

Abstract
Primary cilia are critical sensory and signaling compartments present on most mammalian cell types. These specialized structures require a unique signaling protein composition relative to the rest of the cell to carry out their functions. Defects in ciliary structure and signaling result in a broad group of disorders collectively known as ciliopathies. One ciliopathy, Bardet–Biedl syndrome (BBS; OMIM 209900), presents with diverse clinical features, many of which are attributed to defects in ciliary signaling during both embryonic development and postnatal life. For example, patients exhibit obesity, polydactyly, hypogonadism, developmental delay and skeletal abnormalities along with sensory and cognitive deficits, but for many of these phenotypes it is uncertain, which are developmental in origin. A subset of BBS proteins assembles into the core BBSome complex, which is responsible for mediating transport of membrane proteins into and out of the cilium, establishing it as a sensory and signaling hub. Here, we describe two new mouse models for BBS resulting from a targeted LacZ gene trap allele (Bbs5−/−) that is a predicted congenital null mutation and conditional (Bbs5flox/flox) allele of Bbs5. Bbs5−/− mice develop a complex phenotype consisting of increased pre-weaning lethality craniofacial and skeletal defects, ventriculomegaly, infertility and pituitary anomalies. Utilizing the conditional allele, we show that the male fertility defects, ventriculomegaly and pituitary abnormalities are only present when Bbs5 is disrupted prior to postnatal day 7, indicating a developmental origin. In contrast, mutation of Bbs5 results in obesity, independent of the age of Bbs5 loss.
Introduction
Primary cilia are microtubule-based structures that emanate from the surface of nearly every mammalian cell type. The ciliary membrane is enriched in a unique set of membrane proteins and signaling components that set it apart from the cell membrane (1). This enrichment cultivates a highly specialized and responsive sensory and signaling hub for the cell. The accumulation of the proper signal transduction components at the ciliary membrane is crucial for cilia function and ultimately depends on the cooperation of several macromolecular machines, one of which is the BBSome. The BBSome is an octameric complex containing BBS1, BBS2, BBS4, BBS5, BBS7, BBS8, BBS9 and BBS18/BBIP10 (2,3). Interactions between intraflagellar transport protein (IFT22) (also known as RABL5) and BBS3 (also known as Arl6) are then responsible for the recruitment of the BBSome to the base of the cilium via interactions with the BBS1 subunit (4–6). The recruitment process is also aided by Rab8, the Rab8-specific guanine nucleotide exchange factor (GEF) Rabin8, and Rab11 (2,3,7). BBS5 is structurally and functionally unique among the BBSome components based on predictions that it may directly mediate membrane interactions through its two plextrin homology (PH) domains capable of binding to phosphoinositides (2). However, based on recent analysis of BBS5’s structure and physical interactions with other BBSome components, it is unlikely that it is actually able to interact with the membrane via these PH domains (8). Thus, the functional role and importance of BBS5 in the BBSome remains poorly understood.
Bardet–Biedl syndrome (BBS) patients exhibit a wide range of highly variable pathologies including, but not limited to, obesity, hypogonadism, polydactyly, cognitive deficits, renal anomalies and retinitis pigmentosa. To date, mutations in 24 different loci (BBS 1–24) have been associated with BBS. Mutations specifically affecting the core BBSome complex represent a large proportion of BBS patients (9), with 2% of the mutations occurring in Bbs5 (10). Previously, congenital mutant mouse models of BBSome components BBS1, BBS2, BBS4, BBS7 and BBS8 have been described and recapitulate several, but not all, of the phenotypes associated with the clinical features of the disorder. For example, renal cysts are a prevalent symptom among BBS patients; however, congenital models of BBS have only presented with renal inflammation and mild glomerular cysts in Bbs4 mutant mice without alterations in renal function based on analysis of blood urea nitrogen (BUN), creatine, Na+ and K+ levels (11,12). In addition, a conditional allele for Bbs1 has been described with renal phenotypes that recapitulate some of the clinical features (13–17). However, work done thus far in Bbs5 models has been limited and only demonstrated minor retinal degeneration (18,19). We sought to assess the pathophysiology of Bbs5 loss of function alleles using congenital and conditional Bbs5 mutant approaches. Our goal was to distinguish between phenotypes that are developmental in origin from those that occur as a consequence of loss of BBS5 functions needed for tissue homeostasis in adults. To accomplish this goal, we analyzed phenotypic consequences of Bbs5 disruption during development and in juvenile and adult stages. We report phenotypes including sub-Mendelian survival ratios, shortened skeletons, craniofacial defects, sterility, obesity, ventriculomegaly, persistence of the buccohypophyseal canal and pituitary gland abnormalities. Out of these, obesity was unique in that it is the only phenotype seen in both the congenital allele and when Bbs5 loss is induced after postnatal day 7 (P7), suggesting roles for Bbs5 in both development and adult stages that can impact energy homeostasis. The phenotypes observed in Bbs5 mutant mice described here are directly related to the pathologies presented by BBS patients, and provide the first whole animal validation of the Bbs5 mutant mouse model as a valuable tool to further understand the molecular mechanisms resulting in the pathologies common to BBS.
Results
Bbs5−/− mice exhibit tissue-specific irregularities in splicing
We first sought to verify that Bbs5 is widely expressed using the LacZ cassette engineered into the Bbs5−/− allele (Fig. 1A). However, we were unable to detect β-galactosidase activity in any tissue. We then investigated the expression of the targeted allele by reverse transcriptase polymerase chain reaction (RT-PCR) in brain, testes and kidney. Using primers located in an exon 5′ of the LacZ cassette and a second reverse primer within the LacZ gene, we were unable to detect a product. We then checked for expression of the Bbs5 transcript using primers located in exons 5′ (exon 2) and 3′ (exon 6) of the LacZ cassette. Although a single band appeared in all tissues except for the testes, we identified several different transcripts by sequencing (Fig. 1C). Interestingly, sequencing indicates that the messenger RNA produced from the targeted LacZ gene trap allele present in Bbs5−/− kidney is spliced normally; however, in the brain and testes, the Bbs5 transcript is interrupted by portions of the engineered targeting construct. In the Bbs5−/− brain, the allele splices from exon 3 into the engineered region of the Engrailed2 alternate exon and subsequently back into a sequence of unknown homology, followed by exon 6. Analysis of isoforms found in the wild-type testes shows two transcripts. The differences between the larger and smaller transcript is inclusion of exon 5 in the larger transcript. In the mutant testes, the two transcripts are larger due to incorporation of a partial region of the LacZ cassette in both transcripts. Importantly, there was no wild-type transcript detected in the brain and testes in Bbs5−/− samples. For both the brain and testes isoforms, the sequence results in premature stop codons in the mutant transcripts (Fig. 1D). These data with unexpected splicing indicate that the Bbs5−/− mouse is a complex allele that may disrupt Bbs5 function only in specific tissues related to how splicing occurs.
Figure 1 .
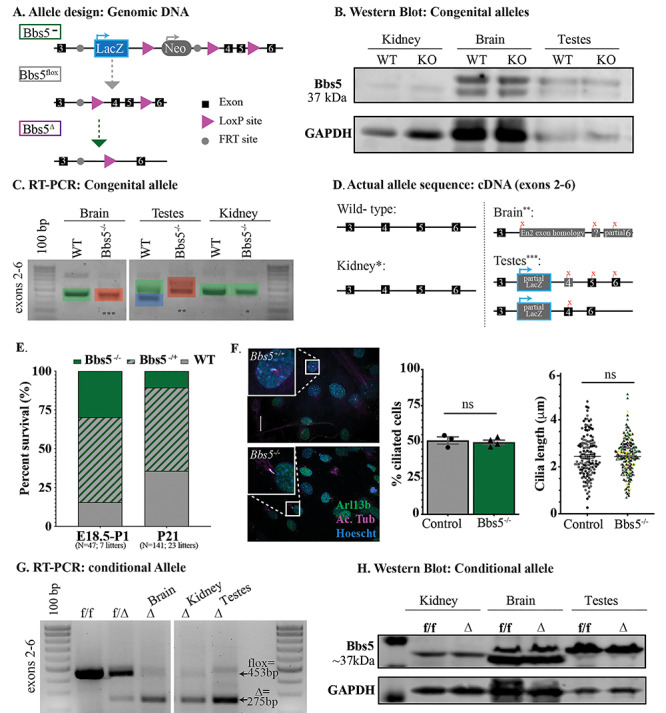
Mouse alleles. (A) The knockout allele construct depicting the genomic DNA in the congenital knock out allele (Bbs5−/−), conditional floxed allele (Bbs5flox/flox) and recombined alleles (Bbs5Δ/Δ). Exons are depicted as black boxes, LoxP sites as purple arrows and FRT sites as gray circles. Gray arrows indicate that an FlpO mouse was used to generate the subsequent allele. Green arrows indicate that a Cre-expressing mouse was used to generate the subsequent allele. (B) Western blot analysis from control (WT) and Bbs5−/− (KO) kidney, brain and testes. (C) RT-PCR analysis of brain, testes and kidney. A forward primer in exon 2 and a reverse primer in exon 6 flank the target region in the KO1st allele. Green shading indicates the wild-type allele, red indicates an aberrant spice isoform in the Bbs5−/− tissues and blue indicates the previously predicted testes-specific isoform. (D) Sequencing of cDNA indicated in (C) reveals unexpected splicing in Bbs5−/− brain and testes and wild-type sequence in cDNA generated from Bbs5−/− kidney. (E) Representation of the percentage of WT, Bbs5−/+ and Bbs5−/− animals at E18.5 [left, χ2(2, N = 47) = 3.09, P > 0.05] and P21 (right, N = 141 progeny from 23 litters) compared with expected frequencies (center). However, Mendelian ratios reflect a significant reduction in the observed number of mutant animals at weening age [χ2(2, N = 141) = 19.93, P < 0.001]. (F) Immunofluorescence on MEFs stained for cilia using acetylated α-tubulin (Ac. Tub, purple) and Arl13b (green) and nuclei using Hoechst (blue). Scale bar = 20 μm. (E) The percentage of cilia compared with nuclei (left) and quantification of cilia length (right) following serum starvation in MEF’s derived from wild-type (N = 3 embryos) and Bbs5−/− (N = 4 embryos) E13.5 embryos. (G) RT-PCR analysis of Bbs5 exons 2–6 generated from brain, kidney and testes cDNA of control (f/f), heterozygous (f/Δ) and Bbs5Δ/Δ mutant animals (Δ). (H) Western blot analysis of lysates generated from cre negative control (f/f) and Bbs5Δ/Δ mutant (Δ) kidney, brain and testes.
