

Abstract
Androgen receptor (AR), a ligand-dependent nuclear transcription factor and a member of steroid hormone receptor family, plays an important role in prostate organogenesis by regulating epithelial differentiation and restricting cell proliferation. Although rarely mutated or amplified in treatment-naïve prostate cancer (PCa), AR signaling drives tumor growth and as a result, therapies that aim to inhibit AR signaling, called ARSIs (AR signaling inhibitors), have been in clinical use for >70 years. Unfortunately, the clinical efficacy of ARSIs is short-lived and the majority of treated patients develop castration-resistant PCa (CRPC). Numerous molecular mechanisms have been proposed for castration resistance; however, the cellular basis for CRPC emergence has remained obscure. One under-appreciated cellular mechanism for CRPC development is the AR heterogeneity that pre-exists in treatment-naive primary tumors, i.e., although most PCa cells express AR (i.e., AR+), there is always a population of PCa cells that express no/low AR (i.e., AR−/lo). Importantly, this AR heterogeneity becomes accentuated during ARSI treatment and highly prominent in established CRPC. Here, we provide a succinct summary of AR heterogeneity across the PCa continuum and discuss its impact on PCa response to treatments. While AR+ PCa cells/clones exhibit exquisite sensitivities to ARSIs, AR−/lo PCa cells/clones, which are greatly enriched in stem cell signaling pathways, display de novo resistance to ARSIs. Finally, we offer several potential combinatorial strategies, e.g., ARSIs with stem cell targeting therapeutics, to co-target both AR+ and AR−/lo PCa cells and metastatic clones.
Keywords: Androgen receptor, Prostate cancer, Cancer cell heterogeneity, Cancer stem cells, Therapy resistance, Castration-resistant prostate cancer
1. Introduction
Androgen receptor (AR), encoded by the NR3C4 gene, is a ligand-regulated nuclear transcription factor (TF) in the steroid hormone receptor superfamily, which also includes estrogen receptor (ER), glucocorticoid receptor (GR) and progesterone receptor (PR) [1,2]. Like all other family members, AR consists of C-terminus ligand-binding domain (LBD), a DNA-binding domain (DBD) and a relatively unstructured N-terminal domain (NTD) where many cofactors bind (Fig. 1A). The FXXLF motif at aa 23–27 mediates highly specific, androgen-dependent interactions between the N-terminus in one AR molecule with the C-terminal LBD in another AR molecule whereas the WXXLF motif (especially WHTLF) at aa 453–457 mediates androgen-independent interactions between two AR molecules [3,4] (Fig. 1A). It’s generally thought that, upon binding of the ligands such as testosterone (T) or dihydrotestosterone (DHT), dimerized AR translocates to the nucleus, interacts with basic transcriptional machinery as well as many other nuclear and chromatin coregulators, and ultimately regulates the transcriptional output (activation or repression) of thousands of target genes involved in cell differentiation and proliferation [5–7]. A recent study, by employing STARRseq (Self-Transcribing Active Regulatory Regions sequencing), mapped genome-wide AR enhancer binding, and the results showed that AR-regulated enhancers often interact with gene-specific promoters and form central chromosomal loops to activate gene transcription [8].
Fig. 1. Domain structure of human AR and its subcellular mobilization.
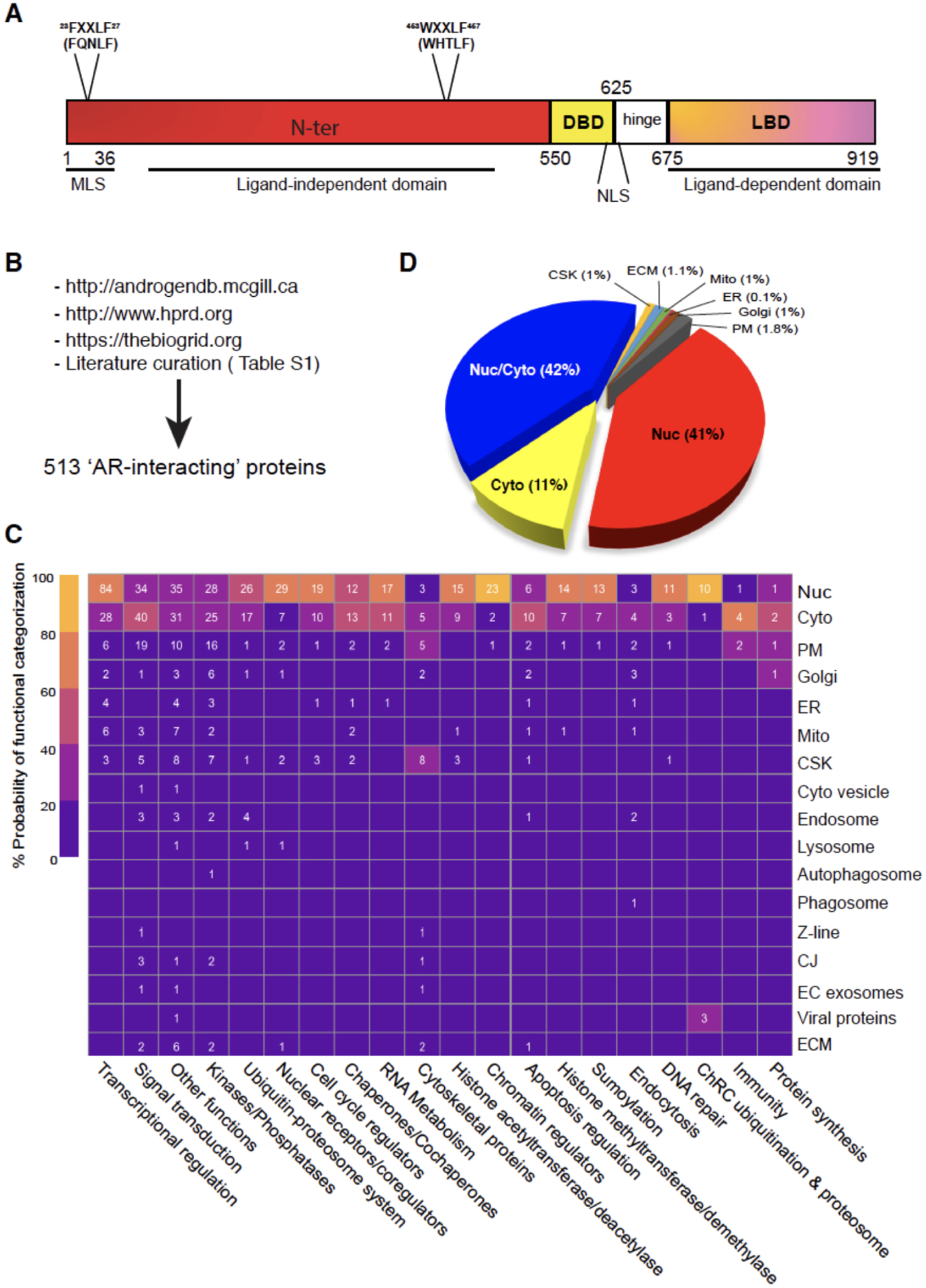
(A) The protein domain structure of human AR. AR is a protein of 919 amino acids (aa) consisting of several functional domains including N-terminal domain (NTD), DNA binding domain (DBD) and ligand binding domain (LBD) at the C-terminus. Also depicted are the FXXLF motif at aa 23–27, the WXXLF motif at aa 453–457, the nuclear localization signal (NLS) and a putative mitochondrial localization signal (MLS).
(B) Pipelines and methods used to curate putative AR-interacting proteins. A total of 513 AR-interacting partners were identified using McGill/HPRAD/Bio-Grid databases as well as extensive literature search and curation (also see Table S1). Note that the AR-interacting proteins encompass those that physically interact with AR (either directly with AR or indirectly through other proteins) as well as those that functionally interact and interface with AR (signaling) but without any physical interactions.
