Genet. 2013;22:3093–3111. [DOI] [PubMed] [Google Scholar]
- [34].Amirouche A, Tadesse H, Miura P, et al. Converging pathways involving microRNA-206 and the RNA-binding protein KSRP control post-transcriptionally utrophin A expression in skeletal muscle. Nucleic Acids Res. 2014;42:3982–3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Al-Rewashdy H, Ljubicic V, Lin W, et al. Utrophin A is essential in mediating the functional adaptations of mdx mouse muscle following chronic AMPK activation. Hum Mol Genet. 2015;24:1243–1255. [DOI] [PubMed] [Google Scholar]
- [36].Gramolini AO, Jasmin BJ. Expression of the utrophin gene during myogenic differentiation. Nucleic Acids Res. 1999;27:3603–3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gramolini AO, Bélanger G, Thompson JM, et al. Increased expression of utrophin in a slow vs. a fast muscle involves posttranscriptional events. Am J Physiol Cell Physiol. 2001;281:C1300–9. [DOI] [PubMed] [Google Scholar]
- [38].Gramolini AO, Angus LM, Schaeffer L, et al. Induction of utrophin gene expression by heregulin in skeletal muscle cells: role of the N-box motif and GA binding protein. Proc Natl Acad Sci USA. 1999;96:3223–3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Chakkalakal JV, Stocksley MA, Harrison M-A, et al. Expression of utrophin A mRNA correlates with the oxidative capacity of skeletal muscle fiber types and is regulated by calcineurin/NFAT signaling. Proc Natl Acad Sci U S A. 2003;100:7791–7796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Chakkalakal JV, Michel SA, Chin ER, et al. Targeted inhibition of Ca2+/calmodulin signaling exacerbates the dystrophic phenotype in mdx mouse muscle. Hum Mol Genet. 2006;15:1423–1435. [DOI] [PubMed] [Google Scholar]
- [41].Chakkalakal JV, Harrison M-A, Carbonetto S, et al. Stimulation of calcineurin signaling attenuates the dystrophic pathology in mdx mice. Hum Mol Genet. 2004;13:379–388. [DOI] [PubMed] [Google Scholar]
- [42].Davies KE, Chamberlain JS. Surrogate gene therapy for muscular dystrophy. Nat Med. 2019;25:1473–1474. [DOI] [PubMed] [Google Scholar]
- [43].Guiraud S, Roblin D, Kay DE. The potential of utrophin modulators for the treatment of Duchenne muscular dystrophy. Expert Opin Orphan Drugs. 2018;6:179–192. [Google Scholar]
- [44].Liew WKM, Kang PB. Recent developments in the treatment of Duchenne muscular dystrophy and spinal muscular atrophy. Ther Adv Neurol Disord. 2013;6:147–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Scully MA, Pandya S, Moxley RT. Review of phase II and phase III clinical trials for Duchenne muscular dystrophy. Expert Opin Orphan Drugs. 2013;1:33–46. [Google Scholar]
- [46].Forcina L, Miano C, Scicchitano BM, et al. Signals from the Niche: insights into the role of IGF-1 and IL-6 in modulating skeletal muscle fibrosis. Cells. 2019;8:232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Smith J, Goldsmith C, Ward A, et al. IGF-II ameliorates the dystrophic phenotype and coordinately down-regulates programmed cell death. Cell Death Differ. 2000;7:1109–1118. [DOI] [PubMed] [Google Scholar]
- [48].Shatsky IN, Terenin IM, Smirnova VV, et al. Cap-independent translation: what’s in a name? Trends Biochem Sci. 2018;43:882–895. [DOI] [PubMed] [Google Scholar]
- [49].Yang Y, Wang Z. IRES-mediated cap-independent translation, a path leading to hidden proteome. J Mol Cell Biol. 2019;11:911–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Shirokikh NE, Preiss T. Translation initiation by cap-dependent ribosome recruitment: recent insights and open questions. WIREs RNA. 2018;9:e1473. [DOI] [PubMed] [Google Scholar]
- [51].Wells SE, Hillner PE, Vale RD, et al. Circularization of mRNA by eukaryotic translation initiation factors. Mol Cell. 1998;2:135–140. [DOI] [PubMed] [Google Scholar]
- [52].Amrani N, Ghosh S, Mangus DA, et al. Translation factors promote formation of two states of the closed loop mRNP. Nature. 2008;453:1276–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Safaee N, Kozlov G, Noronha AM, et al. Interdomain allostery promotes assembly of the Poly(A) mRNA complex with PABP and eIF4G. Mol Cell. 2012;48:375–386. [DOI] [PubMed] [Google Scholar]
- [54].Bogorad AM, Lin KY, Marintchev A. Novel mechanisms of eIF2B action and regulation by eIF2α phosphorylation. Nucleic Acids Res. 2017;45:11962–11979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Gradi A, Imataka H, Svitkin YV, et al. A novel functional human eukaryotic translation initiation factor 4G. Mol Cell Biol. 1998;18:334–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Müller D, Lasfargues C, El Khawand S, et al. 4E-BP restrains eIF4E phosphorylation. Translation (Austin). 2013;1:e25819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Svitkin YV, Gradi A, Imataka H, et al. Eukaryotic initiation factor 4GII (eIF4GII), but not eIF4GI, cleavage correlates with inhibition of host cell protein synthesis after human rhinovirus infection. J Virol. 1999;73:3467–3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Graber TE, Holcik M. Cap-independent regulation of gene expression in apoptosis. Mol Biosyst. 2007;3:825–834. [DOI] [PubMed] [Google Scholar]
- [59].Leppek K, Das R, Barna M. Functional 5′ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat Rev Mol Cell Biol. 2018;19:158–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Pickering BM, Mitchell SA, Spriggs KA, et al. Bag-1 internal ribosome entry segment activity is promoted by structural changes mediated by poly(rC) binding protein 1 and recruitment of polypyrimidine tract binding protein 1. Mol Cell Biol. 2004;24:5595–5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Cobbold LC, Spriggs KA, Haines SJ, et al. Identification of internal ribosome entry segment (IRES)-trans-acting factors for the Myc family of IRESs. Mol Cell Biol. 2008;28:40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Hunt SL, Jackson RJ. Polypyrimidine-tract binding protein (PTB) is necessary, but not sufficient, for efficient internal initiation of translation of human rhinovirus-2 RNA. RNA. 1999;5:344–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Hunt SL, Hsuan JJ, Totty N, et al. unr, a cellular cytoplasmic RNA-binding protein with five cold-shock domains, is required for internal initiation of translation of human rhinovirus RNA. Genes Dev. 1999;13:437–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Pilipenko EV, Pestova TV, Kolupaeva VG, et al. A cell cycle-dependent protein serves as a template-specific translation initiation factor. Genes Dev. 2000;14:2028–2045. [PMC free article] [PubMed] [Google Scholar]
