

Abstract
Klebsiella pneumoniae is a common cause of Gram-negative pneumonia. The spread of antibiotic-resistant and hypervirulent strains has made treatment more challenging. This study sought to determine the immunomodulatory, antibacterial, and therapeutic potential of purified murine stem cell antigen-1+ lung mesenchymal stem cells (Sca-1+ LMSCs) using in vitro cell culture and an in vivo mouse model of pneumonia caused by K. pneumoniae. Sca-1+ LMSCs are plastic adherent, possess colony forming capacity, express mesenchymal stem cell markers, differentiate into osteogenic and adipogenic lineages in vitro, and exhibit a high proliferative capacity. Further, these Sca-1+ LMSCs are morphologically similar to fibroblasts but differ ultrastructurally. Moreover, Sca-1+ LMSCs have the capacity to inhibit lipopolysaccharide-induced secretion of inflammatory cytokines by bone marrow-derived macrophages and neutrophils in vitro. Sca-1+ LMSCs inhibit the growth of K. pneumoniae more potently than do neutrophils. Sca-1+ LMSCs also possess the intrinsic ability to phagocytize and kill K. pneumoniae intracellularly. While the induction of autophagy promotes bacterial replication, inhibition of autophagy enhances the intracellular clearance of K. pneumoniae in Sca-1+ LMSCs during the early time of infection. Adoptive transfer of Sca-1+ LMSCs in K. pneumoniae-infected mice improved survival, reduced inflammatory cells in bronchoalveolar lavage fluid, reduced inflammatory cytokine levels and pathologic lesions in the lung, and enhanced bacterial clearance in the lung and in extrapulmonary organs. These results together illustrate for the first time the protective role of LMSCs in bacterial pneumonia.
Introduction
Bacterial pneumonia remains a major global disease burden (1). Klebsiella pneumoniae, the Gram-negative bacterium, induces severe pneumonia associated with substantial parenchymal damage in the lungs. In recent years, the spread of carbapenem-resistant K. pneumoniae strains, which cause ≥50% mortality, has made treatment more difficult (2, 3). Moreover, K. pneumoniae is known to cause life-threatening lung infections in patients with diabetes and in alcohol consumers (4-6). Thus, the emergence of multidrug resistant K. pneumoniae strains necessitate the investigation of alternative therapeutic options.
Mesenchymal stem cells (MSCs), adult multipotent stromal cells, have shown potential as therapeutic agents with significant clinical application towards a variety of diseases, including those affecting the lung. MSCs are a favored cell type in pre-clinical and clinical research due to their distribution in numerous organs, convenient isolation and propagation under in vitro conditions, extensive proliferative and differentiable capabilities, paracrine functions, and immunosuppressive activities (7-14).
Bone marrow-derived MSCs (BM-MSCs) have been widely studied due to their potential as a cell-based immunotherapy for a broad range of diseases including autoimmune diabetes, severe graft versus host disease, myocardial infarction, Crohn’s disease, pulmonary hypertension, hepatic failure, acute renal failure, endotoxin-induced acute lung injury, sepsis, acute respiratory distress syndrome, bleomycin-induced lung injury and fibrosis, chronic obstructive pulmonary disease, allergic asthma, bronchiolitis obliterans, bronchopulmonary dysplasia, and bacterial pneumonia (9, 11-16). Recent studies using pre-clinical animal models have demonstrated that BM-MSCs have potent beneficial effects for lung injury caused by infection, which are attributed to a number of MSC properties including their immunomodulation, secretion of various reparative growth factors and cytokines, and augmentation of host defense mechanisms (11, 17, 18). In contrast to the well-established use of BM-MSCs in cellular therapy, much less is known about the reparative and paracrine functions of lung mesenchymal stem cells (LMSCs). Moreover, while cells with mesenchymal stem cell characteristics (19-27) have been isolated from the lungs of rodents, sheep and humans, few studies have tested the therapeutic potential of these cells in animal models of lung diseases including elastase-induced pulmonary emphysema (23, 24, 28), allergic asthma (29), and LPS-induced acute respiratory distress syndrome (30).
To our knowledge, the role of LMSCs in bacterial pneumonia has not been investigated. Therefore, we evaluated the therapeutic potential of purified Sca-1+ LMSCs in a mouse model of bacterial pneumonia using the Gram-negative bacterium, K. pneumoniae. In the current study, we cultured total lung cell digests from C57BL/6J mice for an extended time to generate plastic adherent cells and then used stem cell antigen 1 (Sca-1) as a phenotypic marker to purify a specific population of LMSCs from the lung cell culture outgrowth with the help of a fluorescence activated cell sorter. Unlike Sca-1+ bronchoalveolar stem cells (31, 32), the Sca-1+ lung mesenchymal stem cells (Sca-1+ LMSCs) purified in this study could be expanded and cultured (without growth factors) in non-coated (not coated with Matrigel or collagen) plastic cell culture dishes under non-differentiating conditions. These Sca-1+ LMSCs exhibit several characteristics typical of classical MSCs (8, 33) and differentiate into adipogenic and osteogenic lineages in vitro. These Sca-1+ LMSCs exhibit antibacterial properties and more potently suppress the growth of K. pneumoniae in vitro than do bone marrow-derived neutrophils (BMDNs). In addition, Sca-1+ LMSCs possess the ability to phagocytose and kill K. pneumoniae in vitro. Adoptive transfer of these cells into a mouse model of K. pneumoniae-induced pneumonia, improved survival, minimized inflammation and lung injury, and augmented bacterial clearance in the lungs and in extrapulmonary organs.
Materials and Methods
Antibodies.
The following antibodies were used to characterize Sca1+ LMSCs as well as for Western blot, immunofluorescence and confocal microscopic studies. These antibodies were obtained from Abcam [anti-GAPDH antibody, K. pneumoniae antibody, and Goat anti-rabbit IgG H&L (Alexa FluorR 594) secondary antibody], Cytoskeleton, Inc., [Acti-stain™ 488 phalloidin], eBioscience [PE conjugated Donkey anti-mouse IgG), FITC conjugated anti-mouse IgG, PE conjugated anti-mouse CD103, anti-mouse CD105 PE , PE conjugated anti-mouse CD123, PE conjugated anti-mouse CD135, Anti-mouse CD117 (c-kit) PE, FITC conjugated anti-mouse CD54, and FITC conjugated anti-mouse/rat ICOS], BD Pharmingen ™ [FITC Rat anti-mouse CD45, FITC Rat anti-mouse CD34, PE Rat anti-mouse CD73, FITC Rat anti-mouse Ly 6-A/E, FITC Rat anti-mouse CD31, PE Rat anti-Mouse I-A/I-E, FITC Hamster anti-Mouse CD11c, FITC Rat anti-mouse CD44, FITC Rat anti-CD11b (clone M1/70), and PE anti-mouse CRIg (clone 17C9)], BioLegend [PE anti-mouse Toll-like receptor 4 and FITC anti-rat CD90/mouse CD90.1 (Thy 1.1)], Cell Signaling Technology [LC3 A/B Rabbit antibody, Beclin-1 Rabbit mAb, ATG7 Rabbit mAb, horseradish peroxidase-conjugated anti-mouse IgG and horseradish peroxidase-conjugated anti-rabbit IgG], Miltenyi Biotech [FcR blocking reagent], and Thermo Fisher Scientific [CD18 (LFA-1 beta) monoclonal antibody (M18/2), PE].
Animals.
Pathogen-free C57BL/6J mice (8 to 12 weeks old male and female; purchased from The Jackson Laboratory, Bar Harbor, Maine) were used in all experiments. C57BL/6J mice were fed mouse chow and water ad libitum and were kept under specific pathogen-free conditions with a 12 h dark and 12 h light cycle. Animal experiments were performed in accordance with National Institute of Health guidelines for the use of animals with the approval of the Louisiana State University Institutional Animal Care and Use Committee (IACUC).
Bacterial preparation.
In brief, K. pneumoniae [strain 43816, serotype 2 (American Type Culture Collection, Manassas, VA)] was grown as previously described (34, 35). The bacterial suspension was then diluted to the desired concentration [1 x 103 CFU/50 μl of sterile PBS/mouse (for acute pneumonia study) or 5 x 103 CFU/75 μl of sterile PBS/mouse (for survival study)].
Sca-1+ LMSCs isolation.
The Sca-1+ LMSCs were isolated from the lungs of 8-week-old male C57BL/6J mice. Lung tissues were digested using the lung digestion methods described in our earlier publication (36). In brief, we perfused the left and right ventricles of the heart with 15 ml of Dulbecco’s phosphate buffered saline [(DPBS; containing 20 IU/ml Heparin sodium (Sigma-Aldrich)]. The trachea and upper airways were carefully removed and five whole lungs were digested with 20 ml of dissociation buffer containing Collagenase Type II (Worthington Biochemical Corporation, Lakewood, NJ), Dispase II (Sigma-Aldrich), and DNase I (Sigma-Aldrich) for 45 minutes at 37° C in a CO2 incubator. The lung suspension was diluted with an equal volume of complete alpha minimum essential medium [CMEM; containing 9% FBS, 9% donor equine serum, L-glutamine (2 mM), penicillin (100 IU/ml), streptomycin (100 μg/ml), and amphotericin B (Sigma-Aldrich; 0.25 μg/ml)]. Next, the lung digest was filtered using a 70 μm nylon cell strainer (Corning, NY), which was washed twice with DPBS, and centrifuged at 1800 rpm for 10 minutes at 4° C. The supernatant was discarded. Erythrocytes were removed by lysing the cell pellet with RBC lysis buffer (eBiosciences, San Diego, CA) for 5 minutes at room temperature. The lysis reaction was quenched by adding 25 ml of CMEM and centrifugation at 1800 rpm for 10 minutes at 4° C. Cells were resuspended in CMEM and cultured in 25 cm2 plastic tissue culture flasks (Genesee Scientific Corporation, El Cajon, CA). The medium was removed and replaced with fresh CMEM every 2- 3 days. After 7-10 days of culture (passage 1), the adherent cells were washed with DPBS and then trypsinized using 0.25% trypsin EDTA solution (Gibco by Life Technologies, Carlsbad, CA) for 2 to 3 minutes at 37° C in a CO2 incubator. The cells were washed with CMEM and centrifuged at 1800 rpm for 10 minutes at 4° C. Lung cells were then resuspended in CMEM and cultured for another 2 X 7-10 days (passages 2 and 3) in 182 cm2 plastic tissue culture flask (Genesee Scientific). After passage 3, the adherent lung cells were trypsinized, washed with DPBS, and centrifuged. This process was repeated once more. The cell pellet was suspended in cell staining buffer (Sigma-Aldrich) and then incubated with FITC-conjugated Ly 6-A/E (Sca-1) anti-mouse antibody or FITC-conjugated IgG control antibody for 30 minutes on ice. The cells were washed with cell staining buffer, centrifuged, and resuspended in 2 ml of cell staining buffer. The Sca-1+ cells were sorted using a FACS Aria II cell sorter (BD Biosciences, San Jose, CA). The flow-sorted Sca-1+ lung cells were washed with CMEM, centrifuged twice, resuspended in CMEM, and cultured in plastic tissue culture flasks for another three (passages 4 - 6) to six passages (passages 7 - 9) to generate more cells. At passage 6, the adherent flow-sorted Sca-1+ cells were washed with DPBS, trypsinized, and analyzed for differentiation capacity as well as the expression of mesenchymal stem cell and other cell surface antigens. These well characterized passage 6 Sca-1+ lung mesenchymal cells (Sca-1+ LMSCs) were used for the in vitro and in vivo experimental analyses.
Flow cytometry.
Flow cytometric analysis was conducted to analyze the phenotypes of sorted Sca-1+ lung cells (passage 6). Flow sorted Sca-1+ lung cells (passage 6) were first blocked with mouse FcR blocking reagent (0.25 μg/ml) for 15 minutes on ice. Cells were then incubated with primary antibodies (1-4 μg/ml for 30 min on ice) conjugated to fluorescein isothiocyanate (FITC), or phycoerythrin (PE). Mouse IgG PE and IgG-FITC antibodies were used as isotype controls. Cells were analyzed using a FACS Calibur analyzer (BD Biosciences, San Jose, CA) with FlowJo software (FlowJo LLC, Ashland, OR). Antibodies used and manufacturers are detailed in the antibodies section. After characterization of these flow-sorted passage 6 Sca-1+ lung cells using differentiation and proliferation assays and phase contrast and electron microscopies (see below), we cultured these Sca-1+ LMSCs for another 3 passages (passage 9). We analyzed the expression of various mesenchymal stem cell and other cell surface antigens on this passage 9 Sca-1+ LMSCs using flow cytometry. We have also characterized the morphology and ultrastructural features of passage 9 Sca-1+ LMSCs using phase contrast microscopy and TEM, respectively.
Colony forming unit assay.
The colony forming capacity (CFU) of purified Sca-1+ cells (passage 6) was determined using the protocol described by Peister et al. (37). Briefly, Sca-1+ cells (in triplicates) were cultured at a density of 1000 cells in 10 ml of CMEM in 100 mm petri dish for 21 days and the medium was replaced every 3 days. On day 21, the plates were rinsed and stained with 3% crystal violet (Sigma-Aldrich) for 20 minutes.
Differentiation assay.
The differentiation potential of purified Sca-1+ cells (passage 6) in response to adipogenic and osteogenic lineages was assessed using the method described by Piester et al. (37). In brief, the cells were cultured (50 cells/cm2) in 58-cm2 dishes and incubated in CEM for 10 days. Osteogenic differentiation was induced by culturing the adherent cells in Iscove modified Dulbecco medium (IMDM) [containing fetal calf serum (10%), horse serum (10%), penicillin (100 U/ml), streptomycin (100 μg/ml), L-glutamine (12 mM), β-glycerol phosphate (20 mM; Sigma-Aldrich), thyroxine (50 ng/ml; Sigma-Aldrich), dexamethasone (1 nM; Sigma-Aldrich), and ascorbate 2-phosphate (0.5 μM; Sigma-Aldrich)]. The medium was changed every 3 days for three weeks. After three weeks, adherent cells were washed with DPBS, fixed with 10% formalin, and then stained with Alizarin Red-S reagent (Sigma-Aldrich).
For adipogenic differentiation, cell cultures in 24-well plates were stimulated with a specific adipogenic-differentiation medium [IMDM containing fetal calf serum (10%), horse serum (10%), penicillin (100 U/ml), streptomycin (100 μg/ml), L-glutamine (12 mM), insulin (5 μg/ml; Sigma-Aldrich), indomethacin (50 μM; Sigma-Aldrich), dexamethasone (1 × 10−6 M; Sigma-Aldrich), and 3-isobutyl-1-methylxanthine (0.5 μM; Sigma-Aldrich)]. The medium was changed every 3 days for three weeks. After 21 days, the adherent cells were washed with DPBS, fixed with 10% formalin, and then stained with 0.5% Oil Red O (Sigma-Aldrich) reagent for 20 minutes at room temperature. Lipid droplets were then visualized using a light microscope.
Proliferation assay.
To assess proliferation, Sca-1+ LMSCs (passage 6) were cultured at a density of 5 X 103 cells or 20 X 103 cells in 2 ml CMEM/well in 12-well plates (Genesee Scientific). Adherent cells from three random wells were trypsinized at the indicated time points, stained with Trypan blue (MP Biomedicals, LLC, Santa Ana, CA), and counted using a hemocytometer. Growth curves were plotted using the mean values. Experiments were performed two times.
Light microscopy.
For light microscopy, we cultured the flow sorted Sca-1+ LMSCs (passages 6 and 9) in CMEM (using 12 well plates) for 1 day, 2 days, and 5 days. Adherent cells were stained using 1% methylene blue (Electron Microscopy Sciences, Hatfield, PA) and examined under a light microscope.
Scanning electron microscopy (SEM).