To further evaluate expression from the Bbs5−/− allele, we also conducted western blot analysis of lysates from kidney, brain and testes using commercial rabbit polyclonal and rabbit monoclonal antibodies (19). In all of these tissues and with both antibodies, we detect the presence of a 37 kDa band in the wild-type but also in the Bbs5−/− lysates (Fig. 1B). Although in the kidney, this could be explained by the splicing around the LacZ cassette generating wild-type transcript, in the case of the brain and testes, this is difficult to rectify with complete absence of a wild-type transcript in these tissues, raising possible concerns about the specificity of the antibody. This is further addressed using the Bbs5 conditional allele below.
Bbs5−/− mice do not have defects in ciliogenesis
By immunostaining for Arl13b and acetylated α-tubulin, there were no overt differences detected in number or length of primary cilia in analyzed tissues, indicating that Bbs5−/− mutant mice do not display a general defect in ciliogenesis. In addition, Bbs5−/− mouse embryonic fibroblasts (MEFs) form cilia at a similar frequency and with similar lengths as controls (Fig. 1F). The lack of an overt cilia morphology defect is consistent with other core BBSome mutant mice.
Bbs5−/− mice have decreased viability
Homozygous Bbs5−/− mutant mice are viable, but exhibit a significantly increased mortality by weaning age (P21) compared with heterozygous and wild-type littermates (Fig. 1E). Our studies indicate that during the final stages of embryonic development, E18.5-birth, all genotypes are present at the ratios expected from heterozygous matings [χ2(2, N = 47; 7 litters) =3.09, P > 0.05] (Fig. 1E). However, Mendelian ratios reflect a significant reduction [χ2(2, N = 141, 23 litters) =19.93, P < 0.001] in the observed number of mutant animals at weaning (Fig. 1E), indicating perinatal lethality. The cause of the increased pre-weaning lethality in Bbs5−/− mutants is not known.
Observationally, perinatal Bbs5−/− mice appear smaller than their littermate controls. Similar to what has been observed in other BBS mouse models, growth retardation occurs during the first 3 weeks in mutant animals, allowing them to be easily distinguished from their littermates; this is possibly caused by the inability to nurse due to anosmia (17) or decreased nutrient absorption or processing (20,21). However, the actual cause for this growth retardation is not known for any of the BBSome core mutant mice.
Perinatal lethality in Bbs5−/− mice is not due to pulmonary abnormalities
BBS patients can present with highly variable phenotypes. This is thought to be related to the modifying effects of individual patients’ different genetic backgrounds. In mice, it has been reported that phenotypes associated with mutations in other Bbs genes are also affected by genetic background that can contribute to pre-weaning lethality (22). Previous reports of background-dependent lethality in BBS mutant mice have been attributed to neural tube closure defects and pulmonary developmental defects (23,24). During embryo isolations, we never observed neural tube closure defects and Mendelian ratios were observed after neural tube closure (E18.5-birth). For these reasons, we went on to assess whether pulmonary developmental defects could be contributing to perinatal lethality in Bbs5−/− mice. Histological analysis of lungs at E18.5 do not show obvious differences in alveolar space or pulmonary interstitium (Supplementary Material, Fig. S1A). Immunofluorescence staining for the alveolar type I cells, vasculature and alveolar type II cells using antibodies against prosurfactant protein C (SPC1), platelet and endothelial cell adhesion molecule 1 (PECAM1) and clara cell secretory protein (CCSP1) also did not reveal differences compared with control littermate lungs (Supplementary Material, Fig. S1B–D). Thus, in contrast to some of the other core BBSome mutant models, perinatal lethality in Bbs5−/− mice is not associated with overt pulmonary defects.
Bbs5Δ/Δ mutant animals exhibit mild kidney abnormalities
The unexpected splicing events observed in Bbs5−/− mutants led to the investigation of the conditional allele. Analysis performed in tissues isolated from the Bbs5f/f; Cagg-CreERT, which had been induced via tamoxifen injection, shows the expected shift in band size from the floxed allele to the recombined delta allele in both genomic DNA (Supplementary Material, Fig. S2A) and complementary DNA (cDNA) (Fig. 1G). There was minimal floxed allele remaining after induction, and we detected very little wild-type transcript in cDNA analysis. Translation of the transcript after deletion of the two exons (exons 4 and 5) results in a premature translation termination signal. However, as we noted in the western analysis in the Bbs5−/− allele, protein analysis in tissues from the Bbs5Δ/Δ allele did not reflect either a loss of protein or a shift in the size of the band observed relative to the flox control (Fig. 1H). Based on this information and our analysis of the Bbs5−/− alelle, we conclude that, in our hands, the antibodies used are not specific to BBS5 and therefore could not be used to draw any conclusions about the protein in the tissues studied.
Structural kidney abnormalities are observed in BBS patients (25). Thus far, most mouse models affecting core BBSome proteins do not exhibit significant renal abnormalities (12). The exception to this are Bbs2 and Bbs4 models that have an increase in inflammatory cells, and Bbs4 mice that develop mild glomerular cysts or dialtions (11). These mutants did not have altered BUN, creatine, or Na+ or K+ levels. Similar to the other mouse core BBSome models, Bbs5−/− mice did not reveal any kidney phenoypte (data not shown). This could be due to the ability to splice around the LacZ cassette in this tissue, as described above. Thus, we analyzed the kidney in the conditional mutants in which the wild-type transcript is deleted. Bbs5f/f; Cagg-CreERT2 mice were induced at P7 and analyzed 15 weeks after induction (Bbs5Δ/Δ). Analysis of the genomic DNA indicates a near complete deletion of the floxed exons. As reported in the Bbs4 mutants, there is a mild, and not fully penetrant, kidney phenotype in the conditional Bbs5Δ/Δ mutants consisting of a slightly enlarged glomerular caspule space (Supplementary Material, Fig. S2B). Based on the this mild phenotype and data from Bbs2 and Bbs4 mutants, we did not analyze whether there were changes in renal function.
Fertility defects in Bbs5 mutant animals
To determine if Bbs5−/− mutant mice were fertile, we performed matings with both homozygous by heterozygous mice. Although both male and female heterozygous mice are fertile, no litters were produced when either the male or female was homozygous for the Bbs5 mutant allele. In other mouse models of BBS, infertility was associated with a lack of flagellated sperm (14). To investigate whether this could be the cause of the infertility in male Bbs5−/− mice, we isolated the testes and performed histological staining. In Bbs5−/− testes, no flagellated sperm were visible (Fig. 2A). Furthermore, extraction of sperm from the epididymis of mutant mice also did not yield flagellated sperm, whereas isolation from wild-type and heterozygous animals did (Supplementary Material, video 1). This could be a result of defects in flagella formation, sperm differentiation or puberty defects related to disruption of the hypothalamic–pituitary–gonadal axis (26,27).
Figure 4 .
