

Abstract
Acetylcholine (ACh) has distinct functional roles in striatum compared with cortex, and imbalance between these systems may contribute to neuropsychiatric disease. Preclinical studies indicate markedly higher ACh concentrations in the striatum. The goal of this work was to leverage positron emission tomography (PET) imaging estimates of drug occupancy at cholinergic receptors to explore ACh variation across the human brain, because these measures can be influenced by competition with endogenous neurotransmitter. PET scans were analyzed from healthy human volunteers (n = 4) and nonhuman primates (n = 2) scanned with the M1-selective radiotracer [11C]LSN3172176 in the presence of muscarinic antagonist scopolamine, and human volunteers (n = 10) scanned with the α4β2* nicotinic ligand (−)-[18F]flubatine during nicotine challenge. In all cases, occupancy estimates within striatal regions were consistently lower (M1/scopolamine human scans, 31 ± 3.4% occupancy in striatum, 43 ± 2.9% in extrastriatal regions, p = 0.0094; nonhuman primate scans, 42 ± 26% vs. 69 ± 28%, p < 0.0001; α4β2*/nicotine scans, 67 ± 15% vs. 74 ± 16%, p = 0.0065), indicating higher striatal ACh concentration. Subject-level measures of these concentration differences were estimated, and whole-brain images of regional ACh concentration gradients were generated. These results constitute the first in vivo estimates of regional variation in ACh concentration in the living brain and offer a novel experimental method to assess potential ACh imbalances in clinical populations.
Keywords: acetylcholine, muscarinic receptors, nicotinic receptors, positron emission tomography, striatum
Introduction
Acetylcholine (ACh) has widespread and varied effects on brain functions including arousal, attention, and memory. Projection neurons originating in the midbrain or basal forebrain are the major source of ACh in most of the brain. As with other neuromodulators such as dopamine, serotonin, and norepinephrine, much of the functional heterogeneity of cholinergic signaling arises from its release by distinct neuronal subpopulations and by binding at a variety of receptor subtypes (Klinkenberg et al. 2011). Notably for ACh, local cholinergic interneurons provide a second major source of the transmitter. The densest population of these is in the striatum, where they are crucial regulators of dopamine signaling in reward and motor circuits (Goldberg et al. 2012). Although considerable interaction and overlap exists, striatal and extrastriatal ACh can be considered partially separable systems both anatomically and functionally. For instance, silencing striatal cholinergic interneurons has prodepressive effects in preclinical behavioral studies, whereas systemic anticholinergic treatment is typically antidepressant (Warner-Schmidt et al. 2012).
In preclinical models, ACh concentration has been found to be markedly higher in the striatum compared with the rest of the brain. Microdialysis and postmortem studies in rodents report ACh levels approximately 2–3 times higher in the caudate and putamen compared with extrastriatal regions including cortex, hippocampus, and cerebellum (Cohen and Wurtman 1976; Modak et al. 1976; Hallak and Giacobini 1986; Ogane et al. 1990; Ichikawa et al. 2002; Wang et al. 2008), but this has never been confirmed in the living human brain. One meta-analysis found that baseline ACh concentration was 0.88 ± 0.04 nM in rat prefrontal cortex compared with 2.71 ± 0.45 nM in rat caudate (Noori et al. 2012), suggesting that basal ACh in the striatum may approach ACh KD for nicotinic receptors (approximately 3 nM; Kellar et al. 1985; Quick and Lester 2002) but not muscarinic receptors (approximately 20 nM; Uchida et al. 1978). Balance between striatal and cortical ACh signaling has been theorized to be important in psychiatric conditions including mood and anxiety disorders, where preclinical studies suggest that increased cortical but impaired striatal cholinergic signaling may contribute to the clinical symptoms (Higley and Picciotto 2014). In neurological disorders such as Parkinson’s disease, reduced cortical ACh may be accompanied in some cases by increased striatal ACh (Calabresi et al. 2006; Brumberg et al. 2017). However, such regional imbalances or deficits in neuromodulatory signaling are challenging to assess in vivo, and so their potential contribution to the development or progression of neuropsychiatric disorders remains unclear.
Positron emission tomography (PET) can be used to quantify several aspects of neurochemical systems in vivo. For the cholinergic system, studies have identified altered nicotinic and muscarinic receptor availability in Alzheimer’s disease (Sabri et al. 2018) and following extended alcohol exposure (Hillmer et al. 2014). PET studies can also measure target engagement of exogenous drugs that compete with a radiotracer with high affinity at the same site, termed receptor occupancy. This has been applied to measure the occupancy of nicotine and other drugs at ACh receptors (Yamamoto et al. 2011; Baldassarri et al. 2018). The sensitivity of nicotinic receptor radiotracers to changes in ACh levels caused by the acetylcholinesterase inhibitor (AChEI) physostigmine has been demonstrated using similar methods, including decreases in the (−)-[18F]flubatine PET signal of up to 6.5% under drug (Esterlis et al. 2013; Hillmer et al. 2016). However, such baseline/challenge comparisons primarily assess response to a given stimulus and do not allow measurement of baseline cholinergic tone across regions. Interestingly, in the dopamine system, high levels of endogenous neurotransmitter have been shown to influence measures of receptor occupancy, suggesting that differences in basal neurochemical tone between groups or regions or over time may be an important consideration for measuring effects of an acute stimulus (Delforge et al. 2001; Narendran et al. 2004; Kegeles et al. 2008). This is a particularly interesting factor in the context of the cholinergic system given both the substantial preclinical evidence for large differences in ACh concentration in striatum relative to the rest of the brain and the clinical use of AChEIs to alter cholinergic tone. The relationship between endogenous neurotransmitter concentration and measured occupancy implies that systematic variation in PET-derived estimates of receptor occupancy may contain important information about the underlying biological system. The objective of this work was to assess regional variation in brain ACh concentration in healthy people using PET occupancy measures at cholinergic receptors. Competition blocking studies were performed using [11C]LSN3172176 to measure binding at M1 muscarinic ACh receptors (mAChRs) (Naganawa et al., in press) and (−)-[18F]flubatine to measure α4β2* nicotinic acetylcholine receptors (nAChRs) (Brust et al. 2008). Regional differences in receptor occupancy were used to quantify relative ACh concentration across the brain.
Materials and Methods
Theory
Influence of Endogenous Neurotransmitter Concentration on Radioligand Binding
In PET studies of receptor proteins, binding potential (BP) is commonly calculated as a measure of specific radioligand binding relative to nondisplaceable ligand (i.e., both free and nonspecifically bound) using a reference region devoid of the target protein. This is denoted BPND and represents the following quantity:
 |
(1) |
where Bavail is the total number of binding sites available to the radioligand, KDtracer is the affinity constant of the radioligand at those sites, and fND is the free fraction of the radioligand in the brain in the absence of specific binding (Innis et al. 2007). Thus, BPND is directly proportional to the number of binding sites available for binding to the radioligand.
PET radioligands are typically administered at tracer doses that occupy a negligible number of the binding sites (typically < 5%). The pool of targets available to be imaged by PET therefore consist of all sites that are not already bound by an endogenous ligand (Endres and Carson 1998; Paterson et al. 2010), that is, Bavail. With greater concentrations of neurotransmitter (NT), a larger proportion of binding sites is occupied by endogenous ligand. The relationship of BPND with endogenous ligand concentrations is:
 |
(2) |
where Bmax is the total number of binding sites (whether unbound and free for the radiotracer to bind or not), [NT] is the concentration of endogenous neurotransmitter in a given region, and KDNT is the dissociation equilibrium constant for the neurotransmitter at the target. This principle underlies studies of endogenous neurotransmitter release by radioligand displacement during pharmacological or behavioral challenges (Hillmer et al. 2016; Erritzoe et al. 2019), as well as studies of endogenous dopamine levels by acute precursor depletion (Laruelle et al. 1997; Abi-Dargham et al. 2000; Verhoeff et al. 2001), where changes in BPND are interpreted to reflect changes in [NT].
Receptor Occupancy Studies
PET scans performed at baseline and in the presence of a competitor drug can measure the proportion of receptor binding sites that are bound by the drug, called receptor occupancy ( ), by quantifying reduced radioligand binding as follows:
), by quantifying reduced radioligand binding as follows:
 |
(3) |
The extent of this competition depends on the relative concentrations and affinities of the competitor drug and neurotransmitter. Assuming constant receptor affinity, BPND in a blocking scan represents:
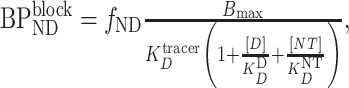 |
(4) |
where [D] is the concentration of the competitor drug and  is its equilibrium dissociation constant for the binding site (Delforge et al. 2001; Gjedde and Wong 2001).
is its equilibrium dissociation constant for the binding site (Delforge et al. 2001; Gjedde and Wong 2001).
