

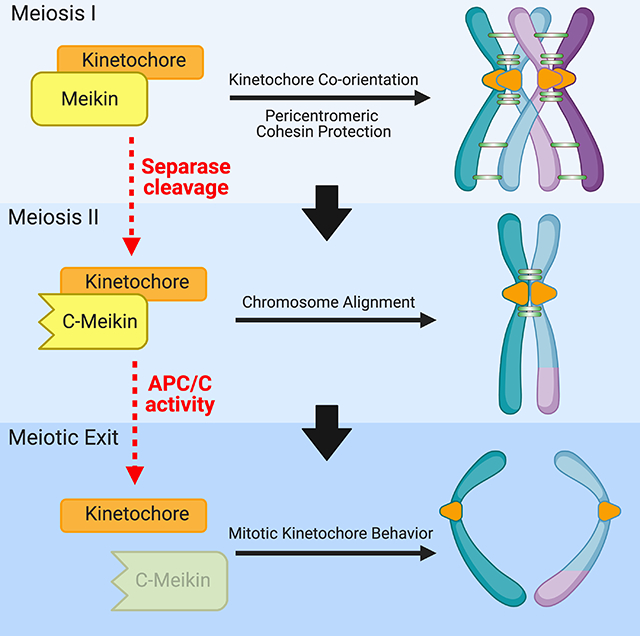
Summary
To generate haploid gametes, germ cells undergo two consecutive meiotic divisions requiring key changes to the cell division machinery. Here, we demonstrate that the protease Separase rewires key cell division processes at the meiosis I/II transition by cleaving the meiosis-specific protein Meikin. Separase proteolysis does not inactivate Meikin but instead alters its function to create a distinct activity state. Full-length Meikin and the C-terminal Meikin Separase-cleavage product both localize to kinetochores, bind to Plk1 kinase, and promote Rec8 cleavage, but our results reveal distinct roles for these proteins in controlling meiosis. Mutations that prevent Meikin cleavage or that conditionally inactivate Meikin at anaphase I result in defective meiosis II chromosome alignment in mouse oocytes. Finally, as oocytes exit meiosis, C-Meikin is eliminated by APC/C-mediated degradation prior to the first mitotic division. Thus, multiple regulatory events irreversibly modulate Meikin activity during successive meiotic divisions to rewire the cell division machinery at two distinct transitions.
Keywords: Meiosis, Kinetochore, Separase, Plk1, Meikin, Cohesin
eTOC Blurb
Meiosis requires the dramatic rewiring of the chromosome segregation machinery at each division. Maier et al find that the critical meiosis-specific kinetochore protein, Meikin, is regulated at key meiotic transitions through Separase-mediated proteolytic cleavage and ubiquitin-mediated degradation, to enable distinct activities during the two meiotic divisions.
Graphical Abstract
Introduction
The generation of haploid germ cells from diploid precursors requires a specialized cell division, termed meiosis, in which two successive rounds of chromosome segregation occur without undergoing intervening DNA replication (Miller et al., 2013). In most organisms, the first round of chromosome segregation during meiosis I reduces the chromosome number by half, requiring three major modifications relative to the mitotic cell division program. First, homologous chromosomes pair and become physically linked through synapsis and recombination. Homolog synapsis and recombination require multiple meiosis-specific proteins, including components of the synaptonemal complex (Cahoon and Hawley, 2016) and the Spo11 endonuclease (Keeney et al., 1997). Second, meiosis requires that sister chromatid cohesion is eliminated in a stepwise manner. Cohesin is a ring-shaped protein complex that acts in mitotic cells to maintain the physical linkage between replicated sister chromatids (Gruber et al., 2003). At anaphase onset, activation of the Anaphase Promoting Complex/Cyclosome (APC/C) E3 ubiquitin ligase results in the activation of the protease Separase, which cleaves the kleisin subunit of cohesin allowing chromosome separation (Kamenz and Hauf, 2017). Cohesin removal is spatially and temporally controlled in a differential manner in the two meiotic divisions. During anaphase I, cohesin on the chromosome arms is removed to allow homolog separation, whereas cohesin complexes at pericentromeric regions are critical for holding sister chromatids together and must be retained until anaphase II (Marston and Amon, 2004). Third, instead of binding to opposing spindle poles as they do in mitosis, the kinetochores of sister chromatids co-orient, fusing into a single microtubule-binding entity, to connect to microtubules from the same spindle pole during meiosis I (Chiang et al., 2010; Goldstein, 1981; Li and Dawe, 2009; Sarangapani et al., 2014).
The meiosis-specific proteins and activities that contribute to protecting pericentromeric cohesin and promoting sister kinetochore co-orientation to enable this alternative division strategy remain important areas of investigation. To achieve these behaviors, meiotic cells establish opposing gradients of kinase and phosphatase activity across the meiosis I chromosome. To mediate pericentromeric cohesion retention during meiosis I, proteins of the Shugoshin/Mei-S332 (Sgo) family localize to pericentromeric regions and bind to Protein Phosphatase 2A (PP2A) (Marston, 2015). The localized activity of PP2A opposes the activity of cohesin kinases by dephosphorylating the meiosis-specific cohesin subunit Rec8, protecting this pericentromeric cohesin population from Separase cleavage during anaphase I (Brar et al., 2006; Ishiguro et al., 2010; Katis et al., 2010; Kudo et al., 2009). In many organisms with regional centromeres, Rec8 containing-cohesin also appears to facilitate sister kinetochore co-orientation (Chelysheva et al., 2005; Chiang et al., 2010; Sakuno et al., 2009; Severson et al., 2009; Watanabe and Nurse, 1999).
As germ cells transition from meiosis I to meiosis II, meiosis I-specific activities must be reversed to allow for sister chromatid segregation during the equational second meiotic division. Sister kinetochore co-orientation must be eliminated such that kinetochores can bi-orient on the meiosis II spindle, and the protection of peri-centromeric cohesion must be relieved to allow for anaphase II chromatid separation. However, the limited time between the two meiotic stages in many organisms (Kishimoto, 2003) restricts the ability of transcriptional or translational control to broadly rewire the cell division apparatus to distinguish these events. Finally, the completion of meiosis is followed by fertilization, and the first mitotic division in the developing embryo requires additional changes to the regulation of the division machinery. How meiotic cells coordinate these rapid and substantial modifications to the cell cycle machinery is a critical unanswered question for our understanding of meiosis.
The vertebrate factor Meikin (meiosis-specific kinetochore protein) plays important roles in meiosis I sister kinetochore co-orientation and the protection of centromeric cohesion (Kim et al., 2015a), with Meikin-null mice exhibiting meiotic defects and sterility. Current functional evidence suggests that Meikin plays an analogous role to that of Spo13 and Moa1 in yeast meiosis (Galander et al., 2019b; Kim et al., 2015a; Miyazaki et al., 2017) and possibly Matrimony (Mtrm) in Drosophila (Bonner et al., 2020; Xiang et al., 2007). Germ cells lacking Meikin fail to properly co-orient sister kinetochores and protect peri-centromeric cohesion. Meikin expression is restricted to meiosis where it localizes to kinetochores and recruits Polo-like kinase 1 (Plk1) (Kim et al., 2015a). However, how Meikin promotes these activities and how these activities are regulated throughout meiosis are unclear. Here, we sought to define the mechanisms that enable the switch between the consecutive meiosis I, meiosis II, and mitotic cell divisions by analyzing the regulatory control of Meikin activity during the different stages of meiosis. Our results demonstrate that Separase cleavage and APC/C-mediated degradation of Meikin act to rewire cell division activities and create distinct behaviors for each meiotic stage.
Results
Meikin is proteolytically cleaved by Separase during anaphase I
To define how germ cells accomplish two distinct meiotic cell divisions in rapid succession, it is critical to determine how the activities of meiosis-specific factors, such as Meikin, are precisely controlled. Although Meikin expression is normally restricted to the germline, we found that ectopically-expressed mNeonGreen-tagged hMeikin localized to kinetochores during both interphase and mitosis in mitotically-dividing human HeLa cells (Figure 1A). The ability of Meikin to localize to mitotic kinetochores suggests that its kinetochore binding partners are at least partially retained between meiosis and mitosis, providing an experimentally-tractable system to analyze Meikin behavior. Consistent with the delocalization of Meikin that occurs during anaphase I of meiosis (Kim et al., 2015a), mNeonGreen-Meikin localization was lost from mitotic kinetochores during anaphase (Figure 1B). Interestingly, as cells progressed into anaphase, Western blotting revealed the formation of a faster migrating form of Meikin, suggestive of proteolytic cleavage (Figure 1C).
Figure 1: Meikin is cleaved by Separase during anaphase.
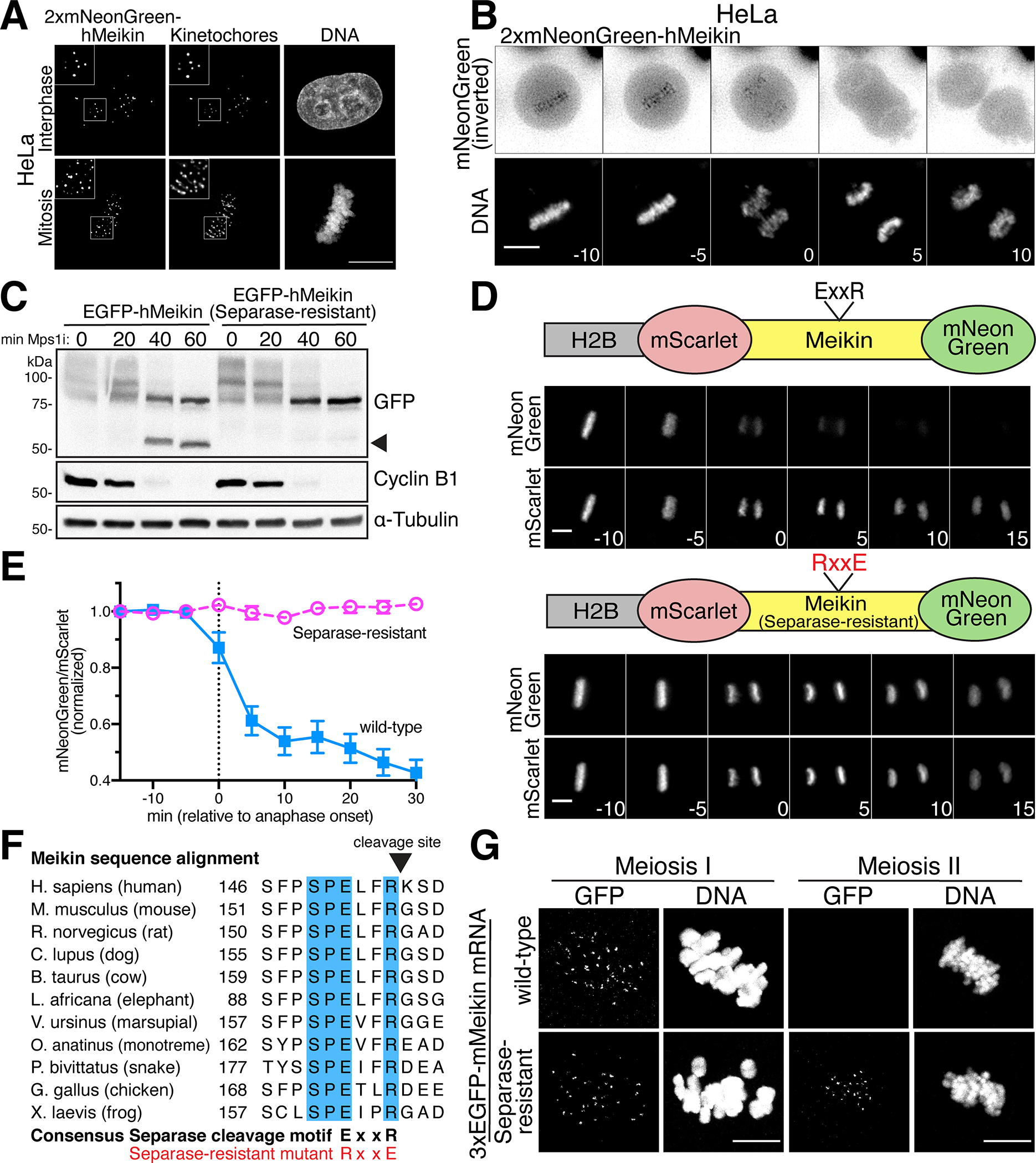
A. Deconvolved immunofluorescence images of HeLa cells expressing hMeikin. Kinetochores are stained with ACA. Images are not scaled equivalently. B. Montage of deconvolved time-lapse images of HeLa cells expressing hMeikin. Numbers indicate minutes relative to anaphase onset. C. Western blot of HeLa cell lysates after Meikin induction. Nocodazole arrested cells were forced into mitotic by treatment with Mps1i for the time indicated. Lower mobility hMeikin phosphorylation bands (see Figure S3) and a high-mobility hMeikin cleavage band are shown (see arrow). Cyclin B1 degradation indicates mitotic exit. D. Time-lapse montage of HeLa cells expressing the schematized cleavage sensor. Numbers indicate minutes relative to anaphase onset. E. Quantification of Meikin cleavage sensor. Error bars indicate 95% confidence interval. 15 cells were analyzed per condition. F. Sequence alignment of the Separase cleavage site in Meikin in selected vertebrates. Fully conserved amino acids are indicated in blue. G. Mouse oocytes injected with the indicated mMeikin mRNA were observed in meiosis I and meiosis II. Localization was consistent across multiple (>5) independent experiments. Number of oocytes analyzed: wild type: MI – 16, MII - 76, Separase-resistant: MI – 19, MII - 84. Images taken at the same timepoint are scaled equivalently. Scale bars, 10 μm. Insets, 5 μm. See also Figure S1.
To determine if Meikin is proteolytically cleaved during anaphase, we adapted a previously described protease cleavage sensor (Shindo et al., 2012) in which we targeted a Meikin fragment lacking the C-terminal kinetochore localization domain (see Figure 2) to chromatin through an N-terminal fusion to histone H2B, with fluorescent proteins placed at the Meikin N- and C-termini (Figure 1D). In this system, Meikin cleavage would result in the delocalization of the mNeonGreen tag, but the chromosomal retention of the H2B-mScarlet tag. Time-lapse imaging revealed that the mNeonGreen tagged Meikin fragment progressively delocalized from chromatin during anaphase (Figure 1D–E). Together, these data indicate that Meikin is proteolytically processed during anaphase when expressed in mitotically-dividing cells.
Figure 2: The Meikin C-terminus is necessary and sufficient for kinetochore localization and CENP-C binding.
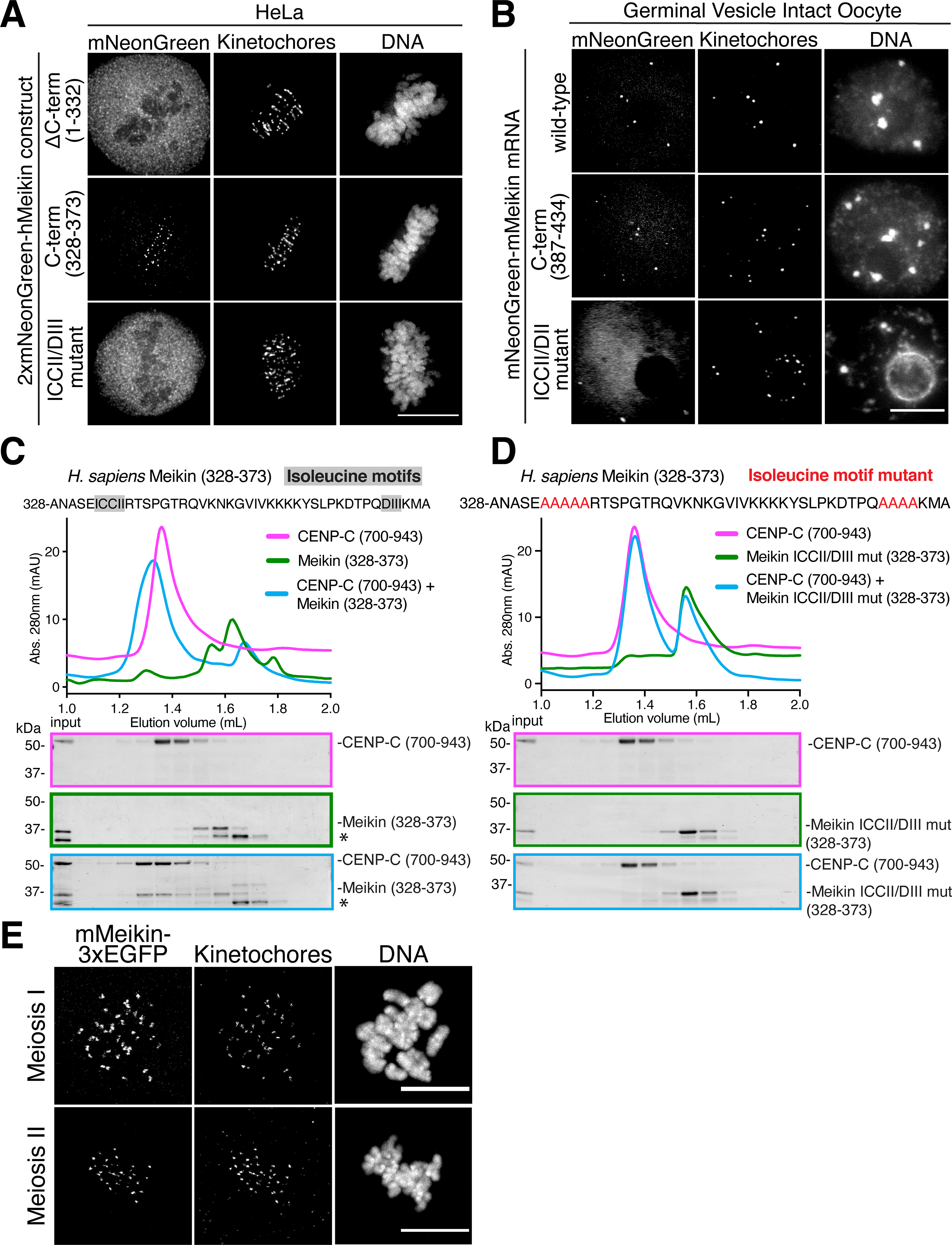
A. Deconvolved immunofluorescence images of HeLa cells expressing the indicated hMeikin constructs. Kinetochores are stained with ACA. Localization was consistent across the cell population in multiple experiments. Images are not scaled equivalently. B. Immunofluorescence images of mouse oocytes injected with the indicated mMeikin mRNA. Kinetochores are stained with mouse CENP-A antibody. Localization was consistent across three experiments. Number of meiosis II oocytes analyzed: wild-type – 19, C-terminus - 30, ICCII/DII - 21. Images are not scaled equivalently. C. Recombinant sfGFP-Meikin and GST-CENP-C protein fragments were bound and complexes analyzed by size exclusion chromatography. Fractions corresponding to elution volumes 1.0 to 2.0 mL were analyzed by SDS-PAGE and Coomassie staining. A contaminating protein in the Meikin prep is indicated with an asterisk. D. Recombinant sfGFP-Meikin containing alanine mutations in the isoleucine motifs and GST-CENP-C fragments were analyzed as above. E. Immunofluorescence images of mouse oocytes injected with mMeikin-3xEGFP mRNA. Kinetochores are stained with mouse CENP-C antibody. Images are scaled equivalently. Localization was consistent across multiple (>5) experiments. Number of oocytes analyzed: meiosis I – 58, meiosis II – 69. Scale bars, 10 μm. See also Figure S2.