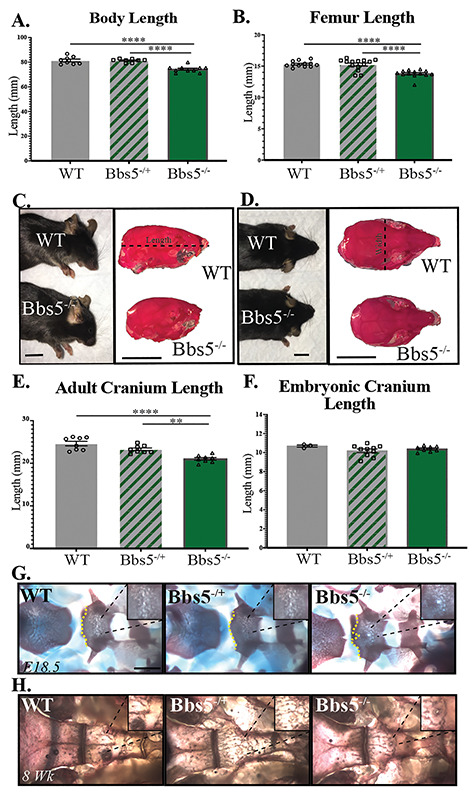
Skeletal analysis. Bbs5−/− mice exhibit craniofacial and skeletal abnormalities. Measurements of (A) skeleton length, N = 8 controls, 8 heterozygotes and 10 mutants, and (B) femur length, N = 12 control, 14 heterozygous and 13 mutant femurs at 8 weeks old. (C) Sideview of WT (top, left) and Bbs5−/− (bottom, left) animals and skulls of WT (top, right) and Bbs5−/− (bottom, right) that have been stained with alizarin red (scale bar = 1 mm). (D) Overhead view of WT (top, left) and Bbs5−/− (bottom, left) animals and skulls of WT (top, right) and Bbs5−/− (bottom, right) that have been stained with alizarin red (scale bar =1 mm). (E) Cranium lengths in 2-month-old WT, Bbs5−/+ and Bbs5−/− animals, N = 8, 8 and 8, respectively. (F) Cranium lengths in E18.5 WT, Bbs5−/+ and Bbs5−/− animals, N = 3, 10 and 8, respectively. Alizarin red and alcian blue staining of WT, Bbs5−/+ and Bbs5−/− cranial base (dorsal aspect) at (G) E18.5 and (H) 2 months old. For measurements, length was measured from the back of the scull to the tip of the nasal bone [dotted line in (C)]. Error bars represent standard error. Significance was determined one-way ANOVA with Tukey’s multiple comparisons test. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Figure 2 .
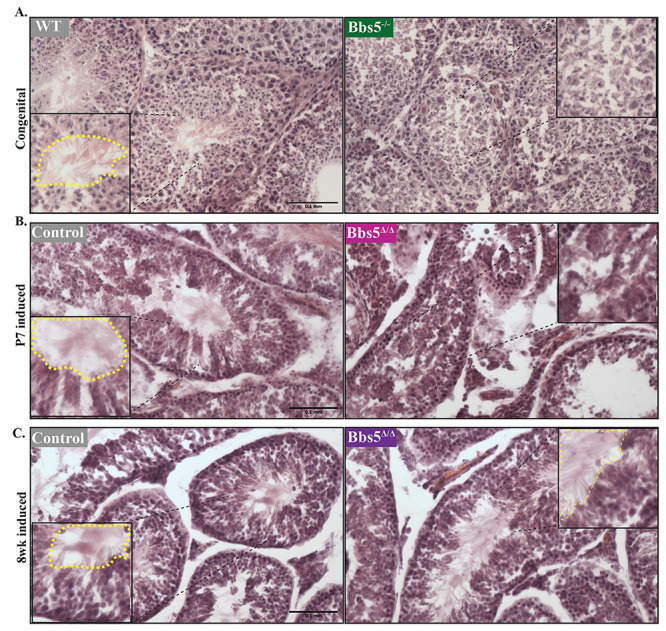
Testes analysis hematoxylin and eosin staining of testes in (A) wild-type and Bbs5−/− mice (congenital), (B) juvenile-induced conditionals (P7 induced) and (C) adult-induced conditionals (8 week induced). Staining shows the presence of flagellated sperm (inside the dotted yellow line) in WT, Bbs5f/f and adult-induced Bbs5Δ/Δ animals versus the lack of flagellated sperm (Bbs5−/−) in Bbs5−/− and juvenile-induced Bbs5Δ/Δ mice (Scale bar 0.1 mm).
To further evalaute whether this is a defect in develoment of the sperm versus flagellar maintanence, we utilized conditional Bbs5flox/flox; Cagg-CreERT2 animals and induced Bbs5 loss at either P7, prior to sexual maturation or after at 8 weeks of age when sexual maturation is complete. Testes isolated from Bbs5Δ/Δ mice induced at P7 and analyzed at least 2 months post-induction showed a variable phenotype, where five out of eight male mice did not develop flagellated sperm and three mice did develop flagellated sperm. In two out of the three Bbs5Δ/Δ mice that produced flagellated sperm, the number of flagellated sperm appeared reduced (Fig. 2B). In the adult-induced (8 weeks) mutants analyzed 10 weeks post-induction, flagellated motile sperm were present in all Bbs5Δ/Δ mice analyzed (N = 5) (Fig. 2C). These data indicate a developmental role for BBS5 during early spermatogenesis events, but not in sperm flagella formation or maintenance.
Bbs5 mutant obesity and neuronal cilia
As indicated, ~1 week following birth, surviving Bbs5−/− animals can be distinguished from their littermates due to their smaller size (data not shown). Over time, the surviving Bbs5−/− mutants not only catch up to their littermates with regards to body weight, but also surpass them and become obese. To determine if the obesity observed in Bbs5−/− mutants was due to a developmental phenotype or a role for BBS5 in adult homeostasis, we utilized the Bbs5 conditional allele (Bbs5flox/flox). Using the near ubiquitously expressed Cagg-CreERT2 allele, which has produced obesity phenotypes in other conditional ciliopathy alleles (28,29), we analyzed adult phenotypes upon the conditional loss of Bbs5 induced at P7 and 8 weeks of age. Both male (blue) and female (red) conditional mutant Bbs5Δ/Δ animals become obese on breeder chow diet (10% crude fat) compared with Cre negative controls (Fig. 3A).
Figure 5 .
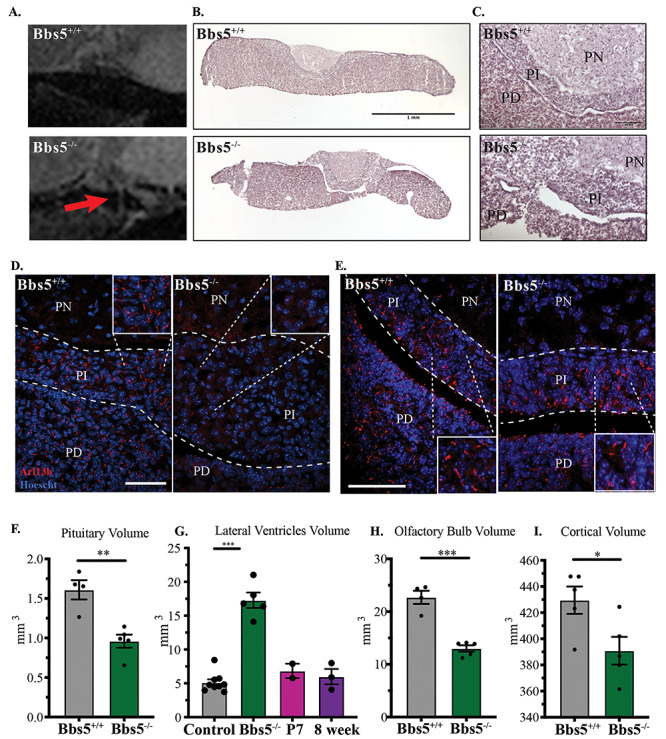
(A) Sagittal cross-section of pituitary MR images reveals structural abnormalities in Bbs5−/− animals compared with controls (red arrows). (B) Hematoxylin and eosin (H&E) histology of the pituitary (scale bar = 1 mm), (C) magnified H&E staining of the PN, PI and PD regions of the pituitary (scale bar =10 μm). Immunofluorescence staining of cilia in adult (D) and P3 (E) pituitaries using the small GTPase Arl13b (scale bar =50 μm). PN, pars nervosa; PI, pars intermedia; PD, pars distalis. Volumetric analysis of MR images shows a significant change in (F) pituitary, (G) lateral ventricles of KO mice compared with control and juvenile- and adult-induced animals: control includes five wild-type and four Bbs5f/f animals, (H) the olfactory bulb, (I) cortex. Significance was determined via unpaired t-test. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Figure 3 .
