

Abstract
Human leukocyte antigen (HLA) genes are among the most polymorphic of our genome, as a likely consequence of balancing selection related to their central role in adaptive immunity. HLA-A and HLA-B genes were recently suggested to evolve through a model of joint divergent asymmetric selection conferring all human populations, including those with severe loss of diversity, an equivalent immune potential. However, the mechanisms by which these two genes might undergo joint evolution while displaying very distinct allelic profiles in populations are still unknown. To address this issue, we carried out extensive data analyses (among which factorial correspondence analysis and linear modeling) on 2,909 common and rare HLA-A, HLA-B, and HLA-C alleles and 200,000 simulated pathogenic peptides by taking into account sequence variation, predicted peptide-binding affinity and HLA allele frequencies in 123 populations worldwide. Our results show that HLA-A and HLA-B (but not HLA-C) molecules maintain considerable functional divergence in almost all populations, which likely plays an instrumental role in their immune defense. We also provide robust evidence of functional complementarity between HLA-A and HLA-B molecules, which display asymmetric relationships in terms of amino acid diversity at both inter- and intraprotein levels and in terms of promiscuous or fastidious peptide-binding specificities. Like two wings of a flying bird, the functional complementarity of HLA-A and HLA-B is a perfect example, in our genome, of duplicated genes sharing their capacity of assuming common vital functions while being submitted to complex and sometimes distinct environmental pressures.
Keywords: HLA diversity, peptide-binding predictions, HLA allele frequencies, functional complementarity, balancing selection, HLA duplicated genes
Introduction
The three classical HLA Class I genes, namely HLA-A, HLA-B, and HLA-C, are the first known genes of the major histocompatibility complex (MHC) in the human genome. A common name (HL-A) together with consecutive numbers were first attributed to the serological specificities detected in the late 1950s and early 1960s (Curtoni et al. 1967) before researchers discovered that these surface antigens were encoded by two different genes, further named HLA-A and HLA-B (Kissmeyer-Nielsen et al. 1968). A third gene found afterward was called HLA-C (Thorsby et al. 1970; Solheim and Thorsby 1973; Thorsby 2009), which displays in many tissues, although not all, lower levels of cell surface expression (Neefjes and Ploegh 1988; Neisig et al. 1998; Kulkarni et al. 2011; Carey et al. 2019).
HLA genes, and more particularly Class I genes, are known to be among the most polymorphic of our genome (Shiina et al. 2009). According to the IPD-IMGT/HLA Database (Release 3.37.0 at https://www.ebi.ac.uk/ipd/imgt/hla/; last accessed December 24, 2020), more than 7,000 alleles have been reported so far for HLA-B and about 6,000 for either HLA-A and HLA-C, which encode altogether almost 12,000 distinct HLA Class I molecules (Robinson et al. 2020). The number of reported HLA alleles does not cease to increase each year, mostly since the advent of high-throughput sequencing technologies, especially Next Generation Sequencing to the HLA region. Nevertheless, these alleles are still attributed to a reduced number of allele families (defined at the first-field level of resolution according to the official HLA nomenclature), often considered as HLA lineages, and generally corresponding to the serological specificities defined decades ago (Marsh et al. 2010).
The extremely high degree of polymorphism in classical HLA Class I genes is usually seen as a consequence of their central role in the regulation of adaptive immunity: the glycoproteins they encode, expressed on the surface of most cell types, bind and present small intracellular peptides, typically 9-mer, to the receptors of CD8+ cytotoxic T lymphocytes, allowing the latter to detect and eliminate virus-infected or tumorous cells (Rudolph et al. 2006). Three forms of pathogen-mediated balancing selection mechanisms were proposed to explain this diversity (Spurgin and Richardson 2010), namely the heterozygote advantage (Doherty and Zinkernagel 1975; Prugnolle et al. 2005; Qutob et al. 2012), the rare-allele advantage (Bodmer 1972; Slade and McCallum 1992), and the fluctuating selection (Hill 1991) models. Besides, directional selection (either negative or positive) on particular HLA alleles associated with susceptibility and resistance to certain pathogens has been suggested (Dendrou et al. 2018; Sanchez-Mazas 2020), although its signatures might be difficult to detect (Penman and Gupta 2018). For instance, studies on malaria have suggested soft selective sweep (Hermisson and Pennings 2005; Messer and Petrov 2013) acting on several HLA-B alleles with moderate protective effects against Plasmodium falciparum and explaining why strong signals of selection may not be demonstrated (Sanchez-Mazas et al. 2017b).
Not surprisingly, the α1 and α2 domains of HLA Class I molecules, which form the peptide-binding groove, became the subject of intensive studies, and a concentration of polymorphic sites was observed in these domains (Hedrick et al. 1991; Robinson et al. 2017; Goeury et al. 2018, although high molecular variation was also recently disclosed in HLA genes’ regulatory regions (Souza et al. 2020). These features of HLA molecules and related peptides may simultaneously affect the binding processes and peptide-binding preferences (Zhang et al. 2017). In case of a 9-mer bound peptide, the p2 (position 2) and p9 (position 9 and C-terminus) residues, corresponding to the B and F pockets within the groove, respectively, were suggested to be the “primary” anchors. These positions might have stronger impact on the peptide-binding specificity (Saper et al. 1991; Madden 1995) compared with the “secondary” anchor positions p1 and p3, and to the other ones (Sidney, Assarsson, et al. 2008; see fig. 1a adapted from Klein and Sato, 2000 and fig. 1b to be presented in later sections). As a result, although each specific HLA molecule is only able to bind a rather limited amount of peptides compared with the quasi unlimited set of peptides that can theoretically be derived from pathogens, the binding repertoires of distinct HLA molecules greatly differ from each other (Falk et al. 1991), providing altogether a remarkable binding potential. This also led to propositions of broad functional groups, often known as “supertypes,” without any consensual definition currently in use (Kangueane et al. 2005; Sidney, Peters, et al. 2008; Wang and Claesson 2014; Mukherjee et al. 2015). Actually, the fraction of bound peptides varies depending on the allele encoding each molecule. Some HLA Class I molecules display larger (or “promiscuous”) repertoires that would protect individuals to a wider variety of pathogens, and thus act as “generalists”; others display narrower but more specific (or “fastidious”) repertoires that would protect individuals to new and likely more virulent pathogens, and thus act as “specialists” (Chappell et al. 2015; Kaufman 2018).
Fig. 1.

Schema showing the anchoring residues (p1–p9) of a 9-mer peptide in the peptide-binding groove (with B and F pockets indicated) of an HLA-B*27:03 molecule taken as an example (a) and its consensus sequence logo chart produced by MHCcluster conveying information about the conservation of a binding motif for each residue (b).
In this context, direct approaches to measure the binding repertoire of HLA molecules using the peptide elution method (Falk et al. 1991) were limited by experimental complexity and labor costs and urged the development of in silico methods predicting HLA-binding specificities (Zhang et al. 2005; Jojic et al. 2006; Jacob and Vert 2008; Hoof et al. 2009). Among these methods, NetMHCpan (Hoof et al. 2009), a neural network-based predictor trained on eluted MHC peptide-binding data contained in the Immune Epitope Database (Vita et al. 2015), was shown to be an effective and state-of-the-art tool in better understanding the evolution of HLA genes (Rasmussen et al. 2014; van Deutekom and Keşmir 2015; Buhler et al. 2016; Pierini and Lenz 2018). Recently, Pierini and Lenz (2018) studied the direct correlation between pairwise sequence divergence and the corresponding peptide-binding repertoire across different HLA genes and supported the divergent allele advantage (known as DAA) as a meaningful quantitative mechanism of pathogen-mediated selection. In another study, Buhler et al. (2016) revealed that the level of functional diversity was maintained in worldwide populations when HLA-A and HLA-B molecules were considered simultaneously, whereas the diversity of HLA-C molecules would not increase significantly the peptide-binding repertoire. The particularity of HLA-C is possibly due both to its lower level of surface expression in many somatic tissues and to the more important role of HLA-C molecules as ligands of killer-cell immunoglobulin-like receptors (KIRs). The authors thus suggested that HLA-A and HLA-B genes coevolved through a model of “joint divergent asymmetric selection” conferring all populations, including those with severe loss of genetic diversity, an equivalent immune potential. However, still unknown are the mechanisms by which these two genes might undergo joint evolution while displaying very distinct allelic profiles in populations.
To address this issue, we studied a large set of HLA Class I alleles by putting together amino acid sequence, peptide-binding affinity and allele frequency data. For 2,909 high-resolution HLA alleles, we extracted their corresponding amino acid sequences and estimated peptide-binding affinities in order to consider the relationships between the primary structure and molecular function, and we gathered their frequencies in 123 human populations. To our knowledge, this is the first study treating simultaneously protein diversity, functional properties, and population profiles of HLA molecules on such a scale. Our results indicate that most selective signals detected for HLA Class I genes at both inter- and intraprotein levels can be explained by functional divergence and complementarity of HLA-A and HLA-B molecules, which then behave, as our factorial correspondence analysis nicely illustrates, like two wings of a flying bird.