Assuming that a system is unperturbed by addition of a competitor drug—that is, [NT], Bmax,  and
and 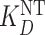 remain unchanged—Equations (2 and 4) can be substituted into Equation (3) to derive the following relationship among measured receptor occupancy, endogenous ligand concentration, and drug concentration:
remain unchanged—Equations (2 and 4) can be substituted into Equation (3) to derive the following relationship among measured receptor occupancy, endogenous ligand concentration, and drug concentration:
 |
(5) |
Thus, increasing concentrations of [NT] reduce the estimated occupancy values by the competitor drug.
Effect of Regional Differences in Neurotransmitter Concentration
Equation (5) relates endogenous neurotransmitter concentration to measured occupancy values. In situations where endogenous neurotransmitter concentration varies across different brain regions during a given scan, using the assumptions (typical in PET studies) of constant KDD and KDNT across the brain, only the term [NT] varies between regions. Therefore, regional differences in endogenous neurotransmitter concentration alter regional occupancy estimates, reducing these values in regions, where [NT] is high (or across the brain, if occupancy is calculated using all regions). From Equation (5), the influence of regional variation in [NT] on estimated occupancy also depends on the competitor drug dose (and thus competitor drug concentration in the brain) such that regional differences in r become smaller as occupancy approaches 0 or 100% in a given region (Supplementary Fig. 4A).
Determination of Regional Differences in Neurotransmitter Concentration
To determine an estimate of true endogenous neurotransmitter concentration differences between brain areas a and b, accounting for these dose effects, we define the quantity ΔNT:
 |
(6) |
Thus, ΔNT represents the difference in concentration of the endogenous neurotransmitter expressed in multiples of NT affinity at the binding site in question. Note therefore that this measure is dependent on target-specific value of KDNT and would differ between different receptors for the same [NT].
From Equations (2–4) and Equation (6), a relationship between  and occupancy estimates can be derived (Supplementary Information 1.1) such that
and occupancy estimates can be derived (Supplementary Information 1.1) such that 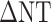 can be determined from
can be determined from  , [D], and
, [D], and  as follows:
as follows:
 |
(7) |
[D] is typically measured in blood or plasma at the time of scan and  and
and  are determined from reduction in radioligand binding under competitor drug. In vivo
are determined from reduction in radioligand binding under competitor drug. In vivo  can be estimated from in vitro studies or determined in separate blocking studies. This method was used to determine ΔACh, the difference in ACh concentrations between regions expressed as a multiple of target KDACh.
can be estimated from in vitro studies or determined in separate blocking studies. This method was used to determine ΔACh, the difference in ACh concentrations between regions expressed as a multiple of target KDACh.
PET Receptor Occupancy Studies
[11C]LSN3172176/scopolamine in Humans
Baseline and blocking PET scans using the selective M1 mAChR radioligand [11C]LSN3172176 (Supplementary Fig. 1) in four healthy volunteers were analyzed (Naganawa et al., in press). Participant and scan details are provided in Supplementary Table 1. The muscarinic antagonist scopolamine (1.5 mg) was administered as a transdermal patch 28.7 ± 0.30 h before the blocking scan for a predicted plasma concentration at the time of scan of approximately 100 pg/mL (Renner et al. 2005; Nachum et al. 2006). Dynamic data were collected on a high-resolution research tomograph (CTI/Siemens, Knoxville, TN) for 120 min. Regional BPND values were determined using the simplified reference tissue model (SRTM, (Lammertsma and Hume 1996)) with cerebellum as reference region, which provided good model fits for baseline and blocking data scans (Naganawa et al., in press). Example time-activity curves are shown in Supplementary Figure 2. Examined regions of interest (ROIs) were broadly divided into two categories: striatal regions, which included caudate, putamen, and ventral striatum, and extrastriatal regions, which included frontal, occipital, parietal, and temporal cortices and hippocampus, amygdala, and thalamus. Whole-brain parametric images of BPND were also generated by SRTM.
[11C]LSN3172176/scopolamine in Monkeys
Regional differences in M1 occupancy by scopolamine were also compared in two male rhesus macaques. Each monkey underwent three PET scans: baseline (no competitor drug) and during constant infusion of two doses of scopolamine, 5 and 20 μg/kg (Supplementary Information 1.3 and Supplementary Table 1). Dynamic PET data were acquired for 120 min on a Focus 220 PET scanner (Siemens/CTI) following previously described procedures (Nabulsi et al. 2019); see Supplementary Information for additional details. Arterial blood samples were acquired and analyzed to measure parent input function, free fraction in plasma fraction, and plasma levels of scopolamine during infusion. Because adequate equilibrium was not achieved in all regions (>15% change/h), regional volume of distribution (VT) values, representing total specific and nonspecific binding and free radioligand in a region, were determined using the one-tissue compartment model (1TCM) rather than as a direct equilibrium ratio between tissue and plasma. Regional BPND values were calculated from VT (BPND = [VTROI—VND]/VND) using VT in the cerebellum as an estimate of nondisplaceable volume of distribution, VND (Nabulsi et al. 2019). Striatal ROIs were caudate, putamen, and ventral striatum; extrastriatal ROIs comprised frontal, occipital, temporal, and cingulate cortices, insula, thalamus, amygdala, and hippocampus.
(−)-[18F]Flubatine/nicotine in Humans
Ten (−)-[18F]flubatine scans collected from six healthy nicotine users (Baldassarri et al. 2018) were analyzed (Supplementary Table 1). (−)-[18F]Flubatine was administered in a bolus/infusion protocol with dynamic data collected 90–210 min after injection. Six 5-min frames covering 90–120 min postinjection were analyzed as baseline. Nicotine was administered in the scanner via electronic cigarette starting at 125 min. After tissue concentration returned to equilibrium, data from 180–210 min after (−)-[18F]flubatine injection were used to determine postdrug radioligand binding. Regional VT values were estimated using equilibrium analysis in each time window as the ratio of mean activity in tissue to metabolite-corrected activity in arterial plasma samples. Nicotine concentrations in e-cigarette fluid were either 8 or 36 mg/mL (both concentrations [separate scans], n = 4; 36 mg/mL only, n = 1; 8 mg/mL only, n = 1). Participants abstained from smoking for at least 5 days prior to each PET scan with abstinence confirmed by scan day measures of CO (6 ± 1 ppm). For seven of the 10 scans, nicotine concentrations were measured in arterial blood samples taken at baseline, which confirmed levels of nicotine and its metabolites below detectable limits, and at intervals following e-cigarette administration to determine mean blood nicotine concentration during each scan. Striatal ROIs were caudate, putamen, and ventral striatum, and extrastriatal ROIs were frontal, occipital, parietal, and temporal cortices, hippocampus, amygdala, and cerebellum. Tracer equilibrium was not achieved in the thalamus, thus VT was not determined and it was excluded from analyses. Regional BPND values were not estimated as no reference region exists for (−)-[18F]flubatine (Hillmer et al. 2016).
Receptor Occupancy
M1 receptor occupancy by scopolamine in humans and monkeys was determined from BPND values for each of the ROIs using Equation (3). Composite striatal and extrastriatal occupancy values were determined as volume-weighted averages of BPND across regions. For visualization, conventional occupancy plots were also constructed in which occupancy can be estimated from the slope of linear regression across regions (Cunningham et al. 2010). ROI occupancy estimates were compared using repeated measures analysis of variance (ANOVA) with region type and, for monkeys, scopolamine dose as within-subject effects. To illustrate the effect of regional variation in neurotransmitter concentration on outcomes in competition binding studies, for scans in monkeys, concentration–occupancy curves were constructed using blood drug concentrations. Striatal and extrastriatal occupancy estimates were fitted separately to a one-site binding model in order to estimate apparent EC50, the plasma drug concentration at which 50% of receptors are occupied for each region type.