(C) Categorization of AR-interacting partners via their functions and subcellular localizations. The heatmap, generated using Morpheus (a Broad Institute’s web-based heatmap builder), presents the 513 AR-interacting proteins according to their potential involvement in the indicated biological processes (x-axis) and subcellular localizations (y-axis). The heatmap legend indicates the relative probability (%) of AR-interacting partners of falling into a specific functional category. For instance, the upper left box with 84 indicates that 84 AR-interacting proteins in the nucleus function, with 60–80% probability (certainty), in ‘Transcriptional regulation’. On the other hand, the upper right box with 1 indicates that 1 AR-interacting protein in the nucleus functions, with 20–40% probability, in ‘Protein synthesis’. Nuc (nucleus), Cyto (cytosol), PM (Plasma membrane), Golgi (Golgi apparatus), ER (endoplasmic reticulum), Mito (mitochondria), CSK (cytoskeleton), Z-line (the dark band in each myofibril where actin and myosin filaments overlap), CJ (cell junction), EC (extracellular), ECM (extracellular matrix).
(D) Pie chart (%) presentation of the 513 AR-interacting proteins according to their subcellular localizations.
AR and AR signaling are important for normal prostate development. Deregulated AR signaling has also been implicated in prostate cancer (PCa) growth and progression, which is why therapies that target androgen production (i.e., androgen-deprivation therapy; ADT) and AR signaling (e.g., enzalutamide; Enza), collectively called AR-signaling inhibitors (ARSIs), have been the mainstay treatment for patients with advanced and metastatic PCa for >70 years. Nevertheless, the therapeutic efficacy of ARSIs is generally short-lived, and the majority of treated PCa become refractory within ~6–20 months. Lack of enduring clinical effects in ARSIs may be intimately linked to cancer cell heterogeneity, an omnipresent feature of most human tumors [9]. Indeed, PCa is very heterogeneous in that one patient may harbor multiple tumor foci at different stages of clonal evolution and manifesting inherently distinct sensitivity to ARSIs [10–12]. At the cellular level, cancer cell heterogeneity may arise from clonal evolution driven by genomic alteration as well as from intra-clonal phenotypic differentiation and functional maturation of stem-like cancer cells or cancer stem cells (CSCs) [13]. These two mechanisms may operate hand-in-hand to create cellular heterogeneity. Yet another way of increasing tumor cell diversity is through cell plasticity, i.e., de-differentiation of ‘mature’ PCa cells back to more primitive cells or trans-differentiation to another cell lineage [9,13]. Herein, we discuss the impact of PCa cell heterogeneity on PCa development and therapy resistance by focusing on the AR heterogeneity from 3 different angles, i.e., AR heterogeneity in its biological functions, subcellular localization, and expression levels.
2. AR functional heterogeneity: Oncogenic vs. tumor-suppressive
In the normal prostate, AR is expressed primarily in differentiated luminal epithelial as well as stromal cells with only low expression in basal cells. The majority (98–99%) of primary PCa at diagnosis are adenocarcinomas in which most PCa cells present as AR+ luminal cells; therefore, AR and AR signaling have been the primary therapeutic target in the clinical treatment of PCa patients [14,15]. As a result, AR has been categorically thought of, by many, as a ‘default’ oncogenic driver of prostate tumorigenesis. In reality, however, AR functions are more complicated and may be context-dependent. Genetic ablation studies in the mouse prostate suggest that, in the normal prostate epithelium, AR cell-autonomously restricts cell proliferation and promotes epithelial differentiation and as such, AR functions as a tumor suppressor [16–21]. Thus, the Ar deficient prostate epithelial cells exhibit increased proliferation with concomitant loss of differentiation forming abnormal glandular structures [16,17]. Of interest, in the mouse prostate with epithelial-specific Ar knockout, there is a significant expansion of the CK5+/CK8+ progenitor (intermediate) cell population accompanied by atrophy of the smooth muscle cell compartment [17–19,21]. Further studies from the Chang group [18,19], comparing primary (TRAMP) tumor development in mouse models where the Ar was knocked out in both prostate epithelium and stroma versus epithelium-specific Ar knockout showed that while Ar knockout in both stroma and epithelia resulted in smaller tumors at early stages, epithelium-specific Ar knockout led to larger tumors with high cell proliferation, suggesting that AR normally behaves as a tumor suppressor in the epithelium but as a tumor promoter in the stroma [20].
The tumor-suppressive functions of AR have also been demonstrated in immortalized prostate epithelial and PCa cells. For example, in immortalized BPH-1 (Benign Prostate Hyperplasia 1) cells that were forced to express AR via lentiviral-mediated transduction, androgen-induced AR signaling led to inhibition of cell proliferation and increased differentiation [22]. Similarly, transfection of PC3 cells with a full-length AR expression vector repressed α6β4 integrin expression, adhesion on laminin, invasion and colony formation [23]. In fact, AR re-expression in PC3 cells led to transcriptionally functional AR protein, which subsequently transactivated p21 and inhibited cell proliferation in vitro and xenograft growth in vivo [24]. Intriguingly, similar tumor-inhibitory effects of restored AR expression were not observed in DU145 cells, presumably due to dysregulation of AR coregulators in DU145 cells [24]. Other studies using AR-re-expressed PC3 cells in tissue recombination assays also revealed inhibitory effects of AR on invasion and metastasis [18,19]. The dichotomous functions of AR as a tumor suppressor vs. a PCa driver are, to a certain degree, also mirrored in AR-specific transgenic mouse models [25,26]. Although transgenic expression of oncogenic AR mutants (e.g., AR-T857A, which confers ligand promiscuity to allow AR activation by different ligands or AR-E231G, which exhibits increased ligand-independent activity) has been reported to drive tumor development in the mouse prostate [26], overexpression of wildtype AR, however, only causes subtle hyperplasia without apparent neoplastic phenotypes [25].
So how is AR turned from a tumor suppressor in normal prostate epithelium to an oncogenic driver in PCa? One potential mechanism could be related to AR functioning not only as a transcriptional activator but also as a transcriptional repressor [27]. For example, in VCaP cells and under androgen-sufficient conditions, the AR, via recruiting LSD1 and H3K4me2, binds to an enhancer in the AR second intron to repress the expression of AR itself as well as many genes involved in DNA synthesis and cell-cycle progression [28]. Intriguingly, under castrated conditions, low levels of androgen stimulate AR activity on enhancer elements leading to increased expression of AR as well as AR-repressed genes [28]. Under supraphysiological androgen levels, AR suppresses the transcription of DNA replication genes by direct association with hypo-phosphorylated retinoblastoma protein (RB) [29].
Another mechanism might be related to the oncogene c-MYC [30,31], which is overexpressed in >80% of early precursor lesions (prostate intraepithelial neoplasia or PIN) and PCa, and is the only oncogene that is sufficient, by itself when overexpressed, to immortalize primary human prostate epithelial cells and to drive full-blown tumor development in the mouse prostate. It is known that androgen-mediated AR signaling within normal prostate epithelial cells results in G0 growth arrest coupled with terminal differentiation into PSA-expressing secretory luminal cells. This AR-driven terminal differentiation requires DNA binding of the AR protein, is associated with decreases in c-MYC mRNA and protein and increases in p21, p27, and SKP2, does not require functional p53, and ultimately leads to down-regulation of cyclin D1 and RB phosphorylation [30,31]. Strikingly, transgenic expression of a constitutive vector encoding c-MYC to prevent c-MYC down-regulation overrides AR-mediated growth arrest in normal prostate epithelial cells, suggesting that AR-induced c-MYC down-regulation is critical in the growth arrest of normal prostate epithelial cells [30,31]. In contrast, in PCa cells, androgen-induced AR signaling up-regulates c-MYC expression. Interestingly, a novel pathway linking AR to PCa cell proliferation has recently been described wherein AR negatively regulates protein synthesis via transcriptional control of the translation inhibitor 4EBP1 [32]. Consequently, low AR allows increased assembly of the eIF4F translation complex, which drives PCa cell proliferation [32].
3. AR signaling heterogeneity: Functions in the nucleus vs. other cellular compartments.
AR, as a TF, has been thought to mostly localize and function in the nucleus, and the nuclear AR, through interactions with a myriad of proteins, regulates genes that are essential to PCa development. Within the nucleus, AR can directly interact with components of the basal transcriptional machinery (e.g., TFIIF and TFIIH) and the RNA polymerase II, as well as with coregulators [5–7]. Furthermore, specific TFs interact with AR in different ways, with some binding the AR directly and influencing its ability to interact with the androgen-responsive elements (AREs) while others competing with AR for the availability of coregulators [5].The FXXLF motifs, commonly found in coactivators, preferentially bind the AR-LBD (Fig. 1A) when the AR is not bound to the DNA.
