

Abstract
Cyclooxygenase-2 (COX-2) is up-regulated by redox imbalance and is considered a target for cancer therapy. The rationale of the COX-2 inhibitor lies in suppressing COX-2 catalyzed peroxidation of omega-6 polyunsaturated fatty acids (PUFAs), which are essential and pervasive in our daily diet. However, COX-2 inhibitors fail to improve cancer patients’ survival and may lead to severe side effects. Here, instead of directly inhibiting COX-2, we utilize a small molecule, iminodibenzyl, which could reprogram the COX-2 catalyzed omega-6 PUFAs peroxidation in lung cancer by inhibiting delta-5-desaturase (D5D) activity. Iminodibenzyl breaks the conversion from dihomo-γ-linolenic acid (DGLA) to arachidonic acid, resulting in the formation of a distinct byproduct, 8-hydroxyoctanoic acid, in lung cancer cells and solid tumors. By utilizing COX-2 overexpression in cancer, the combination of DGLA supplementation and iminodibenzyl suppressed YAP1/TAZ pathway, decreasing the tumor size and lung metastasis in nude mice and C57BL/6 mice. This D5D inhibition-based strategy selectively damaged lung cancer cells with a high COX-2 level, whereas it could avoid harassing normal lung epithelial cells. This finding challenged the COX-2 redox basis in cancer, providing a new direction for developing omega-6 (DGLA)-based diet/regimen in lung cancer therapy.
Keywords: omega-6 polyunsaturated fatty acids, cyclooxygenase-2, lung cancer, dihomo-γ-linolenic acid peroxidation, delta-5-desaturase, 8-hydroxyoctanoic acid
1. Introduction
Lung cancer is the second most common cancer and the leading cause of death worldwide [1]. The occurrence of lung cancer has been found associated with several risk factors, including cigarette smoking, pollution, and unhealthy diets [2]. Although cigarette smoking is still the leading risk factor of lung cancer, many shreds of evidence indicated that unbalanced fatty acid uptake also could significantly raise the risk of lung cancer by inducing chronic inflammation and oxidative stress [3–5]. The excessive uptake of omega-6 polyunsaturated fatty acids (PUFAs) has been recognized as an inflammation and cancer inducer [6]. Arachidonic acid (AA) is one of the omega-6 PUFAs that can be catalyzed by cyclooxygenase-2 (COX-2) to form prostaglandin E2 (PGE2), resulting in increased migration and invasion of cancer cells. Therefore, the overexpressed COX-2 is considered as the target for a variety of cancer therapy [7]. However, current direct COX-2 small molecule inhibitors fail to improve the overall survival of cancer patients in clinical studies [8–10]. Furthermore, celecoxib (a classical COX-2 inhibitor) has been reported that could significantly increase cardiovascular risk in cancer patients [11]. The undesirable response of the COX-2 inhibitor on cancer patients ignites the enthusiasm of researchers to zero in on new approaches for regulating the overexpressed COX-2 in cancer.
Instead of using celecoxib or other selective COX-2 inhibitors, we have established a new strategy that redirects COX-2 catalyzed dihomo-γ-linolenic acid (DGLA) peroxidation by knocking down delta-5-desaturase (D5D) in cancer cells. DGLA is one of the downstream omega-6 PUFAs of linoleic acid, which is an essential fatty acid that highly abundant in most Western diets. Genetically knocking down D5D (via siRNA or shRNA) leads to less formation of AA and PGE2 from DGLA, whereas it promotes the production of a higher content of 8-hydroxyoctanoic acid (8-HOA) [12–17]. We have previously shown that 8-HOA could inhibit colon, pancreatic, and breast cancers in which COX-2 overexpression is prominent [12,13,18]. Similarly, studies have demonstrated that more than half of cases of lung cancer patients have been detected with overexpression of COX-2 [9]. Thus, we hypothesis that lung cancer will also be susceptible to D5D inhibition. Therefore, continued research into a small molecule for suppressing D5D activity in lung cancer is essential to bolster the clinical benefit of this novel therapeutical strategy to patients.
In this study, we evaluated the effect of iminodibenzyl (10,11-dihydro-5H-dibenz[b,f]azepine) on lung cancer in vitro and in vivo. Iminodibenzyl is a pharmaceutical intermediate for synthesizing carbamazepine, which is a tricyclic antidepressant. However, iminodibenzyl itself is only considered as an impurity without specific pharmacological activity [19]. The results of the present study for the first time revealed the effect of iminodibenzyl on COX-2 catalyzed DGLA peroxidation in lung cancer. We showed that iminodibenzyl inhibited AA formation, while it promoted endogenous 8-HOA formation through subverting DGLA peroxidation in lung cancer. Consistent with the effect of exogenous 8-HOA, iminodibenzyl along with DGLA significantly suppressed lung tumor growth and metastasis by regulating cancer cell apoptosis, proliferation, and migration. This study provided a new concept of COX-2 biology in lung cancer therapy by taking advantage of the fact that lung cancer cells overexpress COX-2, and thus are more susceptible to the strategy of pharmacological D5D inhibition and omega-6 fatty acid manipulation. It is an innovative attempt to develop a small molecule therapy and omega-6 (DGLA)-based regimen for lung cancer as omega-6 fatty acids are essential and pervasive in our daily diet.
2. Materials and methods
2.1. Materials, reagents, and cell lines
A549 (CCL-18, non-small cell lung cancer cell line, human), Lewis lung carcinoma (LLC, CRL-1642), and BEAS-2B (CRL-9609, human normal bronchial epithelium) were purchased from American Type Culture Collection (ATCC, VA, USA). Iminodibenzyl (10,11-Dihydro-5H-dibenz[b,f]azepine, Catalog: I1308, CAS Number: 494–19-9) and 8-HOA (Catalog: 55999, CAS Number: 764–89-6) were obtained from Sigma-Aldrich (MO, USA). AA, DGLA, corresponding internal standards (AA-d8, DGLA-d6), and DGLA ethyl ester (#0531920–3), were acquired from Cayman Chemical (MI, USA). Primary antibodies for Western analysis were acquired from Cell Signaling (MA, USA). Goat anti-Rabbit/-Mouse IgG Secondary Antibodies (IRDye 800 CW) were acquired from LI-COR Biosciences (NE, USA). HyClone Dulbecco’s Modified Eagle’s Medium (DMEM) and Cytiva fetal bovine serum (FBS) were obtained from Hyclone Laboratories (GE Healthcare, IL, USA). Antibodies for immunofluorescence analysis were bought from Abcam (MA, USA). C18 ODS reverse phase SPE cartridge (SampliQ Silica) was purchased from Agilent Technology (CA, USA). DAPI with Fluoro-Gel II was acquired from Electron Microscopy Sciences (PA, USA). RIPA lysis buffer (REF: 89900) and BCA protein assay kit (REF: 23225) were obtained from Thermo Fisher Scientific (IL, USA). 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT, L11939) was purchased from Alfa Aesar (MA, USA).
2.2. Determining cell viability by MTT assay
The concentration and treatment period of 8-HOA and the combination of DGLA and iminodibenzyl for the cell study were optimized by MTT assay. Briefly, about 8000 cells were seeded to 96-well plates for overnight culture. 8-HOA (0.01 to 100 μM), DGLA (10 to 100 μM), and iminodibenzyl (1 to 100 μM) were then added to A549 cells for the different period as the assigned group. At end of the treatment, each well was added with 10 μL 0.5% (w/v) MTT for 3 h. Then, the MTT solution was removed and replaced with DMSO. The cell viability in each group was normalized by reading at 570/650 nm in the vehicle group as we previously described [15].
2.3. Evaluating cell survival by colony formation assay
About 1,000 A549 cells were seeded into the 6-well plates and randomly assigned into groups treated with 8-HOA (1 μM), cisplatin (1.5 μM), DGLA (100 μM), and iminodibenzyl (10 μM). After treatment, cells were replaced into the fresh DMEM medium for another 10 days. The colonies were stained by crystal violet at end of incubation. The survival fraction was determined by the established protocol [12,15].
2.4. Transwell assay
At end of the treatment, 5 × 104 cells were collected into the Costar transwell chamber (6.5 mm insert, 8.0 μm polycarbonate membrane) overnight. The upper chamber was then filled with FBS-free DMEM medium, whereas the lower chamber was filled with DMEM with 10% (v/v) FBS. After 48 h, cells were fixed and then stained in crystal violet solution as described [13]. The colonies in the inner side of the upper chamber were gently wiped out by cotton sticks. The number of migrated colonies were counted under Lecia Microsystems. The migration rate can be determined by comparing the cells in the treatment group over cells in the vehicle group) [15].
2.5. Wound healing assay
The wound on each well was created by gently scratching with a sterile pipette in 6 well plates. The dislodged cells were removed and replaced by the DMED with 1% (v/v) FBS as described in the previous study [13]. Wells of each plate were randomly assigned to the treatment of 8-HOA (1 μM), cisplatin (1.5 μM), DGLA (100 μM), and iminodibenzyl (10 μM). The wound area of each well was measured by using ImageJ at 0, 24, and 48 h of post-treatment, respectively. The wound healing rate was determined by the wound area at different time points versus the wound area at 0 h from the same group [13].
2.6. Mouse tumor models to assess the effect of iminodibenzyl
Nude mice (nu/nu, 20–25 g, six weeks) and C57BL/6 mice were obtained from the Jackson Laboratory. Once mice arrived, they were immediately housed in a pathogen-free IVC System (San Diego, CA) as we described before [20]. All the animal studies were approved by the NDSU IACUC (Institutional Animal Care and Use Committee). About 2 × 106 A549 cells (or 5 × 105 LLC cells) were injected into the hind flank of the nude or C57BL/6 mice. The mice were then randomly assigned for treatments: vehicle, DGLA (5 mg/mouse, oral gavage, 20ga polypropylene feeding tubes), iminodibenzyl (15 mg/kg, intraperitoneal injection), and DGLA+ iminodibenzyl for once per day. Depending on the speed of tumor growth, the treatment begins at two weeks of injection of A549 cells in nude mice and one week of injection of LLC cells in C57BL/6 mice. The body weight was recorded weekly since the treatment beginning period. At the end of the treatment, hematoxylin and eosin (H&E) staining was conducted by Advanced Imaging & Microscopy Laboratory at North Dakota State University. The images were acquired from Leica Microsystems as we described [16].
2.7. Monitoring tumor size
The tumor size was measured twice a week. The relative tumor volume was calculated by the formula: L x W2/2 (L: longest axis; W: shortest axis). Vevo 3100 VisualSonics Imaging System was employed to scan the tumor area. At end of the treatment period, blood, tumors, and lungs were isolated from executed mice for further analysis [20].
