

Abstract
Fascin is a pro-metastatic actin-bundling protein that is upregulated in all metastatic carcinomas. Fascin promotes cancer cell migration and invasion by facilitating membrane protrusions, such as filopodia and invadopodia. Aerobic glycolysis is a key feature of cancer metabolism and provides critical intermediate metabolites for tumor growth. Here, we report that fascin increases glycolysis in lung cancer to promote tumor growth and metastasis. Fascin promotes glycolytic flux by increasing the expression and activities of phosphofructose-kinases 1 and 2 (PFK1 and 2). Fascin mediates glycolytic functions via activation of yes-associated protein 1 (YAP1) through its canonical actin-bundling activity by promoting the binding of YAP1 to a TEAD1/4 binding motif located 30 bp upstream of the PFKFB3 transcription start site to activate its transcription. Examination of the TCGA database suggests that the fascin-YAP1-PFKFB3 axis is likely conserved across different types of cancers. Importantly, pharmacological inhibitors of fascin suppressed YAP1-PFKFB3 signaling and glycolysis in cancer cell lines, organoid cultures, and xenograft metastasis models. Taken together, our data reveal that the glycolytic function of fascin is essential for the promotion of lung cancer growth and metabolism, and suggest that pharmacological inhibitors of fascin may be used to reprogram cancer metabolism in lung and potentially other cancers with fascin upregulation.
Keywords: YAP1, metabolism, fascin inhibitor
1. Introduction
The actin cytoskeleton is frequently dysregulated in metastatic cancer cells and promotes cell migration, invasion, and metastatic dissemination. Fascin is a monomeric actin-bundling protein that is upregulated in essentially all metastatic carcinomas [1–3]. Fascin crosslinks actin filaments into straight and stiff bundles through its positively charged actin-binding surfaces [1, 4, 5]. In addition to its well-known role in metastatic dissemination, fascin also increases metabolic stress resistance, chemoresistance, and cancer cell stemness [6–8]. The mechanisms underlying these non-canonical functions of fascin are not well understood.
Metastasis is responsible for more than 90% of cancer-related deaths, and there are few effective therapies for metastatic cancer [9]. Fascin has emerged as an attractive target for the development of anti-metastasis therapeutics for several reasons [7, 10–13]. First, in adult animals, fascin expression is absent in most tissues, except in neuronal tissues [1–3]. Second, fascin expression is highly upregulated in transformed cancer cells, especially in metastatic cancers, and plays a causative role in promoting cancer metastasis [1, 2]. Third, adult mice with homozygous deletion of fascin appeared to be healthy and fertile [14], which suggests that inhibitors specifically targeting fascin are likely to be well tolerated in patients with cancer. Several small molecules, including migrastatin analogs and G2 derivatives, have been identified as fascin inhibitors through chemical biology approaches and high-throughput screening [12, 15, 16]. Some FDA-approved drugs also show inhibitory activity toward fascin [17]. These small molecules inhibit cell migration and invasion in cell culture and block metastatic dissemination in animal models. However, the effectiveness of blocking metastatic dissemination alone in the clinic has been questioned [1, 9], as in most patients with metastatic cancer, the cancer cells have already disseminated at the time of diagnosis.
Cancer cells undergo profound metabolic reprogramming during tumor initiation and progression [18]. In metastatic cancer cells, metabolic reprogramming is essential for the colonization of distant organs, as distant sites have drastically different nutrient and metabolic environments when compared to the primary tumor [18]. It is well documented that cancer cells prefer glycolysis for glucose catabolism even under oxygen-replete conditions (the Warburg effect) [19]. Although this phenomenon was initially thought to be due to defective mitochondria in cancer cells, mitochondrial respiration is functional in most cancer cells; both oxidative phosphorylation (OXPHOS) and glycolysis are essential for tumor growth [19]. Glycolytic metabolism provides crucial intermediate metabolites for the synthesis of nucleotides, amino acids, and lipids, which are building blocks required for the growth and proliferation of cancer cells [19]. Several glycolytic enzymes such as aldolase are actin-binding proteins, and their activities could be regulated by remodeling of the actin cytoskeleton [20]. However, the metabolic function of actin cytoskeleton dysregulation in metastatic cancer cells remains poorly understood.
Targeting deregulated cancer metabolism has emerged as an attractive new approach for the development of cancer therapeutics. A challenge in targeting specific metabolic pathways is the intrinsic metabolic flexibility of cancer cells, as they can alternate between glycolysis and mitochondrial OXPHOS in response to metabolic stress [21]. Therapeutic modalities that simultaneously target glycolysis and mitochondrial OXPHOS have improved efficacy [21]. We previously reported that fascin augments mitochondrial oxidative phosphorylation in lung cancer by remodeling mitochondrial actin filaments and promoting biogenesis of respiratory complex I [7]. Here, we show that fascin upregulates phosphofructose-kinases (PFKs) to promote glycolysis in lung cancer. Our data, together with our earlier findings, suggest that fascin may be targeted to suppress both glycolysis and mitochondrial OXPHOS. Therefore, pharmacological inhibitors of fascin could be useful in preventing lung cancer metastasis by reducing the metabolic flexibility in lung cancer cells.
2. Materials and Methods
2.1. Cell culture
The cancer cell lines (H1650, A549, H292, H23, LLC, DLD1, SW480, HUH-7, and HEPG2) were obtained from the ATCC and authenticated using short tandem repeat profiling. All cell lines were free of microbial contamination, including mycoplasma. H1650, H292, H23, DLD1, and SW480 cells were cultured in RPMI 1640 (HyClone, SH30027.FS) medium supplemented with 10% fetal bovine serum (FBS) (Atlanta Biological, S11150) and 1% penicillin-streptomycin (P/S) (Gibco, 15140163). A549, LLC, HUH-7, and HEPG2 were cultured in Dulbecco’s modified Eagle’s medium (DMEM) medium (HyClone, SH30243.FS) supplemented with 10% FBS and 1% P/S. All cells were maintained at 37 °C in a humidified incubator with 5% CO2.
2.2. Animal experiments
All animal experiments were performed according to protocols approved by Institutional Animal care and use Committee of the Penn State College of Medicine. Seven-week-old female nude mice were purchased from Charles River and housed in a pathogen-free room with a 12 h light/dark cycle. For H1650 lung colonization experiments, 5 × 106 luciferase-labeled H1650 cells were resuspended in 60 μL RPMI medium with 5% Matrigel (serum-free) and inoculated into 7-week-old female nude mice via lung orthotopic injection as previously described [22]. Noninvasive bioluminescence imaging and analysis were performed as described previously using IVIS Lumina Series III (PerkinElmer).
The Lewis lung cancer (LLC) allograft tissues treated with G2 (Enamine, EN300–246105) were collected as described in a previous study on metastatic colonization [7]. Luciferase-labeled LLC cells (5 × 105) stably expressing luciferase were resuspended in 200 μL of serum-free DMEM and injected via tail vein into 7 week-old Albino BL6 female mice. The mice were randomly assigned to two groups (10 mice each, female). Forty-eight hours following the tail-vein injection, the mice were intraperitoneally injected with 100 mg/kg G2 or vehicle control. Mice were euthanized when they reached the humane endpoint, and the lungs were collected for further analysis.
2.3. KrasLSL-G12D/Trp53fl/fl lung cancer model and lung organoid culture
KrasLSL-G12D (Stock No: 008179) and Trp53fl/fl (Stock No: 008462) mice were obtained from the Jackson Laboratory and crossed to generate KP mice (KrasLSL-G12D/Trp53fl/fl) as previously described [23]. The formation of lung adenocarcinoma was induced by intratracheal administration of adenovirus-Cre (1 × 107) following the protocols of DuPage et al. [24]. For lung intratracheal instillation, 9 weeks old KP mice were injected with 50 μL of the virus mix (10 mM CaCl2, 1 × 107 VVC-U-Ad5CMVCre (Carver College of Medicine) diluted in Opti-MEM). Three months after the viral infection, the mice were euthanized and lung tumors were harvested for the preparation of organoid culture. Tumors were washed thrice with ice-cold DMEM and cut with sterile scissors to approximately 1 mm3 pieces. The tissues were digested with collagenase (Sigma, C0130) at a final concentration of 2 mg/mL in serum-free DMEM/F12 media for 60 min at 37 °C. After centrifugation at 200 × g for 5 min, the pellet was washed with medium (DMEM with 10% FBS) five times to rinse off the collagenase. The pellet was resuspended in 300 μL of Matrigel (Corning, 354277) and plated in 24 well plate (50 μL/well). The gel was allowed to solidify at 37 °C for 15 min, and then 500 μL of warm organoid culture medium was added to each well (DMEM/12 500 ml, 10% FBS, 1% P/S, 5 ml ITS-G [Gibco, 41400045], 10 μL A83-01 [25 mM stock, Sigma, SML0788], and 100 μL Y-27632 [50mM stock, Biogems, 1293823]).
2.4. Inhibitor treatment
Unless otherwise indicated, cells and organoids were incubated with medium containing 40 μM G2 (Xcessbi, M60269) at 37 °C for 48 h in a 5% CO2 incubator prior to use in the experiments.
2.5. Plasmids
FSCN1 knockouts (KOs) were generated using pLenti CRISPR V2 vector (Addgene, 52961) encoding single guide (sgRNA) targeting FSCN1 (human FSCN1 sgRNA 1, GAAGAAGCAGATCTGGACGC; human FSCN1 sgRNA 2, CACCTTGAACCCGAACGCCT; mouse FSCN1 sgRNA 1, TCGCTACCTGGCCGCCGACA; mouse Fscn1 sgRNA 2, AGCCGAGGCGTTCGGGTTCA). To rescue FSCN1 knockdown (KD) or overexpression of FSCN1 proteins, wild-type or mutant FSCN1 (S39E and 149–151A mutants) cDNAs were subcloned into pLenti.CMV.blasticidin vector (Addgene, 17486) (Campeau et al., 2009). YAP 6SA expression plasmid was obtained from Addgene (Addgene # 42562). PFKFB3 shRNAs were obtained from the Sigma shRNA library (sh1, TRCN0000314746; sh2, TRCN0000314747). The retroviral and lentiviral particles were packaged in HEK293 cells using the PEI transfection method and concentrated as previously described [25].
2.6. Western blotting
Western blotting was performed as described previously (Lin. et al., 2019). Cells were lysed in sodium dodecyl sulfate (SDS)-NP40 buffer (50 mM Tris [pH 8.0], 150 mM NaCl, 1% NP40, 1% SDS, 1 mM protease inhibitor cocktail) on ice for 1 min. Cells were scraped from the plate and subjected to three cycles of sonication of 5 s each. The lysates were heated at 95 °C for 5 min and centrifuged at 15,000 ×g at 4 °C for 10 min. To prepare cell lysates for the detection of protein phosphorylation, cells were lysed in Triton X-100 buffer (50 mM Tris [pH 7.4], 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, 1 mM phosphatase inhibitor cocktail [Thermo Scientific, 88667], 1 mM protease inhibitor cocktail [Roche, 04693159001]) on ice for 15 min. The proteins (50 μg) were separated by SDS-PAGE and transferred onto polyvinylidene difluoride (PVDF, Millipore, IPVH00010) membranes. The membranes were incubated in blocking buffer (5% w/v nonfat dry milk in Tris-buffered saline, 0.05% Tween-20 [TBS-T]) for 30 min at room temperature (RT), and then incubated with primary antibodies overnight at 4 °C. Next, the membranes were incubated with secondary antibodies for 60 min at RT.