On the other hand, AR may also function outside of nucleus. Indeed, the AR-interacting proteins and AR coregulators can be localized in several different cellular compartments (Fig. 1B–D), and key interactions characterize every step of the AR life cycle and functional role, from the basal cytoplasmic state to ligand binding, from nuclear translocation to the transcription of target genes, and from the disassembly of the transcriptional complex to AR degradation. By combining in silico analysis of publicly available databases with literature search and curation, we have identified 513 potential ‘AR-interacting’ proteins and coregulators, which may physically interact with AR (either directly with AR or indirectly through other proteins) or functionally interact and interface with AR but without any physical relationship (Fig. 1B; Table S1). By interrogating the Human Protein Atlas (HPA) and Uniprot databases, we observed that these AR-interacting proteins, as expected, are mostly localized in the nucleus; however, many AR interactors are also localized in the cytosol or both nucleus and cytosol (Fig. 1C–D). Intriguingly, AR-interacting proteins have also been reported on the plasma membrane (PM) and many other cellular compartments (Fig. 1C–D; Table S1). The ‘primary’ functions of these AR-interacting proteins range from transcriptional regulation, chromatin remodeling and histone modifications (acetyltransferases, deacetylases, methyltransferases, demethylases), to ubiquitination/proteasome pathway, splicing and RNA metabolism, DNA repair, signal transduction, cell cycle, apoptosis, chaperone dynamics, and cytoskeleton regulation (Fig. 1C). In principle, AR interactors or coregulators in different cellular compartments could be involved in regulating the ill-characterized non-nuclear (i.e., non-genomic) functions of AR and in mediating castration resistance.
Compared to our understanding of the genomic (nuclear) actions of AR, the molecular events governing AR localization to other cellular compartments and the downstream AR signaling events remain much less understood. A recent study showed that AR could translocate to PM through interactions with the microtubule-based motor protein kinesin 5B (KIF5B), and disruption of KIF5B functions interfered with AR-PM association and AR signaling [33]. Of interest, the heat-shock protein 27 (HSP27) was activated by membrane-associated AR and, upon activation, HSP27 indirectly potentiated the AR transcriptional activity by mediating membrane-to-nuclear signal transduction [33]. The study suggests that the PM-associated AR may play a role in sustaining AR signaling in CRPC through enhancing nuclear AR transcriptional activity in the presence of castration levels of androgens [33]. Similarly, AR has been reported to localize in the mitochondria and regulate mitochondrial functions [34]. The study revealed that ligand-independent AR, through a potential mitochondrial localization signal (MLS) located at the N-terminus of the AR protein (Fig. 1A), could translocate to the mitochondria in both cell lines and primary prostate tissues, and the mitochondrially located AR controlled expression of many nuclear DNA (nDNA)-encoded mitochondrial oxidative phosphorylation subunits and downregulated the mitochondrial DNA (mtDNA) content [34]. Of note, the PC3-AR PCa cells showed significantly reduced mtDNA content and mtDNA-encoded COX II protein due to the mitochondrial translocation of AR [34]. Intriguingly, the mtDNA content in prostate tumors of African Americans (AA) is >6 times less than in PCa in Caucasian-Americans (CA), and bioinformatics analyses in more than 6000 AA and 33,000 CA prostate tumors revealed that the AR missense mutations in the NTD and MLS domains were present only in AA PCa patients, potentially contributing to deregulated mitochondrial functions and PCa aggressiveness in the AA patient tumors [34].
4. AR expression heterogeneity: AR+ vs. AR−/lo.
4.1. AR heterogeneity in treatment-naïve, castration-resistant, and metastatic PCa.
Why and how would ADT and antiandrogens such as Enza gradually lose their therapeutic efficacy and patients develop what’s called castration-resistant PCa or CRPC? The therapeutic premise of ADT/antiandrogens is that the targeted PCa cell population expresses the target, AR, and relies on AR/AR signaling for their survival. In reality, however, PCa cells that express little AR, i.e., AR−/lo, have been reported for decades across the spectrum of PCa development and progression [35–47]. For instance, all treatment-naïve primary prostate tumors have been shown to harbor not only the bulk AR+ PCa cell population but also AR−/lo cells with varying abundance [e.g., 37–41]. Besides the heterogeneity in AR expression within primary tumors, several studies have revealed an inverse correlation between AR expression and tumor aggressiveness, with AR highly expressed in low grade and more differentiated tumors [38,39]. AR expression varies in hormone-refractory PCa as well, with most cells being AR+ in some CRPC but most cells being AR−/lo in other hormone-refractory tumors [e.g., 36, 37,42–47]. Indeed, tissue microarray analysis of CRPC specimens reveals regions of AR+ cells, AR−/lo cells or a mixed population. By screening 195 CRPC cores and whole-mount sections from 89 patients’ CRPC, we have recently reported 4 patterns of AR expression in treatment-failed PCa: AR−/lo (27%), nuclear AR+/hi (25%), mixed AR+/hi and AR−/lo (8.7%), and mixed nuclear/cytoplasmic AR (39.3%) [36].
AR heterogeneity has also been observed in PCa metastases to other organs such as lymph nodes, bone, lung and the liver. Immunohistochemical (IHC) analysis AR in metastatic lesions of CRPC patients revealed wide variations in AR expression, with 41.5% of the samples showing <10% AR+ cells [42]. Moreover, Davis et al. reported that the percentage of AR+ cells and AR staining intensity are much lower in CRPC metastases compared to benign tissues [45]. Labracque and colleagues recently characterized CRPC metastases by IHC, RNA-seq, and gene set enrichment analysis (GSEA), defining 5 phenotypes based on the expression of AR and neuroendocrine (NE) marker expression: AR-high (AR+/NE−), AR-low (ARlo/NE–), double-positive (AR+/NE+), double-negative (AR–/NE–), and small cell or neuroendocrine PCa (AR–/NE+) [47]. These discussions highlight the presence of AR−/lo PCa cells across the PCa spectrum, i.e., treatment-naïve tumors, CRPC and PCa metastases.
4.2. AR alterations in castration resistance.
Both ADT and antiandrogens are very efficient AR-directed therapies that target the AR+ PCa cells. The normal serum levels of T in 60-year-old men are ~500 ng/dl and ADT is capable of reducing T levels to ≤50 ng/dL (i.e., the castrate levels of T), and most advanced/metastatic PCa patients initially respond to ADT (using gonadotropin-releasing hormone (GnRH) agonists or antagonists) very well by manifesting dramatic tumor-debulking effects. Numerous molecular mechanisms have been uncovered and proposed for the development of CRPC [15,35], most of which, understandably, surround altered AR expression and dysregulation in AR signaling, since the AR+ PCa cells represent the bulk cell population in treatment-naïve tumors. First of all, ADT, though powerfully inhibiting testicular androgen synthesis (normally representing >90% of T in circulation) and reducing systemic levels of T, does not completely wean tumor cells of androgens because PCa cells can employ alternative substrates and metabolic pathways to generate AR ligands within tumors, a phenomenon dubbed intracrine androgen production [48–50]. Following ADT, despite the prominent reduction of circulating T, a significant amount of intratumor androgens is still detected in localized PCa as well as in metastatic CRPC [48–50]. The adrenal androgens dehydroepiandrosterone (DHEA) and androstenedione (AD) are products of de novo steroidogenesis in the adrenal gland beginning with cholesterol (note that adrenal androgens normally represent only a small fraction of the total androgens in circulation). Both DHEA and AD can be converted to T/DHT in the prostate (tumors). Secondly, castration may push AR+ PCa cells to activate, and rely on, signaling from other steroid hormone receptors such as GR [51,52] and PR [53,54].
