

Abstract
The klotho gene, named after a Greek goddess who spins the thread of life, was identified as a putative ‘ageing-suppressor’ gene. Klotho-deficient mice exhibit complex ageing-like phenotypes including hypogonadism, arteriosclerosis (vascular calcification), cardiac hypertrophy, osteopenia, sarcopenia, frailty, and premature death. Klotho protein functions as the obligate co-receptor for fibroblast growth factor-23 (FGF23), a bone-derived hormone that promotes urinary phosphate excretion in response to phosphate intake. Thus, Klotho-deficient mice suffer not only from accelerated ageing but also from phosphate retention due to impaired phosphate excretion. Importantly, restoration of the phosphate balance by placing Klotho-deficient mice on low phosphate diet rescued them from premature ageing, leading us to the notion that phosphate accelerates ageing. Because the extracellular fluid is super-saturated in terms of phosphate and calcium ions, an increase in the phosphate concentration can trigger precipitation of calcium-phosphate. In the blood, calcium-phosphate precipitated upon increase in the blood phosphate concentration is adsorbed by serum protein fetuin-A to form colloidal nanoparticles called calciprotein particles (CPPs). In the urine, CPPs appear in the renal tubular fluid when FGF23 increases phosphate load excreted per nephron. CPPs can induce cell damage, ectopic calcification, and inflammatory responses. CPPs in the blood can induce arteriosclerosis and non-infectious chronic inflammation, whereas CPPs in the urine can induce renal tubular damage and interstitial inflammation/fibrosis. Thus, we propose that CPPs behave like a pathogen that accelerates ageing and should be regarded as a novel therapeutic target against age-related disorders including chronic kidney disease.
Keywords: ageing, calciprotein particles, chronic kidney disease, fibroblast growth factors, Klotho, phosphate
Introduction
Phosphorus is one of the six elements essential for life (hydrogen, carbon, nitrogen, oxygen, sulfur, and phosphorus) [1]. In vivo, phosphorus exists in the form of phosphate, which is not only a major constituent of cell membrane (e.g. phospholipids) and nucleic acids but also a critical functional molecule for energy metabolism (e.g. ATP) and cell signaling (e.g. protein phosphorylation). Phosphate is so fundamental in the structure and function of organisms that a behavior termed ‘phosphate appetite’ has evolved. For instance, animals under phosphate-deficient state instinctively seek for phosphate-rich foods, leading to osteophagic behavior (lick/eat bones of dead animals) even in herbivores [2]. The instinctive behavior to seek selectively for a lacking nutrient is termed specific appetite or specific hunger, and observed only for a limited number of essential nutrients such as sodium, indicating how important phosphate is for life. However, unlike the wild life, we barely fall into a situation of phosphate deficiency. The phosphate content in diet is known to correlate positively with the protein content [3]. Therefore, the western-style diet characterized by high intake of meats and dairy products is rich in phosphate. In addition, some food additives and preservatives contain a large amount of phosphate [4,5]. Consequently, most people on the western diet take in far more phosphate than necessary. How does the excessive phosphate intake affect our health?
Physiological responses to dietary phosphate intake
Oral gavage of phosphate in mice quickly raises their blood phosphate levels, whereas their blood calcium levels are decreased reciprocally [6], although the mechanism is not clear. However, because the blood is a supersaturated solution regarding phosphate and calcium ions, even a slight increase in the phosphate concentration can trigger precipitation of calcium-phosphate and contribute to the decrease in blood calcium levels upon phosphate intake. A decrease in the blood concentration of calcium ions inactivates calcium-sensing receptor (CaSR) expressed on the surface of parathyroid chief cells and induces secretion of parathyroid hormone (PTH) [7]. PTH has the activity that increases blood calcium levels and urinary phosphate excretion, through which blood calcium and phosphate levels are restored to the basal levels. This negative feedback to maintain the phosphate homeostasis takes place within a few hours after dietary phosphate intake [6].
When mice are placed on high phosphate diet for a few days or longer, circulating levels of fibroblast growth factor-23 (FGF23) are increased [6]. FGF23 is a hormone secreted from osteocytes and osteoblasts when they sense phosphate intake [8]. FGF23 circulates in the blood and acts on the kidney to increase urinary phosphate excretion through suppressing phosphate resorption in renal proximal tubules, thereby increasing phosphate excretion per nephron [9]. Thus, two phosphaturic hormones, PTH and FGF23, promote urinary phosphate excretion in response to phosphate intake to maintain the phosphate homeostasis in a short-term (hours) and in a long-term (days), respectively.