Results
General Pattern of Functional Relationships among HLA Class I Molecules
We performed a factorial correspondence analysis (FCA) to display the functional relationships between the 2,909 HLA-A, HLA-B, and HLA-C molecules along with the 200,000 simulated peptides for which the binding affinity was estimated (fig. 2). These relationships show a flying bird-like pattern along the first three axes of the FCA (fig. 2a and d, online tool S1 at https://hla-net.eu/interactive/HLA_wings/tool_S1/ (last accessed December 25, 2020) for a 3D plot and online tool S2 at https://hla-net.eu/interactive/HLA_wings/tool_S2/ providing a user interactive plot; fig. 2e–h to be presented in later sections). The bird’s two “wings” are represented, respectively, by HLA-A and HLA-B molecules, its “head” by additional HLA-B molecules, its “tail” by additional HLA-A molecules, and its tiny “claws” by HLA-C molecules, all surrounded by a cloud of high-affinity peptides. The transition between these extending parts is continuous and is composed of a heterogeneous set of HLA-A, HLA-B, and HLA-C molecules. In general, the principal functional divergence is observed between HLA-A and HLA-B molecules, but the divergence between molecules of the same gene is also considerable in some cases (fig. 2a and b). Despite this large range of peptide-binding affinities, some molecules do appear to be functionally very similar to each other (gray framed parts in fig. 2a and b zoomed in fig. 2c and d, respectively). As for HLA-C molecules, they concentrate substantially more in the center of the plot, indicating that HLA-C is functionally much more homogeneous than HLA-A and HLA-B in view of its peptide-binding affinities (fig. 2a–d).
Fig. 2.

FCA of peptide-binding affinity data for 2,909 HLA Class I molecules using 200,000 simulated peptides with two of the first three correspondences visualized, respectively, representing together ∼50% of the total variance. Both HLA molecules and peptides are plotted, distinguished by colors (red for HLA-A, blue for HLA-B, yellow for HLA-C, and gray for peptides). Only the most common molecule belonging to each HLA-A and HLA-B lineage is labeled by the first-field names (a, b), then the gray framed central part zoomed in with one molecule for each HLA-A, HLA-B, and HLA-C lineage labeled similarly (c, d), and next, for each HLA molecule, the plot size is proportional to the numbers of populations in which its corresponding allele was observed (e, f), and finally, as an example of their population distribution, the HLA molecules are highlighted by labels of different sizes proportional to the allele frequencies they correspond in an Australian Aboriginal population from Cape York Peninsula (g, h).
Going into details, HLA-A and HLA-B molecules can be loosely clustered into three main divergent functional groups, each of them being characterized by relatively unique peptide-binding affinities. The first group (group 1) includes HLA-A*03, *11, *31, *33, *68, and *74 molecules, the second one (group 2) HLA-B*18, *37, *40, *41, *44, *45, and *50 molecules, and the third one (group 3, mostly visible in fig. 2b) HLA-A*02 molecules (note that in some cases, molecules belonging to a same lineage, e.g., HLA-A*24 molecules, cluster in different groups). All the other HLA-A and HLA-B molecules (among which HLA-B*15 encoded by the most polymorphic HLA lineage) constitute a fourth group (group 4) of HLA-A and HLA-B intermixed with HLA-C molecules in the center of the FCA (fig. 2c and d and online tool S2 at https://hla-net.eu/interactive/HLA_wings/tool_S2/, option “Labeling all molecules encoded by alleles of a same lineage”).
When the binding affinity data are checked for each peptide using the threshold for weak binders (ELrank < 2) to identify its HLA binders, a total of 57.6% of the 200,000 peptides are predicted to bind at least one of the 2,909 HLA molecules (fig. 3a), whereas the other 42.4% are not expected to bind any HLA molecule (as shown by gray dots positioned far from any HLA molecule in fig. 2a and b). A quarter (25.5%) of the total peptides are predicted to bind molecule(s) encoded by a same gene, mostly HLA-A or HLA-B (9.5% by HLA-A molecules only, 15.4% by HLA-B only, and merely 0.6% by HLA-C only). Surprisingly, more peptides (32.1%) are predicted to bind HLA molecules encoded by either two or three HLA Class I genes (7.4% by HLA A&B, 1.6% by HLA A&C, 4.7% by HLA B&C, and 18.4% by HLA A&B&C). As expected, when the threshold for strong binders (ELrank < 0.5) is applied, only 37.4% of the 200,000 peptides are predicted to bind HLA molecule(s) (fig. 3b–e to be presented in later sections), and the majority of them (21.7%) are predicted to bind molecule(s) encoded by a same gene (7.8% by HLA-A only, 13.0% by HLA-B only, and merely 0.9% by HLA-C only), whereas the proportion of peptides that are predicted to bind HLA molecules encoded by different genes is severely reduced (15.7%: with 4.2% by HLA A&B, 1.4% by HLA A&C, 3.6% by HLA B&C, and 6.5% by HLA A&B&C). These results confirm that HLA-A and HLA-B molecules display both a main functional divergence and specific peptide-binding affinities, which is not the case for HLA-C, though the sharing of specific peptides among HLA molecules of different genes is definitively not a rare phenomenon.
Fig. 3.

Venn diagrams showing, with two upper thresholds of the rank of predicted binding score (ELrank < 2, for weak binders and ELrank < 0.5 for strong binders), the absolute proportion of peptides, among 200,000 random ones, which are predicted to bind at least one of the 2,909 HLA-A, HLA-B, or HLA-C molecules, respectively, and the proportion of peptides that might bind HLA molecules of two (A&B, A&C, and B&C) or three (A&B&C) genes (a: weak binders; b: strong binders). The proportion of peptides estimated similarly for different categories of HLA molecules characterized by the range of distribution in populations are further summarized by line charts (c, e: weak binders; d, f: strong binders).
Interprotein Relationships: Sequence Variation and Functional Divergence of HLA Class I Molecules
A pairwise divergence matrix of the amino acid sequences of all the 2,909 HLA Class I molecules was used to plot sequence divergence histograms for molecule pairs related either to a single gene or to two different genes, that is, HLA A~A, B~B, and C~C for one-gene molecule pairs and HLA A~B, A~C, and B~C for two-gene molecule pairs (fig. 4a, b, and c-l to be presented in later sections). More than 91% of the one-gene molecule pairs differ by 5–25 amino acid residues, within a range of 0–30. HLA-A and HLA-B show greater proportions of divergent pairs (A~A: mean: 16.33, standard deviation [SD]: 6.71, B~B: mean: 15.88, SD: 5.24) compared with HLA-C (C~C: mean: 11.06, SD: 4.08). Moreover, contrary to HLA B~B pairs that show a symmetric distribution, the distributions for HLA A~A and C~C pairs are somewhat asymmetric and bimodal, with more pairs displaying lower (0–5) and (for A~A pairs) higher (20–25) amino acid differences (fig. 4a). Two-gene pairs differ by much more amino acid residues, within a range of 10–42. Most HLA A~B and A~C pairs differ by more than 20 amino acid residues (A~B: mean: 32.45, SD: 3.61, A~C: mean: 31.30, SD: 3.07), which is more prominent compared with HLA B~C pairs (mean: 23.94, SD: 4.16). The three two-gene pair distributions are more or less symmetric (fig. 4b).
Fig. 4.

Histograms of amino acid sequence differences (α1 and α2 domains encoded by exons 2 and 3) and density distributions of pairwise functional distances between one gene, that is, HLA A~A (red), B~B (blue), and C~C (yellow), and two gene, that is, HLA A~B (violet), A~C (orange), and B~C (green) molecule pairs, which concern all possible combinations of the 2,909 HLA molecules (a–d); or those of the 240 common HLA molecules (e–h); or those of the 31 most common molecules (i–l).
However, the density distributions of functional distances between the HLA molecules based on peptide-binding affinity differences are not all in close accordance with those of their amino acid sequence differences (fig. 4c and d), as confirmed by heterogeneous and sometimes low correlation coefficients (R) between sequence differences and functional distances (R: 0.81 for HLA A~A, 0.65 for B~B, 0.71 for C~C, 0.14 for A~B, −0.23 for A~C, and 0.43 for B~C pairs; see online tool S3 at https://hla-net.eu/interactive/HLA_wings/tool_S3/, panel “Correlation coefficients” with “Minimum number of populations” at zero). For one-gene pairs, much greater proportions of high values (0.7–0.9) are observed for HLA A~A (mean: 0.65, SD: 0.27) and B~B (mean: 0.65, SD: 0.23) pairs compared with C~C ones (mean: 0.44, SD: 0.21) (fig. 4c). Large functional differences have apparently been maintained for most HLA A~A and B~B pairs, which are comparable with the range of functional distances between two-gene pairs (fig. 4d). Contrary to sequence differences, HLA A~C (mean: 0.75, SD: 0.12) and B~C (mean: 0.75, SD: 0.13) pairs show similar distributions of functional distances, less prominent than HLA A~B pairs (mean: 0.80, SD: 0.10). In consequence, asymmetric distributions were observed for all pairs, and the distribution for HLA A~A pairs is again bimodal, with larger proportions of both extremely divergent (0.75–0.9) and extremely similar (<0.1) pairs.