- [65].Bushell M, Stoneley M, Kong YW, et al. Polypyrimidine tract binding protein regulates IRES-mediated gene expression during apoptosis. Mol Cell. 2006;23:401–412. [DOI] [PubMed] [Google Scholar]
- [66].Cornelis S, Tinton SA, Schepens B, et al. UNR translation can be driven by an IRES element that is negatively regulated by polypyrimidine tract binding protein. Nucleic Acids Res. 2005;33:3095–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Costa-Mattioli M, Svitkin Y, Sonenberg N. La autoantigen is necessary for optimal function of the poliovirus and hepatitis C virus internal ribosome entry site in vivo and in vitro. Mol Cell Biol. 2004;24:6861–6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Damiano F, Rochira A, Tocci R, et al. hnRNP A1 mediates the activation of the IRES-dependent SREBP-1a mRNA translation in response to endoplasmic reticulum stress. Biochem J. 2013;449:543–553. [DOI] [PubMed] [Google Scholar]
- [69].Dave P, George B, Sharma DK, et al. Polypyrimidine tract-binding protein (PTB) and PTB-associated splicing factor in CVB3 infection: an ITAF for an ITAF. Nucleic Acids Res. 2017;45:9068–9084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Jo OD, Martin J, Bernath A, et al. Heterogeneous nuclear ribonucleoprotein A1 regulates cyclin D1 and c-myc internal ribosome entry site function through Akt signaling. J Biol Chem. 2008;283:23274–23287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Kim J-K, Kim I, Choi K, et al. Poly(rC) binding protein 2 acts as a negative regulator of IRES-mediated translation of Hr mRNA. Exp Mol Med. 2018;50:e441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Shi Y, Yang Y, Hoang B, et al. Therapeutic potential of targeting IRES-dependent c-myc translation in multiple myeloma cells during ER stress. Oncogene. 2016;35:1015–1024. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [73].Tinton SA, Schepens B, Bruynooghe Y, et al. Regulation of the cell-cycle-dependent internal ribosome entry site of the PITSLRE protein kinase: roles of Unr (upstream of N-ras) protein and phosphorylated translation initiation factor eIF-2alpha. Biochem J. 2005;385:155–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Moldave K. Progress in nucleic acid research and molecular biology, Volume 72. Elsevier; Academic Press, San Diego, California, USA, 2002. [Google Scholar]
- [75].Plank T-DM, Whitehurst JT, Kieft JS. Cell type specificity and structural determinants of IRES activity from the 5′ leaders of different HIV-1 transcripts. Nucleic Acids Res. 2013;41:6698–6714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Lewis SM, Holcik M. For IRES trans-acting factors, it is all about location. Oncogene. 2008;27:1033–1035. [DOI] [PubMed] [Google Scholar]
- [77].Birnkrant DJ, Bushby K, Bann CM, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018;17:251–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Birnkrant DJ, Bushby K, Bann CM, et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol. 2018;17:347–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Buddhe S, Cripe L, Friedland-Little J, et al. Cardiac management of the patient with Duchenne muscular dystrophy. Pediatrics. 2018;142:S72–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Lo Mauro A, Aliverti A. Physiology of respiratory disturbances in muscular dystrophies. Breathe (Sheff). 2016;12:318–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Rosenberg AS, Puig M, Nagaraju K, et al. Immune-mediated pathology in Duchenne muscular dystrophy. Sci Transl Med. 2015;7:299rv4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Law ML, Cohen H, Martin AA, et al. Dysregulation of calcium handling in duchenne muscular dystrophy-associated dilated cardiomyopathy: mechanisms and experimental therapeutic strategies. J Clin Med. 2020;9:520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Mayer OH, Finkel RS, Rummey C, et al. Characterization of pulmonary function in Duchenne muscular dystrophy. Pediatr Pulmonol. 2015;50:487–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Ryder S, Leadley RM, Armstrong N, et al. The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: an evidence review. Orphanet J Rare Dis. 2017;12:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Guiraud S, Chen H, Burns DT, et al. Advances in genetic therapeutic strategies for Duchenne muscular dystrophy. Exp Physiol. 2015;100:1458–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Doorenweerd N, Mahfouz A, van Putten M, et al. Timing and localization of human dystrophin isoform expression provide insights into the cognitive phenotype of Duchenne muscular dystrophy. Sci Rep. 2017;7:12575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Feener CA, Koenig M, Kunkel LM. Alternative splicing of human dystrophin mRNA generates isoforms at the carboxy terminus. Nature. 1989;338:509–511. [DOI] [PubMed] [Google Scholar]
- [88].Tang J, Song X, Ji G, et al. A novel DMD splicing mutation found in a family responsible for X-linked dilated cardiomyopathy with hyper-CKemia. Medicine (Baltimore). 2018;97:e11074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Muntoni F, Wilson L, Marrosu G, et al. A mutation in the dystrophin gene selectively affecting dystrophin expression in the heart. J Clin Invest. 1995;96:693–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Boyce FM, Beggs AH, Feener C, et al. Dystrophin is transcribed in brain from a distant upstream promoter. Proc Natl Acad Sci U S A. 1991;88:1276–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Górecki DC, Monaco AP, Derry JM, et al. Expression of four alternative dystrophin transcripts in brain regions regulated by different promoters. Hum Mol Genet. 1992;1:505–510. [DOI] [PubMed] [Google Scholar]