Nicotine occupancy at α4β2* nAChRs was determined for striatal and extrastriatal regions from VT values using weighted maximum likelihood estimation (MLE), which reduces bias compared with conventional methods (Naganawa et al. 2019). VT in a given region during blocking scans represents a combination of nonspecific binding (assumed to be unchanged during blocking or across regions) and specific binding, VS, in that region, taken as the produce of baseline VS multiplied by the proportion of unoccupied receptors,  . In the present analyses, a two occupancy model was used in which occupancy was estimated separately in prespecified striatal regions (j = 1,2,3) and extrastriatal regions (k = 1,…,10) with VND constant across regions. The following equations were fit to baseline and blocking VT values simultaneously across ROIs using weighted least squares:
. In the present analyses, a two occupancy model was used in which occupancy was estimated separately in prespecified striatal regions (j = 1,2,3) and extrastriatal regions (k = 1,…,10) with VND constant across regions. The following equations were fit to baseline and blocking VT values simultaneously across ROIs using weighted least squares:
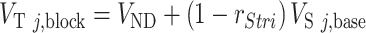 |
(8) |
 |
(9) |
Weights were the inverse of standard error of VT estimates in each ROI. Occupancy was thus estimated for both extrastriatal and striatal regions, along with VS for each ROI and VND. Note that independent estimates of receptor occupancy at the level of individual ROIs could not be determined using this method.
Composite occupancy estimates were compared across subjects in a repeated measures ANOVA with region type and nicotine dose as within-subject effects. Concentration–occupancy curves were constructed for nicotine as in the [11C]LSN3172176 monkey study. For comparison with (−)-[18F]flubatine results, striatal and extrastriatal occupancies were also estimated from VT values for [11C]LSN3172176 scans in humans (Supplementary Table 5) and monkeys (Supplementary Table 6).
Regional Differences in ACh Concentration, ΔACh
Estimates of ΔACh in striatum relative to the rest of the brain were computed for each subject using drug concentration at the time of scan and estimated striatal and extrastriatal occupancy values [Equation (7)]. For the human [11C]LSN3172176 study, an average scopolamine concentration of 100 pg/mL at 24 h after administration of a 1.5 mg transdermal patch (Renner et al. 2005; Nachum et al. 2006) was used as an approximation of plasma drug concentration, adjusted by subject weight (i.e., 100 pg/mL per 70 kg body weight). Scopolamine KD at M1 mAChRs was estimated to be 0.43 nM, an average of prior literature values (Hulme et al. 1978; Larson et al. 1991; Whiting et al. 1991; Mogg et al. 2018). To visualize variation in relative ACh concentrations across the brain, voxel-wise maps were also generated from parametric BPND images. First, whole-brain occupancy images were created [Equation (3)], then Equation (7) was applied to these images using the subject’s occipital lobe occupancy values from ROI analyses as the comparator. This produced estimates of ΔACh between each voxel and occipital lobe (i.e., ΔACh = [ACh]voxel/KDACh—[ACh]occipital/KDACh). For monkeys, ΔACh values between striatal and extrastriatal regions were determined using measured plasma concentration of scopolamine during each scan. For nicotine/α4β2* studies, measured drug concentrations in blood and a literature KD estimate of 3 nM for nicotine at α4β2* nAChRs (Whiting et al. 1991; Quick and Lester 2002) were used. Plasma drug concentrations were not adjusted for unbound fraction or brain penetrance (see Supplementary Information).
Results
Measured Occupancy by Scopolamine at M1 mAChRs is Lower in Striatum in Humans
M1 mAChR occupancy by scopolamine was determined from regional [11C]LSN3172176 BPND estimates in human volunteers (Supplementary Table 1 and Fig. 1A–D). [11C]LSN3172176 BPND at baseline is shown in Figure 1A, representing specific binding at the target. In an occupancy plot based on tracer VT, which illustrates fractional occupancy as approximately the slope of the regression line, striatal regions (caudate, putamen, and ventral striatum) deviate from the regression line fit to the remaining regions (Fig. 1B). Mean M1 receptor occupancy determined from BPND values was 31 ± 3.4% in striatal regions and 42 ± 2.9% in extrastriatal regions (main effect of region type, F1,10 = 10.3, P = 0.0094; Fig. 1C). On average, measured occupancy was 10 ± 3.1 percentage points lower in the striatum, a difference of 25%. At the level of individual ROIs, estimates of M1 receptor occupancy were highest in thalamus and occipital cortex and lowest in caudate, putamen, and ventral striatum (Fig. 1D).
Figure 1 .

Estimates of M1 mAChRs occupancy by scopolamine as determined by [11C]LSN3172176 PET in humans (A–D) and monkeys (E–I). A and E, baseline [11C]LSN3172176 BPND maps in representative subjects; B and F, occupancy plots for representative subjects with regression line fitted to extrastriatal regions only (for illustrative purposes only; see text for methods used to estimate occupancy). C and G, extrastriatal and striatal occupancy estimates from each scan; D and H, occupancy estimates in each ROI; I, occupancy-concentration curve for extrastriatal and striatal regions. Error bars in C represent standard error of the estimate. In G and H, open symbols and pale bars correspond to the 5 μg/kg dose, closed symbols and bright bars to 20 μg/kg.
Measured Occupancy by Scopolamine at M1 mAChRs is Lower in Striatum in Monkeys
As in humans, M1 receptor occupancy by scopolamine was lower in striatal compared with extrastriatal regions in monkeys (Fig. 1E–I). In all cases, scopolamine reached steady plasma concentrations by the time of scan start of 1.5 ± 0.19 ng/mL following a 5 μg/kg dose and 4.9 ± 0.48 ng/mL following a 20 μg/kg dose (Supplementary Fig. 3). At the low dose, mean occupancy was 19% in striatal regions and 45% in extrastriatal regions. With 20 μg/kg scopolamine, occupancy was 62% in the striatum compared with 90% elsewhere (Fig. 1G). Regional patterns were similar in monkeys as in humans, with lower occupancy in caudate, putamen, and ventral striatum (effect of dose, F1,1 = 89.3, P = 0.067; effect of region type, F1,38 = 49.7, P < 0.0001, interaction not significant; Fig. 1H), though the magnitude of difference was higher. Apparent EC50 for scopolamine at M1 receptors in nonhuman primates was 3.8 ng/mL standard error (SE) 1.1 ng/mL) in striatum and 1.2 ng/mL standard error (SE 0.34 ng/mL) in extrastriatal regions (Fig. 1I).
Measured Occupancy by Nicotine at α4β2* nAChRs is Lower in Striatum in Humans
Nicotine occupancy at α4β2* nAChRs was assessed in a separate group of volunteers using (−)-[18F]flubatine (baseline binding distribution, Fig. 2A). Though ROI-level occupancy estimates could not be determined from these data, a consistent pattern of lower occupancy in all striatal versus extrastriatal areas was observed (dose effect, F1,8 = 8.15, P = 0.021, region type effect, F1,8 = 13.3, P = 0.0065; interaction n.s.; Fig. 2B). Occupancy was 56 ± 15% in the striatum and 64 ± 17% in extrastriatal regions following a low dose of nicotine (difference of 12 ± 14%). At the higher dose, these values were 78 ± 4.5% and 84 ± 5.3%, respectively (difference of 7.6 ± 5.1%) (Fig. 2C).
Figure 2 .

Estimates of α4β2* nAChR occupancy by nicotine as determined by (−)-[18F]flubatine PET. A, (−)-[18F]flubatine VT at baseline in a representative subject; B, occupancy plot for a single subject with regression line fitted to extrastriatal regions only (for illustration; see text for methods used to estimate occupancy); C, extrastriatal (blue) and striatal (red) occupancy estimates in six subjects administered e-cigarettes with nicotine concentration of 8 mg/mL (pale bars) or 36 mg/mL (bright bars); error bars show standard error of each occupancy estimate. D, occupancy in extrastriatal and striatal regions versus blood nicotine concentration in 7 scans.
Thus, a consistent pattern of lower estimated receptor occupancy in the striatum versus extrastriatum was observed in both muscarinic and nicotinic receptor systems, supporting the interpretation that differences arise from variation in endogenous ACh levels rather than from characteristics of the receptors or exogenous ligands. Apparent nicotine EC50 was determined to be 1.5 ng/mL (SE 0.57 ng/mL) for striatal regions (Fig. 2D) and 1.0 ng/mL (SE 0.28 ng/mL) in extrastriatal regions.
Relative ACh Concentration Across the Brain
Occupancy estimates from each of the three studies were used to compute ΔACh, defined as the difference in ACh concentration between regions scaled to the affinity of the target for ACh [see Theory, Equation (6)]. Mean ΔACh from [11C]LSN3172176 scans in humans was 0.62 ± 0.23. ΔACh was markedly higher and more variable in monkeys (Table 1), with values of 53 and 17 from the low scopolamine dose scan and 13 and 24 after the 20 μg/kg dose. From (−)-[18F]flubatine scans, mean ΔACh was 1.22 ± 0.60 (Table 1). Given the higher KDACh at mAChRs compared with nAChRs (Uchida et al. 1978; Quick and Lester 2002), these values reflect similar ACh concentration differences across samples. In the nicotine study, ΔACh values showed good agreement between high- and low-dose scans in two of three subjects for whom plasma drug levels (and therefore ΔACh) were available, though estimates tended to be more variable in the high-dose scans (see Discussion).