2.8. Western blot analysis
The cell lysate was prepared in RIPA buffer with the addition of phosphatase/protease inhibitors. The protein concentration was determined by BCA Protein Assay Kit (Thermo Fisher). By mixing with the Laemmli sample buffer, about 80 μg proteins were denatured at 95 °C for 5 min and loaded in 4–15% (v/v) TGX Gels (Bio-Rad) with a constant voltage of 80 V. The gel was then transferred to PVDF membrane in a 30 min standard program in the Turbo Transfer System. The membrane was blocked by 5% (v/v) non-fat dry milk. Then it was incubated with primary antibody overnight at 4°C and subjected to corresponding secondary antibodies for 1 h. Protein signal was captured by Li-Cor Odyssey XL System. ImageJ or Image Studio v.5.2 software was used for densitometry analysis [15].
2.9. LC-MS measured DGLA and AA
Extraction:
Cells and 1 mL cell culture medium (or 1 mL tissue lysate) were harvested and mixed with methanol to generate a 15% (v/v) methanol solution. The DGLA-d6 and AA-d8 were employed as internal standards. The samples were then vortexed and centrifuged (3000 rpm for 15 min) twice to separate the supernatant. The pH of the supernatant was adjusted to 3 by using hydrochloric acid and then subjected to solid-phase extraction under SampliQ Silica C18 ODS cartridge as previously described [13,14,17,21]. Free fatty acids were washed out by 2 mL ethyl acetate, dried in a vacufuge concentrator, and then reconstituted with 100 μL ethanol.
LC-MS analysis:
100 μL samples were injected into the Agilent 1200 series HPLC system with LC-MSD SL to determine the concentration of DGLA and AA. The internal standard curve was made from a series of internal standards in different concentrations. Samples (5 μL) were separated in LC with the Zorbax Eclipse-XDR C18 column (3.5 μm, 4.6 × 75 mm) at a flow rate of 0.8 mL/min. The mobile phase (A: water with 0.01% (v/v) acetic acid; B: acetonitrile with 0.01% (v/v) acetic acid): 68% A and 32% B for 0–12 min (isocratic), 68 to 44% A and 32 to 56% B for 12–14 min, 44% A and 56% B for 14–28 min (isocratic), 44 to 14% A and 56 to 86% B for 28–30 min, 14 to 4% A and 86 to 95% B for 30–38 min, 5% A and 95% B for 38–44 min (isocratic). The MS setting consisted of electrospray (negative mode), total ion current ion in full mass scan mode (m/z 50 to 600), dry temperature (325°C), gas flow (5 L/min), compound stability (20%), nebulizer press (15 psi), number of scans (50). The peak area of DGLA-d6, AA-d8, DGLA, and AA were determined at extracted ion current with m/z 311, 311, 30be5, and 303, respectively. The concentration of DGLA and AA were assessed by comparing the ratio of sample peak area/ internal standard peak area with the internal standard curve [14,15].
2.10. GC-MS for 8-HOA analysis
Cells and 1 mL medium (or 1 mL tissue lysate for in vivo sample) were harvested and mixed with 500 uL methanol with hexanoic acid (as internal standard), 50 uL 1 N hydrochloric acid, and 3 mL dichloromethane. The sample solution was vortexed and centrifuged twice to separate the lower organic layer. The dichloromethane layer was dried in a vacufuge concentrator. Samples were then reconstituted in diisopropylethylamine (50 uL, 1.0% (v/v)), and incubated with PFB-bromide in acetonitrile (1.0% (v/v)) for 30 min at 37 °C. 2 μL sample was injected to Agilent 7890A gas chromatograph (GC oven temperature: 60 to 300°C at 25°C/min; injector and transfer line: 280°C). The source temperature of the Mass selective detector is 230°C. The internal standard curve was made by preparing mixtures of 8-HOA and hexanoic acid in a series of concentrations. Extracted ion current with m/z 181 was used for assessing the peak area of both hexanoic acid and 8-HOA PFB derivatives. The concentration of 8-HOA was determined by comparing the ratio of 8-HOA peak area/ hexanoic acid peak area with the internal standard curve as previously described [13–17,21].
2.11. Immunofluorescence analysis
Cells were seeded to μ-Slide 8 Well (ibidi) with the treatment for 48 h, washed with PBS, and then fixed by using 4% (v/v) paraformaldehyde for 20 min. 0.15% (v/v) Triton X-100 was used to permeabilize the cell. The blocking was conducted by incubating cells with BSA for blocking. Then cells were incubated with primary antibodies, then corresponding secondary antibodies for 1 h. Actin filaments were stained by Phalloidin 488. DAPI was added for staining nuclear. The signals were detected by using Carl Zeiss LSM900 confocal microscopy with Airyscan 2. The tissues were made into the paraffin section as established protocol [15,16]. The sections were then subjected to xylene and ethanol in gradient concentrations for deparaffinization and rehydration. Antigen retrieval was conducted by boiling sections in the sodium citrate buffer. Permeabilization buffer containing 1% (v/v) serum and 0.4% (v/v) Triton X-100 was added for 30 min. Then sections were incubated with primary antibody overnight at 4°C and subjected to corresponding secondary antibodies for 1 h. DAPI was added for staining nuclear. The signals in tissues were detected by using a Zeiss Axio Imager M2 microscope (20x/0.75). The positive rate of Ki67 and cleaved caspase-3 was calculated in the formula of the number of Ki67 or cleaved caspase-3 positive cells over the number of total cells. The relative intensity of COX-2 and D5D in tissues was evaluated by Image-Pro software (Media Cybernetics) [15,16].
2.12. Quantification of ALT and AST activity
ALT and AST activity in mouse serum were measured by AST or ALT activity assay kit (CAT# K752–100, K753–100, BioVision). The mice serum was acquired by centrifuging blood (2500 rpm, 10 min). The production of pyruvate (catalyzed by ALT) or glutamate (catalyzed by AST) was detected at λ = 570 nm as the instruction manual.
2.13. Statistical analysis
Statistical analyses were conducted using Prism 5 (GraphPad Software). The complete randomized design was applied for grouping in vitro and in vivo. Means ± standard error of the mean (SEM) was used to present all the data. Statistical hypothesis tests were done by Student’s t-test or one-way ANOVA test in Tukey’s method. Statistical significance was indicated by differences with the minimum of P < 0.05.
3. Results
3.1. 8-HOA regulated A549 lung cancer cell growth, migration, and apoptosis
In the initial analysis, we examined the effect of exogenous 8-HOA on lung cancer cells to assure the efficacy of the D5D inhibition-based strategy. To optimize the dose and treatment period of 8-HOA on lung cancer cells, we used MTT assay to determine the cell viability of A549 cells by treating them with 0.01, 0.1, 1, 10, 100 μM 8-HOA for 6, 12, 24, and 48 h, respectively. We observed significantly lower cell viability on A549 cells treated with 1 μM 8-HOA for 48 h (p<0.05, Fig. 1A). However, increasing the dose of 8-HOA from 1 μM to 100 μM did not significantly improve the inhibitory effect of 8-HOA on A549 cell viability (Fig. 1B). To achieve the maximum anti-cancer effect of 8-HOA and avoid overdose, we utilized 1 μM 8-HOA for 48 h for all the experimental procedures. To evaluate the effect of 8-HOA on lung cancer cell survival, we performed the colony formation assay in A549 cells incubated with 8-HOA and/or cisplatin (first-line standard chemotherapy) [22]. In contrast to the vehicle group, 8-HOA (1 μM) alone or combined with cisplatin (1.5 μM) significantly suppressed the survival fraction in A549 cells (p<0.01, Fig. 1C). Additionally, the combination of 8-HOA and cisplatin resulted in the formation of fewer colonies of A549 cells than 8-HOA or cisplatin alone (Fig. 1C). Based on the similar effect shown in Fig. S1A (in different dose combinations), we calculated the Combination index based on dose (CId) to evaluate the combined effect of cisplatin and 8-HOA on the survival fraction of A549 cells. As a summary in Fig. S1B, both of CId for 8-HOA and cisplatin were larger than 1, indicating the effect of 8-HOA and cisplatin on survival fraction was synergistic. Additionally, to elucidate the effect of 8-HOA on lung cancer cell migration, we also performed the transwell migration assay and wound healing assay on A549 cells. After 48 h treatment of 8-HOA and cisplatin, less than 50% of A549 cells have been observed that could cross the membrane on the bottom of the transwell chamber, suggesting a significantly lower migration rate compare to A549 cells treated with DGLA or cisplatin alone (p<0.01, Fig. 1D). Moreover, from wound healing assay, we observed significantly slower healing speed when the A549 cells were treated with 8-HOA, suggesting the inhibition of cell migration by 8-HOA (p<0.01, Fig. 1E). Meanwhile, the combination of 8-HOA and cisplatin resulted in a significantly larger wound area at 24 and 48 h than the treatment of 8-HOA or cisplatin alone, indicating that 8-HOA could improve the effectiveness of cisplatin on suppressing lung cancer cell proliferation (p<0.01, Fig. 1E).
Fig. 1. 8-hydroxyoctanoic acid (8-HOA) inhibited the growth of A549 lung cancer cells and enhanced the effectiveness of cisplatin.