- [92].Holder E, Maeda M, Bies RD. Expression and regulation of the dystrophin Purkinje promoter in human skeletal muscle, heart, and brain. Hum Genet. 1996;97:232–239. [DOI] [PubMed] [Google Scholar]
- [93].Flanigan KM, Dunn DM, von Niederhausern A, et al. Mutational spectrum of DMD mutations in dystrophinopathy patients: application of modern diagnostic techniques to a large cohort. Hum Mutat. 2009;30:1657–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Lee BL, Nam SH, Lee JH, et al. Genetic analysis of dystrophin gene for affected male and female carriers with Duchenne/Becker muscular dystrophy in Korea. J Korean Med Sci. 2012;27:274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Traverso M, Malnati M, Minetti C, et al. Multiplex real-time PCR for detection of deletions and duplications in dystrophin gene. Biochem Biophys Res Commun. 2006;339:145–150. [DOI] [PubMed] [Google Scholar]
- [96].Ankala A, da Silva C, Gualandi F, et al. A comprehensive genomic approach for neuromuscular diseases gives a high diagnostic yield. Ann Neurol. 2015;77:206–214. [DOI] [PubMed] [Google Scholar]
- [97].Bladen CL, Salgado D, Monges S, et al. The TREAT-NMD DMD global database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat. 2015;36:395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Bushby K, Finkel R, Birnkrant DJ, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010;9:77–93. [DOI] [PubMed] [Google Scholar]
- [99].Hoffman EP, Brown RH, Kunkel LM. Dystrophin: the protein product of the duchenne muscular dystrophy locus. Cell. 1987;51:919–928. [DOI] [PubMed] [Google Scholar]
- [100].Koenig M, Kunkel LM. Detailed analysis of the repeat domain of dystrophin reveals four potential hinge segments that may confer flexibility. J Biol Chem. 1990;265:4560–4566. [PubMed] [Google Scholar]
- [101].Ervasti JM, Ohlendieck K, Kahl SD, et al. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature. 1990;345:315–319. [DOI] [PubMed] [Google Scholar]
- [102].Omairi S, Hau K-L, Collins-Hooper H, et al. Regulation of the dystrophin-associated glycoprotein complex composition by the metabolic properties of muscle fibres. Sci Rep. 2019;9:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Crist C. Emerging new tools to study and treat muscle pathologies: genetics and molecular mechanisms underlying skeletal muscle development, regeneration, and disease. J Pathol. 2017;241:264–272. [DOI] [PubMed] [Google Scholar]
- [104].Danisovic L, Culenova M, Csobonyeiova M. Induced pluripotent stem cells for Duchenne muscular dystrophy modeling and therapy. Cells. 2018;7:253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Benedetti S, Hoshiya H, Tedesco FS. Repair or replace? Exploiting novel gene and cell therapy strategies for muscular dystrophies. Febs J. 2013;280:4263–4280. [DOI] [PubMed] [Google Scholar]
- [106].Gawlik KI. At the crossroads of clinical and preclinical research for muscular dystrophy—are we 919 closer to effective treatment for patients? Int J Mol Sci. 2018;19:1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Dai A, Baspinar O, Yeşilyurt A, et al. Efficacy of stem cell therapy in ambulatory and nonambulatory children with Duchenne muscular dystrophy - Phase I-II. Degener Neurol Neuromuscul Dis. 2018;8:63–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Amoasii L, Hildyard JCW, Li H, et al. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science. 2018;362:86–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Bengtsson NE, Hall JK, Odom GL, et al. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat Commun. 2017;8:14454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Duchêne BL, Cherif K, Iyombe-Engembe J-P, et al. CRISPR-induced deletion with SaCas9 restores dystrophin expression in dystrophic models in vitro and in vivo. Mol Ther. 2018;26:2604–2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Hakim CH, Wasala NB, Nelson CE, et al. AAV CRISPR editing rescues cardiac and muscle function for 18 months in dystrophic mice. JCI Insight. 2018;3:e124297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Ifuku M, Iwabuchi KA, Tanaka M, et al. Restoration of dystrophin protein expression by exon skipping utilizing CRISPR-Cas9 in myoblasts derived from DMD patient iPS cells. Methods Mol Biol. 2018;1828:191–217. [DOI] [PubMed] [Google Scholar]
- [113].Long C, McAnally JR, Shelton JM, et al. Prevention of muscular dystrophy in mice by CRISPR/Cas9–mediated editing of germline DNA. Science. 2014;345:1184–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Nelson CE, Hakim CH, Ousterout DG, et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science. 2016;351:403–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Xu L, Gao Y, Lau YS, et al. Adeno-associated virus-mediated delivery of CRISPR for cardiac gene editing in mice. J Vis Exp. 2018;138:57560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Kodippili K, Hakim CH, Pan X, et al. Dual AAV gene therapy for Duchenne muscular dystrophy with a 7-kb mini-dystrophin gene in the canine model. Hum Gene Ther. 2018;29:299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Wang Z, Storb R, Halbert CL, et al. Successful regional delivery and long-term expression of a dystrophin gene in canine muscular dystrophy: a preclinical model for human therapies. Mol Ther. 2012;20:1501–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Wasala NB, Shin J-H, Lai Y, et al. Cardiac-specific expression of ΔH2-R15 mini-dystrophin normalized all electrocardiogram abnormalities and the end-diastolic volume in a 23-month-old mouse model of duchenne dilated cardiomyopathy. Hum Gene Ther. 2018;29:737–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Lu Q-L, Cirak S, Partridge T. What can we learn from clinical trials of exon skipping for DMD? Mol Ther Nucleic Acids. 2014;3:e152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Goyenvalle A, Griffith G, Babbs A, et al. Functional correction in mouse models of muscular dystrophy using exon-skipping tricyclo-DNA oligomers. Nat Med. 2015;21:270–275. [DOI] [PubMed] [Google Scholar]