Intraprotein Relationships: Sequence Variation and Functional Divergence between α1 and α2 Domains
The specificity of different anchoring pockets within the HLA peptide-binding groove was investigated in relation to the predicted bound peptides. High-binding specificity of a pocket would result in little variation of the amino acid residues at the corresponding position on the bound peptides, which corresponds to a low value of Simpson’s diversity reciprocal index (SRI). In terms of peptide-binding consensus sequence logo charts, this would mean that a lower SRI corresponds to a larger binding motif (fig. 1b).
As expected, residues at the primary anchor positions (i.e., p2 and p9 corresponding to B and F pockets of the HLA peptide-binding groove, respectively) show much lower SRI values than other residues, for the three genes (fig. 5a–c). However, a striking difference was found between HLA-A and HLA-B: position p9 shows a lower SRI compared with position p2 for the majority of HLA-A molecules, whereas the reverse can be seen for HLA-B (fig. 5d). This suggests that F pocket is more decisive for the binding affinity of HLA-A molecules, whereas B pocket has a greater effect on HLA-B binding preference. This distinctive pattern of affinity determination is also visible in the FCA charts (online tool S2 at https://hla-net.eu/interactive/HLA_wings/tool_S2/, option “Highlighting all the peptides with a specific residue at the positions p2 or p9”): the distribution of peptides with a same p2 residue varies mainly depending on HLA-B molecules, whereas that of peptides with a same p9 residue is mainly related to HLA-A molecules. In reference to the concepts characterizing the binding repertoire of HLA molecules (Chappell et al. 2015; Kaufman 2018), F pocket would thus be more fastidious (specialist) and B pocket more promiscuous (generalist) for HLA-A molecules, whereas B pocket would be more fastidious and F pocket more promiscuous for HLA-B molecules. Very similar results were obtained by using thresholds of ELrank to define bound peptides instead of the proportion of BAscore (results now shown). As of HLA-C molecules, the difference of SRI between p2 and p9 is not obvious.
Fig. 5.

Violin charts of SRI of amino acid residues at each position (p1–p9) of the top 1% among the 200,000 9-mer peptides showing the highest binding affinity (BAscore) to 2,909 HLA-A (a), HLA-B (b), and HLA-C (c) molecules as well as a Box plot synthesizing the results at p2 and p9 for the three genes (d); a plot of SRI values at p2 against SRI values at p9, for 2,909 HLA-A (red), HLA-B (blue), and HLA-C (yellow) molecules (e); and plot of log(SRI) at p2 against log(SRI) at p9 , for 194 HLA-A (red) and HLA-B (blue) common molecules (f), in the latter two the symbol sizes representing the range of their distribution in populations.
When SRI values at p2 are plotted against those at p9 for the 2,909 HLA molecules, a reverse relationship is strongly suggested (fig. 5e;fig. 5f to be presented in later sections). Linear models (lm1–lm5 in table 1) indicate a significantly negative correlation by including “gene” as interaction (lm1, with adjusted R2: 0.4254, P < 0.001). The model can be further improved by using log(SIR) at p2 against log(SRI) at p9, removing all the HLA-C molecules, and including SRI at the two secondary anchor positions, p1 and p3, as interactions (lm5, adjusted R2: 0.6750, P < 0.001). This relationship confirms the complementary role played by the B and F pockets and reveals an asymmetric relationship between HLA-A and HLA-B molecules.
Table 1.
Procedure of Linear Modeling of SRI Values between p2 and p9 and Results of Analysis of Variance (F statistics) for Pairwise Model Comparison(All the values of Coefficient of Determination R2 and F statistics are very Significant with P < 0.001), and the Models lm2 and lm6 Are Retained.
| Models | Description | Adjusted R2 | F Statistics |
|---|---|---|---|
| lm1 | p9 ∼ p2 × gene | 0.4254 | — |
| lm2 | p9 ∼ (p1 + p2 + p3) × gene | 0.5033 | lm2 vs. lm1:76.879 |
| lm3 | log(p9) ∼ log(p2) × gene | 0.5091 | — |
| lm4 | log(p9) ∼ (log(p1) + log(p2) + log(p3)) × gene | 0.6346 | lm4 vs. lm3:167.120 |
| lm5 |
log(p9) ∼ (log(p1) + log(p2) + log(p3)) × gene HLA-A and HLA-B molecules only |
0.6750 | — |
| lm6 |
log(p9) ∼ (log(p1) + log(p2) + log(p3)) × gene common HLA-A and HLA-B molecules only |
0.7309 | — |
Functional Distances between HLA Molecules in Relation to Their Distribution in Populations
In our analyses, we systematically considered the different categories of HLA molecules defined according to the range of their geographic distribution (table 2). Starting from the FCA charts, the 2,909 HLA molecules are represented by different sizes proportional to the numbers of populations in which they are observed (fig. 2e and f). The pattern of the four functional groups of HLA molecules (group 1–group 4) defined above and the functional divergence among them are perfectly maintained when only the common molecules (observed in at least five populations), or even only the most common molecules (observed in at least 75 populations), are taken into account. For each studied population, HLA molecules are labeled proportionally to the frequencies of their corresponding allele on the FCA (an example given in fig. 2g and h, and online tool S2 at https://hla-net.eu/interactive/HLA_wings/tool_S2/ for all populations, option “Labeling only the molecules observed in one population”). Most interestingly, these plots show the presence of HLA molecules belonging to different functional groups within each population, despite different frequency patterns. Actually, most populations exhibit alleles corresponding to the molecules of all four functional groups: either many alleles at very low (e.g., Sudanese and other African populations) or uneven (e.g., Albanian and other European populations; Japanese and other East Asian populations) frequencies or fewer alleles at relatively even (e.g., Australian Aborigines) or uneven (e.g., Navajo and other Native Americans; Taiwanese Aborigines) frequencies. The only exceptions are a few Papuan populations (Abelam, Pawaia, and Rabaul) which lack alleles encoding group 3 (i.e., HLA-A*02) molecules. This means that HLA molecules displaying different functional properties have been kept in almost all populations and likely play a complementary role in their immune defense.
Table 2.
Numbers of HLA-A, HLA-B, and HLA-C Alleles/Molecules of the Categories Characterized by the Minimum Numbers of Populations of Our Data Set in Which They Were Observed.
| Allele/Molecule Categories | HLA-A | HLA-B | HLA-C | Total |
|---|---|---|---|---|
| Estimated for binding affinity | 894 | 1,413 | 622 | 2,909 |
| Observed in at least one population | 213 | 350 | 153 | 716 |
| Observed in at least 5 populations (common) | 67 | 127 | 46 | 240 |
| Observed in at least 10 populations | 47 | 86 | 37 | 170 |
| Observed in at least 25 populations | 32 | 52 | 24 | 108 |
| Observed in at least 50 populations | 14 | 26 | 20 | 60 |
| Observed in at least 75 populations (most common) | 9 | 9 | 13 | 31 |
Null alleles, which are not suitable for peptide-binding affinity estimation, were not included.
When looking at the sharing of bound peptides by the 240 common HLA molecules, the proportion of the peptides that are expected to bind at least one HLA molecule drops only from 57.6% to 50.7% for weak binders and from 37.4% to 30.3% for strong binders compared with the results observed for the total set of 2,909 HLA molecules (fig. 3c and d). Eventually, the 31 most common HLA molecules, with only 9 HLA-A, 9 HLA-B, and 13 HLA-C molecules, still cover about 39.0% of the total peptides as weak binders (fig. 3c). The peptide coverage is thus more or less maintained by the common, or even the most common molecules, which confirms the pattern displayed by the FCA. Interestingly, the proportion of peptides predicted to bind HLA-C molecules seems independent to the range of their distribution in populations (fig. 3e and f).
At the interprotein level, the histograms of pairwise sequence differences remain similar when considering the different categories of HLA molecules (fig. 4e and f for the common molecules and fig. 4i and j for the most common molecules; online tool S3 at https://hla-net.eu/interactive/HLA_wings/tool_S3/, panel “Amino acid sequence differences”). In contrast, the patterns of density charts of pairwise functional distances change substantially when only the common HLA molecules are kept: In these cases, the proportion of functionally similar pairs decreases and the proportion of pairs with intermediate functional distances increases; this is true for HLA A~A, B~B, C~C, A~B, and A~C pairs (fig. 4g and h for the common molecules and fig. 4k and l for the most common molecules; online tool S3, panel “Functional distances”). Actually, the more widely distributed the HLA molecules (see table 2 and supplementary table S1, Supplementary Material online), the more marked this tendency, which was confirmed by the Kolmogorov–Smirnov test (result not shown). HLA A~A pairs also appear to be more sensitive to these changes than B~B and C~C pairs. Intriguingly, when considering the most common HLA molecules, the density distributions of A~A and B~B pairwise functional distances become very similar and the Kolmogorov–Smirnov test is no longer able to distinguish them (fig. 4k and online tool S3).