Pathology induced by excess phosphate intake
It has been known for decades that mice and rats develop renal tubular damage and interstitial fibrosis when placed on high phosphate diet for a few months [10,11]. In rats, the severity of the kidney damage was shown to correlate with phosphate excretion per nephron [10]. However, it should be noted that the kidney damage was not observed when the phosphate excretion per nephron was below ∼1.0 μg per day, indicating that a threshold exists for induction of the kidney damage [10,12].
In animals that maintain phosphate homeostasis, the amount of phosphate adsorbed from the gastrointestinal tract should be equal to the amount of phosphate excreted into urine. Therefore, even when placed on diet containing the same amount of phosphate, animals with lower nephron number should have higher phosphate excretion per nephron and thus develop severer kidney damage. Vice versa, the amount of phosphate intake that induces kidney damage should be different from individual to individual, depending on their residual nephron number.
Although there is a large individual and species difference in the nephron number and the amount of urinary phosphate excretion, normal young adults have ∼1 million nephrons per kidney and excrete ∼1 g of phosphate into urine on average per day [13,14]. Thus, phosphate excretion per nephron is roughly estimated as ∼0.5 μg per day in normal adults. The nephron number is decreased progressively during the course of ageing. People in their sixties or seventies are reported to have ∼50% less nephrons than people in their twenties [14]. Assuming that elderly people whose nephron number has been decreased to ∼0.5 million per kidney are on the same diet when they were young, their phosphate excretion per nephron may reach 1.0 μg per day, the level that potentially causes renal tubular damage and interstitial fibrosis. These histological changes are observed not only in patients with kidney diseases but also in elderly people without any particular disorders. The renal pathology universally observed in the aged is composed of renal tubular damage/atrophy, interstitial fibrosis, arteriosclerosis, and glomerulosclerosis among others, which are collectively designated as ‘ageing kidney’ [15,16]. Hence, dietary phosphate ingested on a daily basis may contribute to acceleration of kidney ageing in individuals with reduced nephron number such as elderly people and patients with chronic kidney disease.
Unlike humans, mice barely develop ageing kidney even when they reach an advanced age. Mice have ∼10,000 nephrons per kidney on average [17] and excrete ∼0.4 mg phosphate on standard chow diet containing 0.35% inorganic phosphate [12]. Thus, the phosphate excretion per nephron is estimated as 0.02 μg per day, which is far below the threshold (1.0 μg per day) to develop kidney damage. However, when mice were placed on diet containing 2.0% inorganic phosphate, their phosphate excretion per nephron reached ∼2.0 μg per day and developed renal tubular damage and interstitial fibrosis [11,12], recapitulating the features of ageing kidney at least in part.
Mechanism of phosphate-induced kidney damage
The molecular mechanism of the chronic kidney damage induced by dietary phosphate load has recently been elucidated [12]. An increase in phosphate intake must be associated with the corresponding increase in urinary phosphate excretion in order to maintain the phosphate balance. This demand is met mainly by increasing circulating FGF23 levels. Because FGF23 suppresses phosphate reabsorption at proximal tubules, it should increase phosphate concentration in the proximal tubular fluid. Once the phosphate concentration exceeds a threshold, calcium-phosphate microcrystals are precipitated in the proximal tubular fluid. These microcrystals bind to toll-like receptor-4 (TLR4) expressed on the apical membrane of proximal tubular cells and removed from the tubular fluid by endocytosis to be transferred to lysosome. However, overload of calcium-phosphate microcrystals elevates the luminal pH of the lysosomes and disturbs lysosomal function and autophagy, which eventually induces proximal tubular cell damage [12,18]. In addition, binding of calcium-phosphate microcrystals to TLR4 activates the p38 and NFκB signaling pathways to induce expression of cytokines and chemokines, including interleukin-6, monocyte chemotactic protein-1, tumor necrosis factor-α, and osteopontin, which potentially contribute to interstitial inflammation [12]. Tubular cell damage and interstitial inflammation eventually lead to fibrosis and kill nephrons. The decrease in the nephron number further boosts FGF23 secretion to maintain the phosphate homeostasis unless phosphate intake is reduced, thereby triggering a deterioration spiral leading to progressive nephron loss and acceleration of kidney ageing (Figure 1).