- [121].Irwin AN, Herink MC. Eteplirsen for the treatment of duchenne muscular dystrophy: quality of evidence concerns-an alternative viewpoint. Pharmacotherapy. 2017;37:e109–11. [DOI] [PubMed] [Google Scholar]
- [122].Niks EH, Aartsma-Rus A. Exon skipping: a first in class strategy for Duchenne muscular dystrophy. Expert Opin Biol Ther. 2017;17:225–236. [DOI] [PubMed] [Google Scholar]
- [123].Charleston JS, Schnell FJ, Dworzak J, et al. Eteplirsen treatment for Duchenne muscular dystrophy: exon skipping and dystrophin production. Neurology. 2018;91:637–637. [DOI] [PubMed] [Google Scholar]
- [124].Baradaran-Heravi A, Balgi AD, Zimmerman C, et al. Novel small molecules potentiate premature termination codon readthrough by aminoglycosides. Nucleic Acids Res. 2016;44:6583–6598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].McDonald CM, Campbell C, Torricelli RE, et al. Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390:1489–1498. [DOI] [PubMed] [Google Scholar]
- [126].Dabrowski M, Bukowy-Bieryllo Z, Zietkiewicz E. Advances in therapeutic use of a drug-stimulated translational readthrough of premature termination codons. Mol Med. 2018;24:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Klamut HJ, Gangopadhyay SB, Worton RG, et al. Molecular and functional analysis of the muscle-specific promoter region of the Duchenne muscular dystrophy gene. Mol Cell Biol. 1990;10:193–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Nudel U, Zuk D, Einat P, et al. Duchenne muscular dystrophy gene product is not identical in muscle and brain. Nature. 1989;337:76–78. [DOI] [PubMed] [Google Scholar]
- [129].Sadoulet-Puccio HM, Kunkel LM. Dystrophin and its isoforms. Brain Pathol. 1996;6:25–35. [DOI] [PubMed] [Google Scholar]
- [130].Blake DJ, Schofield JN, Zuellig RA, et al. G-utrophin, the autosomal homologue of dystrophin Dp116, is expressed in sensory ganglia and brain. Proc Natl Acad Sci U S A. 1995;92:3697–3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Burton EA, Tinsley JM, Holzfeind PJ, et al. A second promoter provides an alternative target for therapeutic up-regulation of utrophin in Duchenne muscular dystrophy. Proc Natl Acad Sci U S A. 1999;96:14025–14030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Weir AP, Burton EA, Harrod G, et al. A- and B-utrophin have different expression patterns and are differentially up-regulated in mdx muscle. J Biol Chem. 2002;277:45285–45290. [DOI] [PubMed] [Google Scholar]
- [133].Baby SM, Bogdanovich S, Willmann G, et al. Differential expression of utrophin-A and -B promoters in the central nervous system (CNS) of normal and dystrophic mdx mice. Brain Pathol. 2010;20:323–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Dennis CL, Tinsley JM, Deconinck AE, et al. Molecular and functional analysis of the utrophin promoter. Nucleic Acids Res. 1996;24:1646–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Nguyen TM, Ellis JM, Love DR, et al. Localization of the DMDL gene-encoded dystrophin-related protein using a panel of nineteen monoclonal antibodies: presence at neuromuscular junctions, in the sarcolemma of dystrophic skeletal muscle, in vascular and other smooth muscles, and in proliferating brain cell lines. J Cell Biol. 1991;115:1695–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Wilson J, Putt W, Jimenez C, et al. Up71 and up140, two novel transcripts of utrophin that are homologues of short forms of dystrophin. Hum Mol Genet. 1999;8:1271–1278. [DOI] [PubMed] [Google Scholar]
- [137].Krag TOB, Bogdanovich S, Jensen CJ, et al. Heregulin ameliorates the dystrophic phenotype in mdx mice. Proc Natl Acad Sci U S A. 2004;101:13856–13860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Moorwood C, Lozynska O, Suri N, et al. Drug discovery for duchenne muscular dystrophy via utrophin promoter activation screening. Plos One. 2011;6:e26169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Tinsley J, Deconinck N, Fisher R, et al. Expression of full-length utrophin prevents muscular dystrophy in mdx mice. Nat Med. 1998;4:1441–1444. [DOI] [PubMed] [Google Scholar]
- [140].Tinsley JM, Potter AC, Phelps SR, et al. Amelioration of the dystrophic phenotype of mdx mice using a truncated utrophin transgene. Nature. 1996;384:349–353. [DOI] [PubMed] [Google Scholar]
- [141].Voisin V, Sébrié C, Matecki S, et al. De la Porte S. L-arginine improves dystrophic phenotype in mdx mice. Neurobiol Dis. 2005;20:123–130. [DOI] [PubMed] [Google Scholar]
- [142].Pearce M, Blake DJ, Tinsley JM, et al. The utrophin and dystrophin genes share similarities in genomic structure. Hum Mol Genet. 1993;2:1765–1772. [DOI] [PubMed] [Google Scholar]
- [143].Pozzoli U, Sironi M, Cagliani R, et al. Comparative analysis of the human dystrophin and utrophin gene structures. Genetics. 2002;160:793–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Clerk A, Morris GE, Dubowitz V, et al. Dystrophin-related protein, utrophin, in normal and dystrophic human fetal skeletal muscle. Histochem J. 1993;25:554–561. [PubMed] [Google Scholar]
- [145].Lin S, Burgunder JM. Utrophin may be a precursor of dystrophin during skeletal muscle development. Brain Res Dev Brain Res. 2000;119:289–295. [DOI] [PubMed] [Google Scholar]
- [146].Helliwell TR, Man NT, Morris GE, et al. The dystrophin-related protein, utrophin, is expressed on the sarcolemma of regenerating human skeletal muscle fibres in dystrophies and inflammatory myopathies. Neuromuscul Disord. 1992;2:177–184. [DOI] [PubMed] [Google Scholar]