We next sought to define the protease responsible for Meikin cleavage. Separase is a cysteine protease that cleaves cohesin to initiate sister chromatid separation (Hauf et al., 2001; Uhlmann et al., 1999; Uhlmann et al., 2000). Separase is specifically activated at the metaphase-anaphase transition, and the behavior of the Meikin cleavage sensor mirrors that of a sensor for the established Separase substrate, Rad21/Scc1 (Shindo et al., 2012) (Figure S1A–B). Indeed, we found that depletion of Separase by RNAi inhibited Meikin proteolysis (Figure S1C). The consensus motif for Separase cleavage is ExxR (Hauf et al., 2001; Uhlmann et al., 1999), and charge-swap mutations (RxxE) of this motif in the cohesin subunit Rad21 eliminate its proteolytic processing (Hauf et al., 2001; Shindo et al., 2012) (Figure S1A–B). Human Meikin contains one conserved ExxR sequence (Figure 1F). We found that charge-swap mutations in this motif eliminated Meikin proteolysis based on analysis of its electrophoretic mobility (Figure 1C) and our H2B-targeted cleavage sensor (Figure 1D–E). For some previously established substrates, Separase recognizes an acidic or phosphorylated residue at the P6 position relative to the cleavage site to enhance proteolysis (Alexandru et al., 2001; Hauf et al., 2001). Meikin contains a conserved serine in the P6 position (human S149, Figure 1F) matching a proline-directed cyclin-dependent kinase (Cdk) phosphorylation motif. Mutation of this residue to alanine reduced Meikin cleavage (Figure S1D). Together, these data indicate that Meikin is cleaved at anaphase onset by Separase when expressed in mitotic cells.
To determine whether Separase also cleaves Meikin during the meiosis I/II transition in germ cells, we next tested the localization of N-terminally tagged murine Meikin in mouse oocytes using mRNA injection. 3xEGFP-Meikin localized to kinetochores in meiosis I but was lost from kinetochores in meiosis II. In contrast, Separase-resistant 3xEGFP-Meikin localized to kinetochores during both meiosis I and meiosis II (Figure 1G). This suggests that cleavage of Meikin by Separase occurs during anaphase I of meiosis. Together, these data demonstrate that Meikin is processed by the Separase protease at anaphase onset in meiotically dividing cells.
The Meikin C-terminus is necessary and sufficient for kinetochore localization
Meikin plays a key role in the meiosis I-specific processes of kinetochore co-orientation and sister chromatid cohesion protection (Kim et al., 2015a), critical activities that must be reversed to enable the events associated with meiosis II. Our discovery that Meikin is specifically targeted by Separase during anaphase I provides an attractive model for how Meikin activity is restricted to meiosis I. Thus, we hypothesized that Separase cleavage would fully inactivate Meikin, similar to the effect of Separase proteolysis to inactivate cohesin complexes and promote sister chromatid separation (Uhlmann et al., 1999; Uhlmann et al., 2000). Alternatively, Meikin cleavage could result in its altered activity or function, but not fully inactivate the protein. To test these hypotheses, we analyzed the consequences of Separase cleavage to Meikin’s known molecular functions. Separase-mediated Meikin cleavage is predicted to produce two protein fragments - N-Meikin (amino acids 1–154) and C-Meikin (amino acids 155–373). Based on the delocalization of the mNeonGreen fluorescence in our cleavage sensor (Figure 1D), these fragments do not remain associated after proteolysis, but whether these fragments retain any activity is unclear.
To understand how this processing event alters Meikin function at the meiosis I/II transition, we sought to define the consequences of Meikin cleavage to its kinetochore localization. By analyzing a series of truncation mutants, we identified a Meikin C-terminal domain (amino acids 328–373) that is both necessary and sufficient for kinetochore localization when expressed ectopically in HeLa cells (Figure 2A). Similarly, we found that the equivalent C-terminal domain of murine Meikin was sufficient to localize to kinetochores in mouse oocytes (Figure 2B). Previous yeast two-hybrid assays (Kim et al., 2015a) and our affinity purifications of Meikin from mitotic cells (Figure S2A; Table S1) both identified interactions with centromere protein C (CENP-C), which is present constitutively at both meiotic and mitotic centromeres (Earnshaw et al., 1989; Kitajima et al., 2011; Tanaka et al., 2009). Meikin also co-localizes with CENP-C to the inner kinetochore (Figure S2B). Using recombinant proteins, we found that the Meikin C-terminal region was sufficient to bind to CENP-C directly in vitro (Figure 2C). Mutation of two C-terminal isoleucine-rich motifs previously implicated in Meikin localization in spermatocytes (Kim et al., 2015a) abolished Meikin’s kinetochore localization in both HeLa cells and mouse oocytes (Figure 2A–B) and its interaction with CENP-C in vitro (Figure 2D). In reciprocal experiments, we identified a minimal C-terminal fragment of CENP-C (amino acids 808–943) that is sufficient for Meikin binding (Figure S2C). As we found that increased salt concentrations promoted Meikin-CENP-C binding (Figure S2D), we tested whether their interface requires hydrophobic interactions. We identified two hydrophobic motifs within CENP-C that are required for Meikin binding (Figure S2E–F), possibly by partnering with the Meikin isoleucine-rich motifs. This binding site for Meikin on CENP-C does not overlap with binding sites for established CENP-C-interaction partners (Klare et al., 2015), suggesting that CENP-C interacts with Meikin without disrupting its other kinetochore interfaces. Thus, the C-terminal Meikin fragment generated by Separase cleavage retains Meikin’s CENP-C binding and kinetochore localization domain.
C-Meikin is retained at meiosis II kinetochores
As the Meikin C-terminus is sufficient for kinetochore localization and its interaction partner CENP-C is present throughout meiosis, this suggests that the C-terminal Meikin fragment generated by Separase cleavage would retain the ability to target to kinetochores. Indeed, although N-terminally tagged Meikin in oocytes is lost from kinetochores in meiosis II (Figure 1G), we found that C-terminally tagged Meikin localizes to kinetochores during both meiosis I and meiosis II (Figure 2E). Thus, while the N-terminal fragment of Meikin is lost from kinetochores following Separase cleavage (Figure 1G), the Meikin C-terminal cleavage fragment remains associated with kinetochores in meiosis II through its interaction with CENP-C, creating the possibility that this fragment could retain some activities at meiosis II kinetochores.
Plk1 displays phospho-dependent binding to the Meikin C-terminal region
Although Meikin kinetochore localization is retained following Separase cleavage, it is possible that other Meikin interaction partners are affected. Prior work suggested that Meikin promotes meiosis I sister kinetochore co-orientation and the protection of centromeric cohesin through its direct interaction with Polo-like kinase 1 (Plk1) (Kim et al., 2015a), an essential cell cycle kinase that controls multiple aspects of mitosis and meiosis (Petronczki et al., 2008). We found that immunoprecipitation of ectopically-expressed Meikin from HeLa cells arrested in mitosis isolated both CENP-C and Plk1 (Figure S2A; Table S1). Plk1-Meikin complexes co-purified from HeLa cells display similar kinase activity on an artificial peptide substrate to Plk1 isolated on its own (Figure S3A). In addition, ectopic Meikin expression in HeLa cells results in increased Plk1 localization to mitotic kinetochores and the corresponding increased phosphorylation of Plk1-dependent kinetochore substrates (Figure S3B–C). The combination of these data suggests that Meikin acts as a targeting subunit for Plk1 to recruit Plk1 to meiotic kinetochores, where it can then phosphorylate downstream targets.
To determine whether Meikin cleavage affects its binding to Plk1, we first sought to define the basis for the Meikin-Plk1 interaction. Plk1 typically binds to its substrates and targeting factors via its phospho-peptide binding domain. We found that Meikin is extensively phosphorylated when expressed in HeLa cells (Figure 1C; Figure S3D) on both Cdk ([pT/pS]-P) and Plk1 ([D/E/N]-x-[pT/pS]) consensus phosphorylation sites (Figure S3E). Chemical inhibition of Plk1 kinase activity eliminates the Meikin-Plk1 interaction (Figure S4A). Human Meikin contains three S-[pT/pS]-P (STP) motifs (T251, T264, T276), which provide potential docking sites for Plk1 (Elia et al., 2003), and which have been implicated previously in Plk1 binding (Kim et al., 2015a). Mutation of all three STP motifs (STP to SAP) reduced but did not eliminate the Meikin-Plk1 interaction (Figure 3A). However, a Meikin mutant that eliminates the STP motifs together with multiple consensus sites for Plk1 (S175, T176, T180, S181, S196), which enhance kinase docking interactions in other substrates (Lee et al., 2008), abrogated Plk1 binding (8A mutant; Figure 3A). Notably, this Meikin-8A mutant is not cleaved efficiently by Separase (Figure S1D), consistent with other Separase substrates that require phosphorylation for their proteolysis (Alexandru et al., 2001; Hauf et al., 2005; Hornig and Uhlmann, 2004; Kim et al., 2015b; Kudo et al., 2009). We conclude that Meikin binding to Plk1requires its phosphorylation at multiple sites.
Figure 3: Full length Meikin and the C-Meikin cleavage fragment bind Plk1 and promote Rec8 cleavage similarly.
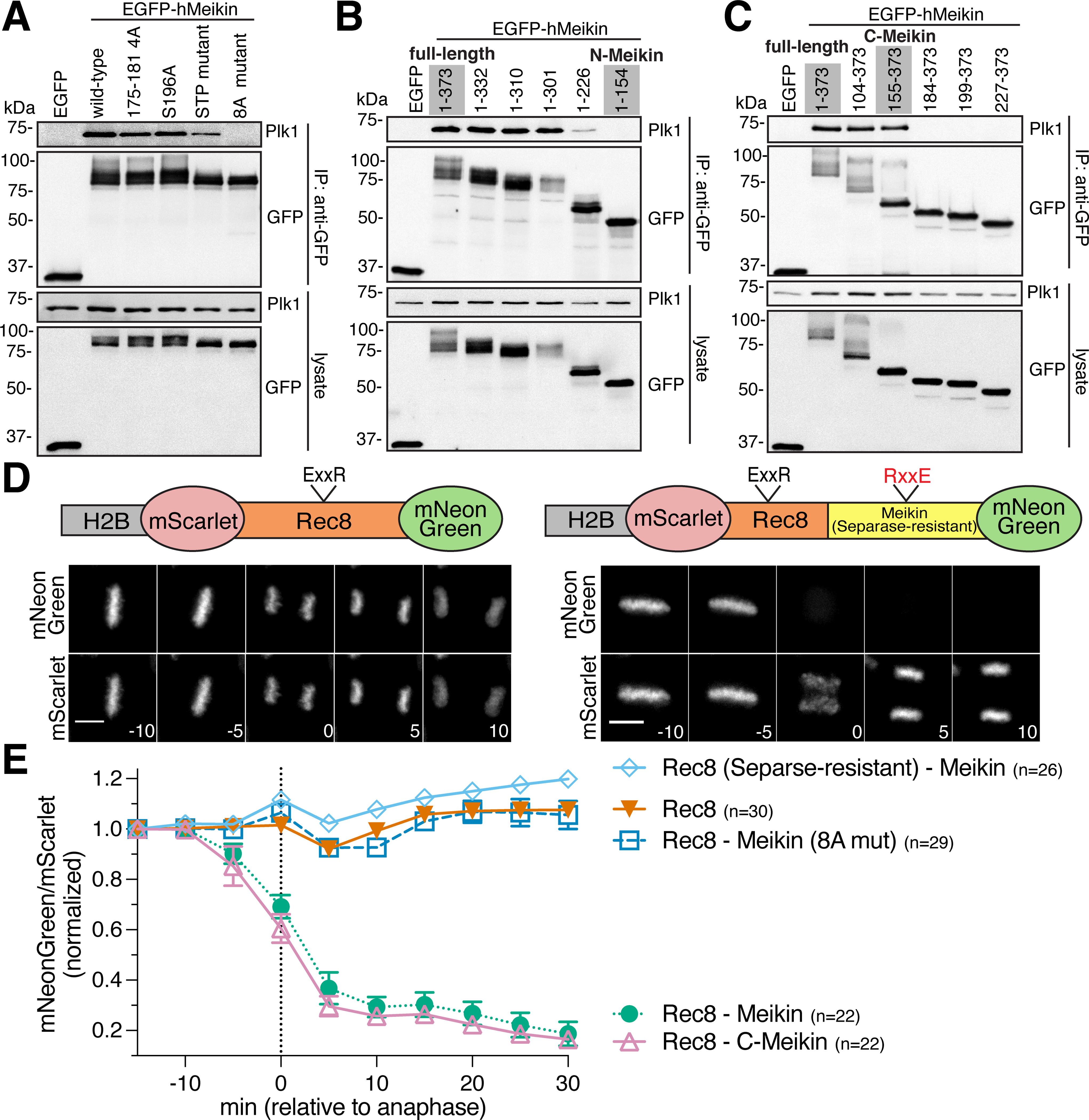
A. Induced HeLa cells were arrested with STLC. GFP-immunoprecipitates were analyzed by Western blot. Specific residues changed to alanine in each mutant are described in the Key Resources Table. B and C. Western blot analysis of GFP immunoprecipitates from cells expressing hMeikin constructs as above. D. Time-lapse montage of HeLa cells expressing the indicated Rec8 cleavage sensor. The sensor contains a fragment of hRec8 including the predicted Separase cleavage sites. Numbers indicate minutes relative to anaphase onset. Scale bars, 10 μm. E. Quantification of Rec8 cleavage sensor analyzed and represented as in Figure 1. n = number of mitoses analyzed. See also Figure S3 and S4.
To determine whether Meikin cleavage affects its binding to Plk1, we next defined the minimal region required for the Meikin-Plk1 interaction. The N-terminal Meikin fragment generated by Separase cleavage (N-Meikin) did not bind to Plk1 (Figure 3B). In contrast, the C-terminal cleavage fragment (C-Meikin) was still capable of interacting with Plk1 (Figure 3C). As an alternative strategy to detect Meikin-Plk1 interactions, we targeted Meikin to chromatin using an H2B fusion. Expression of H2B fusions with either full length Meikin or C-Meikin recruited endogenous Plk1 to chromatin (Figure S4B). Together, these data suggest that Separase cleavage does not prevent the binding of C-Meikin to Plk1, although cleavage may modulate Meikin-Plk1 interactions (see below).
Meikin-Plk1 complexes promote Rec8 cleavage when present in close proximity
We next assessed whether Meikin cleavage affects its downstream activities. Current models suggest that Meikin-Plk1 complexes regulate key features of both kinetochore co-orientation and the protection of pericentric cohesin (Kim et al., 2015a). To define the substrates responsible for these activities, we analyzed the meiosis-specific kleisin subunit of cohesin, Rec8 (Buonomo et al., 2000; Tachibana-Konwalski et al., 2010). Centromere-localized Rec8 has been proposed to play a critical role in promoting kinetochore co-orientation in many organisms (Chelysheva et al., 2005; Sakuno et al., 2009; Severson et al., 2009; Watanabe and Nurse, 1999). In this model, the centromeric Rec8 population would need to be eliminated at the meiosis I/II transition to allow for proper chromosome alignment at metaphase II. In contrast, the pericentric population of Rec8-containing cohesin must be protected from Separase cleavage to ensure that sister chromatids remain associated until anaphase II. Indeed, recent work suggests that the centromeric, pericentric, and chromosome arm populations of Rec8 are differentially regulated at the meiosis I/II transition in mammalian oocytes (Ogushi et al., 2020).