2.7. Immunofluorescent staining and microscopy
Immunofluorescent staining was performed as described previously [7, 26]. A total of 4 × 104 cells were plated onto a laminin-coated coverslip in a 24-well plate and cultured overnight. Cells were then fixed with 4% fresh paraformaldehyde (Sigma, 158127) in PBS at RT for 20 min followed by incubation with primary antibodies diluted in antibody dilution buffer (2% BSA and 0.1% Triton X-100 in TBS) overnight at 4 °C. The cells were then incubated with secondary antibodies for 60 min at RT, followed by staining with DAPI (Thermo Fisher, D1306) at RT for 10 min. The cells were washed extensively with TBS between each step for 5 min each. After a final wash with TBS, the coverslips were mounted with Fluoromount (Sigma, F4680).
For lung organoid staining, the assay was performed as previously described with a few modifications [22]. Matrigel (Corning, 354277) was thawed at 4 °C overnight and used for the organoid culture. A total of 1 × 104 cells were resuspended in 100 μL Matrigel and plated in a glass bottom plate. After incubating at 37 °C for 20 min to allow the Matrigel to solidify, 2 mL of organoid culture medium was added to the plate. After 3 days, the organoids were incubated with vehicle control or G2 (40 μM) for 48 h. Organoids were fixed with 4% fresh paraformaldehyde in PBS at RT for 20 min and then incubated with YAP1 antibody diluted in antibody dilution buffer (2% BSA and 0.1% Triton X‐100 in PBS) at 4 °C overnight. After sequential incubation with secondary antibody (ThermoFisher, A11008) and phalloidin (ThermoFisher, A12381) for 3 h, followed by DAPI for 30 min, the coverslips were mounted with Fluoromount. Extensive washing with TBS was performed between each step (20 min each). Images were captured using a Leica SP8 confocal microscope.
2.8. Mitochondrial stress test
The oxygen consumption rate (OCR) was determined using a Seahorse XF96 Extracellular Flux Analyzer (Agilent Seahorse Bioscience) as previously described [22]. A total of 1 × 104 cells were plated in each well of the XF96 microplates. Cells were maintained in a non-buffered assay medium (Agilent) in a non-CO2 incubator for 60 min before the assay. The XF Cell Mito Stress Test Kit (Agilent, 103015) was used for the assay. The baseline recordings were followed by sequential injection of oligomycin (1 μM), FCCP (1 μM), and rotenone (1 μM)/antimycin A (20 μM).
2.9. Glycolysis stress test
Extracellular acidification rate (ECAR) was determined using a seahorse XF24 Extracellular Flux Analyzer (Agilent Seahorse Bioscience) following the manufacturer’s protocols. A total of 5 × 104 cells were plated in each well of the XF24 microplates. Cells were maintained in a non-buffered assay medium (Agilent) in a non-CO2 incubator for 60 min before the assay. The XF Cell glycolysis Test Kit (Agilent, 103020) was used for the assay. The baseline recordings were followed by sequential injection of the following reagents: glucose (10 mM), oligomycin (1 μM), or 2-DG (50 mM).
2.10. PKM2 crosslinking
A total of 3 × 106 cells were placed in a 10 cm plate and washed with 5 mL ice-cold PBS thrice. Cells were scraped from the plate in 1 ml of ice-cold PBS. After centrifugation at 500 × g for 5 min at 4 °C, the pellet was resuspended in ice-cold PBS (0.5 ml ice-cold PBS). Cells were frozen in lipid nitrogen and thawed in a 37°C water bath three times to lyse the cells. After centrifugation at 10,000 × g for 10 min at 4 °C, formaldehyde (Amresco, M134) was added to the supernatant to a final concentration of 1%. The solution was mixed and incubated at RT for 20 min. Total proteins (50 μg) were resolved on a 8% non-reducing SDS-PAGE gel and immune blotted with PKM2 antibody (Cell Signaling, 3198s).
2.11. FBP level measurement
FBP levels were determined using a kit from BioVision (BioVision, K2036–100) following the manufacturer’s instructions. A total of 2 × 106 cells were lysed with 500 μL of ice-cold frustose-1, 6-biphosphate (F1, 6BP) assay buffer and placed on ice for 10 min. After centrifugation at 10,000 × g at 4 °C for 10 min, the supernatant was transferred to a new microcentrifuge tube and used for the assay. Fluorescence was measured at Ex/Em = 535/587 nm in endpoint mode using FlexStation 3 (Molecular Devices).
2.12. PFK activity assay
PFK activity was determined using a kit from BioVision (BioVision, K776) following the manufacturer’s instructions. A total of 2 × 106 cells were lysed with 200 μL ice-cold assay buffer on ice. After centrifugation at 14,000 × g for 5 min, the supernatant was collected for protein assays. Protein (100 μg in 20 μL) solution was used for the assay. The optical density was measured at 450 nm using a FlexStation 3 (Molecular Devices).
2.13. Immunohistochemistry (IHC)
IHC staining for fascin and YAP1 was performed in specimens from a cohort of 113 patients with lung adenocarcinoma in two separate studies, as previously described [7, 22]. The intensity of the staining was evaluated by two independent investigators blinded to the clinical pathological data of patients using the following criteria: 0, negative; 1, low; 2, medium; and 3, high. The extent of staining was scored as 0, 0% stained; 1, 1% to 25% stained; 2, 26% to 50% stained; 3, 51% to 100% stained. Five random fields (20 × magnification) were evaluated under a light microscope. The final scores were calculated by multiplying the scores of the intensity with those of the extent of staining and the samples were divided into four grades: 0, negative (−); 1 to 2, low staining (+); 3 to 5, medium staining (++); 6 to 9, high staining (+++). The correlation between fascin and YAP1 staining intensities was determined using Spearman’s rank correlation test.
For IHC staining of YAP1, PFKFB3, and Ki67 in xenograft tissues, paraffin-embedded tissue sections were deparaffinized and then heated in a pressure pot for 3 min to retrieve the antigens. The sections were then incubated with primary antibodies (1:200) overnight at 4 °C. Antibody binding was detected by incubating with a peroxidase-conjugated secondary antibody at 37 °C for 30 min. A DAB substrate kit was used to perform the chromogenic reaction.
2.14. Chromatin-immunoprecipitation (ChIP) assay
ChIP was performed using a kit from Abcam (catalog # ab500) following the manufacturer’s protocol. Cells were grown to 90% confluence in a 10 cm plate. A total of 1 × 106 cells were used for each reaction. After sonication, YAP1 antibody (Cell Signaling, 14074S) was used for immunoprecipitation overnight with rotation at 4 °C. After DNA purification, quantitative PCR was performed using the following primers:
Human PFKFB3
hPFKFB3_YAP1ChIP_FW: GGGATAACAGCAGCCAGGA
hPFKFB3_YAP1ChIP_RV: CTTCCACGTGGGGAGGAG
Murine Pfkfb3
mPfkfb3_Yap1ChIP_FW: CTGCTCTCGTCCACCCGCAG
mPfkfb3_Yap1ChIP_RV: CTCCTCTCCAGTTCTGTCGG
2.15. Dual-luciferase reporter assay
Dual-luciferase reporter assay was performed using a kit (Promega, E1910) following the manufacturer’s protocol. Promoter sequences from murine Pfkfb3 (350 bp, 500 bp, 1000 bp, and 6000 bp) gene was subcloned into the pGL3 vector. The plasmids were transiently transfected into 293T cells growing in a 12-well plate using Lipofectamine 2000 (ThermoFisher, 11668019) (1 μg reporter plasmid and 1 μg Renilla luciferase). After 48 h, the cells were lysed with 100 μL PLB buffer from the kit. Cell lysate (20 μL) was transferred to a 96-well reading plate and mixed with 100 μL LARII solution. Luciferase signals were read using FlexStation 3 (Molecular Devices).
2.16. Gene expression profiling interactive analysis (GEPIA)
TCGA data analysis was performed using the GEPIA website, as previously described [27].
2.17. Metabolic flux analysis
For metabolic flux assays of glycolysis intermediates, the cells were incubated with 25 mM U-13C6 glucose in DMEM or control medium for the indicated durations. After washing twice with PBS, 1 mL of 80% methanol/water (−80 °C cooled) was added to quench the reaction and the metabolites were extracted. The cells were incubated in a −80 °C freezer for at least 15 min, scraped into 1.7 mL tubes, and centrifuged at 15000 rpm for 10 min. The supernatant was collected and dried using SpeedVac and re-dissolved in 50 μL of methanol/water (50/50) for LC-MS/MS analysis. LC-MS/MS analysis was performed with a Waters ACQUITY UPLC systems connected to a Xevo TQ-S mass spectrometer using selective reaction monitoring (SRM) methods, as described previously [28, 29]. Chromatographic separation was performed on a Waters UPLC BEH Amide column (2.1 × 150 mm, 1.7 μm). The percentage contributions were calculated based on the mass isotopomer measurements. FluxFix was used for the correction of natural isotope abundance [30].
3. Results
3.1. Fascin promotes glycolysis in non-small cell lung cancer (NSCLC) cells through its actin crosslinking activity
We previously reported that knockdown (KD) or knockout (KO) of fascin in NSCLC inhibits OXPHOS [7]. In response to the inhibition of OXPHOS, cells generally increase glycolytic metabolism as a compensatory response [21, 31]. However, we noted that fascin KO in NSCLC cells reduced both lactate production and glucose uptake by 20%–30% (Fig. 1A–1B, Fig. S1A–S1B), suggesting the inhibition of glycolysis in these cells. Conversely, ectopic expression of fascin in these cells increased both lactate production and glucose uptake (Fig. 1A–1B, Fig. S1A–S1B). The measurement of the extracellular acidification rate (ECAR) confirmed that fascin KD or KO decreased ECAR (Fig. 1C–1D and Fig. S1C), while fascin overexpression (OE) increased ECAR (Fig. 1E–1F and Fig. S1D). This finding suggests that fascin activates glycolysis in addition to its previously reported function of augmenting mitochondrial OXPHOS.
Figure 1. Fascin promotes glycolysis in NSCLC cells through its actin crosslinking activity.