Within a population, we also notice that, for two different HLA molecules, the more functionally similar they are, the lower the expected chance that both are carried by an individual (online tool S4 at https://hla-net.eu/interactive/HLA_wings/tool_S4/). In other words, for a given HLA A~A (nonidentical pairs, i.e., without pairs consisting of identical molecules, e.g., A*01:01~A*01:01), HLA B~B (same remark, e.g., B*07:02~B*07:02) or A~B molecule pair, high frequency can be rarely reached in a population if its functional divergence is low. However, in populations with reduced diversity, some molecules may reach very high frequencies, which inevitably increases the number of homozygotes (identical pairs, online tool S4, dots at the y-axis). For such individuals, a high functional diversity is nevertheless maintained by HLA A~B pairs. This effect is true for all populations we studied and is often more pronounced at HLA-A than at HLA-B.
At the intraprotein level, the differences observed between the anchoring B and F pockets of HLA-A and HLA-B molecules become even clearer when only common molecules are considered (fig. 5f and online tool S5 at https://hla-net.eu/interactive/HLA_wings/tool_S5/). The best linear model describing the negative correlation between log(SRI) values measured for p2 and p9 is finally achieved by only including the common HLA molecules (the lm6 model in table 1, for which adjusted R2: 0.7309, P < 0.001). This relationship is further supported by the amino acid diversity of the α1 and α2 domains observed in each of the 123 populations (fig. 6): in most cases, HLA-A molecules show higher amino acid diversity in α2 than in α1, whereas the reverse is observed for HLA-B molecules. In comparison, HLA-C molecules exhibit lower and more similar amino acid diversity at both domains.
Fig. 6.
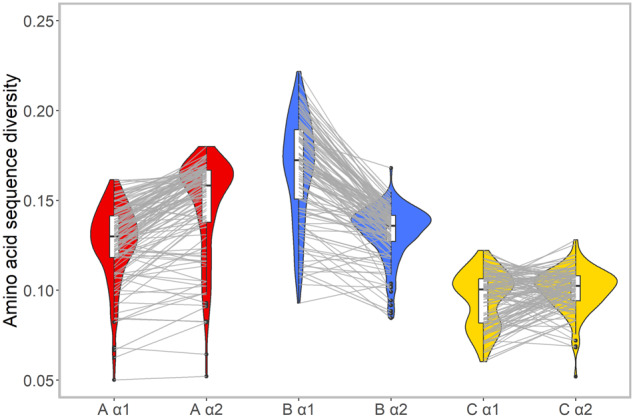
Comparison of amino acid diversity between the α1 and α2 domains of HLA-A (red), HLA-B (blue), and HLA-C (yellow) molecules in 123 populations. For each domain of each gene, the density distribution of the HLA acid amino density estimated for all populations is visualized by a Violin chart and the interdomain relationship of the diversity for each population is further indicated using gray lines linking the corresponding value points.
Ways of Complementary Peptide-Binding Function between HLA Molecules
Finally, by looking at the predicted bound peptides shared by HLA molecules, we synthesized, but not exhaustively, three ways in which HLA molecules, in particular, HLA-A and HLA-B ones, may exert a complementary peptide-binding function. First, HLA molecules with moderate fastidiousness at both B and F pockets (molecules represented in the central part of the plot shown in fig. 5e and f) display shared residue preferences at both p2 and p9 positions and are thus predicted to bind some identical peptides. This means that if such promiscuous HLA-A molecules were missing in a given population, they would be functionally replaced by HLA-B (or HLA-C) molecules sharing similar promiscuous characteristics and vice versa. Second, for HLA molecules with either an extremely fastidious B pocket or an extremely fastidious F pocket (molecules in the two extremities of the plot shown in fig. 5e), the other pocket is expected to be extremely promiscuous; then a given 9-mer peptide with a p9 residue anchoring into the fastidious F pocket of an HLA-A molecule might happen to have its p2 residue anchoring into the fastidious B pocket of an HLA-B molecule. Here again, if one or several HLA-A molecules specific to such a peptide were missing in a given population, specialist HLA-B molecules would be able to bind the same peptide and vice versa. Third, a long peptide may be cleaved differently during the degradation of a pathogenic antigen; hence, a p9 residue of a given 9-mer peptide, which would anchor into the fastidious F pocket of an HLA-A molecule, might also occur at position p2 of another 9-mer peptide of the same pathogenic antigen and anchor into the fastidious B pocket of an HLA-B molecule. One example of each of these three ways of sharing the peptide-binding function is given in fig. 7.
Fig. 7.

Three examples of HLA-A and HLA-B molecules exerting a complementary peptide-binding function, all the six molecules being predicted as strong binders (ELrank < 0.5) and with consensus sequence logo charts shown. First, the peptide FMNHISPKL predicted to bind HLA-A*02:05 and HLA-B*13:01 molecules, both with moderate fastidiousness at both B and F pockets (a). Second, the peptide AEMKSIFQK predicted to bind HLA-A*11:06 molecule with a fastidious F pocket and HLA-B*40:03 molecule with a fastidious B pocket (b). Third, the peptides ALFAVPFVR and VRFFLINRF from ALFAVPFVRFFLINRF predicted to bind HLA-A*31:01 and HLA-B*27:03 molecules, respectively (c).
Discussion
In this study, we analyzed together the amino acid sequences and functional properties of 2,909 HLA-A, HLA-B, and HLA-C molecules (supplementary table S1, Supplementary Material online) and we related them to the distribution of their corresponding HLA alleles in a large set of 123 human populations from all continents (supplementary table S2, Supplementary Material online). By applying several complementary statistical approaches and computer tools, our goal was to better understand the evolutionary mechanisms that shaped the patterns of HLA Class I gene diversity in populations across the world.
Using FCAs, we first explored the functional relationships of these HLA molecules along with 200,000 simulated 9-mer peptides they were predicted to bind (fig. 2a and b and online tool S2 at https://hla-net.eu/interactive/HLA_wings/tool_S2/). The flying bird-like pattern observed in the FCA charts indicates a huge divergence of HLA-A and HLA-B molecules regarding their peptide-binding properties, whereas HLA-C molecules are much less variable in this respect. As a result, three main “functional groups” of HLA-A (groups 1 and 3) and HLA-B (group 2) molecules were defined based on their likely unique and very divergent peptide-binding affinities (i.e., the bird’s wings and tail). In addition, the remaining HLA-A and HLA-B, together with the HLA-C molecules constitute a fourth, more heterozygous group (group 4) that shows intermediate functional distances among the other three groups (i.e., the bird’s body and tiny claws; fig. 2c and d). The pattern is confirmed by the proportion of peptides predicted to bind HLA-A and/or HLA-B molecules compared with those only expected to bind HLA-C molecules (fig. 3a and b). Interestingly, plenty of common HLA molecules (i.e., present in at least 5 populations worldwide) can be found in each of the four groups (fig. 2e and f). By labeling the HLA-A and HLA-B molecules proportionally to their corresponding allele frequencies in each population, we observed the existence of molecules belonging to the four groups in almost all populations of our data set (fig. 2g and h and online tool S2). Indeed, the proportion of predicted bound peptides is more or less maintained for the 194 common or the 18 most common HLA-A and HLA-B molecules (fig. 3c–f).
By investigating more in depth the sequence and functional diversity of the 2,909 HLA molecules, we found that functional distance distributions are asymmetric compared with sequence difference distributions (fig. 4). In general, HLA C~C pairs show both lowest sequence differences and functional distances, whereas A~B pairs show both highest ones. HLA A~A, B~B, and B~C pairs show comparable distributions of sequence differences, much less prominent than HLA A~B and A~C pairs (fig. 4a and b), which is barely changed using the common and most common HLA molecules (fig. 4e, f, i, and j). In contrast, the distributions of functional distances for all these pairs display a shift to higher values (fig. 4c and d), which becomes more pronounced using the common and most common HLA molecules (fig. 4g, h, k, and l). Heterogeneous correlation coefficients were thus obtained between sequence differences and functional distances, which are relatively high for one-gene molecule pairs (A~A, B~B, and C~C pairs) and low for two-gene molecules pairs (A~B, A~C, and B~C pairs). Last, extreme and identical patterns were observed for A~A and B~B pairs when they were composed of the most common HLA molecules (fig. 4k). Such patterns of functional distances suggest that natural selection prevents functionally similar A~B, A~A, and B~B pairs to prevail in populations, which leads to increase the frequencies of functionally distinct HLA-A and HLA-B molecules. Similar, if not so pronounced selective forces might have been acting on A~C and B~C pairs but not on C~C pairs. In consequence, the probability of observing several frequent HLA alleles coding for molecules with similar peptide-binding affinities, either from the same gene or across the two genes HLA-A and HLA-B, would be very low within populations (online tool S4 at https://hla-net.eu/interactive/HLA_wings/tool_S4/). In some populations, certain alleles of one HLA gene occasionally reach very high frequencies due to strong genetic drift or directional selection, resulting in high numbers of homozygous individuals; in these cases the general HLA functional divergence would be more or less maintained by molecule pairs encoded by different HLA genes (online tool S4).