For SEM studies, cells in CMEM [5 X 104 cells in 0.5 ml of CMEM; for Sca-1+ LMSCs (passage 6)] or Dulbecco’s modified eagle medium [DMEM; containing 10% FBS, penicillin (100 IU/ml), and streptomycin (100 μg/ml); for 3T3 mouse embryonic fibroblasts (3T3 MEFs WT ; CRL-2752™, purchased from American Type Culture Collection, Manassas, VA)] were cultured on Thermanox™ coverslips in 24 well plates. After 5 days of culture, the coverslips containing the adherent cells were washed with sterile DPBS and fixed with electron microscope (EM) fixative for 2 h. Cells were then washed with distilled water, dehydrated through an ethanol series, and dried using a Critical Point Drier (Polaron E3000, USA). The dried samples were then coated with gold-palladium in EMS 550X Sputter Coater and examined using a JEOL JSM-6610LV SEM (JEOL USA Inc., Peabody, MA).
Transmission electron microscopy (TEM).
For TEM studies, Sca-1+ LMSCs (passages 6 and 9) and 3T3 MEFs were cultured for 5 days in 182 cm2 flasks using CMEM, as described above for the SEM study. On day 5, adherent cells were trypsinized, washed several times with DPBS, and centrifuged (2000 rpm for 8 min at 4° C). Cell pellets were fixed in primary electron microscope fixative (pH 7.4; 1.6% paraformaldehyde, 2.5% glutaraldehyde, 0.03% CaCl2 in 0.05 M cacodylate buffer) for 2 h, washed with 0.1 M cacodylate buffer supplemented with sucrose, and embedded in 3% agarose. Agarose blocks were cut into 1 mm3 cubes, washed in 0.1 M cacodylate buffer, and fixed in 1% osmium tetroxide (in 0.1 M cacodylate buffer) for 1 h. Samples were then washed in water and stained with 2% uranyl acetate (in 0.2 M sodium acetate buffer, pH 3.5) for 2 h, dehydrated in ascending ethanol concentrations (30-100%) and propylene oxide, and fixed in Epon-Araldite mixture. Blocks were then sectioned using Ultratome Leica EM UC7 (Leica Microsystems Inc. Buffalo Groove, IL). Ultrathin sections (80 nm) were stained with lead citrate for 5 minutes and examined using a JEOL JEM-1400 TEM (JEOL USA Inc., Peabody, MA). All reagents for SEM and TEM studies were purchased from Electron Microscopy Sciences (Hatfield, PA).
Co-culture of Sca-1+ LMSCs with bone marrow-derived macrophages and neutrophils.
We generated macrophages by culturing bone marrow-derived (from 8 to 12-week-old female C57BL/6J mice) cells with recombinant murine M-CSF [PeproTech, Rocky Hill, NJ; 50 ng/ml in DMEM containing 10% fetal bovine serum (FBS)] for 8 days (34). On day 8, we trypsinized the adherent bone marrow derived macrophages (BMDMs) and cultured them with Sca-1+ LMSCs (total of 0.5 million passage 6 Sca-1+ LMSCs/0.5 ml of DMEM containing 10% FBS in 24-well plates) at different ratios (Sca-1+ LMSCs to macrophages at 1:1 to 1:4 ratios) in the presence or absence of lipopolysaccharide [LPS, 055:B5 (Sigma-Aldrich); 500 ng/ml] for 24 h. The levels of TNFα and IL-1β in the culture supernatants were measured using Ready-SET GO™ ELISA kits (eBiosciences, San Diego, CA). To determine whether Sca-1+ LMSCs were able to regulate the production of proinflammatory cytokines by LPS-stimulated neutrophils in a cell-cell contact-dependent manner, we purified neutrophils from bone marrow (BMDNs) of female C57BL/6J mice using the EasySep™ mouse neutrophil enrichment kit (STEMCELL™ Technologies Inc, Cambridge, MA). Then, we cultured the Sca-1+ LMSCs with BMDNs (total of 0.5 million cells/0.5 ml of DMEM containing 10% FBS in 24 well plate) at different ratios (Sca-1+ LMSCs to BMDNs at 1:2 and 1:4 ratios) in the presence or absence of LPS (500 ng/ml) for 8 h. The level of TNFα in the culture supernatants was measured by ELISA (eBiosciences).
Extracellular bacterial killing assay.
The antimicrobial activity of Sca-1+ LMSCs and BMDNs against K. pneumoniae was determined in vitro at different time points. First, we treated the Sca-1+ LMSCs (passage 6) or BMDNs (each 2.5 X 105 cells/ 0.2 ml of DMEM supplemented with 10% FBS) with cytochalasin D (10 μg/ml; Sigma-Aldrich) for 20 min to block phagocytosis prior to infection. K. pneumoniae (in 50 μl sterile PBS) was added [1 multiplicity of infection (MOI) or (10 MOI)] to the wells containing Sca-1+ LMSCs or BMDNs and the cell culture plates were incubated at 37° C for various time periods (1 h to 6 h). The ratios of bacterium to Sca-1+ LMSCs or BMDNs were 1:1 and 10:1. In some experiments, we added K. pneumoniae (10 MOI) to the wells containing LMSCs supernatant (0.1 ml supernatant and 0.1 ml DMEM supplemented with 10% FBS /well) alone or wells containing both LMSCs supernatant (0.1 ml/well) and BMDNs (2.5 X 105 cells/ 0.1 ml of DMEM supplemented with 10% FBS). At designated time points, the plates were centrifuged at 600 g for 4 minutes, supernatants were collected, diluted in sterile PBS, plated onto MacConkey agar plates, and incubated overnight at 37° C.
Quantification of antimicrobial compounds.
To investigate whether infection with K. pneumoniae can induce the secretion of antimicrobial compounds, we cultured the Sca-1+ LMSCs [1 million cells/ml DMEM (containing 10% FBS)] with 20 MOI of opsonized K. pneumoniae for 4 h. We centrifuged the culture supernatants at 2000 rpm for 8 minutes at 4° C and quantified the antimicrobial compounds Cathelicidin-related antimicrobial peptide (CRAMP; MyBioSource.com, San Diego, CA), Leukotriene B4 (LTB4; Cayman Chemical, Ann Arbor, Michigan), and Lipocalin-2/NGAL (R&D Systems, Inc. Minneapolis, MN) in the culture supernatants using ELISA.
Examination of bactericidal activity of CRAMP in vitro.
To investigate whether treatment with CRAMP can inhibit the extracellular growth of K. pneumoniae (38), we incubated the bacteria in tryptic soy agar broth (5 x 10 3 CFU/100 μl/well in 96 well plates) in the presence or absence of murine CRAMP (Chi Scientific, Inc., Maynard, MA) (20 μg/well) for 2 h at 37° C. Then, the bacterial cultures were serially diluted in sterile PBS and plated onto MacConkey agar plates.
Quantification of extracellular H2O2.
The extracellular hydrogen peroxide (H2O2) was quantified using Fluoro H2O2™ kit (Cell Technology, Inc., Hayward, CA). Briefly, the passage 6 LMSCs (1 x 105 cells/well/in 100 μl of Hanks blank salt solution using 96 well cell culture plate) were infected with 20 MOI of opsonized K. pneumoniae for 30 min. Following infection, 50 μl of reaction cocktail was added to each well and incubated for 10 min at room temperature, in the dark. H2O2 was used as the standard. The plates were read at excitation and emission wavelengths of 530 and 570 nm using a fluorescence plate reader.
Opsonophagocytic killing assay.
Opsonophagocytic killing assay was conducted as described by Davis et al. (39) with modifications. In brief, K. Pneumoniae was grown to mid-log phase, PBS-washed, and resuspended in +++ solution. The bacteria (200 x 107 CFU in 400 μl +++ solution) were pre-opsonized with 1600 μl of 3-4 week rabbit complement (Pel-Freez, Rogers, AR) for 30 min at 37° C with constant shaking. The opsonized K. pneumoniae (1 x 107 CFU in 50 μl) were added to wells containing Sca-1+ LMSCs (passage 6) or BMDMs (each 0.5 x 106 cells in 450 μl of DMEM containing 10% FBS/well in 12 well plates) and the plates were incubated for 1 h at 37° C in a CO2 incubator. Cell culture plates were centrifuged at 600 g for 4 minutes at 4° C, the medium was removed, and the extracellular bacteria were removed by washing the plates three times with sterile PBS, as well as by centrifuging the plates at 600 g. Then the cells were cultured with 0.5 ml DMEM [containing 10% FBS and gentamicin sulfate (final concentration 250 μg/ml); Sigma-Aldrich] for 1 h. After 1 h, the medium was removed and the cells were cultured with fresh DMEM (0.5 ml/well) containing 10% FBS and gentamicin sulfate (final concentration 125 μg/ml) for another 2 h (4 h time point) at 37° C in a CO2 incubator. After each time point, the antibiotic was removed by serial (three times) washing with PBS. Adherent cells were lysed with 0.1% Triton X100 (Sigma-Aldrich; 120 μl/well) for 10 min, serially diluted in PBS, and plated onto MacConkey agar plates. The number of viable bacteria were determined after incubating the plates at 37° C for 16 h.
Immunofluorescence labeling and microscopy.
We also confirmed the uptake of live K. pneumoniae by Sca-1+ LMSCs using immunofluorescence microscopy and immunofluorescence confocal microscopy. We opsonized the live K. pneumoniae with rabbit compliment by using the protocol described above. Then, we inoculated the Sca-1+ LMSCs (passage 6) with 20 MOI of opsonized K. pneumoniae and cultured the infected cells in Millicell® EZ slide (EMD Millipore Corporation, Burlington, MA) at 37° C in a CO2 incubator for 3 h. Plates were centrifuged and adherent cells were washed three times with PBS. The adherent cells were fixed with 2% paraformaldehyde overnight, washed thrice with PBS, permeabilized with 0.1% Triton™ X-100 for 10 minutes, and blocked with 1% BSA (in PBS) for 1 h. To localize K. pneumoniae in Sca-1+ LMSCs, cells were incubated with anti-K. pneumoniae antibody (2 μg/ml in PBS containing 0.1% BSA) overnight at 4° C. Cells were washed three times with PBS containing 0.02% Tween 20 and then incubated with pre-adsorbed Goat anti-rabbit IgG H&L (Alexa Fluor® 594) secondary antibody (2 μg/ml in PBS containing 0.1% BSA) for 1 h at room temperature. The cells were washed three times with PBS and stained with Acti-stain™ 488 phalloidin by using the protocol developed by the manufacturer (Cytoskeleton, Inc., Denver, CO). Cells were counterstained with DAPI (VECTASHIELD Mounting Medium with DAPI; Vectar Laboratories, Inc., Burlingame, CA). Images were acquired using a Carl Zeiss immunofluorescence microscope (Carl Zeiss Microscopy, LLC, Thornwood, NY) and Leica TCS SP8 confocal microscope (Leica Microsystems Inc., Buffalo Grove, IL) with a 488-nm argon laser and a 594-nm laser.
Phagocytosis assay using heat-inactivated and fluorescence-labeled K. pneumoniae.
Heat inactivation of K. pneumoniae (strain 43816) was accomplished by incubating 3 x 10 9 CFU of bacteria in 3 ml of sterile PBS for 1 h at 70° C using a water bath (40). BMDMs and Sca-1+ LMSCs (1 million cells/0.5 ml of DMEM containing 10% FBS) were cultured (in 12 well plate) separately either with medium or with 20 MOI and 50 MOI of heat-inactivated and FITC-labeled K. pneumoniae (HI Kp-FITC) for 1 h at 37° C in a CO2 incubator. After 1 h, the plates were centrifuged and washed two times with PBS. The adherent cells were trypsinized, washed three times with PBS and resuspended in PBS. One hundred microliters of filtered (using 0.22 μm filter) Trypan blue was added to 1 ml of cell suspension before measurement using a flow cytometer. Sca-1+ LMSCs cultured for 1 h with DMEM (containing 10% FBS) and then quenched with Trypan blue were used as negative control. The percent uptake of HI Kp-FITC by Sca-1+ LMSCs and BMDMs was analyzed using flow cytometry.
Analysis of expression of complement receptors in Sca-1+ LMSCs.
Sca-1+ LMSCs were labeled with isotype IgG control antibodies (PE conjugated anti-mouse IgG; FITC conjugated anti-mouse IgG), or complement 3 receptor (CR3) antibodies [FITC Rat Anti-CD11b (clone M1/70); CD18 (LFA-1 beta) monoclonal antibody (M18/2), PE] or complement receptor of immunoglobulin superfamily (CRIg) antibody [PE anti-mouse CRIg] for 30 minutes on ice. The cells were washed with PBS, fixed in 2% paraformaldehyde, and analyzed using a flow cytometer.
Measurement of intracellular reactive oxygen species.
Total intracellular reactive oxygen species (ROS) were measured using Cell Meter™ Fluorimetric Intracellular Total ROS Activity assay kit *Deep Red Fluorescence* (AAT Bioquest, Inc. Sunnyvale, CA). Briefly, the LMSCs were seeded in Costar black wall/clear bottom 96 well plates at a density 5 X 104 cells (in 100 μl DMEM containing 10% FBS). The cells were infected with 20 MOI of opsonized K. pneumoniae for 15 and 30 minutes. At the end of each time point, ROS Brite™ 670 working solution (100 μl/well) was added to the wells and incubated for 30 minutes. Increase in fluorescence was measured at excitation and emission wavelengths of 650 and 675 nm using a fluorescence plate reader.
Induction and inhibition of autophagy during opsophagocytosis of K. pneumoniae by LMSCs.
We cultured the cells (0.5 million cells/0.5 ml DMEM containing 10% FBS/well in 12 well plates) in the presence or absence of 5 mM 3-Methyladenine (3-MA; MilliporeSigma, St. Louis, MO) for 1 h as reported (41) . Then, the cells were infected with 20 MOI of opsonized K. pneumoniae for 1 h and 4 h. The plates were centrifuged and washed 3 times with sterile PBS. The adherent cells were lysed with 0.1% Triton X 100, serially diluted and plated onto MacConkey agar plates. The levels of different autophagy-associated proteins in the 3-ME treated LMSCs/3-ME treated and K. pneumoniae infected LMSCs (3 million cells/3 ml DMEM containing 10% FBS/well in 6 well plates) were analyzed using Western blot. LMSCs cultured with medium alone were used as controls. For Western blot analysis, the adherent cells were lysed with NP-40 cell lysis buffer (Invitrogen, Frederick, MD), and centrifuged at 2000 rpm for 8 minutes at 4°C using an Eppendorf centrifuge. The protein concentration in the lysates was quantified using Pierce™ BCA Protein Assay kit (Thermo Scientific, Rockford, IL) to ensure that equal amounts of proteins were loaded onto 12% SDS-PAGE. Then the proteins were electrophoretically transferred onto a PVDF membrane (Millipore) and membranes were incubated with appropriate primary antibodies (LC3 A/B antibody, Beclin-1 Rabbit mAb, ATG7 Rabbit mAb, anti-GAPDH antibody) and then with appropriate secondary antibodies (horseradish peroxidase-conjugated to either anti-mouse IgG or anti-rabbit IgG) linked to HRP conjugates. Blots were developed using an enhanced chemiluminescence kit (Amersham Biosciences UK Ltd, Buckinghamshire, U.K.). For autophagy induction experiments, cells (0.5 million cells/well in 0.5 ml of 10% FBS) were cultured in the presence or absence of 80 μg/ml of Rapamycin (InvivoGen, San Diego, CA) for 2 h before infecting them with 20 MOI of opsonized K. pneumoniae.
K. pneumoniae infection and treatment with Sca-1+ LMSCs.
We infected female C57BL/6J mice with K. pneumoniae as previously described (34). At 4 h after infection with K. pneumoniae, mice were anaesthetized and given either passage 6 Sca-1+ LMSCs (750,000 cells in 50 μl sterile DPBS/mouse/by i.t.) or 3T3 MEFs (750,000 cells in 50 μl sterile DPBS/mouse/by i.t. as a control for LMSCs) or sterile DPBS (50 μl/mouse/by i.t. as a control for bacteria). The mice were sacrificed at 48 h post-infection and the samples were collected from each mouse for bronchoalveolar lavage fluid (BALF) and phenotyping, lung histopathology, as well as to enumerate the bacterial burden in different organs.