To monitor Rec8 cleavage, we adapted the H2B-based cleavage sensor in HeLa cells. In contrast to the Rad21 cleavage sensor (Figure S1B), a Rec8 fragment containing its established Separase cleavage sites did not show any proteolysis upon anaphase onset in mitotic cells (Figure 3D–E). To test whether Meikin-Plk1 complexes could promote Rec8 cleavage when present in close proximity, we created an in-frame Rec8-Meikin fusion. Strikingly, fusion of Meikin to the Rec8 fragment resulted in efficient Rec8 cleavage (Figure 3D–E). Charge-swap mutation of the conserved Separase cleavage site in Rec8 (E401R, R404E) (Kudo et al., 2009) eliminated this proteolysis (Figure 3E). Rec8 cleavage was also dependent on the interaction between Meikin and Plk1, as a Meikin mutant that does not bind Plk1 (8A mutant; Figure 3A) does not potentiate Rec8 cleavage (Figure 3E). This suggests that kinetochore-targeted Meikin-Plk1 complexes could phosphorylate Rec8 to promote the cleavage of adjacent centromere-proximal Rec8-cohesin at anaphase I. Consistent with this, prior work found that Plk1 phosphorylation of mammalian Rec8 enhances Separase-mediated cleavage in vitro (Kudo et al., 2009). These data suggest that Meikin-Plk1 complexes activate proximal Rec8 for cleavage by Separase, providing a mechanism to reverse kinetochore co-orientation following anaphase I.
To determine whether Separase-cleavage inhibits Meikin’s ability to promote Rec8 proteolysis, we next created an in-frame fusion between the C-Meikin cleavage product and Rec8 in our H2B sensor. Similarly to full length Meikin, C-Meikin induced efficient cleavage of the Rec8 sensor (Figure 3E). This suggests that retention of C-Meikin at kinetochores would further promote the elimination of centromeric Rec8 during late anaphase, ensuring complete reversal of kinetochore co-orientation before meiosis II chromosome alignment. Collectively, these data indicate that full length Meikin and the C-Meikin Separase cleavage product display similar activities with respect to their kinetochore localization, Plk1 binding, and ability to promote Rec8 cleavage. Thus, in contrast to other substrates, Separase cleavage does not fully inactivate Meikin function.
A sensitized assay for Plk1 interactions reveals distinct behaviors for full-length and C-Meikin
Given the similar activities observed for full-length Meikin and C-Meikin in the assays described above, we sought to generate a sensitized assay for Meikin activity. Stable expression of Meikin at low levels in mitotically dividing cells results in a modest increase in misaligned chromosomes (Figure S5A). In contrast, increased Meikin expression (under the control of a doxycycline-inducible promoter), caused a potent mitotic arrest with misaligned chromosomes and monopolar spindle structures (Figure S5A–B), phenotypes that are consistent with an effect on Plk1 activity such as its titration away from endogenous targets. This overexpression phenotype is inherently non-physiological as it occurs in mitotic cells that normally do not express Meikin. However, monitoring the mitotic arrest phenotype provides a sensitized assay to detect robust Meikin-Plk1 interactions. Indeed, although individual Meikin phosphorylation site mutants were able to co-immunoprecipitate with Plk1, these mutants did not promote a mitotic arrest (Figure S5C), suggesting that they display reduced Plk1 interactions. Utilizing this assay, we defined a minimal domain (amino acids 124–332) spanning the N- and C-Meikin Separase cleavage fragments that is sufficient to induce a mitotic arrest (Figure S5C). Importantly, neither the N- nor C-terminal Separase cleavage fragment of Meikin was sufficient to generate a mitotic arrest, despite the ability of C-Meikin to immunoprecipiate Plk1 (Figure 3C). Based on these data, we conclude that full-length Meikin and C-Meikin both localize to kinetochores and bind to Plk1 but display different efficiency or properties for their Plk1 interactions in this ectopic system. Thus, Separase cleavage has the potential to differentially control Meikin-Plk1 interactions at the meiosis I/II transition.
Separase cleavage of Meikin is required for chromosome alignment during meiosis II
Based on the results described above, we sought to directly test whether Separase-mediated Meikin proteolysis is required for the proper execution of meiosis. To determine the functional consequences of Meikin cleavage, we ectopically expressed the Separase-resistant Meikin mutant in mouse oocytes, causing full length Meikin to persist at kinetochores into meiosis II (Figure 1G; Figure 4A). As Meikin must localize to centromeres to impart its established function, sufficient expression of the transgene would allow a mutant Meikin protein to compete with the endogenous protein for a limited number of available CENP-C binding sites at kinetochores, resulting in a dominant phenotype. Indeed, ectopic expression of Separase-resistant Meikin, causing persistence of full-length Meikin at meiosis II kinetochores, led to severe chromosome alignment defects during the second meiotic division (Figure 4B–C). Ectopic expression of Separase-resistant Meikin also resulted in increased kinetochore levels of Plk1 and Bub1 at meiosis II kinetochores (Figure 4D–E) compared to ectopic expression of wild-type Meikin to similar levels (Figure S5D). The increase in kinetochore Plk1 levels in oocytes that retain full-length Meikin during meiosis II (Figure 4D–E) supports our results in HeLa cells that full-length Meikin and cleaved C-Meikin have different affinities for Plk1 (Figure S5C), such that the full length is more potent in Plk1 recruitment. As Plk1 and Bub1 regulate kinetochore-microtubule interactions (Combes et al., 2017; Marston and Wassmann, 2017), improper Plk1 and Bub1 kinetochore localization in meiosis II oocytes expressing Separase-resistant Meikin likely contributes to the observed chromosome alignment defects (Figure 4B–C). Thus, our work indicates that Meikin cleavage during anaphase I is critical to enable a proper meiosis II division.
Figure 4: Separase cleavage of Meikin is required for proper meiosis II chromosome alignment.
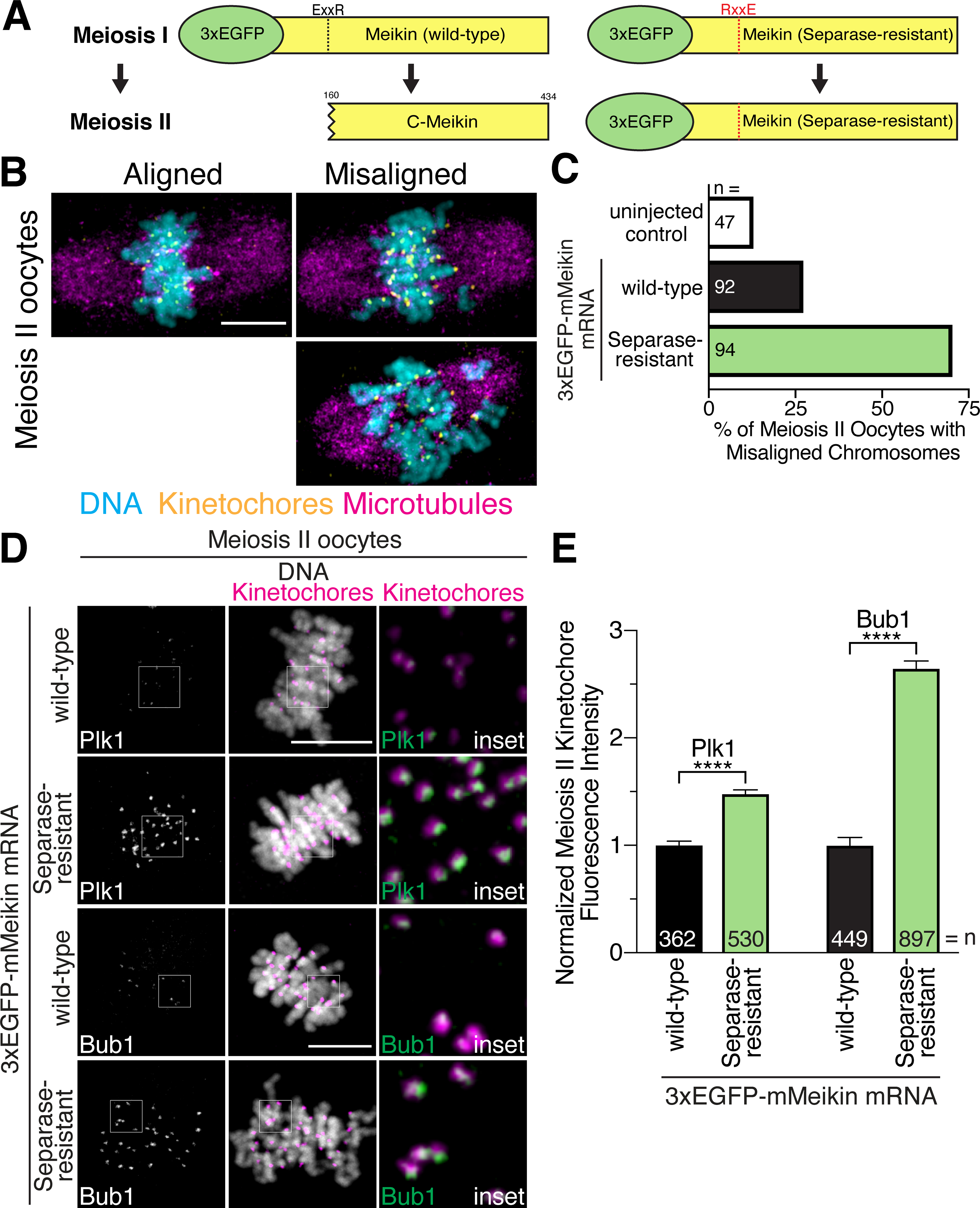
A. Schematic of mMeikin constructs and their expected behaviors at meiosis II. B. Representative immunofluorescence images of chromosome misalignment defects observed in meiosis II oocytes. C. Quantification of defects observed in injected oocytes. n = number of oocytes analyzed. D. Mouse oocytes injected with the indicated Meikin mRNA and stained for Plk1 or Bub1 at meiosis II. Images of similarly stained cells are scaled identically. E. Quantification of kinetochore intensity of Plk1 and Bub1 in meiosis II oocytes injected with the indicated mMeikin mRNA. Values from two experiments were pooled. n = number of kinetochores analyzed. Means and 95% confidence intervals are presented. ****P < 0.0001, two-tailed t-test. Kinetochores are stained with mouse CENP-C antibody. Scale bars, 10 μm. Insets, 5 μm. See also Figure S5.
C-Meikin is required during meiosis II for proper chromosome alignment
Our results indicate that the failure to cleave Meikin results in a defective meiosis II, but that cleaved C-Meikin retains some activity based on its kinetochore localization, at least partial Plk1 binding, and its ability to promote Rec8 cleavage. Thus, instead of abolishing Meikin function, our work suggests that Separase processing modulates Meikin activity at anaphase I to create altered Plk1 binding and promote additional functions during meiosis II. To test this, we sought to disrupt Meikin function by creating dominant-negative Meikin alleles. For these experiments, we first expressed Meikin mutants that retain its CENP-C interaction (allowing them to displace endogenous Meikin at kinetochores) but are defective for Plk1 binding (Figure 5A). Oocytes ectopically expressing wild-type Meikin using mRNA microinjection progressed to metaphase II similarly to mock-injected oocytes. In contrast, expression of the minimal C-terminal murine Meikin kinetochore targeting domain, which lacks Plk1 binding (Figure 3C), caused premature-sister chromatid separation during meiosis II (Figure 5B–C). Similarly, expression of full length Meikin with a mutation in the STP motif also caused premature-sister chromatid separation during meiosis II (Figure 5B–C). These phenotypes suggest that the Meikin-Plk1 interaction acts during meiosis I to protect the pericentric cohesion that holds sister chromatids together until anaphase II, and that mutant versions of Meikin containing its C-terminus can effectively compete with the endogenous protein by displacing it from kinetochores.
Figure 5: The C-Meikin cleavage fragment is required for meiosis II chromosome alignment.
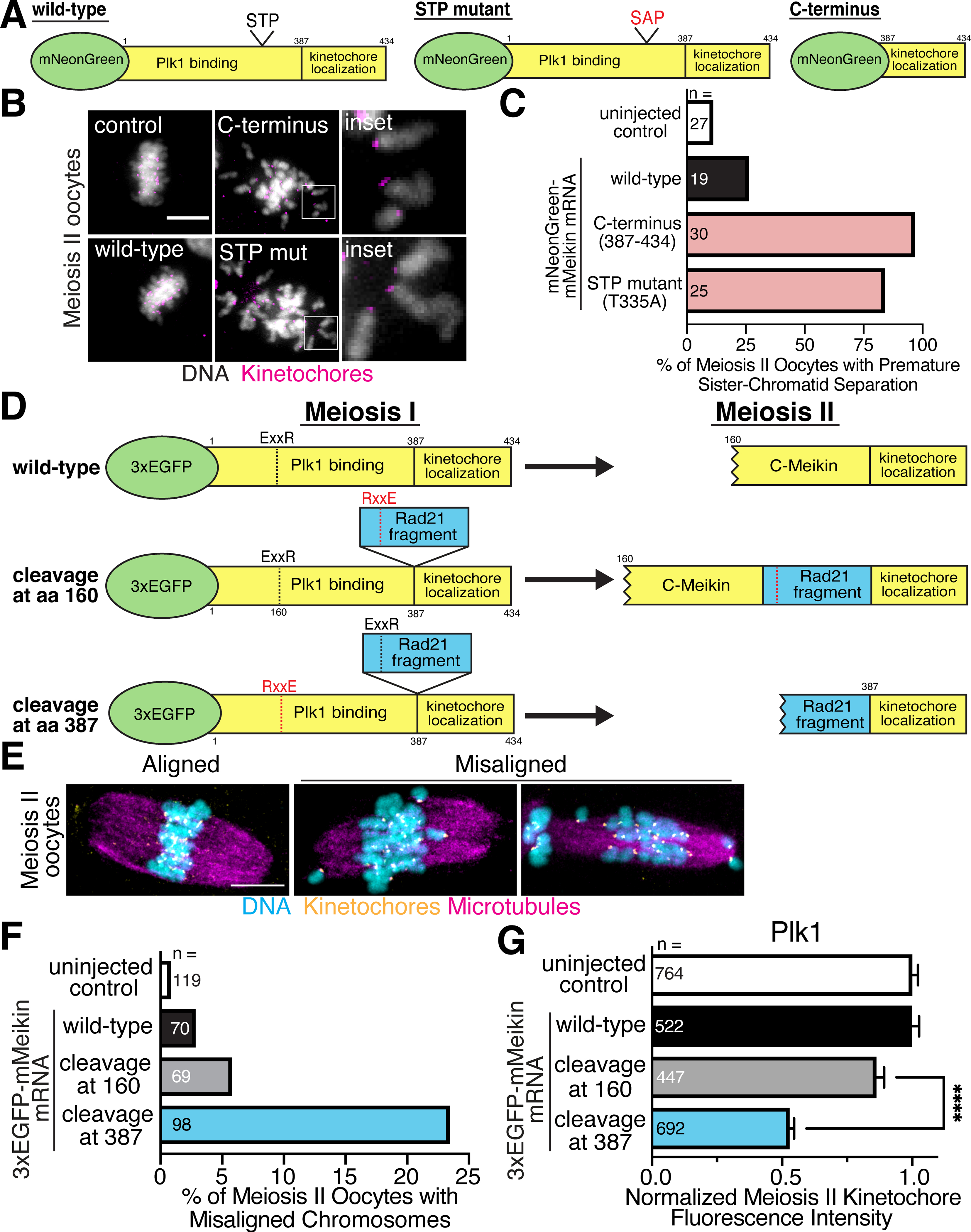
A. Schematic of mMeikin constructs. B. Representative immunofluorescence images of oocytes injected with the indicated Meikin mRNA and matured to meiosis II. Insets show separated chromatids. Kinetochores are stained with Hec1 antibody. C. Quantification of premature sister chromatid separation defects observed in meiosis II oocytes expressing the indicated Meikin mRNA. Kinetochores are stained with Hec1 antibody. D. Schematic of mMeikin constructs and their expected behaviors at meiosis II. A fragment of hRad21 (amino acids 142–275) containing the conserved Separase cleavage site was used. A fragment of hRad21 (amino acids 142–275) was inserted between the Plk1 binding and kinetochore localization domains of Meikin. Charge-swap mutations in the Separase-cleavage sites of Meikin (E156R, R159E) or Rad21 (E169R, R172E) were used to direct the location of Separase cleavage during anaphase I. E. Representative immunofluorescence images of chromosome misalignment defects observed in meiosis II oocytes. Kinetochores are stained with mouse CENP-C antibody. F. Quantification of meiosis II chromosome misalignment defects observed in oocytes injected with the indicated mMeikin construct. n = number of oocytes analyzed. G. Quantification of Plk1 kinetochore intensity in meiosis II oocytes injected with the indicated mMeikin mRNA. Means and 95% confidence intervals from pooled results of two experiments are presented. n = number of kinetochores analyzed. ****P < 0.0001, two-tailed t-test. Scale bars, 10 μm. Insets, 8 μm. See also Figure S6.