Thirdly, under the selective pressure from ADT/antiandrogens, AR is well-known to undergo genomic amplifications that result in AR overexpression and AR hyperactivity, or sustain point mutations in the LBD that confer ligand promiscuity [55,56]. Indeed, amplification of the AR locus has been reported in ~30% of CRPC, and AR overexpression is sufficient to confer resistance to ARSIs. Also, at least 4 major point mutations in the LBD, i.e., L702H, W742C, H875Y, and T878A, have been reported, which occur in 15–20% of CRPC and often turn AR antagonists to agonists within PCa cells. Furthermore, two recent studies [57,58] identified recurrent structural alterations in the AR gene enhancers that became duplicated or amplified and contributed to increased AR expression and activity in CRPC. One study uncovered amplification of a somatically acquired intergenic AR enhancer to be a major driver of therapeutic resistance in advanced PCa, as disruption of this enhancer region decreased proliferation by suppressing AR levels whereas insertion of an additional copy of this region was sufficient to increase proliferation under low-androgen conditions and decreased sensitivity to Enza [57]. Epigenetic data generated in localized PCa and benign prostate specimens suggests that the region spanning the AR intergenic regulatory elements may represent a developmental enhancer [57]. In the other study, whole genome sequencing in 23 mCRPC biopsies and cell-free DNA sequencing data from 86 mCRPC patients showed that in addition to frequent rearrangements in known PCa driver genes, complex rearrangements of the AR locus were also observed in most CRPC [58]. Notably, 70–87% of CRPC cases, compared to <2% of primary PCa, showed highly recurrent tandem duplications of an upstream enhancer of AR, and a subset of patient cases also displayed both AR and/or MYC enhancer duplications [58]. Notably, nearly all these AR mutations and structural alterations are present only in CRPC but not in primary tumors, suggesting that the genomic alterations in AR are mostly induced by AR-targeting therapies.
Finally, persistent castration may select for AR splicing variants (AR-Vs) that completely lack the LBD, conferring ligand-independent AR activities in PCa cells [59–63]. Most of the LBD-less AR variants retain an intact and somewhat functional nuclear localization signal, allowing their constitutive nuclear localization independently of androgens. Knockdown of AR-Vs, but not full-length AR, inhibited androgen-independent PCa cell growth under bicalutamide or Enza treatment [63], suggesting that AR-Vs play an important role in developing castration resistance. In some CRPC, both AR-V7 and AR-V9 were co-expressed to promote resistance to AR-targeted therapies in an androgen-depleted environment [62].
4.3. AR−/lo PCa cells and de novo castration resistance.
What about AR−/lo PCa cells/clones that preexist in treatment-naïve tumors and frequently become enriched during tumor progression and ADT/antiandrogen treatment? By prediction, such PCa cells would not be susceptible to ARSIs. Our recent systematic work has validated this prediction [36]. By establishing isogenic LNCaP cell clones that homogenously express RFP-tagged AR (by using the ZFN-mediated genomic integration) and the clones that lack functional full-length AR (by using CRISPR/Ca9-mediated knockout), we have demonstrated that, although the AR/RFP LNCaP cells require androgens for their in vitro and in vivo growth and are exquisitely sensitive to castration/Enza, the AR-KO LNCaP clones are resistant to these ARSIs de novo [36]. Importantly, the AR-KO LNCaP cells possess unique intrinsic biological properties that allow their aggressive growth and confer a significant competitiveness (over the AR/RFP cells) in androgen-ablated hosts [36]. The de novo castration resistance in AR−/lo PCa cells has also been observed in our paired androgen-dependent (AD) and androgen-independent (AI) xenograft models. When we serially propagated 4 xenograft AD tumors, i.e., LNCaP, LAPC4, VCaP and LAPC9, in castrated mice, these tumors, in a time- and passage-dependent manner, manifested model-specific changes in AR expression and subcellular distribution during their progression to the AI state [36]. Specifically, the LNCaP AI tumors evolved into the AR+/hi phenotype and LAPC9 AI tumors the AR−/lo phenotype whereas the LAPC4 and VCaP AI tumors the mixed nuclear/cytoplasmic AR, much like the AR expression patterns observed in patient CRPC [36]. While all 3 AI models that retained AR expression responded to Enza, the AR−/lo LAPC9 AI tumors were resistant to Enza de novo [36]. Expression profiling revealed that the AR−/lo LAPC9 CRPC is greatly enriched in gene signatures associated with stem cells, neurogenesis, lipid metabolism and immune responses [36]. Critically, our analysis in whole-mount CRPC specimens revealed prominently increased abundance of AR−/lo cells in patients’ CRPC [36].
Studies from others also demonstrated intrinsic castration resistance in AR−/lo PCa cells [46]. It is worthwhile to point out that the AR−/lo PCa cell populations in treatment-naïve tumors vs. ADT-treated and CRPC will likely possess different transcriptomic profiles and epigenetic landscapes despite that they share similarities in phenotype and therapy resistance [13,36].
5. AR heterogeneity and potential therapeutic strategies
The AR−/lo PCa cells generally represent the minority in treatment-naïve tumors but significantly increase in castration-resistant tumors but all CRPC harbor both AR−/lo and AR+ PCa cells [35–37]. Therefore, it’s reasonable to speculate that AR−/lo PCa cells may be constantly interacting with, and cross-talking to, the AR+ cells within the same tumor. Understanding the cross-interactions and cross-regulations between the two PCa cell populations may shed new biological light on CRPC evolution and uncover novel therapeutic targets in both pre-existent and therapy-induced AR−/lo PCa cells. On the other hand, as de novo NEPC are mostly AR-negative and ARSIs frequently cause relapsed PCa with much reduced and heterogeneous AR expression [64–66], perhaps we can learn from these PCa subtypes about the signaling and pro-survival mechanisms in the AR−/lo PCa cells/clones. Below, we highlight a few signaling pathways and molecules that could represent potential therapeutic targets in tackling PCa cell heterogeneity.
5.1. Role of RB and TP53 and lineage plasticity.
As discussed above, AR is a tumor suppressor and pro-differentiation TF in normal prostate epithelial cells. Hence, from a cell biology perspective, AR+ cells are differentiated and able to produce PSA whereas the AR−/lo prostate epithelial and PCa cells are less mature and undifferentiated. In support, our single cell video-microscopy studies using a lentiviral-based reporter have revealed that the undifferentiated (PSA−/lo)AR−/lo PCa cells are not only resistant, de novo, to ARSIs, chemotherapeutic drugs and radiation but also possess stem cell traits being able to undergo asymmetric cell division and differentiate into AR+(PSA+) PCa cells [13,37]. Of interest, the tumor-suppressive functions of AR involve recruitment of RB to the target genes and chromatin [29], and loss of RB and/or TP53 is common to de novo NEPC, small cell carcinoma and other AR−/lo aggressive PCa variants [64,66]. In fact, genetic deletion of Rb and/or p53 is sufficient to cause aggressive prostate tumors with reduced and heterogeneous AR expression and frequently with features of NEPC [67–69], a phenomenon called lineage plasticity. Thus, deletion of Pten in the mouse prostate induces rather indolent adenocarcinomas that are homogenously AR+ and progresses very slowly; however, co-deletion of Pten and Rb1 causes the so-called double knockout (DKO) tumors that are much more aggressive and also show rather heterogenous AR expression [69]. The DKO tumors are initially castration-sensitive but gradually evolve into castration-resistant that are lethal to the hosts. Strikingly, the castration-refractory DKO prostate tumors have largely lost AR assuming an AR−/lo phenotype [69]. Simultaneous deletion of all 3 tumor suppressors, i.e., Pten, Rb1 and p53, led to AR-negative de novo NEPC [69]. These elegant genetic mouse studies [69] highlight the dynamic changes in AR+ and AR−/lo PCa cell populations and distinct PCa subtypes imposed by the loss of critical prostate tumor suppressors and raise the possibility that restoration of the tumor suppressor functions could potentially curb the lineage plasticity (i.e., the conversion of AR+ to AR−/lo PCa cells) and sensitize PCa to ARSIs.