BAL and phenotyping.
The trachea was cannulated using a 20-gauge shielded intravenous catheter (42). BALF was collected four times from each mouse by instilling 0.8 ml of sterile PBS [containing 0.2 U of Heparin and protease inhibitor cocktail (Sigma-Aldrich; one tablet/10 ml)]. To quantify the total white blood cells (TWBC), 3 ml of BALF (from each mouse) was centrifuged at 1800 rpm for 8 minutes at 4° C. The cell pellet obtained from the BALF of each mouse was resuspended in 3 ml of PBS and the TWBC were enumerated using a hemocytometer. To analyze different inflammatory cell populations in the BALF, BALF was centrifuged onto Superfrost/plus microscopic slides (Fisher Scientific Inc., Pittsburgh, PA) using a Wescor Cytopro® Cytocentrifuge (Hyland Scientific, Stanwood, WA). The slides were stained using Quick-Dip kit (Mercedez Scientific, Lakewood Ranch, FL). Differential counts were performed on 300 cells, as described in standard cytologic techniques (43).
Quantification of cytokines/chemokines/growth factors in lung supernatants.
Different cytokines, chemokines and growth factors in the lung supernatants were measured using Milliplex MAP mouse cytokine/chemokine magnetic bead assay (EMD Millipore Corporation) and Milliplex MAP mouse angiogenesis and growth factor magnetic bead assay (EMD Millipore Corporation).
Histology.
The lungs were inflated with 1% low-melting agarose (Sigma-Aldrich) and fixed in 4% phosphate buffered formalin for 18 to 24 h. The whole lung was embedded in paraffin and sections (5 μm thickness) were stained with hematoxylin and eosin (H&E). The H&E stained lung sections in each group were coded, and representative images (whole lung) were acquired with a scanner (NanoZoomer 2.0-HT slide scanner, Hamamatsu Photonics) using NDP scan v3.1.7 software (lens magnification, ×40). These lung sections were scored for the extent of inflammation/injury by a veterinary pathologist in a blinded fashion. We used the following scoring system (scores 0-3) to determine inflammation/injury in the lungs: 0, no inflammation in lung tissue; 1, <10% of the lung tissue was infiltrated by inflammatory cells; 2, about 10-50% of the lung tissue was infiltrated by inflammatory cells; 3, >50% of the lung tissue was infiltrated by inflammatory cells.
Retention of Sca-1+ LMSCs in the lungs.
We labeled the passage 6 Sca-1+ LMSCs with 10 μM Carboxyfluorescein succinimidyl ester (CFSE; Thermo Fisher Scientific-Invitrogen, Waltham, MA) according to the manufacturer’s protocol. We administered either PBS or CFSE-labeled Sca-1+ LMSCs (750,000 cells in 50 μl sterile DPBS/mouse by i.t) into the lungs of K. pneumoniae (1 x 103 CFU/50 μl of sterile PBS/mouse by i.t.) infected mice at 4 h post-infection. We sacrificed the mice 4 h (Group I: Kp + PBS; Kp + CFSE-Sca-1+ LMSCs) and 48 h (Group II: Kp + PBS; Kp + CFSE-Sca-1+ LMSCs) post-infection and inflated the lungs with 1% molten agarose. We fixed the lungs in 4% buffered formalin and embedded them in paraffin. Images (20 per lung section) of the unstained lung sections (5 μm thickness) were acquired using a Leica DM6 B (Allendale, NJ) fluorescence microscope (lens magnification, x20). In each image, the number of CFSE positive cells (green) was counted manually.
Enumeration of bacteria.
Mouse organs [lungs, spleen, and liver (left lower lobe)] were harvested and homogenized in 1 ml of sterile PBS using a Retsch Tissue Lyser (Vender Scientific, Inc. Newtown, PA) for 2 minutes. Aliquots of homogenates and BALF were serially diluted in sterile PBS and plated onto MacConkey agar plates. The viable counts of K. pneumoniae were enumerated after incubating the plates overnight at 37° C.
Survival.
For the survival study, we infected female C57BL/6J mice with K. pneumoniae [5 x 103 CFU in 75 μl sterile DPBS/mouse by oropharyngeal (o.p.) route] and then these mice were treated either with passage 6 Sca-1+ LMSCs (1 x 106 cells in 75 μl sterile DPBS/mouse/by o.p.) or with 3T3 MEFs (1 x 106 cells in 75 μl sterile DPBS/mouse/by o.p.), or with sterile DPBS (75 μl/mouse/by o.p.) at 4 h and 24 h post infection. Survival in each group was recorded every 12 h for up to 15 days. Survival experiments were performed twice.
Statistics.
Statistical analysis was performed using GraphPad Prism 8.3.0 (GraphPad Software, La Jolla, CA). Specific statistical testing is described in the accompanying figure legends. The error bar represents mean ± SEM. A value of p < 0.05 was considered statistically significant. Significant differences are indicated by * (p < 0.05), ** (p < 0.01), *** (p < 0.001), and **** (p < 0.0001).
Results
Isolation and characterization of Sca-1+ LMSCs.
We digested lung tissues using the protocol described in our previous publication (36) and sorted Sca-1+ cells using a fluorescence activated cell sorter. The Sca-1+ cells constituted as much as 20.3% of the total adherent lung cell population (Supplemental Fig. S1C, right panel). Flow cytometric analysis revealed that the purified and expanded passage 6 Sca-1+ cells strongly express mesenchymal stem/stromal cell antigens CD44, CD73, CD105, CD106, and CD123; although the expression of mesenchymal stem/stromal cell antigen CD90.1 (Thy 1.1), as well as CD34, and Toll like receptor 4 (TLR4) antigens was weak (Supplemental Fig. S1D). In contrast, the purified Sca-1+ cells were negative for endothelial marker CD31 and hematopoietic cell markers CD11b and CD45 (Supplemental Fig. S1D). In addition, these cells were negative for MHC class II, CD11c, CD54, CD103, CD117 (c-kit), ICOS, and CD135 antigens (Supplemental Fig. S1D). Similar to passage 6 Sca-1+ LMSCs (Supplemental Fig. S1D), passage 9 Sca-1+ LMSCs expressed higher levels of mesenchymal stem/stromal cell antigens CD105, and CD106; low levels/weak expression of mesenchymal stem/stromal cell antigens CD44, CD73, and CD123, as well as CD34 and TLR4 antigens on their surface (Supplemental Fig. S2). The expression of mesenchymal stem/stromal cell marker CD90.1, reached to control level in passage 9 Sca-1+ LMSCs (Supplemental Fig. S2). Passage 9 Sca-1+ LMSCs were also negative for MHC class II, CD31, CD45, CD54, CD11c, CD11b, CD103, CD117 (c-kit), ICOS, and CD135 antigens (Supplemental Fig. S2).
As indicated in Supplemental Fig. 3, the flow-sorted and expanded Sca-1+ lung cells (passage 6) formed colonies (Supplemental Fig. S3A) and differentiated into alizarin red S-positive osteocytes (Supplemental Fig. S3B, bottom left panel) or oil red ‘O’-positive adipocytes (Supplemental Fig. S3B, bottom right panel) when cultured in differentiation medium. Control cells grown in CMEM did not stain with either dye (Supplemental Fig. S3B, upper panels).
Morphological and ultrastructural features of Sca-1+ LMSCs.
We cultured the flow-sorted Sca-1+ LMSCs (passage 6) in CMEM (for 1 day, 2 days, and 5 days) and stained the plastic adherent cells using methylene blue (Fig. 1A, bottom panels). Within 24 h of initial plating, the purified Sca-1+ LMSCs were able to attach to the plastic and appeared as spindle-shaped cells arranged in parallel or whorls (Fig. 1A, upper panels). Moreover, these cells exhibited high proliferative potential in culture (Fig. 1B). We further characterized (after five days of culture in CMEM) the morphology and ultrastructural features of Sca-1+ LMSCs and 3T3 MEFs (as control) using scanning electron microscopy (SEM) and transmission electron microscopy (TEM). Sca-1+ LMSCs (passage 6) are, as shown by SEM, to be flattened cells with lamellipodia and many thin filopodia (Fig. 1C, i) similar in morphology to cultured 3T3 MEFs (Fig. 1D, i). TEM images revealed the following ultrastructural features: cell bodies that are either rounded, oval or polygonal, and nuclei that are centric or eccentric in both Sca-1+ LMSCs (Fig. 1C) and 3T3 MEFs (Fig. 1D). The nuclei are large, pale, mostly euchromatic, and occupy most of the cell body of Sca-1+ LMSCs (Fig. 1C) when compared to 3T3 MEFs (Fig. 1D). In both Sca-1+ LMSCs and 3T3 MEFs, a) the nuclei contain clefts and invaginations of the nuclear envelope, b) the cell membranes are irregular with lamellipodia and thin filopodia, c) the cellular organelles are distributed throughout the cytoplasm, d) the rough endoplasmic reticulum (rer) is prominent, containing moderately electron-dense material, e) mitochondria are numerous, appearing to be mostly elongated, and occasionally organized in small groups of 3-4 elements. Another frequent finding was the presence of a large number of autophagosomes, endosomes, lysosomes, and vesicles in the cytoplasmic compartment of Sca-1+ LMSCs (Fig. 1C) compared to 3T3 MEFs (Fig. 1D). To investigate whether Sca-1+ LMSCs were able to maintain such morphological and ultrastructural features after subsequent passages, we cultured the passage 9 Sca-1+ LMSCs in CMEM for 5 days and characterized them using phase contrast microscopy and TEM. Similar to passage 6 Sca-1+ LMSCs, the passage 9 Sca-1+ LMSCs are plastic adherent and spindle-shaped cells (Supplemental Fig. S4A). TEM images (Supplemental Fig. S4B) show that passage 9 Sca-1+ LMSCs are ultrastructurally similar to passage 6 Sca-1+ LMSCs (Fig. 1C) except that i) their nuclei are mostly heterochromatic in nature, and ii) they contain relatively fewer numbers of lysosomes in their cytoplasm.
FIGURE 1. Morphology and ultrastructural features of Sca1+ LMSCs.
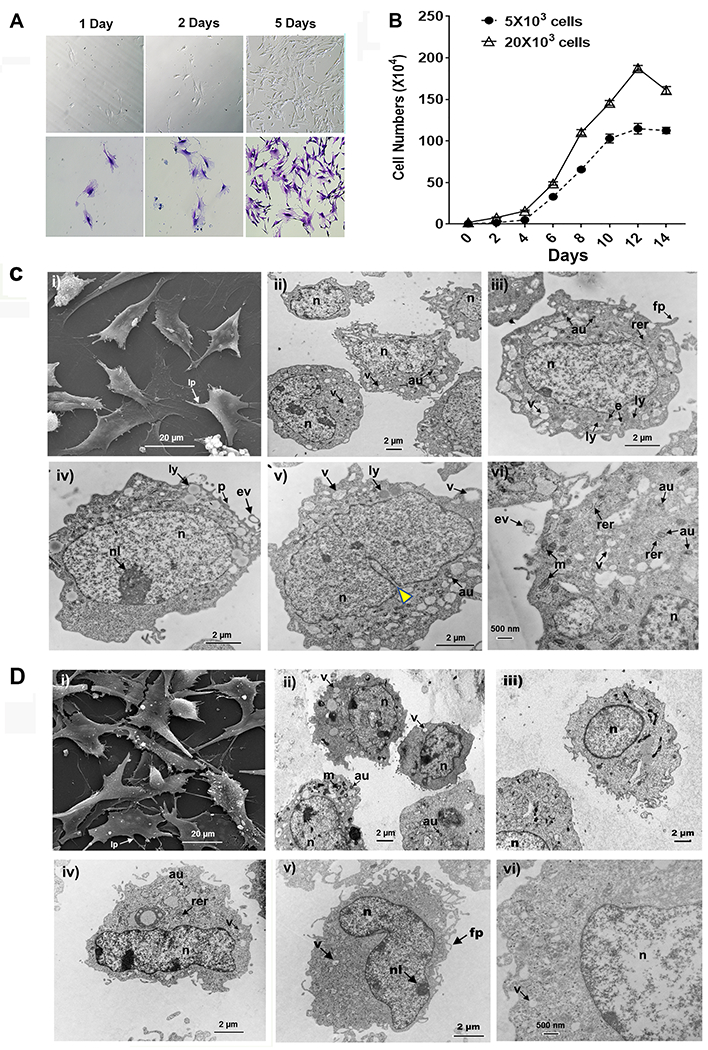
(A) Sca-1+ LMSCs (passage 6) grown on plastic at low density in CMEM for 1 day, 2 days, and 5 days. Adherent cells were stained with methylene blue. Images of the plastic adherent cells (A, upper panel) and methylene blue-stained cells (A, bottom panel) were taken using a conventional optical microscope. (B) Growth curves of Sca-1+ LMSCs. Results are the mean ± SEM from 2 independent experiments. (C) Scanning electron micrograph (i) of Sca-1+ LMSCs grown on a plastic surface (Theranox™ plastic coverslips) for 5 days. Note the flattened cell with lamellipodia and many thin filopodia as well as the fibroblast-like morphology. Magnification, x3000. Scale bar = 20 μm. Transmission electron micrographs (ii – vi) of cultured Sca-1+ LMSCs at (ii) magnification x8000, scale bar = 2 μm, (iii, iv, v) magnification x12,000, scale bars = 2 μm, and (vi) magnification x20,000, scale bar = 500 nm. D) Scanning electron micrograph (i) of 3T3 MEFs grown on a plastic surface (Theranox™ plastic coverslips) for 5 days. Note the flattened cells with processes spreading outward. Magnification, x2500, scale bar = 20 μm. Transmission electron micrographs (ii – vi) of 3T3 MEFs grown in CMEM for 5 days (ii) magnification x8000, (iii) magnification x12,000, (iv) magnification x12,000, (v) magnification x12,000, scale bars = 2 μm, and (vi) magnification x20,000, scale bar = 500 nm. au, autophagosome; e, endosome; ev, extracellular vesicle; fp, filopodia; lp, lamellipodia; ly, lysosome; m, mitochondria; n, nucleus; nl, nucleoli; p, plasma membrane; rer, rough endoplasmic reticulum; v, vesicle; yellow Δ, invagination of nuclear envelope.
FIGURE 3. Effect of Sca-1+ LMSCs and neutrophils on the growth of K pneumoniae.