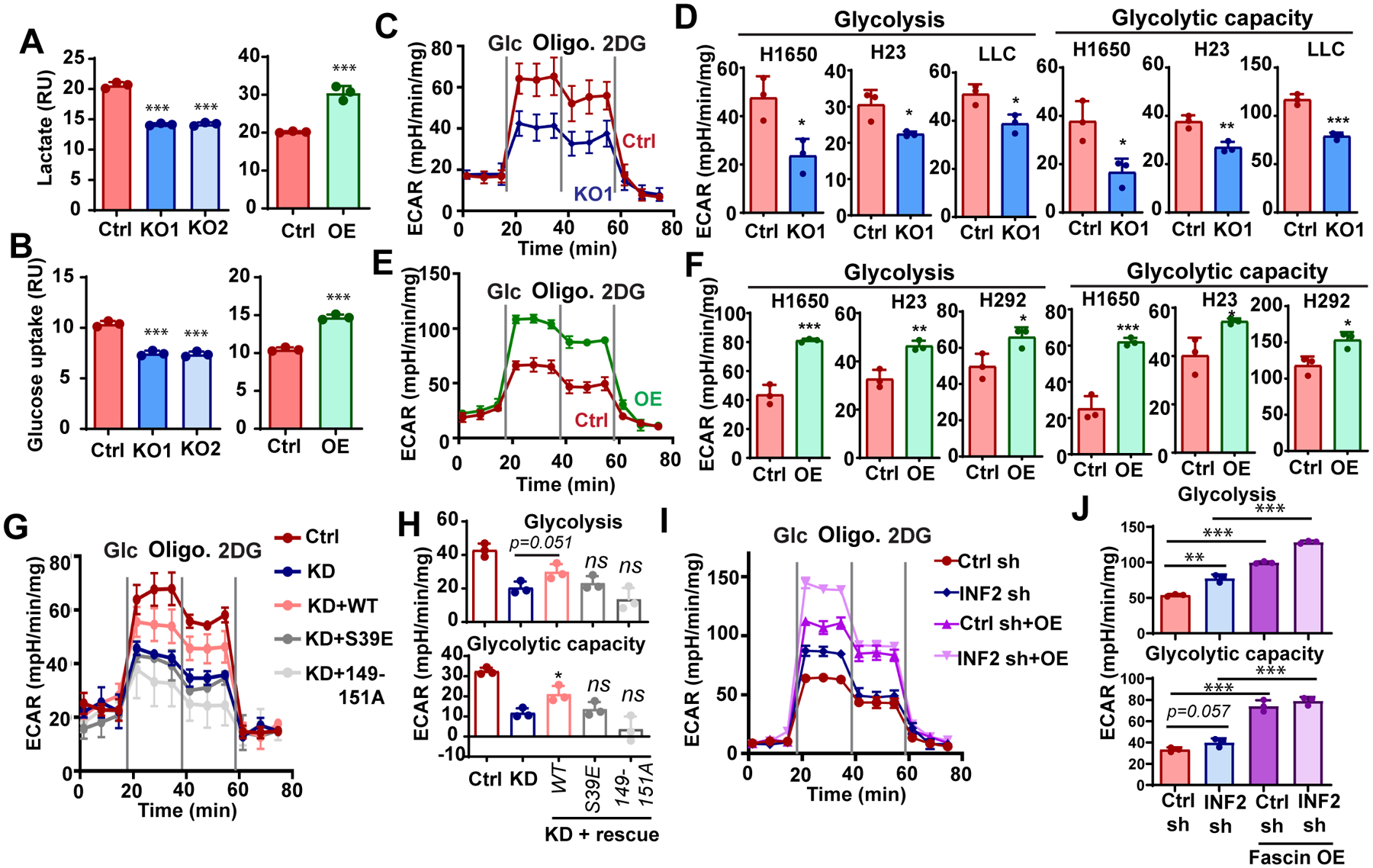
A and B, the effect of fascin KO or OE on lactate production (A) or glucose uptake in H1650 cells.
C and D, the effects of fascin KO (C) or OE (D) on extracellular acidification rate (ECAR), as determined by glycolysis stress test in H1650 cells.
E and F, quantitation of the effects of fascin KO (E) or OE (F) on glycolysis and glycolytic capacity in different NSCLC cell lines.
G and H, the effect of fascin knockdown (KD) or rescue with ectopic expression of wild-type (WT) fascin or bundling defective mutants (S39E and 149–151A) on ECAR in H1650 cells. The ectopic WT fascin, but not the bundling defective mutants, was able to rescue ECAR, including glycolysis and glycolytic capacity, in fascin KD cells. H is the quantitation of glycolysis and glycolytic capacity for G.
I and J, the effect of fascin overexpression on ECAR in H1650 cells stably expressing control shRNA (Ctrl sh) or INF2-CAAX shRNA (INF2 sh). The ectopic expression of wild-type fascin in INF2-CAAX knockdown H1650 cells was able to further increase the ECAR, including glycolysis and glycolytic capacity (J).
Data in A, B, E, F, H and J were analyzed by two-tailed, two-sample unpaired Student’s test. Results from at least three independent experiments are shown. *, ** and **** indicated p<0.05, 0.01 and 0.001, respectively. ns, not significant.
Next, we investigated whether fascin regulates glycolysis in NSCLC through its canonical actin-bundling activity. We previously demonstrated that mutations at the two positively charged actin-binding surfaces abrogated fascin crosslinking activities without causing significant conformational changes [4, 32]. Therefore, we ectopically expressed wild-type fascin (WT) or two bundling-defective mutants (S39E and 149–151A) in fascin KD cells. As shown in Fig. 1G and 1H, ectopic expression of WT fascin, but not the bundling-defective mutants, was able to partially restore ECAR in fascin KD cells. Since fascin controls mitochondrial OXPHOS by remodeling mitochondrial actin filaments (mtF-actin), also in an actin-bundling activity-dependent manner, we next investigated whether mtF-actin is involved in fascin-mediated glycolysis. We disrupted mtF-actin by knocking down INF2-CAAX, the endoplasmic membrane actin nucleator involved in the polymerization of mtF-actin [33]. INF2-CAAX KD abrogates the accumulation of mtF-actin and fascin-mediated enhancement of mitochondrial OXPHOS in lung cancer cells [7]. As shown in Fig. 1I–1J, INF2-CAAX KD modestly increased ECAR in H1650 cells, likely due to the compensatory response to the decrease in mitochondrial OXPHOS in these cells [7]. The overexpression of fascin in INF2-CAAX KD cells increased ECAR to an extent similar to that in control shRNA cells (Fig. 1I–1J), suggesting that mtF-actin is not required for fascin to promote glycolysis. Taken together, our data support that fascin promotes glycolysis in addition to its previously reported role in augmenting mitochondrial OXPHOS. The glycolytic function of fascin requires its actin-bundling activity, but is independent of mtF-actin remodeling.
3.2. Fascin enhances the glycolytic flux to F1, 6BP by upregulating phosphofructokinases (PFKs)
To understand the mechanism by which fascin regulates glycolysis, we used 13C-labeled glucose to investigate the effect of fascin KO on glycolytic flux in H1650 cells. Cells were incubated with U-13C-Glucose for 0, 2, 15, 30, 60, or 360 min, and the incorporation of 13C into several glycolytic intermediate metabolites was analyzed using LC-MS/MS. As shown in Fig. 2A, the levels of 13C-labeled glucose-6-phosphate/fructose-6-phosphate (G6P/F6P), 2/3-phosphoglyceric acid (2PG/3PG), phosphoenolpyruvic acid (PEP), and fructose bisphosphate (FBP) were mostly lower in fascin KO cells than in control H1650 cells at earlier time points (2–60 min) (Fig. 2A). After 360 min, most of the 13C-labeled glycolysis metabolites in KO cells returned to levels comparable to that (or in the case of G6P/F6P higher than) in control cells, except for FBP (Fig. 2A). FBP was markedly lower in KO cells, even at 6 h. F1, 6BP is a product of the phosphorylation of F6P by PFK1. Along with hexokinase (HK) and pyruvate kinase (PK), PFK1 catalyzes one of three irreversible and rate-limiting steps in the glycolysis cascade [19]. The activity of PFK1 is allosterically regulated by fructose-2,6-bisphosphate, a product of PFK2. We confirmed that the levels of fructose-1, 6-bisphosphate was decreased in fascin KO and increased in fascin OE H1650 and H292 cells (Fig. 2B and 2C). Fructose-1, 6-bisphosphate allosterically activates PKM2 (Fig. 2A, left panel), the pyruvate kinase isoform upregulated in cancer, by promoting its tetramerization [34]. As shown in Fig. 2D and 2E, fascin KO or KD decreased the levels of PKM2 tetramers and dimers, but increased the levels of the monomeric proteins. Ectopic expression of WT fascin, but not the bundling-defective mutants, restored the levels of PKM2 tetramer in fascin KD cells. These data are consistent with the notion that fascin KO reduces the glycolytic flux to F1, 6BP.
Figure 2. Fascin enhances the glycolytic flux to fructose-1, 6-bisphosphate.
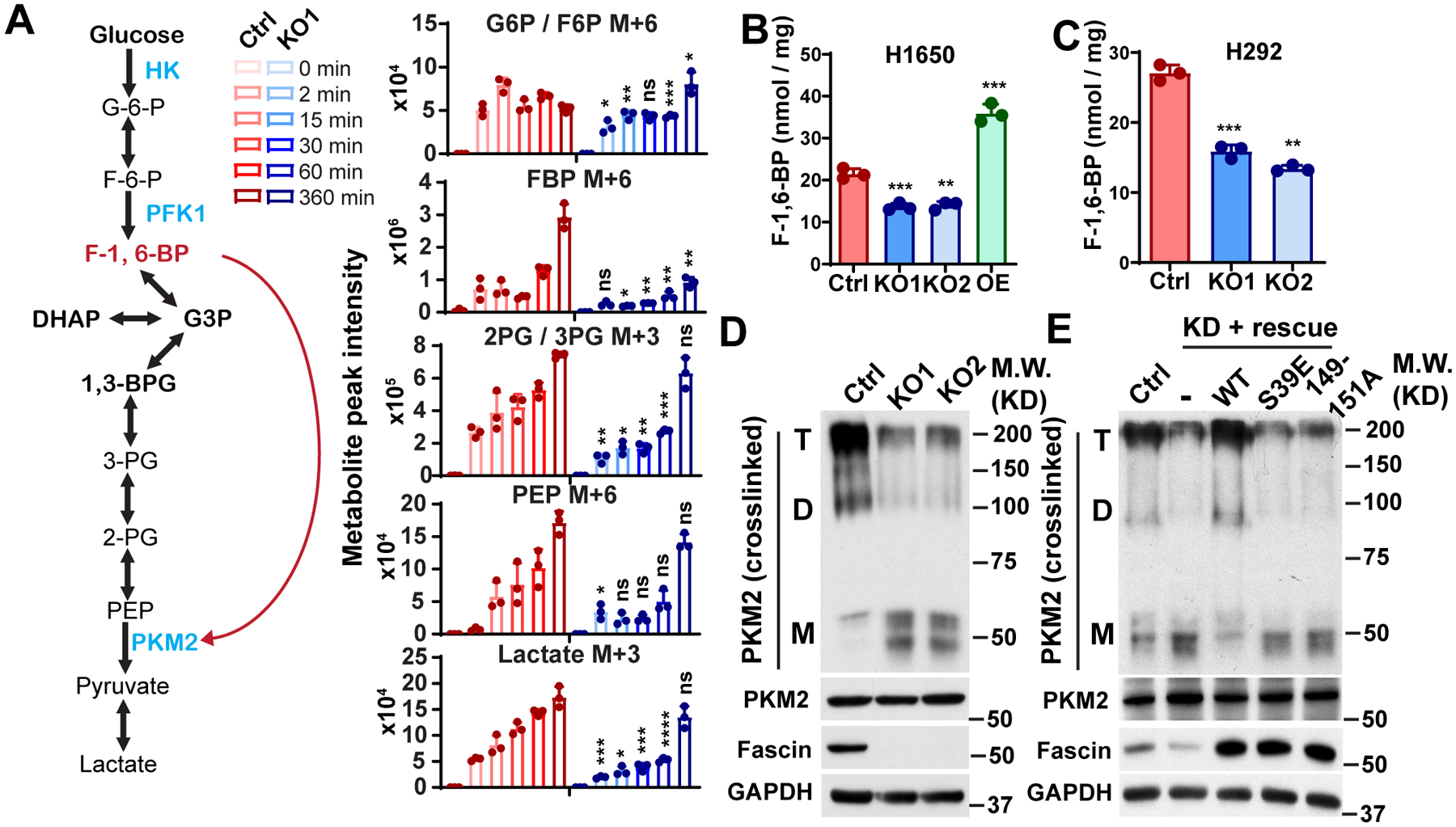
A, left panel, the schematic illustration of glycolysis pathways. The red arrow indicates F-1,6-BP can bind to PKM2 to activate its tetramerization and catalytic activity. Right panel, the relative level of 13C-labeled glycolytic metabolites in control or fascin KO H1650 cells after incubation with U-13C-Glucose for indicated time. M+x indicates the number of 13C-labeled carbon in respective metabolites.