Figure 1. A deterioration spiral triggered by excess phosphate intake.
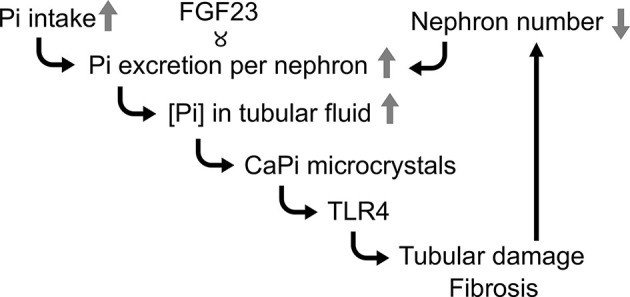
See the text. Serum FGF23 levels correlate with phosphate excretion per nephron; Pi, phosphate. Modified from [12].
It should be noted that the serum phosphate level does not contribute to this mechanism. The critical factor is not the phosphate level in the blood but the phosphate level in the proximal tubular fluid. In clinical settings, however, it is virtually impossible to harvest the renal tubular fluid from proximal tubules by micropuncture and measure the phosphate concentration. Therefore, we have established a method for estimating the phosphate concentration in the renal tubular fluid at the distal portion of proximal tubules from the data obtained by regular blood and urine tests [12]. The phosphate concentration in the glomerular filtrate should be equal to the serum phosphate concentration. Phosphate is then reabsorbed while the glomerular filtrate flows down in the proximal tubule. Phosphate reabsorption takes place almost exclusively at proximal tubules [19]. Thus, the phosphate concentration in the tubular fluid at the distal portion of proximal tubules should be equal to the product of serum phosphate concentration and fractional excretion of phosphate (FEp) unless water is not reabsorbed. However, ∼70% water is actually reabsorbed at proximal tubules [20], resulting in concentrating all the solutes by 3.33-fold. Therefore, the estimated phosphate concentration at the distal portion of proximal tubules (ePTFp) can be calculated by the following equation:
where Sp, Up, Scr, and Ucr represent the concentration of serum phosphate, urine phosphate, serum creatinine, and urine creatinine, respectively. We confirmed that ePTFp indeed served as approximation of the actual phosphate concentration in the proximal tubular fluid harvested by micropuncture in rats [12,21].
Relation between ePTFp, renal tubular damage, and FGF23 was explored in mice (Figure 2A) [12]. Double logarithmic plots of FGF23 and osteopontin against ePTFp revealed the presence of a threshold (Figure 2B). Namely, neither renal tubular damage (i.e., increase in osteopontin expression) nor increase in FGF23 was observed when ePTFp was below ∼5 mg/ml, but once ePTFp exceeded this threshold, renal tubular damage was induced, probably because calcium-phosphate microcrystals were precipitated in the tubular fluid. In addition, the fact that a linear regression between ePTFp and tubular damage became evident in double logarithmic plot is consistent with the fact that calcium-phosphate microcrystals are the true culprit of tubular damage, because it indicates the power function. Specifically,
where Kd indicates the dissociation constant. Ca, Pi, and CamPin indicate calcium, phosphate, and calcium-phosphate, respectively. The equation above shows that the regression between the calcium-phosphate concentration [CamPin] and the phosphate concentration [Pi] should be linear in the double logarithmic plot.
Figure 2. Relation between ePTFp, FGF23, and renal tubular damage.

See the text. (A) The experimental design. Uninephrectomy was performed to reduce the nephron number to one-half. (B) The double logarithmic plots between ePTFp and relative mRNA levels of osteopontin in the kidney (upper panel) and between ePTFp and serum FGF23 levels (lower panel) fitted with two-segmented linear regression with the slope of the first segment being zero. The threshold values of ePTFp and FGF23 were indicated. Modified from [12].
