

Abstract
Metabolic dysfunction and exhaustion in tumor-infiltrating T cells have been linked to ineffectual anti-tumor immunity and the failure of immune checkpoint inhibitor therapy. We report here that chronic stress plays a previously unrecognized role in regulating the state of T cells in the tumor microenvironment (TME). Using two mouse tumor models, we found that blocking chronic adrenergic stress signaling using the pan β-blocker propranolol or by using mice lacking the β2-adrenergic receptor (β2-AR), results in reduced tumor growth rates with significantly fewer infiltrating T cells that express markers of exhaustion, with a concomitant increase in progenitor exhausted T cells. We also report that blocking β-AR signaling in mice increases glycolysis and oxidative phosphorylation in tumor-infiltrating lymphocytes (TILs), which associated with increased expression of the costimulatory molecule CD28 and increased anti-tumor effector functions, including increased cytokine production. Using T cells from Nur77-GFP reporter mice to monitor T-cell activation, we observed that stress-induced β-AR signaling suppresses T-cell receptor (TCR) signaling. Together, these data suggest that chronic stress-induced adrenergic receptor signaling serves as a “checkpoint” of immune responses and contributes to immunosuppression in the TME by promoting T-cell metabolic dysfunction and exhaustion. These results also support the possibility that chronic stress, which unfortunately is increased in many cancer patients following their diagnoses, could be exerting a major negative influence on the outcome of therapies that depend upon the status of TILs and support the use of strategies to reduce stress or β-AR signaling in combination with immunotherapy.
Keywords: Adrenergic stress, Tumor immunology, Tumor microenvironment, T-cell exhaustion, T-cell activation, Metabolic reprogramming
Introduction
For decades, the relationships between stressful events and immunity have been studied by researchers in the field of psychoneuroimmunology (PNI). A new spotlight has now been placed on this field with more recent revelations that tumor growth itself may depend upon neurogenesis and sympathetic nervous system (SNS) activity (1–4). Nerves of the SNS have been found to infiltrate the tumor microenvironment (TME)(5–7), providing a direct conduit by which stress, nervous stimulation, and release of catecholamines, such as norepinephrine (NE), could influence the TME. Studies have also shown that sympathetic nerves innervate primary and secondary lymphoid organs (8), and there is evidence in non-oncological settings that β-AR signaling can directly suppress CD8+ T-cell activation and effector function (9) or can act indirectly through dendritic cells, reducing their ability to prime T cells (10). Overall, these studies generate concern related to how stress and the nervous system might be affecting the immunological balance within the TME; in particular, how does stress signaling influence the development and strength of anti-tumor immune responses and efficacy of immunotherapy?
This is important because many cancer patients experience an increase in chronic stress (11), which could influence the outcome of treatment, including immunotherapies. We have previously shown that adrenergic stress results in immunosuppression in graft-versus-host disease (GvHD) and cancer, both of which depend on a complex balance between immunostimulatory and immunosuppressive activities (12–14). Our previous observations show that mice housed at their standard (IACUC-mandated) housing temperature (ST, 22–23°C) experience mild, but chronic, adrenergic stress that is mediated by the SNS in response to the need for increased metabolic heat production (a process dependent on the adrenergic stress response and NE production)(15–17). Housing mice at thermoneutrality (TT, 30–31°C), blocking β2-adrenergic receptor (β-AR) signaling with β-blockers in mice housed under standard cool conditions, or using mice lacking β2-AR (14,15,18) provide very convenient and physiologically relevant opportunities to study the impact of adrenergic stress on various aspects of the immune system. We previously found that reducing thermal stress or blocking β-ARs in stressed mice enhances anti-tumor immune responses by reducing the suppressive function of myeloid-derived suppressor cells (MDSCs) and increasing the frequency and function of intra-tumoral CD8+ T cells (12,14,15). The improved tumor control is lost when CD8+ T cells are depleted. We also found that reducing cold stress or blocking β-AR signaling significantly improves the efficacy of immune checkpoint immunotherapy, chemotherapy, and radiation (15,17,19). Although these data suggest the potential for stress to inhibit anti-tumor immunity, the mechanisms involved are not known.
Our prior work showed that chronic adrenergic stress increases programmed cell death-1 (PD-1) expression on TILs(15). In a subsequent in vitro analysis, β-AR signaling was observed to suppress metabolic reprogramming during CD8+ T-cell activation (18). Here, we report that chronic stress in mice has the potent ability to shape both the metabolic profile and the status of exhaustion in T cells in vivo within the TME. Most prior research on T-cell exhaustion has focused on identifying the subsets of cells involved, their differentiation, the role of chronic T-cell receptor (TCR) stimulation, and the role of T-cell exhaustion in the overall response to checkpoint inhibitor therapies (20,21). In this study, we asked whether physiological chronic stress drives T-cell dysfunction or exhaustion, characterized by reduced effector function, limited cytokine secretion, and expression of inhibitory receptors such as PD-1 in the TME. We observed that blocking β-AR signaling in mice experiencing mild chronic stress reduced the percentage of exhausted T cells, while simultaneously increasing progenitor exhausted T cells, a condition known to result in better tumor control and response to anti-PD-1 therapy (22). We also found that blocking β-AR signaling increased the metabolic activity and function of TILs, whereas conversely, both in vitro and in vivo data showed that increased β-AR signaling decreased T-cell activation by inhibiting TCR signaling. Together these data provide mechanistic support for the idea that the chronic stress alters the immune contexture of tumors and suppresses the activity of TILs, most likely through local secretion of NEby nerve endings in the TME. These findings also suggest that chronic stress experienced by cancer patients (11,23,24) has the potential to limit response to therapies, particularly immunotherapies, that depend on anti-tumor immune responses.
Materials and methods
Mice
Female C57BL/6NCr (C57BL/6) and Balb/cAnNcr (BALB/c) were purchased from Charles River. Female β2-AR−/− mice on a BALB/c background were originally provided by Dr. David Farrar (University of Texas Southwestern Medical Center, Dallas, TX) and subsequently bred at Roswell Park Comprehensive Cancer Center (RPCCC). Female β2-AR−/− mice on a C57BL/6 background were made by CRISPR technique in RPCCC. Guide RNA was designed, validated, and synthesized by IVT for the Adbr2 gene in C57BL/6J background by the Washington University of St. Louis CRISPR/Cas9 core facility. Briefly, algorithms predicting guide cut sites in Adbr2 were analyzed for off-target effects as well as the double-stranded DNA break leading to a nonsense mediated decay of the DNA which resulted in indels and thus knockouts in repair via non-homologous end joining repair pathway (NHEJ). Guide RNA and Cas9 mRNA were synthesized and sent to the Roswell Park Cancer Institute’s Gene Targeting and Transgenic Shared Resource for injection. The guide plus Cas9 mRNA were injected into the pronuclei of C57BL/6J fertilized one-cell embryos (final concentrations were as follows: IVT gRNA+Cas9 mRNA = 5 ng/µL Cas9, 2.5 ng/µL gRNA). Guide sequence was directed at exon 1: 5’ TCTGGCGCTCGGCTTCCGTT NGG 3’. The injected embryos were transferred into pseudopregnant CD-1 foster mothers and pups were subsequently born. At weaning, tail biopies were taken and screened by WUSTL by targeted next generation sequencing (NGS). It was found that 11 of the animals harbored deletions in their genomes which resulted in homozygous knockout of the Adbr2 gene in these animals. Primers were acquired from WUSTL to maintain genotyping and breeding at Roswell Park. Three independent “founder” lines were chosen that harbored deletions of 5bp, 102bp, and 71bp. These lines were bred with C57BL/6NCr counterparts in order to breed out any off-target effects that may still be present in the early generations of mice, and this was maintained as stock. Once several generations of breeding were done, intercrossing to re-create the homozygous knockout was performed and phenotype confirmed. Female Nur77 GFP reporter mice on a C57BL/6 background were originally purchased from the Jackson Laboratory and maintained in-house. All mice used were at 8–12 weeks old. All mice were maintained in specific pathogen-free facilities, and all studies were conducted following protocols approved by the Institutional Animal Care and Use Committee (IACUC) at RPCCC.
Manipulation of ambient temperature and stress
Mice were housed at either standard (IACUC-required) temperature (ST, 22–23°C) or thermoneutral temperature (TT, 30–31°C) as described previously (12,15). In this model, the control, untreated groups all experience chronic physiological cold stress, which activates the SNS to regulate generation of heat through adaptive thermogenesis (16).
β-adrenergic receptor (β-AR) blocker treatment
To block β-adrenergic signaling, stressed mice (housed at ST) received daily, intraperitoneal injections of 200 μg propranolol (P0884, Sigma-Aldrich) in 100 μL PBS, beginning 7 days prior to, or 7 days after, tumor cell implantation (described below). Treatments continued until end of the experiment. Mice in the control (stressed) group received 100 μL PBS injections only.
Cell culture and tumor models
The B16-OVA melanoma cell line was provided by Dr. Protul Shrikant and has been genetically authenticated by our laboratory. Cell lines were confirmed to be mycoplasma negative (Mycoplasma Plus PCR Primer Set, Aligent Technologies, 302008). The CT26.CL25 colon cancer cell line was purchased from and authenticated by ATCC in 2017. These cells were cultured as described previously (15,19). Both cell lines were passaged twice before tumor implantation after being thawed. A total of 2 ×106 B16-OVA were injected subcutaneously into the lower left abdomen of C57BL/6, C57BL/6 β2-AR−/−, and Nur77GFP mice. 2 ×106 CT26.CL25 cells were injected subcutaneously into the lower left abdomen of BALB/c, BALB/c β2-AR−/− mice. Both cell lines werein 50 μL PBS. In each animal experiment, mice were randomly assigned to each group. Tumor monitoring started 5 days after implantation, and perpendicular diameters (width/length) were measured every 2–3 days until the end of the experiment. Tumor volume was calculated by the formula: Volume (mm3) =W2 L/2. Tumor growth in the CT26 model was blindly measured by a member of the laboratory staff (Ms. Li Feng), who is not an author on this study. Experiments were terminated by day 17–19 after tumor implantation, except for the kinetic study (Day 11 and Day 14). Tumors, spleens, and draining lymph nodes were collected at the endpoint.
CD8+ T-cell isolation and culture
8–12 weeks old BALB/c or Nur77-GFP reporter mice were sacrificed, and spleen and inguinal and axillary lymph nodes were collected. Spleen and lymph nodes were crushed and filtered through a 70 μm nylon cell strainer (Corning). Red blood cells were lysed by ACK lysing buffer (A1049201, Thermo Fisher) CD8+ T cells were isolated using a negative CD8α+ T cell isolation kit (Cat#130–104-075, Miltenyi Biotec) and then cultured at 1 ×106/mL in the presence of plate-coated anti-CD3/anti-CD28 crosslinking antibodies (anti-CD3 2 μg/mL; anti-CD28 2 μg/mL; Supplementary Table S1) in the presence or absence of 10 μM isoproterenol during activation (Sigma) as described previously (18). For immunoblotting (described below), CD8+ T cells were cultured at 2 ×106/mL in the presence of 10 uL anti-CD3/anti-CD28 activation beads (Cat#11452D, ThermoFisher) and harvested at the indicated time points.