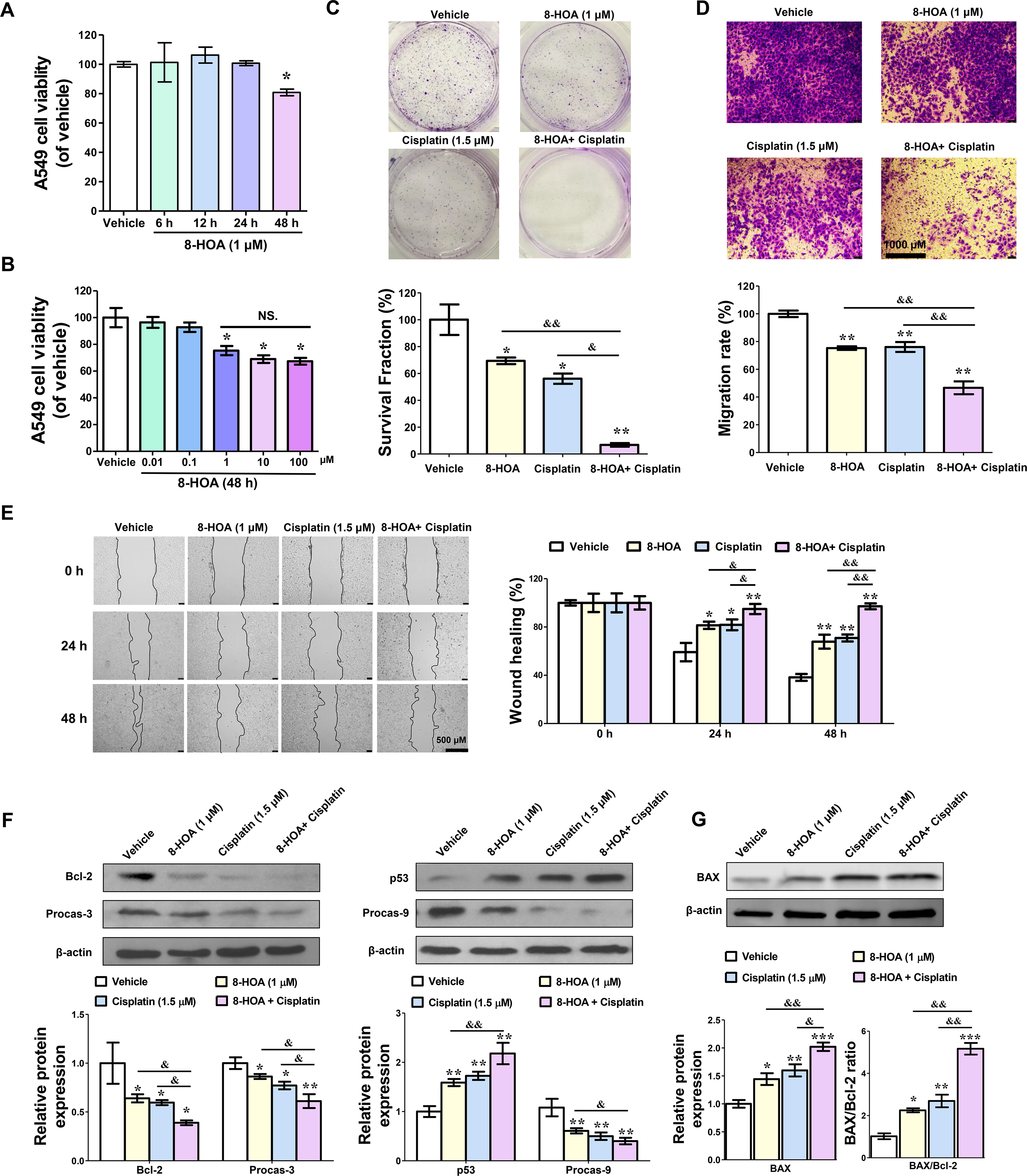
(A) The cell viability of A549 cells treated with 1 μM 8-HOA for 6, 12, 24, and 48 h. (B) The cell viability of A549 cells treated with 0.01, 0.1, 1, 10, and 100 μM 8-HOA for 48 h. (C) Colony formation of A549 cells treated with 1 μM 8-HOA and 1.5 μM cisplatin. The survival fraction (%) of A549 cells was normalized by vehicle group. (D) Transwell migration assay of A549 cells treated with 1 μM 8-HOA and 1.5 μM cisplatin. The migration rate (%) of A549 cells was normalized by vehicle group. (E) Wound healing assay of A549 cells treated with 8-HOA and cisplatin. The healing rate (%) of A549 cells was normalized by vehicle group. (F) Bcl-2, Procaspase-3, p53, and Procaspase-9 protein expression in A549 cells treated with 8-HOA and cisplatin for 48 h. Relative protein expression was normalized with β-actin. Data represent mean ± SEM for n=6. (G) BAX protein expression in A549 cells after treatment (n=3). The ratio of expression of BAX to Bcl-2 was normalized with the vehicle group with the corresponding β-actin. ***P<0.001, **P<0.01, *P<0.05 vs vehicle group; &&P<0.01, &P<0.05 vs 8-HOA+ Cisplatin group; NS.= not significant.
We have previously demonstrated that 8-HOA could induce apoptosis in HCA-7 colony 29 colon cancer cells [18,20]. The platinum-based chemotherapy, cisplatin, could also mediate apoptosis through DNA damage pathways, such as p53 dependent apoptosis pathway [22–25]. Therefore, we hypothesis that the synergy effect of cisplatin and 8-HOA on lung cancer could be attributed to their concerted effect on apoptosis. To evaluate the lung cancer cell apoptosis, we measured the apoptosis-related protein expression by Western analysis after administration of 8-HOA. According to our previous study, the effect of 8-HOA is highly associated with tumor suppressor p53 in colon cancer therapy [17]. p53 has been reported that can inhibit the expression of Bcl-2 (an anti-apoptotic regulator) and promote the cleavage of caspase-3 and caspase-9, resulting in apoptosis in cancers [24,26–30]. By Western analysis, we observed that the protein expression of Bcl-2, procaspase-9, and procaspase-3 was significantly decreased in A549 lung cancer cells treated with 8-HOA or 8-HOA+ cisplatin (p<0.05, Fig. 1F). Additionally, the Bcl-2-associated X protein (BAX) expression and BAX/Bcl-2 ratio was significantly increased by 8-HOA (p<0.05) or 8-HOA+ cisplatin (p<0.001, Fig. 1G). However, the p53 protein expression was significantly increased in A549 cells treated with 8-HOA, indicating the up-regulation of the p53-dependent apoptosis pathway (p<0.01, Fig. 1F). Additionally, higher protein expression of p53 was also found in lung cancer cells treated with the 8-HOA+ cisplatin than cells treated with 8-HOA or cisplatin alone (p<0.01, Fig. 1F). Moreover, the kinetic apoptosis assay has been performed on A549 cells by using polarity sensitive indicator of viability & apoptosis (pSIVA) probe and propidium iodide (PI) live staining. 8-HOA and 8-HOA+ cisplatin significantly increased the percentage of pSIVA positive A549 cells (p<0.01, Fig. S4), indicating the activation of apoptosis by 8-HOA and 8-HOA+cisplatin treatment. Furthermore, 8-HOA could enhance the sensitivity of A549 lung cancer cells to cisplatin through amplifying the apoptosis pathway.
3.2. Iminodibenzyl suppressed A549 lung cancer cell growth via promoting 8-HOA formation
Although exogenous 8-HOA suppressed lung cancer growth, we are still not clear whether iminodibenzyl could inhibit cancer development via inducing D5D inhibition and endogenous 8-HOA formation. To optimize the dose and treatment period of iminodibenzyl in vitro, we conducted MTT assay on A549 cells with different dose combinations of iminodibenzyl and DGLA. We found that the combination of 100 μM DGLA and 10 μM iminodibenzyl significantly suppressed lung cancer growth as the cell viability showed in Fig. 2A and 2B (p<0.05). To determine the effect of iminodibenzyl on DGLA peroxidation, we treated A549 cells with iminodibenzyl and DGLA for 48 h. At the end of the treatment, the DGLA, AA, and 8-HOA concentrations were measured by using LC-MS or GC-MS. We observed a low DGLA versus AA ratio and 8-HOA level in the vehicle and DGLA treated A549 cells, indicating A549 cells tend to produce more AA but less 8-HOA from COX-2 catalyzed DGLA peroxidation (Fig. 2C and D). However, the ratio between DGLA and AA in A549 cells was remarkably increased by the addition of iminodibenzyl to DGLA supplementation (p<0.001, Fig. 2C). This shift of the DGLA peroxidation pattern suggested that iminodibenzyl could suppress D5D activity in lung cancer cells, resulting in the formation of 8-HOA (p<0.001, Fig. 2D). Additionally, the combination of DGLA and iminodibenzyl significantly promoted lactate dehydrogenase (LDH) release from A549 cells, suggesting higher cytotoxicity of iminodibenzyl on lung cancer cells (p<0.001, Fig. S2A). To further evaluate the effect of iminodibenzyl on lung cancer cell survival and migration, we performed colony formation, transwell migration, and wound healing assays on A549 cells. The combination of 100 μM DGLA and 10 μM iminodibenzyl significantly inhibited survival fraction (p<0.001) and migration rate (p<0.05) in A549 cells (Fig. 2E and F). The remarkable change of survival fraction, migration rate, and wound healing rate indicated that the combination of DGLA and iminodibenzyl could suppress lung cancer growth and development. However, DGLA or iminodibenzyl alone showed a negligible effect on A549 cell survival and migration, indicating DGLA is essential in iminodibenzyl based lung cancer (Fig. 2E, F, and G). This phenomenon indicates that 8-HOA, an anti-cancer byproduct, could be produced from COX-2 catalyzed PUFA peroxidation when D5D activity was suppressed by iminodibenzyl in lung cancer cells.
Fig. 2. Iminodibenzyl suppressed A549 lung cancer cell growth via promoting 8-HOA formation from dihomo-γ-linolenic acid (DGLA) peroxidation.

(A) The cell viability of A549 cells treated with 10 μM iminodibenzyl and 100 μM DGLA for 6, 12, 24, and 48 h. (B) The cell viability of A549 cells treated with different concentrations of iminodibenzyl and DGLA for 48 h. (C) DGLA/arachidonic acid (AA) ratio of iminodibenzyl treated A549 cells. A549 cells were treated with DGLA (100 μM), iminodibenzyl (10 μM), and DGLA plus iminodibenzyl for 48 h. (D) GC-MS quantification of 8-HOA from cell suspension containing 1.0 × 106 A549 cells after iminodibenzyl and DGLA treatment for 48 h. (E) Colony formation of A549 cells treated with iminodibenzyl and DGLA for 48 h. The survival fraction of A549 cells was normalized by vehicle group from colony formation assay. (F) Transwell migration assay of A549 cells treated with iminodibenzyl and DGLA. The migration rate (%) of A549 cells was normalized by vehicle group from the transwell migration assay. (G) Wound healing assay of A549 cells treated with iminodibenzyl and DGLA. The healing rate (%) of A549 cells was normalized by vehicle group from wound healing assay. Data represent mean ± SEM for n=6. ***P<0.001, **P<0.01, *P<0.05 vs vehicle group; &&&P<0.001, &&P<0.01, &P<0.05 vs DGLA group.
In contrast to COX-2 overexpression in A549 lung cancer cells, BEAS-2B is a normal human lung epithelial cell line with low to no COX-2 expression as shown in the Western analysis (p<0.001, Fig. S3A). To better understand the role of iminodibenzyl in COX-2 catalyzed DGLA peroxidation, we also employed the BEAS-2B cell as a negative control. Unsurprisingly, we did not find any significant effect of iminodibenzyl and/or DGLA (same dose as administrated to A549) on cytotoxicity, cell viability, and survival fraction of BEAS-2B cell (Fig. S3B to D). This result suggested that COX-2 overexpression is a required condition to exhibit the effectiveness of iminodibenzyl on lung cancer cells. Considering the differential effects of iminodibenzyl on A549 and BEAS-2B cells, we may conclude that not only could iminodibenzyl specifically suppress the growth of lung cancer cells with COX-2 overexpression by promoting 8-HOA formation, but also it would avoid directly affecting normal lung cells, which normally have less COX-2 expression.