These key results indicate that the divergent functional properties of HLA-A and HLA-B molecules taken together cover a large and stable peptide-binding repertoire; they play an instrumental and complementary role in giving the immune protection of human populations, in close agreement with the results presented by Buhler et al. (2016) through a different approach. On the other hand, these results contrast with the apparently more marginal role of HLA-C in peptide-binding and presentation, as, compared with HLA-A and HLA-B molecules, a very low proportion of peptides would uniquely bind HLA-C molecules (fig. 3a, b, e, and f). Actually, distinct kinds (e.g., for HLA-C) and/or intensities (e.g., for HLA-A and HLA-B) of selection may have acted on the different genes, which would have caused the differences we observed between one-gene and two-gene molecule pairs. It also makes great sense in the context of HLA molecular evolution, where HLA-C likely emerged from a duplication of HLA-B (Kulski et al. 1997). This hypothesis may explain its sequence and functional similarity with HLA-B (B~C molecule pairs with higher correlation coefficient compared with A~B and A~C pairs, online tool S3), here shown by FCA charts (fig. 2c and d), Venn charts (fig. 3a and b), and amino acid difference histograms (fig. 4b). Thanks to a possible relaxation of pathogen-driven selection compared with other, still unknown, selective constraints, the “novel” HLA-C gene would have allowed itself to assume other specific functions, in particular, as ligands of KIRs, with unique presence on trophoblast cells and essential role of KIR/HLA-C interactions in pregnancy complications (Chazara et al. 2011; Colucci 2017).
Based on these observations, we went a step further by exploring how HLA Class I genes could play a complementary role in peptide-binding despite the immense functional divergence between them (as shown by the high proportion of peptides that are predicted to bind molecules of different HLA genes, fig. 3a and b). Actually, we showed that the binding preference of a large number of HLA-A and HLA-B molecules is determined by only one of the two B and F pockets within the peptide-binding groove (fig. 5 and online tool S5 at https://hla-net.eu/interactive/HLA_wings/tool_S5/). In terms of physicochemical properties, B pocket of most HLA-A molecules recognizes small and aliphatic peptide residues, whereas F pocket of most HLA-B molecules recognizes aromatic, aliphatic, and hydrophobic ones. In contrast, F pocket of HLA-A molecules anchors mostly either aromatic, basic, or aliphatic residues, whereas B pocket of HLA-B molecules anchors either aromatic, basic, acidic, or aliphatic residues (Sidney et al. 2008). For the majority of HLA-A molecules, B pocket is thus expected to be more promiscuous compared with the more fastidious pocket F, and the reverse is predictable for HLA-B molecules. As the B and F pocket are roughly contained in the α1 and α2 domains, such differences satisfactorily explain our results of amino acid diversity in most populations: The HLA-A α2 domain displays a higher diversity than the HLA-A α1 domain, whereas HLA-B α1 and α2 domains show reverse pattern (fig. 6). Furthermore, SRI at primary anchor positions p2 and p9 (with SRI at secondary anchor positions p1 and p3 as interactions) are negatively correlated to each other and the linear model is significantly improved by including only the common HLA-A and HLA-B molecules (table 1). All these results disclose the secret of HLA Class I functional diversity: natural selection is not in favor of molecules with excessively large or excessively narrow repertoires (i.e., those with either fastidious or promiscuous pockets on both sides of the peptide-binding groove), as shown by the lack of points at the bottom-left and top right corners of figure 5e and f. Rather, an extremely fastidious pocket of an HLA molecule at one side, ensuring a strong binding specificity, usually requests a highly promiscuous pocket on the other side, maintaining a reasonable repertoire size (fig. 5e and f). Given the three ways of sharing bound peptides that we illustrated in fig. 7, HLA molecules encoded by different genes, in particular, HLA-A and HLA-B, would thus complement each other by being capable of anchoring overlapping peptides derived from the same pathogens, despite the distinct residue preferences and fastidiousness at their B and F pockets.
It is now worthy to reconsider the HLA broad functional groups, in particular, the HLA-A and HLA-B “supertypes,” previously defined by other authors with the objective of better understanding the functional complexity of HLA alleles (Kangueane et al. 2005; Sidney, Peters, et al. 2008; Wang and Claesson 2014; Mukherjee et al. 2015). Based on physicochemical properties of pockets B and F, these supertypes were claimed to persist in most human populations, which was not confirmed, at least for HLA-A, in a previous study (Dos Santos Francisco et al. 2015). Here, we provide a different and more complex view of HLA functional groups, which, unlike the supertype definition, does not treat them separately from each other without mutual functional overlap. We did define three HLA protein groups that displayed marked functional differences (mostly between groups 1 and group 2, fig. 2a and b), but also one additional group (group 4) that was intermediate from a functional point of view and composed of mixed HLA-A, HLA-B, and HLA-C molecules (fig. 2c and d). We suggest that the peptide-binding function of an HLA molecule that is occasionally absent in some populations showing a reduced genetic diversity would be complemented to a certain extent by another molecule of either the same or a different HLA gene. This idea is compatible with the model of joint asymmetric selection proposed by Buhler et al. (2016) for HLA Class I genes to explain why all populations likely exhibit comparable potential to present pathogen-derived peptides through their HLA-A and HLA-B complexes taken together, even though the peptide coverage of either of them is substantially depleted.
In our analyses, we have considered different categories of HLA molecules defined by the range of their geographic distribution (table 2). All patterns of functional divergence and complementarity revealed for HLA Class I genes are well represented by common and most common HLA-A and HLA-B molecules (figs. 2e, 2f, 3c–f, 4e–l, 5e, and 5f). As to rare molecules, they merely appear to add some “noise.” Indeed, by including all the 2,909 HLA molecules, a substantial number of HLA A~A pairs exhibit very close functional properties (i.e., low functional distances), whereas slightly more HLA B~B pairs display intermediate functional distances (fig. 4c). These patterns may be explained by different mechanisms generating the diversity of HLA-A and HLA-B molecules. Actually, more than 80% of the HLA Class I alleles are very rare (Robinson et al. 2017). They merely differ, at HLA-A, by one point mutation from older and more common alleles, whereas HLA-B has been shown to be much more affected by gene conversion and recombination events (Buhler and Sanchez-Mazas 2011; Robinson et al. 2017), which partly explains differences in diversity levels among these genes (Vangenot et al. 2020). For example, several HLA-B alleles are hybrids of other HLA-B alleles, for example, B*15:59 (Magira et al. 2000), B*35:31 (Elsner et al. 2002), B*53:01 (Allsopp et al. 1991), B*53:31 (Adamek et al. 2015), which is much less commonly the case for HLA-A alleles (Robinson et al. 2017). Interestingly, even single substitutions would more strongly affect the peptide-binding repertoire of HLA-B than they would do for HLA-A molecules (van Deutekom and Kesmir 2015). This may have created greater functional differences between new and preexisting HLA-B molecules compared with HLA-A, explaining the patterns observed in figure 3d. Given the disadvantage of functional similarity, new HLA-A alleles would thus have more chance to be swept by natural selection than new HLA-B alleles. An alternative interpretation to the bimodal distribution of the functional distances between HLA A~A pairs (fig. 4a), with both extremely similar and extremely divergent pairs, might be the possible existence of at least two functional groups that are both very homogeneous and distant from each other. These groups might have evolved under distinct selective pressures and/or at different times. This can be related to what has been described for HLA-DRB1, where a model of DAA was only sustained for one of two distinct allelic groups, that is, group B but not group A (Lau et al. 2015).
A main task in the future is, of course, to confirm how close our results based on in silico predicted data and random peptides are representing the success or failure of the peptide-binding function in an individual, which may further be complicated considering the surface expression level of HLA molecules, and the composition and type of pathogen proteins. Nevertheless, recent studies showed that the ability to predict peptide-binding affinity has been considerably improved by the immense amount of high-quality mass spectrometry data of eluted HLA ligands (Gfeller and Bassani-Sternberg 2018).
In conclusion, our results disclose both important structural and functional divergence between the molecules encoded by the three classical HLA Class I genes and—what is completely new in this study—robust evidence of functional complementarity at both inter- and intraprotein levels between HLA-A and HLA-B genes, which satisfactorily explains the joint divergent asymmetric selective model previously proposed by Buhler et al. (2016). Intriguingly, the complementarity between HLA-A and HLA-B molecules, which maintains an efficient overall peptide-binding repertoire in populations, is still reflected today by the mutually exclusive first-field numbers assigned to HLA-A and HLA-B allele families during the early years of the long history of HLA study.