Flow cytometry
At the experimental endpoint, tumors were removed and cut into 2–3 mm pieces, then transferred into a gentle MACS C tube (Miltenyi Biotec). Enzymes from Murine Tumor Dissociation Kit (Cat#130–096-730, Miltenyi Biotec) were added and processed through a gentleMACS Dissociator (130–093-235). Single-cell suspensions from tumors or tumor-draining lymph nodes (TDLNs) were obtained by passing dissociated tumors through 70 μm nylon cell strainers (ThermoFisher). Cells were washed with flow running buffer (0.1% BSA in PBS, Thermo Scientific) and incubated with anti-CD16/CD32 (Fc receptor blocker, 1:200, BD) at 4 °C for 10 minutes.
For cell surface staining, cells were stained with following antibodies (Supplementary Table S1): anti-CD45 conjugated to BV605, anti-CD3 conjugated to APC-Cy7, anti-CD8α conjugated to BUV395, anti-CD4 conjugated to BV786, anti-LAG3 conjugated to BV421, anti-CD28 conjugated to Ax647, anti-PD-1 conjugated to BV711, anti-Tim3 conjugated to PE-Cy7, anti-FasL conjugated to PE-Cy7, and anti-β2-adrenergic receptor conjugated to FITC. Live/Dead Fixable violet and aqua (Thermo Fisher) were used to gate out dead cells. For intracellular staining, cell surface markers and Live/Dead Fixable dyes were stained as described above, and then cells were fixed and permeabilized using the FoxP3/Transcription Factor Staining Buffer Set (Cat#00–5523-00, ThermoFisher) following the manufacturer’s protocol. Cells were then stained with anti-IFN-γ conjugated to BV421, anti-TNF-α conjugated to PerCP-Cy5.5, anti-IL2 conjugated with BV711, and anti-TCF1 conjugated to PE (Supplementary Table S1). For staining with mitochondrial dye, cells were first stained with cell surface markers and Live/Dead Fixable dyes, then incubated with 30 mM MitoTracker Green FM (mitochondrial mass; Cat#M7514, Thermo Fisher) in RPMI 1640 at 37 ˚C for 30 minutes. For in vitro studies, CD8+ T cells (see “CD8+ T-cell isolation and culture”) were harvested at either 4 hours or 24 hours after activation and washed twice with flow running buffer. Cells were stained with cell surface markers anti-CD69 conjugated to PerCP-Cy5.5 and Live/Dead Fixable dyes. GLUT-1 (conjugated to Ax647) intracellular staining was performed following the protocol described above (Supplementary Table S1). All flow data was collected using an LSR Fortessa flow cytometry (BD biosciences) and analyzed with FlowJo (RRID: SCR_008520) software v10.
Western blotting
CD8+ T cells (see “CD8+ T-cell isolation and culture”) were harvested at different time points (5, 10, and 20 minutes), washed with PBS twice, and stored at −80°C. Protein was extracted using lysis buffer consisting of RIPA Buffer (Pierce, catalog 89900), protease and phosphatase inhibitor mini tablets (Pierce, A32961), and 0.1 M PMSF (ThermoFisher Scientific). Protein concentration was determined using the BCA Protein Assay Kit (Pierce). Protein resolution was achieved by SDS-PAGE, transferred to a polyvinylidene difluoride membrane (Millipore), and blocked with 5% nonfat milk or 5% BSA (ThermoFisher Scientific) in Tris-buffered saline (Bio-Rad) with 0.1% Tween 20 (Bio-Rad). Membranes were incubated overnight at a concentration of 1:1000 for phospho-Zap-70 (Tyr 319)/Syk (Tyr352), Zap-70, and GAPDH (Supplementary Table S1). Membranes were washed with Tris-buffered saline with Tween 20. Anti-rabbit and anti-mouse horseradish peroxidase–conjugate secondary antibodies were used at a concentration of 1:3000 in 5% nonfat milk. Membranes were developed with ECL-substrate (Bio-Rad), and images were captured using the LI-COR Odyssey Fc (OFC-0756).
Metabolic assays
Single cell suspensions from tumors, spleens, and tumor-draining lymph nodes (TDLNs) were obtained as described above. CD8+ T cells were isolated with a CD8+ T-cell isolation kit (TIL, 130–116-478, Miltenyi Biotec) and plated on cell-tak coated Seahorse XF96 cell culture microplates at a density of 1×105 cells per well. A Mitochondrial Stress Test and Glycolytic Stress Test were performed as described previously (18).
Nanostring
CD8+ T cells were isolated from single-cell suspensions (described above) of B16-OVA tumors by FACS (FACSAriaII). Cell were stained with following antibodies (Supplementary Table S1): anti-CD45 conjugated to FITC, anti-CD3 conjugated to APC-Cy7, anti-CD8α conjugated to APC, anti-CD4 conjugated to PE. RNA was isolated from cells using the RNeasy Plus Mini kit (Qiagen). Equal volumes of reporter codeset and hybridization buffer are mixed together. 20 µL of this mastermix was then aliquoted per tube, followed by addition of 5 µL of total RNA (100 ng) per sample. 5 µL of Capture probeset was then added to each tube, samples are gently mixed by inversion, spun down, and incubated at 65 °C for at least 12 hours (maximum of 30 hours). Immediately after incubation, post-hybridization processing using the nCounter Prep station was carried out. Following the manufacturer’s instructions, the nCounter Prep station was loaded with the hybridized samples and the sample cartridge. Using the automated prompts, hybridized samples were loaded on the cartridge. Subsequent data collection of the sample cartridge was then carried out with the nCounter Digital Analyzer, starting with creation of the cartridge definition file then scanning of the cartridge following manufacturer’s settings and instructions. Analysis was performed with the NanoString Technologies nCounter Analysis System. The nCounter Mouse Immunology Kit (NanoString Technologies), which includes 561 immunology-related mouse genes, was used. Data analysis of created .csv files was then performed with Nanostring’s nSolver software.
Statistical Analysis
All data were presented as mean±standard deviation (SD) using Graphpad Prism 7 software (RRID: SCR_002798). Two-way ANOVA with Tukey analysis was used to compare tumor growth between groups, and an unpaired Student t-test was used to compare data between two groups.
Results
TILs express β2-AR and reducing β-AR signaling in stressed mice decreases tumor growth
In this study, we exploited the fact that standard housing temperature results in chronic activation of the SNS and NE-driven adrenergic receptor signaling (16). Using two different murine tumor models (a melanoma model, B16-OVA, in C57BL/6 mice and a colon cancer model, CT26.CL25, in BALB/c mice), we first confirmed our earlier observations that chronic adrenergic stress in mice accelerated tumor growth rates compared to mice housed at thermoneutral temperatures (Supplementary Fig. S1A-B). Our previous study in the 4T1 murine breast cancer model shows that the effect of chronic adrenergic stress is through β2-AR signaling and that CD8+ T cells are the major cell type involved in anti-tumor immunity (15). We next compared tumor growth rates in wild-type (WT) mice and β2-AR−/− mice, both housed under standard housing temperature (ST). We observed that in both tumor models, tumor growth was slower in β2-AR−/− mice than in WT mice (Fig. 1A-B), confirming the role for host β2-AR in mediating the tumor enhancing effect of chronic stress. We examined expression of the β2-AR on CD8+ T cells from TDLNs and the TME of untreated mice housed at ST by flow cytometry (gating strategy, Supplementary Fig. S1C). Tumor-infiltrating CD8+ T cells had higher expression of β2-AR than CD8+ T cells from the TDLNs (Fig. 1C-F). A higher frequency of β2-AR+CD4+ T cells in the TME compared to the TDLNs was also found (Supplementary Fig. S1D-E). These data together showed that reducing β2-AR signaling in the host decreases tumor growth rate and that a higher frequency of TILs express β2-AR than T cells from the TDLNs, suggesting the possibility that TILs could be more responsive to adrenergic stress-induced signals than those in other normal tissues.
Figure 1: TILs express β2-AR, and reducing β-AR signaling decreases tumor growth.
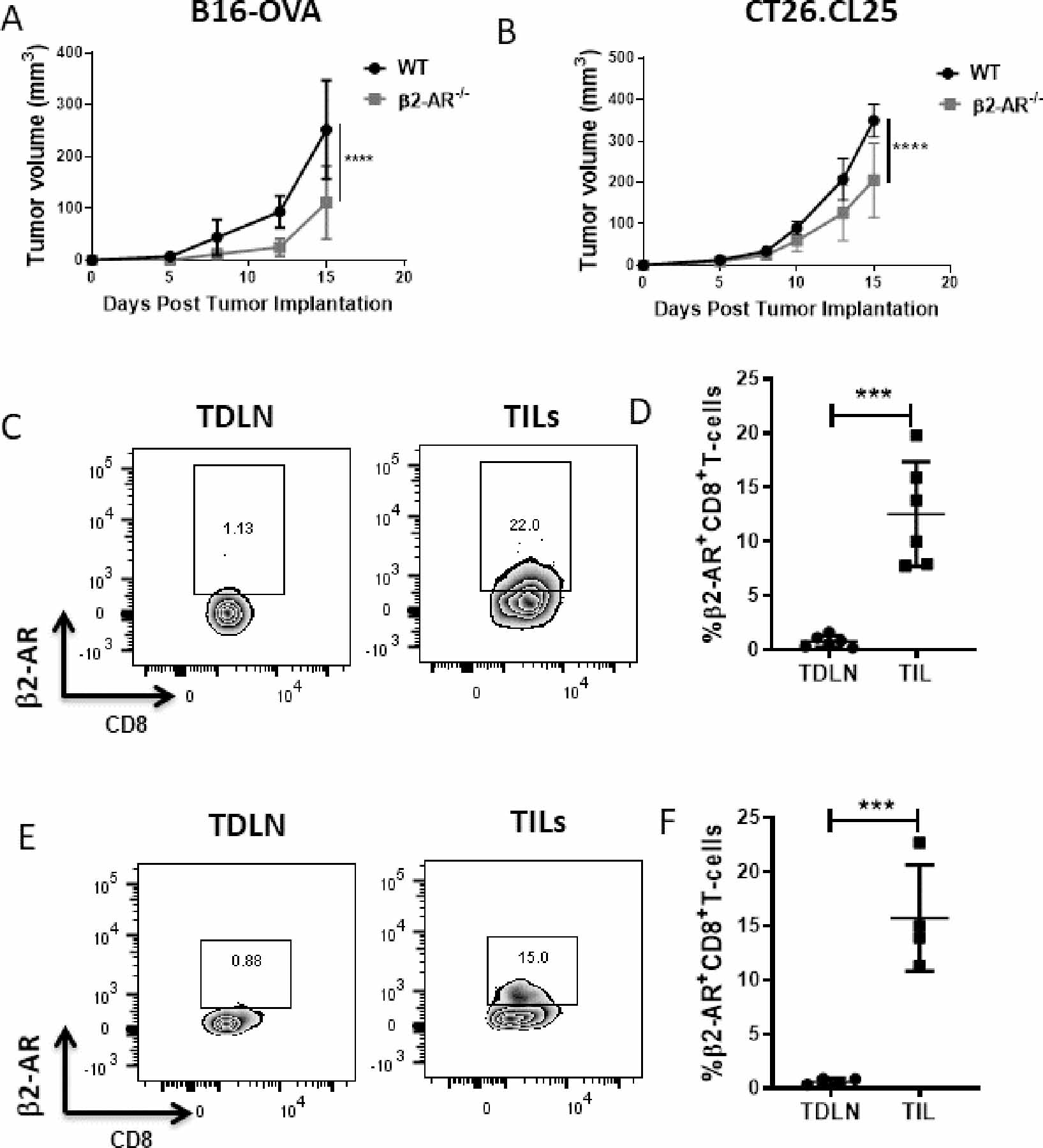
(A) 2×105 B16-OVA cells or (B) CT26.CL25 were injected into WT and β2-AR−/− C57BL/6 mice or WT and β2-AR−/− Balb/c mice, respectively. Tumor growth was monitored every 2–3 days. n=5–7/group, and tumor growth was compared using two-way ANOVA with Tukey analysis. Data are presented as mean±SD; one of two independent experiments. (C, E) Representative flow plots of β2-AR expression on CD8+ T cells from TDLNs and the TME of untreated WT mice in (C) B16-OVA and (E) CT26 models. (D, F) Frequency of β2-AR+CD8+ T cells from TDLNs and the TME in (D) B16-OVA and (F) CT26 models. n=4–6/group; data are presented as mean±SD; one of two independent experiments. Data were analyzed using unpaired Student’s t-test, ***p<0.001, ****p<0.0001.