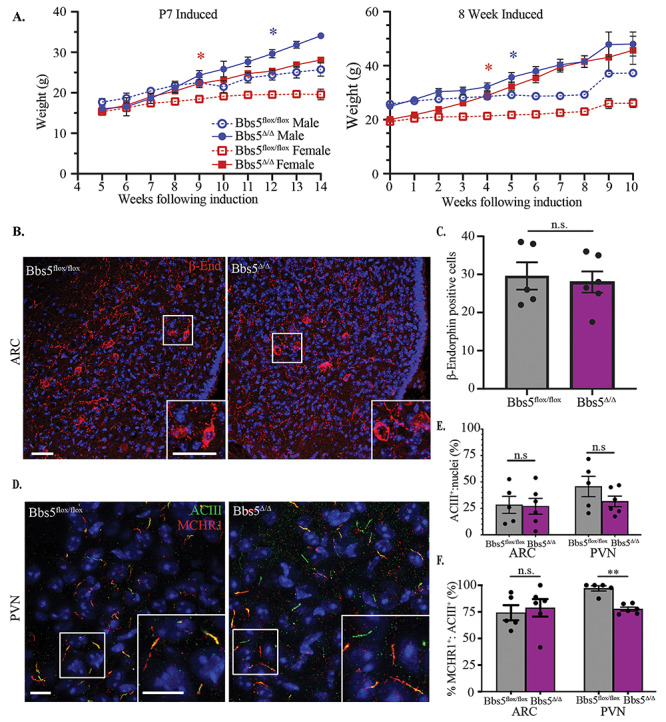
Obesity and neuronal cilia. (A) Body weight measurement after conditional loss of Bbs5. (Left graph) Weights of male (blue) and female (red) Bbs5f/f and Bbs5Δ/Δ mice following induction on postnatal day 7, N = 4 ♂ and 3♀ controls and 3♂ and 2♀ mutants. (Right graph) Weights following adult induction at 8 weeks old (adult induced), N = 9♂ and 7♀ controls and 8♂ and 12♀ mutants. Asterisks represent initial significant differences (after which, significance is maintained) between male Bbs5flox/flox and male Bbs5Δ/Δ (blue asterisk) or female Bbs5flox/flox and female Bbs5Δ/Δ (red asterisk) (P < 0.05) using a mixed-effects analysis with multiple comparisons. Error bars represent SEM. (B) POMC neuron immunofluorescence in the arcuate nucleus (ARC) for β-endorphin (β-end, red) in control and adult Bbs5Δ/Δ male and female mice (right graph). (C) Number of β-endorphin-positive cells per section of ARC was not significantly different (n.s.) between genotypes using a Student’s t-test. Scale bar 10 μ, N = 3♂ and 2♀ control and 3♂ and 3♀ mutant ♂. (D) Primary cilia immunofluorescence for cilia marker adenylate cyclase III (ACIII, green) and cilia GPCR, melanin concentrating hormone receptor 1 (MCHR1, white) in the paraventricular nucleus (PVN). (E) Quantification of the ratio of ACIII-positive cilia to nuclei in the ARC and PVN revealed no significant differences (n.s.). (F) Quantification of ACIII and MCHR1-double positive cilia in ARC and PVN revealed no significant differences in the ARC (n.s.), but reduced double positive cilia in PVN were observed using Student’s t-test (P < 0.01). Scale bar = 10 μm, N = 3 ♂ and 2 ♀ controls and 3 ♂ and 3 ♀ mutants. All Hoechst-stained nuclei blue. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Other congenital BBS mutant mouse models develop obesity and display loss of proopiomelanocortin (POMC) neuron labeling within the arcuate nucleus (ARC) of the hypothalamus. This is consistant with either a loss of proopiomelanocortin (POMC) neurons or a defect in leptin responsiveness in these mutants (30). However, in Bbs5Δ/Δ mutant mice, immunoflourescence for the POMC neuronal marker β-endorphin did not reveal differences between controls and Bbs5Δ/Δ mutants in cell numbers (Fig. 3B and C), suggesting that the POMC neuronal population is intact and that there are no changes in cell number following the onset of obesity (31).
Both BBS2 and BBS4 are important for proper ciliary localization of G-protein coupled receptors like melanin-concentrating hormone receptor 1 (MCHR1), which plays a role in feeding behavior and metabolism (32). Surprisingly, unlike Bbs2 and Bbs4 congenital knockout mice, Bbs5Δ/Δ mice still localize MCHR1 to the cilium in the hypothalamus (Fig. 3D, channel separated Supplementary Material, Fig. S3A) (32). The number of adenylate cyclase III (ACIII)-positive cilia relative to nuclei in the paraventricular nucleus (PVN) and ARC is not significantly different (Fig. 3E). However, in the ARC, MCHR1 is found in cilia at comparable levels with controls, whereas MCHR1:ACIII-double positive cilia are significantly reduced in the PVN of Bbs5Δ/Δ mice compared with controls (P < 0.001) (Fig. 3F). When separated by sex, there are no significant differences between male and female cilia number (Supplementary Material, Fig. S3B). We did not observe overt differences in the frequency of cilia in the Bbs5Δ/Δ mice compared with controls. Together, these data suggest that changes in ciliary composition and subsequent signaling may initiate the obesity phenotype in adults, and it is not solely due to developmental patterning of the hypohthalmaus in this ciliopathy model.
Decreased endochondral bone length in Bbs5 mutant mice
Surviving Bbs5−/− congenital mutant mice displayed skeletal abnormalities as measured by an overall decrease in skeletal length from the tip of the nasal bone to the pubic symphysis (Fig. 4A). This difference is present in both Bbs5−/− (P < 0.0001) and Bbs5−/+ (P < 0.0001) mice compared with wild-type littermates. A decrease in the length of long bones, as represented by shortened femurs (Fig. 4B), follows a similar trend in both Bbs5−/− (P < 0.0001) and Bbs5−/+ (P < 0.0001) mice compared with wild-type littermates. During our gross inspection of the skeletons, we also noted that none of the Bbs5−/− mutant mice analyzed exhibited polydactyly. Although this phenotype is commonly observed in human BBS patients, it has not been observed in the BBS mutant mouse models to date (13–15).
Bbs5−/− animals develop shortened craniofacial bones postnatally
Craniofacial abnormalities in adult Bbs5−/− mice were also observed (lateral view Fig. 4C, overhead view Fig. 4D). Skull length in adult mice measured from the tip of the nasal bone to the back of the skull is significantly different among the genotypes. For example, the distance is shorter in congenital Bbs5−/− animals compared with wild-type Bbs5+/+ mice (P < 0.0001). We also observe significant differences between Bbs5−/+ and Bbs5−/− (P = 0.0019) (Fig. 4E). These phenotypes were not present in E18.5 skulls analyzed, suggesting a role for BBS5 in later stages of craniofacial development and growth (Fig. 4F). These data are similar to previous reports of craniofacial abnormalities in other BBSome mutant animals (15,33).
Skeletal analysis also revealed a persistence of the buccohypophyseal canal in the basisphenoid bone at the base of the skull in E18.5 Bbs5−/− embryos (Fig. 4G) and in adult Bbs5−/− animals (Fig. 4H). These phenotypes are not observed in Bbs5−/+ or wild-type mice. The buccohypophyseal canal is an ancestral vertebrate structure that typically disappears in mammals during development to generate a barrier between the pituitary gland and the oral cavity. The persistence of this canal was also described in Gas1 knockout animals, along with pituitary development abnormalities in Ift88 conditional (Wnt-1Cre), Ofd1 and Kif3a cilia mutant mice. In these mutants, the phenotype was attributed to altered regulation of the hedgehog (Hh) signaling pathway in the midline of the embryos (34). Until now, the only other reported case of basisphenoid abnormalities in BBS mice has been in BBS3/Arl6 congenital mutant models, which is not a member of the core BBSome complex (22).
Brain and pituitary abnormalities in Bbs5 mutant mice
A potential cause for the smaller size of the Bbs5−/− mutant mice, along with defects in sperm production and abnormal bone length, could be pituitary gland dysfunction (35). This possibility is supported by the persistence of the buccohypophyseal canal in Bbs5−/− mice as well as BBS patients presenting with pituitary abnormalities (36). To assess changes in the pituitary gland in the Bbs5−/− mice, we performed magnetic resonance imaging (MRI) on heads of control and congenital mutants as well as conditional mutants where BBS5 loss was induced early (P7) and in adults (8 week old) (Supplementary Material, video 2). Sagital cross-sections of MRI images indicate that the Bbs5−/− mutant pituitary glands exhibit abnormal morphology with ectopic expansions caudally in three out of five mutant animals (Fig. 5A) that were never observed in wild-type control littermates. Importantly, histological analysis of sections through the pituitary gland in Bbs5−/− mutants that appeared normal by MRI revealed cellular abnormalities such as irregular boundaries and hyperplastic expansion between the pars intermedia (PI) and pars distalis (PD) regions (Fig. 5B and C). These abnormalities would not be identifiable by MRI analysis. Immunofluorescence staining using an antibody to the cilia-localized GTPase Arl13b indicates that the wild-type pars nervosa region is sparsely ciliated or lacks Arl13b-positive cilia, but the PI and PD are heavily ciliated (Fig. 5D). This phenomenon does not occur in the PI of perinatal Bbs5−/− mice. In P3 Bbs5−/− mice, the PI region exhibits Arl13b-positive cilia (Fig. 5E), indicating the cilia are lost or Arl13b trafficking to cilia is lost as the mutants age. The PD region of Bbs5−/− mutants by MRI analysis indicates that the pituitary glands in Bbs5−/− mutant mice are also significantly smaller (Fig. 5F, P < 0.01). Future studies of these pituatary abnormalities may reveal how they contribute to the clinical features of BBS.