- [147].Karpati G, Carpenter S, Morris GE, et al. Localization and quantitation of the chromosome 6-encoded dystrophin-related protein in normal and pathological human muscle. J Neuropathol Exp Neurol. 1993;52:119–128. [DOI] [PubMed] [Google Scholar]
- [148].Khurana TS, Watkins SC, Chafey P, et al. Immunolocalization and developmental expression of dystrophin related protein in skeletal muscle. Neuromuscul Disord. 1991;1:185–194. [DOI] [PubMed] [Google Scholar]
- [149].Blake DJ, Love DR, Tinsley J, et al. Characterization of a 4.8kb transcript from the Duchenne muscular dystrophy locus expressed in Schwannoma cells. Hum Mol Genet. 1992;1:103–109. [DOI] [PubMed] [Google Scholar]
- [150].Love DR, Hill DF, Dickson G, et al. An autosomal transcript in skeletal muscle with homology to dystrophin. Nature. 1989;339:55–58. [DOI] [PubMed] [Google Scholar]
- [151].Tinsley JM, Blake DJ, Roche A, et al. Primary structure of dystrophin-related protein. Nature. 1992;360:591–593. [DOI] [PubMed] [Google Scholar]
- [152].Guiraud S, Edwards B, Babbs A, et al. The potential of utrophin and dystrophin combination therapies for Duchenne muscular dystrophy. Hum Mol Genet. 2019;28:2189–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Ishikawa-Sakurai M, Yoshida M, Imamura M, et al. ZZ domain is essentially required for the physiological binding of dystrophin and utrophin to β-dystroglycan. Hum Mol Genet. 2004;13:693–702. [DOI] [PubMed] [Google Scholar]
- [154].Kramarcy NR, Sealock R. Syntrophin isoforms at the neuromuscular junction: developmental time course and differential localization. Mol Cell Neurosci. 2000;15:262–274. [DOI] [PubMed] [Google Scholar]
- [155].Perkins KJ, Davies KE. The role of utrophin in the potential therapy of Duchenne muscular dystrophy. Neuromuscul Disord. 2002;12:S78–89. [DOI] [PubMed] [Google Scholar]
- [156].Peters MF, Sadoulet-Puccio HM, Mark Grady R, et al. Differential membrane localization and intermolecular associations of α-dystrobrevin isoforms in skeletal muscle. J Cell Biol. 1998;142:1269–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Winder SJ, Gibson TJ, Kendrick-Jones J. Dystrophin and utrophin: the missing links! FEBS Lett. 1995;369:27–33. [DOI] [PubMed] [Google Scholar]
- [158].Percival JM. nNOS regulation of skeletal muscle fatigue and exercise performance. Biophys Rev. 2011;3:209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Percival JM, Anderson KNE, Huang P, et al. Golgi and sarcolemmal neuronal NOS differentially regulate contraction-induced fatigue and vasoconstriction in exercising mouse skeletal muscle. J Clin Invest. 2010;120:816–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Stamler JS, Meer G. Physiology of nitric oxide in skeletal muscle. Physiol Rev. 2001;81:209–237. [DOI] [PubMed] [Google Scholar]
- [161].Lai Y, Thomas GD, Yue Y, et al. Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J Clin Invest. 2009;119:624–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Li D, Bareja A, Judge L, et al. Sarcolemmal nNOS anchoring reveals a qualitative difference between dystrophin and utrophin. J Cell Sci. 2010;123:2008–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Belanto JJ, Mader TL, Eckhoff MD, et al. Microtubule binding distinguishes dystrophin from utrophin. Proc Natl Acad Sci USA. 2014;111:5723–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Prins KW, Humston JL, Mehta A, et al. Dystrophin is a microtubule-associated protein. J Cell Biol. 2009;186:363–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Randazzo D, Giacomello E, Lorenzini S, et al. Obscurin is required for ankyrinB-dependent dystrophin localization and sarcolemma integrity. J Cell Biol. 2013;200:523–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Arechavala-Gomeza V, Kinali M, Feng L, et al. Immunohistological intensity measurements as a tool to assess sarcolemma-associated protein expression. Neuropathol Appl Neurobiol. 2010;36:265–274. [DOI] [PubMed] [Google Scholar]
- [167].Schofield JN, Górecki DC, Blake DJ, et al. Dystroglycan mRNA expression during normal and mdx mouse embryogenesis: a comparison with utrophin and the apo-dystrophins. Dev Dyn. 1995;204:178–185. [DOI] [PubMed] [Google Scholar]
- [168].Fisher R, Tinsley JM, Phelps SR, et al. Non-toxic ubiquitous over-expression of utrophin in the mdx mouse. Neuromuscul Disord. 2001;11:713–721. [DOI] [PubMed] [Google Scholar]
- [169].Kennedy TL, Guiraud S, Edwards B, et al. Micro-utrophin improves cardiac and skeletal muscle function of severely affected D2/mdx mice. Mol Ther Methods Clin Dev. 2018;11:92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Cerletti M, Negri T, Cozzi F, et al. Dystrophic phenotype of canine X-linked muscular dystrophy is mitigated by adenovirus-mediated utrophin gene transfer. Gene Ther. 2003;10:750–757. [DOI] [PubMed] [Google Scholar]
- [171].Amann KJ, Guo AW-X, Ervasti JM. Utrophin lacks the rod domain actin binding activity of dystrophin. J Biol Chem. 1999;274:35375–35380. [DOI] [PubMed] [Google Scholar]
- [172].Lai Y, Zhao J, Yue Y, et al. α2 and α3 helices of dystrophin R16 and R17 frame a microdomain in the α1 helix of dystrophin R17 for neuronal NOS binding. Proc Natl Acad Sci U S A. 2013;110:525–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Galvagni F, Capo S, Oliviero S. Sp1 and Sp3 physically interact and co-operate with GABP for the activation of the utrophin promoter. J Mol Biol. 2001;306:985–996. [DOI] [PubMed] [Google Scholar]
- [174].Galvagni F, Cantini M, Oliviero S. The utrophin gene is transcriptionally up-regulated in regenerating muscle. J Biol Chem. 2002;277:19106–19113. [DOI] [PubMed] [Google Scholar]
- [175].Gramolini AO, Dennis CL, Tinsley JM, et al. Local transcriptional control of utrophin expression at the neuromuscular synapse. J Biol Chem. 1997;272:8117–8120. [DOI] [PubMed] [Google Scholar]