β-AR blockade in stressed mice reduces checkpoint receptors and increases CD28 on TILs
Next, we used a pharmacologic method of blocking β-AR signaling and compared stressed mice with those treated with the pan-β-AR antagonist propranolol to further assess the impact of adrenergic stress signaling on T cells in the TME. Consistent with previous findings, tumor growth was slowed (Fig. 2A-B). Previous work from our lab shows that blockade of β-AR signaling improves the efficacy of immune checkpoint inhibitors (15). T-cell resistance to immune checkpoint blockade occurs by upregulation of multiple alternative immune checkpoint molecules, a state characteristic of T-cell exhaustion (25–28). We sought to determine whether the expression of molecular markers of dysfunctional, exhausted T cells was influenced by chronic stress. In the B16 model, blocking β-AR signaling in stressed mice associated with a decreased expression of PD-1 (15), LAG-3, and Tim3 on CD8+ TILs (Fig. 2C-D). The frequency of triple-positive PD-1+Tim3+LAG3+CD8+ TILs was also decreased by β-AR blockade (Fig. 2E-F). This data demonstrates that blockade of β-AR signaling counteracts the influence of chronic stress on expression of exhaustion markers on CD8+ TILs. This result was confirmed by comparing CD8+ TILs from WT and β2-AR−/− mice, where we found fewer exhausted CD8+ T cells in the TME of β2-AR−/− mice (Supplementary Fig. S2A-C). CD4+ TILs showed a similar phenotypic change when β-AR signaling was blocked using propranolol or in β2-AR−/−mice (Supplementary Fig. S2D-E). The reduction in frequency of exhausted T cells in the TME when β-AR signaling was blocked in mice housed at ST was also observed in CD8+ T cells within CT26 tumors (Supplementary Fig. S2F-H). Studies show that progenitor exhausted cells retain polyfunctionality and are better able to control tumor growth than terminally exhausted T cells in response to anti-PD-1 therapy (22).
Figure 2: Blockade of adrenergic signaling in stressed mice reduces immune checkpoint receptor expression on TILs.
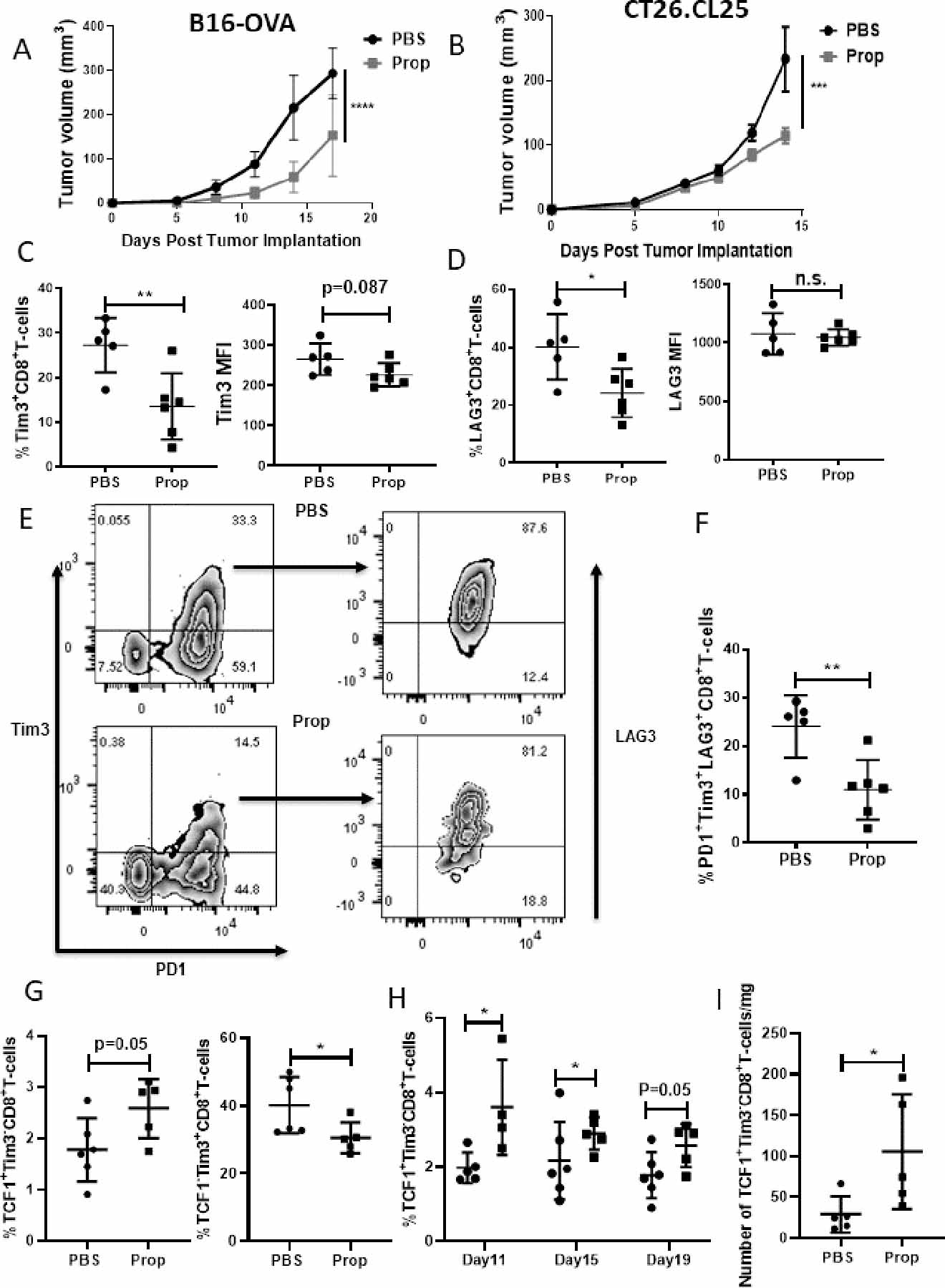
(A) 2×105 B16-OVA or (B) CT26.CL25 cells were injected into C57BL/6 mice or Balb/c mice, respectively. Mice were treated with either PBS or propranolol (Prop, 200μg) daily starting 5 days before tumor implantation until the end of experiment, and tumor growth was monitored every 2–3 days. n=5–7/group; data are presented as mean±SD; one of two independent experiments. Tumor growth was compared using two-way ANOVA with Tukey analysis. (C-I) Single-cell suspensions were made from B16-OVA tumors of mice treated with PBS or propranolol, and TILs were analyzed by flow cytometry. (C-D) Frequency and mean fluorescence intensity (MFI) of (C) Tim3+ and (D) LAG3+ CD8+ TILs. (E) Representative flow plots of PD1+Tim3+LAG3+CD8+ TILs. (F) Frequency of PD1+Tim3+LAG3+CD8+ TILs; n=5–6/group; data are presented as mean±SD; one of two independent experiments. (G) Frequency of progenitor (gated by CD8+>PD-1+CD44+>TCF1+Tim3-) and terminally exhausted CD8+ TILs (gated by CD8+>PD-1+CD44+>TCF1-Tim3+) from B16-OVA tumors. (H) Frequency of progenitor exhausted CD8+ TILs from B16-OVA tumors at day 11, 15, 19 after tumor implantation. (I) Absolute number of progenitor exhausted CD8+ TILs from mice treated with PBS or propranolol 7 days after tumor implantation. n=4–6/group; data are presented as mean±SD. Data were analyzed using unpaired Student’s t-test, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
We used the transcription factor, T-cell factor 1 (TCF1), to distinguish progenitor and terminally exhausted T cells (gating strategy, Supplementary Fig. S3A). Blocking β-AR signaling increased the frequency of progenitor exhausted T cells and decreased terminally exhausted T cells in the two tumor models (Fig. 2G, Supplementary Fig. S3B). We also performed a kinetic analysis of exhausted T cells in the B16-OVA model and found that blocking β-AR signaling increased progenitor exhausted T cells at three different time points, even when tumors were small (Fig. 2H). Because we observed the heterogeneity of β2-AR expression on TILs, we next compared the frequency of progenitor and terminally exhausted T cells in β2-AR+ and β2-AR– TILs. A high frequency of terminally exhausted T cells in β2-AR+ TILs was seen (Supplementary Fig. S3C), again indicating that adrenergic receptor signaling is associated with the exhaustion status of TILs. In these experiments, we initiated the blockade of β-AR signaling started before tumor implantation in our tumor models. Therefore, we also analyzed the phenotype of exhausted T cells when β-AR signaling blockade began after tumor establishment, which is more clinically relevant. Although no significant difference in tumor growth control (Supplementary Fig. S3D) or frequency of progenitor exhausted T cells (Supplementary Fig. S3E) was seen, the absolute number of progenitor exhausted T cells/mg of tumor was increased by β-AR signaling blockade (Fig. 2I). These data together suggest that β-AR signaling blockade decreases exhausted T cells in the TME; however, an increase in progenitor exhausted T cells is also induced by blockade of β-AR signaling and could be the reason that β-AR signaling blockade increases anti-PD-1 therapy efficacy.