Similar to other BBS models (14), adult (2–4 months old) Bbs5−/− mice exhibit the characteristic ventriculomegaly with an increase in the volume of the lateral ventricles (P < 0.001). Interestingly, conditional mice that have been induced at either juvenile or adult timepoints and imaged 2 or 4 months following Cre induction, respectively, ventriculomegaly was not observed (Fig. 5G). In Bbs5−/− mutant mice, the overall volume of the olfactory bulb (Fig. 5H, P < 0.001) and cortex (Fig. 5I,P < 0.05) are reduced. These data suggest that some of the neural antaomical phenotypes observed in BBS, such as ventriculomegaly, are due to their roles in early postnatal development and not in adult homeostasis.
Discussion
Our analysis of the Bbs5−/− mutant mouse unexpectedly revealed it is not likely a true systemic null, but rather an incomplete loss of function allele affecting specific tissues depending on how splicing occurs within the engineered cassette. Interestingly, this still results in an interrupted transcript in the brain and testes and seemingly wild-type cDNA in the kidney. This data points to potentially complex and tissue-specific splicing in the Bbs5 gene. Furthermore, it argues that to study a complete null allele of Bbs5, one would need to use the conditional or delta allele. Nonetheless, this study offers an informative comparative analysis between the two alleles. Furthermore, our results bring about concerns regarding the specificity of the antibody commonly utilized to detect Bbs5. In our hands, we fail to see a change in the protein amount or size compared with control in both the Bbs5−/− or the Bbs5Δ/Δ tissue lysates. The effectiveness of this antibody has also been assayed in MEF isolated from wild-type, Bbs5−/−, Bbs5flox/flox and Bbs5Δ/Δ embryos in which no observable flox band remained by genotyping. Similarly, protein lysates from these cells showed bands that did not change in intensity or size in mutant samples (data not shown).
Classic BBS-associated obesity is observed in all of the Bbs5 mutant mice we analyzed. Most BBS obesity studies have been performed in congenital models. However, this study specifically utilizes a conditional allele for neuronal receptor localization studies. This allows for the interpretation of the consequences of BBS5 loss independent of developmental defects. Due to the fact that obesity occurs when induced at both juvenile and adult timepoints, it can be determined that obesity is driven by a process that occurs throughout the lifespan of the animal. Furthermore, the observations that POMC neuron number remains normal, cilia number is unaffected and MCHR1 trafficking appears normal (except minor defects in the PVN) in the hypothalamus distinguishes this conditional model from most other BBS congenital mutant models (30,32). These data suggest that obesity may be driven by alternative mechanisms than what have been proposed previously. Follow up studies will focus on comparing the congenital Bbs5 mutant obesity with the conditional Bbs5 mutant obesity to determine if loss of BBS5 drives the obesity phenotype through the same mechanism at both ages.
Congenital loss of BBS5 consistently results in a lack of flagellated sperm. Similarly, loss of Bbs5 in a juvenile mouse shows a mixed impact on sperm flagellation. In contrast, disruption of BBS5 in adults has no impact on spermatogenesis. Based on these data, the fertility defects observed in male mutant mice are likely to be the consequence of developmental abnormalities. Spermatogenesis is dependent on several mechanisms including, but not limited to, proper neuronal signaling to coordinate gonadatropin-releasing hormone (GnRH) release followed by follicle-stimulating hormone (FSH) and the proper function of the hypothalmus–pituitary–gonadal axis for tissue autonomous regulation of FSH and androgens (37). Cilia have been shown to play a role in regulating neuronal activity of GnRH neurons. The cilia on these neurons express the kisspeptin receptor (Kiss1r), which is responsible for responding to kisspeptin and regulating the onset of puberty (26). The resulting animals that form flagellated sperm may be a result of the timing and efficiency of induction during a critical window in the initial wave of spermatogenesis. The high turnover rate of sperm production would indicate that, if BBS5 is necessary for spermatogenesis, its loss should affect sperm formation regardless of age of induction. Instead, we note that loss of BBS5 in adult animals does not affect sperm production. This supports a role for BBS5 during initial spermatogenesis but not directly in flagella formation. In female mice, reduced fecundity may be caused by multiple factors including, but not limited to, behavioral abnormalities, hormonal irregularities or abnormalities in the female reproductive tract. These observations regarding fertility, paired with the skeletal abnormalities at the cranial base, and pituitary abnormalities point to hormonal dysregulation as a potential culprit driving the phenotypes observed in Bbs5 mutant mice.
In addition to being smaller in size, pituitary glands in three out of five Bbs5 mutant mice have defects that are visible by MRI analysis. The remaining two have defects visible by histological analysis. These results point to the possibility that pituitary dysfunction may be a result of defects in the developmental process itself. The observation that the primary cilia in Bbs5−/− pituitaries are also affected indicates that there may be further hindrance of pituitary function that is a direct result of ciliary signaling dysfunction, although this awaits more detailed analysis.
Overall, our studies highlight several requirements for BBS5 in regulating the development of the axial and craniofacial skeleton. Although craniofacial abnormalities have been reported in mouse models of BBS (15,33), the only other reported case of basisphenoid abnormalities, as we observe in Bbs5 mutants, is in Bbs3/Arl6 congenital mutant models (22). This canal is hypothesized to be reminiscent of the transient developmental structure, Rathke’s pouch. During mammalian pituitary development, the basal diencephalon gives rise to neuroectoderm, which along with oral epithelium, migrates via Rathke’s pouch through the developing palatine bone to form the anterior pituitary. In contrast, the posterior pituitary is derived from the neural ectoderm (38). In addition, the observation that the pituitary in Bbs5−/− mutant mice is structurally compromised compared with wild-type animals points to possible hormonal dysregulation in these mutant mice. Defects in pituitary hormonal regulation could also underlie the developmental defects such as bone length and reproductive abnormalities observed in Bbs5−/− mutant mice. Furthermore, pituitary abnormalities such as hypoplasia, small Rathke’s cleft cyst and pituitary enlargement have recently been reported in the BBS patient population (39). Thus, the Bbs5 mutants described here will be a good model in which to explore the hypothalmus–pituitary–gonadal axis defects associated with disruption of the BBSome and in ciliopathies.
The development of the pituitary is an event that requires the tightly regulated synchronization of interactions between and migration of both the neural ectoderm and Rathke’s pouch derived from the oral ectoderm. It has been shown that abnormalities in the development of the pituitary can result in the persistence of the buccohypophyseal canal (34). In work done by the Dupé laboratory, there is a similar persistence of the buccohypophyseal canal in mice that are haploinsufficient for Sonic Hedgehog (Shh) (40). This becomes more severe in animals that are heterozygous for both Shh and the Notch pathway gene, Rbpj. These data indicate a requirement for both Shh and Notch signaling in closing of the buccohypophyseal canal pointing to the developing pituitary as a unique region within the embryo that is sensitive to the level of activity of the Shh and Notch pathways combined. It is widely accepted that canonical Hh signaling is dependent on the presence of the primary cilium. Bbs5 mutant animals do not exhibit classic Hh signaling defects (e.g. dorsal ventral neural tube patterning defects, polydactyly), suggesting that it is largely unaffected in most of the embryo. This study suggests that the loss of BBS5 specifically in the developing pituitary may be just enough to predispose animals to subtle Hh-associated pituitary abnormalities. This is further supported by disruption of Arl13b signaling in the intermediate region of mutant pituitaries, as Arl13b is also known to regulate Shh signaling events (41,42). Of course, this result does not indicate whether cilia are still present, but unable to traffic Arl13b, or that cilia are absent altogether from the PI in mutant mice. Attempts to answer this question included using traditional ciliary markers for ACIII, IFT components, and acetylated α-tubulin were unsuccessful due to lack of expression of ACIII in the pituitary and difficulty getting the remaining antibodies to work in neuronal tissues. Based on the current understanding of BBSome function, it would be unlikely that cilia are not present. Alternatively, a loss of cilia in the PI could be a result of cell differentiation abnormalities, which may cause variability in cell types that may or may not be ciliated normally. Further investigation into the role of the primary cilium and the BBSome in pituitary development is necessary to definitively answer these questions.
By performing MRIs on congenital and conditional Bbs5 mutant mice, we were not only able to identify structural abnormalities in the pituitary, but also to further expand on the classic BBS phenotype, ventriculomegaly. Based on the MRI data, the Bbs5−/− mice also have a reduction in cortical and olfactory bulb volume. MRIs performed on both juvenile- and adult-induced conditional Bbs5 mutant animals addressed whether these phenotypes are a result of developmental consequence of loss of BBS5 or a requirement for BBS5 in normal tissue function. Conditional ablation of Bbs5 at both juvenile and adult stages does not result in enlarged ventricles.