5.2. Cancer stem cell (CSC) signaling
Interestingly, the above-mentioned DKO tumors, especially the AR−/lo castration-resistant DKO tumors, exhibited significantly increased stemness as evidenced by upregulation of numerous genes normally involved in stem cell regulation and neurogenesis such as SOX2 and EZH2 [69]. In LNCaP human PCa cells, similar deletion of RB1 and TP53 also caused lineage plasticity by turning these cells into basal-like prostate cells with increased expression of SOX2 and other stemness genes and resistant to ARSIs [68]. Indeed, the AR−/lo PCa cells seem to preferentially employ stem cell/CSC signaling to support their survival and resistance to various therapies [13,36,37,70]. For example, the AR-negative prostate tumors have been shown to upregulate β-catenin signaling [71], a well-established developmental and stem cell signaling pathway. The (PSA−/lo)AR−/lo PCa cell population, mentioned earlier, is highly enriched in authentic CSCs that can undergo asymmetric cell division, possess stem cell gene expression profiles, and are inherently resistant to ARSIs and many other PCa therapies [13,36,37,70]. The PSA−/lo PCa cell population is yet heterogeneous harboring more tumorigenic subsets, e.g., the ALDHhiCD44+α2β1+ (or Triple Marker+/TM+) subpopulation that can both initiate and propagate CRPC in fully castrated hosts at as few as 100 cells [70]. Our focused cell biology, mechanistic and in vivo therapeutic studies in these well-defined PCSC (prostate cancer stem cell) populations have identified several CSC-enriched therapeutic targets in AR−/lo PCa cells, including CD44, OPN (osteopontin) and NANOG [13]; IGF-1R [13,37]; SOX9, RegIV, and ALDH1A1 [70]; and integrin α2, MYC, and, importantly, BCL-2 [13,36,37,70; see below].
5.3. BCL-2
BCL-2 is a well-studied pro-survival BCL-2 family member that has been implicated in facilitating castration resistance (reviewed in [36]). Our early studies indicated that in (PSA−/lo)AR−/lo PCa cells, BCL-2 is preferentially associated with the bivalent (i.e., H3K4me3 and H3K27me3) chromatin occupancy [37]. In the LNCaP AD → AI progression model, castration slightly upregulated BCL-2 in the ADT-resistant tumors and ADT/Enza led to further substantially increased BCL-2 [36]. Thus, combination of Enza and the BCL-2 antagonist, ABT-199, inhibited the emergence of Enza-resistant AR+/hi CRPC by 75%, and ABT-199 alone also inhibited the growth of AR−/lo LAPC9 AI tumors [36]. Significantly, BCL-2, but not BCL-xL and MCL-1, is exclusively upregulated in PCa patients who have been treated with neoadjuvant ADT [36]. These recent results [36] have led to an ongoing phase Ib/II clinical trial of treating Enza-naïve metastatic PCa patients with a combination of Enza and Venetoclax, the FDA-approved BCL-2 inhibitor (NCT03751436). Notably, BCL-2 has recently been reported to be a potential therapeutic target in AR−/lo NEPC and small-cell carcinoma [72]. RNA-seq data, collected from AR-active PCa (ARPC), small-cell neuroendocrine PCa (SCNPC) and two NEPC, were cross-compared to identify conserved druggable targets. This analysis revealed BCL-2 as highly upregulated in SCNPC compared to ARPC, and treatment with the BCL-2 inhibitor Navitoclax reduced SCNPC PDX tumor growth in vivo [74].
5.4. FGF signaling
Bluemn and colleagues recently reported [46] that the percentage of AR-negative tumors in patients with mCRPC has increased significantly from 11% (during 1998–2011) to 36% (during 2012–2016) since the introduction of potent AR antagonist such as Enza and abiraterone. These AR-negative mCRPC also lack the NE phenotype and are thus called double-negative PCa (DNPC; [46]). Interestingly, the DNPC displayed elevated FGF and MAPK pathway activity, which was inversely associated with AR activity. In DNPC, FGF appears to activate MAPK signaling and bypasses a requirement for androgens and AR dependence [46]. Thus, FGFR inhibition abrogated DNPC growth in model systems, and FGF/MAPK blockade has been proposed as a therapeutic strategy to counteract the growth of AR-null PCa cells in patients stratified by AR activity status [46].
5.5. Polycomb repressor complex signaling
A recent study has implicated the PRC1 (Polycomb Repressor Complex 1) signaling in regulating the DNPC [73]. The authors showed that PRC1 was required for tumor initiation and metastasis in DNPC by promoting the expression of various pro-metastatic genes. CCL2 was identified as the major target, and the cytokine coded by this gene functions in an autocrine fashion to promote the self-renewal of DNPC cells and in a paracrine fashion to recruit M2-like tumor-associated macrophages and regulatory T cells, fostering immunosuppression and neo-angiogenesis. Thus, targeting PRC1 in DNPC represents an innovative strategy to reverse stemness and immune evasion, which may sensitize mCRPC to immunotherapy [73].
6. Conclusions and perspectives
The androgen biosynthesis and AR functions in the nucleus have been the prime targets of PCa therapy, and ARSIs, especially the new generation of antiandrogens Enza, Apalutamide and Abiraterone, are ‘wonder’ drugs that manifest remarkable tumor-debulking effects in virtually all PCa patients. Unfortunately, much like all other targeted anti-cancer therapies, ARSIs gradually lose their ‘charm’ and patients develop CRPC. Both intrinsic PCa cell heterogeneity, which preexists in treatment-naïve tumors, and PCa cell plasticity, caused by genetic mutations in key tumor suppressors such as RB1 and TP53 as well as by epigenetic mechanisms and treatments, may play intimate roles in CRPC development. One significant aspect of PCa cell heterogeneity is reflected in the co-existence of both AR+ and AR−/lo PCa cells in primary tumors (Fig. 2A). These two cell populations, during months to years of ADT, gradually evolve into (metastatic) CRPC that harbors both the AR+/hi population and a much expanded AR−/lo PCa cell population (Fig. 2A; [36]). Work from our lab [13,36,37,70] and from other groups [46,71–73] suggests that such ADT-failed tumors could potentially be treated with a combination of the next-generation antiandrogens with inhibitors against CSC factors such as BCL-2, WNT and FGFR (Fig. 2A). Our ongoing phase Ib/II clinical trial (NCT03751436), based on preclinical modeling [36], represents such a combination and aims to treat Enza-naïve mCRPC patients with Enza and the BCL-2 inhibitor Venetoclax. Such a combinatorial approach may be especially applicable to mCRPC patients at higher risk of developing AR-negative disease such as those with RB1 and/or TP53 mutations. Similar type of combinatorial strategies may also be conceptualized for the treatment of primary PCa, in which the AR+ PCa cells will be targeted by standard-of-care ADT and Enza whereas the minor AR−/lo population of PCa cells by CSC inhibitors (Fig. 2B). In theory, such combinations should significantly delay and even prevent CRPC emergence and progression (Fig. 2B). Other potential CSC targets may include molecules such as SOX2 and EZH2, which frequently become upregulated in CRPC and uniquely enriched in AR−/lo cells under androgen depleted conditions [68,69]. Further investigation is needed to elucidate the role of critical AR coregulators and downstream AR targets under castrated conditions in AR−/lo PCa cells.
Fig 2. Potential therapeutic strategies to target PCa cell heterogeneity and plasticity.