B and C, the effect of fascin KO on F-1,6-BP levels in H1650 and H292 cells.
D, Western blotting showing that fascin KO reduced the levels of PKM2 dimer and tetramer without affecting the expression of total PKM2 protein levels in H1650 cells. T, D, M indicated tetramer, dimer and monomer.
E, the ectopic expression of wild-type fascin, but not the bundling defective mutants (S39E and 149–151A), in fascin knockdown H1650 cells was able to rescue PKM2 dimer and tetramer level in H1650 cells. T, D, M indicated tetramer, dimer and monomer.
Data in A– C were analyzed by two-tailed, two-sample unpaired Student’s test. Representative results from at least three (B–E) or two (A) independent experiments are shown. *, **, *** and **** indicated p<0.05, 0.01, 0.001 and 0.0001, respectively. ns, not significant.
To understand the mechanism by which fascin regulates FBP production, we examined the effect of fascin KO on the expression levels of the three isoforms of PFK1 (PFKP, PFKM, and PFML) and PFKFB3, the PFK2 isoform that is frequently upregulated in cancer. As shown in Fig. 3A, fascin KO significantly reduced the protein levels of PFKP, PFKM, and PFKFB3, but not that of PFKL. Consistent with the reduction in PFK protein levels, PFK activity was also lower in NSCLC cells with fascin KO (Fig. 3B). Conversely, ectopic expression of fascin increased the protein levels of PFKFB3, PFKP, and PFKM, but not that of PFKL (Fig. 3C). Interestingly, the overexpression of fimbrin, another actin-bundling protein, had no effect on PFK protein upregulation (Fig. S2A). To determine the role of PFK2 and PFK1 in fascin-mediated glycolysis, we ectopically expressed PFKFB3 and PFKP in fascin KO cells (Fig. S2B). As shown in Fig. 3D and S2C–S2D, ectopic PFKFB3 partially restored glycolysis and lactate production in fascin KO cells. Ectopically expressed PFKP had no effect on ECAR or lactate in fascin KO cells (Fig. 3D and S2C–S2D). Ectopic expression of PFKFB3 restored PFK activity in fascin KO cells to a level similar to that in control cells (Fig. 3E). In contrast, the effect of ectopic PFKP on PFK activity was marginal (Fig. 3E). To further determine whether PFKFB3 is required for fascin-mediated glycolysis, we used shRNA to KD PFKFB3 in H1650 cells (Fig. S2E). As shown in Fig. 3G and Fig. S2F, PFKFB3 KD abrogated the ability of fascin to increase FBP production and markedly reduced the ability of fascin to increase ECAR and lactate. Taken together, our data suggest that fascin promotes glycolysis in NSCLC by upregulating PFKs, and PFKFB3 is critical for fascin to promote glycolytic metabolism.
Figure 3. PFKFB3 is required for fascin to promote glycolysis.
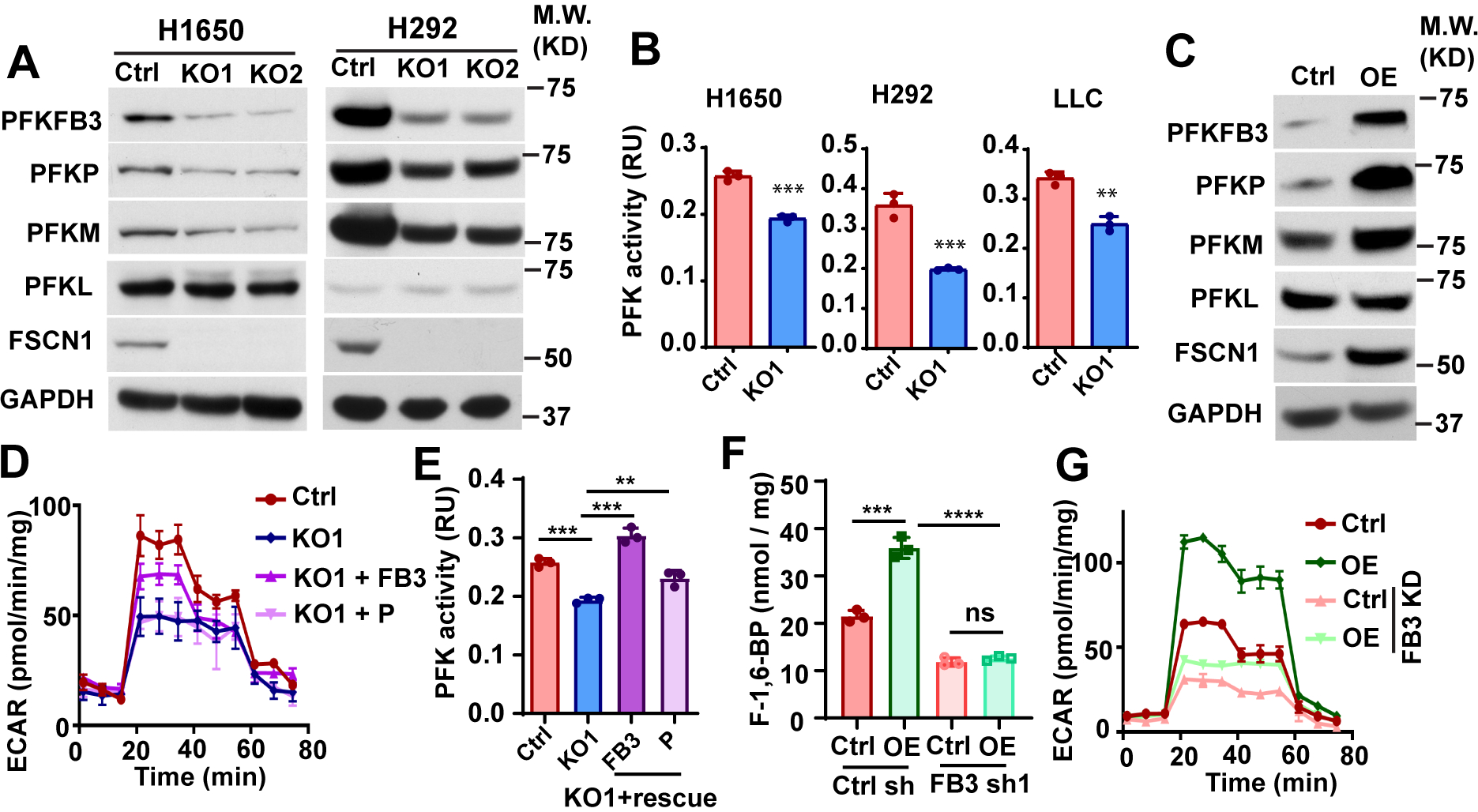
A and B, the effects of fascin KO on PFK protein expression levels (A) and PFK activities in NSCLC cells.
C, the effect of fascin overexpression (OE) on PFK protein levels in H1650 cells.
D, PFKFB3 or PFKP were ectopically expressed in fascin KO H1650 cells and the effect of ectopic PFKs on ECAR was determined. The ectopic expression of PFKFB3, but not PFKP could partially rescue ECAR in fascin KO cells.
E and F, the effect of ectopic PFKFB3 (FB3) or PFKP (P) on PFK activities (E) and F-1,6-BP levels in fascin KO H1650 cells.
G, the effects of PFKFB3 knockdown (FB3 KD) on ECAR in H1650 cells stably expressing control vector (Ctrl) or wild type fascin (OE).
Data in B, E and F were analyzed by two-tailed, two-sample unpaired Student’s test. Representative results from at least three independent experiments are shown. **, *** and **** indicated p<0.01, 0.001 and 0.0001, respectively. ns, not significant.
3.3. Fascin activates the transcription of PFKFB3 through a YAP1/TEAD-binding site on its promoter
The stability of PFK1 and PFK2 is regulated through proteasomal degradation [35]. However, we found that treatment of control and fascin KO cells with the proteasome inhibitor MG132 had no effect on PFKFB3 levels in H1650 cells (Fig. S3A). Instead, we noted upregulation and downregulation of PFKFB3 mRNA in fascin OE and KO cells, respectively (Fig. S3B). Therefore, we hypothesized that fascin may promote glycolysis through the transcriptional activation of PFKFB3.
Recently, the Hippo pathway transcription co-activator YAP1 has emerged as an important sensor of actin cytoskeleton tension and a regulator of glycolysis in cancer [36]. Remodeling of the actin cytoskeleton activates YAP1 and its paralog TAZ through mechanisms that remain unclear [37]. We observed that fascin KO decreased the protein levels of YAP1 (Fig. 4A and Fig. S3C), whereas it increased the levels of pS127-YAP1 (Fig. 4A). Fascin KO also increased the levels of active LATS1, the serine/threonine kinase responsible for phosphorylation of YAP1 at S127 and promoting its degradation (Fig. 4A), and suppressed the nuclear localization of YAP1 (Fig. 4B–4C). The expression of TAZ was not affected by fascin KO (Fig. S3C). Fascin KD decreased the expression of YAP1 and PFKFB3, which was restored by WT, but not by the bundling-defective mutants of fascin (Fig. 4D). TAZ protein levels were not affected by fascin KD or rescue treatment (Fig. 4D). Taken together, our data suggest that overexpression of fascin in NSCLC activates YAP1 and increases PFKFB3 expression through its actin crosslinking activity, likely through LATS1.
Figure 4. Fascin promote the transcription of PFKFB3 by activating YAP1.
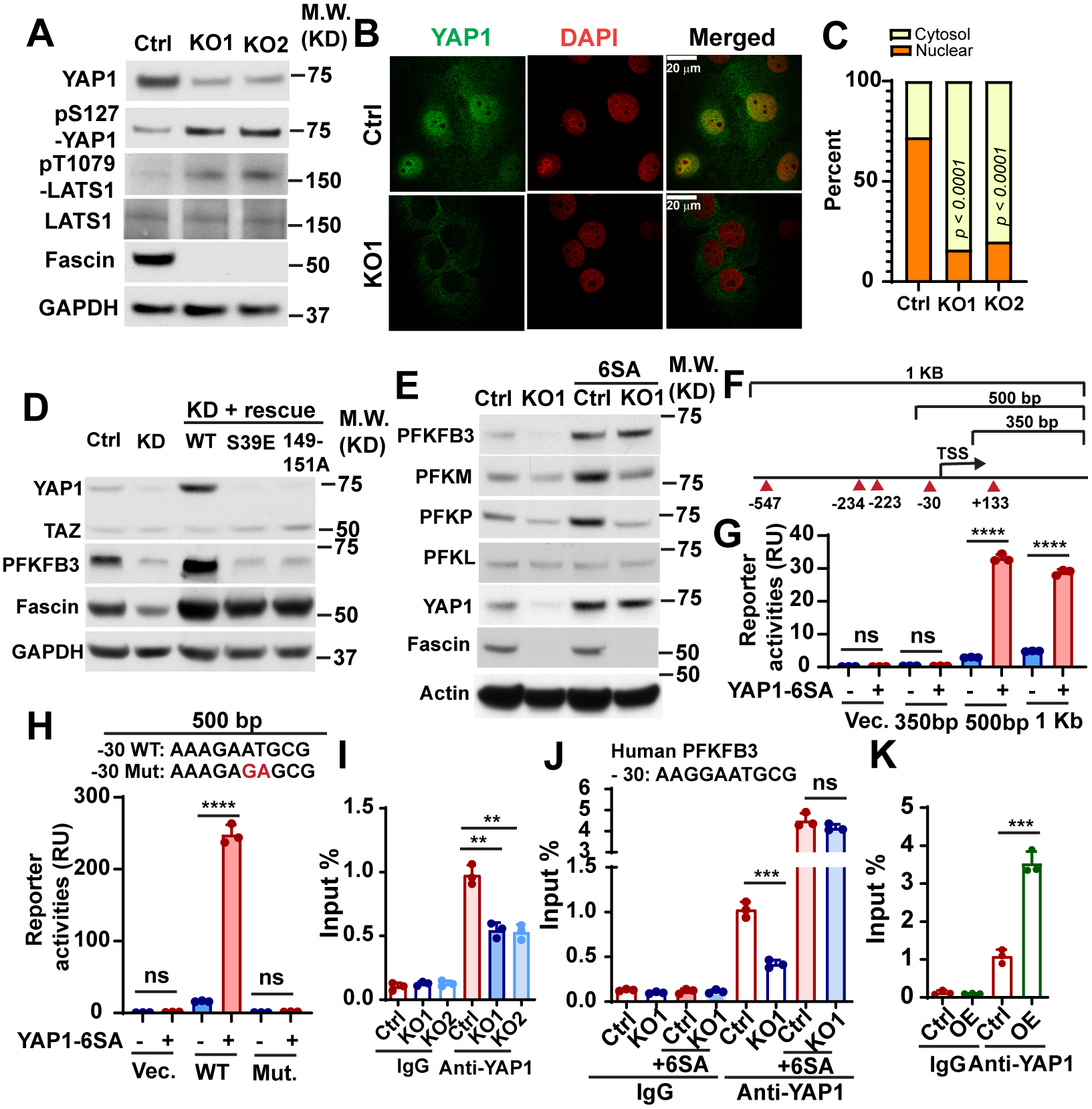
A, Western blotting showing the effect of fascin KO on the levels of total YAP1 protein, phosphorylated YAP1 (pS127), phosphorylated LATS1 (pT1079) and total LATS1 in H1650 cells.