In summary, the Bbs5 mutant mice described here will be a good model to evaluate multiple phenotypes associated with BBS patients, although caution must be taken with the Bbs5−/− allele. Importantly, this includes pituitary defects. Pituitary abnormalities have been reported in both BBS and Joubert syndrome (OMIM 213300) ciliopathy patients (39,43). This study is the first to show defects in pituitary development in a BBS mouse model. Although not yet considered one of the classic pathologies associated with BBS or other ciliopathies, perhaps some of the underlying pathologies in patients are driven by a dysfunctional pituitary. This could also explain why mutation of Bbs5 results in tissue-specific phenotypes, which is unexpected, given that Bbs5 is thought to be expressed in all ciliated cells. Differences between this model and other mouse models of BBS may provide evidence that mutations to Bbs5 specifically target the pituitary. This evidence provides valuable insight into the mechanisms driving the disease state and may provide critical opportunities for pituitary-focused clinical intervention.
Materials and Methods
Generation of Bbs5 mutant alleles
All animal studies were conducted in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee at the University of Alabama at Birmingham. Mice were maintained on LabDiet® JL Rat and Mouse/Irr 10F 5LG5 chow. Bbs5 knockout first (Bbs5tm1a(EUCOMM)Wtsi/+; Bbs5−/+) embryonic stem cells, from C57BL/6NTac background mice, were obtained from Eucomm and injected into C57BL/6 J (JAX Stock No: 000058) blastocysts to establish the Bbs5−/− (tm1a) line. The allele was then maintained on the C57BL/6 J strain. Tm1c conditional allele mice were generated by mating tm1a to FlpO recombinase mice (C57BL/6 J), thus removing the LacZ and Neo cassettes and generating a conditional allele (tm1c; Bbs5flox/flox). Progeny that contained the recombined allele were crossed off of the FlpO line and bred to Cagg-CreERT2 males (C57BL/6 J) to generate the tm1d (Bbs5Δ/Δ) allele. Here, we refer to these alleles as Bbs5−/− (tm1a), Bbs5flox/flox (tm1c) and Bbs5Δ/Δ (tm1d) alleles (Fig. 1A). Primers used for genotyping are as follows for the Bbs5−/− allele: 5′-TTCAGTTGGTCAGTTTTGTATCGT-3′, 5′-TCAGCACCGGATAACAGAGC-3′ and 5′-CATAGTTGGCAGTGTTTGGGG-3′, and for the Bbs5flox/flox and Bbs5Δ/Δ alleles: 5′-TGTTTTGTTGGTAGATGATGCATGGG-3′, 5′-CAGAGAAGCATTGGTAATAACCGAGC-3′ and 5′-TGAGGGTAGGAACGGAGCTCAGAG-3′. Primers used for RT-PCR analysis: 5′-AAACAAGACCCGGGGAAGTCCTC-3′ and 5′- GGTCGCTGGACAGATTCCATCG-3′.
Embryo isolation
Timed pregnancies using Bbs5+/− animals were established with embryonic timepoint of E0.5 being noted at noon on the morning of observing the copulatory plug. To isolate embryos, pregnant females were anesthetized using isoflurane followed by cervical dislocation. Embryonic tissues or whole embryos were isolated and fixed in 4% paraformaldehyde (PFA, Sigma, 158127) in phosphate-buffered saline (PBS).
Generation of cDNA from tissues
RNA was isolated from wild-type and mutant brain, testes and kidney via Trizol extraction. cDNA was generated from RNA using SuperScript IV reverse transcriptase as per the manufacturer’s instructions.
MEF isolation
Embryos were isolated at E13.5. Following the removal of the liver and head, embryos were mechanically dissociated and cultured in Dulbecco's Modified Eagle Medium (DMEM) (Gibco, 11 039-021) supplemented with 10% fetal bovine serum (FBS), 1× penicillin and streptomycin, 0.05% primocin, 3.6 μl/0.5 L β-mercaptoethanol. Cells were grown to confluency at which time media was changed to DMEM containing 0.5% FBS to induce cilia formation.
Tissue isolation and histology
Mice were anesthetized with 0.1 ml/10 g of body weight dose of 2.0% tribromoethanol (Sigma Aldrich, St. Louis, MO) and transcardially perfused with PBS followed by 4% PFA (Affymetrix Inc., Cleveland, OH). Tissues were post-fixed in 4% PFA overnight at 4°C and then cryoprotected by submersion in 30% sucrose in PBS for 16–24 h, embedded in optimal cutting temperature (OCT), then cryosectioned for immunofluorescence and hematoxylin (Fisher Chemical, SH26-500D) and eosin (Sigma Aldrich, HT110132-1 L) staining was performed.
Immunofluorescence microscopy
Tissue sections of 10 μm thicknes (brain sections were 35 μm) were used for immunofluorescence microscopy. For staining MEFs, cells were grown on glass cover slips treated with 0.1% gelatin until confluent, then serum starved using DMEM containing 0.5% FBS for 24 h to induce cilia formation (44). Sections were fixed with 4% PFA for 10 min, permeabilized with 0.1% Triton X-100 in PBS for 8 min and then blocked in a PBS solution containing 1% bovine serum albumin, 0.3% Triton X-100, 2% (vol/vol) normal donkey serum and 0.02% sodium azide for 1 h at room temperature. Primary antibody incubation was performed in blocking solution overnight at 4°C. Primary antibodies include acetylated α-tubulin (Sigma, T7451) direct conjugated to Alexa 647 (Invitrogen, A20186) and used at 1:1000, ACIII (Encor, CPCA-ACIII, 1:1000), Arl13b (Proteintech, 1771-1AP, 1:500), BBS5 (Proteintech, 14 569-1-AP, 1:300), β-endorphin (Phoenix Pharmaceuticals, Inc., 1:200), CCSP1 (Abcam, ab40873, 1:250), glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Abcam, ab9483, 1:500), MCHR1 (Invitrogen, 711649, 1:1000), PECAM1 (Abcam, ab7388, 1:250) and SPC1 (Millipore Corp, AB3786, 1:250). Cryosections were then washed with PBS three times for 5 min at room temperature. Secondary antibodies diluted in blocking solution were added for 1 h at room temperature. Secondary antibodies included donkey-conjugated Alexa Fluor 647, 488 and 594 (Invitrogen, 1:1000). Samples were then washed in PBS and stained with Hoechst nuclear stain 33258 (Sigma Aldrich) for 5 min at room temperature. Cover slips were mounted using SlowFade Diamond Antifade Mountant (Life Technologies) for PVN and ARC sections and Immu-Mount (Thermo Scientific) for all others. Brain sections were imaged on a Leica SP8 confocal using 60× objective (NA = 1.4). All other fluorescence images were captured on Nikon spinning-disk confocal microscope with Yokogawa X1 disk, using Hamamatsu flash4 sCMOS camera; 60× Apo-TIRF (NA = 1.49) or 20× Plan Flour Multi-immersion (NA = 0.8) objectives were used. Images were processed using Nikon’s elements or Fiji software.
Skeletal preparations and bone measurements
The skin and internal organs (except brain) of 2-month-old mice were removed and the skeletons were submerged in 1% KOH overnight at room temperature. Skeletons were rinsed and cleaned of further excess tissue and fresh KOH solution added. Skeletons were left in KOH solution until sufficient tissue could be removed. Skeletons were rinsed with water and placed in a solution of 1.6% KOH and 0.004% alizarin red for 2 days. Skeletons were rinsed with water and placed in clearing solution (2 volumes glycerol; 2 volumes 70% ethanol; 1 volume benzyl alcohol). Skeletons were then stored in 100% glycerol and imaged using a Nikon SMZ800 stereo microscope.
Tamoxifen Cre induction
Recombination of the Bbs5flox/flox allele was induced in juvenile Bbs5flox/flox; CAGG-creERT2 mice at P7 by a single intraperitoneal (IP) injection of 9 mg tamoxifen (Millipore Sigma, T5648) per 40 g body weight. Tamoxifen was dissolved in corn oil. Adult animals were induced at 8 weeks old by IP injections of 6 mg/40 g (body weight) tamoxifen, administered once daily for three consecutive days.
Sequencing
Fluorescence-based Sanger sequencing using the Illumina NextSeq500 Next Generation Sequencing instrument at the Heflin Center for Genomic Sciences was performed on cDNA generated from brain, heart, lung, kidney, testes and retinal extract in wild-type and Bbs5−/− mice.
MRI imaging
MRI (9.4 T) of post-mortem brains was conducted using T2 weighting (echo time (TE): 36 repetition time (TR): 1800). Imaging was performed on adult mice at 2 months of age. All imaging was performed at the UAB Small Animal Imaging Shared Facility. Images were analyzed using Horos and ImageJ software.