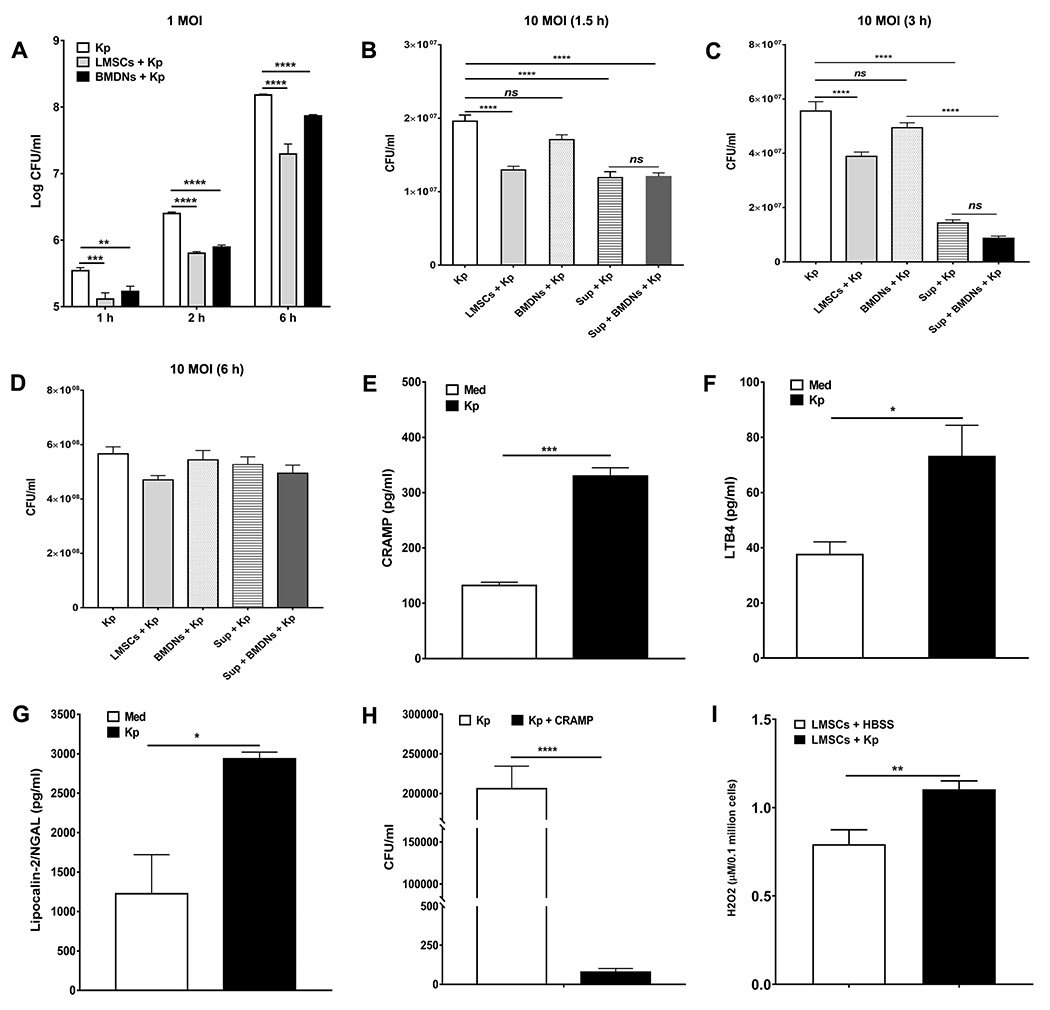
Both Sca-1+ LMSCs and BMDNs showed similar killing effects at all time points tested when infected with low MOI (1 MOI) of K. pneumoniae (A). **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 for the indicated comparisons. Data are mean ± SEM from three separate experiments (quadruplicate samples). Data were analyzed using one-way ANOVA with Turkeys multiple comparisons test at each time point. However, the killing effects of Sca-1+ LMSCs and LMSC-derived supernatants were remarkably higher at 1.5 h (B) and 3 h (C) post-infection than that of BMDNs when infected with a higher MOI (10 MOI) of K. pneumoniae. Both Sca-1+ LMSCs and their supernatants as well as BMDNs failed to inhibit the growth of K. pneumoniae (10 MOI) at 6 h post-infection (D). Results are the mean ± SEM from three independent experiments. ****, p < 0.0001 for the indicated comparisons. Data were analyzed using one-way ANOVA with Tukey’s multiple comparisons test. Infection with K. pneumoniae induces the secretion of CRAMP (E), LBT4 (F) and lipocalin-2/NGAL (G) in Sca-1+ LMSCs. Results are the mean ± SEM of triplicate samples from each group. *, p < 0.05; ***, p < 0.001 by unpaired t test. (H) CRAMP is potent for killing K. pneumoniae in vitro. Results are the mean ± SEM from three independent experiments. Each experimental condition was carried out using triplicate samples. ****, p < 0.0001 for the indicated comparison. Data were analyzed using unpaired t test. (I) LMSCs secrete H2O2 following infection with K. pneumonia. Results are the mean ± SEM from three separate experiments. **, p < 0.01 for the indicated comparison. Data were analyzed using unpaired t test.
Production of LPS-induced inflammatory cytokines by macrophages and neutrophils is modulated by Sca-1+ LMSCs.
Sca-1+ LMSCs exhibit an immunomodulatory effect on innate immune cells in response to LPS challenge in vitro. We co-cultured the Sca-1+ LMSCs with BMDMs for 24 h or with BMDNs for 8 h in the presence or absence of LPS (500 ng/million cells/ml) at different ratios (1:1, 1:2, or 1:4 LMSCs to BMDMs or BMDNs) and quantified inflammatory cytokines in culture supernatants. Inflammatory cytokine production was attenuated in both BMDMs and BMDNs in the presence of LMSCs (Fig. 2A-C). Treatment with LMSCs remarkably inhibited LPS-induced secretion of TNFα (Fig. 2A) and IL-1β (Fig. 2B) by BMDMs and TNFα by BMDNs (Fig. 2C) at all ratios tested. Neither BMDMs nor BMDNs secreted TNFα or IL-1β under unstimulated conditions (data not shown). Further, Sca-1+ LMSCs also did not secrete TNFα and IL-1β under unstimulated conditions or following stimulation with LPS (data not shown).
FIGURE 2. Role of Sca-1+ LMSCs in the production of inflammatory cytokines by macrophages and neutrophils.
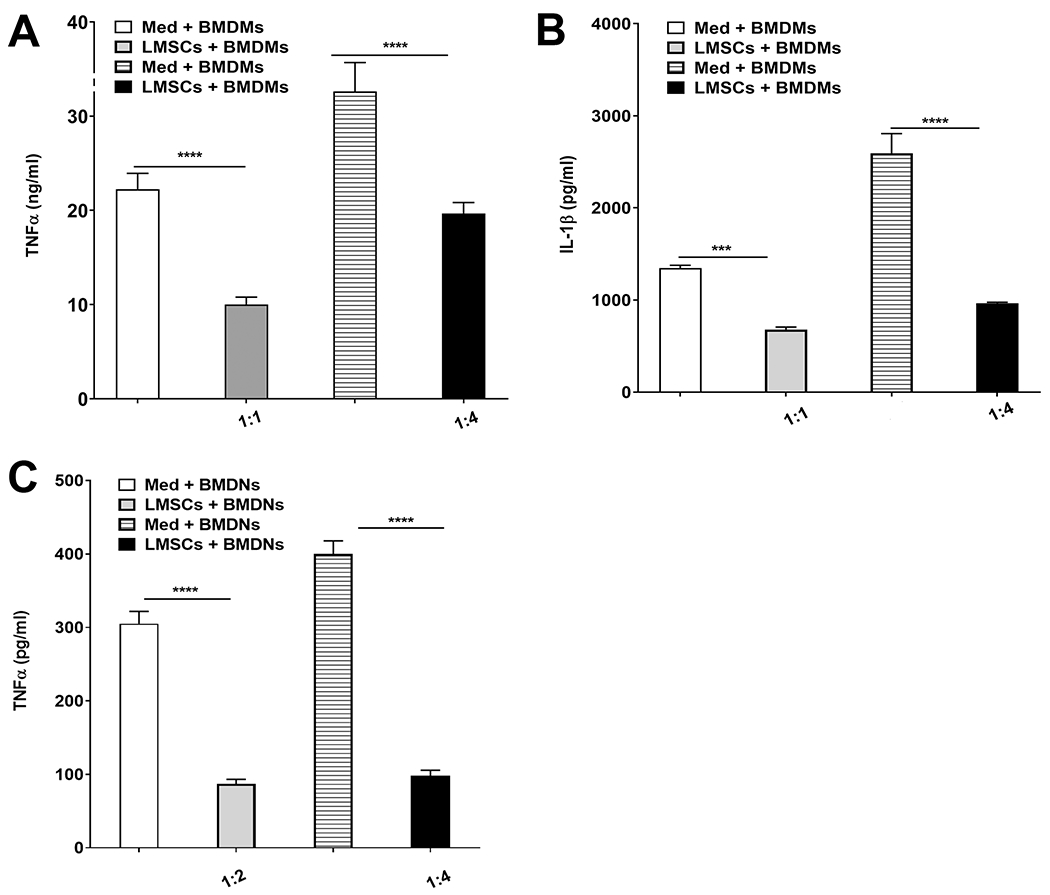
Purified Sca-1+ LMSCs were co-cultured separately with BMDMs and BMDNs at different ratios in the presence or absence of LPS and TNFα and IL-1β in culture supernatants were quantified using ELISA. LMSCs inhibited the LPS-induced secretion of TNFα (A) and IL-1β (B) in BMDMs and TNFα (C) in BMDNs at all ratios tested. Data are mean ± SEM from three independent experiments. ***, p < 0.001; ****, p < 0.0001 for the indicated comparisons. Data were analyzed using one-way ANOVA followed by Tukey’s multiple comparisons test. Med, medium.
Both Sca-1+ LMSCs and neutrophils inhibit the growth of K pneumoniae in vitro.
Bacterial killing by neutrophils, macrophages, mast cells, and gamma delta T cells plays a critical role in clearance of K. pneumonia (35, 44-46). First, we performed extracellular killing assays by infecting cytochalasin D-treated Sca-1+ LMSCs and BMDNs with K. pneumoniae [1 and 10 MOI] for various time periods. Both Sca-1+ LMSCs and BMDNs showed similar rates of killing when inoculated with a low MOI (1 MOI) of K. pneumoniae at all time points examined (Fig. 3A). However, only Sca-1+ LMSCs and Sca-1+ LMSCs-derived supernatants were efficient at killing K. pneumoniae at 1.5 h (Fig. 3B) and 3 h (Fig. 3C) time points when cultured with a higher MOI (10 MOI) of K. pneumoniae. Interestingly, the Sca-1+ LMSCs-derived supernatant exhibited more potent killing of K. pneumoniae than Sca-1+ LMSCs at 3 h (Fig. 3C). Intriguingly, the combination of Sca-1+ LMSCs-derived supernatant with BMDNs increased the extracellular killing effect of BMDNs at the 3 h time point (Fig. 3C), although BMDNs did not appear to have potent killing ability and it is mostly due to LMSCs-derived supernatant. At 6 h, both Sca-1+ LMSCs and Sca-1+ LMSCs-derived supernatant, as well as BMDNs failed to inhibit the growth of K. pneumoniae (Fig. 3D). To investigate whether increased bacterial killing by Sca-1+ LMSCs was associated with enhanced production of antimicrobial compounds, we cultured the Sca-1+ LMSCs with 20 MOI of opsonized K. pneumoniae for 4 h and quantified antimicrobial compounds (CRAMP, LTB4, and lipocalin-2/NGAL) in the culture supernatants using ELISA. As shown in Figure 3, Sca-1+ LMSCs secrete CRAMP (Fig. 3E), LTB4 (Fig. 3F), and lipocalin-2/NGAL (Fig. 3G) under unstimulated conditions, all of them increased at 4 h post-infection with K. pneumoniae. Importantly, treatment with murine CRAMP remarkably inhibited the extracellular growth of K. pneumoniae in vitro (Fig. 3H). In addition, infection with K. pneumoniae also induced the secretion of H2O2 by Sca-1+ LMSCs (Fig. 3I).
Both Sca-1+ LMSCs and BMDMs possess phagocytic and intracellular killing abilities.
Next, we compared the phagocytic uptake and intracellular bacterial killing abilities of Sca-1+ LMSCs and BMDMs using an opsonophagocytic assay. We performed the intracellular killing assay using 20 MOI of opsonized K. pneumoniae for 1 h and 4 h and found that BMDMs were more efficient in the phagocytotic uptake of K. pneumoniae than Sca-1+ LMSCs at the 1 h time point (Fig. 4A). However, Sca-1+ LMSCs displayed more efficient intracellular killing of K. pneumoniae compared to that of BMDMs at 4 h post-infection (Fig. 4A). We also confirmed the presence of K. pneumoniae in Sca-1+ LMSCs using both confocal and fluorescence microscopies. Imaging using confocal (Fig. 4B, indicated by yellow arrow in right panel) and immunofluorescence microscopy (Fig. 4C, indicated by yellow arrows) shows the presence of this Gram-negative bacterium in Sca-1+ LMSCs infected with opsonized K. pneumoniae. These findings prompted us to further investigate the phagocytotic capacity of LMSCs by flow cytometry using HI Kp-FITC. Representative flow cytometric analyses for the percent uptake of HI Kp-FITC by Sca-1+ LMSCs and BMDMs are depicted in Fig. 4D and Fig. 4E, respectively. Similar to the opsonophagocytosis assay using live K. pneumoniae (Fig. 4A), the percent of BMDMs involved in phagocytosis after 1 h of HI Kp-FITC incubation was significantly higher when compared to Sca-1+ LMSCs (Fig. 4F). Next, we evaluated the surface expression of CR3 (CR3; CD11b/CD18) and CRIg in Sca-1+ LMSCs, which are known to be involved in the phagocytosis of complement opsonized bacteria (47, 48). Flow cytometric analysis revealed the expression of CRIg in Sca-1+ LMSCs (Fig. 4G) while LMSCs were negative for CR3 (Fig. 4G). Quantification of intracellular ROS using Cell Meter™ Fluorimetric ROS Activity assay kit also shows increased production of intracellular ROS by LMSCs at 15 minutes post-infection with opsonized K. pneumoniae over that of control LMSCs (Fig. 4H).
FIGURE 4. Phagocytotic uptake and intracellular killing of K. pneumoniae by Sca1+ LMSCs and BMDMs.
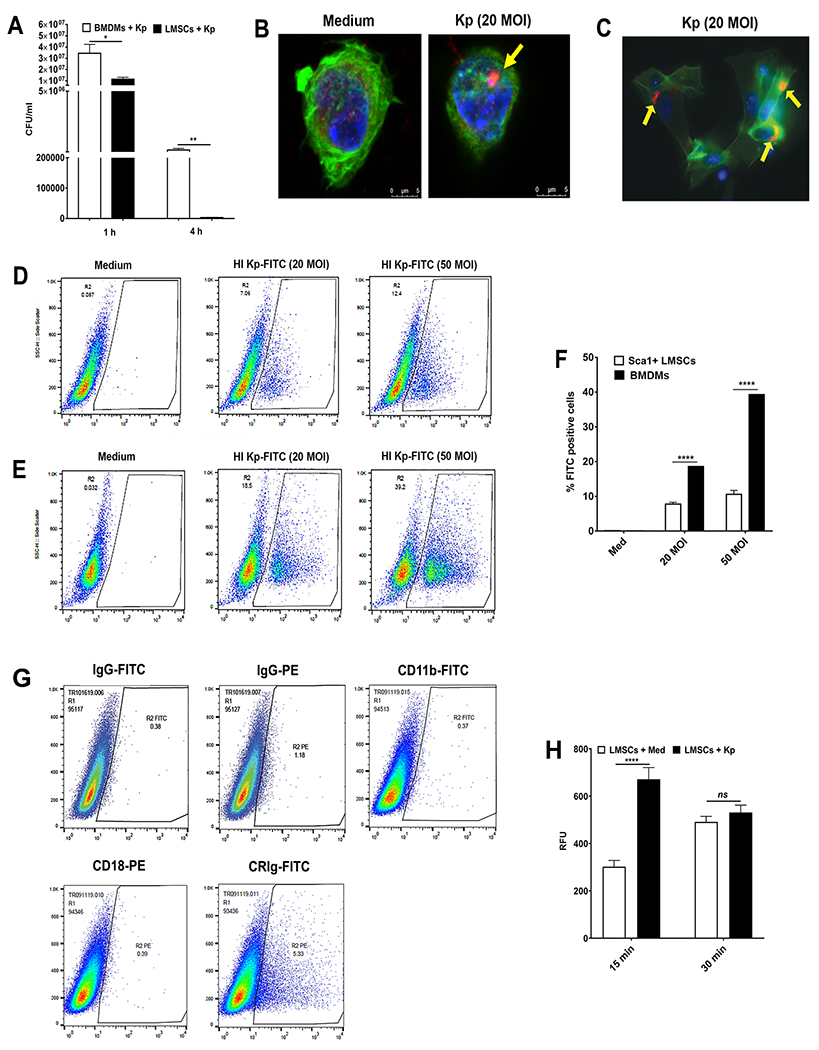
(A) Bacterial uptake and intracellular killing abilities of Sca-1+ LMSCs and BMDNs was assessed at 1 h and 4 h by measuring intracellular live bacteria in these cells, as described in the Methods section. Data are expressed as the mean ± SEM from three independent experiments. *, p < 0.05; **, p < 0.01 as compared with LMSCs by multiple t tests. (B) Fluorescence confocal microscopy of Sca-1+ LMSCs cultured with medium (B, left panel) or cultured with 20 MOI of opsonized K. pneumoniae (B, right panel) for 3 h. K. pneumoniae (red) and F-actin (green) were stained as described in the Methods section. A representative confocal section through the middle of Sca-1+ LMSCs is shown for observation of intracellular K. pneumoniae (B, indicated by yellow arrow in right panel). Magnification x60. (C) A representative immunofluorescence microscopy image shows the presence of K. pneumoniae (C, indicated by yellow arrows) in infected Sca-1+ LMSCs. Magnification x40. (D-F) Examination of phagocytic capacity of BMDMs and Sca-1+ LMSCs by flow cytometry. Representative dot plots of Sca-1+ LMSCs (D) and BMDMs (E) cultured separately either with medium or with 20 or 50 MOI of opsonized HI Kp-FITC. (F) The percentage Sca1+ LMSCs and BMDMs positive for HI Kp-FITC are shown as a bar graph. The results are mean ± SEM from two independent experiments (3-4 samples in each group). ****, p < 0.0001 by multiple t-tests. (G) Analysis of expression of CR3 (CD11b/CD18) and CRIg in Sca-1+ LMSCs using flow cytometer. Representative dot plots of Sca-1+ LMSCs stained separately with isotype control antibodies or fluorescence conjugated CR3 (CD11b-FITC/CD18-PE) and anti-CRIg (CRIg-FITC) antibodies are shown. n = 2 independent experiments. (H) Sca-1+ LMSCs produce intracellular ROS upon infection with K. pneumoniae. Data are mean ± SEM from three independent experiments (triplicate samples). ****, p < 0.0001 by multiple t tests.