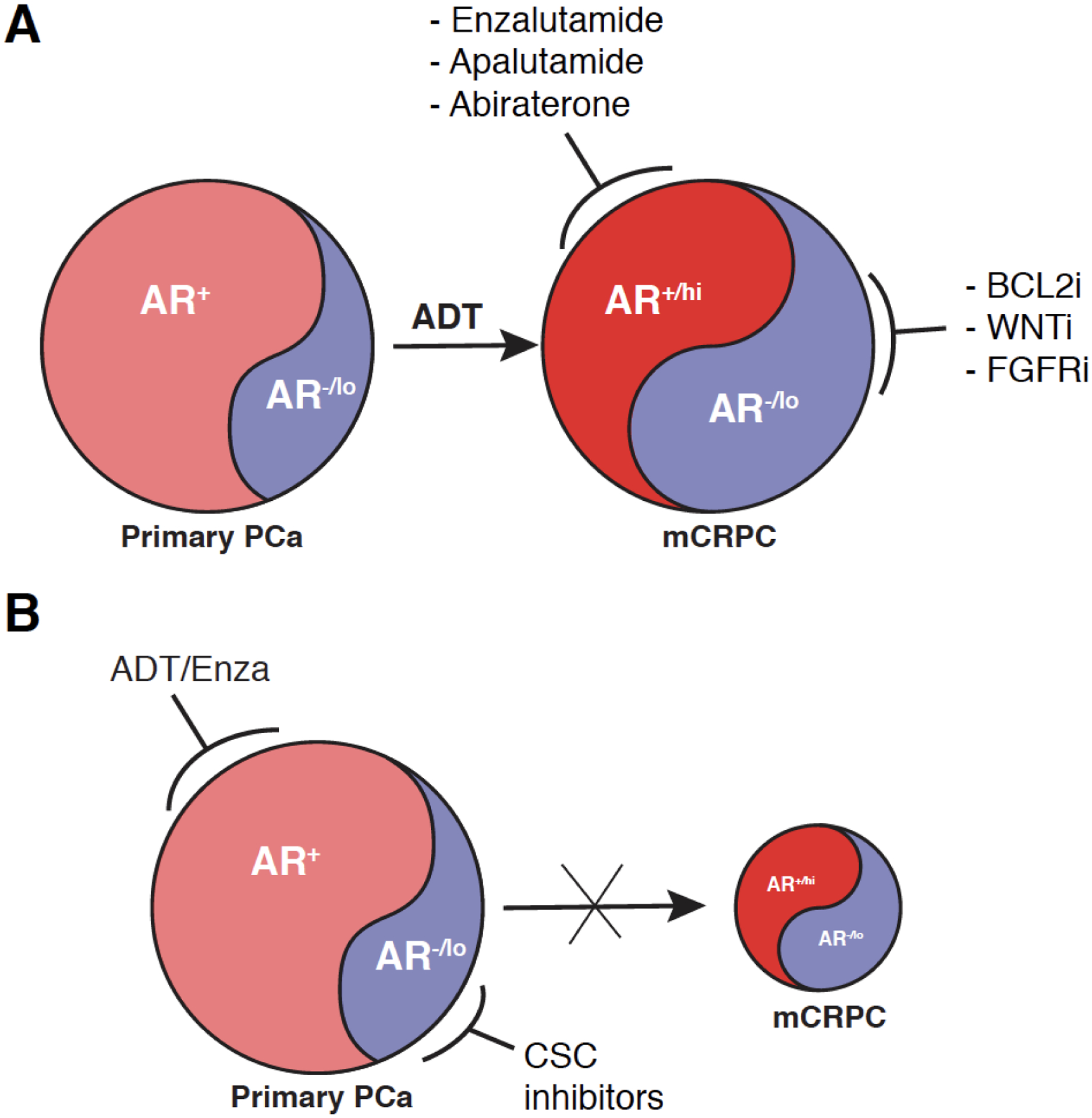
(A) Combinatorial strategies in the mCRPC setting.
(B) Combinatorial strategies in the primary PCa setting.
Please see Text for discussions. Note that the AR−/lo PCa cell populations in treatment-naïve tumors (left) vs. ADT-treated and CRPC (right) will likely possess different transcriptomic profiles and epigenetic landscapes despite that they share similarities in phenotype and therapy resistance [13,36]. Also, the cartoon (adapted from ref. 36 with permission) was not intended to imply that the AR+/hi and AR−/lo cell populations in mCRPC are directly derived from the AR+ and AR−/lo cell populations in treatment-naïve primary tumors, respectively. This is because ADT-induced cancer cell plasticity may inter-convert the two cell populations, but the proposed combinatorial strategies may simultaneously target both PCa cell heterogeneity and plasticity in established CRPC (A) or significantly delay and inhibit the emergence of CRPC when used to treat primary tumors (B).
As we discussed above, the AR heterogeneity may also be reflected at its functional levels and its subcellular localizations. For example, there is evidence supporting non-genomic AR activity mediated by membrane-associated AR. The membrane translocation appears to help sustain AR activity in CRPC by enhancing the AR nuclear activity in the presence of low levels of androgen and by promoting cell proliferation, invasion, and survival through AKT/ERK pathways [33]. Therefore, targeting AR membrane transport might also represent a novel strategy to fully suppress AR signaling under androgen depleted conditions in CRPC. Likewise, the AR functional heterogeneity in being both a tumor promoter and tumor suppressor poses a challenge to standard-of-care ADT that systematically depletes the androgen and consequently, may suppress both functions of AR. The ideal therapeutic approach would be to selctively target the AR oncogenic functions without impacting its repression of DNA replication and cell-cycle genes [27–31]. To develop such novel therapeutics, we need to understand how in different contexts, AR dynamically recruits the repertoire of unique coregulators involved in functional and cellular responses to androgen depletion.
Supplementary Material
Highlights:
Discussed the dichotomous AR biological functions in normal prostate epithelium (as a tumor suppressor) vs. in prostate cancer cells (as an oncogenic driver)
Presented an updated list of ‘AR-interacting’ proteins
Focused on AR heterogeneity in subcellular localizations and expression levels
Discussed the impact of AR heterogeneity on therapy response/resistance
Highlighted several signaling pathways and offered potential therapeutic strategies in the AR−/lo PCa
Acknowledgements
Work in the authors’ lab was supported, in part, by grants from the U.S National Institutes of Health (NIH) National Cancer Institute (NCI) R01CA237027, R01CA240290, R21CA237939, and R21CA218635, and Department of Defense (W81XWH-16-1-0575) (to DGT), and by the Roswell Park Comprehensive Cancer Center (RPCCC) and NCI center grant P30CA016056. We acknowledge the support of RPCCC Shared Resources (SR) including BSGSR, ETM, FICSR, GSR, LASR, and PNSR. We also thank other Tang lab members for contributions to the AR projects and for helpful discussions.
Abbreviations
- AA
African American
- AD
Androgen-dependent
- ADT
Androgen deprivation therapy
- AI
Androgen-independent
- AR
Androgen receptor
- ARE
Androgen responsive elements
- AR-KO
AR knockout
- ARSIs
AR signaling inhibitors
- AR-Vs
AR splice variants
- CA
Caucasian American
- CRPC
Castration-resistant prostate cancer
- CSCs
Cancer stem cells
- CTD
C-terminal domain
- DBD
DNA-binding domain
- DKO
Double knockout (i.e., Pten−/l−Rb1−/−)
- DNPC
Double negative prostate cancer
- DHT
5α-dihydrotestosterone
- DHEA
Dehydroepiandrosterone
- eIF4F
Eukaryotic initiation factor 4F
- Enza
Enzalutamide
- GR
Glucocorticoid receptor
- LBD
Ligand binding domain
- MLS
Mitochondrial localization signal
- mCRPC
Metastatic castration resistant prostate cancer
- mtDNA
Mitochondrial DNA content
- NEPC
Neuroendocrine prostate cancer
- NLS
Nuclear localization signal
- NTD
N-terminal domain
- nDNA
Nuclear DNA
- PCa
Prostate cancer
- PCSCs
Prostate cancer stem cells
- PSA
Prostate specific antigen
- TF
Transcription factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interest Statement:
The authors declare no conflict of interests.