3.3. Iminodibenzyl and DGLA promoted lung cancer cell apoptosis through downregulating the YAP1/TAZ pathway
Since iminodibenzyl could trigger the formation of 8-HOA as shown in Fig. 2D, it is plausible that apoptosis will also be promoted in lung cancer cells in the presence of iminodibenzyl and DGLA. To evaluate the effect of iminodibenzyl and DGLA on apoptosis, we measured the expression of the apoptosis-related protein in A549 cells by Western analysis. The protein expression of Bcl-2, procaspase-3, and procaspase-9 was significantly suppressed in A549 cells incubated with DGLA and iminodibenzyl, whereas p53 was significantly upregulated, indicating the combination of DGLA and iminodibenzyl could activate the p53 dependent apoptosis pathway in lung cancer cells (p<0.05, Fig. 3A). In the progression of apoptosis, the cleavage of procaspase-3 to cleaved caspase-3 is the crucial step of apoptosis activation [31,32]. To determine the role of iminodibenzyl in lung cancer apoptosis, the cleaved caspase-3 positive A549 cells were quantified by immunofluorescence analysis. We observed that the percentage of cleaved caspase-3 positive A549 cells significantly increased to more than 45% (versus ~1% in the vehicle group) after the treatment of DGLA plus iminodibenzyl (p<0.001, Fig. 3B). To explore the effect of iminodibenzyl on ferroptosis, we assessed the mRNA and protein expression of glutathione peroxidase 4 (GPX4) and cystine/glutamate antiporter xCT (SLC7A11) in A549 cells by qPCR and Western analysis. The combination of DGLA and iminodibenzyl did not change the mRNA expression of GPX4 and SLC7A11 in A549 cells (Fig. S6A and B). Interestingly, DGLA alone significantly decreased the protein expression of GPX4 (p<0.05, Fig. S6C) and SLC7A11 (p<0.001, Fig. S6D). However, the addition of iminodibenzyl significantly restored the expression of these two ferroptosis markers (p<0.01, Fig. S6C and D), indicating that the effect of iminodibenzyl on lung cancer is ferroptosis independent. The possible ferroptosis mechanism is shown in Fig. S6E.
Fig. 3. Iminodibenzyl and DGLA promoted apoptosis through downregulating yes-associated protein 1/transcriptional coactivator with PDZ-binding motif (YAP1/TAZ) pathway in A549 lung cancer cell.

(A) Bcl-2, Procaspase-9, Procaspase-3, and p53 protein expression in A549 cells treated with 10 μM iminodibenzyl and 100 μM DGLA for 48 h. Relative protein expression was normalized with β-actin. (B) Immunofluorescence images and quantification of cleaved caspase-3 relative intensity in A549 cells were obtained by confocal microscopy. Expression of cleaved caspase-3 was stained in green, and cell nuclei were counter-stained with DAPI. (C) Protein expression of YAP1 and TAZ in A549 cells treated with iminodibenzyl and DGLA for 48 h. Relative expression of proteins to β-actin in vehicle group was normalized to 1. (D) Protein expression of connective tissue growth factor (CTGF) and cysteine-rich angiogenic inducer 61 (Cyr61) in A549 cells treated with iminodibenzyl and DGLA. Relative expression of proteins to β-actin in vehicle group was normalized to 1. (E) Confocal microscopy images of YAP1 expression in A549 cells treated with iminodibenzyl and DGLA. Data represent mean ± SEM for n=3. ***P<0.001, **P<0.01, *P<0.05 vs vehicle group.
In our previous study, we found that 8-HOA serves as a novel histone deacetylase (HDAC) inhibitor in cancer therapy [15]. Therefore, the inhibition of HDAC activity in A549 cells also could evident the formation of 8-HOA in A549 cells treated with DGLA and iminodibenzyl (Fig. S2B). A study has demonstrated that several classical HDAC inhibitors could activate apoptotic pathways and inhibit the protein/gene expression of yes-associated protein 1/transcriptional coactivator with PDZ-binding motif (YAP1/TAZ), which is a critical pathway for cancer development [33]. The activated YAP1/TAZ could be translocated into nuclei, resulting in the promotion of cancer cell proliferation and survival [34,35]. To determine the effect of DGLA and iminodibenzyl on the YAP1/TAZ pathway, we performed Western and immunofluorescence analysis on A549 lung cancer cells treated with DGLA and iminodibenzyl. The Western analysis suggested that the combination of DGLA and iminodibenzyl could significantly affect the protein expression of YAP1/TAZ (p<0.05, Fig. 3C), phospho-YAP1 (p-YAP1) (p<0.05, Fig. S5), and its downstream molecules connective tissue growth factor (CTGF) and cysteine-rich angiogenic inducer 61 (Cyr61) in A549 cells (p<0.05, Fig. 3D). Additionally, reduced nuclear translocation of YAP1 in DGLA+ iminodibenzyl treated lung cancer cells was confirmed by the immunofluorescence analysis in confocal microscopy (Fig. 3E). Hence, the suppression of the YAP1/TAZ pathway may attribute to the anti-apoptosis effect of DGLA and iminodibenzyl on lung cancer cells.
3.4. Iminodibenzyl and DGLA supplementation suppressed A549 xenograft tumor growth
To evaluate the effect of iminodibenzyl and DGLA on the xenograft lung tumor, we established the subcutaneous lung tumor model by injecting A549 cells to the flank of nude mice (see scheme in Fig. 4A). At the end of the 4 weeks of the treatment, we observed a significant reduction of tumor size in mice treated with iminodibenzyl and the combination of DGLA and iminodibenzyl (p<0.001, Fig. 4B and D). The ultrasound images suggested that the speed of tumor growth was abated since the administration of DGLA and iminodibenzyl (Fig. 4C). To investigate the safety of iminodibenzyl, we monitored the bodyweight and measured the change of aspartate/alanine aminotransferase (AST/ALT) level at the end of the 4 weeks of treatment. We did not observe any toxic effect of iminodibenzyl and DGLA during 4 weeks of treatment on nude mice (Fig. 4E, F, and G). Additionally, Table S1 displayed the concentration of iminodibenzyl in blood, organs, and tumor from nude mice at the end of the treatment.
Fig. 4. Iminodibenzyl and DGLA supplementation suppressed A549 xenograft tumor growth.

(A) Study design for evaluating the anti-cancer effect of iminodibenzyl and DGLA supplementation on nude mice. (B) Relative tumor volume measured twice a week using a digital caliper during the DGLA and iminodibenzyl treatment. (C) Ultrasound images of tumor growth in mice before and after 4-week treatments. Note, day 14 indicates the first day of the treatment after cancer cell implantation. (D) Images of harvested tumors at the end of 4 weeks of treatment. (E) Aspartate aminotransferase (AST) activity colorimetric assay quantification of AST level from the blood. (F) Alanine aminotransferase (ALT) activity colorimetric assay quantification of ALT level from the blood. (G) Bodyweight of nude mice from starting of treatment (14 days) to end of treatment (42 days). Data represent mean ± SEM for n=6. ***P<0.001, *P<0.05 vs vehicle group.
3.5. Iminodibenzyl induced 8-HOA formation in lung tumor tissues
Based on our in vitro results, the continuous production of endogenous 8-HOA required high COX-2 expression in lung cancer cells. Therefore, for in vivo study, we first evaluated the COX-2 and D5D protein expression in lung tumor tissues from mice treated with DGLA and/or iminodibenzyl. In the nude mice model, the Western and immunofluorescence analysis suggested that iminodibenzyl, DGLA, and the combination of them did not change the COX-2 and D5D protein expression levels in tumors (Fig. 5A and B). However, the DGLA versus AA ratio and 8-HOA were significantly up-regulated in tumors from mice treated with the combination of DGLA and iminodibenzyl (p<0.01, Fig. 5C and D). The shift of the DGLA peroxidation manner indicated that iminodibenzyl could inhibit D5D activity in lung tumor tissues without altering the D5D and COX-2 protein expression. To determine the effect of iminodibenzyl on lung cancer proliferation, we conducted immunofluorescence analysis on tumor tissues to count the positive rate of Ki67. Unlike high Ki67 expression in untreated tumors, we observed a significantly lower Ki67 positive rate in tumor tissues from mice treated with iminodibenzyl alone and combined with DGLA, indicating the inhibition of cancer cell proliferation by iminodibenzyl (p<0.001, Fig. 5E). Consistent with the in vitro result, the combination of DGLA and iminodibenzyl also significantly increased the protein expression of p53 (p<0.01), whereas it decreased the expression of Bcl-2, procaspase-9, and procaspase-3 (p<0.05, Fig. 5F). Additionally, the HDAC activity significantly decreased in tumor tissues from the combination of DGLA and iminodibenzyl treated mice (p<0.05, Fig. S2C). The promotion of apoptosis and inhibition of HDAC activity in lung tumor tissues could evident the formation of 8-HOA from COX-2 catalyzed DGLA peroxidation. However, histone acetyltransferases (HAT) activity in tumor tissues did not significantly change by DGLA and iminodibenzyl, indicating that the unbalanced status of histone acetylation and deacetylation was resulted by newly formed 8-HOA in tumors (Fig. S2D).
Fig. 5. Iminodibenzyl and DGLA supplementation induced apoptosis via promoting 8-HOA formation in A549 xenograft tumor from nude mice.
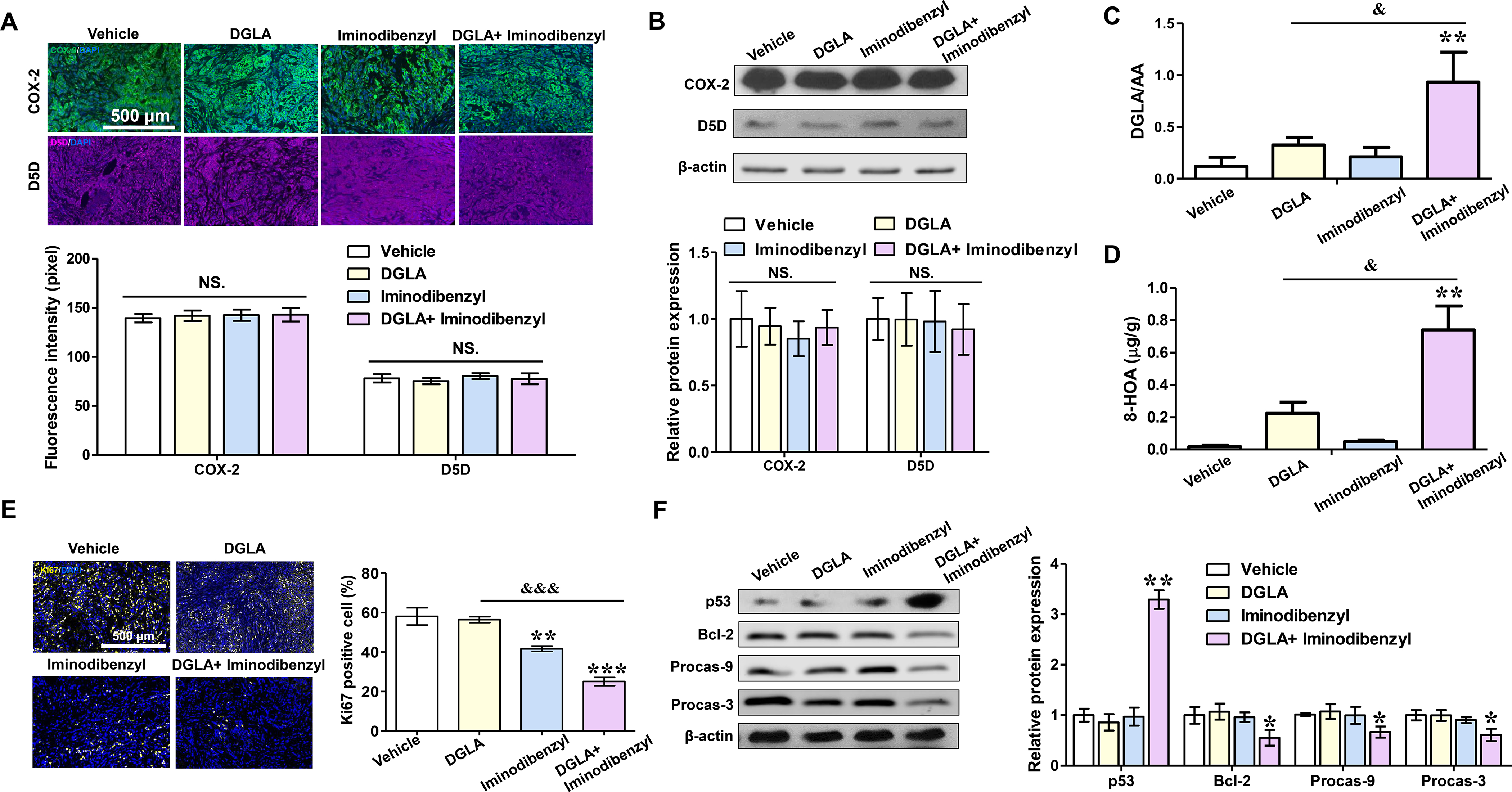
(A) Immunofluorescence analysis of COX-2 and D5D relative intensity in tumor tissues from nude mice treated with DGLA and iminodibenzyl for 4 weeks. Expression of COX-2 was in green, D5D in violet, and cell nuclei were counter-stained with DAPI. (B) Protein expression of COX-2 and D5D in tumor tissues. Relative expression of proteins to β-actin in vehicle group was normalized to 1. (C) DGLA/AA ratio in lung tumor tissues treated with iminodibenzyl and DGLA. (D) Quantification of 8-HOA in tumor tissues from nude mice. (E) Immunofluorescence images and quantification of Ki67 in tumor tissues after 4 weeks of treatment of DGLA and iminodibenzyl. Expression of Ki67 was stained in yellow, and cell nuclei were counter-stained with DAPI. (F) Expression of p53, procaspase-9, procaspase-3, and Bcl-2 in A549 tumors treated with DGLA and iminodibenzyl. Relative protein expression was normalized with β-actin. Data represent mean ± SEM for n=3. ***P<0.001, **P<0.01, *P<0.05 vs vehicle group; &&&P<0.001, &P<0.05 vs DGLA group; NS.= not significant.