B and C, representative images (B) and quantitation (C) showing YAP1 nuclear translocation were inhibited in H1650 fascin KO cells. n=50 cell per group.
D, the effect of fascin knockdown (KD) or the rescue of the knockdown (with ectopic wild-type fascin (WT) or the bundling defective mutants (S39E and 149–151A)) on YAP1 and PFKFB3 protein levels in H1650 cells.
E, the effect of constitutively active YAP1 mutant (6SA) on the protein expression levels of PFKFB3, PFKM, PFKP and PFKL in control or fascin KO1 H1650 cells.
F, schematic illustration of Pfkfb3 promoter reporter constructs and the position of TEAD binding motifs in the promoter.
G and H, the effect of YAP1–6SA mutant on the transcription of luciferase when cotransfected with indicated luciferase reporters, as determined by the dual-luciferase reporter assay. The ability of YAP1–6SA to promote luciferase transcription was abolished by the GA mutations in the 500 bp reporter construct (H).
I– J, Chromatin-immunoprecipitation (ChIP) assay indicates fascin KO could decrease the binding of YAP1 to murine (I) or human (J) PFKFB3 / Pfkfb3 promoter in LLC or H1650 cells. The binding of YAP1–6SA mutant to PFKFB3 promoter was not affected by fascin KO (J).
K, ChIP assay showing that fascin overexpression could increase the YAP1 binding with PFKFB3 promoter in H1650 cells.
Data in G-K were analyzed by two-tailed, two-sample unpaired Student’s test. Data in C were analyzed by two-tailed Fisher’s exact test. Representative results from at least three independent experiments are shown. **, *** and **** indicated p<0.01, 0.001 and 0.0001, respectively. ns, not significant.
To determine whether fascin regulates PFKs through YAP1, we expressed YAP1-6SA, a constitutively active mutant of YAP1, in control or fascin KO cells. As shown in Fig. 4E, YAP1-6SA increased the protein levels of PFKFB3, PFKM, and PFKP in control cells and restored the expression of PFKFB3 in KO cells to levels comparable to that in control-6SA cells (Fig. 4E). However, the protein levels of PFKM and PFKP in KO1-6SA cells were still considerably lower than that in control-6SA cells (Fig. 4E). These data suggest that fascin upregulates PFKFB3 expression through YAP1, but the upregulation of PFKM and PFKP by fascin likely also involves a YAP1-independent mechanism.
YAP1 regulates the transcription of its target genes by forming a complex with TEAD transcription factors [36]. Our survey of the JASPAR database [38] indicated the existence of multiple TEAD-binding motifs in the proximal promoter region of both murine and human PFKFB3 gene. To determine whether YAP1 directly activates the transcription of PFKFB3, we constructed several luciferase reporters containing different promoter regions from the Pfkfb3 gene (Fig. 4F). As shown in Fig. 4G, the ectopic expression of YAP1–6SA robustly activated the transcription of both the 1 kb and 500 bp reporter, but not that of the 350 bp construct (Fig. 4G). This led us to further examine a single TEAD1/4 binding motif located 30 bp upstream of the transcription start site (TSS). Mutation of this motif from AAAGAATGCG to AAAGAGAGCG completely abrogated the ability of YAP1–6SA to activate luciferase transcription in the 500 bp reporter (Fig. 4H). The −30 bp TEAD1/4 binding motif is conserved between humans and mice (AAGGAATGCG in humans). To determine whether fascin promotes the binding of YAP1 to this TEAD1/4 binding motif, we used ChIP assay to examine the binding of YAP1 to this site in control and KO cells. As shown in Fig. 4I–4J, fascin KO decreased the binding of YAP1 to this site in the promoter of both murine and human PFKFB3 genes. In contrast, the binding of ectopically expressed YAP1–6SA mutant was not affected by fascin KO (Fig. 4J). Conversely binding of YAP1 to the PFKFB3 promoter increased more than 3-fold when fascin was overexpressed in H1650 cells (Fig. 4K). Taken together, our data suggest that PFKFB3 is a direct YAP1/TEAD target gene and that fascin activates the transcription of PFKFB3 through a YAP1/TEAD-binding site in its promoter.
3.4. Fascin upregulation correlates with YAP1 activation and PFKFB3 overexpression in cancer patients across different types of cancer
We previously examined the expression of fascin and YAP1 through IHC staining in the same cohort of 113 patients with lung adenocarcinoma in two separate studies [7, 22]. A review of these staining results indicated that patients with lung cancer with high expression levels of fascin tended to have strong nuclear staining of YAP1 (r = 0.497, p < 0.001, n = 113, Spearman’s rank correlation test) (Fig. 5A–5B), which indicated the activation of YAP1 by fascin in patients with lung cancer. To further evaluate the role of fascin in regulating the YAP1-PFKFB3 circuit to promote glycolysis in patients with cancer, we examined the correlation between multiple PFK1 and PFK2 genes and two well established YAP1 target genes (CTGF and CYR61) in the TCGA RNA sequencing database (Fig. 5C–5D, Fig. S4, and Table S1). Fascin mRNA levels were significantly correlated with PFKFB3 and PFKP in lung cancer (LUAD, lung adenocarcinoma and LUSC, lung squamous cell carcinoma) and many other types of cancer, most notably in liver hepatocellular carcinoma (LIHC), colon adenocarcinoma (COAD), and rectum adenocarcinoma (Fig. 5C, Fig. S4A–S4B and Table S1). Significant correlation between fascin and YAP1 target genes were also observed in LUAD, READ, LIHC, and COAD (but not in LUSC), among other cancers (Fig. 5D, Fig. S4C–S4D; Table S1). In the cancer cell lines used in this study, the overall expression levels of PFKFB3 and YAP1 (but not TAZ) also appeared to correlate with the levels of fascin (Fig. S4E). Taken together, our data indicate that fascin may promote glycolysis and activate YAP1 signaling through a conserved mechanism across different cancers.
Figure 5. Fascin expression correlates with PFKs and YAP1 target genes in cancer patients.
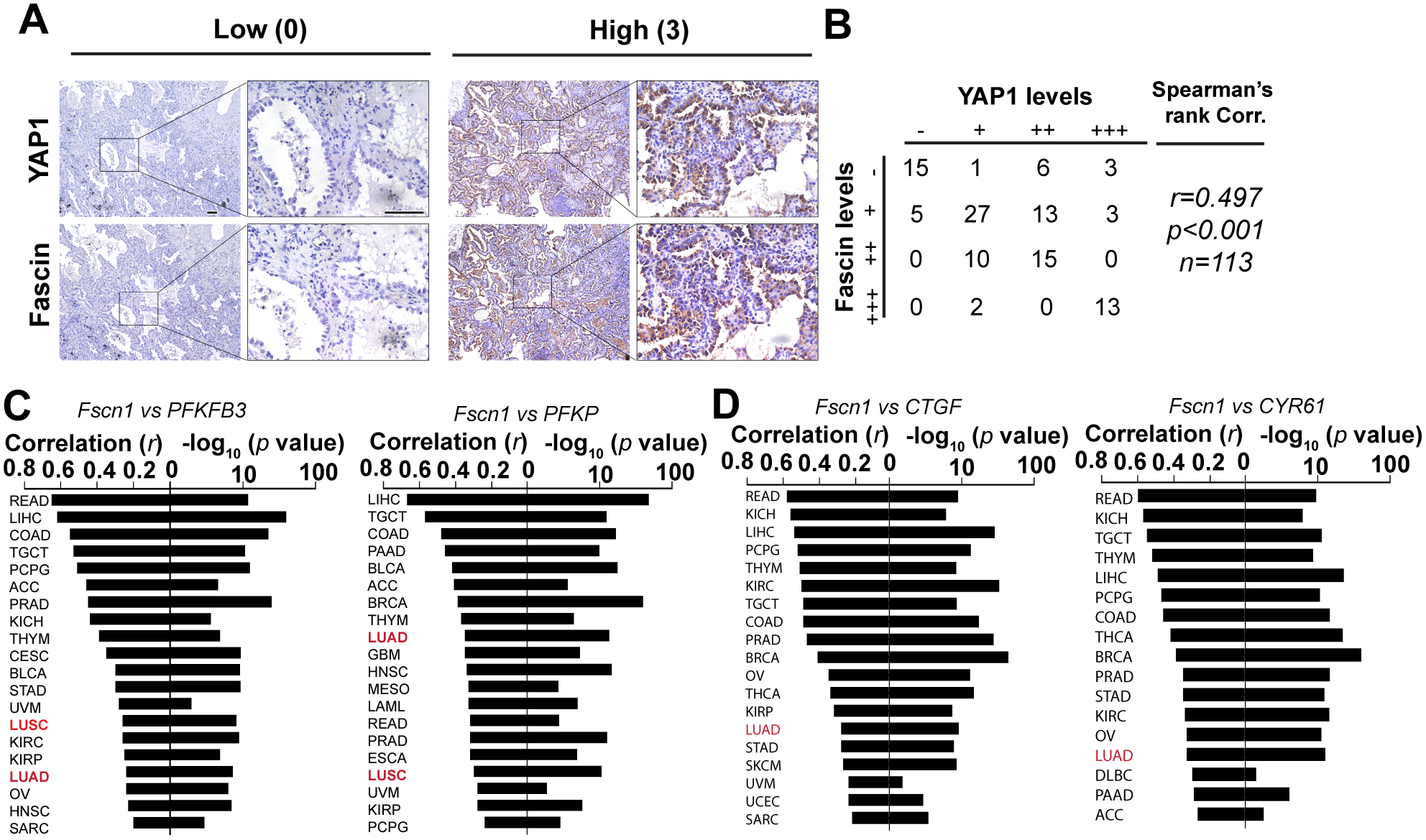
A, representative immunohistochemistry staining images showing expression levels of YAP1 and fascin in lung adenocarcinoma patients.