Autophagy is essential for the intracellular replication of K. pneumoniae during early time of infection in Sca-1+ LMSCs.
To investigate whether autophagy is associated with intracellular survival of K. pneumoniae in Sca-1+ LMSCs, we first pre-treated the Sca-1+ LMSCs either with 3-MA or with rapamycin and then performed an opsonophagocytosis assay using 20 MOI of opsonized K. pneumoniae. We found that pre-treatment of Sca-1+ LMSCs with the autophagy inhibitor, 3-MA, remarkably suppressed the intracellular replication of K. pneumoniae at 1 h post-infection (Supplemental Fig. S5A). In contrast, pre-treatment with the autophagy inducer, rapamycin, significantly enhanced the intracellular replication of K. pneumoniae during early time of infection (1 h) in Sca-1+ LMSCs (Supplemental Fig. S5B). However, in both groups at 4 h pos-infection, Sca-1+ LMSCs were able to clear most of the phagocytosed K. pneumoniae (Supplemental Fig. S5A & B). No live intracellular K. pneumoniae was detected in Sca-1+ LMSCs pre-treated with 3-MA (Supplemental Fig. S5A) and few live K. pneumoniae were observed in lysates from Sca-1+ LMSCs pre-treated with rapamycin at 4 h post infection (Supplemental Fig. S5B).
To determine whether autophagy inhibition results in decreased levels of key autophagy-associated proteins, we extracted proteins from control Sca-1+ LMSCs, K. pneumoniae-infected Sca-1+ LMSCs, and 3-MA pre-treated and K. pneumoniae infected Sca-1+ LMSCs and carried out immunoblotting with antibodies against Beclin-1, ATG7, and LC3. All three of these autophagy-associated proteins were abundantly expressed in Sca-1+ LMSCs at baseline (Supplemental Fig. S5C). Infection with K. pneumoniae further increased the levels of Beclin-1, ATG7, and LC3 at 1 h post-infection in Sca-1+ LMSCs (Supplemental Fig. S5C). However, pre-treatment with 3-MA decreased the levels of these autophagy associated-proteins in Sca-1+ LMSCs at 1 h post-infection (Supplemental Fig. S5C). These results suggest activation of the autophagy pathway occurs following opsonophagocytic uptake of K. pneumoniae by Sca-1+ LMSCs.
Retention of Sca-1+ LMSCs in the lungs of K. pneumoniae infected mice.
Retention of CFSE-labeled Sca-1+ LMSCs in the lungs of K. pneumoniae-infected mice was evaluated using fluorescence microscopy. The numbers of CFSE-labeled Sca-1+ LMSCs were comparatively higher in the lung sections from the mice that were sacrificed at 4 h post-LMSC administration (2.26 ± 0.26 cells/image) than those mice that were sacrificed at 44 h post-LMSC transplantation (1.69 ± 0.23 cells/image) (Supplemental Fig. S6A&B). Moreover, Sca-1+ LMSCs formed contiguous clusters of 4 - 7 cells (appearing as green in the Supplemental Fig. S6A&B) interspersed throughout the lung parenchyma of the K. pneumoniae infected mice.
Sca-1+ LMSCs attenuate K. pneumoniae-induced lung inflammation.
To determine the effect of treatment with Sca-1+ LMSCs on K. pneumoniae-induced lung inflammation, TWBC as well as different inflammatory cell populations were analyzed in BALF from mice 48 h after K. pneumoniae infection/Sca-1+ LMSC-treatment. Treatment with Sca-1+ LMSCs markedly decreased inflammatory cell infiltration into the BALF of mice infected with K. pneumoniae (Fig. 5A). The TWBC number in BALF at 48 h after K. pneumoniae infection of mice treated with PBS was significantly higher than those treated with Sca-1+ LMSCs (Fig. 5A). In both K. pneumoniae-infected and K. pneumoniae-infected/Sca-1+ LMSCs-treated groups, the vast majority of infiltrating cells in the BALF were neutrophils. Treatment with Sca-1+ LMSCs reduced the infiltrating neutrophils in the K. pneumoniae-infected group. In contrast, macrophages were the major inflammatory cell population in the BALF of the infection control (PBS) group (Fig. 5A). However, administration of 3T3 MEFs had no significant effect on total WBC as well as different inflammatory cell populations in the BALF of K. pneumoniae infected mice (Supplemental Fig. S7A). To determine the regulatory effect of Sca-1+ LMSCs on inflammatory cytokine and chemokine responses in the lung, we analyzed the protein levels of different cytokines, chemokines, and growth factors in lung supernatants. As expected, lung supernatants from the infection control (PBS) mice showed very low levels of inflammatory markers, while lung supernatants from those infected with K. pneumoniae showed higher levels of cytokines (IL-6, TNFα, IL-1α, and IL-1β) and chemokines (MIP-1α, MIP-2, MCP-1, and KC) (Fig. 5B). Treatment with Sca-1+ LMSCs significantly reduced the levels of IL-6, TNFα, IL-1α, IL-1β, MIP-1α, MIP-2, MCP-1, and KC in the lungs of the K. pneumoniae-infected group (Fig. 5B). However, treatment with Sca-1+ LMSCs did not alter the levels of LIX, GM-CSF, IFNγ, IL-10, Angiopoietin-2, or SDF1 (Fig. 5B). Next, we measured the extent of inflammation in H&E-stained lung sections from K. pneumoniae-infected (Fig. 5C, upper left panel) and Sca1+ LMSC-treated (Fig. 5C, bottom left panel) mice. We found that the lung pathology scores were lower in the lung sections from K. pneumoniae-infected and Sca1+ LMSC-treated mice when compared to K. pneumoniae-infected mice that received PBS (Fig. 5D). Hematoxylin and eosin-stained lung sections from K. pneumoniae-infected and Sca1+ LMSC-treated mice show attenuated inflammatory foci (Fig. 5C, bottom left panel) containing lower numbers of inflammatory cells (Fig. 5C, bottom magnified image) as well as granular microcolonies of bacteria (indicated by arrows in Fig. 5C) when compared to the K. pneumoniae-infected mice that received PBS (Fig. 5C, upper panels).
FIGURE 5. Effect of Sca1+ LMSCs on K. pneumoniae-induced inflammation.
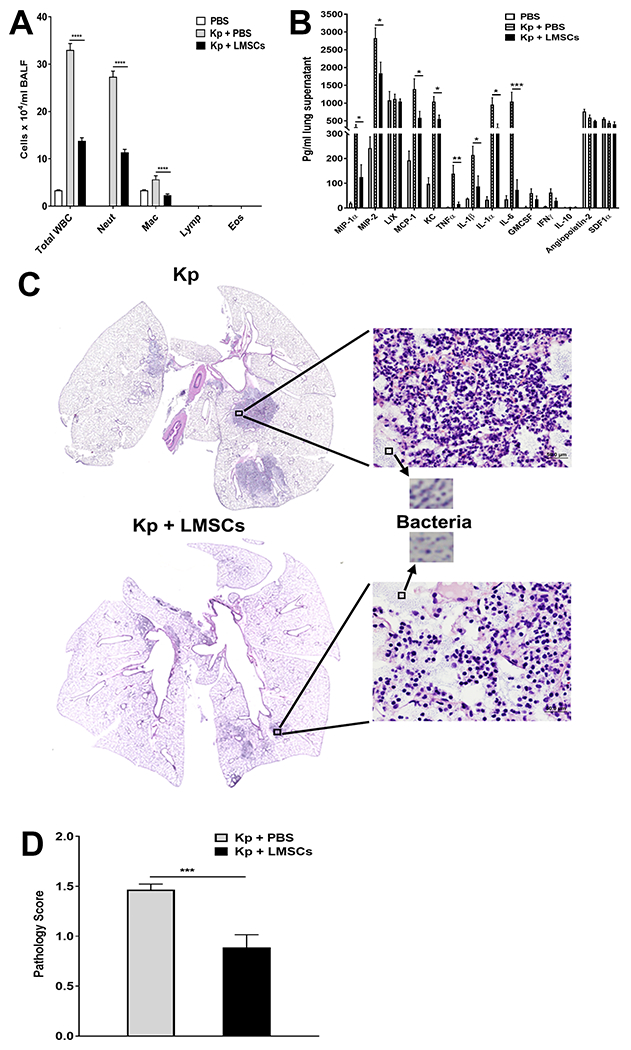
(A) K. pneumoniae-infected mice treated with Sca-1+ LMSCs show reduced total white blood cells and predominantly reduced levels of neutrophils and macrophages in the BALF when compared to K. pneumoniae-infected and PBS-treated mice. Data are mean ± SEM from three independent experiments (n = 5 - 6 mice/group). ****, p < 0.0001 for the indicated comparisons. Data (for each inflammatory cell type) were analyzed using one-way ANOVA with Tukey’s multiple comparisons test. WBC, white blood cells; Neut, neutrophils; Mac, macrophages; Lymph, lymphocytes; Eos, eosinophils. (B) Analysis of cytokines, chemokines, and growth factors in the lung supernatants using the Millipore magnetic bead assay. Data are mean ± SEM of 5=6 mice/group. *, p < 0.05; **, p < 0.01; ***, p < 0.001 for the indicated comparisons. Data were analyzed using multiple t tests. (C) Treatment with Sca-1+ LMSCs reduced K. pneumoniae-induced pathologic lesions in the lungs of mice. Representative photomicrographs of H&E-stained lung sections (magnification, x40) from K. pneumoniae-infected (top left panel)/K. pneumoniae-infected and Sca-1+ LMSC-treated mice are shown (bottom left panel). Magnified images (x200) show the presence of inflammatory cells (top and bottom right panels) and granular microcolonies of bacteria (indicated by arrows) in the lungs of K. pneumoniae-infected/K. pneumoniae-infected and Sca-1+ LMSC-treated mice. D) Lung injury scores. Data are mean ± SEM from two independent experiments (n = 5-6 mice/group). ***, p < 0.001. Data were analyzed using unpaired t test.
Sca-1+ LMSCs augment bacterial clearance and improve survival in K. pneumoniae-infected mice.
To evaluate the effect of Sca-1+ LMSCs on bacterial burden in the lungs and in extrapulmonary organs, we determined the viable bacterial counts in the lungs, BALF, spleen, and liver. Interestingly, we found reduced bacterial burden in the lungs (Fig. 6A) and BALF (Fig. 6B) of Sca-1+ LMSC-treated mice when compared to the K pneumoniae-infected and PBS-treated mice. Consistent with the findings in the lungs, Sca-1+ LMSC-treated mice also showed reduced dissemination to the spleen (Fig. 6C) and liver (Fig. 6D). Similar to 3T3 mouse lung fibroblast-mediated effects observed during E. coli induced pneumonia (49), transplanted 3T3 MEFs did not significantly influence the bacterial burden in the lungs, BALF, and in extrapulmonary organs of K. pneumoniae infected mice (Supplemental Fig. S7B-E).
FIGURE 6. Effect of Sca1+ LMSCs on bacterial growth and survival in K. pneumoniae-infected mice.
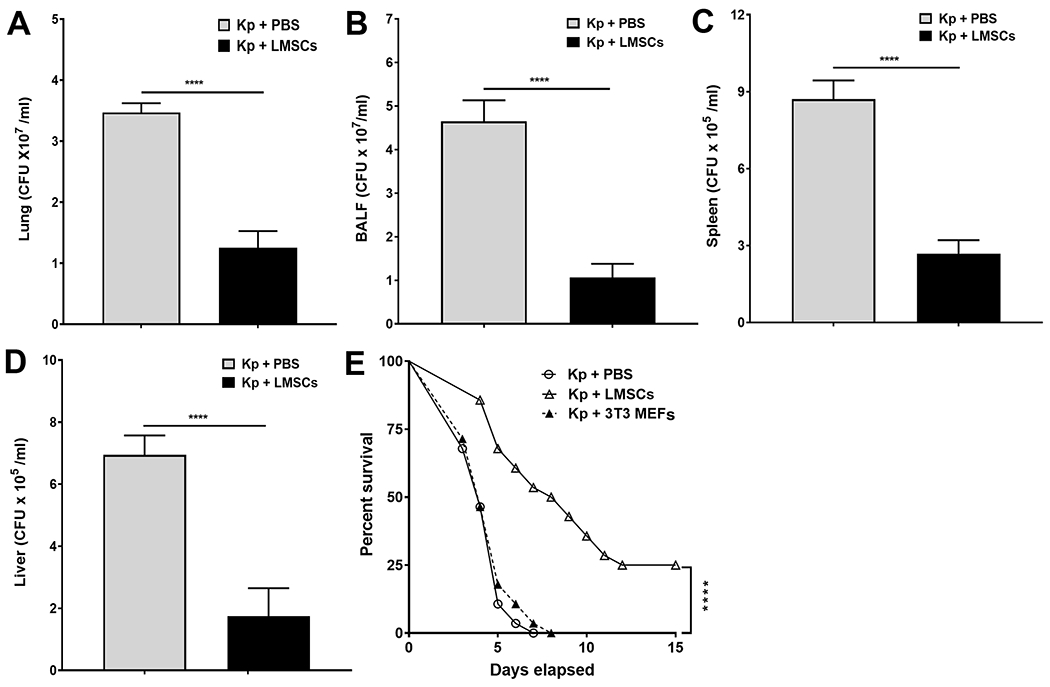
Intratracheal administration of Sca1+ LMSCs 4 h after K. pneumoniae-infection reduced K. pneumoniae CFU in mouse lung (A), BALF (B), spleen (C), and liver (D) by 48 h after infection. Data are mean ± SEM from three separate experiments (n = 5 - 6 mice/group). ****, p < 0.0001 by Mann-Whitney test. (E) Mice were treated with PBS, LMSCs, or 3T3 MEFs 4 h and 24 h after infection with K. pneumoniae. Survival was monitored every 12 h for up to 15 days. Data are the mean of two pooled experiments (n = 28 mice/group) and are shown as percent survival. Significance between groups was examined by Log-rank (Mantel-Cox) test and asterisk indicates the difference between PBS and LMSC-administered mice. ****, p < 0.0001.
We next explored if the enhanced bacterial clearance in LMSC-treated mice contributes to survival during bacterial pneumonia caused by K. pneumoniae. At an inoculum of 0.5 x 104 CFU, about 50% of PBS-treated mice as well as 3T3 MEF-treated mice (as control) succumbed to infection at 96 h (Fig. 6E). Analysis by log-rank (Mantel-Cox) test showed enhanced survival in Sca-1+ LMSC-treated mice compared to PBS and 3T3 MEF-treated mice (Fig. 6E). Furthermore, the median survival for Sca-1+ LMSC-treated mice was 204 h (8.5 days) compared to 96 h (4 days) for PBS- and 3T3 MEF-treated mice (Fig. 6E).