3.6. Iminodibenzyl and DGLA supplementation suppressed syngeneic Lewis lung carcinoma (LLC) tumor growth and metastasis in C57BL/6 mice
To understand the role of iminodibenzyl in lung cancer metastasis, we employed the syngeneic lung cancer model by injecting LLC cells to C57BL/6 mice. After injection of highly tumorigenic LLC cells, the tumor formed on the site of injection in a week and then migrate to the lung in several weeks. The tumor growth curve in Fig. 6A indicated that LLC tumor size in vehicle mice was sharply increased at 24 days after cell injection. However, the speed of tumor growth was significantly slower in mice treated with iminodibenzyl alone (p<0.01) or combined with DGLA (p<0.001, Fig. 6A). Additionally, the tumors in DGLA+ iminodibenzyl treated mice were much smaller when we collected them at end of 4 weeks of treatment, indicating the effectiveness of the combination of DGLA and iminodibenzyl on immunocompetent mice (Fig. 6B). In contrast to the A549 xenograft model on nude mice, the LLC model developed tumor nodules (~2 nodules per lung in the vehicle group) on the lung as shown in isolated lung images and H&E stained lung sections (Fig. 6C). Not only the inhibition of primary tumor growth, but also the suppression of metastasized nodules were found in the lungs from the mice administrated with DGLA and iminodibenzyl together. To understand the mechanism of iminodibenzyl on the LLC model, we analyzed the DGLA peroxidation pattern of LLC tumors and lungs, by LC-MS and GC-MS. The DGLA versus AA ratio and 8-HOA concentration significantly increased in DGLA and iminodibenzyl treated tumors and lung tissues, indicating the decrease of D5D activity in both tumors and lungs (p<0.01, Fig. 6D to G). Not only did iminodibenzyl shifted DGLA peroxidation in vivo, but it significantly increased 8-HOA formation in DGLA treated LLC cells in vitro (p<0.01, Fig. S2F). Additionally, significant suppression of HDAC activity was found in tumor tissues treated with iminodibenzyl (~84% of the vehicle) or combined iminodibenzyl with DGLA (~63% of the vehicle) compared to tumors from mice treated with DGLA alone (p<0.05, Fig. S2E). Both results of HDAC activity assays for the LLC syngeneic lung cancer model and the A549 xenograft lung cancer model indicated that endogenous 8-HOA induced by iminodibenzyl could be served as an HDAC inhibitor for suppressing lung tumor growth. Additionally, Table S1 displayed the concentration of iminodibenzyl in blood, organs, and tumor from C57BL/6 mice at the end of the treatment.
Fig. 6. Iminodibenzyl and DGLA supplementation suppressed syngeneic Lewis lung carcinoma (LLC) tumor growth and metastasis in C57BL/6 mice.

(A) Relative tumor volume measured twice a week using a digital caliper during the DGLA and iminodibenzyl treatment. Note, day 7 indicates the first day of the treatment after cancer cell implantation. (B) Images of harvested tumors at the end of 4 weeks of treatment. (C) Images of lungs (upper panel) and Hematoxylin and eosin (H&E) staining of harvested lungs (lower panel) at the end of 4 weeks of treatment. Arrows indicated the area of tumor nodules on lung tissues. (D) DGLA/AA ratio in tumor tissues from C57BL/6 mice. (E) DGLA/AA ratio in lung tissues from C57BL/6 mice. (F) 8-HOA formation in tumor tissues from C57BL/6 mice treated with DGLA and iminodibenzyl. (G) 8-HOA formation in lung tissues from C57BL/6 mice treated with DGLA and iminodibenzyl. Data represent mean ± SEM for n=6. ***P<0.001, **P<0.01, vs vehicle group; &&&P<0.001, &P<0.05 vs DGLA group.
4. Discussion
Cancer cells are the vital sources of free radicals and inflammatory mediators, such as AA, prostaglandins (PGs), and cytokines. Elevated inflammatory mediators could further lead to cancer proliferation, migration, and invasion [36–41]. COX-2 is a membrane-bound and rate-limiting enzyme that triggers inflammation via catalyzing PUFAs peroxidation and PGs formation [42–45]. Many studies suggested PGs in cancer, especially PGE2, could bind to the PGE2 receptor, resulting in inhibition of apoptosis, promotion of migration, angiogenesis, and invasion through PI3K/AKT, Rac, matrix metalloproteinases (MMPs), and coactivation of epidermal growth factor receptor (EGFR) signaling in cancer cells [46–49]. All these studies highlight the role of COX-2 in inflammatory cancer cells and indicate the pertinent clinical impact of COX-2 inhibition therapies.
However, several COX-2 inhibitors have not achieved the desired clinical effect on cancers [8,10,11]. Although high COX-2 expression has been identified in most non-small cell lung cancer patients, adding celecoxib to standard chemotherapy does not improve the patient’s survival, irrespective of COX-2 expression [9]. The failure is likely due to the activation of the feedback loop of the COX-2 pathway. As an inducible form of COX, COX-2 might be upregulated by the COX-2 inhibitor itself through a negative feedback loop, resulting in the opposite effect of COX-2 inhibitors [47,50]. Another concern is the dose of celecoxib in clinical trials. A phase I study suggested that only a high dose of celecoxib (600 to 800 mg) could suppress the concentration of PGE-M (a urinary metabolite of PGE2) in cancer patients. Conversely, 400 mg is considered as the maximum dose of celecoxib for pain control in inflammatory diseases [9,51]. Furthermore, studies suggested that the high dose of COX-2 inhibitor may lead to severe cardiovascular side effects, such as myocardial infarction [11].
To avoid the feedback upregulation and side effects of COX-2 inhibitor in cancer therapy, we have demonstrated a novel strategy that could reprogram COX-2 catalyzed lipid peroxidation, resulting in a decrease of AA formation in colon, pancreatic, breast, and lung cancers. Instead of directly inhibit COX-2, we previously found that genetically knocking down D5D via siRNA, shRNA, or RNA nanoparticle could break the conversion of DGLA to AA in cancer cells [12,14–16,52]. Interestingly, COX-2 could directly catalyze DGLA to form an anti-cancer byproduct 8-HOA, leading to suppression of cancer cell proliferation, migration, invasion, but induction of apoptosis [18]. Further, we have demonstrated that exogenous 8-HOA also could enhance the efficacy of chemotherapy (fluorouracil, gemcitabine, and cisplatin) for colon and pancreatic cancers in previous studies [18,21,52], and lung cancer in the current study. However, some limitations of RNAi-based therapy are worth noting; competition with endogenous RNA, invocation of innate immune responses, and off-target silencing [53–56]. Hence, to improve the effectiveness of the D5D inhibition strategy, in this study, we identified a small molecule D5D inhibitor (iminodibenzyl) as an alternative for the RNAi-based approach.
Iminodibenzyl is the material and impurity in the synthesis of carbamazepine, which is an anticonvulsant medication. Unlike carbamazepine, iminodibenzyl cannot act on voltage-gated sodium channels for epilepsy and neuropathic pain [19,57]. The biological activity and pharmacokinetics/pharmacodynamics profile of iminodibenzyl are still underexplored. In this study, we tested the effect of iminodibenzyl on COX-2 catalyzed DGLA peroxidation in lung cancer cells. The change of DGLA, AA, and 8-HOA profiles suggested that iminodibenzyl could serve as a promising D5D inhibitor for cancer therapy. Additionally, we observed that the effect of iminodibenzyl on lung cancer is closely associated with the COX-2 status in cells. Iminodibenzyl barely influenced the survival and function of normal lung epithelial cells with low COX-2 expression, whereas it significantly produced toxic effects in lung cancer cells with high COX-2 expression. This characteristic of iminodibenzyl could allow us to expand its clinical benefit to cancer therapy and avoid unwanted side effects, which we believe is the drawback of traditional COX-2 inhibitors.