Table 1.
ΔACh values representing the difference in ACh concentration between striatal and extrastriatal regions relative to receptor KDACh
| Humans | Monkeys | |||||
|---|---|---|---|---|---|---|
| Study | Subj | Scopolamine | Nicotine | Scopolamine | ||
| Low | High | Low | High | |||
| Human M1 | 1 | 0.31 | ||||
| 2 | 0.39 | |||||
| 3 | 0.51 | |||||
| 4 | 0.73 | |||||
| Human α4β2* | 1 | 1.81 | 1.44 | |||
| 2 | 1.15 | 0.01 | ||||
| 3 | 1.19 | 1.17 | ||||
| 4 | 1.77 | |||||
| Monkey M1 | 1 | 52.7 | 12.8 | |||
| 2 | 16.7 | 24.2 | ||||
| Mean ± SD | 0.49 ± 0.18 | 1.39 ± 0.37 | 1.10 ± 0.75 | 34.7 | 18.5 | |
Note: ΔACh was computed from Equation (7) and expresses the difference in ACh concentration in multiples of receptor KDACh, that is,  . Values were determined using regional occupancy values and plasma drug levels: estimated scopolamine plasma concentration in humans, measured blood nicotine concentrations available in seven of 10 scans, or measured plasma scopolamine levels in monkeys.
. Values were determined using regional occupancy values and plasma drug levels: estimated scopolamine plasma concentration in humans, measured blood nicotine concentrations available in seven of 10 scans, or measured plasma scopolamine levels in monkeys.
To visualize whole-brain ACh tone, voxel-wise images of ΔACh were generated from [11C]LSN3172176/scopolamine scans using occipital lobe occupancy estimate from ROI analyses as the comparator rather than all extrastriatal regions. Figure 3 shows the pattern of average relative ACh concentration across the four human subjects. Higher ACh concentration in caudate and putamen are evident, particularly in the ventral striatum. ACh concentration also appears to be high in temporal regions and higher in frontal cortex compared with parietal and occipital cortices.
Figure 3 .

Mean ΔACh across the brain compared with occipital lobe (i.e., concentration difference between each voxel and occipital lobe mean, scaled to ACh affinity) in four subjects determined in a receptor occupancy study with [11C]LSN3172176 PET. ΔACh was calculated by comparing voxel-wise scopolamine occupancy estimates from BPND images to mean occupancy in occipital lobe from ROI analyses using Equation (7). Images were masked to exclude voxels with low specific binding (BPND < 1).
Discussion
Our primary finding is that ACh concentration is markedly higher in the striatum compared with extrastriatal regions. This is evident in consistent patterns across two studies in humans and one in monkeys in which PET occupancy estimates at muscarinic M1 mAChRs and nicotinic α4β2* nAChRs were significantly lower in striatum compared with the rest of the brain. From these observed differences, we generated subject-level estimates of regional ACh concentration differences and images of relative ACh concentration throughout the brain, to our knowledge the first such estimates in living humans.
Regional Differences in Occupancy
The observed regional variations in measured receptor occupancy reflect the influence of endogenous ACh levels on PET measurements. Endogenous neurotransmitter present in high concentrations can occupy available binding sites, reducing PET binding availability measures (Endres and Carson 1998; Abi-Dargham et al. 2000; Verhoeff et al. 2001; Narendran et al. 2004; Paterson et al. 2010; Hillmer et al. 2014; Erritzoe et al. 2019), and, in the case of blocking studies, lowering estimates of target occupancy. This effect may be negligible if endogenous transmitters occupy a small proportion of binding sites. Similarly, when neurotransmitter concentrations are relatively constant across regions, study populations, or time, occupancy may be underestimated but with a consistent bias that may not be practically important. In the present data, occupancy estimates were 10–15 percentage points lower in the striatum across receptor types, consistent with regional variations in the ACh system seen in preclinical models. Since (−)-[18F]flubatine exhibits excellent selectivity for α4β2* nAChRs, and scopolamine is a nonselective muscarinic antagonist expected to affect [11C]LSN3172176 binding in a uniform manner, these spatial patterns are unlikely to result from binding of the tracers at other sites (see Supplementary Information 2.1). (−)-[18F]flubatine exhibits reduced specific binding following elevated cholinergic tone from AChEIs (Hillmer et al. 2016), supporting sensitivity of this radioligand to ACh competition. In vitro data indicate similar sensitivity for [11C]LSN3172176, and similar human studies are ongoing to confirm this behavior in vivo. Notably, the regional variation presented here reflects an effect of endogenous ACh concentration on PET measurements rather than true differences in competitor drug binding to the target. The 10–15% difference in regional occupancy may therefore represent a meaningful variation given the narrow therapeutic window for some neuropsychiatric drugs, such as the commonly cited target range of 65–80% occupancy at D2 receptors for antipsychotic medications (Farde et al. 1992; Nordstrom et al. 1993). Thus, research on drugs targeting cholinergic receptors should carefully consider regional variation in ACh concentration in study design and interpretation.
Quantitative estimates of ΔACh
ΔACh, defined as the difference in ACh concentration between striatal and extrastriatal regions expressed relative to KDACh [see Equations (6 and 7)], was determined from the regional differences in measured occupancy. We estimate ΔACh values of approximately 1.22 for nAChRs and 0.62 for mAChRs in humans. Given that KDACh is up to 10 times higher for mAChRs compared with nAChRs (Uchida et al. 1978; Kellar et al. 1985; Quick and Lester 2002), this suggests that ΔACh values reflect comparable but slightly lower regional differences in ACh concentration in the nicotine study compared with the scopolamine. An important caveat for these values is the reliance on literature estimates of scopolamine and nicotine KD values (Supplementary Table 7) and the use of plasma drug concentrations as estimates of concentration in brain (Supplementary Information 2.2.5). Other pharmacological influences may also contribute to quantitative differences between the studies (see Supplementary Information, 2.2). Importantly, occupancy differences from scopolamine and nicotine studies appear to follow the same theoretically predicted relationship with respect to KD-normalized dose (Supplementary Fig. 2), consistent with similar relative ACh concentrations. In contrast, striatal/extrastriatal ACh differences were markedly higher in monkeys compared with humans (Table 1) and the dose-occupancy relationship was shifted (Supplementary Fig. 4). These differences are likely due to a confluence of species and experimental differences such as the effects of anesthesia on drug delivery, free plasma fractions, ACh levels, or receptor affinity (see Supplementary Information 2.2.6). Using measured drug concentrations in the (−)-[18F]flubatine study, it can be estimated that ΔACh values reflect ACh concentrations 2.6 ± 1.8 times higher in striatum (Supplementary Information 1.2 and Supplementary Table 2). This is slightly lower than findings from microdialysis studies in rodents, which are typically 3–4 times higher with estimates ranging as high as a 12-fold difference (Cohen and Wurtman 1976; Modak et al. 1976; Hallak and Giacobini 1986; Ogane et al. 1990; Ichikawa et al. 2002; Wang et al. 2008). To our knowledge, there are no published studies assessing regional ACh concentration in primate brain. The relative ACh concentrations here can be considered an index of availability at cholinergic binding sites or “effective” concentration rather than true physical concentration. In contrast, microdialysis measurements reflect concentrations in extracellular fluid and may be less sensitive to signaling within synapses (in addition to being heavily influenced by experimental factors (Konig et al. 2018)). Volume transmission (acting at extrasynaptic receptors) may be more prominent within the striatum, and so the larger magnitude of regional differences in microdialysis studies may reflect, in part, the particular importance of cholinergic volume transmission in those regions (Descarries and Mechawar 2000; Abudukeyoumu et al. 2019; Sarter and Lustig 2020). That is, methods used here may be more sensitive to the point-to-point synaptic transmission that predominates in cortex. However, volume transmission could also lead to greater tracer displacement at extrasynaptic sites in the striatum compared with other regions, where extrasynaptic receptors exist but may not be routinely activated (Sarter and Lustig 2020). Although nAChRs and mAChRs are expressed in both synaptic and extrasynaptic compartments, this could be another source of differences in quantitative outcomes between tracers with different targets if the relative density of each receptor in synaptic versus extrasynaptic spaces is different. Thus, although the results provide confidence that the pattern of regional ACh differences is consistent across species, quantitative comparisons across studies should be interpreted with caution.