Next, we compared the gene expression of other inhibitory molecules in CD8+ TILs from PBS-treated (stressed) and propranolol-treated B16-OVA tumor-bearing mice by Nanostring analysis. Blockade of β-AR signaling resulted in a decrease in expression of checkpoint genes (Btla, Cd274, Tigit, Ceacam1, and CD160) and T-cell activation and function suppression genes (Lair, Klar7, Pteger4) in CD8+ TILs in the propranolol-treated compared to PBS-treated tumor-bearing mice (Fig. 3A), which correlates with the flow cytometry data showing reduced expression of other checkpoint markers when adrenergic signaling was blocked. Together, these data suggest that β-AR signaling increases expression of multiple checkpoint inhibitory molecules in CD8+ TILs and promotes a T-cell exhausted phenotype in the TME.
Figure 3: Blockade of adrenergic signaling in stressed mice reduces immune checkpoint receptor expression and increases CD28 expression on TILs.
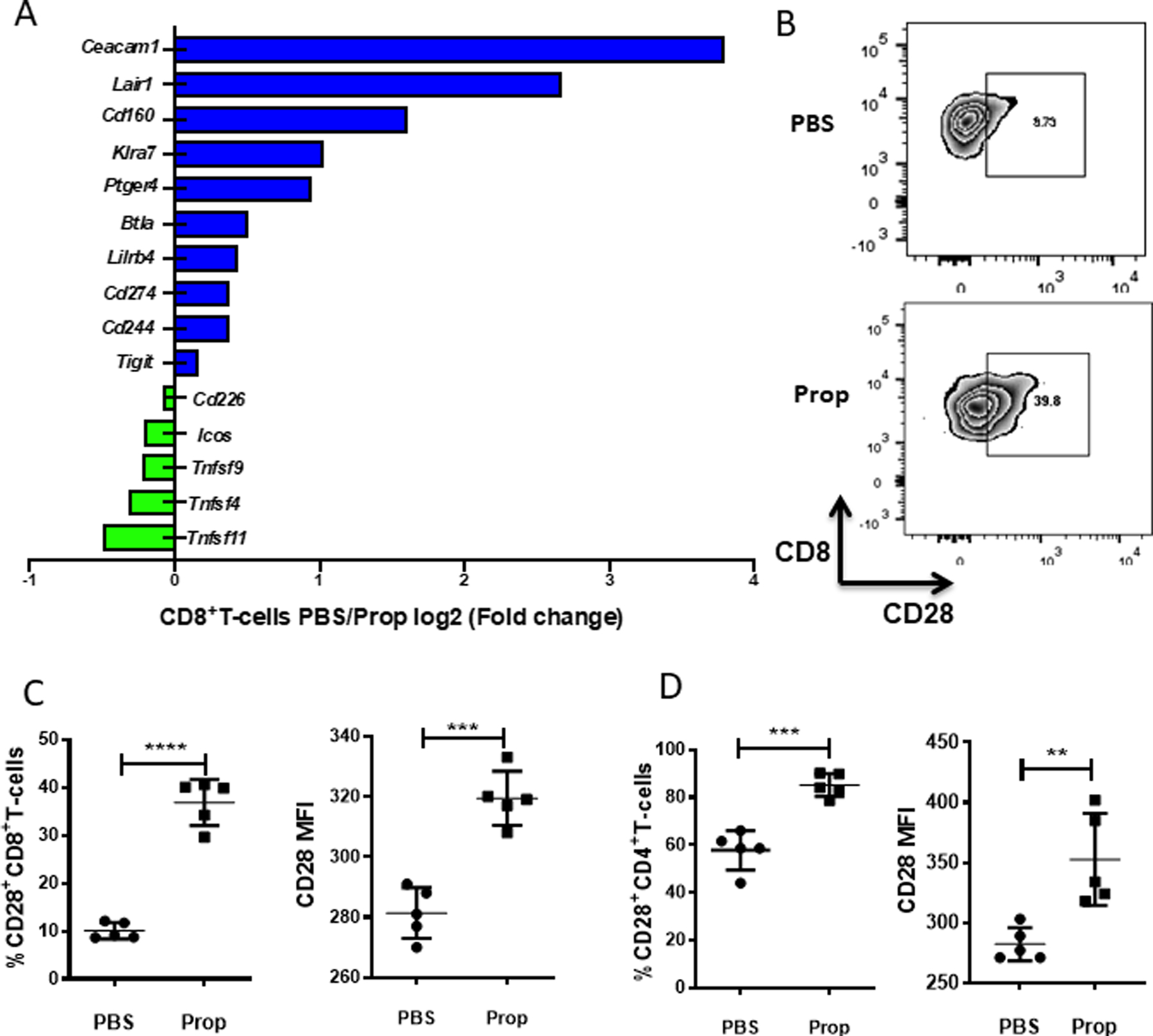
Single-cell suspensions were made from B16-OVA tumors harvested from mice treated with PBS or propranolol (Prop). (A) Single-cell suspensions of B16-OVA tumors from PBS- or propranolol-treated mice (CD8+ T cells were pooled from 5 mice/group) were prepared, and tumor infiltrating CD8+ T cells were sorted, lysed, and RNA was extracted for Nanosting nCounter microarray analysis. Graph shows co-stimulatory and inhibitor molecules by log2 (fold-change of PBS/Prop). Blue shows high in PBS group; green shows high in Prop group. (B) Representative flow plots of CD28 expression on CD8+ T cells from B16-OVA tumors. (C) Frequency and mean fluorescence intensity (MFI) of CD28 expression on CD8+ T cells from B16-OVA tumors. (D) Frequency and MFI of CD28 expression on CD4+ T cells from B16-OVA tumors. n = 5/group. (B-D) Data are presented as mean±SD; one of two independent experiments; data were analyzed using unpaired Student’s t test, **p<0.01, ***p<0.001, ****p<0.0001
It has been reported that CD28 expression is required for reversing CD8+ T-cell exhaustion by anti-PD-1 therapy (32,33). Therefore, we quantified the expression of CD28 on TILs from stressed mice treated with either PBS or propranolol and found that in both melanoma (Fig. 3B-C) and colon cancer (Supplementary Fig. S4A) models, blockade of β-AR signaling increased CD28 expression on CD8+ TILs. Similar results were also observed in CD4+ TILs in melanoma (Fig. 3D) and colon cancer (Supplementary Fig. S4B) models. Consistent with our findings that the costimulatory molecule CD28 was upregulated, gene expression data showed that other costimulatory molecules (Cd226, Icos, and Tnfrsf9) were also upregulated in CD8+ T cells when β-AR signaling was blocked (Fig. 3A).
β-AR blockade in stressed mice increases anti-tumor cytokines and proteins in TILs
Thus far, we have shown that blockade of β-AR signaling in stressed mice decreased the percentage of exhausted TILs. Next, we sought to determine whether blockade of β-AR signaling could improve effector functions of TILs. We isolated CD8+ TILs from PBS- or propranolol-treated B16 melanoma tumor-bearing mice (housed at ST) and analyzed selected immune function-related gene expression by Nanostring. Expression of several cytokine genes was significantly altered by blocking β-AR signaling, with an increased expression of effector molecules Ifng, Gzmb, and Il12a and a decreased expression of pro-inflammatory cytokines Il1b, Il4, Il6, and Il10 (Fig. 4A). The cytokine expression by TILs from B16 tumors was also measured by flow cytometry and showed that the frequencies of IFNγ+, TNFα+, and double-positive IFNγ+TNFα+CD8+ T-cells were enhanced by blocking β-AR signaling. IL2 expression by CD8+ TILs were also elevated in the propranolol group (Fig. 4B-E). In the CT26.CL25 model, the frequencies of IFNγ+ (Supplementary Fig. S5A) and FasL+CD8+ T cells (Supplementary Fig. S5B) were also increased in the TME. CD4+ TILs also showed a similar alteration of effector phenotype in the melanoma (Fig. 4F-G) and colon cancer models (Supplementary Fig. S5C-E). These data together suggest that blocking β-AR signaling in stressed mice increases the expression of anti-tumor cytokines and cytotoxic proteins in TILs.
Figure 4: Blocking β-AR signaling in stressed mice increases expression of anti-tumor cytokines and cytotoxic proteins in TILs.
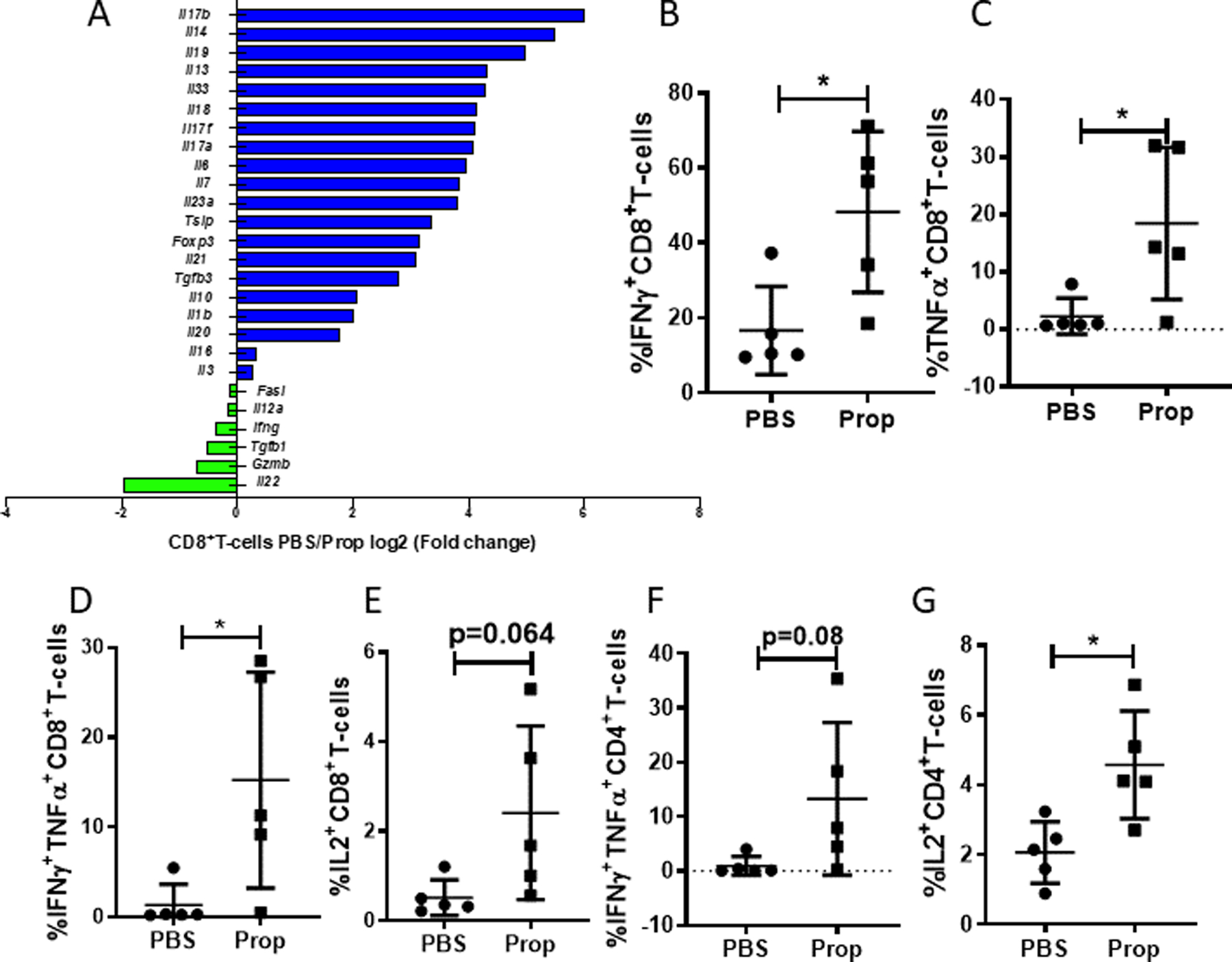
Single-cell suspensions were made from B16-OVA tumors of mice treated with PBS or propranolol (Prop). (A) Single-cell suspensions of B16-OVA tumors of PBS- or propranolol-treated mice (CD8+ T cells were pooled from 5 mice/group) were prepared, and tumor infiltrating CD8+ T cells were sorted, lysed, and RNA was extracted for Nanosting nCounter microarray analysis. Graph shows cytokines by log2 (fold-change of PBS/Prop). Blue shows high in PBS group; green shows high in Prop group (B-E) Frequency of (B) IFNγ, (C) TNFα, (D) IFNγ+TNFα+, and (E) IL2 expression in CD8+ T cells from B16-OVA tumors. (F-G) Frequency of (F) IFNγ+TNFα+ and (G) IL2 expression in CD4+ T cells from B16-OVA tumors. n = 5. (B-G) Data are presented as mean±SD; one of two independent experiments; data were analyzed using unpaired Student’s t test, *p<0.05.