The similar correlation between ePTFp and FGF23 was observed in CKD patients as well (Figure 3A) [12]. The threshold values of ePTFp and FGF23 in humans were 2.32 and 53 pg/ml, respectively. A prospective cohort study (5039 participants) indicated that patients with FGF23 ≥ 53 pg/ml developed kidney events (initiation of chronic dialysis or serum creatinine doubling) within 5 years more frequently than patients with FGF23 < 53 pg/ml, independently of serum creatinine levels (Figure 3B) [12]. It should be noted that approximately 1/4 of ‘healthy’ adults aged 45 years or older and 2/3 of stage 3 CKD patients are estimated to have higher serum FGF23 levels than 53 pg/ml [22,23] and thus may have already developed renal tubular damage caused by calcium-phosphate microcrystals in the renal tubular fluid.
Figure 3. Clinical study.

(A) Relation between ePTFp and serum FGF23 levels in 148 CKD patients at various stages. The threshold values of ePTFp and FGF23 were indicated. (B) Patients participated in the EMPATHY study were stratified into two groups by 53 pg/ml of FGF23 and the cumulative incidence of renal events were compared. Modified from [12].
The phosphate-centric view of chronic kidney disease
Chronic kidney disease (CKD) is a relatively new disease entity established a few decades ago and defined as a state of any abnormality in renal function and/or structure persisting for 3 months or longer [24]. In most cases, CKD ensues when the age-associated decrease in the nephron number is accelerated by kidney diseases (e.g. acute kidney injury, glomerulonephritis, polycystic kidney) and disorders causing renal complications, most notably hypertension and diabetes mellitus. Thus, CKD is very prevalent in the ageing society and affects more than 10% of the total population [25,26]. CKD is classified into five stages based on the estimated glomerular filtration rate (eGFR) that can be calculated from the serum creatinine level [26]. In order of progression, Stage 1: eGFR 90 ml/min/1.72m2 or greater, Stage 2: eGFR between 60 and 89, Stage 3: eGFR between 30 and 59, Stage 4: eGFR between 15 and 29, Stage 5: eGFR less than 15. Once CKD advances to stage 5, renal replacement therapy (dialysis or renal transplantation) should be considered.
One of the earliest signs of CKD includes an increase in FGF23 [23]. Serum FGF23 levels start increasing as early as stage 2-3, which is regarded as a physiological response to compensate for an decrease in the nephron number by increasing phosphate excretion per nephron by FGF23 to maintain phosphate homeostasis. However, the increase in FGF23 can increase the risk for formation of calcium-phosphate microcrystals in the tubular fluid and induce renal tubular damage and interstitial fibrosis [12]. Once these renal damages reduce the functional nephron number, FGF23 should be further increased to trigger the deterioration spiral towards acceleration of CKD progression as shown in Figure 1 [12]. Besides functioning as a phosphaturic hormone, FGF23 functions as a counter-regulatory hormone for vitamin D [9]. FGF23 lowers circulating levels of active vitamin D (1,25-dihydroxyvitamin D3) by down-regulating expression of 1α-hydroxylase necessary for synthesis of active vitamin D and up-regulating expression of 24-hydroxylase necessary for inactivation of active vitamin D in renal proximal tubular cells. A decrease in active vitamin D increases PTH, because a potent negative feedback loop exists between PTH and active vitamin D, in which PTH raises serum active vitamin D levels, whereas active vitamin D lowers serum PTH levels [27]. Thus, the increase in FGF23 induces secondary hyperparathyroidism through suppressing serum levels of active vitamin D. Indeed, increase in FGF23, decrease in active vitamin D, and increase in PTH occur in this order during the course of CKD progression [23]. Once the residual nephron number is decreased to the level that cannot maintain the phosphate balance by increasing FGF23 any longer, phosphate retention and hyperphosphatemia ensues. Accordingly, increase in serum phosphate is observed only in advanced CKD patients at stage 4-5 (end-stage renal disease; ESRD) (Figure 4). Besides hyperphosphatemia, hypocalcemia ensues due to low active vitamin D [28]. The disturbed phosphate/calcium homeostasis and the changes in the hormone levels (high FGF23, low active vitamin D, and high PTH) that occur during CKD progression are usually associated with bone mineral loss and reciprocal mineralization in extra-osseous tissues (e.g. vascular calcification), and designated as CKD-MBD (mineral and bone disorder) [29].