To understand the role of iminodibenzyl in cancer therapy, we evaluated the effect of iminodibenzyl on the intrinsic pathway of apoptosis and the YAP1/TAZ pathway in A549 lung cancer cells. Intrinsic apoptosis could be triggered by many internal stimuli, such as genetic damage, hypoxia, and oxidative stress in cancers [31,58]. The suppression and resistance of intrinsic apoptosis in cancer cells contribute to carcinogenesis by upregulating anti-apoptotic protein (e.g. Bcl-2) and reducing caspase function [32,59–61]. In this study, both in vivo and in vitro results supported that iminodibenzyl and iminodibenzyl induced 8-HOA could induce apoptosis in lung cancer cells via restoring the balance of Bcl-2 and BAX, increasing the expression of p53, and promoting the cleavage of caspase-3. YAP1 and its coactivator TAZ are the downstream effectors of the Hippo pathway [62]. Several studies have demonstrated that apoptosis could be inhibited by YAP1, which is extensively expressed in tumors and correlated with poor survival [33]. In cancer cells, YAP1/TAZ is translocated to the nuclear and activated when the Hippo pathway was off, resulting in cancer cell proliferation, metastasis, survival, and inhibition of apoptosis [35,63,64]. Our previous study has demonstrated that the 8-HOA, which derived from DGLA by genetically knocking down D5D expression, could suppress the activation of the YAP1/TAZ pathway via inhibiting HDAC activity (possibly class I, II, or IV) [15]. Analogous to the effect of D5D knocking down, our results suggested that iminodibenzyl could significantly downregulate the HDAC activity in lung cancer cells. In addition to HDAC, many studies suggested that the balance between protein acetylation and de-acetylation also could be influenced by other enzymes, such as HAT [65]. However, our results implicated that iminodibenzyl did not significantly change HAT activity, specifying that the effect of iminodibenzyl on de-/acetylation may be primarily through HDAC regulation. Many HDAC inhibitors, including 8-HOA, have been found that they are closely associated with the YAP1/TAZ pathway [15,33]. Therefore, not only HDAC inhibitor modifies cancer epigenetics, but it also suppresses cancer development via deactivation of the YAP1/TAZ pathway. The decrease of expression of p-YAP1 and YAP1/TAZ and nuclear translocation further resulted in decreasing the expression of its downstream molecules CTGF and Cyr61. Both CTGF and Cyr61 play a vital role in regulating metastasis and invasion of human lung adenocarcinoma [66,67]. This helps us to explain the effect of iminodibenzyl on suppression of migration in A549 cells and inhibition of nodules formation in the syngeneic LLC tumor model observed in our study. Although we observed the similar effect of exogenous 8-HOA and endogenous 8-HOA (triggered by iminodibenzyl and DGLA), iminodibenzyl may also play a role in other pathways for cancer inhibition. Therefore, it is critical to continuously explore the molecular mechanism of iminodibenzyl on cancer treatment by considering other aspects, including tumor microenvironment, redox homeostasis, and cancer epigenetics.
PUFA synthesis and peroxidation also can regulate ferroptosis, which is the iron-dependent programmed cell death pathway [68]. AA has recently been identified as the key mediator of ferroptosis in gastric cancer. D5D could serve as the checkpoint in the ferroptosis pathway by catalyzing AA formation from DGLA [69]. This is consistent with our observation in which DGLA alone could decrease the protein expression of SLC7A11 and its downstream molecule GPX4, indicating the promotion of ferroptosis. However, as the scheme in Fig. S6E, the addition of iminodibenzyl reversed the effect of DGLA on ferroptosis in lung cancer cells. Therefore, the anti-cancer effect of DGLA and iminodibenzyl may mainly depend on 8-HOA-mediated apoptosis, not AA-mediated ferroptosis.
To better assess the role of iminodibenzyl in lung cancer, we employed both the xenograft nude mice model and the syngeneic LLC tumor model for investigating the effect of iminodibenzyl on lung tumor growth and metastasis in the different murine system. The xenograft model on nude mice could represent the complexities and growth pattern of human cancer [70,71]. However, the syngeneic LLC tumor model allows us to evaluate the effect of iminodibenzyl on metastasis under an immunocompetent murine background [72,73]. Interestingly, we do observe the differences between the xenograft nude mice model and the syngeneic LLC tumor model in primary tumor growth pattern and response to iminodibenzyl. The speed of tumor growth in the LLC model is remarkably higher than the nude mice model. We observed that injection of 2 × 106 A549 cells resulted in a ~30-fold change on relative tumor volume in 42 days, whereas 5 × 105 LLC cells resulted in the same change just within 28 days. In the LLC model, iminodibenzyl alone displayed a higher inhibitory effect on tumor growth than the nude mice model. Furthermore, we observed that 8-HOA was significantly increased in the LLC mice treated with iminodibenzyl in absence of DGLA. Conversely, we previously demonstrated that the therapeutical threshold level of 8-HOA only can be achieved by supplementing DGLA, which is an essential precursor for 8-HOA formation in cancer cells [17,20,21]. So why does iminodibenzyl alone could affect tumor growth in the LLC model? The paracrine of 8-HOA in LLC tumor tissues may matter. The LLC mouse showed a larger tumor than the nude mouse after the same period of cancer cell injection. Thus, for the LLC model, without DGLA supplementation, even though the ability of the production of 8-HOA by a single cancer cell is negligible, the aggregation of cancer cells might create an environment in which the 8-HOA level could reach the threshold concentration and elicit an anti-cancer effect. This distinct characteristic of the iminodibenzyl-based strategy may provide a safer outcome compare to traditional COX-2 inhibitor, which is usually requiring a high dose for cancer therapy that is unfortunately associated with an increase of cardiovascular risk in cancer patients [9,11,51].
Another possible explanation is the differentiation of the absorption and distribution of iminodibenzyl in different animal models. We observed that the concentration of iminodibenzyl in blood, tissues, and tumor from the LLC C57BL/6 mice model was higher than the concentration of iminodibenzyl in the nude mice model (Table S1). It may explain the better effectiveness of iminodibenzyl (without DGLA) on the LLC model compare to the nude mice model. We hypothesis that iminodibenzyl in high concentration might suppress lung cancer growth and metastasis by multiple mechanisms rather than exclusively reprogramming COX-2 catalyzed DGLA peroxidation. For instance, one recent study has reported that D5D could promote the development of laryngeal squamous cell carcinoma by activating AKT/mTOR pathway [74]. Hence, future work should therefore include follow-up work designed to investigate other possible mechanisms of iminodibenzyl on cancer. Additionally, this phenomenon also might be reasoned by the differences in physiology properties between these two distinct animal models. Although further investigation on pharmacokinetics and enzyme kinetics are required warranty to understand the role of iminodibenzyl in fatty acid peroxidation, this D5D inhibitor may serve as a promising game-changer that could subvert the redox basis of COX-2 in cancer therapy.
In the current study, we observed that the combination of DGLA and iminodibenzyl resulted in a significant increase of 8-HOA concentration in lung cancer cells (~0.8 μM in 106 cells) and in tumor tissues (~0.7 μg/g tumor). According to reports, a tumor reaching the size of 1 cm3 (approximately 1 g wet weight) is commonly assumed to contain 3 ×107 to 1 × 109 cells [75]. Although the same weight of tumor tissues have more cells than in vitro sample, we observed that the 8-HOA level in tumor tissues was slightly lower than endogenous levels of 8-HOA generated in A549 cells. The relatively lower 8-HOA level in tumor tissues may be due to the comprehensive metabolism and elimination system presenting in the animal model. 8-HOA is an omega-hydroxy fatty acid. It is plausible that the hydroxyl group of 8-HOA may facilitate its biotransformation and further excretion in the animal rather than in the cell model. Therefore, the continuous treatment of iminodibenzyl and DGLA (every day dosing in this study) to ensure the 8-HOA level above threshold in tumor tissues is necessary for animal study. Although it is less likely to directly compare 8-HOA generation in vitro and in vivo, we may conclude from the current data that the exposure time and endogenous level of 8-HOA are both essential for inhibiting tumor growth.
In summary, we demonstrated that iminodibenzyl could reprogram the COX-2 catalyzed fatty acid peroxidation in lung cancer by inhibiting D5D activity. Notably, this is the first study to our knowledge to investigate the effectiveness of iminodibenzyl in COX-2 catalyzed DGLA peroxidation. We found that iminodibenzyl could suppress AA production via inhibiting the activity of D5D, resulting in the formation of 8-HOA in lung cancer cells. As an anti-cancer free radical byproduct, 8-HOA led to a decrease in cell proliferation, migration, survival, but induction of apoptosis in lung cancer cells through downregulating the YAP1/TAZ pathway and activating p53 dependent intrinsic apoptotic pathway. Additionally, 8-HOA could enhance the efficacy of cisplatin on lung cancer in a synergistic pattern. In addition to in vitro study, we found that iminodibenzyl and DGLA supplementation also could significantly inhibit tumor growth and metastasis in both xenografted nude mice and syngeneic C57BL/6 mice models. This D5D inhibition-based strategy might selectively toxic to lung cancer cells with a high COX-2 level, whereas it could avoid harassing the growth of normal lung epithelial cells with low COX-2 expression. Taken together, our findings strongly suggested that iminodibenzyl may hold a great promise to generate a more effective and safer outcome in cancer therapy by shifting COX-2 catalyzed DGLA peroxidation pattern in cancer cells.
Supplementary Material
Acknowledgments
This work is dedicated to the memory of Dr. Steven Y. Qian, who passed away last year. The authors would like to thank Ashish Kumar for his technical help in revising the manuscript.
Funding
This work was supported by the National Institutes of Health grant [1R15CA195499–01A1] to Dr. Qian; the Dakota cancer collaborative on translational activity [U54GM128729].
Abbreviations
- PUFAs
polyunsaturated fatty acids
- D5D
delta-5-desaturase
- DGLA
dihomo-γ-linolenic acid
- COX-2
cyclooxygenase-2
- PGs
prostaglandins
- PGE2
prostaglandin-E2
- AA
arachidonic acid
- 8-HOA
8-hydroxyoctanoic acid
- RNAi
RNA interference
- HDAC
histone deacetylases
- YAP1
Yes-associated protein-1
- TAZ
transcriptional coactivator with PDZ-binding motif
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- PBS
phosphate buffer saline
- Bax
Bcl-2-associated X protein
- MMP
Matrix metalloproteinase
- CTGF
connective tissue growth factor
- Cyr61
cysteine-rich angiogenic inducer 61
- H&E
hematoxylin and eosin
- CId
combination index based on dose
Footnotes
Declaration of interests
S.Q. is an inventor on a patent (US-2019070193-A1) related to this work filed by NDSU Research Foundation. The authors declare no other competing interests.