References
- 1.Tsai MJ, O’Malley BW, molecular mechanisms of action of steroid/thyroid receptor superfamily members, Annu. Rev. Biochem 63 (1994) 451–486. [DOI] [PubMed] [Google Scholar]
- 2.Auwerx J, Baulieu E, Beato M, Becker-Andre M, Burbach PH, Camerino G, et al. , A unified nomenclature system for the nuclear receptor superfamily, Cell 97 (1999) 161–163. [DOI] [PubMed] [Google Scholar]
- 3.Centenera M, Harris JM, Tilley WD, Butler LM, The contribution of different androgen receptor domains to receptor dimerization and signaling, Mol. Endocrinol 22 (2008) 2373–2382. [DOI] [PubMed] [Google Scholar]
- 4.van Royen ME, van Cappellen WA, de Vos C, Houtsmuller AB, Trapman J, Stepwise androgen receptor dimerization, J. Cell Sci 125 (2012) 1970–1979. [DOI] [PubMed] [Google Scholar]
- 5.Shang Y, Myers M, Brown M, Formation of the androgen receptor transcription complex, Mol. Cell 9 (2002) 601–610. [DOI] [PubMed] [Google Scholar]
- 6.Heemers HV, Tindall DJ, Androgen receptor (AR) coregulators: A diversity of functions converging on and regulating the AR transcriptional complex, Endocr. Rev 28 (2007) 778–808. [DOI] [PubMed] [Google Scholar]
- 7.Mcewan IJ, Gustafsson JÅ, Interaction of the human androgen receptor transactivation function with the general transcription factor TFIIF, Proc. Natl. Acad. Sci. U. S. A 94 (1997) 8485–8490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.F Huang CC, Lingadahali S, Morova T, Ozturan DD, Hu E, Yu IPL, et al. , Functional mapping of androgen receptor enhancer activity. Genome Biol 22 (2021) 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Visvader JE, Cells of origin in cancer. Nature 469 (2011) 314–322. [DOI] [PubMed] [Google Scholar]
- 10.Sowalsky AG, Kissick HT, Gerrin SJ, Schaefer RJ, Xia Z, Russo JW, et al. , Gleason 7 prostate cancers emerge through branched evolution of clonal Gleason pattern 3 and 4, Clin Cancer Res 23 (2017) 3823–3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilkinson S, Harmon FA, Terrigino NT, Karzai F, Pinto PA, Madan RA et al. , A case report of multiple primary prostate tumors with different drug sensitivity. Nat Commun 11 (2020) 837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilkinson S, Ye H, Karzai F, Harmon FA, Terrigino NT, VanderWeele DJ, et al. , Nascent prostate cancer heterogeneity drives evolution and resistance to intense hormonal therapy. Eur. Urol. March (2021) Online publication ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qin J, Liu X, Laffin B, Chen X, Choy G, Jeter CR, et al. , The PSA(−/lo) prostate cancer cell population harbors self-renewing long-term tumor-propagating cells that resist castration. Cell Stem Cell 10 (2012) 556–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen CD, Welsbie DS, Train C, Baek SH, Chen R, Vessella R, et al. , Molecular determinants of resistance to antiandrogen therapy. Nature Med 10 (2004) 33–39. [DOI] [PubMed] [Google Scholar]
- 15.Watson PA, Arora VK, Sawyers CL. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer 15(12):701–11, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simanainen U, Allan CM, Lim P, McPherson S, Jimenez M, Zajac JD, et al. , Disruption of prostate epithelial andrigen receptor impedes prostate lobe-specific growth and function. Endocrinol, 148 (2007) 2264–2272. [DOI] [PubMed] [Google Scholar]
- 17.Wu CT, Altuwaijri S, Ricke WA, Huang SP, Yeh S, Zhang C, et al. , Increased prostate cell proliferation and loss of cell differentiation in mice lacking prostate epithelial androgen receptor. Proc. Natl. Acad. Sci. USA, 104 (2007) 12679–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Niu Y, Altuwaijri S, Lai K, Wu C, Ricke WA, et al. , Androgen receptor is a tumor suppressor and proliferator in prostate cancer. Proc Natl Acad Sci U S A 105 (2008) 12182–12187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Niu Y, Altuwaijri S, Yeh S, Lai KP, Yu S, Chuang KH, et al. , Targeting the stromal androgen receptor in primary prostate tumors at earlier stages, Proc. Natl. Acad. Sci. U. S. A 105 (2008) 12188–12193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Niu Y, Chang TM, Yeh S, Ma WL, Wang YZ, Chang C, Differential androgen receptor signals in different cells explain why androgen-deprivation therapy of prostate cancer fails, Oncogene 29 (2010) 3593–604. [DOI] [PubMed] [Google Scholar]
- 21.Niu Y, Wang J, Shang Z, Huang SP, Shyr CR, Yeh S, et al. , Increased CK5/CK8-positive intermediate cells with stromal smooth muscle cell atrophy in the mice lacking prostate epithelial androgen receptor. PLoS One 6(2011) e20202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu S, Wang MW, Yao X, Chan FL, Establishment of a novel immortalized human prostatic epithelial cell line stably expressing androgen receptor and its application for the functional screening of androgen receptor modulators. Biochem Biophys Res Commun 382 (2009) 756–761. [DOI] [PubMed] [Google Scholar]
- 23.Bonaccorsi L, Carloni V, Muratori M, Salvadori A, Giannini A, Carini M, et al. , Androgen receptor expression in prostate carcinoma cells suppresses α6β4 integrin-mediated invasive phenotype. Endocrinol 141 (2000) 3172–3182. [DOI] [PubMed] [Google Scholar]
- 24.Litvinov IV, Antony L, Dalrymple SL, Becker R, Cheng L & Isaacs JT, PC3 but not DU145, human prostate cancer cells retain the coregulators required for tumor suppressor ability of androgen receptor, Prostate 66 (2006) 1329–1338. [DOI] [PubMed] [Google Scholar]
- 25.Stanbrough M, Leav I, Kwan PWL, Bubley GJ, Balk SP, Prostatic intraepithelial neoplasia in mice expressing an androgen receptor transgene in prostate epithelium, Proc. Natl. Acad. Sci. U. S. A 98 (2001) 10823–10828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han G, Buchanan G, Ittmann M, Harris JM, Yu X, DeMayo FJ, et al. , Mutation of the androgen receptor causes oncogenic transformation of the prostate, Proc. Natl. Acad. Sci. U. S. A 102 (2005) 1151–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao JC, Yu J, Runkle C, Wu L, Hu M, Wu D, et al. , Cooperation between Polycomb and androgen receptor during oncogenic transformation, Genome Res 22 (2012) 322–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cai C, He HH, Chen S, Coleman I, Wang H, Fang Z, et al. , Androgen receptor gene expression in prostate cancer is directly suppressed by the androgen receptor through recruitment of lysine-specific demethylase 1, Cancer Cell 20 (2011) 457–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao S, Gao Y, He HH, Han D, Han W, Avery A, et al. , Androgen Receptor Tumor Suppressor Function Is Mediated by Recruitment of Retinoblastoma Protein, Cell Rep 17 (2016) 966–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vander Griend DJ, Litvinov IV, Isaacs JT, Conversion of androgen receptor signaling from a growth suppressor in normal prostate epithelial cells to an oncogene in prostate cancer cells involves a gain of function in c-Myc regulation, Int. J. Biol. Sci 10 (2014) 627–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lizzam A, Schoor FVD, Dalrymple SL, Issac JT, Androgen receptor (AR) suppresses normal human prostate epithelial cell proliferation via AR/β-catenin/TCF-4 complex inhibition of c-MYC transcription, Prostate 74 (2014) 1118–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu Y, Horn JL, Banda K, Goodman AZ, Lim Y, Jana S, et al. , The androgen receptor regulates a druggable translational regulon in advanced prostate cancer, Sci. Transl. Med 11 (2019) eaaw4993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li J, Fu X, Cao S, Li J, Xing S, Li D, et al. , Membrane-associated androgen receptor (AR) potentiates its transcriptional activities by activating heat shock protein 27 (HSP27) J Biol Chem, 293 (2018) 12719–12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bajpai P, Koc E, Sonpavde G, Singh R, Sing KK, Mitochondrial localization, import, and mitochondrial function of the androgen receptor, J. Biol. Chem, 294 (2019) 6621–6634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deng Q & Tang DG Androgen receptor and prostate cancer stem cells: Biological mechanisms and clinical implications, Endocr-Relat Cancer 22 (2015) T209–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li Q, Deng Q, Chao HP, Liu X, Lu Y, Lin K, et al. , Linking prostate cancer cell AR heterogeneity to distinct castration and enzalutamide responses, Nat. Commun 9 (2018) 3600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu X, Chen X, Rycaj K, Chao HP, Deng Q, Jeter C, et al. Systematic dissection of phenotypic, functional, and tumorigenic heterogeneity of human prostate cancer cells, Oncotarget 6 (2015) 23959–23986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ruizeveld De Winter JA, Janssen PJA, Sleddens HMEB, Verleun-Mooijman MCT, Trapman J, Brinkmann AO, et al. , Androgen receptor status in localized and locally progressive hormone refractory human prostate cancer, Am. J. Pathol 144 (1994) 735–746. [PMC free article] [PubMed] [Google Scholar]