B, correlation between fascin and YAP1 staining intensities in a cohort of 113 lung adenocarcinoma patient.
C, waterfall plot showing the correlation between fascin and PFKFB3 or PFKP in different cancers from the TCGA RNA sequencing database.
D, waterfall plot showing the correlation of fascin and YAP1 target genes (CTGF and CYR61) in different cancers from the TCGA RNA sequencing database.
Correlation coefficient (r) and p value in B–D were determined using Spearman’s rank test.
PFKFB3
3.5. PFKFB3 is required for fascin-mediated promotion of lung cancer growth and metastasis
To understand the glycolytic functions of fascin in lung cancer metastasis, we knocked down PFKFB3 in control or fascin OE H1650 cells. Luciferase-labeled control or fascin OE cells expressing control shRNA or PFKFB3 shRNA were orthotopically injected into the left lung of nude mice. Tumor growth was monitored weekly by bioluminescence imaging (BLI) (Fig. 6A and 6B). Thirty days after implantation, the mice were euthanized and the development of metastasis was evaluated through ex vivo BLI imaging (Fig. 6C–6H). Fascin OE in control shRNA H1650 cells increased the BLI signal by more than 2-fold (Fig. 6A and 6B). Knockdown of PFKFB3 inhibited the BLI signal by more than 95%, and fascin OE did not increase the BLI signal in the PFKFB3 shRNA group (Fig. 6A–6B). Ex vivo BLI imaging further confirmed that PFKFB3 was required for fascin to promote H1650 tumor growth and metastasis (Fig. 6C–6H). Fascin overexpression increased the BLI signals of the primary tumor (left lung) (Fig. 6C–6D) and metastasis to the contralateral lung (right lung) (Fig. 6E–6F) by approximately 3- and 6-fold, respectively. PFKFB3 KD decreased ex vivo BLI signals in both lungs by more than 99% when compared to the control shRNA group, and ectopic expression of fascin failed to increase BLI signals in either primary tumor or contralateral lung metastases in H1650 tumors expressing PFKFB3 shRNA (Fig. 6C–6F). We also observed chest wall metastases in 100% or 50% of the control shRNA H1650 group with or without fascin overexpression, respectively (Fig. 6G and 6H). No chest wall metastases were observed in any of the mice in the two groups expressing PFKFB3 shRNA (Fig. 6H). Many of these chest wall metastases, especially those in the fascin OE group, were visible to the naked eye (Fig. S5A, yellow arrow), and was confirmed by hematoxylin and eosin (H&E staining; Fig. S5B).
Figure 6. PFKFB3 is required for fascin to promote tumor growth and metastasis in lung cancer.
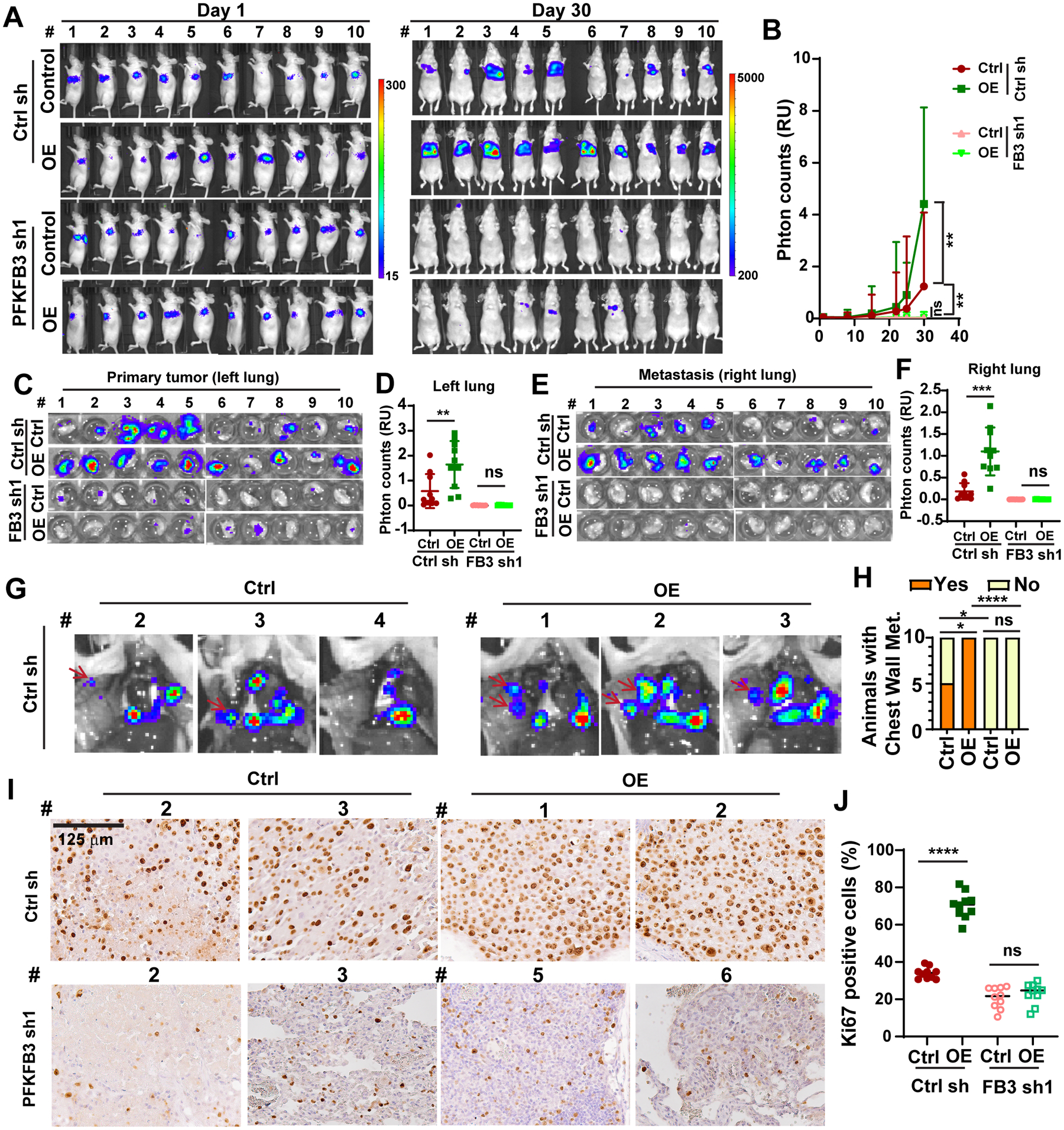
A and B, the effects of fascin overexpression (OE) and / or PFKFB3 knockdown on orthotopic lung cancer growth and metastasis. A, BLI images of all the mice on day 1 and day 30 after orthotopic injection. B, quantitation of BLI imaging data from orthotopic experiment in A. n=10 female nude mice per group.
C–F, ex vivo bioluminescence imaging of extracted left lungs (C and D) or right lungs (E and F). D and F are quantitation of imaging data in C and E, respectively.
G, representative ex vivo bioluminescence imaging of the opened chest (G) showing metastases lesion on the chest wall (red arrow) in control shRNA group mice with or without fascin overexpression.
H, summary of the number of mice in each group with chest wall metastases. n = 10 mice per group.
I and J, representative Ki67 immunohistochemistry staining images (I) and quantitation of Ki67 positive cells (J) showing that fascin overexpression increased the proportion of Ki67 positive cells, which was abrogated by the shRNA knockdown of PFKFB3. n = 10 mice per group.
Data in B were analyzed using two-tailed two-way ANOVA. D, F, and J were analyzed by two-tailed, two-sample Mann Whitney test. Data in H was analyzed using Chi-square test. *, **, *** and **** indicated p<0.05, 0.01, 0.001 and 0.0001, respectively. ns, not significant.
We and others have previously reported that patients with NSCLC with elevated levels of fascin also had larger tumor size and higher percentage of Ki67 staining, a marker for proliferating cells [7, 39]. Therefore, we evaluated the effect of fascin overexpression and PFKFB3 KD on Ki67 staining in the orthotopic xenograft tumors. As shown in Fig. 6I and 6J, fascin overexpression increased Ki67 positive cells from 34% to 70%. PFKFB3 KD decreased the number of Ki67 positive cells to 21%, and fascin OE in PFKFB3 shRNA tumors did not increase the proportion of proliferating cells in the orthotopic tumors. Taken together, our data support that PFKFB3 is required for fascin to promote lung cancer growth and metastasis.
3.6. Pharmacological inhibition of fascin may be used to suppress YAP1-PFKFB3 signaling and glycolysis in NSCLC
As fascin-mediated upregulation of YAP1 and PFKFB3 depends on actin-bundling activity, we reasoned that pharmacological inhibitors of fascin such as G2 may be used to suppress glycolysis in lung cancer. As shown in Fig. 7A and 7B, G2 markedly reduced the levels of PFKFB3 and YAP1 in lung cancer cells at 20–40 μM. G2 treatment had no effect on the protein levels of either PFKFB3 or YAP1 in fascin KO H1650 cells, which indicated that the G2 effect was specifically due to its inhibition of fascin (Fig. S6A). As fascin expression strongly correlated with PFKFB3 and YAP1 target genes in liver and colorectal cancers in the TCGA database, we also examined the effect of G2 in these cells. As shown in Fig. 7C, G2 treatment robustly suppressed the expression of YAP1 and PFKFB3 in liver and colorectal cancer cells. G2 treatment also inhibited both ECAR and OCR in H1650 cells (Fig. 7D–7G), which recapitulated the effects of fascin KO or KD in these cells.
Figure 7. Pharmacological inhibition of fascin could be employed to suppress YAP1 and PFKFB3 in lung cancer.