Statistical analysis
Calculations were performed using Graphpad Prism and Microsoft Excel. Specific tests used are indicated in figure legends with significance indicated as follows: *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Abbreviations
MEF, mouse embryonic fibroblasts; ARC, arcuate nucleus; BBS, Bardet–Biedl syndrome; IFT, intraflagellar transport; SHH, sonic hedgehog; ACIII, adenylate cyclase III; MCHR1, melanin-concentrating hormone receptor 1; PVN, paraventricular nucleus; PI, pars intermedia; PD, pars distalis; MRI, magnetic resonance imaging; GnRH, gonadatropin-releasing hormone; FSH, follicle-stimulating hormone; PFA, paraformaldehyde; PH, plextrin homology; P7, postnatal day 7; cDNA, complementary DNA; PBS, phosphate-buffered saline; FBS, fetal bovine serum; IP, intraperitoneal.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
The authors would like to thank the members of Dr Bradley K. Yoder’s and Dr Nicolas F. Berbari’s laboratories for intellectual and technical support on the project; Dr John Totenhagen, Dr Anna Sorace and the support of the Small Animal Imaging Shared Facility (UAB); Dr Sally Camper and her laboratory in the Department of Human Genetics at The University of Michigan for guidance and expertise in the area of pituitary gland development; the National Institute of Diabetes and Digestive and Kidney Diseases and the National Heart, Lung, and Blood Institute for financial support of these studies.
Conflict of Interest statement. None declared.
Contributor Information
Melissa R Bentley-Ford, Department of Cell, Developmental and Integrative Biology, University of Alabama at Birmingham, Birmingham, AL 35294, USA.
Staci E Engle, Department of Biology, Indiana University-Purdue University Indianapolis, Indianapolis, IN 46202, USA.
Kelsey R Clearman, Department of Cell, Developmental and Integrative Biology, University of Alabama at Birmingham, Birmingham, AL 35294, USA.
Courtney J Haycraft, Department of Cell, Developmental and Integrative Biology, University of Alabama at Birmingham, Birmingham, AL 35294, USA.
Reagan S Andersen, Department of Cell, Developmental and Integrative Biology, University of Alabama at Birmingham, Birmingham, AL 35294, USA.
Mandy J Croyle, Department of Cell, Developmental and Integrative Biology, University of Alabama at Birmingham, Birmingham, AL 35294, USA.
Addison B Rains, Department of Cell, Developmental and Integrative Biology, University of Alabama at Birmingham, Birmingham, AL 35294, USA.
Nicolas F Berbari, Department of Biology, Indiana University-Purdue University Indianapolis, Indianapolis, IN 46202, USA.
Bradley K Yoder, Department of Cell, Developmental and Integrative Biology, University of Alabama at Birmingham, Birmingham, AL 35294, USA.
Funding
National Institutes of Health (2R01DK065655, 1U54DK126087 and 3P30DK074038 to B.K.Y, F31HL150898 and 5T32HL007918-20 to M.R.B., R01DK114008 to N.F.B.).
References
- 1.Garcia, G., 3rd, Raleigh, D.R. and Reiter, J.F. (2018) How the ciliary membrane is organized inside-out to communicate outside-in. Curr. Biol., 28, R421–R434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nachury, M.V., Loktev, A.V., Zhang, Q., Westlake, C.J., Peränen, J., Merdes, A., Slusarski, D.C., Scheller, R.H., Bazan, J.F., Sheffield, V.C. and Jackson, P.K. (2007) A core complex of BBS proteins cooperates with the GTPase Rab 8 to promote ciliary membrane biogenesis. Cell, 129, 1201–1213. [DOI] [PubMed] [Google Scholar]
- 3.Loktev, A.V., Zhang, Q., Beck, J.S., Searby, C.C., Scheetz, T.E., Bazan, J.F., Slusarski, D.C., Sheffield, V.C., Jackson, P.K. and Nachury, M.V. (2008) A BBSome subunit links ciliogenesis, microtubule stability, and acetylation. Dev. Cell, 15, 854–865. [DOI] [PubMed] [Google Scholar]
- 4.Xue, B., Liu, Y.X., Dong, B., Wingfield, J.L., Wu, M., Sun, J., Lechtreck, K.F. and Fan, Z.C. (2020) Intraflagellar transport protein RABL5/IFT22 recruits the BBSome to the basal body through the GTPase ARL6/BBS3. Proc. Natl. Acad. Sci. U.S.A., in press; 117, 2496–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jin, H., White, S.R., Shida, T., Schulz, S., Aguiar, M., Gygi, S.P., Bazan, J.F. and Nachury, M.V. (2010) The conserved Bardet-Biedl syndrome proteins assemble a coat that traffics membrane proteins to cilia. Cell, 141, 1208–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mourao, A., Nager, A.R., Nachury, M.V. and Lorentzen, E. (2014) Structural basis for membrane targeting of the BBSome by ARL6. Nat. Struct. Mol. Biol., 21, 1035–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seo, S., Zhang, Q., Bugge, K., Breslow, D.K., Searby, C.C., Nachury, M.V. and Sheffield, V.C. (2011) A novel protein LZTFL1 regulates ciliary trafficking of the BBSome and smoothened. PLoS Genet., 7, e1002358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang, S., Bahl, K., Chou, H.T., Woodsmith, J., Stelzl, U., Walz, T. and Nachury, M.V. (2020) Near-atomic structures of the BBSome reveal the basis for BBSome activation and binding to GPCR cargoes. elife, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Manara, E., Paolacci, S., D’Esposito, F., Abeshi, A., Ziccardi, L., Falsini, B., Colombo, L., Iarossi, G., Pilotta, A., Boccone, L.et al. (2019) Mutation profile of BBS genes in patients with Bardet-Biedl syndrome: an Italian study. Ital. J. Pediatr., 45, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li, J.B., Gerdes, J.M., Haycraft, C.J., Fan, Y., Teslovich, T.M., May-Simera, H., Li, H., Blacque, O.E., Li, L., Leitch, C.C.et al. (2004) Comparative genomics identifies a flagellar and basal body proteome that includes the BBS5 human disease gene. Cell, 117, 541–552. [DOI] [PubMed] [Google Scholar]
- 11.Guo, D.F., Beyer, A.M., Yang, B., Nishimura, D.Y., Sheffield, V.C. and Rahmouni, K. (2011) Inactivation of Bardet-Biedl syndrome genes causes kidney defects. Am. J. Physiol. Renal Physiol., 300, F574–F580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marchese, E., Ruoppolo, M., Perna, A., Capasso, G. and Zacchia, M. (2020) Exploring key challenges of understanding the pathogenesis of kidney disease in Bardet-Biedl syndrome. Kidney Int. Rep., 5, 1403–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kulaga, H.M., Leitch, C.C., Eichers, E.R., Badano, J.L., Lesemann, A., Hoskins, B.E., Lupski, J.R., Beales, P.L., Reed, R.R. and Katsanis, N. (2004) Loss of BBS proteins causes anosmia in humans and defects in olfactory cilia structure and function in the mouse. Nat. Genet., 36, 994–998. [DOI] [PubMed] [Google Scholar]
- 14.Davis, R.E., Swiderski, R.E., Rahmouni, K., Nishimura, D.Y., Mullins, R.F., Agassandian, K., Philp, A.R., Searby, C.C., Andrews, M.P., Thompson, S.et al. (2007) A knockin mouse model of the Bardet-Biedl syndrome 1 M390R mutation has cilia defects, ventriculomegaly, retinopathy, and obesity. Proc. Natl. Acad. Sci. U. S. A., 104, 19422–19427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nishimura, D.Y., Fath, M., Mullins, R.F., Searby, C., Andrews, M., Davis, R., Andorf, J.L., Mykytyn, K., Swiderski, R.E., Yang, B.et al. (2004) Bbs 2-null mice have neurosensory deficits, a defect in social dominance, and retinopathy associated with mislocalization of rhodopsin. Proc. Natl. Acad. Sci. U.S.A., 101, 16588–16593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang, Q., Seo, S., Bugge, K., Stone, E.M. and Sheffield, V.C. (2012) BBS proteins interact genetically with the IFT pathway to influence SHH-related phenotypes. Hum. Mol. Genet., 21, 1945–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tadenev, A.L., Kulaga, H.M., May-Simera, H.L., Kelley, M.W., Katsanis, N. and Reed, R.R. (2011) Loss of Bardet-Biedl syndrome protein-8 (BBS8) perturbs olfactory function, protein localization, and axon targeting. Proc. Natl. Acad. Sci. U.S.A., 108, 10320–10325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kretschmer, V., Patnaik, S.R., Kretschmer, F., Chawda, M.M., Hernandez-Hernandez, V. and May-Simera, H.L. (2019) Progressive characterization of visual phenotype in Bardet-Biedl syndrome mutant mice. Invest. Ophthalmol. Vis. Sci., 60, 1132–1143. [DOI] [PubMed] [Google Scholar]