To test Meikin function during meiosis II, we next developed a conditional strategy to eliminate Meikin activity at anaphase I onset, taking advantage of mutant Meikin displacing endogenous Meikin from kinetochores. This strategy allowed us to circumvent the consequences of the dominant negative Meikin mutants that disrupt meiotic behaviors in meiosis I (Figure 5A–C). To do this, we altered the position of Separase proteolysis by inserting the Rad21 Separase cleavage site between the Meikin Plk1 binding and kinetochore targeting domains defined by our analysis (Figure 2A–B, Figure 3B–C), forming a Meikin allele with two potential cleavage sites (Figure 5D). Introducing inactivating mutations at either the endogenous Meikin cleavage site or the inserted cleavage site enabled us to control the position of Separase proteolysis to generate either a product equivalent to C-Meikin or a product completely lacking the Plk1 binding domain. Importantly, both alleles retain full Meikin activity during meiosis I, allowing us to determine the functional consequences of specifically inactivating Meikin during meiosis II. Expression of the control Meikin construct that is cleaved at the endogenous Meikin proteolysis site (amino acid 160) resulted in normal meiosis II oocytes (Fig 5E–F). In contrast, Separase cleavage at the introduced downstream site (amino acid 387) to eliminate Meikin-Plk1 interactions during meiosis II resulted in chromosome alignment defects in meiosis II oocytes (Fig 5E–F). We find that oocytes expressing mutant Meikin that is cleaved at the introduced downstream site had significantly lower levels of Plk1 at meiosis II kinetochores (Figure 5G, Figure S6A). Thus, C-Meikin recruits an intermediate level of Plk1 to promote chromosome alignment during meiosis II, consistent with the roles for Plk1 in regulating kinetochore microtubule-attachments (Combes et al., 2017). These data indicate that C-Meikin is not simply an inactive form of Meikin, but is required for proper chromosome alignment after anaphase I. Therefore, instead of Meikin acting as a meiosis I-specific factor as proposed by prior work (Kim et al., 2015a), Meikin remains active throughout meiosis with Separase cleavage at anaphase I generating a distinct form of Meikin with different activity critical to enable meiosis II events.
C-Meikin is targeted for proteosome degradation by the APC/C during anaphase II
Our data demonstrate that C-Meikin is retained at meiotic kinetochores to enable meiosis II chromosome alignment. However, Meikin is not expressed in mitotic cells and is not required for mitotic cell division (Kim et al., 2015a). We found that C-terminally tagged Meikin-EGFP is removed from kinetochores upon completion of meiosis during anaphase II (Figure 6A), providing a potential mechanism to eliminate Meikin activity to return to a mitotic state. To define the behavior of Meikin at meiotic exit, we considered additional levels of regulatory control that could act to remove or degrade C-Meikin prior to the first mitotic division of the fertilized zygote. Meikin contains a non-canonical D-box motif (Figure 6B), which is conserved among placental mammals and could serve for recognition by the APC/C E3-ubiquitin ligase (Davey and Morgan, 2016). The APC/C is specifically activated during anaphase onset leading to the ubiquitination and proteasomal degradation of critical anaphase substrates, such as Securin and cyclin B (Sivakumar and Gorbsky, 2015). Strikingly, mutation of the Meikin D-box motif to alanine stabilized Meikin-EGFP at anaphase II kinetochores (Figure 6A). This suggests that C-Meikin is subject to APC/C-mediated degradation during anaphase II.
Figure 6: C-Meikin is targeted for degradation by the APC/C during anaphase II.
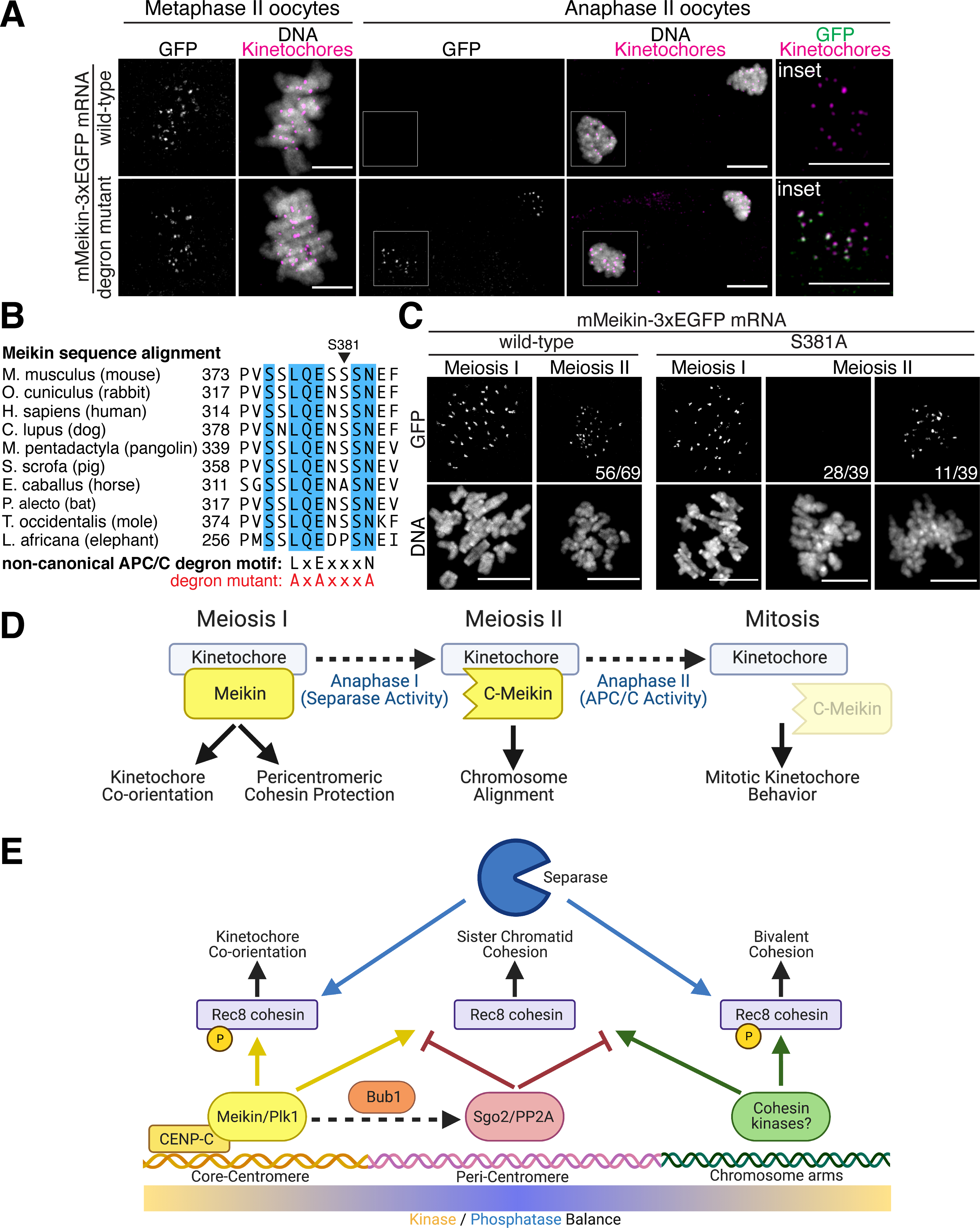
A. Immunofluorescence images of oocytes injected with indicated mMeikin mRNA and fixed at the indicated stage. Kinetochores are stained with mouse CENP-C antibody. Localization was consistent across two experiments. Number of oocytes analyzed: wild-type: Metaphase II – 18, Anaphase II - 21, degron mutant: Metaphase II – 20, Anaphase II - 38. Images of oocytes at the same stage are scaled equivalently. B. Sequence alignment of the non-canonical APC/C D-box motif in Meikin from selected placental mammals. Fully conserved amino acids are indicated in blue. Critical residues changed to alanine in the mutant alleles are indicated. C. Immunofluorescence images of oocytes injected with the indicated mMeikin mRNA and fixed at the indicated stage. Localization was consistent across multiple experiments. The number of oocytes analyzed: wild-type (>5 independent experiments, also see Figure 2E): MI – 58, MII – 69, S381A (two independent experiments): MI – 27, MII – 39. The fraction of oocytes with the displayed meiosis II localization pattern is indicated. Images of meiosis II oocytes expressing the S381A mutant are scaled equivalently. D. Model for differential Meikin activity at distinct stages of meiosis. E. Model for spatial control of Rec8 phosphorylation and cleavage during meiosis I. Created with Biorender.com. Scale bars, 10 μm.
Although we find that Meikin contains a conserved APC/C degron that is functional during anaphase II, the APC/C is also activated during anaphase I (Homer, 2013). Thus, eliminating Meikin specifically at meiotic exit would require an additional strategy to restrict Meikin degradation to the second meiotic division. Some APC/C degrons can be masked by the phosphorylation of neighboring residues (Davey and Morgan, 2016). The Meikin APC/C degron surrounds a serine residue (mouse S381) that matches a potential Plk1 consensus phosphorylation motif (Figure 6B). Interestingly, we found that mutation of this serine to alanine resulted in the premature degradation of Meikin during anaphase I in the majority of oocytes tested (Figure 6C). This suggests that Plk1 mediated phosphorylation of Meikin inhibits APC/C-mediated degradation during meiosis I.
Together, we find that Meikin is subject to multiple levels of post-translational regulation during the two distinct meiotic divisions. Full length Meikin acts to enable meiosis I events, such as kinetochore co-orientation and the protection of pericentric cohesin (Kim et al., 2015a). During anaphase I, Separase-mediated cleavage of Meikin generates a truncated form of the protein with altered activity that is required for proper meiosis II chromosome alignment. Finally, during anaphase II, APC/C-mediated degradation of Meikin allows for a return to the mitotic cell division state (Figure 6D).
Discussion
Our data reveal the intricate regulatory control by which the meiotic divisions are coordinated, providing a mechanism to rewire the cell division machinery between meiosis I and II. We find that the protease Separase acts as a molecular “scalpel” to precisely and irreversibly modulate Meikin activity between the two meiotic divisions. Instead of proteolytic cleavage completely inactivating the protein, full-length Meikin and the C-Meikin Separase cleavage product both localize to kinetochores but differ in their functional activities. Importantly, the activity of C-Meikin during meiosis II is required for proper chromosome alignment and cannot be replaced with full-length Meikin. Both the failure to cleave Meikin (Figure 4A–C) or the complete inactivation of Meikin at anaphase I (Figure 5D–F) result in defective chromosome alignment during meiosis II. Thus, Separase cleavage of Meikin is critical for mammalian meiosis.
Our data also demonstrate that Meikin is a substrate for APC/C-mediated degradation (Figure 6A). Importantly, this degradation only occurs during anaphase II. Our data suggest that there is feedback between Plk1 phosphorylation and APC/C degradation, such that strong Meikin-Plk1 interactions during meiosis I suppress APC/C degradation through phosphorylation of the degron motif (Figure 6C) to allow for C-Meikin specific activities during meiosis II. Meikin cleavage then leads to a reduction in Plk1 binding and phosphorylation allowing for APC/C degradation during the second meiotic anaphase. Thus, temporal ordering of proteolytic cleavage and degradation events regulates Meikin activity states throughout meiosis. Notably, Meikin counterparts in other organisms also contain APC/C recognition sites (Galander and Marston, 2020). Budding yeast Spo13 is degraded by the APC/C at anaphase I, but blocking this degradation has little effect on meiotic progression (Sullivan and Morgan, 2007). Drosophila Mtrm is also subject to APC/C mediated degradation during late meiosis, and this degradation is critical for proper embryonic development (Whitfield et al., 2013). The consequence of inappropriate retention of an APC/C Meikin mutant on early embryogenesis has not yet been determined. However, our experiments in HeLa cells show that Meikin can cause chromosome alignment and spindle orientation defects when present during mitosis (Figure S5A).
Our data support a model in which kinetochore-localized Meikin establishes a gradient of Plk1 activity during meiosis I centered on the kinetochore, which leads to different fates for cohesin complexes based on their location within the gradient (Figure 6E). Meikin is targeted to kinetochores through its binding to CENP-C (Figure 2A–D), allowing Meikin to recruit Plk1 (Figure S3B–C). In this model, centromere-localized Meikin-Plk1 complexes differentially regulate the centromeric and pericentromeric populations of Rec8 cohesin during meiosis, with opposing effects on these two pools. By increasing the concentration of centromere-localized Plk1, Meikin-Plk1 complexes direct phosphorylation of centromere-bound Rec8 to promote its efficient cleavage during anaphase I (Figure 3D–E), thereby reversing meiosis I kinetochore coorientation. In parallel, Meikin-Plk1 activity recruits Bub1 (Figure 4D–E), which in turn recruits Sgo2-PP2A complexes to the pericentromere to reverse Rec8 phosphorylation and protect pericentromeric Rec8 from Separase cleavage (Figure 5A–C) (Galander et al., 2019a; Galander et al., 2019b; Marston, 2015; Miyazaki et al., 2017). Thus, during anaphase I, Separase cleaves centromere-proximal and phosphorylation-primed Rec8, but not pericentromeric Rec8, eliminating kinetochore co-orientation while maintaining sister chromatid cohesion. At anaphase I, Separase also cleaves Meikin (Figure 1G), resulting in a C-terminal Meikin fragment that remains associated with kinetochores (Figure 2E). However, while retaining key activities, this cleaved version is less potent in its ability to bind to or regulate Plk1 (Figure S5C), leading to reduced Plk1 at kinetochores in meiosis II compared to meiosis I (Figure 4D–E) (Kim et al., 2015a). This reduced Plk1 activity leads to a corresponding reduction in pericentromeric Bub1 and Sgo2, allowing for Rec8 phosphorylation and cleavage at anaphase II. This model is consistent with recent findings that Separase activity at meiosis I is required for the deprotection of peri-centromeric cohesin and individualization of sister kinetochores during anaphase I (Gryaznova et al., 2021; Ogushi et al., 2020). Finally, as germ cells exit meiosis, Meikin is completely eliminated through APC/C-mediated degradation (Figure 6A), returning kinetochore regulation to a mitotic state.
In conclusion, Meikin is a novel Separase substrate that is cleaved specifically during anaphase of meiosis I. Despite the identification of Separase more than two decades ago and extensive efforts to identify additional targets, the number of confirmed Separase substrates remains low. Our data reveal that Meikin cleavage by Separase in anaphase I does not inactivate the protein, but rather modulates its activity to allow Meikin to transition from meiosis I to meiosis II specific functions. Finally, APC/C-mediated degradation specifically eliminates Meikin during anaphase II resetting the cell division machinery to a mitotic state. This provides a mechanism to rapidly reverse the meiosis I-specific modifications to the cell cycle machinery, coordinate the sequential meiotic divisions, and establish the correct conditions for the first mitotic division.
Limitations of the Study
Previous work visualizing the endogenous Meikin protein by immunofluorescence suggested that Meikin localization and function was specific to meiosis I (Kim et al., 2015a). In contrast, using ectopic expression of fluorescently tagged proteins, we find that a substantial portion of Meikin is retained beyond the first meiotic division. Distinct detection strategies likely explain these differing results. We were unable to clearly detect endogenous Meikin by Western blotting using the previously published antibody (Kim et al., 2015a) with a readily obtainable number of staged meiosis I or meiosis II oocytes. Thus, limited antibody accessibility or affinity, or the loss of N-terminal epitopes following Separase cleavage of Meikin, may have prevented detection of Meikin during meiosis II. However, it remains possible that ectopically expressed, tagged Meikin may behave differently than endogenous Meikin. Further experiments using new antibody reagents for Meikin detection or combining depletion or knockout of endogenous Meikin with expression of the mutants identified here will be required to resolve these differing results.
Our functional studies rely on the ability of ectopically expressed Meikin to displace the endogenous protein from kinetochores. Using this strategy, our data demonstrate that both too much and too little Meikin-Plk1 during meiosis II have the potential to cause substantial chromosome alignment defects. We propose that both phenotypes result from mis-regulation of kinetochore-microtubule interactions due to an imbalance in kinetochore kinase-phosphatase activity. Future experiments utilizing complete genetic replacements of Meikin may reveal additional distinguishing features of these phenotypes. Similarly, our experiments focused on the roles of Meikin after oocytes exit their prophase I arrest. Previous genetic evidence indicates that Meikin plays a role in establishing kinetochore co-orientation (Kim et al., 2015a). This process likely requires Meikin-Plk1 activity during loading of core centromeric Rec8-cohesin complexes in meiotic prophase before our manipulations in this study. Future work will be required to define how sister kinetochore co-orientation is established prior to the first meiotic division.
STAR Methods
Resource Availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Iain Cheeseman (icheese@wi.mit.edu).
Materials Availability
Select plasmids used in this study have been deposited with Addgene. Additional plasmids and cell lines generated in this study are available upon request from the lead contact.
Data and Code Availability
Original Western blot images and mass-spectrometry data have been deposited at Mendeley Data and are publicly available as of the date of publication. The DOI is listed in the key resources table. All other data, including microscopy data reported in this paper will be shared by the lead contact upon request.