β-AR blockade in stressed mice alleviates mitochondrial dysfunction in CD8+ TILs
The data indicate that reducing stress signaling by blocking β-AR signaling in stressed mice led to a more activated and less exhausted phenotype of TILs. T-cell exhaustion is associated with mitochondrial dysfunction (29–31,34–36), and our previous work shows that β-AR signaling impairs mitochondrial function (i.e., impairs upregulation of glycolysis and oxidative phosphorylation) during activation in vitro (18). Therefore, we next asked whether β-AR signaling also impaired metabolism of CD8+ T cells in vivo. First, we assessed mitochondrial mass by staining TILs with MitoTracer Green FM. Consistent with a previous report (35), CD4+ and CD8+ T cells from the TME had lower mitochondrial mass than those from the TDLNs (35)(Supplementary Fig. S6A-B). However, the mitochondrial mass of CD8+ TILs from propranolol-treated mice was increased compared to CD8+ TILs from PBS-treated stressed mice, indicating that blocking β-AR signaling in stressed mice can partially rescue the mitochondrial loss in CD8+ TILs (Fig. 5A-B). This phenotype was also observed in the CT26 model (Supplementary Fig. S6C-D). In contrast, propranolol treatment of mice did not increase mitochondrial mass in CD8+ T cells from TDLNs, which expressed only low levels of β2-AR (Supplementary Fig. S6E). The increase in mitochondrial mass with β-AR blockade was also seen in CD4+ TILs (Fig. 5C, Supplementary Fig. S6F). Because loss of mitochondrial mass is associated with mitochondrial dysfunction (37), we next isolated CD8+ T cells from spleens, TDLNs, or tumors (Fig. 5D, Supplementary Fig. S7A-B) of PBS-treated and propranolol-treated stressed mice. We used the Seahorse Extracellular Flux Analyzer to assess oxidative phosphorylation (oxygen consumption rate, OCR). There was no significant difference in baseline OCR or spare respiratory capacity of CD8+ T cells from spleens or TDLNs (Supplementary Fig. S7A-B), which was consistent with the similar mitochondrial mass present in these cells. However, we did see a significant increase in baseline OCR (Fig. 5E), maximum OCR (Fig. 5F), and spare respiratory capacity (Fig. 5G) of CD8+ TILs from propranolol-treated tumor-bearing mice (Fig. 5D). This phenotype was also found in the CT26 model (Supplementary Fig. S7C-F. Together, these data suggest that blocking β-AR signaling in mice experiencing chronic stress results in increased mitochondrial mass and improved metabolic function of TILs, which may lead to an increased functional phenotype.
Figure 5: Blocking β-AR signaling in stressed mice alleviates mitochondrial dysfunction of CD8+ T cells in the TME.
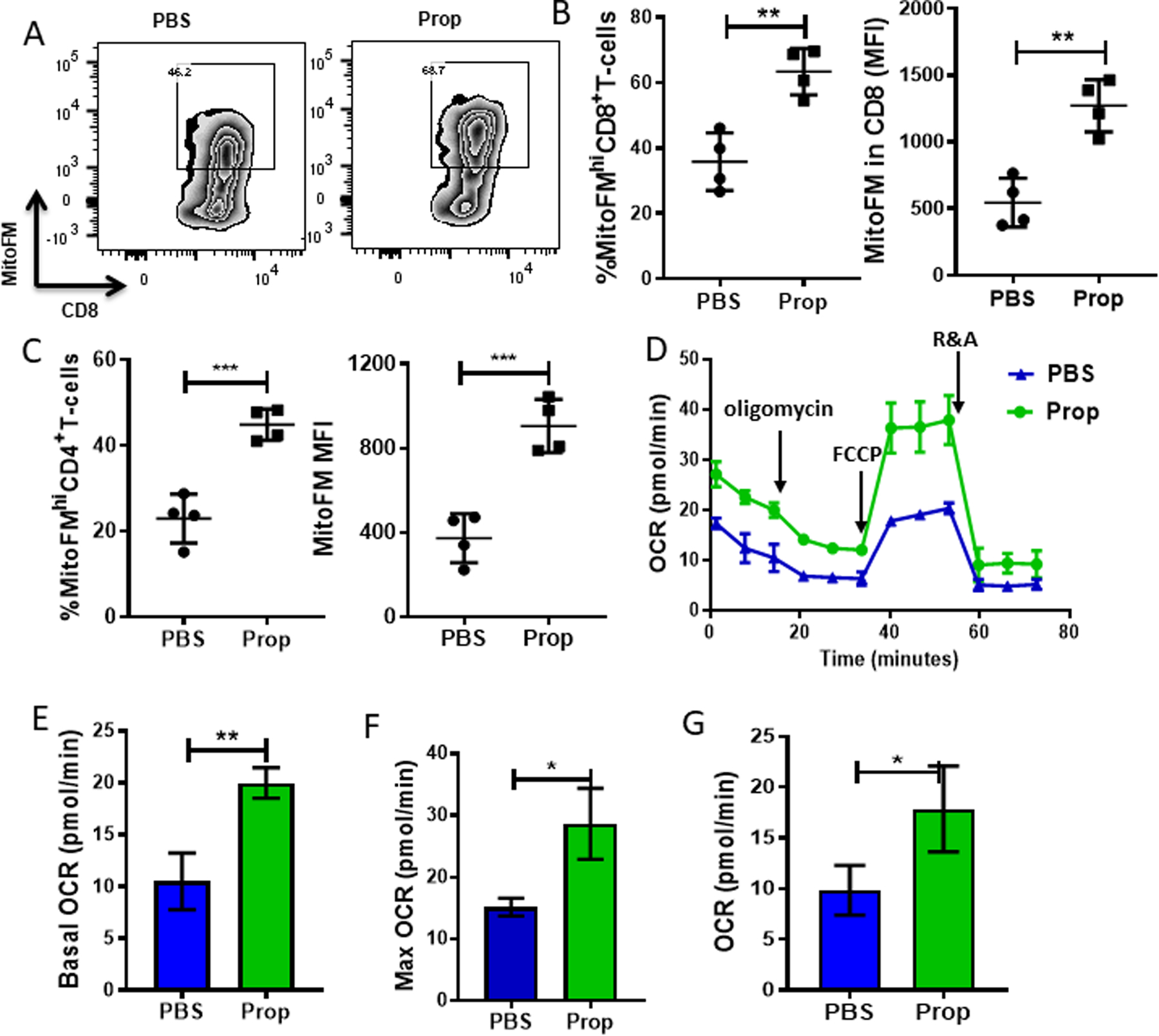
Single-cell suspensions were made from B16-OVA tumors harvested from mice treated with PBS or propranolol (Prop), and CD8+ T cells (pooled from 5 mice/group) were isolated, and oxygen consumption rate (OCR) was measured in 1×105 CD8+ T cells using the Seahorse Extracellular Flux Analyzer. (A) Representative flow plots of mitochondrial mass of CD8+ TILs from B16-OVA tumors. (B-C) Frequency and mean fluorescence intensity (MFI) of mitochondrial mass of (B) CD8+ and (C) CD4+ TILs. (D) OCR of CD8+ T cells from B16-OVA tumors. Addition of reagents indicated by arrows: (1) oligomycin; (2) FCCP; and (3) antimycin A and rotenone. (E) Basal OCR of CD8+ TILs. (F) Maximum OCR of CD8+ TILs. (G) Spare respiratory capacity (SRC) of CD8+ TILs. n = 3–5. Data are presented as mean±SD; one of two independent experiments. Data were analyzed using unpaired Student’s t-test, *p<0.05, **p<0.01, ***p<0.001,
Blocking β-AR signaling in stressed mice increases glycolysis in CD8+ T cells in the TME
TCR-induced activation drives T-cell metabolic reprogramming to meet the cellular energetic and biosynthetic demands of activation, differentiation, and effector function. In the TME, the low nutrient and high acidity conditions induce metabolic competition between immune cells and tumor cells. In the TME, impaired glycolytic function of CD8+ T cells associates with low IFNγ expression (38), and the insufficiency of glycolytic metabolites leads to defects in TCR-induced Ca2+ and activation (39). β-AR signaling reduces glucose transporter-1 (GLUT1) expression on CD8+ TILs and decreases glycolysis in CD8+ T cells during activation in vitro (18). Therefore, we speculated that the glycolysis in CD8+ TILs was also impaired by β-adrenergic signaling. To test this idea, we isolated CD8 +T cells from spleens, TDLNs, and tumors from stressed mice treated with PBS or propranolol and used the Seahorse Extracellular Flux Analyzer to analyze glycolysis via measuring the extracellular acidification rate (ECAR)(Fig. 6A, Supplementary Fig. S7G-H). Because CD8+ T cells from spleens or TDLNs have a very low level of glycolysis, no significant difference in ECAR of CD8+ T cells from spleens or TDLNs was observed between the two groups (Supplementary Fig. S7C-D). However, we observed a significant increase in baseline ECAR (Fig. 6B) and maximal ECAR (Fig. 6C) of CD8+ TILs from propranolol-treated mice (Fig. 6A), indicating that an increase in glycolytic function occurs following β-AR blockade.
Figure 6: Blocking β-AR signaling in stressed mice increases glycolysis in CD8+ T cells in the TME.