More importantly, like two wings of a flying bird, the joint asymmetric relationship between HLA-A and HLA-B in terms of amino acid sequence diversity and peptide-binding specificity is a perfect example, in our genome, of duplicated genes sharing their capacity of assuming common vital functions while being submitted to complex and sometimes distinct evolutionary mechanisms. Such mechanisms include the ways of accumulating molecular diversity through either point mutations or recombination events, selective pressures, and random variation due to genetic drift and other processes related to the history of populations. A key issue that remains to be investigated is whether such a functional complementarity between different HLA genes is also reflected in the maintenance of long, multilocus HLA haplotypes in distinct populations. We hope that further international collaboration helping to gather substantial sets of high-quality HLA multilocus genotype data will soon allow to address this, and other, crucial questions in a future extension of our work.
Materials and Methods
HLA Functional Data Analyses: Prediction of HLA Peptide-Binding Affinity
We applied the netMHCpan 4.0 (Jurtz et al. 2017) program for the estimation of HLA Class I peptide-binding affinity. In order to quantify the functional relationship between HLA molecules, we used MHCcluster 2.8, a tool to predict peptide-binding specificity and functionally cluster Class I molecules (Thomsen et al. 2013). All the 2,909 HLA Class I molecules currently available in both programs were included. The binding affinity of each molecule was estimated to a set of 200,000 random natural 9-mer peptides.
With netMHCpan, a binding affinity score (BAscore) varying between zero and one was estimated, and the percentile rank of the predicted binding score (%Rank EL or ELrank) was also reported to determine whether the protein was expected to bind a peptide, with 2 and 0.5 as upper thresholds for weak and strong binders, respectively. In the majority of cases, ELrank achieves improved predictive performance compared with raw prediction scores (Jurtz et al. 2017).
With MHCcluster, the peptide-binding similarity s between two molecules was estimated by the correlation between the top 10% peptides with strongest affinity to each of them. Then the pairwise functional distance (DF) was computed as follows:
which was standardized to fall into the range between zero and one (Hoof et al. 2009). Based on these estimations, a logo chart of binding motifs (as in fig. 1b) was provided for each HLA molecule and a matrix of functional distances between each pair of HLA molecules was created using the Seq2Logo service (Thomsen and Nielsen 2012). Finally, MHCcluster created an unrooted phylogenetic tree visualizing the functional relationships between the HLA molecules.
However, such a phylogenetic tree is not able to show the relationships between random peptides and HLA molecules, nor the geographic distributions of the latter. We thus performed FCA using the whole set of BAscore data, without setting any a priori threshold to evaluate the HLA peptide-binding affinities. As an extension of principal component analyses to categorical data, FCAs provide a solution for summarizing and visualizing bivariate relationships between pairs of variables in multidimensional plots. Using an algorithm implemented in the R package FactoMineR (Le et al. 2008), we managed to plot simultaneously the HLA molecules and peptides. A chi-square statistic was computed to test the robustness of the FCA results.
The distribution of pairwise functional distances between HLA molecules was further visualized by using the kernel density function implemented in R package ggplot2 (Wickham 2016). To compare these results, we applied the Kolmogorov–Smirnov test that determines if two samples of data follow the same distribution (Marsaglia et al. 2003). This nonparametric test is entirely agnostic to what this distribution actually is. A two-tail test was performed, with the null hypothesis of no difference between the empirical distribution of the two the samples, by the ks.test function implemented in R.
HLA Sequence Data Analyses: Estimation of Sequence Divergence among HLA Molecules
To better understand the link between their peptide-binding functions and amino acid sequences, the consensual sequence data for all the 2,909 HLA molecules were retrieved from IPD-IMGT/HLA Database (Release 3.37.0 at https://www.ebi.ac.uk/ipd/imgt/hla/). We focused on the α1 and α2 domains encoded by exons 2 and 3 of each HLA Class I gene, respectively, to take into account their essential role in the peptide-binding function. For each pair of HLA molecules, sequence divergence was estimated by counting the number of different amino acid residues in the α1 and α2 domains.
To estimate the degree of diversity at each residue of the 9-mer peptides showing high-binding affinity (top 1% highest BAscore) to a given HLA molecule, we computed an SRI as follows:
where fi is the fraction of amino acid residue i at that specific position (Simpson 1949). The SRI varies between 1 and 20 (i.e., the number of distinct residues) and defines a weighted number for the amount of different residues observed at a specific position. A higher SRI value at a given residue position means a lower diversity, also reflecting a higher fastidiousness (or lower promiscuity) of the corresponding pocket within the HLA peptide-binding groove. The SRI values computed for different HLA molecules were summarized by using the Violin and Box plot functions implemented in R package ggplot2 (Wickham 2016).
Moreover, relationship between SRI values estimated for positions p1–p9 was assessed by means of linear modeling, and backward stepwise regression was used to test if other independent variables such as gene and geographic distribution could be retained in the final model (Venables and Ripley 2013).
HLA Population Data Analyses: Allele Ranges and Estimation of Allele Pair Frequencies
A large set of HLA Class I frequency data were collected from both the literature (published between 1992 and 2020) and the reports of the 11th–16th International HLA and Immunogenetics Workshops. We defined the following criteria to control for the quality of the data:
Samples typed at high resolution, that is, second-field, third-field, or fourth-field levels for HLA-A, HLA-B, and HLA-C genes (alleles defined at the third- and fourth-field levels were recoded and combined to alleles defined at the second-field level since these resolution levels correspond to identical sequences at the peptide-binding groove level of the protein);
Populations not known to have undergone recent admixture or gene flow;
Frequency of “blank” (i.e., the sum of undefined alleles) not exceeding 5% for any of the three genes;
No deviation from Hardy–Weinberg equilibrium reported.
Our final data set consists of 123 population samples typed for HLA-A, HLA-B, and HLA-C genes. All population information is available in supplementary table S2, Supplementary Material online.
A total of 744 nonnull HLA alleles were observed in our population data set, the binding affinities of which were all estimated by using netMHCpan, whereas 18 null alleles were excluded because they were not suitable for functional analyses, and 28 alleles were not available in MHCcluster (supplementary table S1a, b, c, Supplementary Material online). However, as the latter mostly appeared with very low frequencies in the populations, their exclusion was not expected to change in any substantial way the results of our analyses.
Based on the distribution of HLA alleles at the global or at regional geographic levels, previous studies defined allele categories reported in the Common and Well-Documented alleles (CWD) 2.0.0 Catalog (Mack et al. 2013), the Common, Intermediate and Well‐Documented alleles 3.0.0 Catalog (Mack et al. 2013; Hurley et al. 2020) and the European CWD Catalog (Sanchez-Mazas et al. 2017a). Only allele frequency data were available for the populations used in this study, which was not suitable for direct allele counting as to classify the alleles in these categories. We thus defined common alleles when they were present in at least 5 populations of our data set, and we applied this criterion to the HLA molecules they encode. This category of common alleles/molecules was further extended by increasing the minimal number of populations in which an allele was observed, that is, 10, 25, 50, and 75, respectively, the last being referred as the most common alleles/molecules. Accordingly, the number of alleles decreases as the range of populations enlarges (table 2 and supplementary table S1, Supplementary Material online). We did not use higher limits such as 100 populations, as they left too few alleles to perform analyses.
Combination of HLA Sequence, Functional and Population Data
For each HLA allele/molecule category we defined (table 2), a summarized functional distance distribution chart was created, and a Box plot chart was also created for the SRI values. In the FCA and other charts plotting HLA molecules, this information is represented by different symbol sizes.
From a functional point of view, given two HLA-A and/or HLA-B nonnull codominant alleles, the probability that both corresponding molecules (an HLA A~A, B~B, or A~B pair) are present on a cell surface of an individual is expected to be the product of the allele frequencies estimated in the population the individual belongs to. This was used to plot pairwise functional distances (DF) between HLA molecules against their expected frequencies in each population.
Moreover, to estimate the degree of polymorphism of each amino acid residue within the α1 and α2 domains of the HLA molecules in a population, we created a set of sequences for all HLA-A, HLA-B, and HLA-C molecules carried by the individuals of each population sample, using allele frequency data. Similar to the nucleotide diversity index pi, an amino acid sequence diversity was computed from these sequences by using the R package PopGenome (Pfeifer et al. 2014) based on a method suggested by Nei (1987).
Other analyses and data visualization were performed in R (R_Core_Team 2018) version 4.0.0 using RStudio (RStudio_Team 2015), with packages including ggplot2 (Wickham 2016), eulerr (Larsson 2020), rworldmap (South 2011), and reshape2 (Wickham 2007). In order to better organize and display the large amount of our supplementary results, we developed several user-interactive online tools using the R Shiny package (Chang et al. 2018). The data underlying this article will be shared on request to the corresponding authors.
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Supplementary Material
Acknowledgments
We thank Dr William Klitz for his useful suggestions. We appreciate the three anonymous reviewers for their very helpful and constructive comments. We are also grateful to Shayire Shoket for her help during data collection.
This work was supported by the Swiss National Science Foundation (Grants Nos. 31003A_144180 and 310030_188820 to A.S.-M.).