Discussion
Although the therapeutic potential of BM-MSCs is currently being examined in clinical trials for lung diseases (clinicaltrials.gov), understanding tissue-resident LMSCs in lung diseases remains elusive. While reports suggest that only a small subset of MSCs is responsible for their therapeutic properties, MSCs have conventionally been studied as a polyclonal product of cell culture. Interestingly, Corselli et al. (50) have suggested that enrichment or purification of well-characterized functional subsets could potentially enhance the efficiency of current clinical trials of MSCs. Therefore, in the present study, we used Sca-1 as a stem cell marker to isolate a subset of MSCs from a mixed population of mouse lung cells (LMSCs). These purified Sca-1+ cells appear to be a homogeneous population of LMSCs that express mesenchymal antigens and differentiate into osteocytes and adipocytes when subjected to specific differentiation conditions. Moreover, these LMSCs are highly replicative in vitro. More importantly, unlike bronchoalveolar stem cells (31, 32), these Sca-1+ LMSCs can be cultured and expanded on non-coated plastic dishes. Purified Sca-1+ LMSCs show similar morphological features and to a certain extent, surface marker profiles as MSCs isolated from bone marrow, spleen, brain, lungs, pancreas, kidneys, muscle, and thymus (8, 23).
There is limited knowledge concerning the morphology and ultrastructural features of LMSCs (26). The most striking features of Sca-1+ LMSCs are that they are rich in autophagosomes, endosomes, lysozomes, vesicular elements, and long mitochondria. A recent study (51) using TEM showed the presence of a large number of autophagosomes in the cytoplasmic compartment of undifferentiated human BM-MSCs. TEM analysis of Sca-1+ LMSCs revealed ultrastructural features common for all cells with high proteosynthetic levels. However, unlike Sca-1+ LMSCs (present study) and human BM-MSCs (51), the cytoplasm of adipose-derived mesenchymal stem cells (52) contains large numbers of small lipid droplets and electron-dense lamellar bodies.
MSCs also exhibit broad immunoregulatory properties and have the ability to influence both innate and adaptive immune responses. The mechanisms by which BM-MSCs and other tissue-derived MSCs exert their immunomodulatory effects are thought to be mediated primarily through the release of soluble factors and/or direct cell-cell contact (15, 53). Much of what is known about the immunoregulatory properties of MSCs has been learned through co-culture of MSCs with innate and adaptive immune cells. Recent reports have shown that MSCs interact with the innate immune system resulting in both anti-inflammatory and pro-inflammatory effects (54). Further, MSCs secrete numerous soluble factors that exert immunosuppressive effects by modulating both innate (macrophages, monocytes, neutrophils, dendritic cells, mast cells, myeloid derived suppressor cells, and natural killer cells) and adaptive (CD4+ T cells, FOXP3 T regulatory cells, CD8+ T cells, B cells, and Th17 cells) immune responses (55-57). However, when compared to MSCs from other tissue sources, very little is known about the immunomodulatory role of LMSCs (29, 30, 58-60). In the current study, we evaluated the immunoregulatory properties of Sca-1+ LMSCs by co-culturing them with BMDMs and BMDNs, two key innate immune cells that play an important role in host protection against bacterial infection. Our experimental results show that treatment with Sca-1+ LMSCs remarkably inhibited the LPS-induced secretion of TNFα and IL-1β from BMDMs and TNFα from BMDNs. Similarly, murine BM-MSCs as well as BM-MSC-derived supernatants inhibited the secretion of inflammatory cytokines TNFα, IL-12p70, IL-6, and interferon-γ by LPS-stimulated peritoneal macrophages (61). Importantly, unlike BM-MSCs (62), Sca-1+ LMSCs did not secrete the inflammatory cytokine TNFα, when left unstimulated or following stimulation with LPS. However, the mechanism(s) by which Sca-1+ LMSCs suppress inflammatory cytokine production by BMDMs and BMDNs remains to be elucidated.
Apart from immunomodulating properties, BM-MSCs are also endowed with the ability to directly affect the infectious agent. These antimicrobial effects are mediated through the secretion of antimicrobial compounds (LL-37, beta defensin-2, lipocalin-2) (49, 63, 64) or through the intensification of phagocytosis of monocytes and macrophages (65). In contrast, nothing is known about the antimicrobial properties of LMSCs. Bacterial killing by neutrophils (35), macrophages (44), mast cells (45), and gamma delta T cells (46) are major contributors to the clearance of K. pneumoniae. Therefore, we compared the bacterial killing ability of Sca-1+ LMSCs with that of BMDNs and BMDMs in vitro. Our results show that both Sca-1+ LMSCs and BMDNs exhibit similar extracellular killing effects when cultured with low MOI of K. pneumoniae. Moreover, when cultured with a higher MOI of bacteria, only LMSCs and LMSC-derived supernatants were able to kill K. pneumoniae at 1.5 and 3 h time points. However, at a later time point (6 h), LMSCs, LMSC-derived supernatant, as well as BMDNs failed to kill K. pneumoniae. Sca-1+ LMSCs secrete large amounts of antimicrobial compounds, such as CRAMP, LTB4, and lipocalin-2, even under resting conditions, an effect that was increased following infection with K. pneumoniae. Intriguingly, treatment with CRAMP peptide inhibited the extracellular growth of K. pneumoniae in vitro. LMSCs also secrete increased amounts of H2O2 following infection with K. pneumoniae, which might have partly contributed to the inhibition (66) of the extracellular growth of this Gram-negative bacterium in vitro.
Although multiple studies have shown the contribution of soluble factors secreted by MSCs to killing of bacterial pathogens, not much is known about the direct interaction and subsequent phagocytic uptake and intracellular killing of infectious agents by MSCs. Recently, two studies have demonstrated binding and phagocytic uptake of Mycobacterium tuberculosis (Mtb) by BM-MSCs (67, 68). Another study identified the direct interaction and internalization of both Gram-negative (Escherichia coli) and Gram-positive (Streptococcus pyogenes and Staphylococcus aureus) bacteria by adipose-derived MSCs (69). In our study, we compared the phagocytotic uptake and intracellular killing abilities of Sca-1+ LMSCs and BMDMs using opsonized, live K. pneumoniae in vitro and found that both Sca-1+ LMSCs and BMDMs were efficient in the phagocytotic uptake of K. pneumoniae. Interestingly, Sca-1+ LMSCs were more effective at controlling the intracellular replication of K. pneumoniae than were BMDMs at 4 h post-infection.
Complement plays an important role in the opsonization of circulating pathogens. CR3, CR4, and CRIg are involved in the phagocytosis of opsonized pathogens (47, 48). The B7 family-related protein, CRIg is selectively expressed on tissue-resident macrophages (liver Kupfer cells and peritoneal macrophages) and dendritic cells (70) and is a major receptor for phagocytosis of complement-opsonized bacteria in tissue-resident macrophages (48). Our data shows increased expression of CRIg in Sca-1+ LMSCs, suggesting the possible involvement of CRIg in the phagocytotic uptake of opsonized K. pneumoniae by Sca-1+ LMSCs.
Both phagocytosis and autophagy are cellular catabolic pathways that use lysosomes to digest particles (71). Recent studies reported interactions between phagosomes formed by phagocytosis and autophagosomes formed by autophagy (71-73). The interactions between these two forms of cellular eating processes represent a new dimension in host defense against both extracellular and intracellular bacterial pathogens. Our study also shows that activation of the autophagic pathway in LMSCs following phagocytotic uptake of the opsonized K. pneumoniae and pre-treatment with the autophagy inhibitor, 3-MA, results in an enhanced ability of cells to control K. pneumoniae intracellular replication 1 h post-infection, as evidenced by lower numbers of visible K. pneumoniae in LMSCs. Conversely, pretreatment with the autophagy inducer rapamycin, impairs the ability of the cell to control K. pneumoniae replication early after (1 h) infection, as evidenced by greater numbers of viable K. pneumoniae in LMSCs. In addition, the autophagy-associated proteins Beclin-1, ATG7, and LC3 are highly expressed in LMSCs and infection with K. pneumoniae further increases the levels of Beclin-1, ATG7, and LC3 at 1 h post-infection. Further, pre-treatment with 3-MA decreases the levels of these autophagy associated-proteins in LMSCs at 1 h post-infection, suggesting that autophagy is activated during the phagocytotic uptake of K. pneumoniae by LMSCs. Most bacterial pathogens are rapidly degraded in the phagolysosome and ROS are critical weapons in the phagocyte arsenal (74). Interestingly, phagocytotic uptake and intracellular killing of K. pneumoniae is clearly associated with increased production of intracellular ROS in LMSCs. Our data also shows that LMSCs are more effective than BMDMs at clearing phagocytosed K. pneumoniae as almost all bacteria were gone by 4 h post-infection.
Next, we further confirmed the ability of Sca-1+ LMSCs to phagocytize live K. pneumoniae in vitro using immunofluorescence microscopy and confocal microscopy. In addition, flow cytometric analysis also revealed the phagocytic uptake of HI Kp-FITC by both Sca-1+ LMSCs and BMDMs. Thus, the phagocytic activity of Sca-1+ LMSCs and BMDMs is likely to contribute to bacterial clearance/elimination during acute K. pneumoniae infection. Interestingly, human BM-MSCs also exhibit marked autophagy, and siRNA knockdown of autophagy inhibitor beclin-1 and inhibition of NO has been found to enhance the intracellular survival of Mtb (67).
Although the existence of region-specific stem or progenitor cells in the murine respiratory tract has been demonstrated (75-78), the identification and localization of LMSCs in different regions of the murine and human lungs has been challenging due to the lack of specific markers. Accordingly, the role of endogenous lung resident MSCs in normal tissue homeostasis and disease settings is still unclear. A recent study (79) demonstrated the presence of abundant numbers of LMSCs in the lung parenchymal tissues of humans. However, sensitization and challenges with chicken ovalbumin (80) increased the numbers of LMSCs in the lungs of mice, suggesting that the allergens may act through lung resident MSCs to promote processes that are involved in the pathogenesis of asthma. Few studies have quantified the efficiency of LMSCs transplantation. Badri et al. (81) demonstrated the in vivo retention and interaction of the PKH-26 or DSRed lentivirus-labeled human LMSCs with alveolar epithelial cells in murine lungs. These human LMSCs were found to persist in murine lungs for up to six months. In another elegant study, Jun et al. (58) have identified ATP-binding cassette super family G member 2 as a LMSC marker to localize and study the immunomodulatory and protective effects of these cells in vivo in both murine and human tissues. Bleomycin treatment induced the loss of these endogenous LMSCs and elicited inflammation, fibrosis and pulmonary arterial hypertension. Rescuing the deficient endogenous LMSC population via intravenous administration of LMSCs attenuated lung injury in mice (58). Aother study (82) using FISH analysis revealed that the primary MSCs in human transplanted lungs are perivascularly located CD90/CD105+ tissue-resident cells. Interestingly, all MSCs samples isolated from biopsies taken as long as sixteen years post-transplantation showed donor sex karyotype (82). This finding contributes further evidence that these transplanted LMSCs are long-lived and/or self-renewing populations of lung stroma. However, it is yet to be shown clinically that improved MSCs persistence leads to enhanced efficacy of MSCs-based therapies.
The observed therapeutic effect of BM-MSCs is mediated by the production of immunomodulatory factors during the initial days following BM-MSCs transplantation (83). A number of growth factors secreted by BM-MSCs and other tissue-derived MSCs are critical for neoangiogenesis (84), and vascular endothelial growth factor has been shown to be a key mobilizer of cardiac stem cells in the infarcted heart (85). BM-MSC administration stimulates the proliferation of cardiac stem cells in the infarcted heart of pigs (86) and increased the proliferation potential of bronchoalveolar stem cells in a mouse model of bronchopulmonary dysplasia (87). However, no prior studies have closely examined LMSCs effect on lung resident stem or progenitor cells in vivo. In our study, we observed the retention of increased numbers of CFSE-labeled Sca-1+ LMSCs in the lung parenchyma of K. pneumoniae-infected mice at 4 h post- administration when compared to the lungs of K. pneumoniae-infected mice that were sacrificed at 44 h post-LMSCs transplantation.
Despite that the role of BM-MSCs and other tissue-derived MSCs in inflammatory and infectious diseases is fairly well established (11, 12, 14), the therapeutic potential of lung-derived stem/mesenchymal stem cells in bacterial infection and endogenous immune responses is unclear. Nonetheless, most studies have used the Gram-negative bacterium, Escherichia coli to infect mice or rats, reporting reduced lung inflammation, pulmonary edema, alveolar epithelial permeability, and enhanced bacterial clearance after the intratracheal (i.t.) or intravenous (i.v.) administration of MSCs (49, 65, 88, 89). E. coli infection in animals is usually associated with acute onset of severe lung inflammation and clearance of bacteria and related more to acute lung injury than pneumonia. However, K. pneumoniae infection in animals induces a gradually evolving inflammatory response with a steadily increasing bacterial load in the lungs and extrapulmonary organs (34, 90, 91), which largely resembles the clinical scenario of bacterial pneumonia. Intravenous infusion has been used as a preferred route of cell delivery for a large number of preclinical studies and is the favored route of administration in human clinical trials as well (11, 53, 92, 93). A major pitfall of i.v. administered MSCs is their inability to migrate across pulmonary capillaries to the lung stroma (94, 95) and these i.v. injected MSCs have short half-lives (94). In our study, we administered a single dose of Sca-1+ LMSCs to mice by intratracheal (i.t.) route for acute bacterial pneumoniae and two doses of Sca-1+ LMSCs by o.p. for the survival studies.
Our study using purified Sca-1+ LMSCs showed multiple beneficial effects of these cells in pneumonia caused by the Gram-negative bacterium, K. pneumoniae. Treatment with Sca-1+ LMSCs reduced the levels of inflammatory cells, predominantly neutrophils and macrophages, in the BALF of K. pneumoniae-infected mice. Interestingly, this effect was clearly associated with reduced monocyte/macrophage/neutrophil attracting chemokines such as MIP-1α, MIP-2, KC, MCP-1, and the inflammatory cytokines TNFα, IL-1β, IL-1α, and IL-6 in the lungs of K. pneumoniae-infected and LMSC-treated mice. Our results are similar to the results obtained using BM-MSCs, which attenuated the level of neutrophils in the BALF of mice infected with E.coli (65), K. pneumoniae (96), and Streptococcus pneumoniae (97). Similar to what was seen with BM-MSCs (96), treatment with LMSCs did not alter the level of the anti-inflammatory cytokine IL-10 in the BALF of K. pneumoniae-infected mice. Interestingly, in the in vivo models of bacterial pneumonia and acute lung injury/acute respiratory distress syndrome induced by live bacteria, BM-MSCs demonstrated the ability not only to attenuate inflammation, but also to improve bacterial clearance (49, 65, 97). The study by Hackstein et al. (96) showed the protective effect of purified murine BM-MSCs in a mouse model of pneumonia caused by hypervirulent K. pneumoniae, which was associated with reduced pulmonary inflammation and improved survival in mice; however, treatment with BM-MSCs did not reduce bacterial growth in the lungs or extrapulmonary organs of the mice. In this study, a single dose of purified PαS MSC was intratracheally administered into K. pneumoniae-infected C57BL/6 mice and the survival of the mice was monitored up to 5 days. Another recent study by Perlee et al. (91) showed profound immunomodulatory effects of intravenously administered human adipose-derived MSCs in the lungs, resulting in reduced lung inflammation and bacterial burden in the lungs and distal organs, but did not influence distal organ injury during pneumosepsis caused by K. pneumoniae. However, this study did not investigate whether i.v. administration of adipose-derived MSCs can protect the mice from death caused by K. pneumoniae-induced pneumosepsis in mice. In our study, administration of Sca-1+ LMSCs was found to not only attenuate pulmonary inflammation, but also promote marked bacterial clearance in the lungs and in extrapulmonary organs, as well as rescue mice from death caused by K. pneumoniae. As anticipated, administration of 3T3 MEF neither decreased the levels of inflammatory cells in the BAL fluid nor attenuated the bacterial growth in the lungs and extrapulmonary organs of K. pneumoniae infected mice. In our study, we monitored the survival of the mice for up to 15 days. Similar to the protective effects obtained using BM-MSCs (96), i.t. delivery of Sca-1+ LMSCs rescued 85% of the mice from death caused by K. pneumoniae at 4 days post-infection whereas 25% of the LMSCs treated mice survived at 15 days post-infection. These different levels of MSCs-mediated protection observed during K. pneumoniae infection in our study and other studies (91, 96) are likely due to the source of MSCs used; dose, route, time, and the frequency of administration of MSCs, and the amount of the inoculum (CFU) of K. pneumoniae used to infect the animals; as well as the days taken to monitor the survival of the mice. Future studies are warranted to investigate whether the immunomodulatory, antibacterial, and reparative/regenerative potential of Sca-1+ LMSCs can be enhanced by altering the dose, route, time, frequency of delivery, as well as by preconditioning of the Sca-1+ LMSCs with bacterial LPS, inflammatory cytokines, or hypoxia (98). Also, additional studies are required to determine whether Sca-1+ LMSCs can induce host protection 1) in the setting of influenza virus-associated bacterial pneumonia (99), as well as 2) in pneumonia caused by the multidrug resistant K. pneumoniae (100) and Gram-positive bacterial species including S. pneumoniae.