Cellprofiler pipelines have been deposited at Mendeley Data. The DOI is listed in the key resources table. All other original code is available from the lead contact upon request.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| α-Tubulin | Sigma-Aldrich, mouse clone DM1A (1:3000 IF) | Cat#T6199 RRID: AB_477583, lot 078M5796V |
| α-Tubulin | Abcam, rabbit clone EP1322Y (1:500 IF) | Cat#ab52866, RRID: AB_869989, lot GR3241238-2 |
| α-Tubulin (HRP-conjugated) | Abcam, mouse clone DM1A (1:5000 WB) | Cat#ab40742, RRID: AB_880625, lot GR3229214-1 |
| β-Tubulin | Sigma Aldrich, mouse clone Tub2.1 (1:500 Oocyte IF) | Cat#T4026 RRID: AB_477577 |
| GFP | Roche, mouse clones 13.1 and 7.1 (1:1000 WB) | Cat#11814460001, RRID: AB_390913, lot 14442000 |
| mNeonGreen | Chromotek, mouse clone 32F6 (1:1000 WB) | Cat#32F6, RRID: AB_2827566, lot 71108021 |
| Anti-centromere (ACA) | Antibodies Inc, human polyclonal (1:100 IF) | Cat#15234, RRID: AB_2687472, lot 1CK37 |
| CENP-A | Abcam, mouse clone 3–19 (1:1000 IF) | Cat#ab13939, RRID: AB_300766, lot GR3265183-3 |
| Mouse CENP-A | Cell Signaling Tech, rabbit clone C51A7 (1:200 oocyte IF) | Cat#2048S, RRID: AB_1147629 |
| CENP-C | Cheeseman lab (Gascoigne et al., 2011), rabbit polyclonal (1 μg/mL IF) | pBB280 |
| Mouse CENP-C | Cheeseman lab (Swartz et al., 2019), rabbit polyclonal (1:1000 oocyte IF) | pKG137 |
| pS311 CENP-C | Cheeseman lab, this study, rabbit polyclonal (1 μg/mL IF) | 85B |
| Plk1 | Santa Cruz Biotech, mouse clone F8 (1:200 IF, WB) | Cat#sc17783, RRID: AB_628157, lot D1219 |
| Plk1 | Sigma Aldrich, mouse clone 35–206 (1:200 oocyte IF) | Cat#05-844, RRID: AB_310836 |
| Ndc80 “Bonsai” complex | Cheeseman lab (Schmidt et al., 2012), rabbit polyclonal (1 μg/mL IF) | Anti-Bonsai |
| Hec1 | Santa Cruz Biotech, mouse clone C11 (1:100 oocyte IF) | Cat#sc515550 |
| Mouse Bub1 | Gift of Yoshinori Watanabe (U Tokyo) (Kawashima et al., 2010), mouse polyclonal (1:100 oocyte IF) | Anti-mouse Bub1 |
| Separase | Abcam, mouse clone XJ11-1B12 (1:500 WB) | Cat#ab16170, RRID: AB_2101815, lot GR44153-1 |
| Cyclin B1 | Cell Signaling Tech, rabbit polyclonal (1:1000 WB) | Cat#4138, RRID: AB_2072132, lot 3 |
| Histone 3 pS10 | Abcam, rabbit polyclonal (1:3000 FC) | Cat#ab5176, RRID: AB_304763, lot GR3217296-1 |
| GFP polyclonal | Cheeseman lab, rabbit (IP) (Cheeseman and Desai, 2005) | Rabbit GFP polyclonal |
| Bacterial and Virus Strains | ||
| E coli: LOBSTR-BL21 (DE3)-RIL | Kerafast (Andersen et al., 2013) | Cat#EC1002 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Doxycycline | Sigma-Aldrich | Cat#D9891, CAS:24390-14-5 |
| S-trityl-L-cysteine (STLC) | Sigma-Aldrich | Cat#164739, CAS:2799-07-7 |
| Nocodazole | Sigma-Aldrich | Cat#M1404, CAS:31430-18-9 |
| AZ-3146 (Mps1i) | Tocris | Cat#3994, CAS:1124329-14-1 |
| BI-2536 (Plk1i) | Fisher Scientific | Cat#508733, CAS:755038-02-9 |
| RO-3306 (Cdk1i) | Sigma-Aldrich | Cat#SML0569, CAS:872573-93-8 |
| Lambda Protein Phosphatase | New England Biolabs | Cat#P0753 |
| Milrinone | Sigma-Aldrich | Cat#M4659 |
| GST-hCENP-C (700–943) | This study | pKM138 |
| GST-hCENP-C (808–943) | This study | pNM221 |
| GST-hCENP-C (775–943, 835–838A) | This study | pNM232 |
| GST-hCENP-C (775–943, 865–867A) | This study | pNM271 |
| GST-sfGFP-hMeikin (328–373) *GST tag removed during purification | This study | pNM190 |
| GST-sfGFP-hMeikin (328–373, ICCII/DIII mutant) *GST tag removed during purification | This study | pNM201 |
| GST-sfGFP-hMeikin (302–373) *GST tag removed during purification | This study | pNM175 |
| Critical Commercial Assays | ||
| PhosphoSens Plk1 activity assay | AssayQuant <assayquant.com> | Cat#CSKS-AQT0691K |
| T7 mScript Standard mRNA Production System | CellScript | Cat#CMSC11610 |
| Deposited Data | ||
| Original CellProfiler pipelines | Mendeley Data | Doi: 10.17632/7gwbym8y9n.1 |
| Mass-spectrometry RAW files | Mendeley Data | Doi: 10.17632/7gwbym8y9n.1 |
| Uncropped Western blot images | Mendeley Data | Doi: 10.17632/7gwbym8y9n.1 |
| Experimental Models: Cell Lines | ||
| Human: HeLa cells | Cheeseman lab stocks | RRID:CVCL_0030 |
| Human: 293GP cells (for retrovirus generation) | Cheeseman lab stocks | RRID:CVCL_E072 |
| Constitutive expression: 2xmNeonGreen-hMeikin | Hela: Retroviral transduction with pNM481, clonal, this study | cNM216-7 |
| Constitutive expression: 2xmNeonGreen-hMeikin (328–373) | Hela: Retroviral transduction with pNM512, clonal, this study | cNM239-14 |
| Constitutive expression: 2xmNeonGreen-hMeikin (1–332) | Hela: Retroviral transduction with pNM513, clonal, this study | cNM240-4 |
| Constitutive expression: 2xmNeonGreen-hMeikin (ICCII/DIII mutant) | Hela: Retroviral transduction with pNM511, clonal, this study | cNM238-3 |
| Constitutive expression: hMeikin-mNeonGreen | Hela: Retroviral transduction with pNM795, clonal, this study | cNM396-15 |
| Doxycycline inducible: EGFP | Hela: AAVS1 Safe-harbor insertion of pNM280, this study | cNM087 |
| Doxycycline inducible: EGFP-hMeikin | Hela: AAVS1 Safe-harbor insertion of pNM299, this study | cNM102 |
| Doxycycline inducible: EGFP-hMeikin (104–373) | Hela: AAVS1 Safe-harbor insertion of pNM480, this study | cNM220 |
| Doxycycline inducible: EGFP-hMeikin (124–373) | Hela: AAVS1 Safe-harbor insertion of pNM549, this study | cNM244 |
| Doxycycline inducible: EGFP-hMeikin (155–373) | Hela: AAVS1 Safe-harbor insertion of pNM603, this study | cNM271 |
| Doxycycline inducible: EGFP-hMeikin (184–373) | Hela: AAVS1 Safe-harbor insertion of pNM335, this study | cNM142 |
| Doxycycline inducible: EGFP-hMeikin (199–373) | Hela: AAVS1 Safe-harbor insertion of pNM336, this study | cNM143 |
| Doxycycline inducible: EGFP-hMeikin (227–373) | Hela: AAVS1 Safe-harbor insertion of pNM325, this study | cNM134 |
| Doxycycline inducible: EGFP-hMeikin (1–332) | Hela: AAVS1 Safe-harbor insertion of pNM310, this study | cNM123 |
| Doxycycline inducible: EGFP-hMeikin (1–310) | Hela: AAVS1 Safe-harbor insertion of pNM338, this study | cNM145 |
| Doxycycline inducible: EGFP-hMeikin (1–301) | Hela: AAVS1 Safe-harbor insertion of pNM311, this study | cNM124 |
| Doxycycline inducible: EGFP-hMeikin (1–226) | Hela: AAVS1 Safe-harbor insertion of pNM502, this study | cNM226 |
| Doxycycline inducible: EGFP-hMeikin (1–154) | Hela: AAVS1 Safe-harbor insertion of pNM680, this study | cNM311 |
| Doxycycline inducible: EGFP-hMeikin (124–332) | Hela: AAVS1 Safe-harbor insertion of pNM611, this study | cNM273 |
| Doxycycline inducible: EGFP-hMeikin (175–181 4A mut) | Hela: AAVS1 Safe-harbor insertion of pNM400, this study | cNM172 |
| Doxycycline inducible: EGFP-hMeikin (S196A) | Hela: AAVS1 Safe-harbor insertion of pNM398, this study | cNM170 |
| Doxycycline inducible: EGFP-hMeikin (STP mutant) | Hela: AAVS1 Safe-harbor insertion of pNM300, this study | cNM119 |
| Doxycycline inducible: EGFP-hMeikin (8A mutant) | Hela: AAVS1 Safe-harbor insertion of pNM604, this study | cNM272 |
| Doxycycline inducible: EGFP-hMeikin (E151R, R154E) | Hela: AAVS1 Safe-harbor insertion of pNM726, this study | cNM321 |
| Doxycycline inducible: EGFP-hMeikin (1–332)-H2B | Hela: AAVS1 Safe-harbor insertion of pNM801, this study | cNM371 |
| Doxycycline inducible: EGFP-hMeikin (1–154)-H2B | Hela: AAVS1 Safe-harbor insertion of pNM888, this study | cNM469 |
| Doxycycline inducible: EGFP-hMeikin (155–332)-H2B | Hela: AAVS1 Safe-harbor insertion of pNM802, this study | cNM366 |
| Doxycycline inducible: EGFP-hMeikin (1–332, STP mutant)-H2B | Hela: AAVS1 Safe-harbor insertion of pNM804, this study | cNM370 |
| Doxycycline inducible: EGFP-hMeikin (1–332, 8A mut)-H2B | Hela: AAVS1 Safe-harbor insertion of pNM803, this study | cNM372 |
| Doxycycline inducible: EGFP-hPlk1 | Hela: AAVS1 Safe-harbor insertion of pNM364, this study | cNM161 |
| Constitutive expression: H2B-mScarlet-hMeikin(1–332)-mNeonGreen | Hela: Retroviral transduction with pNM848, fluorescence enriched, this study | cNM426e |
| Constitutive expression: H2B-mScarlet-hMeikin(1–332, E151R, R154E)-mNeonGreen | Hela: Retroviral transduction with pNM849, fluorescence enriched, this study | cNM427e |
| Constitutive expression: H2B-mScarlet-hMeikin(1–332, S149A)-mNeonGreen | Hela: Retroviral transduction with pNM850, fluorescence enriched, this study | cNM428e |
| Constitutive expression: H2B-mScarlet-hMeikin(1–332, 8A mut)-mNeonGreen | Hela: Retroviral transduction with pNM852, fluorescence enriched, this study | cNM430e |
| Constitutive expression: H2B-mScarlet-hRad21(142–476)-mNeonGreen | Hela: Retroviral transduction with pNM853, fluorescence enriched, this study | cNM431e |
| Constitutive expression: H2B-mScarlet-hRad21(142–476, E169R, R172E, E447R, R450E, E457R, R460E)-mNeonGreen | Hela: Retroviral transduction with pNM854, fluorescence enriched, this study | cNM432e |
| Constitutive expression: H2B-mScarlet-hRec8(297–506)-mNeonGreen | Hela: Retroviral transduction with pNM937, fluorescence enriched, this study | cNM478e |
| Constitutive expression: H2B-mScarlet-hRec8(297–506)-hMeikin(1–332, E151R, R154E)-mNeonGreen | Hela: Retroviral transduction with pNM869, fluorescence enriched, this study | cNM486e |
| Constitutive expression: H2B-mScarlet-hRec8(297–506, E401R, R404E)-hMeikin(1–332, E151R, R154E)-mNeonGreen | Hela: Retroviral transduction with pNM972, fluorescence enriched, this study | cNM500e |
| Constitutive expression: H2B-mScarlet-hRec8(297–506)-hMeikin(155–332)-mNeonGreen | Hela: Retroviral transduction with pNM870, fluorescence enriched, this study | cNM487e |
| Constitutive expression: H2B-mScarlet-hRec8(297–506)-hMeikin(1–332, E151R, R154E, 8A mut)-mNeonGreen | Hela: Retroviral transduction with pNM946, fluorescence enriched, this study | cNM493e |
| Experimental Models: Organisms/Strains | ||
| Mouse: NSA(CF-1) | Envigo | N/A |
| Oligonucleotides | ||
| siRNA targeting ESPL1: GCUUGUGAUGCCAUCCUGAUU | Dharmacon (Waizenegger et al., 2002) | N/A |
| siRNA control pool | Dharmacon | Cat#D-001810-10 |
| Primers used for plasmid cloning | Eurofins Genomics | See Table S2 |
| Recombinant DNA | ||
| 3xEGFP-mMeikin | This study (for oocyte injection) | pNM530 |
| 3xEGFP-mMeikin (E156R, R159E) | This study (for oocyte injection) | pNM606 |
| 3xEGFP-mMeikin(1–391, E156R, R159E) - hRad21(142–275) - mMeikin(387–434) | This study (for oocyte injection) | pNM962 |
| 3xEGFP-mMeikin(1–391) - hRad21(142–275, E169R, R172E) - mMeikin(387–434) | This study (for oocyte injection) | pNM963 |
| mMeikin-3xEGFP | This study (for oocyte injection) | pNM739 |
| mMeikin-3xEGFP (L377A, E379A, N383A) | This study (for oocyte injection) | pNM1051 |
| mMeikin-3xEGFP (S381A) | This study (for oocyte injection) | pNM1089 |
| mNeonGreen-mMeikin | This study (for oocyte injection) | pNM474 |
| mNeonGreen-mMeikin (387–434) | This study (for oocyte injection) | pNM477 |
| mNeonGreen-mMeikin (392ICCII, 425DII → 392AAAAA, 425AAA) | This study (for oocyte injection) | pNM476 |
| mNeonGreen-mMeikin (T335A) | This study (for oocyte injection) | pNM475 |
| Software and Algorithms | ||
| Fiji (ImageJ) | NIH (Schindelin et al., 2012) | 2.0.0-rc-69/1.52p |
| CellProfiler | Broad Institute (McQuin et al., 2018) | v3.1.9 |
| Graphpad Prism | Graphpad Software | v8.4.3 |
| ProteomeDiscoverer | ThermoFisher | v2.4 |
| FACSDiva | BD Biosciences | v6.1.2 |
Experimental Model and Subject Details
Cell Culture
HeLa cells (transformed human female cervical epithelium) were cultured in Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum, 100 U/mL penicillin and streptomycin, and 2 mM L-glutamine at 37°C with 5% CO2. Doxycycline inducible cell lines were cultured in medium containing FBS certified as tetracycline free and were induced by addition of doxycycline to 1 μg/mL for 16 hr. Other drugs used on human cells were kinesin Eg5 inhibitor (S-trityl-L-cysteine, STLC, 10 μM), nocodazole (0.3 μM), AZ-3146 (Mps1i, 3 μM), BI-2536 (Plk1i, 10 μM), RO-3306 (Cdk1i, 10 μM). Hela cells were regularly monitored for mycoplasma contamination using commercial detection kits.
Mouse oocyte collection and culture
8–14 week-old female mice used in this study were purchased from Envigo (Strain: NSA(CF-1)). All animal experiments were approved by the University of Pennsylvania Institutional Animal Care and Use Committee and were consistent with the National Institutes of Health guidelines. Female mice were hormonally primed with 5U of Pregnant Mare Serum Gonadotropin (PMSG, Calbiochem, cat# 367222) 44–48 hr prior to oocyte collection. Germinal vesicle intact oocytes were collected in M2 medium (Sigma, M7167), denuded from cumulus cells, and cultured in CZB medium (Sigma, MR-019-D) covered with mineral oil (Sigma, M5310) in a humidified atmosphere of 5% CO2 in air at 37°C. During collection, meiotic resumption was inhibited by addition of 2.5 μM milrinone (Sigma, M4659).
Method Details
Plasmid cloning
hMeikin, mMeikin, and E. coli codon-optimized hCENP-C genes were synthesized at Genewiz. Mammalian Gene Collection (MGC Project Team, 2004) cDNAs of hRec8 (clone ID 3535425) and hPlk1 (clone ID 2822226) were obtained from Horizon Discovery. hRad21 gene sequence was obtained from plasmid NS205 and NS207 (Shindo et al., 2012). Plasmids were assembled by polymerase chain reaction (PCR) amplification of inserts with Q5 DNA Polymerase followed by digestion with restriction enzymes and ligation by T4-ligase (New England Biolabs) into vector backbones described below. Specific mutations were generated by overlap-extension PCR. Primers used are listed in Table S2. Ligation mixes were transformed into Mach1 E-coli (ThermoFisher Scientific) and colonies screened by Sanger-sequencing.