References
- [1].Howlader N, Noone AM, Krapcho M, Miller D, Brest A, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, SEER Cancer Statistics Review, 1975–2016, National Cancer Institute. Bethesda, MD, National Cancer Institute. Bethesda, MD, 2019. [Google Scholar]
- [2].Molina JR, Yang P, Cassivi SD, Schild SE, Adjei AA, Non-Small Cell Lung Cancer: Epidemiology, Risk Factors, Treatment, and Survivorship, Mayo Clinic Proceedings. 83 (2008) 584–594. 10.4065/83.5.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Esme H, Cemek M, Sezer M, Saglam H, Demir A, Melek H, Unlu M, High levels of oxidative stress in patients with advanced lung cancer, Respirology. 13 (2008) 112–116. 10.1111/j.1440-1843.2007.01212.x. [DOI] [PubMed] [Google Scholar]
- [4].Filaire E, Dupuis C, Galvaing G, Aubreton S, Laurent H, Richard R, Filaire M, Lung cancer: What are the links with oxidative stress, physical activity and nutrition, Lung Cancer. 82 (2013) 383–389. 10.1016/j.lungcan.2013.09.009. [DOI] [PubMed] [Google Scholar]
- [5].Azad N, Rojanasakul Y, Vallyathan V, Inflammation and Lung Cancer: Roles of Reactive Oxygen/Nitrogen Species, Journal of Toxicology and Environmental Health, Part B. 11 (2008) 1–15. 10.1080/10937400701436460. [DOI] [PubMed] [Google Scholar]
- [6].Calder PC, Polyunsaturated fatty acids and inflammation, Prostaglandins, Leukotrienes and Essential Fatty Acids. 75 (2006) 197–202. 10.1016/j.plefa.2006.05.012. [DOI] [PubMed] [Google Scholar]
- [7].Subbaramaiah K, Dannenberg AJ, Cyclooxygenase 2: a molecular target for cancer prevention and treatment, Trends Pharmacol. Sci. 24 (2003) 96–102. 10.1016/S0165-6147(02)00043-3. [DOI] [PubMed] [Google Scholar]
- [8].Groen HJ, Sietsma H, Vincent A, Hochstenbag MM, van Putten JW, van den Berg A, Dalesio O, Biesma B, Smit HJ, Termeer A, Randomized, placebo-controlled phase III study of docetaxel plus carboplatin with celecoxib and cyclooxygenase-2 expression as a biomarker for patients with advanced non–small-cell lung cancer: the NVALT-4 study, J. Clin. Oncol. 29 (32) (2009) 4320–4326. [DOI] [PubMed] [Google Scholar]
- [9].Gulyas M, Mattsson JSM, Lindgren A, Ek L, Lamberg Lundström K, Behndig A, Holmberg E, Micke P, Bergman B, Group SLCS, COX-2 expression and effects of celecoxib in addition to standard chemotherapy in advanced non-small cell lung cancer, Acta Oncologica. 57 (2018) 244–250. [DOI] [PubMed] [Google Scholar]
- [10].Groen HJM, Socinski MA, Grossi F, Juhasz E, Gridelli C, Baas P, Butts CA, Chmielowska E, Usari T, Selaru P, Harmon C, Williams JA, Gao F, Tye L, Chao RC, Blumenschein GR, A randomized, double-blind, phase II study of erlotinib with or without sunitinib for the second-line treatment of metastatic non-small-cell lung cancer (NSCLC), Ann Oncol. 24 (2013) 2382–2389. 10.1093/annonc/mdt212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Solomon SD, McMurray JJ, Pfeffer MA, Wittes J, Fowler R, Finn P, Anderson WF, Zauber A, Hawk E, Bertagnolli M, Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention, New England Journal of Medicine. 352 (2005) 1071–1080. [DOI] [PubMed] [Google Scholar]
- [12].Xu Y, Yang X, Wang T, Yang L, He Y-Y, Miskimins K, Qian SY, Knockdown delta-5-desaturase in breast cancer cells that overexpress COX-2 results in inhibition of growth, migration and invasion via a dihomo-γ-linolenic acid peroxidation dependent mechanism, BMC Cancer. 18 (2018) 330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Yang X, Xu Y, Wang T, Shu D, Guo P, Miskimins K, Qian SY, Inhibition of cancer migration and invasion by knocking down delta-5-desaturase in COX-2 overexpressed cancer cells, Redox Biol. 11 (2017) 653–662. 10.1016/j.redox.2017.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Xu Y, Pang L, Wang H, Xu C, Shah H, Guo P, Shu D, Qian SY, Specific delivery of delta-5-desaturase siRNA via RNA nanoparticles supplemented with dihomo-γ-linolenic acid for colon cancer suppression, Redox Biol. 21 (2019) 101085. 10.1016/j.redox.2018.101085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Pang L, Shah H, Wang H, Shu D, Qian SY, Sathish V, EpCAM-Targeted 3WJ RNA Nanoparticle Harboring Delta-5-Desaturase siRNA Inhibited Lung Tumor Formation via DGLA Peroxidation, Molecular Therapy - Nucleic Acids. 22 (2020) 222–235. 10.1016/j.omtn.2020.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Shah H, Pang L, Wang H, Shu D, Qian SY, Sathish V, Growth inhibitory and anti-metastatic activity of epithelial cell adhesion molecule targeted three-way junctional delta-5-desaturase siRNA nanoparticle for breast cancer therapy, Nanomedicine: Nanotechnology, Biology and Medicine. 30 (2020) 102298. 10.1016/j.nano.2020.102298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Xu Y, Yang X, Zhao P, Yang Z, Yan C, Guo B, Qian SY, Knockdown of delta-5-desaturase promotes the anti-cancer activity of dihomo-γ-linolenic acid and enhances the efficacy of chemotherapy in colon cancer cells expressing COX-2, Free Radical Biology and Medicine. 96 (2016) 67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Xu Y, Qi J, Yang X, Wu E, Qian SY, Free radical derivatives formed from cyclooxygenase-catalyzed dihomo-γ-linolenic acid peroxidation can attenuate colon cancer cell growth and enhance 5-fluorouracil׳ s cytotoxicity, Redox Biology. 2 (2014) 610–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Džodić PL, Ivanovi LJ, Proti AD, Zeevi ML, Joci BM, Determination of Carbamazepine and Its Impurities Iminostilbene and Iminodibenzyl in Solid Dosage Form by Column High-Performance Liquid Chromatography, J AOAC Int. 93 (2010) 1059–1068. 10.1093/jaoac/93.4.1059. [DOI] [PubMed] [Google Scholar]
- [20].Xu Y, Yang X, Gao D, Yang L, Miskimins K, Qian SY, Dihomo-γ-linolenic acid inhibits xenograft tumor growth in mice bearing shRNA-transfected HCA-7 cells targeting delta-5-desaturase, BMC Cancer. 18 (2018) 1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Yang X, Xu Y, Gao D, Yang L, Qian SY, Dihomo-γ-linolenic acid inhibits growth of xenograft tumors in mice bearing human pancreatic cancer cells (BxPC-3) transfected with delta-5-desaturase shRNA, Redox Biology. 20 (2019) 236–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Cisplatin-Based Adjuvant Chemotherapy in Patients with Completely Resected Non–Small-Cell Lung Cancer, New England Journal of Medicine. 350 (2004) 351–360. 10.1056/NEJMoa031644. [DOI] [PubMed] [Google Scholar]
- [23].Wang Y, Blandino G, Oren M, Givol D, Induced p53 expression in lung cancer cell line promotes cell senescence and differentially modifies the cytotoxicity of anti-cancer drugs, Oncogene. 17 (1998) 1923–1930. 10.1038/sj.onc.1202113. [DOI] [PubMed] [Google Scholar]
- [24].Han J-Y, Chung Y-J, Park SW, Kim JS, Rhyu M-G, Kim H-K, Lee KS, The relationship between cisplatin-induced apoptosis and p53, bcl-2 and bax expression in human lung cancer cells, Korean J Intern Med. 14 (1999) 42–52. 10.3904/kjim.1999.14.1.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Matsumoto M, Nakajima W, Seike M, Gemma A, Tanaka N, Cisplatin-induced apoptosis in non-small-cell lung cancer cells is dependent on Bax- and Bak-induction pathway and synergistically activated by BH3-mimetic ABT-263 in p53 wild-type and mutant cells, Biochemical and Biophysical Research Communications. 473 (2016) 490–496. 10.1016/j.bbrc.2016.03.053. [DOI] [PubMed] [Google Scholar]
- [26].Chen D, Zheng X, Kang D, Yan B, Liu X, Gao Y, Zhang K, Apoptosis and expression of the Bcl-2 family of proteins and P53 in human pancreatic ductal adenocarcinoma, Medical Principles and Practice: International Journal of the Kuwait University, Health Science Centre. 21 (2012) 68–73. 10.1159/000332423. [DOI] [PubMed] [Google Scholar]
- [27].Laudanski J, Niklinska W, Burzykowski T, Chyczewski L, Niklinski J, Prognostic significance of p53 and bcl-2 abnormalities in operable nonsmall cell lung cancer, The European Respiratory Journal. 17 (2001) 660–666. 10.1183/09031936.01.17406600. [DOI] [PubMed] [Google Scholar]
- [28].Lee HK, Lee HS, Yang H-K, Kim WH, Lee KU, Choe KJ, Kim J-P, Prognostic significance of Bcl-2 and p53 expression in gastric cancer, International Journal of Colorectal Disease. 18 (2003) 518–525. 10.1007/s00384-003-0491-2. [DOI] [PubMed] [Google Scholar]
- [29].Linjawi A, Kontogiannea M, Halwani F, Edwardes M, Meterissian S, Prognostic significance of p53, bcl-2, and Bax expression in early breast cancer, Journal of the American College of Surgeons. 198 (2004) 83–90. 10.1016/j.jamcollsurg.2003.08.008. [DOI] [PubMed] [Google Scholar]
- [30].Palmer JE, Sant Cassia LJ, Irwin CJ, Morris AG, Rollason TP, P53 and bcl-2 assessment in serous ovarian carcinoma, International Journal of Gynecological Cancer: Official Journal of the International Gynecological Cancer Society. 18 (2008) 241–248. 10.1111/j.1525-1438.2007.01000.x. [DOI] [PubMed] [Google Scholar]
- [31].Wong RS, Apoptosis in cancer: from pathogenesis to treatment, J Exp Clin Cancer Res. 30 (2011) 87. 10.1186/1756-9966-30-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ghobrial IM, Witzig TE, Adjei AA, Targeting apoptosis pathways in cancer therapy, CA Cancer J Clin. 55 (2005) 178–194. 10.3322/canjclin.55.3.178. [DOI] [PubMed] [Google Scholar]