Supplementary Material
Key points.
A homogeneous population of LMSCs is purified from mouse lungs using Sca-1.
LMSCs modulate neutrophil and macrophage functions, and shows bacterial killing.
LMSCs improve survival, minimizes inflammation, and enhances bacterial clearance
Acknowledgments.
The authors thank Marilyn Dietrich at Louisiana State University (LSU) School of Veterinary Medicine (SVM) for helping with flow cytometry and Yuliya Sokolova and Xiaochu Wu at LSU Shared Instrumentation Facility for their assistance with Light and Electron Microscopic studies. We thank Mariano Carossino at LSU-SVM for helping with NanoZoomer-HT.
Footnote.
This work was supported by NIH grants (R01 HL-091958, R01 AI-113720, R01 AI-140500, R21 AI-133681, P20 GM13055) to SJ, R01 HL133336 to SC, NIH Pilot Grants (P30 GM110760 and P20 GM13055) to TR, and by AHA 18POST34080070 Grant to LJ.
Abbreviations used in this article.
- Sca-1+ LMSCs
stem cell antigen-1+ lung mesenchymal stem cells
- BM-MSCs
bone marrow-derived mesenchymal stem cells
- K. pneumoniae
Klebsiella pneumoniae
- BALF
bronchoalveolar lavage fluid
- CFU
colony forming unit
- MOI
multiplicity of infection
- CRAMP
cathelicidin-related antimicrobial peptide
- BMDMs
bone marrow-derived macrophages
- BMDNs
bone marrow-derived neutrophils
- CFSE
carboxyfluorescein succinimidyl ester
- CMEM
complete alpha minimum essential medium
- LPS
lipopolysaccharide
- 3T3 MEFs
3T3 mouse embryonic fibroblasts
- SEM
scanning electron microscope
- TEM
transmission electron microscope
- LTB4
leukotriene B4
- 3-MA
3-Methyladenine
- H2O2
hydrogen peroxide
- FITC
fluorescein-5-isothiocyanate
- CRIg
complement receptor of the immunoglobulin superfamily
- ROS
reactive oxygen species
- TWBC
total white blood cells
Footnotes
Disclosures
The authors have no conflict of interest.
References
- 1.Craig A, Mai J, Cai S, and Jeyaseelan S. 2009. Neutrophil recruitment to the lungs during bacterial pneumonia. Infect Immun 77: 568–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Endimiani A, Depasquale JM, Forero S, Perez F, Hujer AM, Roberts-Pollack D, Fiorella PD, Pickens N, Kitchel B, Casiano-Colon AE, Tenover FC, and Bonomo RA. 2009. Emergence of blaKPC-containing Klebsiella pneumoniae in a long-term acute care hospital: a new challenge to our healthcare system. J Antimicrob Chemother 64: 1102–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schwaber MJ, Lev B, Israeli A, Solter E, Smollan G, Rubinovitch B, Shalit I, Carmeli Y, and Israel G Carbapenem-Resistant Enterobacteriaceae Working. 2011. Containment of a country-wide outbreak of carbapenem-resistant Klebsiella pneumoniae in Israeli hospitals via a nationally implemented intervention. Clin Infect Dis 52: 848–855. [DOI] [PubMed] [Google Scholar]
- 4.Jong GM, Hsiue TR, Chen CR, Chang HY, and Chen CW. 1995. Rapidly fatal outcome of bacteremic Klebsiella pneumoniae pneumonia in alcoholics. Chest 107: 214–217. [DOI] [PubMed] [Google Scholar]
- 5.Saibal MA, Rahman SH, Nishat L, Sikder NH, Begum SA, Islam MJ, and Uddin KN. 2012. Community acquired pneumonia in diabetic and non-diabetic hospitalized patients: presentation, causative pathogens and outcome. Bangladesh Med Res Counc Bull 38: 98–103. [DOI] [PubMed] [Google Scholar]
- 6.Balamayooran G, Batra S, Fessler MB, Happel KI, and Jeyaseelan S. 2010. Mechanisms of neutrophil accumulation in the lungs against bacteria. Am J Respir Cell Mol Biol 43: 5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Friedenstein AJ, Deriglasova UF, Kulagina NN, Panasuk AF, Rudakowa SF, Luria EA, and Ruadkow IA. 1974. Precursors for fibroblasts in different populations of hematopoietic cells as detected by the in vitro colony assay method. Exp Hematol 2: 83–92. [PubMed] [Google Scholar]
- 8.da Silva Meirelles L, Chagastelles PC, and Nardi NB. 2006. Mesenchymal stem cells reside in virtually all post-natal organs and tissues. J Cell Sci 119: 2204–2213. [DOI] [PubMed] [Google Scholar]
- 9.Lee JW, Fang X, Gupta N, Serikov V, and Matthay MA. 2009. Allogeneic human mesenchymal stem cells for treatment of E. coli endotoxin-induced acute lung injury in the ex vivo perfused human lung. Proc Natl Acad Sci U S A 106: 16357–16362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bernardo ME, and Fibbe WE. 2013. Mesenchymal stromal cells: sensors and switchers of inflammation. Cell Stem Cell 13: 392–402. [DOI] [PubMed] [Google Scholar]
- 11.Lee JW, Fang X, Krasnodembskaya A, Howard JP, and Matthay MA. 2011. Concise review: Mesenchymal stem cells for acute lung injury: role of paracrine soluble factors. Stem Cells 29: 913–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weiss DJ 2013. Stem cells, cell therapies, and bioengineering in lung biology and diseases. Comprehensive review of the recent literature 2010-2012. Ann Am Thorac Soc 10: S45–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matthay MA, Anversa P, Bhattacharya J, Burnett BK, Chapman HA, Hare JM, Hei DJ, Hoffman AM, Kourembanas S, McKenna DH, Ortiz LA, Ott HC, Tente W, Thebaud B, Trapnell BC, Weiss DJ, Yuan JX, and Blaisdell CJ. 2013. Cell therapy for lung diseases. Report from an NIH-NHLBI workshop, November13-14, 2012. Am J Respir Crit Care Med 188: 370–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matthay MA, Pati S, and Lee JW. 2017. Concise Review: Mesenchymal Stem (Stromal) Cells: Biology and Preclinical Evidence for Therapeutic Potential for Organ Dysfunction Following Trauma or Sepsis. Stem Cells 35: 316–324. [DOI] [PubMed] [Google Scholar]
- 15.Lau AN, Goodwin M, Kim CF, and Weiss DJ. 2012. Stem cells and regenerative medicine in lung biology and diseases. Mol Ther 20: 1116–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matthay MA, Thompson BT, Read EJ, McKenna DH Jr., Liu KD, Calfee CS, and Lee JW. 2010. Therapeutic potential of mesenchymal stem cells for severe acute lung injury. Chest 138: 965–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mei SH, Haitsma JJ, Dos Santos CC, Deng Y, Lai PF, Slutsky AS, Liles WC, and Stewart DJ. 2010. Mesenchymal stem cells reduce inflammation while enhancing bacterial clearance and improving survival in sepsis. Am J Respir Crit Care Med 182: 1047–1057. [DOI] [PubMed] [Google Scholar]
- 18.Gupta N, Sinha R, Krasnodembskaya A, Xu X, Nizet V, Matthay MA, and Griffin JH. 2018. The TLR4-PAR1 Axis Regulates Bone Marrow Mesenchymal Stromal Cell Survival and Therapeutic Capacity in Experimental Bacterial Pneumonia. Stem Cells 36: 796–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sabatini F, Petecchia L, Tavian M, Jodon de Villeroche V, Rossi GA, and Brouty-Boye D. 2005. Human bronchial fibroblasts exhibit a mesenchymal stem cell phenotype and multilineage differentiating potentialities. Lab Invest 85: 962–971. [DOI] [PubMed] [Google Scholar]
- 20.Martin J, Helm K, Ruegg P, Varella-Garcia M, Burnham E, and Majka S. 2008. Adult lung side population cells have mesenchymal stem cell potential. Cytotherapy 10: 140–151. [DOI] [PubMed] [Google Scholar]
- 21.McQualter JL, Brouard N, Williams B, Baird BN, Sims-Lucas S, Yuen K, Nilsson SK, Simmons PJ, and Bertoncello I. 2009. Endogenous fibroblastic progenitor cells in the adult mouse lung are highly enriched in the sca-1 positive cell fraction. Stem Cells 27: 623–633. [DOI] [PubMed] [Google Scholar]
- 22.Hua J, Yu H, Dong W, Yang C, Gao Z, Lei A, Sun Y, Pan S, Wu Y, and Dou Z. 2009. Characterization of mesenchymal stem cells (MSCs) from human fetal lung: potential differentiation of germ cells. Tissue Cell 41: 448–455. [DOI] [PubMed] [Google Scholar]
- 23.Hoffman AM, Paxson JA, Mazan MR, Davis AM, Tyagi S, Murthy S, and Ingenito EP. 2011. Lung-derived mesenchymal stromal cell post-transplantation survival, persistence, paracrine expression, and repair of elastase-injured lung. Stem Cells Dev 20: 1779–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ingenito EP, Tsai L, Murthy S, Tyagi S, Mazan M, and Hoffman A. 2012. Autologous lung-derived mesenchymal stem cell transplantation in experimental emphysema. Cell Transplant 21: 175–189. [DOI] [PubMed] [Google Scholar]
- 25.Hu P, Pu Y, Li X, Zhu Z, Zhao Y, Guan W, and Ma Y. 2015. Isolation, in vitro culture and identification of a new type of mesenchymal stem cell derived from fetal bovine lung tissues. Mol Med Rep 12: 3331–3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rolandsson Enes S, Andersson Sjoland A, Skog I, Hansson L, Larsson H, Le Blanc K, Eriksson L, Bjermer L, Scheding S, and Westergren-Thorsson G. 2016. MSC from fetal and adult lungs possess lung-specific properties compared to bone marrow-derived MSC. Sci Rep 6: 29160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zulueta A, Colombo M, Peli V, Falleni M, Tosi D, Ricciardi M, Baisi A, Bulfamante G, Chiaramonte R, and Caretti A. 2018. Lung mesenchymal stem cells-derived extracellular vesicles attenuate the inflammatory profile of Cystic Fibrosis epithelial cells. Cell Signal 51: 110–118. [DOI] [PubMed] [Google Scholar]
- 28.Cappetta D, De Angelis A, Spaziano G, Tartaglione G, Piegari E, Esposito G, Ciuffreda LP, Liparulo A, Sgambato M, Russo TP, Rossi F, Berrino L, Urbanek K, and D’Agostino B. 2018. Lung Mesenchymal Stem Cells Ameliorate Elastase-Induced Damage in an Animal Model of Emphysema. Stem Cells Int 2018: 9492038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abreu SC, Antunes MA, Xisto DG, Cruz FF, Branco VC, Bandeira E, Zola Kitoko J, de Araujo AF, Dellatorre-Texeira L, Olsen PC, Weiss DJ, Diaz BL, Morales MM, and Rocco PRM. 2017. Bone Marrow, Adipose, and Lung Tissue-Derived Murine Mesenchymal Stromal Cells Release Different Mediators and Differentially Affect Airway and Lung Parenchyma in Experimental Asthma. Stem Cells Transl Med 6: 1557–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Silva JD, Lopes-Pacheco M, Paz AHR, Cruz FF, Melo EB, de Oliveira MV, Xisto DG, Capelozzi VL, Morales MM, Pelosi P, Cirne-Lima E, and Rocco PRM. 2018. Mesenchymal Stem Cells From Bone Marrow, Adipose Tissue, and Lung Tissue Differentially Mitigate Lung and Distal Organ Damage in Experimental Acute Respiratory Distress Syndrome. Crit Care Med 46: e132–e140. [DOI] [PubMed] [Google Scholar]
- 31.Kim CF, Jackson EL, Woolfenden AE, Lawrence S, Babar I, Vogel S, Crowley D, Bronson RT, and Jacks T. 2005. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell 121: 823–835. [DOI] [PubMed] [Google Scholar]
- 32.Qian S, Ding JY, Xie R, An JH, Ao XJ, Zhao ZG, Sun JG, Duan YZ, Chen ZT, and Zhu B. 2008. MicroRNA expression profile of bronchioalveolar stem cells from mouse lung. Biochem Biophys Res Commun 377: 668–673. [DOI] [PubMed] [Google Scholar]
- 33.Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S, and Marshak DR. 1999. Multilineage potential of adult human mesenchymal stem cells. Science 284: 143–147. [DOI] [PubMed] [Google Scholar]
- 34.Cai S, Batra S, Del Piero F, and Jeyaseelan S. 2016. NLRP12 modulates host defense through IL-17A-CXCL1 axis. Mucosal Immunol 9: 503–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sharma A, Steichen AL, Jondle CN, Mishra BB, and Sharma J. 2014. Protective role of Mincle in bacterial pneumonia by regulation of neutrophil mediated phagocytosis and extracellular trap formation. J Infect Dis 209: 1837–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Williams MA, Rangasamy T, Bauer SM, Killedar S, Karp M, Kensler TW, Yamamoto M, Breysse P, Biswal S, and Georas SN. 2008. Disruption of the transcription factor Nrf2 promotes pro-oxidative dendritic cells that stimulate Th2-like immunoresponsiveness upon activation by ambient particulate matter. J Immunol 181: 4545–4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peister A, Mellad JA, Larson BL, Hall BM, Gibson LF, and Prockop DJ. 2004. Adult stem cells from bone marrow (MSCs) isolated from different strains of inbred mice vary in surface epitopes, rates of proliferation, and differentiation potential. Blood 103: 1662–1668. [DOI] [PubMed] [Google Scholar]
- 38.Kovach MA, Ballinger MN, Newstead MW, Zeng X, Bhan U, Yu FS, Moore BB, Gallo RL, and Standiford TJ. 2012. Cathelicidin-related antimicrobial peptide is required for effective lung mucosal immunity in Gram-negative bacterial pneumonia. J Immunol 189: 304–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davis KM, Akinbi HT, Standish AJ, and Weiser JN. 2008. Resistance to mucosal lysozyme compensates for the fitness deficit of peptidoglycan modifications by Streptococcus pneumoniae. PLoS Pathog 4: e1000241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang L, Shen D, Wu H, and Ma Y. 2017. Resistance of hypervirulent Klebsiella pneumoniae to both intracellular and extracellular killing of neutrophils. PLoS One 12: e0173638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schnaith A, Kashkar H, Leggio SA, Addicks K, Kronke M, and Krut O. 2007. Staphylococcus aureus subvert autophagy for induction of caspase-independent host cell death. J Biol Chem 282: 2695–2706. [DOI] [PubMed] [Google Scholar]
- 42.Jeyaseelan S, Chu HW, Young SK, and Worthen GS. 2004. Transcriptional profiling of lipopolysaccharide-induced acute lung injury. Infect Immun 72: 7247–7256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rangasamy T, Cho CY, Thimmulappa RK, Zhen L, Srisuma SS, Kensler TW, Yamamoto M, Petrache I, Tuder RM, and Biswal S. 2004. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J Clin Invest 114: 1248–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Broug-Holub E, Toews GB, van Iwaarden JF, Strieter RM, Kunkel SL, Paine R 3rd, and Standiford TJ. 1997. Alveolar macrophages are required for protective pulmonary defenses in murine Klebsiella pneumonia: elimination of alveolar macrophages increases neutrophil recruitment but decreases bacterial clearance and survival. Infect Immun 65: 1139–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sutherland RE, Olsen JS, McKinstry A, Villalta SA, and Wolters PJ. 2008. Mast cell IL-6 improves survival from Klebsiella pneumonia and sepsis by enhancing neutrophil killing. J Immunol 181: 5598–5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moore TA, Moore BB, Newstead MW, and Standiford TJ. 2000. Gamma delta-T cells are critical for survival and early proinflammatory cytokine gene expression during murine Klebsiella pneumonia. J Immunol 165: 2643–2650. [DOI] [PubMed] [Google Scholar]