Cell line generation
The cell lines used in this study are described in the Key Resources Table. pBABE derivatives were transfected with Effectene (Qiagen) according to the manufacturer’s protocol along with VSVG packaging plasmid into 293-GP cells for generation of retrovirus as described (Morgenstern and Land, 1990). Supernatant-containing retrovirus was sterile filtered, supplemented with 20 μg/mL polybrene (Millipore) and used to transduce HeLa cells. Doxycycline-inducible cell lines were generated by homology-directed insertion into the AAVS1 “safe-harbor” locus. Donor plasmid containing selection marker, the tetracycline-responsive promoter, the transgene, and reverse tetracycline-controlled transactivator flanked by AAVS1 homology arms (Qian et al., 2014) was transfected using Effectene with a pX330-based plasmid (Cong et al., 2013) expressing both spCas9 and a guide RNA specific for the AAVS1 locus (pNM220, gRNA sequence – 5’-GGGGCCACTAGGGACAGGAT). Two days post-transfection or transduction, cells were selected with the appropriate antibiotic (puromycin at 0.5 μg/mL or blasticidin at 2 μg/mL, Life Technologies). Where indicated, clonal lines were obtained by fluorescence activated cell-sorting single cells into 96 well plates. Fluorescence enriched lines were generated by bulk sorting a polyclonal population for fluorescence positive cells.
Immunofluorescence and microscopy of mitotic cells
Cells for immunofluorescence were seeded on poly-L-lysine (Sigma-Aldrich) coated coverslips and fixed in PBS plus 4% formaldehyde for 10 min unless otherwise noted in figure legends. Coverslips were washed with PBS plus 0.1% Triton X-100 and blocked in Abdil (20 mM Tris-HCl, 150 mM NaCl, 0.1% Triton X-100, 3% bovine serum albumin, 0.1% NaN3, pH 7.5). Primary antibodies used in this study are described in the Key Resources Table and were diluted in Abdil. Dilutions are listed in the Key Resources Table (IF = immunofluorescence, WB = Western blot, IP = immunoprecipitation, FC = flow cytometry). Cy3- and Cy5-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories) were diluted 1:300 in PBS plus 0.1% Triton X-100. DNA was stained with 1 μg/mL Hoechst-33342 (Sigma-Aldrich) in PBS plus 0.1% Triton X-100 for 10 min. Coverslips were mounted using PPDM (0.5% p-phenylenediamine, 20 mM Tris-HCl, pH 8.8, 90% glycerol). Images were acquired on a DeltaVision Core deconvolution microscope (Applied Precision) equipped with a CoolSnap HQ2 charge-coupled device camera and deconvolved where appropriate. All images are maximal projections in z. Image analysis was performed in Fiji (ImageJ, NIH) (Schindelin et al., 2012).
Integrated fluorescence intensity of mitotic kinetochores was measured with a custom CellProfiler pipeline (McQuin et al., 2018). The median intensity of a 5-pixel wide region surrounding each kinetochore was used to background subtract each measurement.
Live-cell imaging of mitotic cells
For live-cell imaging, cells were seeded into 8-well glass-bottomed chambers (Ibidi) and moved into CO2-independent media (Life Technologies) before imaging at 37°C. For certain movies, DNA was stained with SiR-DNA (Cytoskeleton Inc) at 0.2 μM. Images were acquired on a DeltaVision Core deconvolution microscope (Applied Precision) equipped with a CoolSnap HQ2 charge-coupled device camera and deconvolved where appropriate. Image analysis was performed in Fiji (ImageJ, NIH). For quantification of H2B cleavage sensors, cells undergoing anaphase were selected in Fiji then analyzed using a custom CellProfiler pipeline (McQuin et al., 2018). The pipeline segmented cells based on SiR-DNA signal, performed background subtraction, and measured mNeonGreen and mScarlet signal in each cell. The mNeonGreen/mScarlet ratio was normalized to the first timepoint (t = −15 min).
RNAi treatment
siRNAs against ESPL1 (5’-GCUUGUGAUGCCAUCCUGAUU) (Waizenegger et al., 2002) and non-targeting control pool (D-001810–10) were obtained from Dharmacon. 5 μL of 20 μM stock siRNA was mixed with 5 μL of Lipofectamine RNAiMax (Life Technologies) and diluted in 500 μL of OptiMem (Life Technologies). The reaction was applied to cells in a 6-well dish. Transfection media was changed after 24 hr.
Mitotic index determination
Meikin expressing cells were induced with 1 μg/mL doxycycline for 24 hr. Cells were collected by incubation for 10 min in PBS + 5 mM EDTA, washed once in PBS, then fixed in PBS + 2% formaldehyde for 10 min at room temperature. Cells were blocked in Abdil for 30 min followed by immunostaining for phosphorylated S10 on histone 3 followed by Cy-5 conjugated secondary antibody. The proportion of GFP-positive single cells also staining positive for H3pS10 was determined on an LSRFortessa (BD Biosciences) flow cytometer and analyzed with FACSDiva software (BD Biosciences). Over 5,000 GFP-positive cells were analyzed per condition.
GFP immunoprecipitation and Mass-spectrometry
IP-MS experiments were performed as described previously (Cheeseman and Desai, 2005). Harvested cells were washed in PBS and resuspended 1:1 in 1X Lysis Buffer (50 mM HEPES, 1 mM EGTA, 1 mM MgCl2, 100 mM KCl, 10% glycerol, pH 7.4) then drop frozen in liquid nitrogen. Cells were thawed after addition of an equal volume of 1.5X lysis buffer supplemented with 0.075% Nonidet P-40, 1X Complete EDTA-free protease inhibitor cocktail (Roche), 1 mM phenylmethylsulfonyl fluoride, 20 mM beta-glycerophosphate, 1 mM sodium fluoride, and 0.4 mM sodium orthovanadate. Cells were lysed by sonication and cleared by centrifugation. The supernatant was mixed with Protein A beads coupled to rabbit anti-GFP antibodies (Cheeseman lab) and rotated at 4°C for 1 hr. Beads were washed five times in Wash Buffer (50 mM HEPES, 1 mM EGTA, 1 mM MgCl2, 300 mM KCl, 10% glycerol, 0.05% NP-40, 1 mM dithiothreitol, 10 μg/mL leupeptin/pepstatin/chymostatin, pH 7.4). After a final wash in Wash Buffer without detergent, bound protein was eluted with 100 mM glycine pH 2.6. Eluted proteins were precipitated by addition of 1/5th volume trichloroacetic acid at 4°C overnight. Precipitated proteins were reduced with TCEP, alkylated with iodoacetamide, and digested with mass-spectrometry grade Lys-C and trypsin (Promega). Digested peptides were cleaned up using C18 spin columns (Pierce) according to the manufacturer’s instructions. Samples were analyzed on an Orbitrap Exploris 480 mass spectrometer connected to an EASY-nLC chromatography system (Thermo Fisher Scientific). Protein identifications were generated in Proteome Discoverer 2.4 (Thermo Fisher Scientific) using Sequest HT and MS Amanda 2.0. Peptide-spectrum matches were validated using Percolator. Phosphorylation sites were identified using IMP-ptmRS in PhosphoRS mode.
GFP immunoprecipitation and Western blot
For IP-Western experiments, cells were harvested, washed once in PBS, then lysed on ice for 15 min in Lysis Buffer (50 mM HEPES, 1 mM EGTA, 1 mM MgCl2, 100 mM KCl, 10% glycerol, 1% Triton X-100, 0.05% NP-40, 1 mM dithiothreitol, 1X Complete EDTA-free protease inhibitor cocktail (Roche), 1 mM phenylmethylsulfonyl fluoride, 20 mM β-glycerophosphate, 1 mM sodium fluoride, and 0.4 mM sodium orthovanadate, pH 7.4). Cellular debris was removed by centrifugation. Protein concentrations in each sample were measured using Bradford reagent (Bio-Rad), and sample concentrations were normalized before addition of Protein A beads (Bio-Rad) coupled to affinity-purified rabbit anti-GFP polyclonal antibodies (Cheeseman lab). After 1 hr incubation at 4°C, beads were washed 3X with Wash Buffer (50 mM HEPES, 1 mM EGTA, 1 mM MgCl2, 100 mM KCl, 10% glycerol, 0.05% NP-40, 1 mM dithiothreitol, 10 μg/mL leupeptin/pepstatin/chymostatin, pH 7.4). Beads were then incubated in an equal volume of Laemmli buffer for 5 min at 95°C to remove bound protein. Samples were analyzed by SDS-PAGE and Western blotting. Samples were separated by SDS-PAGE and semidry transferred to nitrocellulose. Membranes were blocked for 1h in Blocking Buffer (5% milk in TBS + 0.1% Tween-20). Primary antibodies were diluted in Blocking Buffer + 0.2% NaN3 and applied to the membrane for 1 hr. HRP-conjugated secondary antibodies (GE Healthcare) were diluted 1:10,000 in TBS + 0.1% Tween-20 and applied to the membrane for 1 hr. After washing in TBS + 0.1% Tween-20, Clarity enhanced chemiluminescence substrate (Bio-Rad) was added to the membrane according to the manufacturer’s instructions. Membranes were imaged with a KwikQuant Imager (Kindle Biosciences). Membranes were stripped (55°C, 1 hr) in stripping buffer (60 mM Tris-HCl, pH 6.8, 2% SDS, 100 mM β-mercaptoethanol) and re-blocked before re-probing. Densitometry analysis was performed in Image J.
Anaphase synchronization and cell lysis
For synchronization in anaphase, cells were arrested by treatment with STLC for 16 hr followed by addition of Mps1i for the times indicated in the figure legends. Cells were harvested and washed once in PBS. Cell pellets were resuspended in radioimmunoprecipitation buffer (RIPA, ThermoFisher) supplemented with 1X Complete EDTA-free protease inhibitor cocktail (Roche), 1 mM phenylmethylsulfonyl fluoride, 20 mM β-glycerophosphate, 1 mM sodium fluoride, and 0.4 mM sodium orthovanadate. Cells were lysed on ice for 15 min, and cellular debris was removed by centrifugation at >10,000 g for 10 min at 4°C. Protein concentrations were measured and normalized using a bicinchoninic protein assay (Pierce). Samples were analyzed by SDS-PAGE and Western blot.
Phosphatase treatment of lysates
Cells treated with the drugs indicated in the Figure legends were collected, washed once with PBS then lysed in Lysis Buffer without phosphatase inhibitors (50 mM HEPES, 1 mM EGTA, 1 mM MgCl2, 100 mM KCl, 10% glycerol, 0.05% NP-40, 1 mM dithiothreitol, 1X Complete EDTA-free protease inhibitor cocktail (Roche), 1 mM phenylmethylsulfonyl fluoride, pH 7.4) for 10 min on ice. Cellular debris was removed by centrifugation and protein concentrations were measured by Bradford assay (Bio-Rad) and normalized. Cell lysates were supplemented with 1X Protein MetalloPhosphatase buffer (New England Biolabs) and 1 mM MnCl2. 1 μL of Lambda Protein Phosphatase (New England Biolabs) or 1 μL of phosphatase inhibitor mix (20 mM β-glycerophosphate, 1 mM sodium fluoride, and 0.4 mM sodium orthovanadate) was added to each 50 μL reaction. After incubation at 30°C for 30 min, reactions were stopped by addition of 2X Laemmli buffer. Samples were analyzed by SDS-PAGE and Western blot.
In vitro Plk1 activity assays
Plk1 and Plk1/Meikin complexes were immunoprecipitated from HeLa cells. Cells were induced to express EGFP-Plk1 or EGFP-Meikin by treatment with doxycycline and arrested in mitosis by STLC for 16 hr. Cells were harvested, lysed and GFP-immunoprecipitated as above for mass-spectrometry. Bound proteins were eluted by addition of recombinant Tobacco Etch Virus (TEV) protease (Cheeseman lab) and incubation at 4°C for 16 hr which leads to cleavage between EGFP and the tagged-protein. The amount of Plk1 in each elution was normalized by quantitative Western blot.
Enzyme kinetic parameters were measured in a continuous read fluorescence assay. A peptide substrate containing the non-natural Sox amino acid utilizes chelation-enhanced fluorescence to report on phosphorylation status (Shults et al., 2006). The proprietary peptide sequence was validated for use with human Plk1 by the manufacturer (PhosphoSens CSKS-AQT0691K, Assay Quant). Assay was performed according to manufacturer directions in a SpectraMax iD3 plate reader (Molecular Devices) using a 96-well format at 30°C with reads every 60 sec. Substrate concentrations were varied from 1–35 μM. Initial reaction rates were fit to the Michaelis-Menten equation in GraphPad Prism.
Recombinant protein expression and purification
Plasmids used for recombinant protein expression were based on the pGEX-6P1 backbone and are described in the Key Resources Table. BL21(DE3) LOBSTR cells (Andersen et al., 2013) carrying the pRARE tRNA plasmid were transformed with the appropriate plasmid and plated on Luria-Bertani (LB)-agar plates containing the appropriate antibiotic. Overnight liquid cultures of LB supplemented with antibiotics and 4% glucose were grown overnight at 37°C from single colonies. The saturated overnight culture was diluted 1:100 and grown to an OD600nm of 0.6–0.7 at 30°C. Cells were shifted to 16°C and induced with 0.3 mM isopropyl β-D-1-thiogalactopyranoside and incubated for 16 hr. Cells were collected by centrifugation, resuspended in lysis buffer, and flash frozen in liquid nitrogen.
Cell pellets were resuspended in Lysis buffer (1X PBS supplemented with 250 mM NaCl, 0.1% Tween-20, 1 mM dithiothreitol, and 1 mM phenylmethylsulfonyl fluoride). Cells were disrupted by sonication, and the lysate was cleared by centrifugation. The lysate was applied to 0.5 mL of glutathione agarose (Sigma-Aldrich) per liter of culture for 1 hr at 4°C. Agarose was washed three times in Lysis Buffer, and proteins were eluted using Elution Buffer (50 mM Tris-HCl, 75 mM KCl, 10 mM reduced glutathione, pH 8.0). For certain proteins, the glutathione Stransferase (GST) purification tag was removed by incubation overnight at 4°C with 1 mg HRV-3C protease (Cheeseman lab) per 50 mL elution fraction. Final polishing was performed by gelfiltration on a Superdex 200 16/60 coupled to an AktaPurifier system (GE Healthcare) into Binding Buffer (50 mM HEPES, 400 mM KCl, 10% glycerol, 0.1% Tween-20, 1 mM EDTA, 1 mM dithiothreitol, pH 7.4). Peak fractions were concentrated in Vivaspin concentrators (GE Healthcare), aliquoted, and snap-frozen in liquid nitrogen.
In vitro binding assays and gel filtration
50 μL binding reactions were prepared with recombinant proteins diluted to 3.5 μM each in Binding Buffer (50 mM HEPES, 400 mM KCl, 10% glycerol, 0.1% Tween-20, 1 mM EDTA, 1 mM dithiothreitol, pH 7.4). Reactions were incubated on ice for 1 hr before clearing by centrifugation at >10,000 g for 10 min. Cleared samples were run over a Superdex 200 3.2/300 column on an Akta Micro FPLC system (GE Healthcare) pre-equilibrated in Binding Buffer. 40 μL fractions were analyzed by SDS-PAGE followed by staining with Acquastain (Bulldog Bio). For examination of the binding interaction under variable salt concentrations, the KCl concentration in the Binding Buffer was altered according to Figure legends.
Phospho-antibody generation
The CENP-C pS311 phosphospecific antibody was generated against a synthesized phosphopeptide with the following amino acid sequence: CNLRNEE(pS)VLLFTQ (New England Peptide; Covance). Peptide was coupled to Sulfolink Coupling Resin (Thermo Fisher Scientific). Serum from immunized rabbit was depleted against the unphosphorylated peptide and affinity purified against the phosphorylated peptide.
Mouse oocyte injection
Germinal vesicle intact oocytes were microinjected with ~5 pL of cRNAs in M2 medium containing milrinone at room temperature with a micromanipulator TransferMan NK 2 (Eppendorf) and picoinjector (Medical Systems Corp.). Injected oocytes were kept in CZB with milrinone for 6–12 hr to allow protein expression, before switching to milrinone-free CZB medium to develop to meiosis I (6.5 hr in vitro maturation) or meiosis II (16 hr in vitro maturation). Oocytes were checked for germinal vesicle breakdown 1.5 hr after milrinone washout, and those that did not breakdown the germinal vesicle were discarded. Meiosis II eggs were activated to progress to anaphase II in CZB medium containing 5mM SrCl2 and 2 mM EGTA for 1 hr as previously described (Kishigami and Wakayama, 2007).
Plasmids for cRNA generation were generated in the pCS2+ backbone (von Dassow et al., 2009) and are described in the Key Resources Table. cRNAs were synthesized using the T7 mScript Standard mRNA Production System (Cell Script) and injected at ~125 ng/μL final concentration.