- [176].Khurana TS, Rosmarin AG, Shang J, et al. Activation of utrophin promoter by heregulin via the ets-related transcription factor complex GA-binding protein alpha/beta. Mol Biol Cell. 1999;10:2075–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Perkins KJ, Burton EA, Davies KE. The role of basal and myogenic factors in the transcriptional activation of utrophin promoter A: implications for therapeutic up-regulation in Duchenne muscular dystrophy. Nucleic Acids Res. 2001;29:4843–4850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Pisani C, Strimpakos G, Gabanella F, et al. Utrophin up-regulation by artificial transcription factors induces muscle rescue and impacts the neuromuscular junction in mdx mice. Biochim Biophys Acta (BBA) - Mol Basis Dis. 2018;1864:1172–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Bonet-Kerrache A, Fortier M, Comunale F, et al. The GTPase RhoA increases utrophin expression and stability, as well as its localization at the plasma membrane. Biochem J. 2005;391:261–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Mishra MK, Loro E, Sengupta K, et al. Functional improvement of dystrophic muscle by repression of utrophin: let-7c interaction. PLoS One. 2017;12:e0182676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Basu U, Lozynska O, Moorwood C, et al. Translational regulation of utrophin by miRNAs. Plos One. 2011;6:e29376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Loro E, Sengupta K, Bogdanovich S, et al. High-throughput identification of post-transcriptional utrophin up-regulators for Duchenne muscle dystrophy (DMD) therapy. Sci Rep. 2020;10:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Pasquini F, Guerin C, Blake D, et al. The effect of glucocorticoids on the accumulation of utrophin by cultured normal and dystrophic human skeletal muscle satellite cells. Neuromuscul Disord. 1995;5:105–114. [DOI] [PubMed] [Google Scholar]
- [184].Roma J, Munell F, Fargas A, et al. Evolution of pathological changes in the gastrocnemius of the mdx mice correlate with utrophin and beta-dystroglycan expression. Acta Neuropathol. 2004;108:443–452. [DOI] [PubMed] [Google Scholar]
- [185].Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Ghosh T, Basu U. Cis-acting sequence elements and upstream open reading frame in mouse utrophin-A 5ʹ-UTR repress cap-dependent translation. Plos One. 2015;10:e0134809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Manzur AY, Kuntzer T, Pike M, et al. Glucocorticoid corticosteroids for Duchenne muscular dystrophy. Cochrane Database Syst Rev. 2008;1:CD003725. [DOI] [PubMed] [Google Scholar]
- [188].Guglieri M, Bushby K, McDermott MP, et al. Developing standardized corticosteroid treatment for Duchenne muscular dystrophy. Contemp Clin Trials. 2017;58:34–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Wehling-Henricks M, Lee JJ, Tidball JG. Prednisolone decreases cellular adhesion molecules required for inflammatory cell infiltration in dystrophin-deficient skeletal muscle. Neuromuscul Disord. 2004;14:483–490. [DOI] [PubMed] [Google Scholar]
- [190].Kissel JT, Burrow KL, Rammohan KW, et al. Mononuclear cell analysis of muscle biopsies in prednisone-treated and untreated Duchenne muscular dystrophy. CIDD study group. Neurology. 1991;41:667–672. [DOI] [PubMed] [Google Scholar]
- [191].Argüelles S, Camandola S, Cutler RG, et al. Elongation factor 2 diphthamide is critical for translation of two IRES-dependent protein targets, XIAP and FGF2, under oxidative stress conditions. Free Radic Biol Med. 2014;67:131–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Kahns S, Lund A, Kristensen P, et al. The elongation factor 1 A-2 isoform from rabbit: cloning of the cDNA and characterization of the protein. Nucleic Acids Res. 1998;26:1884–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Ruest L-B, Marcotte R, Wang E. Peptide elongation factor eEF1A-2/S1 expression in cultured differentiated myotubes and its protective effect against caspase-3-mediated apoptosis. J Biol Chem. 2002;277:5418–5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Lopez-Valenzuela JA, Gibbon BC, Hughes PA, et al. eEF1A isoforms change in abundance and actin-binding activity during maize endosperm development. Plant Physiol. 2003;133:1285–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Mateyak MK, Kinzy TG. eEF1A: thinking outside the ribosome. J Biol Chem. 2010;285:21209–21213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Knudsen SM, Frydenberg J, Clark BF, et al. Tissue-dependent variation in the expression of elongation factor-1 alpha isoforms: isolation and characterisation of a cDNA encoding a novel variant of human elongation-factor 1 alpha. Eur J Biochem/FEBS. 1993;215:549–554. [DOI] [PubMed] [Google Scholar]
- [197].Lee S, Francoeur A M, Liu S, Wang E.. Tissue-specific expression in mammalian brain, heart, and muscle of S1, a member of the elongation factor-1 alpha gene family. J Biol Chem. 1992;267:24064–24068. [PubMed] [Google Scholar]
- [198].Briguet A, Courdier-Fruh I, Foster M, et al. Histological parameters for the quantitative assessment of muscular dystrophy in the mdx-mouse. Neuromuscul Disord. 2004;14:675–682. [DOI] [PubMed] [Google Scholar]
- [199].Flanigan KM, Dunn DM, von Niederhausern A, et al. DMD Trp3X nonsense mutation associated with a founder effect in North American families with mild Becker muscular dystrophy. Neuromuscul Disord. 2009;19:743–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Ikeda T, Fujinaka H, Goto K, et al. Becker muscular dystrophy caused by exon 2-truncating mutation of DMD. Hum Genome Var. 2019;6:1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Baird SD, Turcotte M, Korneluk RG, et al. Searching for IRES. RNA. 2006;12:1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Pawlikowski B, Vogler TO, Gadek K, et al. Regulation of skeletal muscle stem cells by fibroblast growth factors. Dev Dyn. 2017;246:359–367. [DOI] [PubMed] [Google Scholar]