References
- Adamek M, Klages C, Bauer M, Kudlek E, Drechsler A, Leuser B, Scherer S, Opelz G, Tran TH.. 2015. Seven novel HLA alleles reflect different mechanisms involved in the evolution of HLA diversity: description of the new alleles and review of the literature. Hum Immunol. 76(1):30–35. [DOI] [PubMed] [Google Scholar]
- Allsopp CE, Hill AV, Kwiatkowski D, Hughes A, Bunce M, Taylor CJ, Pazmany L, Brewster D, McMichael AJ, Greenwood BM.. 1991. Sequence analysis of HLA-Bw53, a common West African allele, suggests an origin by gene conversion of HLA-B35. Hum Immunol. 30(2):105–109. [DOI] [PubMed] [Google Scholar]
- Bodmer WF.1972. Evolutionary significance of the HL-A system. Nature 237(5351):139–145 passim. [DOI] [PubMed] [Google Scholar]
- Buhler S, Nunes JM, Sanchez-Mazas A.. 2016. HLA class I molecular variation and peptide-binding properties suggest a model of joint divergent asymmetric selection. Immunogenetics 68(6–7):401–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhler S, Sanchez-Mazas A.. 2011. HLA DNA sequence variation among human populations: molecular signatures of demographic and selective events. PLoS One 6(2):e14643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey BS, Poulton KV, Poles A.. 2019. Factors affecting HLA expression: a review. Int J Immunogenet. 46(5):307–320. [DOI] [PubMed] [Google Scholar]
- Chang W, Cheng J, Allaire J, Xie Y, McPherson J. 2020. Shiny: Web Application Framework for R. R package version 1.0.3. Available from: https://CRAN.R-project.org/package=shiny. [Google Scholar]
- Chap pell P, Meziane el K, Harrison M, Magiera L, Hermann C, Mears L, Wrobel AG, Durant C, Nielsen LL, Buus S, et al. 2015. Expression levels of MHC class I molecules are inversely correlated with promiscuity of peptide binding. Elife. 4:e05345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chazara O, Xiong S, Moffett A.. 2011. Maternal KIR and fetal HLA-C: a fine balance. J Leukoc Biol. 90(4):703–716. [DOI] [PubMed] [Google Scholar]
- Colucci F.2017. The role of KIR and HLA interactions in pregnancy complications. Immunogenetics 69(8–9):557–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtoni ES, Mattiuz PL, Tosi RM.. 1967. The workshop data and nomenclature: HLA. In: Curtoni ES, Mattiuz PL, Tosi RM, editors. Histocompatibility testing. Copenhagen (Denmark: ): Munksgaard. p. 435–449. [Google Scholar]
- Dendrou CA, Petersen J, Rossjohn J, Fugger L.. 2018. HLA variation and disease. Nat Rev Immunol. 18(5):325–339. [DOI] [PubMed] [Google Scholar]
- Doherty PC, Zinkernagel RM.. 1975. A biological role for the major histocompatibility antigens. Lancet 305(7922):1406–1409. [DOI] [PubMed] [Google Scholar]
- Dos Santos Francisco R, Buhler S, Nunes JM, Bitarello BD, Franca GS, Meyer D, Sanchez-Mazas A.. 2015. HLA supertype variation across populations: new insights into the role of natural selection in the evolution of HLA-A and HLA-B polymorphisms. Immunogenetics 67(11–12):651–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsner HA, Himmel A, Steitz M, Hammer P, Schmitz G, Ballas M, Blasczyk R.. 2002. HLA-B3531, a hybrid of B35 and B61, implications for diagnostic approaches to alleles with complex ancestral compositions. Tissue Antigens 60(1):95–97. [DOI] [PubMed] [Google Scholar]
- Falk K, Rötzschke O, Stevanovié S, Jung G, Rammensee H-G.. 1991. Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. Nature 351(6324):290–296. [DOI] [PubMed] [Google Scholar]
- Gfeller D, Bassani-Sternberg M.. 2018. Predicting antigen presentation—what could we learn from a million peptides? Front Immunol. 9:1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GoeuryT, CrearyLE, BrunetL, GalanM, PasquierM, KervaireB, LanganeyA, TiercyJ-M, Fernández-ViñaMA, Nunes JM, . et al. 2018. Deciphering the fine nucleotide diversity of full HLA class I and class II genes in a well-documented population from sub-Saharan Africa. HLA. 91(1):36–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrick PW, Whittam TS, Parham P.. 1991. Heterozygosity at individual amino acid sites: extremely high levels for HLA-A and -B genes. Proc Natl Acad Sci U S A. 88(13):5897–5901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermisson J, Pennings PS.. 2005. Soft sweeps: molecular population genetics of adaptation from standing genetic variation. Genetics 169(4):2335–2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill AVS.1991. HLA associations with malaria in Africa: some implications for MHC evolution. In: Klein J, Klein D, editors. Molecular evolution of the major histocompatibility complex. Berlin (Germany: ): Springer. [Google Scholar]
- Hoof I, Peters B, Sidney J, Pedersen LE, Sette A, Lund O, Buus S, Nielsen M.. 2009. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics 61(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley CK, Kempenich J, Wadsworth K, Sauter J, Hofmann JA, Schefzyk D, Schmidt AH, Galarza P, Cardozo MBR, Dudkiewicz M, et al. 2020. Common, intermediate and well-documented HLA alleles in world populations: CIWD version 3.0.0. HLA 95(6):516–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob L, Vert JP.. 2008. Efficient peptide-MHC-I binding prediction for alleles with few known binders. Bioinformatics 24(3):358–366. [DOI] [PubMed] [Google Scholar]
- Jojic N, Reyes-Gomez M, Heckerman D, Kadie C, Schueler-Furman O.. 2006. Learning MHC I-peptide binding. Bioinformatics 22(14):e227–e235. [DOI] [PubMed] [Google Scholar]
- Jurtz V, Paul S, Andreatta M, Marcatili P, Peters B, Nielsen M.. 2017. NetMHCpan-4.0: improved peptide-MHC Class I interaction predictions integrating eluted ligand and peptide binding affinity data. J Immunol. 199(9):3360–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kangueane P, Sakharkar MK, Rajaseger G, Bolisetty S, Sivasekari B, Zhao B, Ravichandran M, Shapshak P, Subbiah S.. 2005. A framework to sub-type HLA supertypes. Front Biosci. 10(1–3):879–886. [DOI] [PubMed] [Google Scholar]
- Kaufman J.2018. Generalists and specialists: a new view of how MHC class I molecules fight infectious pathogens. Trends Immunol. 39(5):367–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kissmeyer-Nielsen F, Svejgaard A, Hauge M.. 1968. Genetics of the human HL-A transplantation system. Nature 219(5159):1116–1119. [DOI] [PubMed] [Google Scholar]
- Klein J, Sato A.. 2000. The HLA system. First of two parts. N Engl J Med. 343(10):702–709. [DOI] [PubMed] [Google Scholar]
- Kulkarni S, Savan R, Qi Y, Gao X, Yuki Y, Bass SE, Martin MP, Hunt P, Deeks SG, Telenti A, et al. 2011. Differential microRNA regulation of HLA-C expression and its association with HIV control. Nature 472(7344):495–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulski JK, Gaudieri S, Bellgard M, Balmer L, Giles K, Inoko H, Dawkins RL.. 1997. The evolution of MHC diversity by segmental duplication and transposition of retroelements. J Mol Evol. 45(6):599–609. [DOI] [PubMed] [Google Scholar]
- Larsson J.2020. eulerr: Area-Proportional Euler and Venn Diagrams with Ellipses (R package). Version 6.1.0. Available from: https://cran.r-project.org/package=eulerr.