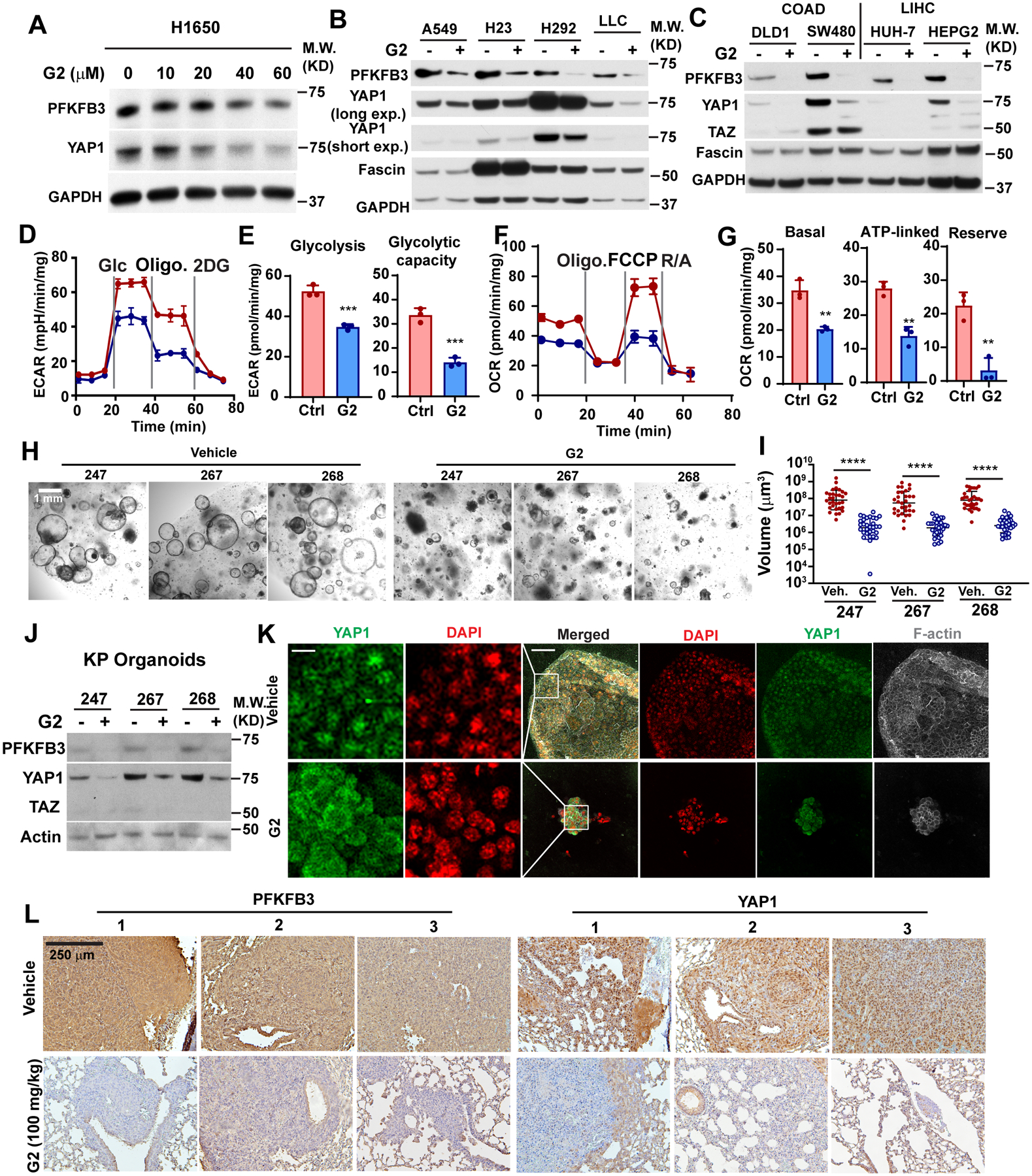
A–C, Western blotting showing the effect of G2 treatment on the protein levels of PFKFB3 and YAP1 protein in NSCLC cells (A and B) or liver and colorectal cancer cells (C).
D–G, the effect of G2 treatment on ECAR (D and E) and OCR (F and G) in H1650 cells. E and G are quantitation of ECAR and OCR measurement in D and F, respectively.
H and I, representative bright field images of tumoroids (H) and quantitation of tumoroid size (I) showing the effects of G2 treatment on tumoroid growth. Tumoroids were derived from lung tumors extracted from 3 KP mice and treated with vehicle or 40 μM G2 for 48 hours before imaging. 30 organoids were measured for each group.
J, western blotting showing the effect of G2 treatment (40 μM, 48 h) treatment with on PFKFB3 and YAP1 protein levels in KP organoids.
K, representative immunofluorescence images showing YAP1 subcellular localization in tumoroids treated with G2 or vehicle control.
L, representative immunohistochemistry staining images showing expression levels of PFKFB3 and YAP1 in LLC tumors in mice treated with vehicle or G2 (100mg/kg daily via i.p.).
Data in E and G were analyzed by two-tailed, two-sample unpaired Student’s test. Data in I were analyzed by Mann Whitney test. Representative results from at least three independent experiments are shown (A-K). *, **, *** and **** indicated p<0.05, 0.01, 0.001 and 0.0001, respectively.
Patient and genetically engineered mouse model (GEMM)-derived organoid cultures recapitulate the characteristics of tumors they were derived from and can be used to predict the response to cancer therapies [40]. The KrasLSL-G12D; p53fl/fl (KP) model faithfully recapitulates the characteristics of LUAD in humans [23]. Therefore, we tested the effect of G2 treatment on the growth of tumoroid cultures prepared from the KP lung cancer model. The formation of lung cancer in KP mice was induced by intratracheal administration of adenoviral Cre-recombinase [23]. Three months following the viral induction, lung cancer tissues were harvested to prepare the tumoroids. G2 treatment markedly reduced the tumoroid volume by 40- to 50-fold (Fig. 7H and 7I) and suppressed the protein levels of PFKFB3 and YAP1 in the tumoroid cultures in all three mice (Fig. 7J). Immunofluorescent staining revealed that G2 treatment induced the translocation of YAP1 from the nucleus to the cytosol in the KP tumoroids (Fig. 7K).
We previously used G2 to treat nude mice in a tail-vein LLC metastasis model [7]. G2 treatment significantly reduced the tumor burden in the lungs of mice injected with LLC cells via the tail vein (Fig. S6B) and improved the survival of these animals (Fig. S6C). IHC staining of tumor tissues harvested from these mice also showed that the LLC tumors from G2-treated mice showed a marked decrease in both YAP1 and PFKFB3 staining (Fig. 7L). Intriguingly, the levels of YAP1 and PFKFB3 in adjacent normal lung tissue appeared to be unaffected, which indicated that G2 can be used to specifically suppress YAP1 signaling and glycolysis in cancer cells with fascin upregulation. Taken together, our data indicated that pharmacological inhibitors of fascin can be used to suppress glycolytic metabolism, tumor growth, and metastasis in lung cancer, and potentially in other types of cancers with high levels of fascin expression.
4. Discussion
Fascin is one of the most frequently upregulated actin-binding proteins in metastatic cancers [1, 2]. The overexpression of fascin in metastatic cancer is a result of inflammatory cytokines in the tumor microenvironment, epithelial-to-mesenchymal transition program, and environmental stresses such as hypoxia and nutrient limitation [1, 41–44]. Although it is believed that fascin promotes tumor progression by increasing tumor cell migration and invasion, fascin expression in NSCLC is correlated with larger tumor size and increased Ki67 staining [7, 39]. Inducible deletion of fascin in lung cancer cells prevents lung colonization by disseminated cells [7]. These findings suggest a role for fascin in regulating cancer cell proliferation and metastatic colonization. Our data, together with our earlier report [7], support a metabolic role of fascin in lung cancer and possibly other types of cancers. By remodeling mitochondrial actin filaments and activating the YAP1-PFKFB3 signaling circuit, fascin augments both mitochondrial OXPHOS and glycolysis to increase metabolic stress resistance and cell proliferation under glucose-limited conditions. It is possible that fascin promotes lung cancer growth and metastatic colonization by increasing metabolic flexibility. Consistent with this notion, fascin overexpression increased the proportion of Ki67 positive cells in an orthotopic tumor model, and fascin-mediated proliferation was abrogated by shRNA-mediated depletion of PFKFB3. Depletion of PFKFB3 also abrogated fascin-mediated tumor growth and metastasis in an orthotopic lung cancer xenograft model.
Remodeling of the actin cytoskeleton in mammalian cells is energetically costly. It is estimated that half of the ATP generated in cells is consumed by actin cytoskeleton remodeling [45]. Many glycolytic enzymes are well-known actin-binding proteins [46]. It is conceivable that dysregulation of the actin cytoskeleton in metastatic cancer cells may affect bioenergetics and cancer metabolism. Consistent with this notion, insulin activates glycolysis by mobilizing the release and activation of actin filament-bound aldolase [20]. Our data demonstrate that fascin promotes glycolysis in NSCLC by transcriptional activation of PFKFB3. We further demonstrated that PFKFB3 is a direct target gene of the YAP1/TEAD complex and fascin overexpression in NSCLC promotes the binding of YAP1 to a conserved TEAD1/4 binding site on the PFKFB3 promoter. Fascin regulates YAP1 or TAZ activation in lung cancer and melanoma cells independent of bundling activity and through direct interaction with the Hippo pathway kinase MST1/2 [47, 48]. Our data demonstrate that the actin-bundling activity is required for fascin to activate the YAP1-PFKFB3 axis, as the actin-bundling defective mutants of fascin were unable to increase the expression of YAP1 or PFKFB3, nor promote glycolysis in lung cancer cells. It is well established that YAP/TAZ activity is regulated by mechanical tension and actomyosin contractility of the actin cytoskeleton [49]. In addition to its well-known role in filopodia formation, fascin has also been reported to regulate focal adhesion dynamics and stress fiber organization [50]. It will be interesting to determine whether fascin controls YAP1-PFKFB3 signaling by modulating focal adhesion turnover and integrin signaling.
There is considerable interest in the development of anti-metastatic therapies targeting fascin. Several small-molecule inhibitors of fascin bundling have been developed [10, 12, 13, 17]. These inhibitors may be used to inhibit tumor growth and metastatic colonization and to reprogram cancer metabolism in lung cancer and potentially other cancers with fascin upregulation.
Supplementary Material
Acknowledgment
We would like to thank Dr. Katherine Aird for assistance with the use of the YSI 2700 Biochemical Analyzer. The research in Shengyu Yang’s lab is supported by grants from the National Institute of Health (R01CA233844), Elsa U. Pardee Foundation, and a bridge grant from the Penn State Cancer Institute.
Footnotes
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have influenced the work reported in this paper.