- [203].Maddaluno L, Urwyler C, Werner S. Fibroblast growth factors: key players in regeneration and tissue repair. Development. 2017;144:4047–4060. [DOI] [PubMed] [Google Scholar]
- [204].Conte C, Ainaoui N, Delluc-Clavières A, et al. Fibroblast growth factor 1 induced during myogenesis by a transcription–translation coupling mechanism. Nucleic Acids Res. 2009;37:5267–5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Oliver L, Raulais D, Vigny M. Acidic fibroblast growth factor (aFGF) in developing normal and dystrophic (mdx) mouse muscles. Distribution in degenerating and regenerating mdx myofibres. Growth Factors. 1992;7:97–106. [DOI] [PubMed] [Google Scholar]
- [206].Neuhaus P, Oustanina S, Loch T, et al. Reduced mobility of fibroblast growth factor (FGF)-deficient myoblasts might contribute to dystrophic changes in the musculature of FGF2/FGF6/mdx triple-mutant mice. Mol Cell Biol. 2003;23:6037–6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Mendell JR, Engel WK, Derrer EC. Duchenne muscular dystrophy: functional ischemia reproduces its characteristic lesions. Science. 1971;172:1143–1145. [DOI] [PubMed] [Google Scholar]
- [208].Mendell JR, Engel WK, Derrer EC. Increased plasma enzyme concentrations in rats with functional ischaemia of muscle provide a possible model of Duchenne muscular dystrophy. Nature. 1972;239:522–524. [DOI] [PubMed] [Google Scholar]
- [209].Hathaway PW, Engel WK, Zellweger H. Experimental myopathy after microarterial embolization; comparison with childhood x-linked pseudohypertrophic muscular dystrophy. Arch Neurol. 1970;22:365–378. [DOI] [PubMed] [Google Scholar]
- [210].Huez I, Créancier L, Audigier S, et al. Two independent internal ribosome entry sites are involved in translation initiation of vascular endothelial growth factor mRNA. Mol Cell Biol. 1998;18:6178–6190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Akiri G, Nahari D, Finkelstein Y, et al. Regulation of vascular endothelial growth factor (VEGF) expression is mediated by internal initiation of translation and alternative initiation of transcription. Oncogene. 1998;17:227–236. [DOI] [PubMed] [Google Scholar]
- [212].Miller DL, Dibbens JA, Damert A, et al. The vascular endothelial growth factor mRNA contains an internal ribosome entry site. FEBS Lett. 1998;434:417–420. [DOI] [PubMed] [Google Scholar]
- [213].Stéphanie B, Leonel P-L, Amandine B, et al. Translational induction of VEGF internal ribosome entry site elements during the early response to ischemic stress. Circ Res. 2007;100:305–308. [DOI] [PubMed] [Google Scholar]
- [214].Shibuya M. Vascular endothelial growth factor (VEGF) and its receptor (VEGFR) signaling in angiogenesis. Genes Cancer. 2011;2:1097–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Messina S, Mazzeo A, Bitto A, et al. VEGF overexpression via adeno-associated virus gene transfer promotes skeletal muscle regeneration and enhances muscle function in mdx mice. Faseb J. 2007;21:3737–3746. [DOI] [PubMed] [Google Scholar]
- [216].Ray PS, Jia J, Yao P, et al. A stress-responsive RNA switch regulates VEGFA expression. Nature. 2009;457:915–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Yablonka-Reuveni Z, Danoviz ME, Phelps M, et al. Myogenic-specific ablation of Fgfr1 impairs FGF2-mediated proliferation of satellite cells at the myofiber niche but does not abolish the capacity for muscle regeneration. Front Aging Neurosci. 2015;28:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Saera-Vila A, Kish PE, Kahana A. Fgf regulates dedifferentiation during skeletal muscle regeneration in adult zebrafish. Cell Signal. 2016;28:1196–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Hantelys F, Godet A-C, David F, et al. Vasohibin1, a new mouse cardiomyocyte IRES trans-acting factor that regulates translation in early hypoxia. eLife. 2019;8:e50094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Saito A, Higuchi I, Nakagawa M, et al. An overexpression of fibroblast growth factor (FGF) and FGF receptor 4 in a severe clinical phenotype of facioscapulohumeral muscular dystrophy. Muscle Nerve. 2000;23:490–497. [DOI] [PubMed] [Google Scholar]
- [221].Bugler B, Amalric F, Prats H. Alternative initiation of translation determines cytoplasmic or nuclear localization of basic fibroblast growth factor. Mol Cell Biol. 1991;11:573–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].Vagner S, Gensac MC, Maret A, et al. Alternative translation of human fibroblast growth factor 2 mRNA occurs by internal entry of ribosomes. Mol Cell Biol. 1995;15:35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Jump SS, Childs TE, Zwetsloot KA, et al. Fibroblast growth factor 2-stimulated proliferation is lower in muscle precursor cells from old rats. Exp Physiol. 2009;94:739–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Chakkalakal JV, Jones KM, Basson MA, et al. The aged niche disrupts muscle stem cell quiescence. Nature. 2012;490:355–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Anderson JE, Mitchell CM, McGeachie JK, et al. The time course of basic fibroblast growth factor expression in crush-injured skeletal muscles of SJL/J and BALB/c mice. Exp Cell Res. 1995;216:325–334. [DOI] [PubMed] [Google Scholar]
- [226].Rao N, Evans S, Stewart D, et al. Fibroblasts influence muscle progenitor differentiation and alignment in contact independent and dependent manners in organized co-culture devices. Biomed Microdevices. 2013;15:161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Hannon K, Kudla AJ, McAvoy MJ, et al. Differentially expressed fibroblast growth factors regulate skeletal muscle development through autocrine and paracrine mechanisms. J Cell Biol. 1996;132:1151–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].DiMario J, Buffinger N, Yamada S, et al. Fibroblast growth factor in the extracellular matrix of dystrophic (mdx) mouse muscle. Science. 1989;244:688–690. [DOI] [PubMed] [Google Scholar]