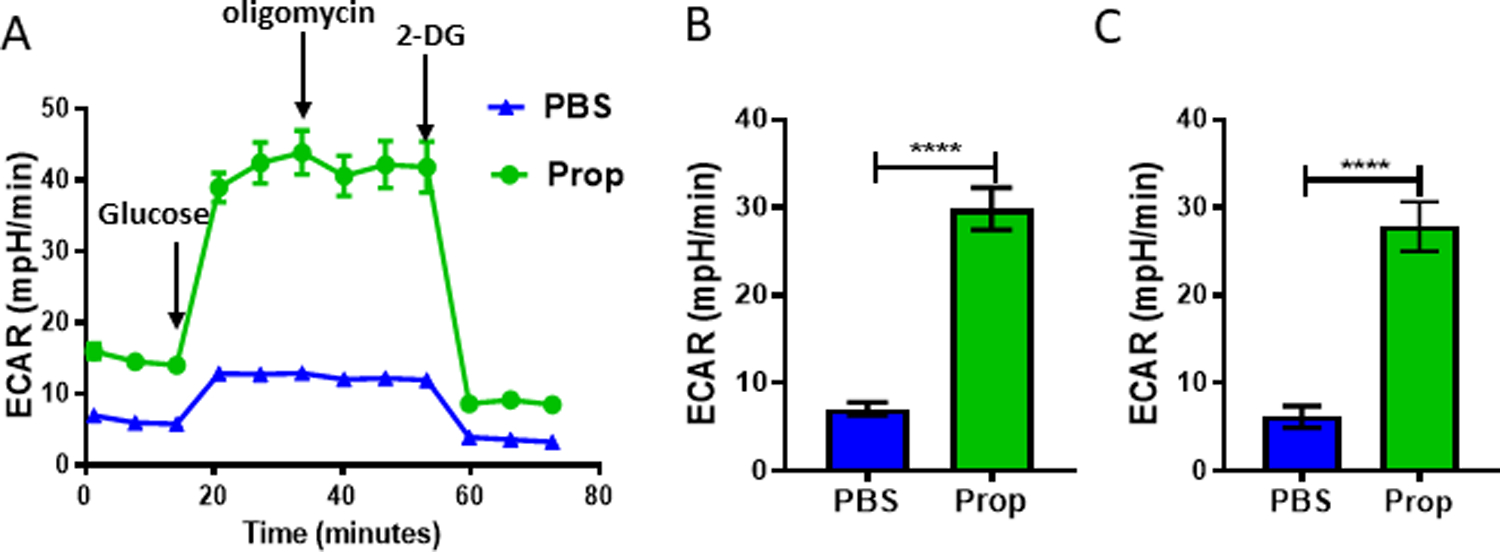
Single-cell suspensions were made from B16-OVA tumors from mice treated with PBS or propranolol (Prop). CD8+ T cells (pooled from 5 mice/group) were isolated, and extracellular acidification rate (ECAR) was measured after adding 1×105 CD8+ T cells to a Seahorse Extracellular Flux Analyzer. (A) ECAR results from CD8+ TILs from B16-OVA tumors. Addition of reagents indicated by arrows: (1) glucose; (2) oligomycin; and (3) 2-DG. (B) Basal ECAR of CD8+ TILs from B16-OVA tumors. (C) Maximum ECAR of CD8+ TILs from B16-OVA tumors. n = 3–5. Data are presented as mean±SD; one of two independent experiments. Data were analyzed using unpaired Student’s t-test, ****p<0.0001
β-AR signaling impairs TCR signaling
The ability of T cells to develop and differentiate into effector or memory T cells relies on multiple signals, among which TCR- and CD28-mediated signals are critical for initiating T-cell metabolic reprogramming during activation. T-cell activation through TCR ligation rapidly induces glycolysis, which is linked to T-cell effector function (40), and glycolysis during early activation of CD8+ T cells in vitro is inhibited by the β-AR agonist isoproterenol (18). Therefore, we speculated that signaling through the β-AR may impair early TCR signaling. Here, we tested this by isolating CD8+ T cells from naïve mice and activating them ex vivo in the presence of isoproterenol (β-AR agonist), added either at the beginning of activation or 2 hours after activation. Treatment with isoproterenol during activation led to impaired T-cell activation (CD69 expression) and GLUT-1 expression compared to controls, but this suppressive effect was lost when isoproterenol was added 2 hours after activation (Fig. 7A-B). This indicates that β-AR signaling inhibits CD8+ T-cell activation at an early stage, likely via the TCR signaling pathway. We collected cells from these two groups at different time points after activation (5, 10, and 20 minutes) and used Western blots to assess the phosphorylation of ZAP-70, the kinase immediately downstream of TCR engagement. We showed that by activating β-AR signaling, the phosphorylation of ZAP-70 was significantly reduced, indicating an impairment of TCR signaling (Fig. 7C). We confirmed this result using CD8+ T-cells from Nur77-GFP reporter mice, in which TCR signaling in T cells triggers the early response gene Nur77 and induces GFP expression. In these cells, GFP expression correlates with the strength of TCR signaling. By activating CD8+ T-cells isolated from Nur77-GFP mice in vitro, with or without isoproterenol, we again observed that adrenergic signaling impaired TCR signaling (Fig. 7D). In the B16 melanoma model, Nur77-GFP reporter mice treated with β-AR blockade had increased TCR signaling in CD8+ TILs in vivo (Fig. 7E). Together these data demonstrate that β-AR signaling inhibits metabolic reprograming of CD8+ T-cell activation through impairment of TCR signaling.
Figure 7: β-AR signaling impairs TCR signaling.
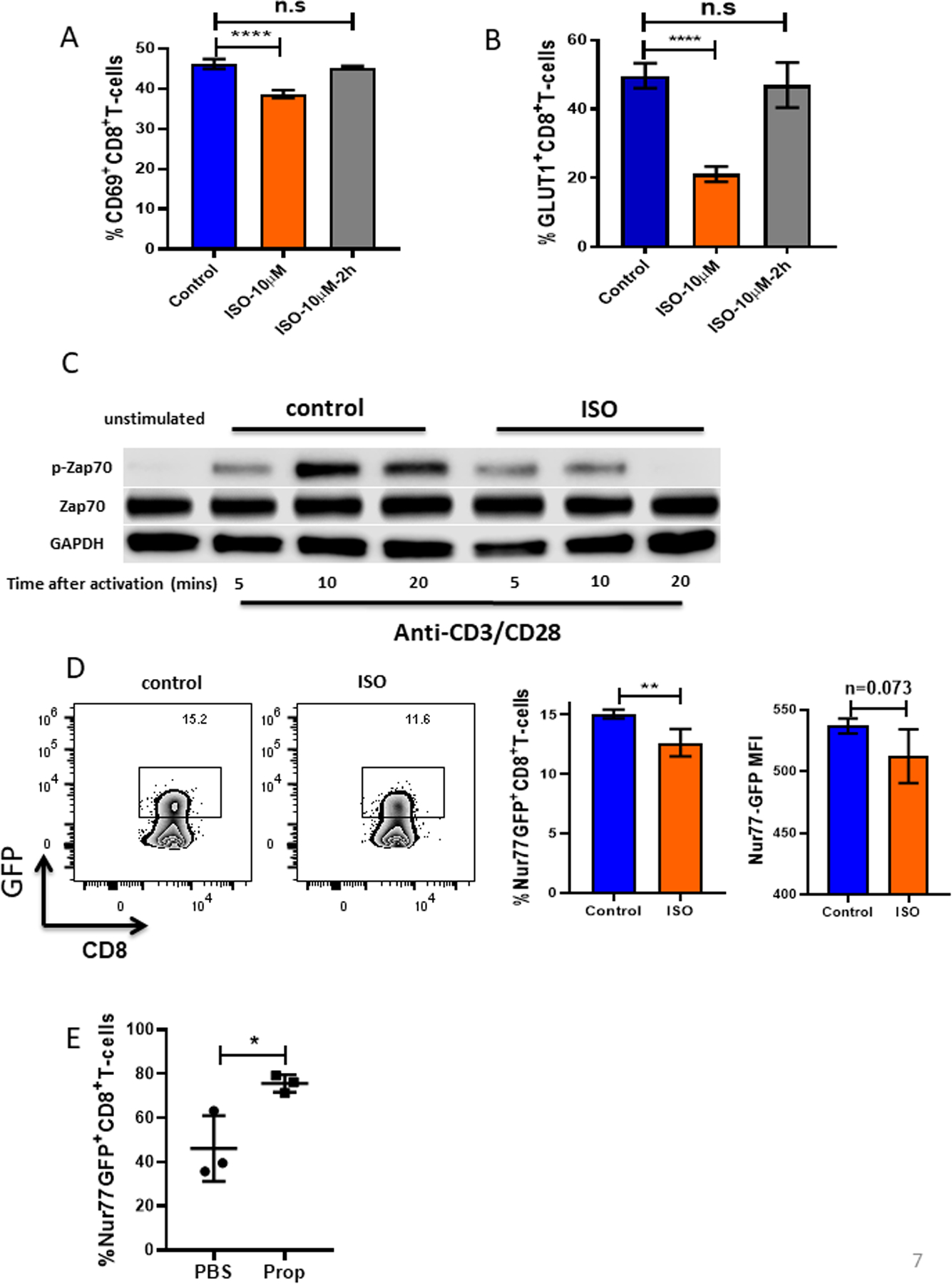
(A-B) CD8+ T cells from BALB/c mice were isolated and purified from lymph nodes and spleens of non-tumor-bearing mice, activated with anti-CD3/CD28 antibodies, and treated with or without isoproterenol (ISO, 10μM). (A) CD69 and (B) GLUT-1 were measured by flow cytometry 24 hours after activation. (C) CD8+ T cells activated and treated with or without ISO were collected and lysed. Total ZAP-70, phosphorylated ZAP-70, and GAPDH protein expression were assessed by Western blot at the indicated times. (D) CD8+ T cells from Nur77-GFP reporter mice were isolated and purified from lymph nodes and spleens of non-tumor-bearing mice and activated with anti-CD3/CD28 antibodies with or without ISO. Nur77, represented by GFP, was measured by flow cytometry 4 hours after activation. MFI: mean fluorescence intensity. (E) 2×105 B16-OVA cells were injected into C57BL/6 mice. Mice were treated daily with either PBS or propranolol (Prop, 200ug) starting 5 days before tumor implantation and continuing through the end of experiment. Single-cell suspensions were made from B16-OVA tumors. Nur77 was represented by GFP and was measured by flow cytometry. n = 3/group; data are presented as mean±SD; one of two independent experiments. Data were analyzed using Student’s t-test, *p<0.05, **p<0.01, ****p<0.0001.
Discussion
The equilibrium established among immune cells within tumors is known to be sensitive to physiological factors, such as hypoxia (41) or nutrient availability, that influence metabolic changes in T cells (38,42) and responses to immune checkpoint therapies (43). Tumors are sites that induce neurogenesis and serve as targets of stress-driven signals via these nerves (4,44,45). Thus, a stressful event, particularly long-term chronic stress, could be directly influencing tumors through neurotransmittors released into the TME in response to stressful stimuli. Here, we demonstrated that chronic stress-induced adrenergic signaling impairs TCR signaling and activation, while increasing the frequency of exhausted T cells in the TME. Conversely, β-AR signaling blockade can remodel the immune contexture of the TME of stressed mice, resulting in fewer exhausted T cells and an increase in effector T-cell numbers compared to untreated stressed mice.