- 39.Masai M, Sumiya H, Akimoto S, Yatani R, Chang C, Liao S, et al. , Immunohistochemical study of androgen receptor in benign hyperplastic and cancerous human prostates, Prostate 17 (1990) 293–300. [DOI] [PubMed] [Google Scholar]
- 40.Chodak GW, Kranc DM, Puy LA, Takeda H, Johnson K, Chang C, Nuclear localization of androgen receptor in heterogeneous samples of normal, hyperplastic and neoplastic human prostate, J. Urol 147 (1992) 798–803. [DOI] [PubMed] [Google Scholar]
- 41.Sadi MV, Walsh PC, Barrack ER, Immunohistochemical study of androgen receptors in metastatic prostate cancer. Comparison of receptor content and response to hormonal therapy, Cancer 67 (1991) 3057–3064. [DOI] [PubMed] [Google Scholar]
- 42.van der Kwast TH, Schalken J, de Winter JAR, van Vroonhoven JCC, Mulder E, Boersma W, Androgen receptors in endocrine-therapy-resistant human prostate cancer, Int. J. Cancer 48 (1991) 189–193. [DOI] [PubMed] [Google Scholar]
- 43.Sadi MV, Barrack ER, Image analysis of androgen receptor immunostaining in metastatic prostate cancer heterogeneity as a predictor of response to hormonal therapy, Cancer 71 (1993) 2574–2580. [DOI] [PubMed] [Google Scholar]
- 44.Shah RB, Mehra R, Chinnaiyan AM, Shen R, Ghosh D, Zhou M, et al. , Androgen-independent prostate cancer is a heterogeneous group of diseases: Lessons from a rapid autopsy program, Cancer Res 64 (2004) 9209–9216. [DOI] [PubMed] [Google Scholar]
- 45.Davis JN, Wojno KJ, Daignault S, Hofer MD, Kuefer R, Rubin MA, et al. , Elevated E2F1 inhibits transcription of the androgen receptor in metastatic hormone-resistant prostate cancer, Cancer Res 66 (2006) 11897–11906. [DOI] [PubMed] [Google Scholar]
- 46.Bluemn EG, Coleman IM, Lucas JM, Coleman RT, Tharakan R, Bianchi-frias D, et al. , Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained through FGF Signaling, Cancer cell 32 (2018) 474–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Labrecque MP, Coleman IM, Brown LG, True LD, Kollath L, Lakely B, et al. , Molecular profiling stratifies diverse phenotypes of treatment-refractory metastatic castration-resistant prostate cancer, J. Clin. Invest 129 (2019) 4492–4505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nishiyama T, Hashimoto Y, Takahashi K, The influence of androgen deprivation therapy on dihydrotestosterone levels in the prostatic tissue of patients with prostate cancer, Clin. Cancer Res 10 (2004) 7121–7126. [DOI] [PubMed] [Google Scholar]
- 49.Titus MA, Schell MJ, Lih FB, Tomer KB, Mohler JL, Testosterone and dihydrotestosterone tissue levels in recurrent prostate cancer, Clin. Cancer Res 11 (2005) 4653–4657. [DOI] [PubMed] [Google Scholar]
- 50.Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS, Maintenance of intratumoral androgens in metastatic prostate cancer: A mechanism for castration-resistant tumor growth, Cancer Res 68 (2008) 4447–4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arora VK, Schenkein E, Murali R, Subudhi SK, Wongvipat J, Balbas MD, et al. , Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade, Cell 155 (2013) 1309–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Isikbay M, Otto K, Kregel S, Kach J, Cai Y, Vander Griend DJ, et al. , Glucocorticoid Receptor Activity Contributes to Resistance to Androgen-Targeted Therapy in Prostate Cancer, Horm Cancer 5 (2014) 72–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grindstad T, Andersen S, Al-Saad S, Donnem T, Kiselev Y, Melbø-Jørgensen CN, et al. , High progesterone receptor expression in prostate cancer is associated with clinical failure, PLoS One 10s(2015) e0116691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bonkhoff H, Fixemer T, Hunsicker I, Remberger K, Progesterone receptor expression in human prostate cancer: Correlation with tumor progression, Prostate 48 (2001) 285–291. [DOI] [PubMed] [Google Scholar]
- 55.Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinänen R, Palmberg C, et al. , In vivo amplification of the androgen receptor gene and progression of human prostate cancer, Nat. Genet 9 (1995) 401–406. [DOI] [PubMed] [Google Scholar]
- 56.Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, et al. , Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer, Nat. Genet 44 (2012) 685–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Takeda DY, Spisak S, Seo J-H, Bell C, O’Connor E, Korthauer K et al. , A Somatically Acquired Enhancer of the Androgen Receptor Is a Noncoding Driver in Advanced Prostate Cancer Cell 12 (2018) 422–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Viswanathan SR, Ha G, Hoff AM, Wala JA, Carrot-Zhang J, Whelan CW, et al. , Structural Alterations Driving Castration-Resistant Prostate Cancer Revealed by Linked-Read Genome Sequencing. Cell 174 (2018) 433–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Watson PA, Chen YF, Balbas MD, Wongvipat J, Socci ND, Viale A, et al. , Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor, Proc. Natl. Acad. Sci. USA, 107 (2010) 16759–16765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ware KE, Garcia-Blanco MA, Armstrong AJ, Dehm SM, Biologic and clinical significance of androgen receptor variants in castration resistant prostate cancer, Endocr. Relat. Cancer, 21 (2014) T87–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nakazawa M, Antonarakis ES, Luo J, Androgen Receptor Splice Variants in the Era of Enzalutamide and Abiraterone, Hormones and Cancer 5 (2014) 265–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kohli M, Ho Y, Hillman DW, Van Etten JL, Henzler C, Yang R, et al. , Androgen Receptor Variant AR-V9 Is Coexpressed with AR-V7 in Prostate Cancer Metastases and Predicts Abiraterone Resistance, Clinical Cancer research 23 (2017) 4704–4715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li Y, Chan SC, Brand LJ, Hwag TH, Silverstein KAT, Dehm SM. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines, Cancer Res 73 (2013) 483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Beltran H, Rickman DS, Park K, Chae SS, Sboner A, MacDonald TY, et al. , Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets, Cancer Discov 1 (2011) 487–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Epstein JI, Amin MB, Beltran H, Lotan TL, Mosquera JM, Reuter VE, et al. , Proposed morphologic classification of prostate cancer with neuroendocrine differentiation, Am. J. Surg. Pathol 38 (2014) 756–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tan HL, Sood A, Rahimi HA, Wang W, Gupta N, Hicks J, et al. , Rb loss is characteristic of prostatic small cell neuroendocrine carcinoma, Clin. Cancer Res 20 (2014) 890–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhou Z, Flesken-Nikitin A, Corney DC, Wang W, Goodrich DW, Roy-Burman P, et al. , Synergy of p53 and Rb deficiency in a conditional mouse model for metastatic prostate cancer, Cancer Res 66 (2006) 7889–7898. [DOI] [PubMed] [Google Scholar]
- 68.Mu P, Zhang Z, Benelli M, Karthaus WR, Hoover E, Chen CC, et al. , SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 355 (2017) 84–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW, et al. , Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 355 (2017) 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen X, Li Q, Liu X, Liu C, Liu R, Rycaj K, et al. , Defining a population of stem-like human prostate cancer cells that can generate and propagate castration-resistant prostate cancer, Clin. Cancer Res 22 (2016) 4505–4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wan X, Liu J, Lu JF, Tzelepi V, Yang J, Starbuck MW, et al. , Activation of β-catenin signaling in androgen receptor-negative prostate cancer cells, Clin. Cancer Res 18 (2012) 726–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Corella AN, A Ordonio MV, Coleman I, Lucas JM, Kaipainen A, Nguyen HM, et al. , Identification of Therapeutic Vulnerabilities in Small-cell Neuroendocrine Prostate Cancer, Clin Cancer Res 26 (2020) 1667–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Su W, Han HH, Wang Y, Zhang B, Zhou B, Cheng Y, et al. , The Polycomb Repressor Complex 1 Drives Double-Negative Prostate Cancer Metastasis by Coordinating Stemness and Immune Suppression, Cancer Cell 36 (2019) 139–155.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.