- [229].Conte C, Riant E, Toutain C, et al. FGF2 translationally induced by hypoxia is involved in negative and positive feedback loops with HIF-1α. PLoS One. 2008;3:e3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Adhikary S, Choudhary D, Tripathi AK, et al. FGF-2 targets sclerostin in bone and myostatin in skeletal muscle to mitigate the deleterious effects of glucocorticoid on musculoskeletal degradation. Life Sci. 2019;229:261–276. [DOI] [PubMed] [Google Scholar]
- [231].Bergman D, Halje M, Nordin M, et al. Insulin-like growth factor 2 in development and disease: a mini-review. Gerontology. 2013;59:240–249. [DOI] [PubMed] [Google Scholar]
- [232].Hughes A, Mohanasundaram D, Kireta S, et al. Insulin-like growth factor-II (IGF-II) prevents proinflammatory cytokine-induced apoptosis and significantly improves islet survival after transplantation. Transplantation J. 2013;95:671–678. [DOI] [PubMed] [Google Scholar]
- [233].Himes BE, Obraztsova K, Lian L, et al. Rapamycin-independent IGF2 expression in Tsc2-null mouse embryo fibroblasts and human lymphangioleiomyomatosis cells. Plos One. 2018;13:e0197105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Walsh JG, Cullen SP, Sheridan C, et al. Executioner caspase-3 and caspase-7 are functionally distinct proteases. Proc Natl Acad Sci USA. 2008;105:12815–12819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Pedersen SK, Christiansen J, Hansen TVO, et al. Human insulin-like growth factor II leader 2 mediates internal initiation of translation. Biochem J. 2002;363:37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [236].Dai N, Rapley J, Angel M, et al. mTOR phosphorylates IMP2 to promote IGF2 mRNA translation by internal ribosomal entry. Genes Dev. 2011;25:1159–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].Giraud S, Greco A, Brink M, et al. translation initiation of the insulin-like growth factor i receptor mrna is mediated by an internal ribosome entry site. J Biol Chem. 2001;276:5668–5675. [DOI] [PubMed] [Google Scholar]
- [238].Higashi Y, Holder K, Delafontaine P. Thiazolidinediones Up-regulate insulin-like growth factor-1 receptor via a peroxisome proliferator-activated receptor γ-independent pathway. J Biol Chem. 2010;285:36361–36368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [239].Meng Z, King PH, Nabors LB, et al. The ELAV RNA-stability factor HuR binds the 5′-untranslated region of the human IGF-IR transcript and differentially represses cap-dependent and IRES-mediated translation. Nucleic Acids Res. 2005;33:2962–2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [240].Figueroa A, Cuadrado A, Fan J, et al. Role of HuR in skeletal myogenesis through coordinate regulation of muscle differentiation genes. Mol Cell Biol. 2003;23:4991–5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [241].Janice Sánchez B, Tremblay A-MK, Leduc-Gaudet J-P, et al. Depletion of HuR in murine skeletal muscle enhances exercise endurance and prevents cancer-induced muscle atrophy. Nat Commun. 2019;10:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [242].Van Pelt DW, Hettinger ZR, Vanderklish PW. RNA-binding proteins: the next step in translating skeletal muscle adaptations? J Appl Physiol. 2019;127:654–660. [DOI] [PubMed] [Google Scholar]
- [243].Komar AA, Hatzoglou M. Exploring internal ribosome entry sites as therapeutic targets. Front Oncol. 2015;5:233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [244].Goel HL, Mercurio AM. VEGF targets the tumour cell. Nat Rev Cancer. 2013;13:871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [245].Kieran MW, Kalluri R, Cho Y-J. The VEGF pathway in cancer and disease: responses, resistance, and the path forward. Cold Spring Harb Perspect Med. 2012;2:a006593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [246].Anand M, Valente L, Carr-Schmid A, et al. Translation elongation factor 1 functions in the yeast Saccharomyces cerevisiae. Cold Spring Harb Symp Quant Biol. 2001;66:439–448. [DOI] [PubMed] [Google Scholar]
- [247].Kawamura M, Endo C, Sakurada A, et al. The prognostic significance of eukaryotic elongation factor 1 alpha-2 in non-small cell lung cancer. Anticancer Res. 2014;34:651–658. [PubMed] [Google Scholar]
- [248].Lee M-H, Surh Y-J. eEF1A2 as a putative oncogene. Ann N Y Acad Sci. 2009;1171:87–93. [DOI] [PubMed] [Google Scholar]
- [249].Pinke DE, Kalloger SE, Francetic T, et al. The prognostic significance of elongation factor eEF1A2 in ovarian cancer. Gynecol Oncol. 2008;108:561–568. [DOI] [PubMed] [Google Scholar]
- [250].Sun Y, Du C, Wang B, et al. Up-regulation of eEF1A2 promotes proliferation and inhibits apoptosis in prostate cancer. Biochem Biophys Res Commun. 2014;450:1–6. [DOI] [PubMed] [Google Scholar]
- [251].Veremieva M, Kapustian L, Khoruzhenko A, et al. Independent overexpression of the subunits of translation elongation factor complex eEF1H in human lung cancer. BMC Cancer. 2014;14:913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [252].Worst TS, Waldbillig F, Abdelhadi A, et al. The EEF1A2 gene expression as risk predictor in localized prostate cancer. BMC Urol. 2017;17:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [253].Korc M, Friesel RE. The role of fibroblast growth factors in tumor growth. Curr Cancer Drug Targets. 2009;9:639–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [254].Spriggs KA, Stoneley M, Bushell M, et al. Re-programming of translation following cell stress allows IRES-mediated translation to predominate. Biol Cell. 2008;100:27–38. [DOI] [PubMed] [Google Scholar]
- [255].Lambrechts D, Carmeliet P. VEGF at the neurovascular interface: therapeutic implications for motor neuron disease. Biochim Biophys Acta (BBA) - Mol Basis Dis. 2006;1762:1109–1121. [DOI] [PubMed] [Google Scholar]
- [256].Lambrechts D, Storkebaum E, Morimoto M, et al. VEGF is a modifier of amyotrophic lateral sclerosis in mice and humans and protects motoneurons against ischemic death. Nat Genet. 2003;34:383–394. [DOI] [PubMed] [Google Scholar]