Whole-Brain ACh Concentration Gradients
Brain maps of difference in ACh concentration from occipital cortex were generated by applying Equation (7) at the voxel level (Fig. 3). These images confirmed a predominant striatal/extrastriatal effect, which supports the “a priori” separation of brain regions into striatal and extrastriatal groups based on the preclinical literature. However, variation in ACh concentration within these sets of regions is also an important consideration. ACh concentration appears to be somewhat higher in ventral striatum compared with dorsal (Figs 1D and H, and 3), in contrast to rodent microdialysis experiments showing higher concentrations in caudate and putamen than in the nucleus accumbens (Ichikawa et al. 2002; Hernandez et al. 2008). On the other hand, hippocampal ACh concentrations in rodents are typically somewhat higher than cortical levels but still well below levels in caudate and putamen (Cohen and Wurtman 1976; Ogane et al. 1990; Wang et al. 2008). This is consistent with the patterns of regional occupancy values in Figure 1D and H, with occupancy estimates in hippocampus and amygdala among the lowest of extrastriatal regions. To explore the influence of variation within other subcortical structures on estimates of relative ACh concentration, ΔACh was recalculated as the difference between striatal and cortical regions only. Values were effectively identical to those presented in Table 1 for humans (reflecting the limited influence of hippocampus and amygdala ROIs in these volume-weighted analyses) and were 1–5% higher for monkeys. Variation within the cortex is also evident in these analyses, including higher ACh concentration in the frontal cortex and insula compared with parietooccipital regions (Fig. 3). These patterns are also consistent with regional distribution of cholinergic terminal markers in humans (Kuhl et al. 1994; Siegal et al. 2004; Albin et al. 2018). We present ΔACh between striatal and extrastriatal regions, as a primary outcome because this difference was predicted to be large and consistent across subjects and was visually clear in occupancy plots (Fig. 1B and F). More regionally specific comparisons may also be useful in specific contexts, such as limbic structures in mood disorders (Picciotto et al. 2015). Overall, these observations highlight the utility of brain maps to visualize relative ACh concentration to identify more subtle patterns of variation.
Limitations
Several key assumptions underlie the interpretation that observed occupancy differences reflect differences in ACh concentration. First, we assume that nondisplaceable volume of distribution (VND) and receptor affinities are constant throughout the brain. Previous blocking studies in nonhuman primates with high doses of competitor drugs (>98% occupancy) resulted in 10% higher VT in the striatum compared with extrastriatal regions for [11C]LSN3172176 (Nabulsi et al. 2019), suggestive of possible VND differences, whereas VT in striatum was negligibly different compared with extrastriatal regions for (−)-[18F]flubatine (Bois et al. 2015). Separate striatal VND values cannot be reliably estimated due to the low number of regions and moderate occupancy levels. Since higher VND in striatal regions could also produce the reported pattern of lower occupancy seen here, data from [11C]LSN3172176 monkey scans were reanalyzed to estimate occupancy from VT values using the MLE method but fixing VND to be 10% higher in striatum than in the rest of the brain. Large differences in occupancy still emerged (Supplementary Table 3), indicating that VND differences do not drive the regional patterns in [11C]LSN3172176 studies. As such differences are also unlikely to be a factor in (−)-[18F]flubatine studies, we do not believe VND differences account for the patterns observed here. Similarly, receptor affinities are assumed to be uniform across striatal and extrastriatal regions. However, it is conceivable that some regional variation exists, particularly for nicotinic receptors, which undergo agonist-induced desensitization, resulting in a state of higher agonist affinity (Calabresi et al. 2006; Higley and Picciotto 2014). Importantly, at tracer doses (−)-[18F]flubatine binding is thought to reflect receptors in the high affinity state only (Brumberg et al. 2017; Sabri et al. 2018). Since PET studies suggest that heavy smokers have a greater proportion of receptors in the desensitized state (Hillmer et al. 2014; Baldassarri et al. 2018; Sabri et al. 2018), the greater variability in occupancy differences in the α4β2*/(−)-[18F]flubatine study compared with the M1/[11C]LSN3172176 may result from the (−)-[18F]flubatine participants consisting of regular tobacco or e-cigarette smokers. The similar regional patterns across mAChRs and nAChRs suggest that regional differences in receptor affinity are unlikely to account for the reported regional differences in occupancy, but quantification of differences in ACh tone (ΔACh) should be considered in the context of these and other factors.
Beyond assumptions of regional homogeneity, comparisons of ΔACh between subjects or time points rely on the additional assumption that changes in VND, KD, or Bmax are either negligible or consistent across scans. In particular, ACh release by competitor drugs would challenge this assumption; both mAChR blockade by scopolamine and nAChR activation by nicotine can increase ACh release across the brain (Toide and Arima 1989; Tani et al. 1998). These effects are seen across cortical and subcortical areas but may vary in magnitude across regions. Some preclinical studies suggest that the magnitude of drug-induced ACh release is low relative to concentration differences between regions (Toide and Arima 1989; Pfister et al. 1994; Tani et al. 1998), others find large though short-lived effects (Day et al. 1991), and comparisons across doses and species are challenging. Although the magnitude of ACh release may be small relative to regional differences in concentration, the ΔACh values presented here likely represent some combination of baseline ACh levels and stimulated release, which may also vary by region. Lower competitor drug dose may help to minimize these effects.
Several methodological points also require consideration. Sensitivity of [11C]LSN3172176 to endogenous ACh concentration has not yet been demonstrated. Studies in vitro indicate direct competition between [11C]LSN3172176 and ACh (Mogg et al. 2018), and human studies with AChEIs are ongoing to assess this in vivo, which would increase confidence in these results. Additionally, estimates of [11C]LSN3172176 BPND using SRTM may be slightly underestimated relative to estimates using the 1TCM (Naganawa et al., in press), particularly in striatal regions. Occupancy differences estimated using 1TCM-derived BPND values were similar to those in primary analyses (Supplementary Table 4). This result confirms that SRTM is an appropriate method, validating that regional ACh comparisons can be performed without the need for arterial blood sampling. Finally, precise occupancy quantification is challenging for (−)-[18F]flubatine, where range of VT values across regions is limited and occupancy estimates are strongly influenced by higher-binding regions. Weighted MLE methods were used to estimate occupancy in an effort to mitigate this and minimize the influence of regions with high uncertainty in VT estimates.
Altogether, despite these caveats for quantification, the pattern of results across nAChR and mAChR systems and different competitor drugs suggests that systematic differences in ACh occupancy estimates reflect regional variation in ACh concentration. The different systems, drugs, and routes of administration used here, as well as the potential added variability associated with smoking status and abstinence in the nicotine study, are limitations, particularly for comparisons between subjects. Optimization of these studies to further characterize quantitative results, explore ΔACh sensitivity and variability, and identify contributions to the large species differences in ΔACh could include the use of different antagonist drugs in a range of doses with direct measurement of plasma concentration (see further suggestions, below). Direct validation of ΔACh measures could be performed in preclinical models by comparing occupancy and microdialysis measurements in nonhuman primates. Effects of regional modulation of cholinergic tone on PET occupancy measurements could also be explored, for example through local injection of an AChEI within the striatum. This would be expected to increase occupancy differences between striatum and cortex compared with a baseline condition. In humans, evidence of variation in ΔACh estimates in clinical populations with known regional cholinergic deficits such as Alzheimer’s disease would also support the interpretation presented here. Such work exploring regional ΔACh variation in different populations and experimental conditions would advance the field’s basic understanding of these methods and systems.
Applications of ΔACh
Given the practical and theoretical limitations with (−)-[18F]flubatine and nAChRs, we propose the use of [11C]LSN3172176 PET with a muscarinic antagonist such as scopolamine as an optimal paradigm for occupancy-based measurements of relative ACh concentrations (see Supplementary Information 2.3). However, an appealing potential application of this measure is in situations where a receptor availability study is planned, and occupancy data could provide complementary information about ACh concentrations with relatively low added burden. In such cases, the choice of target or radiotracer may be dictated by primary study questions, but competitor drug and dosing can still be chosen carefully. In the nAChR study, among the four subjects who underwent (−)-[18F]flubatine scans with both nicotine concentrations, higher striatal/extrastriatal regional occupancy differences were observed at the lower nicotine dose. Mean occupancy differences between regions were 11 ± 2.8% at 8 mg/mL, when average extrastriatal occupancy was 68%, which decreased to 5.9 ± 4.8% at 36 mg/mL when extrastriatal occupancy was over 85%. This observation is consistent with the predicted (Supplementary Fig. 4A) and observed (Supplementary Fig. 4B–C) relationships within and across studies in which regional differences in occupancy decrease as occupancy itself approaches 100% (i.e., as drug dose increases). Although the ΔACh measure is intended to account for this dose relationship when comparing regional differences quantitatively, competitor dose can be selected to ensure that such differences are detected reliably, since estimates of ΔACh are more variable at high drug occupancy levels. Peak effects will vary somewhat with magnitude of regional ACh differences, but in these studies differences of 10% or higher were consistently observed at extrastriatal occupancy levels of approximately 40–70%, suggesting a relatively broad range of effective doses. Because minimizing pharmacological effects of the competitor drug is also an important consideration, a dose near or slightly below ED50 may be ideal to probe these relationships. Further work will be critical to assess the sensitivity and stability of the ΔACh measure in research applications.