- Lau Q, Yasukochi Y, Satta Y.. 2015. A limit to the divergent allele advantage model supported by variable pathogen recognition across HLA-DRB1 allele lineages. Tissue Antigens 86(5):343–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le S, Josse J, Husson F.. 2008. FactoMineR: an R package for multivariate analysis. J Stat Soft. 25(1):18. [Google Scholar]
- Mack SJ, Cano P, Hollenbach JA, He J, Hurley CK, Middleton D, Moraes ME, Pereira SE, Kempenich JH, Reed EF, et al. 2013. Common and well-documented HLA alleles: 2012 update to the CWD catalogue. Tissue Antigens 81(4):194–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madden DR.1995. The three-dimensional structure of peptide-MHC complexes. Annu Rev Immunol. 13(1):587–622. [DOI] [PubMed] [Google Scholar]
- Magira E, Beznik-Cizman B, Monos D.. 2000. HLA-B1559: a hybrid allele including exon 2 of HLA-B35 and exon 3 of HLA-B15 and serologically typed as B35. Tissue Antigens 56(5):460–462. [DOI] [PubMed] [Google Scholar]
- Marsaglia G, Tsang WW, Wang J.. 2003. Evaluating Kolmogorov’s distribution. J Stat Soft. 8(18):1–4. [Google Scholar]
- Marsh SG, Albert ED, Bodmer WF, Bontrop RE, Dupont B, Erlich HA, Fernandez-Vina M, Geraghty DE, Holdsworth R, Hurley CK, et al. 2010. Nomenclature for factors of the HLA system, 2010. Tissue Antigens 75(4):291–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messer PW, Petrov DA.. 2013. Population genomics of rapid adaptation by soft selective sweeps. Trends Ecol Evol. 28(11):659–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S, Warwicker J, Chandra N.. 2015. Deciphering complex patterns of class-I HLA-peptide cross-reactivity via hierarchical grouping. Immunol Cell Biol. 93(6):522–532. [DOI] [PubMed] [Google Scholar]
- Neefjes JJ, Ploegh HL.. 1988. Allele and locus-specific differences in cell surface expression and the association of HLA class I heavy chain with beta 2-microglobulin: differential effects of inhibition of glycosylation on class I subunit association. Eur J Immunol. 18(5):801–810. [DOI] [PubMed] [Google Scholar]
- Nei M.1987. Molecular evolutionary genetics. New York: Columbia University Press. [Google Scholar]
- Neisig A, Melief CJ, Neefjes J.. 1998. Reduced cell surface expression of HLA-C molecules correlates with restricted peptide binding and stable TAP interaction. J Immunol. 160(1):171–179. [PubMed] [Google Scholar]
- Penman BS, Gupta S.. 2018. Detecting signatures of past pathogen selection on human HLA loci: are there needles in the haystack? Parasitology 145(6):731–739. [DOI] [PubMed] [Google Scholar]
- Pfeifer B, Wittelsburger U, Ramos-Onsins SE, Lercher MJ.. 2014. PopGenome: an efficient Swiss army knife for population genomic analyses in R. Mol Biol Evol. 31(7):1929–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierini F, Lenz TL.. 2018. Divergent allele advantage at human MHC genes: signatures of past and ongoing selection. Mol Biol Evol. 35(9):2145–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prugnolle F, Manica A, Charpentier M, Guegan JF, Guernier V, Balloux F.. 2005. Pathogen-driven selection and worldwide HLA class I diversity. Curr Biol. 15(11):1022–1027. [DOI] [PubMed] [Google Scholar]
- Qutob N, Balloux F, Raj T, Liu H, Marion de Proce S, Trowsdale J, Manica A.. 2012. Signatures of historical demography and pathogen richness on MHC class I genes. Immunogenetics 64(3):165–175. [DOI] [PubMed] [Google Scholar]
- R_Core_Team 2018. R: a language and environment for statistical computing. Vienna (Austria: ): R Foundation for Statistical Computing. Available from: https://www.R-project.org/. [Google Scholar]
- Rasmussen M, Harndahl M, Stryhn A, Boucherma R, Nielsen LL, Lemonnier FA, Nielsen M, Buus S.. 2014. Uncovering the peptide-binding specificities of HLA-C: a general strategy to determine the specificity of any MHC class I molecule. J Immunol. 193(10):4790–4802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson J, Barker DJ, Georgiou X, Cooper MA, Flicek P, Marsh SGE.. 2020. IPD-IMGT/HLA Database. Nucleic Acids Res. 48(D1):D948–D955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson J, Guethlein LA, Cereb N, Yang SY, Norman PJ, Marsh SGE, Parham P.. 2017. Distinguishing functional polymorphism from random variation in the sequences of >10,000 HLA-A, -B and -C alleles. PLoS Genet. 13(6):e1006862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RStudio_Team 2015. RStudio: integrated development for R. Boston: RStudio, Inc. [Google Scholar]
- Rudolph MG, Stanfield RL, Wilson IA.. 2006. How TCRs bind MHCs, peptides, and coreceptors. Annu Rev Immunol. 24(1):419–466. [DOI] [PubMed] [Google Scholar]
- Sanchez-MazasA, NunesJM, MiddletonD, SauterJ, BuhlerS, McCabeA, HofmannJ, BaierDM, SchmidtAH, Nicoloso G, . et al. 2017a. Common and well-documented HLA alleles over all of Europe and within European sub-regions: A catalogue from the European Federation for Immunogenetics. HLA. 89(2):104–113. [DOI] [PubMed] [Google Scholar]
- Sanchez-MazasA, ČernýV, DiD, BuhlerS, PodgornáE, ChevallierE, BrunetL, WeberS, KervaireB, Testi M, . et al. 2017. b. The HLA-B landscape of Africa: Signatures of pathogen-driven selection and molecular identification of candidate alleles to malaria protection. Mol Ecol. 26(22):6238–6252. [DOI] [PubMed] [Google Scholar]
- Sanchez-Mazas A.2020. A review of HLA allele and SNP associations with highly prevalent infectious diseases in human populations. Swiss Med Wkly. 150:w20214. [DOI] [PubMed] [Google Scholar]
- Saper MA, Bjorkman PJ, Wiley DC.. 1991. Refined structure of the human histocompatibility antigen HLA-A2 at 2.6 A resolution. J Mol Biol. 219(2):277–319. [DOI] [PubMed] [Google Scholar]
- Shiina T, Hosomichi K, Inoko H, Kulski JK.. 2009. The HLA genomic loci map: expression, interaction, diversity and disease. J Hum Genet. 54(1):15–39. [DOI] [PubMed] [Google Scholar]
- Sidney J, Assarsson E, Moore C, Ngo S, Pinilla C, Sette A, Peters B.. 2008. Quantitative peptide binding motifs for 19 human and mouse MHC class I molecules derived using positional scanning combinatorial peptide libraries. Immunome Res. 4(1):2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidney J, Peters B, Frahm N, Brander C, Sette A.. 2008. HLA class I supertypes: a revised and updated classification. BMC Immunol. 9(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson E.1949. Measurement of diversity. Nature 163(4148):688. [Google Scholar]
- Slade RW, McCallum HI.. 1992. Overdominant vs. frequency-dependent selection at MHC loci. Genetics 132(3):861–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solheim BG, Thorsby E.. 1973. Evidence of a third HL-A locus. Transplant Proc. 5(4):1579–1580. [PubMed] [Google Scholar]
- South A.2011. rworldmap : a new R package for mapping global data. R J. 3(1):35. [Google Scholar]
- Souza AS, Sonon P, Paz MA, Tokplonou L, Lima THA, Porto IOP, Andrade HS, Silva N, Veiga-Castelli LC, Oliveira MLG, et al. 2020. Hla-C genetic diversity and evolutionary insights in two samples from Brazil and Benin. HLA 96(4):468–486. [DOI] [PubMed] [Google Scholar]
- Spurgin LG, Richardson DS.. 2010. How pathogens drive genetic diversity: MHC, mechanisms and misunderstandings. Proc Biol Sci. 277(1684):979–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen M, Lundegaard C, Buus S, Lund O, Nielsen M.. 2013. MHCcluster, a method for functional clustering of MHC molecules. Immunogenetics 65(9):655–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen MC, Nielsen M.. 2012. Seq2Logo: a method for construction and visualization of amino acid binding motifs and sequence profiles including sequence weighting, pseudo counts and two-sided representation of amino acid enrichment and depletion. Nucleic Acids Res. 40(Web Server issue):W281–W287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsby E.2009. A short history of HLA. Tissue Antigens. 74(2):101–116. [DOI] [PubMed] [Google Scholar]
- Thorsby E, Sandberg L, Lindholm A, Kissmeyer-Nielsen F.. 1970. The HL-A system: evidence of a third sub-locus. Scand J Haematol. 7(3):195–200. [DOI] [PubMed] [Google Scholar]
- van Deutekom HWM, Keşmir C.. 2015. Zooming into the binding groove of HLA molecules: which positions and which substitutions change peptide binding most? Immunogenetics 67(8):425–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vangenot C, Nunes JM, Doxiadis GM, Poloni ES, Bontrop RE, de Groot NG, Sanchez-Mazas A.. 2020. Similar patterns of genetic diversity and linkage disequilibrium in Western chimpanzees (Pan troglodytes verus) and humans indicate highly conserved mechanisms of MHC molecular evolution. BMC Evol Biol. 20(1):119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venables WN, Ripley BD.. 2013. Modern applied statistics with S-PLUS. New York: Springer Science & Business Media. [Google Scholar]
- Vita R, Overton JA, Greenbaum JA, Ponomarenko J, Clark JD, Cantrell JR, Wheeler DK, Gabbard JL, Hix D, Sette A, et al. 2015. The immune epitope database (IEDB) 3.0. Nucleic Acids Res. 43(D1):D405–D412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Claesson MH.. 2014. Classification of human leukocyte antigen (HLA) supertypes. Methods Mol Biol. 1184:309–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H.2007. Reshaping Data with the reshape Package. J Stat Soft. 21(12):1–20. [Google Scholar]
- Wickham H.2016. ggplot2: elegant Graphics for Data Analysis. New York: Springer-Verlag. [Google Scholar]
- Zhang GL, Khan AM, Srinivasan KN, August JT, Brusic V.. 2005. MULTIPRED: a computational system for prediction of promiscuous HLA binding peptides. Nucleic Acids Res. 33(Web Server):W172–W179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YH, Xing Z, Liu C, Wang S, Huang T, Cai YD, Kong X.. 2017. Identification of the core regulators of the HLA I-peptide binding process. Sci Rep. 7(1):42768. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.