- [33].Han H, Yang B, Nakaoka HJ, Yang J, Zhao Y, Le Nguyen K, Bishara AT, Mandalia TK, Wang W, Hippo signaling dysfunction induces cancer cell addiction to YAP, Oncogene. 37 (2018) 6414–6424. 10.1038/s41388-018-0419-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hong W, Guan K-L, The YAP and TAZ transcription co-activators: Key downstream effectors of the mammalian Hippo pathway, Seminars in Cell & Developmental Biology. 23 (2012) 785–793. 10.1016/j.semcdb.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Plouffe SW, Hong AW, Guan K-L, Disease implications of the Hippo/YAP pathway, Trends in Molecular Medicine. 21 (2015) 212–222. 10.1016/j.molmed.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Nam T, Lipid Peroxidation and Its Toxicological Implications, Toxicol Res. 27 (2011) 1–6. 10.5487/TR.2011.27.1.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Erejuwa OO, Sulaiman SA, Ab Wahab MS, Evidence in Support of Potential Applications of Lipid Peroxidation Products in Cancer Treatment, Oxid Med Cell Longev. 2013 (2013). 10.1155/2013/931251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Banerjee K, Mandal M, Oxidative stress triggered by naturally occurring flavone apigenin results in senescence and chemotherapeutic effect in human colorectal cancer cells, Redox Biology. 5 (2015) 153–162. 10.1016/j.redox.2015.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Görlach A, Dimova EY, Petry A, Martínez-Ruiz A, Hernansanz-Agustín P, Rolo AP, Palmeira CM, Kietzmann T, Reactive oxygen species, nutrition, hypoxia and diseases: Problems solved?, Redox Biology. 6 (2015) 372–385. 10.1016/j.redox.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Xu H, Li C, Mozziconacci O, Zhu R, Xu Y, Tang Y, Chen R, Huang Y, Holzbeierlein JM, Schöneich C, Huang J, Li B, Xanthine oxidase-mediated oxidative stress promotes cancer cell-specific apoptosis, Free Radical Biology and Medicine. 139 (2019) 70–79. 10.1016/j.freeradbiomed.2019.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Peña-Oyarzun D, Bravo-Sagua R, Diaz-Vega A, Aleman L, Chiong M, Garcia L, Bambs C, Troncoso R, Cifuentes M, Morselli E, Ferreccio C, Quest AFG, Criollo A, Lavandero S, Autophagy and oxidative stress in non-communicable diseases: A matter of the inflammatory state?, Free Radical Biology and Medicine. 124 (2018) 61–78. 10.1016/j.freeradbiomed.2018.05.084. [DOI] [PubMed] [Google Scholar]
- [42].Rockwell P, Martinez J, Papa L, Gomes E, Redox regulates COX-2 upregulation and cell death in the neuronal response to cadmium, Cellular Signalling. 16 (2004) 343–353. 10.1016/j.cellsig.2003.08.006. [DOI] [PubMed] [Google Scholar]
- [43].Wang X, Lin H, Gu Y, Multiple roles of dihomo-γ-linolenic acid against proliferation diseases, Lipids in Health and Disease. 11 (2012) 25. 10.1186/1476-511X-11-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Calder PC, Campoy C, Eilander A, Fleith M, Forsyth S, Larsson P-O, Schelkle B, Lohner S, Szommer A, van de Heijning BJM, Mensink RP, A systematic review of the effects of increasing arachidonic acid intake on PUFA status, metabolism and health-related outcomes in humans, The British Journal of Nutrition. 121 (2019) 1201–1214. 10.1017/S0007114519000692. [DOI] [PubMed] [Google Scholar]
- [45].Ma P, L M, Sj D, Jl W, Dietary Lipids Induce Ferroptosis in Caenorhabditiselegans and Human Cancer Cells., Dev Cell. 54 (2020) 447–454.e4. 10.1016/j.devcel.2020.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Li S, Jiang M, Wang L, Yu S, Combined chemotherapy with cyclooxygenase-2 (COX-2) inhibitors in treating human cancers: Recent advancement, Biomedicine & Pharmacotherapy. 129 (2020) 110389. 10.1016/j.biopha.2020.110389. [DOI] [PubMed] [Google Scholar]
- [47].Hashemi Goradel N, Najafi M, Salehi E, Farhood B, Mortezaee K, Cyclooxygenase-2 in cancer: A review, J Cell Physiol. 234 (2019) 5683–5699. 10.1002/jcp.27411. [DOI] [PubMed] [Google Scholar]
- [48].B Y, O B, C C, S P, Combinational Treatment Effect of Tetrahydrocurcumin and Celecoxib on Cervical Cancer Cell-Induced Tumor Growth and Tumor Angiogenesis in Nude Mice., J Med Assoc Thai. 99Suppl 4 (2016) S23–31. [PubMed] [Google Scholar]
- [49].Abdallah FM, Helmy MW, Katary MA, Ghoneim AI, Synergistic antiproliferative effects of curcumin and celecoxib in hepatocellular carcinoma HepG2 cells, Naunyn Schmiedebergs Arch Pharmacol. 391 (2018) 1399–1410. 10.1007/s00210-018-1557-6. [DOI] [PubMed] [Google Scholar]
- [50].Noda M, Tatsumi Y, Tomizawa M, Takama T, Mitsufuji S, Sugihara H, Kashima K, Hattori T, Effects of etodolac, a selective cyclooxygenase-2 inhibitor, on the expression of E-cadherin-catenin complexes in gastrointestinal cell lines, J Gastroenterol. 37 (2002) 896–904. 10.1007/s005350200151. [DOI] [PubMed] [Google Scholar]
- [51].Reckamp KL, Krysan K, Morrow JD, Milne GL, Newman RA, Tucker C, Elashoff RM, Dubinett SM, Figlin RA, A phase I trial to determine the optimal biological dose of celecoxib when combined with erlotinib in advanced non-small cell lung cancer, Clin Cancer Res. 12 (2006) 3381–3388. 10.1158/1078-0432.CCR-06-0112. [DOI] [PubMed] [Google Scholar]
- [52].Yang X, Xu Y, Brooks A, Guo B, Miskimins KW, Qian SY, Knockdown delta-5-desaturase promotes the formation of a novel free radical byproduct from COX-catalyzed omega-6 peroxidation to induce apoptosis and sensitize pancreatic cancer cells to chemotherapy drugs, Free Radical Biology and Medicine. 97 (2016) 342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Aagaard L, Rossi JJ, RNAi therapeutics: principles, prospects and challenges, Advanced Drug Delivery Reviews. 59 (2007) 75–86. 10.1016/j.addr.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Grimm D, Streetz KL, Jopling CL, Storm TA, Pandey K, Davis CR, Marion P, Salazar F, Kay MA, Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways, Nature. 441 (2006) 537–541. 10.1038/nature04791. [DOI] [PubMed] [Google Scholar]
- [55].Hornung V, Guenthner-Biller M, Bourquin C, Ablasser A, Schlee M, Uematsu S, Noronha A, Manoharan M, Akira S, de Fougerolles A, Endres S, Hartmann G, Sequence-specific potent induction of IFN-alpha by short interfering RNA in plasmacytoid dendritic cells through TLR7, Nat Med. 11 (2005) 263–270. 10.1038/nm1191. [DOI] [PubMed] [Google Scholar]
- [56].Jackson AL, Bartz SR, Schelter J, Kobayashi SV, Burchard J, Mao M, Li B, Cavet G, Linsley PS, Expression profiling reveals off-target gene regulation by RNAi, Nat Biotechnol. 21 (2003) 635–637. 10.1038/nbt831. [DOI] [PubMed] [Google Scholar]
- [57].Gierbolini J, Giarratano M, Benbadis SR, Carbamazepine-related antiepileptic drugs for the treatment of epilepsy - a comparative review, Expert Opinion on Pharmacotherapy. 17 (2016) 885–888. 10.1517/14656566.2016.1168399. [DOI] [PubMed] [Google Scholar]
- [58].Elmore S, Apoptosis: A Review of Programmed Cell Death, Toxicol Pathol. 35 (2007) 495–516. 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Tsujimoto Y, Finger LR, Yunis J, Nowell PC, Croce CM, Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation, Science. 226 (1984) 1097–1099. 10.1126/science.6093263. [DOI] [PubMed] [Google Scholar]
- [60].Reed JC, Bcl-2 family proteins: regulators of apoptosis and chemoresistance in hematologic malignancies, Semin Hematol. 34 (1997) 9–19. [PubMed] [Google Scholar]
- [61].Gross A, McDonnell JM, Korsmeyer SJ, BCL-2 family members and the mitochondria in apoptosis, Genes Dev. 13 (1999) 1899–1911. 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- [62].Zhao B, Lei Q-Y, Guan K-L, The Hippo–YAP pathway: new connections between regulation of organ size and cancer, Current Opinion in Cell Biology. 20 (2008) 638–646. 10.1016/j.ceb.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Calses PC, Crawford JJ, Lill JR, Dey A, Hippo Pathway in Cancer: Aberrant Regulation and Therapeutic Opportunities, Trends in Cancer. 5 (2019) 297–307. 10.1016/j.trecan.2019.04.001. [DOI] [PubMed] [Google Scholar]
- [64].Maugeri-Saccà M, De Maria R, The Hippo pathway in normal development and cancer, Pharmacology & Therapeutics. 186 (2018) 60–72. 10.1016/j.pharmthera.2017.12.011. [DOI] [PubMed] [Google Scholar]
- [65].Marks PA, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK, Histone deacetylases and cancer: causes and therapies, Nature Reviews Cancer. 1 (2001) 194–202. 10.1038/35106079. [DOI] [PubMed] [Google Scholar]
- [66].Piccolo S, Dupont S, Cordenonsi M, The biology of YAP/TAZ: hippo signaling and beyond, Physiol Rev. 94 (2014) 1287–1312. 10.1152/physrev.00005.2014. [DOI] [PubMed] [Google Scholar]
- [67].Chang C-C, Shih J-Y, Jeng Y-M, Su J-L, Lin B-Z, Chen S-T, Chau Y-P, Yang P-C, Kuo M-L, Connective tissue growth factor and its role in lung adenocarcinoma invasion and metastasis, J Natl Cancer Inst. 96 (2004) 364–375. 10.1093/jnci/djh059. [DOI] [PubMed] [Google Scholar]
- [68].Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR, Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis, PNAS. 113 (2016) E4966–E4975. 10.1073/pnas.1603244113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Lee J-Y, Nam M, Son HY, Hyun K, Jang SY, Kim JW, Kim MW, Jung Y, Jang E, Yoon S-J, Kim J, Kim J, Seo J, Min J-K, Oh K-J, Han B-S, Kim WK, Bae K-H, Song J, Kim J, Huh Y-M, Hwang G-S, Lee E-W, Lee SC, Polyunsaturated fatty acid biosynthesis pathway determines ferroptosis sensitivity in gastric cancer, PNAS. 117 (2020) 32433–32442. 10.1073/pnas.2006828117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Kellar A, Egan C, Morris D, Preclinical Murine Models for Lung Cancer: Clinical Trial Applications, BioMed Research International. 2015 (2015) e621324. 10.1155/2015/621324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Steiner P, Joynes C, Bassi R, Wang S, Tonra JR, Hadari YR, Hicklin DJ, Tumor Growth Inhibition with Cetuximab and Chemotherapy in Non–Small Cell Lung Cancer Xenografts Expressing Wild-type and Mutated Epidermal Growth Factor Receptor, Clin Cancer Res. 13 (2007) 1540–1551. 10.1158/1078-0432.CCR-06-1887. [DOI] [PubMed] [Google Scholar]
- [72].Sakai Y, Sasahira T, Ohmori H, Yoshida K, Kuniyasu H, Conjugated linoleic acid reduced metastasized LL2 tumors in mouse peritoneum, Virchows Arch. 449 (2006) 341–347. 10.1007/s00428-006-0249-7. [DOI] [PubMed] [Google Scholar]
- [73].Papageorgiou A, Stravoravdi P, Sahpazidou D, Natsis K, Chrysogelou E, Toliou T, Effect of Navelbine on Inhibition of Tumor Growth, Cellular Differentiation and Estrogen Receptor Status on Lewis Lung Carcinoma, CHE. 46 (2000) 188–194. 10.1159/000007277. [DOI] [PubMed] [Google Scholar]
- [74].Zhao R, Tian L, Zhao B, Sun Y, Cao J, Chen K, Li F, Li M, Shang D, Liu M, FADS1 promotes the progression of laryngeal squamous cell carcinoma through activating AKT/mTOR signaling, Cell Death & Disease. 11 (2020) 1–14. 10.1038/s41419-020-2457-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Monte UD, Does the cell number 109 still really fit one gram of tumor tissue?, Cell Cycle. 8 (2009) 505–506. 10.4161/cc.8.3.7608. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.