An in vivo measure of relative ACh concentration across the brain has important potential applications in studies of neurological or psychiatric disease. Regional deficits in cholinergic signaling, such as that occur in Alzheimer’s and Parkinson’s diseases (Coyle et al. 1983; Durany et al. 2000; Gil-Bea et al. 2005; Conti et al. 2018), would be expected to influence occupancy differences to the extent that they reflect changes in the concentration of ACh itself. Note that the presented occupancy-based measures are independent of changes in receptor availability, thus cholinergic deficits could be identified whether or not total receptor number is altered. Independent or opposing regional differences in cholinergic signaling have also been demonstrated in mood disorders and psychosis (Ichikawa et al. 2002; Jones et al. 2012; Saricicek et al. 2012; Hannestad et al. 2013; Lewis and Picciotto 2013) and could be identified or tracked the same way. For example, preclinical studies showing opposing regional effects of chronic stress on ACh activity (Pullia et al. 1996; Higley and Picciotto 2014) predict smaller differences between regions (lower ΔACh) in individuals with depression compared with healthy people. The methods described here may offer the opportunity to explore such relationships in certain situations while simultaneously using procedures common in clinical research and drug development (i.e., drug occupancy scans). Taking into consideration the recommendations described above—and in particular constraining quantitative comparisons to be within the same study and holding key variables, such as drug dose and KD estimates, constant—this approach may offer a valuable, though indirect, research tool.
Conclusion
This work demonstrates regional variations in brain ACh concentration in humans for the first time, in particular markedly higher concentrations in the striatum. We illustrate a novel approach to image differences in endogenous ACh concentration across brain regions in living people, yielding a new tool to investigate regionally disrupted cholinergic signaling in the context of neuropsychiatric disorders.
Supplementary Material
Contributor Information
Kelly Smart, Yale PET Center, Yale School of Medicine, New Haven, CT 06510, USA; Department of Radiology & Biomedical Imaging, Yale School of Medicine, New Haven, CT 06520, USA.
Mika Naganawa, Yale PET Center, Yale School of Medicine, New Haven, CT 06510, USA; Department of Radiology & Biomedical Imaging, Yale School of Medicine, New Haven, CT 06520, USA.
Stephen R Baldassarri, Department of Internal Medicine, Section of Pulmonary, Critical Care, and Sleep Medicine, Yale School of Medicine, New Haven, CT 06510, USA.
Nabeel Nabulsi, Yale PET Center, Yale School of Medicine, New Haven, CT 06510, USA; Department of Radiology & Biomedical Imaging, Yale School of Medicine, New Haven, CT 06520, USA.
Jim Ropchan, Yale PET Center, Yale School of Medicine, New Haven, CT 06510, USA; Department of Radiology & Biomedical Imaging, Yale School of Medicine, New Haven, CT 06520, USA.
Soheila Najafzadeh, Yale PET Center, Yale School of Medicine, New Haven, CT 06510, USA.
Hong Gao, Yale PET Center, Yale School of Medicine, New Haven, CT 06510, USA.
Antonio Navarro, Eli Lilly and Co., Indianapolis, IN 46225, USA.
Vanessa Barth, Eli Lilly and Co., Indianapolis, IN 46225, USA.
Irina Esterlis, Department of Psychiatry, Yale School of Medicine, New Haven, CT 06511, USA.
Kelly P Cosgrove, Department of Radiology & Biomedical Imaging, Yale School of Medicine, New Haven, CT 06520, USA; Department of Psychiatry, Yale School of Medicine, New Haven, CT 06511, USA.
Yiyun Huang, Yale PET Center, Yale School of Medicine, New Haven, CT 06510, USA; Department of Radiology & Biomedical Imaging, Yale School of Medicine, New Haven, CT 06520, USA.
Richard E Carson, Yale PET Center, Yale School of Medicine, New Haven, CT 06510, USA; Department of Radiology & Biomedical Imaging, Yale School of Medicine, New Haven, CT 06520, USA; Department of Biomedical Engineering, Yale University, New Haven, CT 06511, USA.
Ansel T Hillmer, Yale PET Center, Yale School of Medicine, New Haven, CT 06510, USA; Department of Radiology & Biomedical Imaging, Yale School of Medicine, New Haven, CT 06520, USA; Department of Psychiatry, Yale School of Medicine, New Haven, CT 06511, USA; Department of Biomedical Engineering, Yale University, New Haven, CT 06511, USA.
Notes
The authors thank Daniel Holden and Krista Fowles for their contributions to the study in nonhuman primates as well as the rest of the staff at the Yale PET Center for their expert assistance. Conflict of interest: A.N. and V.B. are employees of Eli Lilly. The other authors declare no competing interests.
Funding
Eli Lilly and Co., the Yale Tobacco Center of Regulatory Science/National Institute on Drug Abuse/Food and Drug Administration Center for Tobacco Products (grant numbers P50DA036151-03S1, R01DA038832-01A1, K02DA031750, K23DA045957); the National Institute of Mental Health (grant number K01MH092681); and the National Institute on Alcohol Abuse and Alcoholism (grant number K01AA024788).
References
- Abi-Dargham A, Rodenhiser J, Printz D, Zea-Ponce Y, Gil R, Kegeles LS, Weiss R, Cooper TB, Mann JJ, Van Heertum RL et al. 2000. Increased baseline occupancy of D2 receptors by dopamine in schizophrenia. Proc Natl Acad Sci U S A. 97:8104–8109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abudukeyoumu N, Hernandez-Flores T, Garcia-Munoz M, Arbuthnott GW. 2019. Cholinergic modulation of striatal microcircuits. Eur J Neurosci. 49:604–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albin RL, Bohnen NI, Muller M, Dauer WT, Sarter M, Frey KA, Koeppe RA. 2018. Regional vesicular acetylcholine transporter distribution in human brain: a [(18) F]fluoroethoxybenzovesamicol positron emission tomography study. J Comp Neurol. 526:2884–2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldassarri SR, Hillmer AT, Anderson JM, Jatlow P, Nabulsi N, Labaree D, Cosgrove KP, O'Malley SS, Eissenberg T, Krishnan-Sarin S et al. 2018. Use of electronic cigarettes leads to significant beta2-nicotinic acetylcholine receptor occupancy: evidence from a PET imaging study. Nicotine Tob Res. 20:425–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bois F, Gallezot JD, Zheng MQ, Lin SF, Esterlis I, Cosgrove KP, Carson RE, Huang Y. 2015. Evaluation of [18F]-(−)-norchlorofluorohomoepibatidine [18F]-(−)-NCFHEB) as a PET radioligand to image the nicotinic acetylcholine receptors in non-human primates. Nucl Med Biol. 42:570–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brumberg J, Kusters S, Al-Momani E, Marotta G, Cosgrove KP, van Dyck CH, Herrmann K, Homola GA, Pezzoli G, Buck AK et al. 2017. Cholinergic activity and levodopa-induced dyskinesia: a multitracer molecular imaging study. Ann Clin Transl Neurol. 4:632–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brust P, Patt JT, Deuther-Conrad W, Becker G, Patt M, Schildan A, Sorger D, Kendziorra K, Meyer P, Steinbach J et al. 2008. In vivo measurement of nicotinic acetylcholine receptors with [18F]norchloro-fluoro-homoepibatidine. Synapse. 62:205–218. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Picconi B, Parnetti L, Di Filippo M. 2006. A convergent model for cognitive dysfunctions in Parkinson's disease: the critical dopamine-acetylcholine synaptic balance. Lancet Neurol. 5:974–983. [DOI] [PubMed] [Google Scholar]
- Cohen EL, Wurtman RJ. 1976. Brain acetylcholine: control by dietary choline. Science. 191:561–562. [DOI] [PubMed] [Google Scholar]