- 47.Ernst JD 1998. Macrophage receptors for Mycobacterium tuberculosis. Infect Immun 66: 1277–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van Lookeren Campagne M, and Verschoor A. 2018. Pathogen clearance and immune adherence “revisited” Immuno-regulatory roles for CRIg. Semin Immunol 37: 4–11. [DOI] [PubMed] [Google Scholar]
- 49.Gupta N, Krasnodembskaya A, Kapetanaki M, Mouded M, Tan X, Serikov V, and Matthay MA. 2012. Mesenchymal stem cells enhance survival and bacterial clearance in murine Escherichia coli pneumonia. Thorax 67: 533–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Corselli M, Crisan M, Murray IR, West CC, Scholes J, Codrea F, Khan N, and Peault B. 2013. Identification of perivascular mesenchymal stromal/stem cells by flow cytometry. Cytometry A 83: 714–720. [DOI] [PubMed] [Google Scholar]
- 51.Nuschke A, Rodrigues M, Stolz DB, Chu CT, Griffith L, and Wells A. 2014. Human mesenchymal stem cells/multipotent stromal cells consume accumulated autophagosomes early in differentiation. Stem Cell Res Ther 5: 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Varga I, Miko M, Oravcova L, Backayova T, Koller J, and Danisovic L. 2015. Ultra-structural morphology of long-term cultivated white adipose tissue-derived stem cells. Cell Tissue Bank 16: 639–647. [DOI] [PubMed] [Google Scholar]
- 53.Zhu YG, Hao Q, Monsel A, Feng XM, and Lee JW. 2013. Adult stem cells for acute lung injury: remaining questions and concerns. Respirology 18: 744–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Keating A 2012. Mesenchymal stromal cells: new directions. Cell Stem Cell 10: 709–716. [DOI] [PubMed] [Google Scholar]
- 55.Duffy MM, Ritter T, Ceredig R, and Griffin MD. 2011. Mesenchymal stem cell effects on T-cell effector pathways. Stem Cell Res Ther 2: 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Le Blanc K, and Davies LC. 2015. Mesenchymal stromal cells and the innate immune response. Immunol Lett 168: 140–146. [DOI] [PubMed] [Google Scholar]
- 57.Fan L, Hu C, Chen J, Cen P, Wang J, and Li L. 2016. Interaction between Mesenchymal Stem Cells and B-Cells. Int J Mol Sci 17. 650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jun D, Garat C, West J, Thorn N, Chow K, Cleaver T, Sullivan T, Torchia EC, Childs C, Shade T, Tadjali M, Lara A, Nozik-Grayck E, Malkoski S, Sorrentino B, Meyrick B, Klemm D, Rojas M, Wagner DH Jr., and Majka SM. 2011. The pathology of bleomycin-induced fibrosis is associated with loss of resident lung mesenchymal stem cells that regulate effector T-cell proliferation. Stem Cells 29: 725–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jarvinen L, Badri L, Wettlaufer S, Ohtsuka T, Standiford TJ, Toews GB, Pinsky DJ, Peters-Golden M, and Lama VN. 2008. Lung resident mesenchymal stem cells isolated from human lung allografts inhibit T cell proliferation via a soluble mediator. J Immunol 181: 4389–4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Khatri M, O’Brien TD, Goyal SM, and Sharma JM. 2010. Isolation and characterization of chicken lung mesenchymal stromal cells and their susceptibility to avian influenza virus. Dev Comp Immunol 34: 474–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Maggini J, Mirkin G, Bognanni I, Holmberg J, Piazzon IM, Nepomnaschy I, Costa H, Canones C, Raiden S, Vermeulen M, and Geffner JR. 2010. Mouse bone marrow-derived mesenchymal stromal cells turn activated macrophages into a regulatory-like profile. PLoS One 5: e9252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kyurkchiev D, Bochev I, Ivanova-Todorova E, Mourdjeva M, Oreshkova T, Belemezova K, and Kyurkchiev S. 2014. Secretion of immunoregulatory cytokines by mesenchymal stem cells. World J Stem Cells 6: 552–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Krasnodembskaya A, Song Y, Fang X, Gupta N, Serikov V, Lee JW, and Matthay MA. 2010. Antibacterial effect of human mesenchymal stem cells is mediated in part from secretion of the antimicrobial peptide LL-37. Stem Cells 28: 2229–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sung DK, Chang YS, Sung SI, Yoo HS, Ahn SY, and Park WS. 2016. Antibacterial effect of mesenchymal stem cells against Escherichia coli is mediated by secretion of beta- defensin- 2 via toll- like receptor 4 signalling. Cell Microbiol 18: 424–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Devaney J, Horie S, Masterson C, Elliman S, Barry F, O’Brien T, Curley GF, O’Toole D, and Laffey JG. 2015. Human mesenchymal stromal cells decrease the severity of acute lung injury induced by E. coli in the rat. Thorax 70: 625–635. [DOI] [PubMed] [Google Scholar]
- 66.Imlay JA 2003. Pathways of oxidative damage. Annu Rev Microbiol 57: 395–418. [DOI] [PubMed] [Google Scholar]
- 67.Khan A, Mann L, Papanna R, Lyu MA, Singh CR, Olson S, Eissa NT, Cirillo J, Das G, Hunter RL, and Jagannath C. 2017. Mesenchymal stem cells internalize Mycobacterium tuberculosis through scavenger receptors and restrict bacterial growth through autophagy. Sci Rep 7: 15010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Naik SK, Padhi A, Ganguli G, Sengupta S, Pati S, Das D, and Sonawane A. 2017. Mouse Bone Marrow Sca-1(+) CD44(+) Mesenchymal Stem Cells Kill Avirulent Mycobacteria but Not Mycobacterium tuberculosis through Modulation of Cathelicidin Expression via the p38 Mitogen-Activated Protein Kinase-Dependent Pathway. Infect Immun 85: e00471–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fiedler T, Salamon A, Adam S, Herzmann N, Taubenheim J, and Peters K. 2013. Impact of bacteria and bacterial components on osteogenic and adipogenic differentiation of adipose-derived mesenchymal stem cells. Exp Cell Res 319: 2883–2892. [DOI] [PubMed] [Google Scholar]
- 70.Small AG, Al-Baghdadi M, Quach A, Hii C, and Ferrante A. 2016. Complement receptor immunoglobulin: a control point in infection and immunity, inflammation and cancer. Swiss Med Wkly 146: w14301. [DOI] [PubMed] [Google Scholar]
- 71.Sanjuan MA, and Green DR. 2008. Eating for good health: linking autophagy and phagocytosis in host defense. Autophagy 4: 607–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, and Deretic V. 2004. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 119: 753–766. [DOI] [PubMed] [Google Scholar]
- 73.Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, and Eissa NT. 2007. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 27: 135–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Craig M, and Slauch JM. 2009. Phagocytic superoxide specifically damages an extracytoplasmic target to inhibit or kill Salmonella. PLoS One 4: e4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Leeman KT, Fillmore CM, and Kim CF. 2014. Lung stem and progenitor cells in tissue homeostasis and disease. Curr Top Dev Biol 107: 207–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Konda B, Mulay A, Yao C, Beil S, Israely E, and Stripp BR. 2020. Isolation and Enrichment of Human Lung Epithelial Progenitor Cells for Organoid Culture. J Vis Exp. 161. [DOI] [PubMed] [Google Scholar]
- 77.Weiss DJ 2014. Concise review: current status of stem cells and regenerative medicine in lung biology and diseases. Stem Cells 32: 16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hoffman AM, and Ingenito EP. 2012. Alveolar epithelial stem and progenitor cells: emerging evidence for their role in lung regeneration. Curr Med Chem 19: 6003–6008. [DOI] [PubMed] [Google Scholar]
- 79.Sinclair KA, Yerkovich ST, Chen T, McQualter JL, Hopkins PM, Wells CA, and Chambers DC. 2016. Mesenchymal Stromal Cells are Readily Recoverable from Lung Tissue, but not the Alveolar Space, in Healthy Humans. Stem Cells 34: 2548–2558. [DOI] [PubMed] [Google Scholar]
- 80.Bentley JK, Popova AP, Bozyk PD, Linn MJ, Baek AE, Lei J, Goldsmith AM, and Hershenson MB. 2010. Ovalbumin sensitization and challenge increases the number of lung cells possessing a mesenchymal stromal cell phenotype. Respir Res 11: 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Badri L, Walker NM, Ohtsuka T, Wang Z, Delmar M, Flint A, Peters-Golden M, Toews GB, Pinsky DJ, Krebsbach PH, and Lama VN. 2011. Epithelial interactions and local engraftment of lung-resident mesenchymal stem cells. Am J Respir Cell Mol Biol 45: 809–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rolandsson S, Andersson Sjoland A, Brune JC, Li H, Kassem M, Mertens F, Westergren A, Eriksson L, Hansson L, Skog I, Bjermer L, Scheding S, and Westergren-Thorsson G. 2014. Primary mesenchymal stem cells in human transplanted lungs are CD90/CD105 perivascularly located tissue-resident cells. BMJ Open Respir Res 1: e000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Prockop DJ 2013. Concise review: two negative feedback loops place mesenchymal stem/stromal cells at the center of early regulators of inflammation. Stem Cells 31: 2042–2046. [DOI] [PubMed] [Google Scholar]
- 84.Wannemuehler TJ, Manukyan MC, Brewster BD, Rouch J, Poynter JA, Wang Y, and Meldrum DR. 2012. Advances in mesenchymal stem cell research in sepsis. J Surg Res 173: 113–126. [DOI] [PubMed] [Google Scholar]
- 85.Tang J, Wang J, Kong X, Yang J, Guo L, Zheng F, Zhang L, Huang Y, and Wan Y. 2009. Vascular endothelial growth factor promotes cardiac stem cell migration via the PI3K/Akt pathway. Exp Cell Res 315: 3521–3531. [DOI] [PubMed] [Google Scholar]
- 86.Hatzistergos KE, Quevedo H, Oskouei BN, Hu Q, Feigenbaum GS, Margitich IS, Mazhari R, Boyle AJ, Zambrano JP, Rodriguez JE, Dulce R, Pattany PM, Valdes D, Revilla C, Heldman AW, McNiece I, and Hare JM. 2010. Bone marrow mesenchymal stem cells stimulate cardiac stem cell proliferation and differentiation. Circ Res 107: 913–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tropea KA, Leder E, Aslam M, Lau AN, Raiser DM, Lee JH, Balasubramaniam V, Fredenburgh LE, Alex Mitsialis S, Kourembanas S, and Kim CF. 2012. Bronchioalveolar stem cells increase after mesenchymal stromal cell treatment in a mouse model of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 302: L829–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kim ES, Chang YS, Choi SJ, Kim JK, Yoo HS, Ahn SY, Sung DK, Kim SY, Park YR, and Park WS. 2011. Intratracheal transplantation of human umbilical cord blood-derived mesenchymal stem cells attenuates Escherichia coli-induced acute lung injury in mice. Respir Res 12: 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Curley GF, Jerkic M, Dixon S, Hogan G, Masterson C, O’Toole D, Devaney J, and Laffey JG. 2017. Cryopreserved, Xeno-Free Human Umbilical Cord Mesenchymal Stromal Cells Reduce Lung Injury Severity and Bacterial Burden in Rodent Escherichia coli-Induced Acute Respiratory Distress Syndrome. Crit Care Med 45: e202–e212. [DOI] [PubMed] [Google Scholar]
- 90.Cai S, Paudel S, Jin L, Ghimire L, Taylor CM, Wakamatsu N, Bhattarai D, and Jeyaseelan S. 2021. NLRP6 modulates neutrophil homeostasis in bacterial pneumonia-derived sepsis. Mucosal Immunol 14: 574–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Perlee D, de Vos AF, Scicluna BP, Mancheno P, de la Rosa O, Dalemans W, Nurnberg P, Lombardo E, and van der Poll T. 2019. Human Adipose-Derived Mesenchymal Stem Cells Modify Lung Immunity and Improve Antibacterial Defense in Pneumosepsis Caused by Klebsiella pneumoniae. Stem Cells Transl Med 8: 785–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kabat M, Bobkov I, Kumar S, and Grumet M. 2020. Trends in mesenchymal stem cell clinical trials 2004-2018: Is efficacy optimal in a narrow dose range? Stem Cells Transl Med 9: 17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Thompson M, Mei SHJ, Wolfe D, Champagne J, Fergusson D, Stewart DJ, Sullivan KJ, Doxtator E, Lalu M, English SW, Granton J, Hutton B, Marshall J, Maybee A, Walley KR, Santos CD, Winston B, and McIntyre L. 2020. Cell therapy with intravascular administration of mesenchymal stromal cells continues to appear safe: An updated systematic review and meta-analysis. EClinicalMedicine 19: 100249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Eggenhofer E, Benseler V, Kroemer A, Popp FC, Geissler EK, Schlitt HJ, Baan CC, Dahlke MH, and Hoogduijn MJ. 2012. Mesenchymal stem cells are short-lived and do not migrate beyond the lungs after intravenous infusion. Front Immunol 3: 297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fischer UM, Harting MT, Jimenez F, Monzon-Posadas WO, Xue H, Savitz SI, Laine GA, and Cox CS Jr. 2009. Pulmonary passage is a major obstacle for intravenous stem cell delivery: the pulmonary first-pass effect. Stem Cells Dev 18: 683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hackstein H, Lippitsch A, Krug P, Schevtschenko I, Kranz S, Hecker M, Dietert K, Gruber AD, Bein G, Brendel C, and Baal N. 2015. Prospectively defined murine mesenchymal stem cells inhibit Klebsiella pneumoniae-induced acute lung injury and improve pneumonia survival. Respir Res 16: 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Asami T, Ishii M, Namkoong H, Yagi K, Tasaka S, Asakura T, Suzuki S, Kamo T, Okamori S, Kamata H, Zhang H, Hegab AE, Hasegawa N, and Betsuyaku T. 2018. Anti-inflammatory roles of mesenchymal stromal cells during acute Streptococcus pneumoniae pulmonary infection in mice. Cytotherapy 20: 302–313. [DOI] [PubMed] [Google Scholar]
- 98.Saeedi P, Halabian R, and Imani Fooladi AA. 2019. A revealing review of mesenchymal stem cells therapy, clinical perspectives and Modification strategies. Stem Cell Investig 6: 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Robinson KM, Kolls JK, and Alcorn JF. 2015. The immunology of influenza virus-associated bacterial pneumonia. Curr Opin Immunol 34: 59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chen K, McAleer JP, Lin Y, Paterson DL, Zheng M, Alcorn JF, Weaver CT, and Kolls JK. 2011. Th17 cells mediate clade-specific, serotype-independent mucosal immunity. Immunity 35: 997–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.