Figure 4. Progression of CKD-MBD.

See the text. Regardless of the underlying disorders, progression of CKD can be viewed as the progressive loss of the functional nephron number. Modified from [8].
Phosphate restriction
Phosphate restriction by reducing dietary phosphate intake and/or by taking phosphate binders is currently applied only for ESRD patients with hyperphosphatemia (stage 4-5), aiming at lowering serum phosphate levels [30]. The expected clinical outcomes include suppression of vascular calcification and cardiovascular events. However, as discussed above, the deterioration spiral leading to progressive nephron loss has already been launched in early stage CKD patients (stage 2-3) when serum FGF23 levels start increasing. Thus, reduction of phosphate excretion per nephron by phosphate restriction should be beneficial not only for ESRD patients with hyperphosphatemia but also for early stage CKD patients with hyper-FGF23-emia but without hyperphosphatemia [8,12,31]. The expected clinical outcome is suppression of nephron loss or CKD progression, but not suppression of vascular calcification and cardiovascular events.
The current diet therapy for hyperphosphatemia is to avoid phosphate-rich foods based on the food composition table, such as meat, fish, and dairy products [32]. Because the phosphate content in foods is positively correlated with the protein content [3], phosphate-restricted diet inevitably means protein-restricted diet. However, recent clinical studies have shown that strict restriction of dietary protein intake can lead to a malnutrition state termed protein-energy wasting (PEW), which is associated with increased mortality [33]. Therefore, ‘phosphate restriction without protein restriction’ is desirable. To attain this goal, the following two approaches may be practical and effective.
First, it is necessary to pay attention not only to the phosphate content but also to the absorption efficiency, namely, how much percent of phosphate in each food ingredient is actually absorbed from intestine. For example, soybeans are a major plant source of protein and listed as an ingredient rich in phosphate in the food composition table. Thus, ESRD patients with hyperphosphatemia may have been instructed to avoid soybean products. However, phosphate in soybeans exists primarily in the form of phytate (inositol hexakisphosphate; IP6), which is not absorbed from the intestine and excreted into feces as is [34]. In plants, phosphate exists mainly as phytate. In order for phosphate in phytate to be absorbed from the gastrointestinal tract, inorganic phosphate must be released from phytate by hydrolyzation of the inositol-phosphate linkages with phytase (Figure 5). Phytase is an enzyme produced by enteric bacteria that reside in the gut of ruminant animals [35]. Therefore, cows and sheep can hydrolyze phytate and utilize soybeans as a source of phosphate but monogastric animals such as pigs and chickens cannot. The fact that pigs and chickens are unable to absorb phosphate from phytate raises two problems [36]. First, inorganic phosphate must be added to their feed to support their growth. Second, phytate excreted into feces can cause soil pollution with phosphorus. To solve these problems, phytase has been added to the feed containing soy flour for pigs and chickens. Because humans cannot utilize phytate as a source of phosphate either, replacement of animal-based protein with plant-based protein is considered as an effective way to reduce phosphate absorption without restricting protein intake [37].
Figure 5. Phytate as a source of phosphate.

See the text. Phytase is necessary to release phosphate from phytate.
Second, it is of critical importance for dietary phosphate restriction to avoid food additives containing inorganic phosphate [5,32]. Unlike phosphate in foods, phosphate in food additives is inorganic and thus adsorbed nearly 100%. Although various kinds of food additives are used in processed foods, it is not compulsory under the current regulations to indicate the amount of phosphate added to the processed foods in the nutrition facts label. Therefore, it is virtually impossible for consumers to know how much phosphate-containing additives has been used. However, it was reported that instructions how to read the nutrition facts labels and avoid phosphate-containing additives when purchasing groceries or visiting fast food restaurants lowered serum phosphate levels in ESRD patients [38]. Substantial contribution of food additives to daily phosphate intake is also endorsed by the fact that the amount of phosphate intake is associated with socio-economic status. Specifically, people in a low socio-economic status may have insufficient money and time to prepare well-balanced homemade meals from fresh ingredients without food additives, and tend to purchase processed foods and cheap junk foods, which in most cases contain a large amount of food additives. Indeed, the socio-economic status has been reported as an independent determinant of serum phosphate levels (i.e. a low socio-economic status is associated with high serum phosphate levels) [39,40].