Oocyte immunocytochemistry and imaging
For immunocytochemistry, samples were fixed in 2% paraformaldehyde in PBS for 30 min at room temperature. The cells were then permeabilized for 15 min in PBS containing 0.2% Triton X-100, blocked in PBS containing 0.2% immunoglobulin G-free bovine serum albumin and 0.01% Tween-20 for 30 min (blocking solution) and then incubated with the primary antibody for 1 hr at room temperature. After four 15 min washings in blocking solution, samples were incubated for 1 hr with either Alexa Flour 594-conjugated (1:500, Invitrogen) or Cy5-conjugated (1:100, Jackson ImmunoResearch) secondary antibody diluted in blocking solution. After an additional three 15-min washings in blocking solution, the samples were mounted in Vectashield mounting solution containing DAPI (Vector Laboratories). Confocal images were collected with a microscope (DMI4000 B; Leica) equipped with a 63× 1.3 NA glycerol-immersion objective lens, an x-y piezo Z stage (Applied Scientific Instrumentation), a spinning disk confocal scanner (Yokogawa Corporation of America), an electron multiplier charge-coupled device camera (ImageEM C9100–13; Hamamatsu Photonics), and an LMM5 laser merge module with 488- and 593-nm diode lasers (Spectral Applied Research) controlled by MetaMorph software (Molecular Devices). Confocal images were collected as z-stacks at 1 μm intervals to visualize the entire meiotic spindle.
To quantify Plk1 and Bub1 signal intensity at kinetochores, CENP-C staining was used to select kinetochores. For each kinetochore, a maximum intensity projection of the optical Z-sections containing CENP-C signal was performed using Fiji/Image J. A circle was drawn around the CENP-C signal, and the same circle was used to quantify Plk1 or Bub1 signal intensity. The mean signal intensity was measured for each circle after subtracting background signal from the surrounding area. Values were normalized to the mean of the wild-type or uninjected control.
To compare the relative expression levels of injected mRNA in mouse oocytes, the average GFP signal intensity across the entire cell volume of injected oocytes was measured by Image J and the background signal of the area outside of the cell was subtracted.
Quantification and statistical analysis
Fiji/ImageJ (NIH) was used for image manipulation and fluorescence quantification. Where indicated in the method details, a custom CellProfiler pipeline was used. Statistical tests and analysis of enzyme kinetics were performed in Graphpad Prism. All statistical details including type of test and exact value of n is included in the figure legends.
Supplementary Material
Table S1. Proteins identified by mass-spectrometry in EGFP-hMeikin immunoprecipitates from HeLa cells. Related toFigure 2. HeLa cells expressing EGFP-hMeikin were lysed and immunoprecipitation with anti-GFP antibody was performed. Immunoprecipitated proteins were identified by mass-spectrometry. Key proteins relevant to this work are highlighted. The sum of multiple experiments is shown. Proteins with low statistical confidence and known contaminants (keratin, tubulin, actin) have been removed from the list.
Table S2. Primers used for plasmid cloning.
Highlights.
The meiosis specific kinetochore protein Meikin is cleaved by the protease Separase
Preventing Meikin cleavage causes meiosis II chromosome alignment defects in oocytes
The C-terminal Meikin fragment retains key activities and is required for meiosis II
Meikin is eliminated by APC/C-mediated degradation at meiotic exit
Acknowledgements
We thank the members of the Cheeseman and Lampson labs for their support and input. This work was supported by grants from The Harold G & Leila Y. Mathers Charitable Foundation, NSF (2029868), and a Pilot award from the Global Consortium for Reproductive Longevity and Equity to IMC, grants from the NIH/NIGMS (R35GM126930 to IMC and GM122475 to MAL), and the Henry and Frances Keany Rickard Fund Fellowship from the MIT Office of Graduate Education to NKM. The graphical abstract was created with Biorender.com.
Footnotes
Declaration of Interests
Iain Cheeseman is a member of the Editorial Advisory Board for Developmental Cell.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexandru G, Uhlmann F, Mechtler K, Poupart MA, and Nasmyth K (2001). Phosphorylation of the cohesin subunit Scc1 by Polo/Cdc5 kinase regulates sister chromatid separation in yeast. Cell 105, 459–472. [DOI] [PubMed] [Google Scholar]
- Andersen KR, Leksa NC, and Schwartz TU (2013). Optimized E. coli expression strain LOBSTR eliminates common contaminants from His-tag purification. Proteins 81, 1857–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashkenazy H, Abadi S, Martz E, Chay O, Mayrose I, Pupko T, and Ben-Tal N (2016). ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res 44, W344–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonner AM., Hughe SE., and Hawle RS. (2020). Regulation of Polo Kinase by Matrimony Is Required for Cohesin Maintenance during Drosophila melanogaster Female Meiosis. Curr Biol 30, 715–722.e713. [DOI] [PubMed] [Google Scholar]
- Brar GA, Kiburz BM, Zhang Y, Kim JE, White F, and Amon A (2006). Rec8 phosphorylation and recombination promote the step-wise loss of cohesins in meiosis. Nature 441, 532–536. [DOI] [PubMed] [Google Scholar]
- Buonomo SB, Clyne RK, Fuchs J, Loidl J, Uhlmann F, and Nasmyth K (2000). Disjunction of homologous chromosomes in meiosis I depends on proteolytic cleavage of the meiotic cohesin Rec8 by separin. Cell 103, 387–398. [DOI] [PubMed] [Google Scholar]
- Cahoon CK, and Hawley RS (2016). Regulating the construction and demolition of the synaptonemal complex. Nat Struct Mol Biol 23, 369–377. [DOI] [PubMed] [Google Scholar]
- Cheeseman IM, and Desai A (2005). A combined approach for the localization and tandem affinity purification of protein complexes from metazoans. Sci STKE 2005, pl1. [DOI] [PubMed] [Google Scholar]
- Chelysheva L, Diallo S, Vezon D, Gendrot G, Vrielynck N, Belcram K, Rocques N, Márquez-Lema A, Bhatt AM, Horlow C, et al. (2005). AtREC8 and AtSCC3 are essential to the monopolar orientation of the kinetochores during meiosis. J Cell Sci 118, 4621–4632. [DOI] [PubMed] [Google Scholar]
- Chiang T, Duncan FE, Schindler K, Schultz RM, and Lampson MA (2010). Evidence that weakened centromere cohesion is a leading cause of age-related aneuploidy in oocytes. Curr Biol 20, 1522–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combes G, Alharbi I, Braga LG, and Elowe S (2017). Playing polo during mitosis: PLK1 takes the lead. Oncogene 36, 4819–4827. [DOI] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey NE, and Morgan DO (2016). Building a Regulatory Network with Short Linear Sequence Motifs: Lessons from the Degrons of the Anaphase-Promoting Complex. Mol Cell 64, 12–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earnshaw WC, Ratrie H 3rd, and Stetten G (1989). Visualization of centromere proteins CENP-B and CENP-C on a stable dicentric chromosome in cytological spreads. Chromosoma 98, 1–12. [DOI] [PubMed] [Google Scholar]
- Elia AE, Cantley LC, and Yaffe MB (2003). Proteomic screen finds pSer/pThr-binding domain localizing Plk1 to mitotic substrates. Science 299, 1228–1231. [DOI] [PubMed] [Google Scholar]
- Galander S, Barton RE, Borek WE, Spanos C, Kelly DA, Robertson D, Rappsilber J, and Marston AL (2019a). Reductional Meiosis I Chromosome Segregation Is Established by Coordination of Key Meiotic Kinases. Dev Cell 49, 526–541.e525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galander S, Barton RE, Kelly DA, and Marston AL (2019b). Spo13 prevents premature cohesin cleavage during meiosis. Wellcome Open Res 4, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galander S, and Marston AL (2020). Meiosis I Kinase Regulators: Conserved Orchestrators of Reductional Chromosome Segregation. Bioessays 42, e2000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gascoigne KE, Takeuchi K, Suzuki A, Hori T, Fukagawa T, and Cheeseman IM (2011). Induced ectopic kinetochore assembly bypasses the requirement for CENP-A nucleosomes. Cell 145, 410–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein LS (1981). Kinetochore structure and its role in chromosome orientation during the first meiotic division in male D. melanogaster. Cell 25, 591–602. [DOI] [PubMed] [Google Scholar]
- Gruber S, Haering CH, and Nasmyth K (2003). Chromosomal cohesin forms a ring. Cell 112, 765–777. [DOI] [PubMed] [Google Scholar]
- Gryaznova Y, Keating L, Touati SA, Cladière D, El Yakoubi W, Buffin E, and Wassmann K (2021). Kinetochore individualization in meiosis I is required for centromeric cohesin removal in meiosis II. Embo j, e106797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauf S, Roitinger E, Koch B, Dittrich CM, Mechtler K, and Peters JM (2005). Dissociation of cohesin from chromosome arms and loss of arm cohesion during early mitosis depends on phosphorylation of SA2. PLoS Biol 3, e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauf S, Waizenegger IC, and Peters JM (2001). Cohesin cleavage by separase required for anaphase and cytokinesis in human cells. Science 293, 1320–1323. [DOI] [PubMed] [Google Scholar]
- Homer H (2013). The APC/C in female mammalian meiosis I. Reproduction 146, R61–71. [DOI] [PubMed] [Google Scholar]
- Hornig NC, and Uhlmann F (2004). Preferential cleavage of chromatin-bound cohesin after targeted phosphorylation by Polo-like kinase. Embo j 23, 3144–3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiguro T, Tanaka K, Sakuno T, and Watanabe Y (2010). Shugoshin-PP2A counteracts casein-kinase-1-dependent cleavage of Rec8 by separase. Nat Cell Biol 12, 500–506. [DOI] [PubMed] [Google Scholar]
- Kamenz J, and Hauf S (2017). Time To Split Up: Dynamics of Chromosome Separation. Trends Cell Biol 27, 42–54. [DOI] [PubMed] [Google Scholar]
- Katis VL, Lipp JJ, Imre R, Bogdanova A, Okaz E, Habermann B, Mechtler K, Nasmyth K, and Zachariae W (2010). Rec8 phosphorylation by casein kinase 1 and Cdc7-Dbf4 kinase regulates cohesin cleavage by separase during meiosis. Dev Cell 18, 397–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima SA, Yamagishi Y, Honda T, Ishiguro K, and Watanabe Y (2010). Phosphorylation of H2A by Bub1 prevents chromosomal instability through localizing shugoshin. Science 327, 172–177. [DOI] [PubMed] [Google Scholar]
- Keeney S, Giroux CN, and Kleckner N (1997). Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell 88, 375–384. [DOI] [PubMed] [Google Scholar]
- Kim J, Ishiguro K, Nambu A, Akiyoshi B, Yokobayashi S, Kagami A, Ishiguro T, Pendas AM, Takeda N, Sakakibara Y, et al. (2015a). Meikin is a conserved regulator of meiosis-I-specific kinetochore function. Nature 517, 466–471. [DOI] [PubMed] [Google Scholar]
- Kim J, Lee K, and Rhee K (2015b). PLK1 regulation of PCNT cleavage ensures fidelity of centriole separation during mitotic exit. Nat Commun 6, 10076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishigami S, and Wakayama T (2007). Efficient strontium-induced activation of mouse oocytes in standard culture media by chelating calcium. J Reprod Dev 53, 1207–1215. [DOI] [PubMed] [Google Scholar]
- Kishimoto T (2003). Cell-cycle control during meiotic maturation. Curr Opin Cell Biol 15, 654–663. [DOI] [PubMed] [Google Scholar]
- Kitajima TS, Ohsugi M, and Ellenberg J (2011). Complete kinetochore tracking reveals error-prone homologous chromosome biorientation in mammalian oocytes. Cell 146, 568–581. [DOI] [PubMed] [Google Scholar]
- Klare K, Weir JR, Basilico F, Zimniak T, Massimiliano L, Ludwigs N, Herzog F, and Musacchio A (2015). CENP-C is a blueprint for constitutive centromere-associated network assembly within human kinetochores. J Cell Biol 210, 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo NR, Anger M, Peters AH, Stemmann O, Theussl HC, Helmhart W, Kudo H, Heyting C, and Nasmyth K (2009). Role of cleavage by separase of the Rec8 kleisin subunit of cohesin during mammalian meiosis I. J Cell Sci 122, 2686–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KS, Park JE, Kang YH, Zimmerman W, Soung NK, Seong YS, Kwak SJ, and Erikson RL (2008). Mechanisms of mammalian polo-like kinase 1 (Plk1) localization: self- versus non-self-priming. Cell Cycle 7, 141–145. [DOI] [PubMed] [Google Scholar]
- Li X, and Dawe RK (2009). Fused sister kinetochores initiate the reductional division in meiosis I. Nat Cell Biol 11, 1103–1108. [DOI] [PubMed] [Google Scholar]
- Marston AL (2015). Shugoshins: tension-sensitive pericentromeric adaptors safeguarding chromosome segregation. Mol Cell Biol 35, 634–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marston AL, and Amon A (2004). Meiosis: cell-cycle controls shuffle and deal. Nat Rev Mol Cell Biol 5, 983–997. [DOI] [PubMed] [Google Scholar]
- Marston AL, and Wassmann K (2017). Multiple Duties for Spindle Assembly Checkpoint Kinases in Meiosis. Front Cell Dev Biol 5, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuin C, Goodman A, Chernyshev V, Kamentsky L, Cimini BA, Karhohs KW, Doan M, Ding L, Rafelski SM, Thirstrup D, et al. (2018). CellProfiler 3.0: Next-generation image processing for biology. PLoS Biol 16, e2005970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MGC Project Team. (2004). The status, quality, and expansion of the NIH full-length cDNA project: the Mammalian Gene Collection (MGC). Genome Res 14, 2121–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MP, Amon A, and Ünal E (2013). Meiosis I: when chromosomes undergo extreme makeover. Curr Opin Cell Biol 25, 687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki S, Kim J, Yamagishi Y, Ishiguro T, Okada Y, Tanno Y, Sakuno T, and Watanabe Y (2017). Meikin-associated polo-like kinase specifies Bub1 distribution in meiosis I. Genes Cells 22, 552–567. [DOI] [PubMed] [Google Scholar]
- Morgenstern JP, and Land H (1990). Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res 18, 3587–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogushi S, Rattani A, Godwin J, Metson J, Schermelleh L, and Nasmyth K (2020). Loss of Sister Kinetochore Co-orientation and Peri-centromeric Cohesin Protection after Meiosis I Depends on Cleavage of Centromeric REC8. bioRxiv, 2020.2002.2006.935171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petronczki M, Lénárt P, and Peters JM (2008). Polo on the Rise-from Mitotic Entry to Cytokinesis with Plk1. Dev Cell 14, 646–659. [DOI] [PubMed] [Google Scholar]
- Qian K, Huang CT, Chen H, Blackbourn L.W.t., Chen Y, Cao J, Yao L, Sauvey C, Du Z, and Zhang SC (2014). A simple and efficient system for regulating gene expression in human pluripotent stem cells and derivatives. Stem Cells 32, 1230–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuno T, Tada K, and Watanabe Y (2009). Kinetochore geometry defined by cohesion within the centromere. Nature 458, 852–858. [DOI] [PubMed] [Google Scholar]
- Sarangapani KK, Duro E, Deng Y, Alves Fde L, Ye Q, Opoku KN, Ceto S, Rappsilber J, Corbett KD, Biggins S, et al. (2014). Sister kinetochores are mechanically fused during meiosis I in yeast. Science 346, 248–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt JC, Arthanari H, Boeszoermenyi A, Dashkevich NM, Wilson-Kubalek EM, Monnier N, Markus M, Oberer M, Milligan RA, Bathe M, et al. (2012). The kinetochore-bound Ska1 complex tracks depolymerizing microtubules and binds to curved protofilaments. Dev Cell 23, 968–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severson AF, Ling L, van Zuylen V, and Meyer BJ (2009). The axial element protein HTP-3 promotes cohesin loading and meiotic axis assembly in C. elegans to implement the meiotic program of chromosome segregation. Genes Dev 23, 1763–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindo N, Kumada K, and Hirota T (2012). Separase sensor reveals dual roles for separase coordinating cohesin cleavage and cdk1 inhibition. Dev Cell 23, 112–123. [DOI] [PubMed] [Google Scholar]
- Shults MD, Carrico-Moniz D, and Imperiali B (2006). Optimal Sox-based fluorescent chemosensor design for serine/threonine protein kinases. Anal Biochem 352, 198–207. [DOI] [PubMed] [Google Scholar]
- Sivakumar S, and Gorbsky GJ (2015). Spatiotemporal regulation of the anaphase-promoting complex in mitosis. Nat Rev Mol Cell Biol 16, 82–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan M, and Morgan DO (2007). A novel destruction sequence targets the meiotic regulator Spo13 for anaphase-promoting complex-dependent degradation in anaphase I. J Biol Chem 282, 19710–19715. [DOI] [PubMed] [Google Scholar]
- Swartz SZ, McKay LS, Su KC, Bury L, Padeganeh A, Maddox PS, Knouse KA, and Cheeseman IM (2019). Quiescent Cells Actively Replenish CENP-A Nucleosomes to Maintain Centromere Identity and Proliferative Potential. Dev Cell 51, 35–48.e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana-Konwalski K, Godwin J, van der Weyden L, Champion L, Kudo NR, Adams DJ, and Nasmyth K (2010). Rec8-containing cohesin maintains bivalents without turnover during the growing phase of mouse oocytes. Genes Dev 24, 2505–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Chang HL, Kagami A, and Watanabe Y (2009). CENP-C functions as a scaffold for effectors with essential kinetochore functions in mitosis and meiosis. Dev Cell 17, 334–343. [DOI] [PubMed] [Google Scholar]
- Uhlmann F, Lottspeich F, and Nasmyth K (1999). Sister-chromatid separation at anaphase onset is promoted by cleavage of the cohesin subunit Scc1. Nature 400, 37–42. [DOI] [PubMed] [Google Scholar]
- Uhlmann F, Wernic D, Poupart MA, Koonin EV, and Nasmyth K (2000). Cleavage of cohesin by the CD clan protease separin triggers anaphase in yeast. Cell 103, 375–386. [DOI] [PubMed] [Google Scholar]
- von Dassow G, Verbrugghe KJ, Miller AL, Sider JR, and Bement WM (2009). Action at a distance during cytokinesis. J Cell Biol 187, 831–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waizenegger I, Giménez-Abián JF, Wernic D, and Peters JM (2002). Regulation of human separase by securin binding and autocleavage. Curr Biol 12, 1368–1378. [DOI] [PubMed] [Google Scholar]
- Watanabe Y, and Nurse P (1999). Cohesin Rec8 is required for reductional chromosome segregation at meiosis. Nature 400, 461–464. [DOI] [PubMed] [Google Scholar]
- Whitfield ZJ, Chisholm J, Hawley RS, and Orr-Weaver TL (2013). A meiosis-specific form of the APC/C promotes the oocyte-to-embryo transition by decreasing levels of the Polo kinase inhibitor matrimony. PLoS Biol 11, e1001648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y, Takeo S, Florens L, Hughes SE, Huo LJ, Gilliland WD, Swanson SK, Teeter K, Schwartz JW, Washburn MP, et al. (2007). The inhibition of polo kinase by matrimony maintains G2 arrest in the meiotic cell cycle. PLoS Biol 5, e323. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Proteins identified by mass-spectrometry in EGFP-hMeikin immunoprecipitates from HeLa cells. Related toFigure 2. HeLa cells expressing EGFP-hMeikin were lysed and immunoprecipitation with anti-GFP antibody was performed. Immunoprecipitated proteins were identified by mass-spectrometry. Key proteins relevant to this work are highlighted. The sum of multiple experiments is shown. Proteins with low statistical confidence and known contaminants (keratin, tubulin, actin) have been removed from the list.