References
- [1].Lin S, Taylor MD, Singh PK, Yang S, How Does Fascin Promote Cancer Metastasis?, Febs J, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Liu H, Zhang Y, Li L, Cao J, Guo Y, Wu Y, Gao W, Fascin actin-bundling protein 1 in human cancer: promising biomarker or therapeutic target?, Mol Ther Oncolytics, 20 (2021) 240–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hashimoto Y, Skacel M, Adams JC, Roles of fascin in human carcinoma motility and signaling: prospects for a novel biomarker?, Int J Biochem Cell Biol, 37 (2005) 1787–1804. [DOI] [PubMed] [Google Scholar]
- [4].Lin S, Lu S, Mulaj M, Fang B, Keeley T, Wan L, Hao J, Muschol M, Sun J, Yang S, Monoubiquitination Inhibits the Actin Bundling Activity of Fascin, J Biol Chem, 291 (2016) 27323–27333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Machesky LM, Li A, Fascin: Invasive filopodia promoting metastasis, Commun Integr Biol, 3 (2010) 263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Barnawi R, Al-Khaldi S, Majed Sleiman G, Sarkar A, Al-Dhfyan A, Al-Mohanna F, Ghebeh H, Al-Alwan M, Fascin is Critical for the Maintenance of Breast Cancer Stem Cell Pool Predominantly via the Activation of the Notch Self-Renewal Pathway, Stem cells, (2016). [DOI] [PubMed] [Google Scholar]
- [7].Lin S, Huang C, Gunda V, Sun J, Chellappan SP, Li Z, Izumi V, Fang B, Koomen J, Singh PK, Hao J, Yang S, Fascin Controls Metastatic Colonization and Mitochondrial Oxidative Phosphorylation by Remodeling Mitochondrial Actin Filaments, Cell reports, 28 (2019) 2824–2836 e2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ghebeh H, Al-Khaldi S, Olabi S, Al-Dhfyan A, Al-Mohanna F, Barnawi R, Tulbah A, Al-Tweigeri T, Ajarim D, Al-Alwan M, Fascin is involved in the chemotherapeutic resistance of breast cancer cells predominantly via the PI3K/Akt pathway, Br J Cancer, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Valastyan S, Weinberg RA, Tumor metastasis: molecular insights and evolving paradigms, Cell, 147 (2011) 275–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Han S, Huang J, Liu B, Xing B, Bordeleau F, Reinhart-King CA, Li W, Zhang JJ, Huang XY, Improving fascin inhibitors to block tumor cell migration and metastasis, Mol Oncol, 10 (2016) 966–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wang Y, Zhang JJ, Huang XY, Anti-Metastasis Fascin Inhibitors Decrease the Growth of Specific Subtypes of Cancers, Cancers (Basel), 12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Francis S, Croft D, Schuttelkopf AW, Parry C, Pugliese A, Cameron K, Claydon S, Drysdale M, Gardner C, Gohlke A, Goodwin G, Gray CH, Konczal J, McDonald L, Mezna M, Pannifer A, Paul NR, Machesky L, McKinnon H, Bower J, Structure-based design, synthesis and biological evaluation of a novel series of isoquinolone and pyrazolo[4,3-c]pyridine inhibitors of fascin 1 as potential anti-metastatic agents, Bioorg Med Chem Lett, 29 (2019) 1023–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Huang FK, Han S, Xing B, Huang J, Liu B, Bordeleau F, Reinhart-King CA, Zhang JJ, Huang XY, Targeted inhibition of fascin function blocks tumour invasion and metastatic colonization, Nature communications, 6 (2015) 7465. [DOI] [PubMed] [Google Scholar]
- [14].Yamakita Y, Matsumura F, Yamashiro S, Fascin1 is dispensable for mouse development but is favorable for neonatal survival, Cell motility and the cytoskeleton, 66 (2009) 524–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Chen L, Yang S, Jakoncic J, Zhang JJ, Huang XY, Migrastatin analogues target fascin to block tumour metastasis, Nature, 464 (2010) 1062–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Huang F-K, Han S, Xing B, Huang J, Liu B, Bordeleau F, Reinhart-King CA, Zhang JJ, Huang X-Y, Targeted inhibition of fascin function blocks tumour invasion and metastatic colonization, Nature communications, 6 (2015) 7465. [DOI] [PubMed] [Google Scholar]
- [17].Alburquerque-Gonzalez B, Bernabe-Garcia A, Bernabe-Garcia M, Ruiz-Sanz J, Lopez-Calderon FF, Gonnelli L, Banci L, Pena-Garcia J, Luque I, Nicolas FJ, Cayuela-Fuentes ML, Luchinat E, Perez-Sanchez H, Montoro-Garcia S, Conesa-Zamora P, The FDA-Approved Antiviral Raltegravir Inhibits Fascin1-Dependent Invasion of Colorectal Tumor Cells In Vitro and In Vivo, Cancers (Basel), 13 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Schild T, Low V, Blenis J, Gomes AP, Unique Metabolic Adaptations Dictate Distal Organ-Specific Metastatic Colonization, Cancer Cell, 33 (2018) 347–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lunt SY, Vander Heiden MG, Aerobic glycolysis: meeting the metabolic requirements of cell proliferation, Annu Rev Cell Dev Biol, 27 (2011) 441–464. [DOI] [PubMed] [Google Scholar]
- [20].Hu H, Juvekar A, Lyssiotis CA, Lien EC, Albeck JG, Oh D, Varma G, Hung YP, Ullas S, Lauring J, Seth P, Lundquist MR, Tolan DR, Grant AK, Needleman DJ, Asara JM, Cantley LC, Wulf GM, Phosphoinositide 3-Kinase Regulates Glycolysis through Mobilization of Aldolase from the Actin Cytoskeleton, Cell, 164 (2016) 433–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Elgendy M, Ciro M, Hosseini A, Weiszmann J, Mazzarella L, Ferrari E, Cazzoli R, Curigliano G, DeCensi A, Bonanni B, Budillon A, Pelicci PG, Janssens V, Ogris M, Baccarini M, Lanfrancone L, Weckwerth W, Foiani M, Minucci S, Combination of Hypoglycemia and Metformin Impairs Tumor Metabolic Plasticity and Growth by Modulating the PP2A-GSK3beta-MCL-1 Axis, Cancer Cell, 35 (2019) 798–815 e795. [DOI] [PubMed] [Google Scholar]
- [22].Lin S, Huang C, Sun J, Bollt O, Wang X, Martine E, Kang J, Taylor MD, Fang B, Singh PK, Koomen J, Hao J, Yang S, The mitochondrial deoxyguanosine kinase is required for cancer cell stemness in lung adenocarcinoma, EMBO molecular medicine, 11 (2019) e10849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Jackson EL, Olive KP, Tuveson DA, Bronson R, Crowley D, Brown M, Jacks T, The differential effects of mutant p53 alleles on advanced murine lung cancer, Cancer Res, 65 (2005) 10280–10288. [DOI] [PubMed] [Google Scholar]
- [24].DuPage M, Dooley AL, Jacks T, Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase, Nature protocols, 4 (2009) 1064–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Yang S, Zhang JJ, Huang XY, Mouse models for tumor metastasis, Methods Mol Biol, 928 (2012) 221–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sun J, Lu F, He H, Shen J, Messina J, Mathew R, Wang D, Sarnaik AA, Chang WC, Kim M, Cheng H, Yang S, STIM1- and Orai1-mediated Ca2+ oscillation orchestrates invadopodium formation and melanoma invasion, J Cell Biol, 207 (2014) 535–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z, GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses, Nucleic acids research, 45 (2017) W98–W102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Gunda V, Yu F, Singh PK, Validation of metabolic alterations in microscale cell culture lysates using hydrophilic interaction liquid chromatography (HILIC)-tandem mass spectrometry-based metabolomics, PloS one, 11 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Yuan M, Breitkopf SB, Yang X, Asara JM, A positive/negative ion–switching, targeted mass spectrometry–based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue, Nature protocols, 7 (2012) 872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Trefely S, Ashwell P, Snyder NW, FluxFix: automatic isotopologue normalization for metabolic tracer analysis, BMC Bioinformatics, 17 (2016) 485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Vander Heiden MG, Cantley LC, Thompson CB, Understanding the Warburg effect: the metabolic requirements of cell proliferation, Science, 324 (2009) 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Yang S, Huang FK, Huang J, Chen S, Jakoncic J, Leo-Macias A, Diaz-Avalos R, Chen L, Zhang JJ, Huang XY, Molecular mechanism of fascin function in filopodial formation, J Biol Chem, 288 (2013) 274–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Korobova F, Ramabhadran V, Higgs HN, An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2, Science, 339 (2013) 464–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ashizawa K, Willingham MC, Liang CM, Cheng SY, In vivo regulation of monomer-tetramer conversion of pyruvate kinase subtype M2 by glucose is mediated via fructose 1,6-bisphosphate, J Biol Chem, 266 (1991) 16842–16846. [PubMed] [Google Scholar]
- [35].Park JS, Burckhardt CJ, Lazcano R, Solis LM, Isogai T, Li L, Chen CS, Gao B, Minna JD, Bachoo R, DeBerardinis RJ, Danuser G, Mechanical regulation of glycolysis via cytoskeleton architecture, Nature, 578 (2020) 621–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Koo JH, Guan KL, Interplay between YAP/TAZ and Metabolism, Cell Metab, 28 (2018) 196–206. [DOI] [PubMed] [Google Scholar]
- [37].Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, Elvassore N, Piccolo S, Role of YAP/TAZ in mechanotransduction, Nature, 474 (2011) 179–183. [DOI] [PubMed] [Google Scholar]
- [38].Fornes O, Castro-Mondragon JA, Khan A, van der Lee R, Zhang X, Richmond PA, Modi BP, Correard S, Gheorghe M, Baranasic D, Santana-Garcia W, Tan G, Cheneby J, Ballester B, Parcy F, Sandelin A, Lenhard B, Wasserman WW, Mathelier A, JASPAR 2020: update of the open-access database of transcription factor binding profiles, Nucleic acids research, 48 (2020) D87–D92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Pelosi G, Pastorino U, Pasini F, Maissoneuve P, Fraggetta F, Iannucci A, Sonzogni A, De Manzoni G, Terzi A, Durante E, Bresaola E, Pezzella F, Viale G, Independent prognostic value of fascin immunoreactivity in stage I nonsmall cell lung cancer, British journal of cancer, 88 (2003) 537–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Tiriac H, Belleau P, Engle DD, Plenker D, Deschenes A, Somerville TDD, Froeling FEM, Burkhart RA, Denroche RE, Jang GH, Miyabayashi K, Young CM, Patel H, Ma M, LaComb JF, Palmaira RLD, Javed AA, Huynh JC, Johnson M, Arora K, Robine N, Shah M, Sanghvi R, Goetz AB, Lowder CY, Martello L, Driehuis E, LeComte N, Askan G, Iacobuzio-Donahue CA, Clevers H, Wood LD, Hruban RH, Thompson E, Aguirre AJ, Wolpin BM, Sasson A, Kim J, Wu M, Bucobo JC, Allen P, Sejpal DV, Nealon W, Sullivan JD, Winter JM, Gimotty PA, Grem JL, DiMaio DJ, Buscaglia JM, Grandgenett PM, Brody JR, Hollingsworth MA, O’Kane GM, Notta F, Kim E, Crawford JM, Devoe C, Ocean A, Wolfgang CL, Yu KH, Li E, Vakoc CR, Hubert B, Fischer SE, Wilson JM, Moffitt R, Knox J, Krasnitz A, Gallinger S, Tuveson DA, Organoid Profiling Identifies Common Responders to Chemotherapy in Pancreatic Cancer, Cancer Discov, 8 (2018) 1112–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Li A, Morton JP, Ma Y, Karim SA, Zhou Y, Faller WJ, Woodham EF, Morris HT, Stevenson RP, Juin A, Jamieson NB, MacKay CJ, Carter CR, Leung HY, Yamashiro S, Blyth K, Sansom OJ, Machesky LM, Fascin is regulated by slug, promotes progression of pancreatic cancer in mice, and is associated with patient outcomes, Gastroenterology, 146 (2014) 1386–1396 e1381–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zhao X, Gao S, Ren H, Sun W, Zhang H, Sun J, Yang S, Hao J, Hypoxia-inducible factor-1 promotes pancreatic ductal adenocarcinoma invasion and metastasis by activating transcription of the actin-bundling protein fascin, Cancer Res, 74 (2014) 2455–2464. [DOI] [PubMed] [Google Scholar]
- [43].Sun J, He H, Pillai S, Xiong Y, Challa S, Xu L, Chellappan S, Yang S, GATA3 transcription factor abrogates Smad4-mediated fascin overexpression, invadopodium formation and breast cancer cell invasion, J Biol Chem, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Sun J, He H, Xiong Y, Lu S, Shen J, Cheng A, Chang WC, Hou MF, Lancaster JM, Kim M, Yang S, Fascin protein is critical for transforming growth factor beta protein-induced invasion and filopodia formation in spindle-shaped tumor cells, J Biol Chem, 286 (2011) 38865–38875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Bernstein BW, Bamburg JR, Actin-ATP hydrolysis is a major energy drain for neurons, J Neurosci, 23 (2003) 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Masters C, Interactions between glycolytic enzymes and components of the cytomatrix, J Cell Biol, 99 (1984) 222s–225s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kang J, Wang J, Yao Z, Hu Y, Ma S, Fan Q, Gao F, Sun Y, Sun J, Fascin induces melanoma tumorigenesis and stemness through regulating the Hippo pathway, Cell Commun Signal, 16 (2018) 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Liang Z, Wang Y, Shen Z, Teng X, Li X, Li C, Wu W, Zhou Z, Wang Z, Fascin 1 promoted the growth and migration of non-small cell lung cancer cells by activating YAP/TEAD signaling, Tumour Biol, 37 (2016) 10909–10915. [DOI] [PubMed] [Google Scholar]
- [49].Totaro A, Panciera T, Piccolo S, YAP/TAZ upstream signals and downstream responses, Nat Cell Biol, 20 (2018) 888–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Elkhatib N, Neu MB, Zensen C, Schmoller KM, Louvard D, Bausch AR, Betz T, Vignjevic DM, Fascin plays a role in stress fiber organization and focal adhesion disassembly, Curr Biol, 24 (2014) 1492–1499. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.