In summary, a practical way to attain ‘dietary phosphate restriction without protein restriction’ would be to replace animal-based protein with plant-based protein and to reduce intake of phosphate-containing food additives.
Phosphate and ageing
Thirty years ago, we reported an obscure mutant mouse strain that exhibited complex phenotypes resembling human ageing [41]. The founder of this strain was a transgenic mouse carrying an insertional mutation caused by transgene integration on the chromosome 5. Homozygotes for the transgene grew normally until weaning, but thereafter developed multiple ageing-like phenotypes including growth arrest, multiple organ atrophy (gonads, thymus, skin), vascular calcification, cardiac hypertrophy, sarcopenia, osteopenia, emphysematous lung, hearing disturbance, cognition impairment, frailty, and died around 2 months of age. The mutant was named klotho after a Greek goddess who spins the thread of life. The gene disrupted by the transgene insertion (the klotho gene) encoded a ∼130 kD type-I single-pass transmembrane protein with a large extracellular domain and a very short intracellular domain composed of only 11 amino acids. It was expressed in limited cell types including renal tubular epithelial cells, choroid plexus epithelial cells, and parathyroid chief cells. The extracellular domain of Klotho was composed of two homologous domains. Each domain had weak homology to the family 1 glycosidases. It is still controversial whether Klotho protein has enzymatic activity as a glycosidase [42–46], because two conserved catalytic glutamate residues essential for the glycosidase activity and thus conserved in all the family 1 glycosidases are replaced with other amino acids in Klotho protein.
The clue for the Klotho protein function was a report on FGF23 knockout mice [47]. As predicted, FGF23 knockout mice failed to increase urinary phosphate excretion in response to phosphate intake and exhibited phosphate retention phenotypes, including hyperphosphatemia and vascular calcification. In addition, they unexpectedly displayed complex ageing-like phenotypes, including growth arrest, osteopenia, and shortened life span. These phenotypes were reminiscent of those seen in klotho mice [41]. The remarkable similarity between FGF23 knockout mice and klotho mice led us to hypothesize that FGF23 and Klotho might function in the same signaling pathway. At that time, FGF23 was supposed to function through binding to fibroblast growth factor receptors (FGFRs). FGFRs are receptor tyrosine kinases encoded by 4 distinct genes (FGFR1-4). The FGFR1, FGFR2, and FGFR3 genes generate multiple isoforms through alternative splicing [48]. However, the affinity of FGF23 to any FGFR isoforms are too low (KD = 200–700 nM) for FGF23 to function at its physiological concentration (1–2 pM) [49]. The answer was that FGF23 requires Klotho to bind to FGFRs. We found that Klotho protein formed constitutive complexes with FGFR1c, FGFR3c, or FGFR4. The FGFR-Klotho complexes had much higher affinity for FGF23 than FGFR alone [48]. The same finding was confirmed later independently by another laboratory [50]. The fact that Klotho functions as the obligate co-receptor for FGF23 clearly explained why FGF23 knockout mice and klotho mice developed very similar phenotypes. In 2018, crystal structure of the Klotho-FGFR1c-FGF23 ternary complex was solved [45]. Like its namesake who spins the thread of life, Klotho protein turned out to have a long ‘thread’ termed ‘receptor binding arm (RBA)’. Although the RBA appeared to have an intrinsically disordered structure, it took a solid structure once it captured FGFR1c to generate a groove into which FGF23 fit perfectly [51].
The true nature of the problem in FGF23 knockout mice and klotho mice is phosphate retention caused by defects in the FGF23-Klotho endocrine axis that lead to impaired phosphate excretion into urine. Indeed, restoration of the phosphate balance by placing FGF23 knockout mice and klotho mice on low phosphate diet rescued them from ageing-like phenotypes [52,53]. Based on these observations, we have reached the notion that phosphate accelerates ageing [31,54].