Table S2. Primers used for plasmid cloning.
Data Availability Statement
Original Western blot images and mass-spectrometry data have been deposited at Mendeley Data and are publicly available as of the date of publication. The DOI is listed in the key resources table. All other data, including microscopy data reported in this paper will be shared by the lead contact upon request.
Cellprofiler pipelines have been deposited at Mendeley Data. The DOI is listed in the key resources table. All other original code is available from the lead contact upon request.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| α-Tubulin | Sigma-Aldrich, mouse clone DM1A (1:3000 IF) | Cat#T6199 RRID: AB_477583, lot 078M5796V |
| α-Tubulin | Abcam, rabbit clone EP1322Y (1:500 IF) | Cat#ab52866, RRID: AB_869989, lot GR3241238-2 |
| α-Tubulin (HRP-conjugated) | Abcam, mouse clone DM1A (1:5000 WB) | Cat#ab40742, RRID: AB_880625, lot GR3229214-1 |
| β-Tubulin | Sigma Aldrich, mouse clone Tub2.1 (1:500 Oocyte IF) | Cat#T4026 RRID: AB_477577 |
| GFP | Roche, mouse clones 13.1 and 7.1 (1:1000 WB) | Cat#11814460001, RRID: AB_390913, lot 14442000 |
| mNeonGreen | Chromotek, mouse clone 32F6 (1:1000 WB) | Cat#32F6, RRID: AB_2827566, lot 71108021 |
| Anti-centromere (ACA) | Antibodies Inc, human polyclonal (1:100 IF) | Cat#15234, RRID: AB_2687472, lot 1CK37 |
| CENP-A | Abcam, mouse clone 3–19 (1:1000 IF) | Cat#ab13939, RRID: AB_300766, lot GR3265183-3 |
| Mouse CENP-A | Cell Signaling Tech, rabbit clone C51A7 (1:200 oocyte IF) | Cat#2048S, RRID: AB_1147629 |
| CENP-C | Cheeseman lab (Gascoigne et al., 2011), rabbit polyclonal (1 μg/mL IF) | pBB280 |
| Mouse CENP-C | Cheeseman lab (Swartz et al., 2019), rabbit polyclonal (1:1000 oocyte IF) | pKG137 |
| pS311 CENP-C | Cheeseman lab, this study, rabbit polyclonal (1 μg/mL IF) | 85B |
| Plk1 | Santa Cruz Biotech, mouse clone F8 (1:200 IF, WB) | Cat#sc17783, RRID: AB_628157, lot D1219 |
| Plk1 | Sigma Aldrich, mouse clone 35–206 (1:200 oocyte IF) | Cat#05-844, RRID: AB_310836 |
| Ndc80 “Bonsai” complex | Cheeseman lab (Schmidt et al., 2012), rabbit polyclonal (1 μg/mL IF) | Anti-Bonsai |
| Hec1 | Santa Cruz Biotech, mouse clone C11 (1:100 oocyte IF) | Cat#sc515550 |
| Mouse Bub1 | Gift of Yoshinori Watanabe (U Tokyo) (Kawashima et al., 2010), mouse polyclonal (1:100 oocyte IF) | Anti-mouse Bub1 |
| Separase | Abcam, mouse clone XJ11-1B12 (1:500 WB) | Cat#ab16170, RRID: AB_2101815, lot GR44153-1 |
| Cyclin B1 | Cell Signaling Tech, rabbit polyclonal (1:1000 WB) | Cat#4138, RRID: AB_2072132, lot 3 |
| Histone 3 pS10 | Abcam, rabbit polyclonal (1:3000 FC) | Cat#ab5176, RRID: AB_304763, lot GR3217296-1 |
| GFP polyclonal | Cheeseman lab, rabbit (IP) (Cheeseman and Desai, 2005) | Rabbit GFP polyclonal |
| Bacterial and Virus Strains | ||
| E coli: LOBSTR-BL21 (DE3)-RIL | Kerafast (Andersen et al., 2013) | Cat#EC1002 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Doxycycline | Sigma-Aldrich | Cat#D9891, CAS:24390-14-5 |
| S-trityl-L-cysteine (STLC) | Sigma-Aldrich | Cat#164739, CAS:2799-07-7 |
| Nocodazole | Sigma-Aldrich | Cat#M1404, CAS:31430-18-9 |
| AZ-3146 (Mps1i) | Tocris | Cat#3994, CAS:1124329-14-1 |
| BI-2536 (Plk1i) | Fisher Scientific | Cat#508733, CAS:755038-02-9 |
| RO-3306 (Cdk1i) | Sigma-Aldrich | Cat#SML0569, CAS:872573-93-8 |
| Lambda Protein Phosphatase | New England Biolabs | Cat#P0753 |
| Milrinone | Sigma-Aldrich | Cat#M4659 |
| GST-hCENP-C (700–943) | This study | pKM138 |
| GST-hCENP-C (808–943) | This study | pNM221 |
| GST-hCENP-C (775–943, 835–838A) | This study | pNM232 |
| GST-hCENP-C (775–943, 865–867A) | This study | pNM271 |
| GST-sfGFP-hMeikin (328–373) *GST tag removed during purification | This study | pNM190 |
| GST-sfGFP-hMeikin (328–373, ICCII/DIII mutant) *GST tag removed during purification | This study | pNM201 |
| GST-sfGFP-hMeikin (302–373) *GST tag removed during purification | This study | pNM175 |
| Critical Commercial Assays | ||
| PhosphoSens Plk1 activity assay | AssayQuant <assayquant.com> | Cat#CSKS-AQT0691K |
| T7 mScript Standard mRNA Production System | CellScript | Cat#CMSC11610 |
| Deposited Data | ||
| Original CellProfiler pipelines | Mendeley Data | Doi: 10.17632/7gwbym8y9n.1 |
| Mass-spectrometry RAW files | Mendeley Data | Doi: 10.17632/7gwbym8y9n.1 |
| Uncropped Western blot images | Mendeley Data | Doi: 10.17632/7gwbym8y9n.1 |
| Experimental Models: Cell Lines | ||
| Human: HeLa cells | Cheeseman lab stocks | RRID:CVCL_0030 |
| Human: 293GP cells (for retrovirus generation) | Cheeseman lab stocks | RRID:CVCL_E072 |
| Constitutive expression: 2xmNeonGreen-hMeikin | Hela: Retroviral transduction with pNM481, clonal, this study | cNM216-7 |
| Constitutive expression: 2xmNeonGreen-hMeikin (328–373) | Hela: Retroviral transduction with pNM512, clonal, this study | cNM239-14 |
| Constitutive expression: 2xmNeonGreen-hMeikin (1–332) | Hela: Retroviral transduction with pNM513, clonal, this study | cNM240-4 |
| Constitutive expression: 2xmNeonGreen-hMeikin (ICCII/DIII mutant) | Hela: Retroviral transduction with pNM511, clonal, this study | cNM238-3 |
| Constitutive expression: hMeikin-mNeonGreen | Hela: Retroviral transduction with pNM795, clonal, this study | cNM396-15 |
| Doxycycline inducible: EGFP | Hela: AAVS1 Safe-harbor insertion of pNM280, this study | cNM087 |
| Doxycycline inducible: EGFP-hMeikin | Hela: AAVS1 Safe-harbor insertion of pNM299, this study | cNM102 |
| Doxycycline inducible: EGFP-hMeikin (104–373) | Hela: AAVS1 Safe-harbor insertion of pNM480, this study | cNM220 |
| Doxycycline inducible: EGFP-hMeikin (124–373) | Hela: AAVS1 Safe-harbor insertion of pNM549, this study | cNM244 |
| Doxycycline inducible: EGFP-hMeikin (155–373) | Hela: AAVS1 Safe-harbor insertion of pNM603, this study | cNM271 |
| Doxycycline inducible: EGFP-hMeikin (184–373) | Hela: AAVS1 Safe-harbor insertion of pNM335, this study | cNM142 |
| Doxycycline inducible: EGFP-hMeikin (199–373) | Hela: AAVS1 Safe-harbor insertion of pNM336, this study | cNM143 |
| Doxycycline inducible: EGFP-hMeikin (227–373) | Hela: AAVS1 Safe-harbor insertion of pNM325, this study | cNM134 |
| Doxycycline inducible: EGFP-hMeikin (1–332) | Hela: AAVS1 Safe-harbor insertion of pNM310, this study | cNM123 |
| Doxycycline inducible: EGFP-hMeikin (1–310) | Hela: AAVS1 Safe-harbor insertion of pNM338, this study | cNM145 |
| Doxycycline inducible: EGFP-hMeikin (1–301) | Hela: AAVS1 Safe-harbor insertion of pNM311, this study | cNM124 |
| Doxycycline inducible: EGFP-hMeikin (1–226) | Hela: AAVS1 Safe-harbor insertion of pNM502, this study | cNM226 |
| Doxycycline inducible: EGFP-hMeikin (1–154) | Hela: AAVS1 Safe-harbor insertion of pNM680, this study | cNM311 |
| Doxycycline inducible: EGFP-hMeikin (124–332) | Hela: AAVS1 Safe-harbor insertion of pNM611, this study | cNM273 |
| Doxycycline inducible: EGFP-hMeikin (175–181 4A mut) | Hela: AAVS1 Safe-harbor insertion of pNM400, this study | cNM172 |
| Doxycycline inducible: EGFP-hMeikin (S196A) | Hela: AAVS1 Safe-harbor insertion of pNM398, this study | cNM170 |
| Doxycycline inducible: EGFP-hMeikin (STP mutant) | Hela: AAVS1 Safe-harbor insertion of pNM300, this study | cNM119 |
| Doxycycline inducible: EGFP-hMeikin (8A mutant) | Hela: AAVS1 Safe-harbor insertion of pNM604, this study | cNM272 |
| Doxycycline inducible: EGFP-hMeikin (E151R, R154E) | Hela: AAVS1 Safe-harbor insertion of pNM726, this study | cNM321 |
| Doxycycline inducible: EGFP-hMeikin (1–332)-H2B | Hela: AAVS1 Safe-harbor insertion of pNM801, this study | cNM371 |
| Doxycycline inducible: EGFP-hMeikin (1–154)-H2B | Hela: AAVS1 Safe-harbor insertion of pNM888, this study | cNM469 |
| Doxycycline inducible: EGFP-hMeikin (155–332)-H2B | Hela: AAVS1 Safe-harbor insertion of pNM802, this study | cNM366 |
| Doxycycline inducible: EGFP-hMeikin (1–332, STP mutant)-H2B | Hela: AAVS1 Safe-harbor insertion of pNM804, this study | cNM370 |
| Doxycycline inducible: EGFP-hMeikin (1–332, 8A mut)-H2B | Hela: AAVS1 Safe-harbor insertion of pNM803, this study | cNM372 |
| Doxycycline inducible: EGFP-hPlk1 | Hela: AAVS1 Safe-harbor insertion of pNM364, this study | cNM161 |
| Constitutive expression: H2B-mScarlet-hMeikin(1–332)-mNeonGreen | Hela: Retroviral transduction with pNM848, fluorescence enriched, this study | cNM426e |
| Constitutive expression: H2B-mScarlet-hMeikin(1–332, E151R, R154E)-mNeonGreen | Hela: Retroviral transduction with pNM849, fluorescence enriched, this study | cNM427e |
| Constitutive expression: H2B-mScarlet-hMeikin(1–332, S149A)-mNeonGreen | Hela: Retroviral transduction with pNM850, fluorescence enriched, this study | cNM428e |
| Constitutive expression: H2B-mScarlet-hMeikin(1–332, 8A mut)-mNeonGreen | Hela: Retroviral transduction with pNM852, fluorescence enriched, this study | cNM430e |
| Constitutive expression: H2B-mScarlet-hRad21(142–476)-mNeonGreen | Hela: Retroviral transduction with pNM853, fluorescence enriched, this study | cNM431e |
| Constitutive expression: H2B-mScarlet-hRad21(142–476, E169R, R172E, E447R, R450E, E457R, R460E)-mNeonGreen | Hela: Retroviral transduction with pNM854, fluorescence enriched, this study | cNM432e |
| Constitutive expression: H2B-mScarlet-hRec8(297–506)-mNeonGreen | Hela: Retroviral transduction with pNM937, fluorescence enriched, this study | cNM478e |
| Constitutive expression: H2B-mScarlet-hRec8(297–506)-hMeikin(1–332, E151R, R154E)-mNeonGreen | Hela: Retroviral transduction with pNM869, fluorescence enriched, this study | cNM486e |
| Constitutive expression: H2B-mScarlet-hRec8(297–506, E401R, R404E)-hMeikin(1–332, E151R, R154E)-mNeonGreen | Hela: Retroviral transduction with pNM972, fluorescence enriched, this study | cNM500e |
| Constitutive expression: H2B-mScarlet-hRec8(297–506)-hMeikin(155–332)-mNeonGreen | Hela: Retroviral transduction with pNM870, fluorescence enriched, this study | cNM487e |
| Constitutive expression: H2B-mScarlet-hRec8(297–506)-hMeikin(1–332, E151R, R154E, 8A mut)-mNeonGreen | Hela: Retroviral transduction with pNM946, fluorescence enriched, this study | cNM493e |
| Experimental Models: Organisms/Strains | ||
| Mouse: NSA(CF-1) | Envigo | N/A |
| Oligonucleotides | ||
| siRNA targeting ESPL1: GCUUGUGAUGCCAUCCUGAUU | Dharmacon (Waizenegger et al., 2002) | N/A |
| siRNA control pool | Dharmacon | Cat#D-001810-10 |
| Primers used for plasmid cloning | Eurofins Genomics | See Table S2 |
| Recombinant DNA | ||
| 3xEGFP-mMeikin | This study (for oocyte injection) | pNM530 |
| 3xEGFP-mMeikin (E156R, R159E) | This study (for oocyte injection) | pNM606 |
| 3xEGFP-mMeikin(1–391, E156R, R159E) - hRad21(142–275) - mMeikin(387–434) | This study (for oocyte injection) | pNM962 |
| 3xEGFP-mMeikin(1–391) - hRad21(142–275, E169R, R172E) - mMeikin(387–434) | This study (for oocyte injection) | pNM963 |
| mMeikin-3xEGFP | This study (for oocyte injection) | pNM739 |
| mMeikin-3xEGFP (L377A, E379A, N383A) | This study (for oocyte injection) | pNM1051 |
| mMeikin-3xEGFP (S381A) | This study (for oocyte injection) | pNM1089 |
| mNeonGreen-mMeikin | This study (for oocyte injection) | pNM474 |
| mNeonGreen-mMeikin (387–434) | This study (for oocyte injection) | pNM477 |
| mNeonGreen-mMeikin (392ICCII, 425DII → 392AAAAA, 425AAA) | This study (for oocyte injection) | pNM476 |
| mNeonGreen-mMeikin (T335A) | This study (for oocyte injection) | pNM475 |
| Software and Algorithms | ||
| Fiji (ImageJ) | NIH (Schindelin et al., 2012) | 2.0.0-rc-69/1.52p |
| CellProfiler | Broad Institute (McQuin et al., 2018) | v3.1.9 |
| Graphpad Prism | Graphpad Software | v8.4.3 |
| ProteomeDiscoverer | ThermoFisher | v2.4 |
| FACSDiva | BD Biosciences | v6.1.2 |