- 19.Bales, K.L., Bentley, M.R., Croyle, M.J., Kesterson, R.A., Yoder, B.K. and Gross, A.K. (2020) BBSome component BBS5 is required for cone photoreceptor protein trafficking and outer segment maintenance. Invest. Ophthalmol. Vis. Sci., 61, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marion, V., Mockel, A., de Melo, C., Obringer, C., Claussmann, A., Simon, A., Messaddeq, N., Durand, M., Dupuis, L., Loeffler, J.P.et al. (2012) BBS-induced ciliary defect enhances adipogenesis, causing paradoxical higher-insulin sensitivity, glucose usage, and decreased inflammatory response. Cell Metab., 16, 363–377. [DOI] [PubMed] [Google Scholar]
- 21.Guo, D.F., Cui, H., Zhang, Q., Morgan, D.A., Thedens, D.R., Nishimura, D., Grobe, J.L., Sheffield, V.C. and Rahmouni, K. (2016) The BBSome controls energy homeostasis by mediating the transport of the leptin receptor to the plasma membrane. PLoS Genet., 12, e1005890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kawasaki, M., Izu, Y., Hayata, T., Ideno, H., Nifuji, A., Sheffield, V.C., Ezura, Y. and Noda, M. (2017) Bardet-Biedl syndrome 3 regulates the development of cranial base midline structures. Bone, 101, 179–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weihbrecht, K., Goar, W.A., Carter, C.S., Sheffield, V.C. and Seo, S. (2018) Genotypic and phenotypic characterization of the Sdccag 8Tn (sb-Tyr)2161B.CA1C2Ove mouse model. PLoS One, 13, e0192755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ross, A.J., May-Simera, H., Eichers, E.R., Kai, M., Hill, J., Jagger, D.J., Leitch, C.C., Chapple, J.P., Munro, P.M., Fisher, S.et al. (2005) Disruption of Bardet-Biedl syndrome ciliary proteins perturbs planar cell polarity in vertebrates. Nat. Genet., 37, 1135–1140. [DOI] [PubMed] [Google Scholar]
- 25.Zacchia, M., Di Iorio, V., Trepiccione, F., Caterino, M. and Capasso, G. (2017) The kidney in Bardet-Biedl syndrome: possible pathogenesis of urine concentrating defect. Kidney Dis. (Basel), 3, 57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koemeter-Cox, A.I., Sherwood, T.W., Green, J.A., Steiner, R.A., Berbari, N.F., Yoder, B.K., Kauffman, A.S., Monsma, P.C., Brown, A., Askwith, C.C. and Mykytyn, K. (2014) Primary cilia enhance kisspeptin receptor signaling on gonadotropin-releasing hormone neurons. Proc. Natl. Acad. Sci. U.S.A., 111, 10335–10340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.d'Anglemont de Tassigny, X., Fagg, L.A., Dixon, J.P., Day, K., Leitch, H.G., Hendrick, A.G., Zahn, D., Franceschini, I., Caraty, A., Carlton, M.B., Aparicio, S.A.J.R. and Colledge, W.H. (2007) Hypogonadotropic hypogonadism in mice lacking a functional Kiss 1 gene. Proc. Natl. Acad. Sci. U. S. A., 104, 10714–10719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davenport, J.R., Watts, A.J., Roper, V.C., Croyle, M.J., van Groen, T., Wyss, J.M., Nagy, T.R., Kesterson, R.A. and Yoder, B.K. (2007) Disruption of intraflagellar transport in adult mice leads to obesity and slow-onset cystic kidney disease. Curr. Biol., 17, 1586–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang, L., de Solis, A.J., Goffer, Y., Birkenbach, K.E., Engle, S.E., Tanis, R., Levenson, J.M., Li, X., Rausch, R., Purohit, M.et al. (2019) Ciliary gene RPGRIP1L is required for hypothalamic arcuate neuron development. JCI Insight, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seo, S., Guo, D.F., Bugge, K., Morgan, D.A., Rahmouni, K. and Sheffield, V.C. (2009) Requirement of Bardet-Biedl syndrome proteins for leptin receptor signaling. Hum. Mol. Genet., 18, 1323–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berbari, N.F., Pasek, R.C., Malarkey, E.B., Yazdi, S.M., McNair, A.D., Lewis, W.R., Nagy, T.R., Kesterson, R.A. and Yoder, B.K. (2013) Leptin resistance is a secondary consequence of the obesity in ciliopathy mutant mice. Proc. Natl. Acad. Sci. U.S.A., 110, 7796–7801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berbari, N.F., Lewis, J.S., Bishop, G.A., Askwith, C.C. and Mykytyn, K. (2008) Bardet-Biedl syndrome proteins are required for the localization of G protein-coupled receptors to primary cilia. Proc. Natl. Acad. Sci. U.S.A., 105, 4242–4246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tobin, J.L., di Franco, M., Eichers, E., May-Simera, H., Garcia, M., Yan, J., Quinlan, R., Justice, M.J., Hennekam, R.C., Briscoe, J.et al. (2008) Inhibition of neural crest migration underlies craniofacial dysmorphology and Hirschsprung's disease in Bardet-Biedl syndrome. Proc. Natl. Acad. Sci. U.S.A., 105, 6714–6719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khonsari, R.H., Seppala, M., Pradel, A., Dutel, H., Clément, G., Lebedev, O., Ghafoor, S., Rothova, M., Tucker, A., Maisey, J.G.et al. (2013) The buccohypophyseal canal is an ancestral vertebrate trait maintained by modulation in sonic hedgehog signaling. BMC Biol., 11, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Andersen, B., Pearse II, R.V., Jenne, K., Sornson, M., Lin, S.C., Bartke, A. and Rosenfeld, M.G. (1995) The Ames dwarf gene is required for Pit-1 gene activation. Dev. Biol., 172, 495–503. [DOI] [PubMed] [Google Scholar]
- 36.Bonfrate, A., Farah, J., De Marzi, L., Delacroix, S., Herault, J., Sayah, R., Lee, C., Bolch, W.E. and Clairand, I. (2016) Influence of beam incidence and irradiation parameters on stray neutron doses to healthy organs of pediatric patients treated for an intracranial tumor with passive scattering proton therapy. Phys. Med., 32, 590–599. [DOI] [PubMed] [Google Scholar]
- 37.O'Shaughnessy, P.J. (2014) Hormonal control of germ cell development and spermatogenesis. Semin. Cell Dev. Biol., 29, 55–65. [DOI] [PubMed] [Google Scholar]
- 38.Larkin, S. and Ansorge, O. (2000) Development and microscopic anatomy of the pituitary gland. In Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., Dungan, K., Grossman, A., Hershman, J.M., Kaltsas, G., Koch, C., Kopp, P.et al. (eds), Endotext. South Dartmouth (MA): MDText.com, Inc. Available from:https://www.ncbi.nlm.nih.gov/books/NBK425703/. [PubMed] [Google Scholar]
- 39.Guran, T., Ekinci, G., Atay, Z., Turan, S., Akcay, T. and Bereket, A. (2011) Radiologic and hormonal evaluation of pituitary abnormalities in patients with Bardet-Biedl syndrome. Clin. Dysmorphol., 20, 26–31. [DOI] [PubMed] [Google Scholar]
- 40.Hamdi-Rozé, H., Ware, M., Guyodo, H., Rizzo, A., Ratié, L., Rupin, M., Carré, W., Kim, A., Odent, S., Dubourg, C.et al. (2020) Disrupted hypothalamo-pituitary axis in association with reduced SHH underlies the pathogenesis of NOTCH-deficiency. J. Clin. Endocrinol. Metab., 105,e3183–e3196. [DOI] [PubMed] [Google Scholar]
- 41.Mariani, L.E., Bijlsma, M.F., Ivanova, A.A., Suciu, S.K., Kahn, R.A. and Caspary, T. (2016) Arl 13b regulates Shh signaling from both inside and outside the cilium. Mol. Biol. Cell, 23: 3780–3790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gigante, E.D. and Caspary, T. (2020) Signaling in the primary cilium through the lens of the 609 Hedgehog pathway. Wiley Interdiscip. Rev. Dev. Biol., 9, e377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Niceta, M., Dentici, M.L., Ciolfi, A., Marini, R., Barresi, S., Lepri, F.R., Novelli, A., Bertini, E., Cappa, M., Digilio, M.C., Dallapiccola, B. and Tartaglia, M. (2020) Co-occurrence of mutations in KIF7 and KIAA0556 in Joubert syndrome with ocular coloboma, pituitary malformation and growth hormone deficiency: a case report and literature review. BMC Pediatr., 20, 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Breslow, D.K. and Nachury, M.V. (2015) Analysis of soluble protein entry into primary cilia using semipermeabilized cells. Methods Cell Biol., 127, 203–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.