- Conti MM, Chambers N, Bishop C. 2018. A new outlook on cholinergic interneurons in Parkinson's disease and L-DOPA-induced dyskinesia. Neurosci Biobehav Rev. 92:67–82. [DOI] [PubMed] [Google Scholar]
- Coyle JT, Price DL, DeLong MR. 1983. Alzheimer's disease: a disorder of cortical cholinergic innervation. Science. 219:1184–1190. [DOI] [PubMed] [Google Scholar]
- Cunningham VJ, Rabiner EA, Slifstein M, Laruelle M, Gunn RN. 2010. Measuring drug occupancy in the absence of a reference region: the Lassen plot re-visited. J Cereb Blood Flow Metab. 30:46–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day J, Damsma G, Fibiger HC. 1991. Cholinergic activity in the rat hippocampus, cortex and striatum correlates with locomotor activity: an in vivo microdialysis study. Pharmacol Biochem Behav. 38:723–729. [DOI] [PubMed] [Google Scholar]
- Delforge J, Bottlaender M, Pappata S, Loc'h C, Syrota A. 2001. Absolute quantification by positron emission tomography of the endogenous ligand. J Cereb Blood Flow Metab. 21:613–630. [DOI] [PubMed] [Google Scholar]
- Descarries L, Mechawar N. 2000. Ultrastructural evidence for diffuse transmission by monoamine and acetylcholine neurons of the central nervous system. Prog Brain Res. 125:27–47. [DOI] [PubMed] [Google Scholar]
- Durany N, Zochling R, Boissl KW, Paulus W, Ransmayr G, Tatschner T, Danielczyk W, Jellinger K, Deckert J, Riederer P. 2000. Human post-mortem striatal α4β2 nicotinic acetylcholine receptor density in schizophrenia and Parkinson's syndrome. Neurosci Lett. 287:109–112. [DOI] [PubMed] [Google Scholar]
- Endres CJ, Carson RE. 1998. Assessment of dynamic neurotransmitter changes with bolus or infusion delivery of neuroreceptor ligands. J Cereb Blood Flow Metab. 18:1196–1210. [DOI] [PubMed] [Google Scholar]
- Erritzoe D, Ashok AH, Searle GE, Colasanti A, Turton S, Lewis Y, Huiban M, Moz S, Passchier J, Saleem A et al. 2019. Serotonin release measured in the human brain: a PET study with [11C]CIMBI-36 and d-amphetamine challenge. Neuropsychopharmacology. 45:804–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esterlis I, Hannestad JO, Bois F, Sewell RA, Tyndale RF, Seibyl JP, Picciotto MR, Laruelle M, Carson RE, Cosgrove KP. 2013. Imaging changes in synaptic acetylcholine availability in living human subjects. J Nucl Med. 54:78–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farde L, Nordstrom AL, Wiesel FA, Pauli S, Halldin C, Sedvall G. 1992. Positron emission tomographic analysis of central D1 and D2 dopamine receptor occupancy in patients treated with classical neuroleptics and clozapine. Relation to extrapyramidal side effects. Arch Gen Psychiatry. 49:538–544. [DOI] [PubMed] [Google Scholar]
- Gil-Bea FJ, Garcia-Alloza M, Dominguez J, Marcos B, Ramirez MJ. 2005. Evaluation of cholinergic markers in Alzheimer's disease and in a model of cholinergic deficit. Neurosci Lett. 375:37–41. [DOI] [PubMed] [Google Scholar]
- Gjedde A, Wong DF. 2001. Quantification of neuroreceptors in living human brain. v. Endogenous neurotransmitter inhibition of haloperidol binding in psychosis. J Cereb Blood Flow Metab. 21:982–994. [DOI] [PubMed] [Google Scholar]
- Goldberg JA, Ding JB, Surmeier DJ. 2012. Muscarinic modulation of striatal function and circuitry. In: Fryer A, Christopoulos A, Nathanson N, editors. Muscarinic Receptors. Handbook of Experimental Pharmacology, vol. 208. Berlin (Germany): Springer. p 223–241. [DOI] [PubMed] [Google Scholar]
- Hallak M, Giacobini E. 1986. Relation of brain regional physostigmine concentration to cholinesterase activity and acetylcholine and choline levels in rat. Neurochem Res. 11:1037–1048. [DOI] [PubMed] [Google Scholar]
- Hannestad JO, Cosgrove KP, DellaGioia NF, Perkins E, Bois F, Bhagwagar Z, Seibyl JP, McClure-Begley TD, Picciotto MR, Esterlis I. 2013. Changes in the cholinergic system between bipolar depression and euthymia as measured with [123I]5IA single photon emission computed tomography. Biol Psychiatry. 74:768–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez LF, Segovia G, Mora F. 2008. Chronic treatment with a dopamine uptake blocker changes dopamine and acetylcholine but not glutamate and GABA concentrations in prefrontal cortex, striatum and nucleus accumbens of the awake rat. Neurochem Int. 52:457–469. [DOI] [PubMed] [Google Scholar]
- Higley MJ, Picciotto MR. 2014. Neuromodulation by acetylcholine: examples from schizophrenia and depression. Curr Opin Neurobiol. 29:88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillmer AT, Esterlis I, Gallezot JD, Bois F, Zheng MQ, Nabulsi N, Lin SF, Papke RL, Huang Y, Sabri O et al. 2016. Imaging of cerebral alpha4beta2* nicotinic acetylcholine receptors with (−)-[18F]Flubatine PET: implementation of bolus plus constant infusion and sensitivity to acetylcholine in human brain. Neuroimage. 141:71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillmer AT, Tudorascu DL, Wooten DW, Lao PJ, Barnhart TE, Ahlers EO, Resch LM, Larson JA, Converse AK, Moore CF et al. 2014. Changes in the α4β2* nicotinic acetylcholine system during chronic controlled alcohol exposure in nonhuman primates. Drug Alcohol Depend. 138:216–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulme EC, Birdsall NJ, Burgen AS, Mehta P. 1978. The binding of antagonists to brain muscarinic receptors. Mol Pharmacol. 14:737–750. [PubMed] [Google Scholar]
- Ichikawa J, Dai J, O'Laughlin IA, Fowler WL, Meltzer HY. 2002. Atypical, but not typical, antipsychotic drugs increase cortical acetylcholine release without an effect in the nucleus accumbens or striatum. Neuropsychopharmacology. 26:325–339. [DOI] [PubMed] [Google Scholar]
- Innis RB, Cunningham VJ, Delforge J, Fujita M, Gjedde A, Gunn RN, Holden J, Houle S, Huang SC, Ichise M et al. 2007. Consensus nomenclature for in vivo imaging of reversibly binding radioligands. J Cereb Blood Flow Metab. 27:1533–1539. [DOI] [PubMed] [Google Scholar]
- Jones CK, Byun N, Bubser M. 2012. Muscarinic and nicotinic acetylcholine receptor agonists and allosteric modulators for the treatment of schizophrenia. Neuropsychopharmacology. 37:16–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kegeles LS, Slifstein M, Frankle WG, Xu X, Hackett E, Bae SA, Gonzales R, Kim JH, Alvarez B, Gil R et al. 2008. Dose-occupancy study of striatal and extrastriatal dopamine D2 receptors by aripiprazole in schizophrenia with PET and [18F]fallypride. Neuropsychopharmacology. 33:3111–3125. [DOI] [PubMed] [Google Scholar]
- Kellar KJ, Martino AM, Hall DP Jr, Schwartz RD, Taylor RL. 1985. High-affinity binding of [3H]acetylcholine to muscarinic cholinergic receptors. J Neurosci. 5:1577–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinkenberg I, Sambeth A, Blokland A. 2011. Acetylcholine and attention. Behav Brain Res. 221:430–442. [DOI] [PubMed] [Google Scholar]
- Konig M, Thinnes A, Klein J. 2018. Microdialysis and its use in behavioural studies: focus on acetylcholine. J Neurosci Methods. 300:206–215. [DOI] [PubMed] [Google Scholar]
- Kuhl DE, Koeppe RA, Fessler JA, Minoshima S, Ackermann RJ, Carey JE, Gildersleeve DL, Frey KA, Wieland DM. 1994. In vivo mapping of cholinergic neurons in the human brain using SPECT and IBVM. J Nucl Med. 35:405–410. [PubMed] [Google Scholar]
- Lammertsma AA, Hume SP. 1996. Simplified reference tissue model for PET receptor studies. Neuroimage. 4:153–158. [DOI] [PubMed] [Google Scholar]
- Larson EW, Pfenning MA, Richelson E. 1991. Selectivity of antimuscarinic compounds for muscarinic receptors of human brain and heart. Psychopharmacology (Berl). 103:162–165. [DOI] [PubMed] [Google Scholar]