In two different models, we found that propranolol given to mice experiencing chronic adrenergic stress significantly reduced the percentage of exhausted T cells expressing immune checkpoint receptors (PD-1, Tim3, and LAG3) in the TME compared to PBS-treated mice. We also showed other genes that are markers of exhaustion, such as CD160, Btla, Ceacam1, and genes related to suppression of T-cell activation and function (lair1, Pteger4) were decreased in CD8+ TILs from mice treated with β-AR blockade. When we further analyzed these TILs, we found that the blockade of adrenergic receptor signaling increased the number of progenitor exhausted T cells, which may explain why β2-AR blockade increases the response to anti-PD-1 therapy. Studies show that exhausted T cells are heterogeneous, and there is evidence that they may be a separate lineage, including self-renewing progenitor and terminally exhausted T cells which respond differently to PD-1 blockade (21,26). Previous work by others has shown that progenitor exhausted T cells, which are polyfunctional, can control tumor growth and response to anti-PD-1 therapy better than terminally exhausted T cells (22). We also observed a high frequency of terminally exhausted T cells in β2-AR+ TILs, indicating that adrenergic receptor signaling impacts the status of exhaustion of TILs.
We found that blockade of adrenergic receptor signaling in mice experiencing chronic stress was associated with an increased percentage of T cells producing effector cytokines such as IFNγ, TNFα, and IL2. In our gene expression data, we found CD8+ TILs from propranolol-treated stressed mice expressed lower levels of the pro-inflammatory genes Il1b, Il4, Il6, and Il10 than T cells from untreated stressed mice. It has been shown previously that in human head and neck squamous cell carcinoma (HNSCC), PD-1+Tim3+CD8+ T cells are immunosuppressive and suppress T-cell proliferation of non-exhausted T cells by producing IL10 and through close cell-cell contact (46). Therefore, in the future, it will be critical to determine whether reducing expression of Il1b, Il4, Il6, and Il10 by exhausted CD8+ T cells with blockade of β-AR signaling is associated with a reduction of immune checkpoint receptor-positive T cells in TME.
Our data suggests that CD28 expression on TILs is also increased by blockade of β-AR signaling in both tumor models. Several studies show that the CD28 signaling pathway is the target of PD-1-recruited Shp2 phosphatase and is required in reversing CD8+ T cells exhaustion by anti-PD-1 therapy (33,47). However, it is still not clear whether there is a relationship between reducing expression of exhaustion markers and increasing expression of CD28 on TILs by blocking β-AR signaling. It is also not known how CD28 is regulated by β-AR signaling and whether the increase of CD28 expression leads to an increase in CD28 signaling. The role of stress in the biology of CD28 expression is therefore a major topic that must be addressed in future studies. We previously showed (18) that β-adrenergic signaling inhibits metabolic reprogramming in CD8+ T cells during activation in vitro. It is known that CD28 regulates T-cell metabolism; however, we still cannot conclude that β-AR inhibits metabolic reprogramming in a CD28-dependent manner in vivo. This needs to be studied in greater detail in future work.
In this study, we observed that propranolol blockade in mice experiencing chronic adrenergic stress increases both glycolysis and oxidative phosphorylation in CD8+ TILs. Previous studies exploring T-cell metabolism have revealed that the functional status of T cells associates with metabolic reprogramming, with naïve and memory T cells primarily undergoing oxidative phosphorylation and effector T cells increasing glycolysis (42). Our previous work shows that increasing β-AR signaling during CD8+ T-cell activation in vitro (using the agonist isoproterenol) impairs metabolic reprogramming, and together these data suggest that β-AR signaling is a significant factor regulating the metabolic status of CD8+ T cells in the TME. Exhausted T cells undergo metabolic insufficiency, with decreased effector function and poor responsiveness to immunotherapies. It has been shown that that PD-1 ligation decreases T-cell glycolysis and promotes lipolysis and fatty acid oxidation (48). The co-expression of immune checkpoint receptors has also been associated with mitochondrial dysfunction (35), and loss of mitochondrial function can result in increased production of reactive oxygen species (ROS), which promotes T-cell exhaustion (36). Therefore, targeting stress may be a good strategy to reverse T-cell exhaustion and improve immunotherapy. However, the specific relationships between T-cell exhaustion, T-cell metabolism, and β-AR signaling remain to be clarified. More study is needed on the impact of stress on altered T-cell metabolism during tumor development, and how this might contribute to suppressed anti-tumor immunity.
The new data presented here show that β-AR signaling suppresses TCR signaling during CD8+ T-cell activation. By using T cells from Nur77-GFP transgenic mice, in which the expression of Nur77 (downstream of TCR signaling) is indicated by GFP fluorescence, we observed that blocking β-AR signaling increased the expression of Nur77-GFP in CD8+ T cells from the TME, with no significant difference in frequency or relative number of antigen specific CD8+ TILs (15), which indicates that this inhibition of TCR signaling by β-AR signaling is also involved in regulation of anti-tumor immunity. Studies have shown that β-AR signaling suppresses CD8+ T-cell cytokine production and cytolytic function in response to TCR activation (9). We speculate that the suppressive effect of β-AR signaling on TCR signaling occurs indirectly through GLUT-1 downregulation. It has been shown that GLUT-1 expression, and the subsequent metabolic increase in glycolysis, play a key role in T-cell activation and function. Studies show that suppression of GLUT-1 and glucose metabolism (by decreased Akt/mTORC1 signaling) drives T-cell impairment (49) and exhaustion (50). We previously published that β2-AR signaling decreases GLUT-1 expression by T cells in vitro and in vivo (18). Therefore, we propose that the β-AR signaling-induced decrease in GLUT-1 expression inhibits TCR signaling and drives T-cell exhaustion, but this must be studied in more detail. It is also important to investigate whether the same phenotype change can be observed in humans, which could explain, in part, why some patients do not respond well to immunotherapy.
The specific effect of adrenergic signaling on CD8+ T cells in tumor models needs to be clarified more completely by generating CD8+ T cell-specific β2-AR knockout mice to further investigate the precise mechanisms of how β2-AR signaling affects CD8+ T-cell phenotype and metabolism. Although here we demonstrated that adrenergic signaling in CD8+ T cells increases exhaustion and prevents metabolic reprogramming and acquisition of effector function, our previous studies have shown that adrenergic signaling also enhances the suppressive function of MDSCs (14). How these effects, and those on other immune cells such as DCs and CD4+ T cells are integrated to regulate the anti-tumor immune response needs to be addressed in future studies.
These findings add to a growing appreciation of the importance of nervous stimulation and chronic stress in shaping tumor-immune contexture. Given the wide effects of adrenergic signaling on immune cells, especially suppression on T cells, we have previously suggested that the β-AR could be another immune checkpoint that can be targeted for improving anti-tumor immune responses (45). In this regard, β-AR antagonists, including propranolol, could be repurposed as a checkpoint inhibitor for immunotherapy. A retrospective study reveals that β-blocker usage for non-oncological purposes, which included β2 but not specific β1-blockers, can significantly improve the outcome of immunotherapy in patients with melanoma (51). A Phase II biomarker clinical trial shows that when patients with breast cancer are given propranolol 7 days prior to surgery, pro-metastatic and proinflammatory gene expression are downregulated, with evidence of increased tumor infiltration of CD68+ macrophages and CD8+ T cells (52). Our group has completed a Phase I trial prospectively combining propranolol with pembrolizumab (anti-PD-1) and show encouraging responses in patients with advanced melanoma (53). The benefit seen in these epidemiological and therapeutic evaluations, combined with the mechanistic data provided here, provide a compelling rationale for combining immunotherapies with stress-reducing strategies or use of pharmacological β-AR blockade.
Supplementary Material
Synopsis:
T-cell exhaustion impacts immunotherapy efficacy. How T-cell exhaustion is regulated remains incompletely understood. Here, the sympathetic stress response is shown to regulate the development of T-cell exhaustion by modulating CD8+ T-cell metabolism and function in the TME.
Acknowledgments
This work was supported by the National Cancer Institute (NCI) Grant R01CA205246; The Breast Cancer Coalition of Rochester, NY, USA; The Roswell Park Alliance Foundation; the Roswell Comprehensive Cancer Center and National Cancer Institute (NCI) P30CA016056 involving the use of Roswell Park Comprehensive Cancer Center’s Flow and Image Cytometry and NIH Grants 1R50CA211108 and S10OD018048 as well as to the Immune Analysis Shared Resources. We thank Jeanne Prendergast and Li Feng for their help in both the planning and management of experiments. We thank Dr. David Farrar (University of Texas Southwestern Medical Center) for the gift of β2-AR−/−mice and Genomics Shared Resource (Roswell Park) for expert support. We thank Dr. Aimee Stablewski for generating β2-AR−/−mice on the C57BL/6 background and Dr. Stephanie Egan for her help in generating Nur77 GFP-reporter mice. We thank Dr. Junko Matsuzaki and Jessie Chiello for their help with the Seahorse analysis and Dr. Kitty De Jong and Xiaojun Liu for advice and help in conducting FACS.
Footnotes
Disclosure of Potential Conflicts of Interest
The authors declare no potential conflicts of interest.