Mechanism by which phosphate accelerates ageing
Consistent with the notion that life is derived from the sea, composition of the sea water and the human body is similar (Table 1) [55]. Indeed, 9 out of the top 10 abundant elements are identical between them. The exceptions are magnesium and phosphorus. Magnesium is among the top 10 elements in the sea water, whereas magnesium is replaced with phosphorus in the human body [55]. This fact may imply that organisms that accumulate phosphorus have emerged at some time point during evolution. It occurred about 400 million years ago when the bony fish appeared. Organisms evolved thereafter accumulate phosphorus in the bone in the form of calcium-phosphate. Some organisms before the bony fish have cartilage or skeletons made of calcium-carbonate. Because calcium-phosphate is physically much harder than calcium-carbonate, the bone made of calcium-phosphate is stronger than the skeletons made of calcium-carbonate. Thus, acquisition of the strong bone made of calcium-phosphate may be a prerequisite for evolution of terrestrial vertebrates, which are required to support their own body weight and move around without the help of water buoyancy [8,56]. The reason that magnesium has been replaced with phosphorus may lie in the fact that magnesium can inhibit formation of calcium-phosphate crystals.
Table 1. The top 10 abundant elements in the sea water and human body.
| Rank | Sea water | Human body |
|---|---|---|
| 1 | H | H |
| 2 | O | O |
| 3 | Na | C |
| 4 | Cl | N |
| 5 | Mg | Na |
| 6 | S | Ca |
| 7 | K | P |
| 8 | Ca | S |
| 9 | C | K |
| 10 | N | Cl |
Modified from [61].
To create the bone made of calcium-phosphate, terrestrial vertebrates maintain the extracellular fluid in a super-saturated condition regarding calcium and phosphate ions and provide a cue for precipitation when and where they want to make the bone. To secure this strategy, they have acquired two systems. First, the FGF23-Klotho endocrine system has evolved to strictly control the extracellular phosphate concentration. It should be noted that the klotho gene orthologs exist only in organisms that have bones made of calcium-phosphate [56], suggesting that the klotho gene may have evolved to maintain the phosphate homeostasis. Second, they have acquired defense systems to prevent growth of calcium-phosphate precipitations if it should occur in extraosseous tissues and extracellular fluid. Formation of calciprotein particles (CPPs) is one of such defense mechanisms.
Calciprotein particles
CPPs are defined as mineral-protein complexes containing solid-phase calcium-phosphate and serum protein fetuin-A [57,58]. Fetuin-A has an ability to adsorb calcium-phosphate precipitates and prevent them from growing to large crystals. The process of CPP formation has been investigated in vitro using a solution containing calcium, phosphate, and serum (Figure 6) [31,59–62]. Once the concentration of phosphate and calcium ions exceeds the solubility limit, tiny amorphous calcium-phosphate precipitates appear in the solution. These precipitates are adsorbed immediately by fetuin-A. As a result, a fetuin-A molecule laden with amorphous calcium-phosphate are generated, which are the most primitive CPPs called calciprotein monomers (CPMs). CPMs have diameter of ∼9 nm and spontaneously undergo aggregation to become primary CPPs with a diameter of less than 100 nm. Primary CPPs undergo further aggregation and transition of the calcium-phosphate from the amorphous phase to the crystalline phase. CPPs containing crystalline calcium-phosphate are called secondary CPPs with a diameter usually larger than primary CPPs [6,58]. This series of events is not a biological process but a physicochemical phenomenon that progresses spontaneously over time.
Figure 6. Process of CPP formation and maturation.

See the text. Gray circles, white circles, and black circles indicate fetuin-A, amorphous calcium-phosphate (CaPi), and crystalline calcium-phosphate, respectively. Modified from [61].