- Laruelle M, D'Souza CD, Baldwin RM, Abi-Dargham A, Kanes SJ, Fingado CL, Seibyl JP, Zoghbi SS, Bowers MB, Jatlow P et al. 1997. Imaging D2 receptor occupancy by endogenous dopamine in humans. Neuropsychopharmacology. 17:162–174. [DOI] [PubMed] [Google Scholar]
- Lewis AS, Picciotto MR. 2013. High-affinity nicotinic acetylcholine receptor expression and trafficking abnormalities in psychiatric illness. Psychopharmacology (Berl). 229:477–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modak AT, Weintraub ST, McCoy TH, Stavinoha WB. 1976. Use of 300-msec microwave irradiation for enzyme inactivation: a study of effects of sodium pentobarbital on acetylcholine concentration in mouse brain regions. J Pharmacol Exp Ther. 197:245–252. [PubMed] [Google Scholar]
- Mogg AJ, Eessalu T, Johnson M, Wright R, Sanger HE, Xiao H, Crabtree MG, Smith A, Colvin EM, Schober D et al. 2018. In vitro pharmacological characterization and in vivo validation of LSN3172176 a novel M1 selective muscarinic receptor agonist tracer molecule for positron emission tomography. J Pharmacol Exp Ther. 365:602–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabulsi NB, Holden D, Zheng MQ, Bois F, Lin SF, Najafzadeh S, Gao H, Ropchan J, Lara-Jaime T, Labaree D et al. 2019. Evaluation of [11C]-LSN3172176 as a novel PET tracer for imaging M1 muscarinic acetylcholine receptors in nonhuman primates. J Nucl Med. 60:1147–1153. [DOI] [PubMed] [Google Scholar]
- Nachum Z, Shupak A, Gordon CR. 2006. Transdermal scopolamine for prevention of motion sickness: clinical pharmacokinetics and therapeutic applications. Clin Pharmacokinet. 45:543–566. [DOI] [PubMed] [Google Scholar]
- Naganawa M, Gallezot JD, Rossano S, Carson RE. 2019. Quantitative PET imaging in drug development: estimation of target occupancy. Bull Math Biol. 81:3508–3541. [DOI] [PubMed] [Google Scholar]
- Naganawa M, Nabulsi N, Henry S, Matuskey D, Lin SF, Slieker L, Schwarz AJ, Kant N, Gao H, Ropchan J et al. In press. First in human assessment of the novel M1 muscarinic acetylcholine receptor PET radiotracer [11C]LSN3172176. J Nucl Med. doi: jnumed.120.246967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendran R, Hwang DR, Slifstein M, Talbot PS, Erritzoe D, Huang Y, Cooper TB, Martinez D, Kegeles LS, Abi-Dargham A et al. 2004. In vivo vulnerability to competition by endogenous dopamine: comparison of the D2 receptor agonist radiotracer (−)-N-[11C]propyl-norapomorphine ([11C]NPA) with the D2 receptor antagonist radiotracer [11C]raclopride. Synapse. 52:188–208. [DOI] [PubMed] [Google Scholar]
- Noori HR, Fliegel S, Brand I, Spanagel R. 2012. The impact of acetylcholinesterase inhibitors on the extracellular acetylcholine concentrations in the adult rat brain: a meta-analysis. Synapse. 66:893–901. [DOI] [PubMed] [Google Scholar]
- Nordstrom AL, Farde L, Wiesel FA, Forslund K, Pauli S, Halldin C, Uppfeldt G. 1993. Central D2-dopamine receptor occupancy in relation to antipsychotic drug effects: a double-blind PET study of schizophrenic patients. Biol Psychiatry. 33:227–235. [DOI] [PubMed] [Google Scholar]
- Ogane N, Takada Y, Iga Y, Kawanishi G, Mizobe F. 1990. Effects of a M1 muscarinic receptor agonist on the central cholinergic system, evaluated by brain microdialysis. Neurosci Lett. 114:95–100. [DOI] [PubMed] [Google Scholar]
- Paterson LM, Tyacke RJ, Nutt DJ, Knudsen GM. 2010. Measuring endogenous 5-HT release by emission tomography: promises and pitfalls. J Cereb Blood Flow Metab. 30:1682–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfister M, Boix F, Huston JP, Schwarting RK. 1994. Different effects of scopolamine on extracellular acetylcholine levels in neostriatum and nucleus accumbens measured in vivo: possible interaction with aversive stimulation. J Neural Transm Gen Sect. 97:13–25. [DOI] [PubMed] [Google Scholar]
- Picciotto MR, Lewis AS, van Schalkwyk GI, Mineur YS. 2015. Mood and anxiety regulation by nicotinic acetylcholine receptors: a potential pathway to modulate aggression and related behavioral states. Neuropharmacology. 96:235–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullia D, D'Amato FR, Mele A, Oliverio A, Zocchi A, Pavone F. 1996. Time-related effects of stress on cholinergic sensitivity. Brain Res. 743:333–336. [DOI] [PubMed] [Google Scholar]
- Quick MW, Lester RA. 2002. Desensitization of neuronal nicotinic receptors. J Neurobiol. 53:457–478. [DOI] [PubMed] [Google Scholar]
- Renner UD, Oertel R, Kirch W. 2005. Pharmacokinetics and pharmacodynamics in clinical use of scopolamine. Ther Drug Monit. 27:655–665. [DOI] [PubMed] [Google Scholar]
- Sabri O, Meyer PM, Graf S, Hesse S, Wilke S, Becker GA, Rullmann M, Patt M, Luthardt J, Wagenknecht G et al. 2018. Cognitive correlates of α4β2 nicotinic acetylcholine receptors in mild Alzheimer's dementia. Brain. 141:1840–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saricicek A, Esterlis I, Maloney KH, Mineur YS, Ruf BM, Muralidharan A, Chen JI, Cosgrove KP, Kerestes R, Ghose S et al. 2012. Persistent β2*-nicotinic acetylcholinergic receptor dysfunction in major depressive disorder. Am J Psychiatry. 169:851–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarter M, Lustig C. 2020. Forebrain cholinergic signaling: wired and phasic, not tonic, and causing behavior. J Neurosci. 40:712–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegal D, Erickson J, Varoqui H, Ang L, Kalasinsky KS, Peretti FJ, Aiken SS, Wickham DJ, Kish SJ. 2004. Brain vesicular acetylcholine transporter in human users of drugs of abuse. Synapse. 52:223–232. [DOI] [PubMed] [Google Scholar]
- Tani Y, Saito K, Imoto M, Ohno T. 1998. Pharmacological characterization of nicotinic receptor-mediated acetylcholine release in rat brain--an in vivo microdialysis study. Eur J Pharmacol. 351:181–188. [DOI] [PubMed] [Google Scholar]
- Toide K, Arima T. 1989. Effects of cholinergic drugs on extracellular levels of acetylcholine and choline in rat cortex, hippocampus and striatum studied by brain dialysis. Eur J Pharmacol. 173:133–141. [DOI] [PubMed] [Google Scholar]
- Uchida S, Takeyasu K, Ichida S, Yoshida H. 1978. Muscarinic cholinergic receptors in mammalian brain: differences between bindings of acetylcholine and atropine. Jpn J Pharmacol. 28:853–862. [DOI] [PubMed] [Google Scholar]
- Verhoeff NP, Kapur S, Hussey D, Lee M, Christensen B, Psych C, Papatheodorou G, Zipursky RB. 2001. A simple method to measure baseline occupancy of neostriatal dopamine D2 receptors by dopamine in vivo in healthy subjects. Neuropsychopharmacology. 25:213–223. [DOI] [PubMed] [Google Scholar]
- Wang XC, Du XX, Tian Q, Wang JZ. 2008. Correlation between choline signal intensity and acetylcholine level in different brain regions of rat. Neurochem Res. 33:814–819. [DOI] [PubMed] [Google Scholar]
- Warner-Schmidt JL, Schmidt EF, Marshall JJ, Rubin AJ, Arango-Lievano M, Kaplitt MG, Ibanez-Tallon I, Heintz N, Greengard P. 2012. Cholinergic interneurons in the nucleus accumbens regulate depression-like behavior. Proc Natl Acad Sci U S A. 109:11360–11365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiting P, Schoepfer R, Lindstrom J, Priestley T. 1991. Structural and pharmacological characterization of the major brain nicotinic acetylcholine receptor subtype stably expressed in mouse fibroblasts. Mol Pharmacol. 40:463–472. [PubMed] [Google Scholar]
- Yamamoto S, Nishiyama S, Kawamata M, Ohba H, Wakuda T, Takei N, Tsukada H, Domino EF. 2011. Muscarinic receptor occupancy and cognitive impairment: a PET study with [11C](+)3-MPB and scopolamine in conscious monkeys. Neuropsychopharmacology. 36:1455–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.