References
- 1.Glaser R. Stress-associated immune dysregulation and its importance for human health: a personal history of psychoneuroimmunology. Brain Behav Immun 2005;19(1):3–11 doi 10.1016/j.bbi.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 2.Glaser R, Kiecolt-Glaser JK. Stress-induced immune dysfunction: implications for health. Nat Rev Immunol 2005;5(3):243–51 doi 10.1038/nri1571. [DOI] [PubMed] [Google Scholar]
- 3.Antoni MH, Lutgendorf SK, Cole SW, Dhabhar FS, Sephton SE, McDonald PG, et al. The influence of bio-behavioural factors on tumour biology: pathways and mechanisms. Nat Rev Cancer 2006;6(3):240–8 doi 10.1038/nrc1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zahalka AH, Frenette PS. Nerves in cancer. Nat Rev Cancer 2020. doi 10.1038/s41568-019-0237-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zahalka AH, Arnal-Estape A, Maryanovich M, Nakahara F, Cruz CD, Finley LWS, et al. Adrenergic nerves activate an angio-metabolic switch in prostate cancer. Science 2017;358(6361):321–6 doi 10.1126/science.aah5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Magnon C, Hall SJ, Lin J, Xue X, Gerber L, Freedland SJ, et al. Autonomic nerve development contributes to prostate cancer progression. Science 2013;341(6142):1236361 doi 10.1126/science.1236361. [DOI] [PubMed] [Google Scholar]
- 7.Kamiya A, Hayama Y, Kato S, Shimomura A, Shimomura T, Irie K, et al. Genetic manipulation of autonomic nerve fiber innervation and activity and its effect on breast cancer progression. Nat Neurosci 2019;22(8):1289–305 doi 10.1038/s41593-019-0430-3. [DOI] [PubMed] [Google Scholar]
- 8.Bellinger DL, Lorton D. Autonomic regulation of cellular immune function. Auton Neurosci 2014;182:15–41 doi 10.1016/j.autneu.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 9.Estrada LD, Agac D, Farrar JD. Sympathetic neural signaling via the beta2-adrenergic receptor suppresses T-cell receptor-mediated human and mouse CD8(+) T-cell effector function. Eur J Immunol 2016;46(8):1948–58 doi 10.1002/eji.201646395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grebe KM, Hickman HD, Irvine KR, Takeda K, Bennink JR, Yewdell JW. Sympathetic nervous system control of anti-influenza CD8+ T cell responses. Proc Natl Acad Sci U S A 2009;106(13):5300–5 doi 10.1073/pnas.0808851106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Andersen BL, Yang HC, Farrar WB, Golden-Kreutz DM, Emery CF, Thornton LM, et al. Psychologic intervention improves survival for breast cancer patients: a randomized clinical trial. Cancer 2008;113(12):3450–8 doi 10.1002/cncr.23969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kokolus KM, Capitano ML, Lee CT, Eng JW, Waight JD, Hylander BL, et al. Baseline tumor growth and immune control in laboratory mice are significantly influenced by subthermoneutral housing temperature. Proc Natl Acad Sci U S A 2013;110(50):20176–81 doi 10.1073/pnas.1304291110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leigh ND, Kokolus KM, O’Neill RE, Du W, Eng JW, Qiu J, et al. Housing Temperature-Induced Stress Is Suppressing Murine Graft-versus-Host Disease through beta2-Adrenergic Receptor Signaling. J Immunol 2015;195(10):5045–54 doi 10.4049/jimmunol.1500700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mohammadpour H, MacDonald CR, Qiao G, Chen M, Dong B, Hylander BL, et al. beta2 adrenergic receptor-mediated signaling regulates the immunosuppressive potential of myeloid-derived suppressor cells. J Clin Invest 2019;129(12):5537–52 doi 10.1172/JCI129502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bucsek MJ, Qiao G, MacDonald CR, Giridharan T, Evans L, Niedzwecki B, et al. beta-Adrenergic Signaling in Mice Housed at Standard Temperatures Suppresses an Effector Phenotype in CD8(+) T Cells and Undermines Checkpoint Inhibitor Therapy. Cancer Res 2017;77(20):5639–51 doi 10.1158/0008-5472.CAN-17-0546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hylander BL, Repasky EA. Thermoneutrality, Mice, and Cancer: A Heated Opinion. Trends Cancer 2016;2(4):166–75 doi 10.1016/j.trecan.2016.03.005. [DOI] [PubMed] [Google Scholar]
- 17.Eng JW, Reed CB, Kokolus KM, Pitoniak R, Utley A, Bucsek MJ, et al. Housing temperature-induced stress drives therapeutic resistance in murine tumour models through beta2-adrenergic receptor activation. Nat Commun 2015;6:6426 doi 10.1038/ncomms7426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qiao G, Bucsek MJ, Winder NM, Chen M, Giridharan T, Olejniczak SH, et al. beta-Adrenergic signaling blocks murine CD8(+) T-cell metabolic reprogramming during activation: a mechanism for immunosuppression by adrenergic stress. Cancer Immunol Immunother 2019;68(1):11–22 doi 10.1007/s00262-018-2243-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen M, Qiao G, Hylander BL, Mohammadpour H, Wang XY, Subjeck JR, et al. Adrenergic stress constrains the development of anti-tumor immunity and abscopal responses following local radiation. Nat Commun 2020;11(1):1821 doi 10.1038/s41467-020-15676-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wherry EJ. T cell exhaustion. Nat Immunol 2011;12(6):492–9 doi 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- 21.Beltra JC, Manne S, Abdel-Hakeem MS, Kurachi M, Giles JR, Chen Z, et al. Developmental Relationships of Four Exhausted CD8(+) T Cell Subsets Reveals Underlying Transcriptional and Epigenetic Landscape Control Mechanisms. Immunity 2020;52(5):825–41e8 doi 10.1016/j.immuni.2020.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, et al. Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol 2019;20(3):326–36 doi 10.1038/s41590-019-0312-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Antoni MH, Dhabhar FS. The impact of psychosocial stress and stress management on immune responses in patients with cancer. Cancer 2019;125(9):1417–31 doi 10.1002/cncr.31943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lutgendorf SK, DeGeest K, Dahmoush L, Farley D, Penedo F, Bender D, et al. Social isolation is associated with elevated tumor norepinephrine in ovarian carcinoma patients. Brain Behav Immun 2011;25(2):250–5 doi 10.1016/j.bbi.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun 2016;7:10501 doi 10.1038/ncomms10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kallies A, Zehn D, Utzschneider DT. Precursor exhausted T cells: key to successful immunotherapy? Nat Rev Immunol 2020;20(2):128–36 doi 10.1038/s41577-019-0223-7. [DOI] [PubMed] [Google Scholar]
- 27.Kurtulus S, Madi A, Escobar G, Klapholz M, Nyman J, Christian E, et al. Checkpoint Blockade Immunotherapy Induces Dynamic Changes in PD-1(−)CD8(+) Tumor-Infiltrating T Cells. Immunity 2019;50(1):181–94e6 doi 10.1016/j.immuni.2018.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Datar I, Sanmamed MF, Wang J, Henick BS, Choi J, Badri T, et al. Expression Analysis and Significance of PD-1, LAG-3, and TIM-3 in Human Non-Small Cell Lung Cancer Using Spatially Resolved and Multiparametric Single-Cell Analysis. Clin Cancer Res 2019;25(15):4663–73 doi 10.1158/1078-0432.CCR-18-4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bengsch B, Johnson AL, Kurachi M, Odorizzi PM, Pauken KE, Attanasio J, et al. Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8(+) T Cell Exhaustion. Immunity 2016;45(2):358–73 doi 10.1016/j.immuni.2016.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vardhana SA, Hwee MA, Berisa M, Wells DK, Yost KE, King B, et al. Impaired mitochondrial oxidative phosphorylation limits the self-renewal of T cells exposed to persistent antigen. Nat Immunol 2020;21(9):1022–33 doi 10.1038/s41590-020-0725-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu YR, Imrichova H, Wang H, Chao T, Xiao Z, Gao M, et al. Disturbed mitochondrial dynamics in CD8(+) TILs reinforce T cell exhaustion. Nat Immunol 2020;21(12):1540–51 doi 10.1038/s41590-020-0793-3. [DOI] [PubMed] [Google Scholar]
- 32.Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 2017;355(6332):1428–33 doi 10.1126/science.aaf1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kamphorst AO, Wieland A, Nasti T, Yang S, Zhang R, Barber DL, et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science 2017;355(6332):1423–7 doi 10.1126/science.aaf0683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Klein Geltink RI, O’Sullivan D, Corrado M, Bremser A, Buck MD, Buescher JM, et al. Mitochondrial Priming by CD28. Cell 2017;171(2):385–97e11 doi 10.1016/j.cell.2017.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC, et al. The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immunity 2016;45(2):374–88 doi 10.1016/j.immuni.2016.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scharping NE, Rivadeneira DB, Menk AV, Vignali PDA, Ford BR, Rittenhouse NL, et al. Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat Immunol 2021. doi 10.1038/s41590-020-00834-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC, et al. The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immunity 2016;45(3):701–3 doi 10.1016/j.immuni.2016.08.009. [DOI] [PubMed] [Google Scholar]
- 38.Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015;162(6):1229–41 doi 10.1016/j.cell.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 2015;162(6):1217–28 doi 10.1016/j.cell.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Menk AV, Scharping NE, Moreci RS, Zeng X, Guy C, Salvatore S, et al. Early TCR Signaling Induces Rapid Aerobic Glycolysis Enabling Distinct Acute T Cell Effector Functions. Cell Rep 2018;22(6):1509–21 doi 10.1016/j.celrep.2018.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hatfield SM, Kjaergaard J, Lukashev D, Schreiber TH, Belikoff B, Abbott R, et al. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci Transl Med 2015;7(277):277ra30 doi 10.1126/scitranslmed.aaa1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Buck MD, O’Sullivan D, Pearce EL. T cell metabolism drives immunity. J Exp Med 2015;212(9):1345–60 doi 10.1084/jem.20151159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med 2018;24(5):541–50 doi 10.1038/s41591-018-0014-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eng JW, Kokolus KM, Reed CB, Hylander BL, Ma WW, Repasky EA. A nervous tumor microenvironment: the impact of adrenergic stress on cancer cells, immunosuppression, and immunotherapeutic response. Cancer Immunol Immunother 2014;63(11):1115–28 doi 10.1007/s00262-014-1617-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qiao G, Chen M, Bucsek MJ, Repasky EA, Hylander BL. Adrenergic Signaling: A Targetable Checkpoint Limiting Development of the Antitumor Immune Response. Front Immunol 2018;9:164 doi 10.3389/fimmu.2018.00164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pfannenstiel LW, Diaz-Montero CM, Tian YF, Scharpf J, Ko JS, Gastman BR. Immune-Checkpoint Blockade Opposes CD8(+) T-cell Suppression in Human and Murine Cancer. Cancer Immunol Res 2019;7(3):510–25 doi 10.1158/2326-6066.CIR-18-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kelly PN. CD28 is a critical target for PD-1 blockade. Science 2017;355(6332):1386 doi 10.1126/science.355.6332.1386-b. [DOI] [PubMed] [Google Scholar]
- 48.Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun 2015;6:6692 doi 10.1038/ncomms7692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Siska PJ, van der Windt GJ, Kishton RJ, Cohen S, Eisner W, MacIver NJ, et al. Suppression of Glut1 and Glucose Metabolism by Decreased Akt/mTORC1 Signaling Drives T Cell Impairment in B Cell Leukemia. J Immunol 2016;197(6):2532–40 doi 10.4049/jimmunol.1502464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gemta LF, Siska PJ, Nelson ME, Gao X, Liu X, Locasale JW, et al. Impaired enolase 1 glycolytic activity restrains effector functions of tumor-infiltrating CD8(+) T cells. Sci Immunol 2019;4(31) doi 10.1126/sciimmunol.aap9520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kokolus KM, Zhang Y, Sivik JM, Schmeck C, Zhu J, Repasky EA, et al. Beta blocker use correlates with better overall survival in metastatic melanoma patients and improves the efficacy of immunotherapies in mice. Oncoimmunology 2018;7(3):e1405205 doi 10.1080/2162402X.2017.1405205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hiller JG, Cole SW, Crone EM, Byrne DJ, Shackleford DM, Pang JB, et al. Preoperative beta-Blockade with Propranolol Reduces Biomarkers of Metastasis in Breast Cancer: A Phase II Randomized Trial. Clin Cancer Res 2020;26(8):1803–11 doi 10.1158/1078-0432.CCR-19-2641. [DOI] [PubMed] [Google Scholar]
- 53.Gandhi S, Pandey MR, Attwood K, Ji W, Witkiewicz AK, Knudsen ES, et al. Phase I Clinical Trial of Combination Propranolol and Pembrolizumab in Locally Advanced and Metastatic Melanoma: Safety, Tolerability, and Preliminary Evidence of Antitumor Activity. Clin Cancer Res 2020. doi 10.1158/1078-0432.CCR-20-2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.