There is a significant difference in the activity between primary CPPs and secondary CPPs. Primary CPPs function as a physiological regulator of FGF23 production and secretion [6]. Regarding the mechanism by which osteoblasts and osteocytes sense phosphate intake, it has been postulated that these cells may express a putative ‘phosphate-sensing receptor’ through which they sense a transient increase in blood phosphate levels after phosphate intake (postprandial hyperphosphatemia) and secrete FGF23. This notion is obviously prompted by the fact that parathyroid chief cells express CaSR through which they sense changes in blood calcium levels and regulate PTH secretion. However, the putative phosphate-sensing receptor has not been identified. Rather, several lines of evidence suggest that FGF23 secretion may not necessarily be increased in response to an increase in serum phosphate levels. For instance, an increase in serum phosphate levels failed to induce FGF23 secretion in the presence of hypocalcemia in mice and rats [63,64]. In addition, an increase in serum calcium levels induced FGF23 secretion, but it did not in the presence of hypophosphatemia [64]. These observations indicate that both calcium and phosphate are necessary for inducing FGF23 secretion, suggesting that it may not be phosphate but CPPs that can induce FGF23 secretion and production. In fact, primary CPPs, but not phosphate, were found as a potent inducer of FGF23 production and secretion in cultured osteoblastic cells [6]. An increase in FGF23 production and secretion by CPPs induces phosphaturia and increases phosphate outflow from the blood. It also lowers the serum active vitamin D level. Because the primary activity of active vitamin D is to increase calcium absorption in the gut, a decrease in the serum active vitamin D level reduces calcium absorption from the intestine and thus calcium inflow into the blood. The increase in phosphate outflow and the decrease in calcium inflow by FGF23 should suppress formation of CPPs in the blood, thereby closing a negative feedback loop (Figure 7) [6]. Thus, CPPs have emerged as a physiological regulator of the FGF23-Klotho endocrine axis. Furthermore, CPPs may function as a carrier that delivers calcium and phosphate absorbed from the gastrointestinal tract directly to the bone. In fact, an in vivo imaging of mice after intravenous injection of fluorescently labeled CPPs detected accumulation of the CPPs on the inner bone surface [6].
Figure 7. The FGF23-Klotho endocrine axis.

See the text. Klotho protein has a long amino-acid stretch designated as the receptor binding arm (RBA) that directly interacts with FGFRs. Modified from [6,8,51].
On the other hand, secondary CPPs have been reported to exert pathogenic activity. Secondary CPPs can induce calcification in cultured vascular smooth muscle cells and innate immune responses in culture macrophages as if they were a pathogen [65–69]. In clinical studies, circulating levels of secondary CPPs were reported to correlate with parameters for inflammation (high sensitive CRP), vascular stiffness (aortic pulse wave velocity), vascular calcification (coronary artery calcification score), and coronary artery plaque thickness [70–72]. A recent clinical study using a novel high sensitive assay for quantification of secondary CPPs in serum and plasma samples identified the serum phosphate level and the age as two independent determinant of plasma secondary CPP levels [73]. Namely, high serum phosphate levels and old age are independently associated with high plasma secondary CPP levels. Based on these observations, we hypothesize that secondary CPPs may be a pro-ageing factor that induces non-infectious chronic inflammation and arteriosclerosis.
Concluding remarks
In mammals, insoluble materials are adsorbed by specific serum proteins and become colloidal particles to be dispersed in the blood and transferred between organs. Lipids and calcium-phosphate are the two major insoluble materials in mammals. Lipids are adsorbed by apoproteins and become colloidal particles called lipoproteins. The activity of colloidal particles depends not only on their composition but also on their colloidal properties, including particle size and density. In fact, low-density lipoprotein (LDL) is pro-atherogenic, whereas high-density lipoprotein (HDL) is anti-atherogenic. Lipids should be eventually stored in adipose tissues, but when targeted ectopically in arteries, arteriosclerosis (atherosclerosis) ensues. When stored in the liver and skeletal muscles, fatty liver and insulin resistance ensues, leading to the metabolic syndrome. Likewise, calcium-phosphate is adsorbed by fetuin-A and become CPPs. The activity of CPPs depends on not only their composition (whether or not they contain crystalline calcium-phosphate) but also their particle size (secondary CPPs are larger than primary CPPs). Calcium-phosphate should be eventually stored in the bone, but when targeted ectopically in arteries, arteriosclerosis (vascular stiffness and calcification) ensues. When targeted in white blood cells, non-infectious chronic inflammation is induced, which may accelerate ageing (Table 2) [8,61]. Although research on CPPs is still in its infancy, we expect that CPPs may become a subject of study as important as lipoproteins in the future.
Table 2. Colloids in the blood.
| Insoluble materials | Lipids | Calcium-phosphate |
|---|---|---|
| Protein | Apoprotein | Fetuin-A |
| Colloid | Lipoprotein | CPP |
| Receptor | LDL receptor | Toll-like receptor-4 Scavenger receptor-A |
| Storage | Fat | Bone |
| Pathology | Arteriosclerosis (athelosclerosis) Metabolic syndrome | Arteriosclerosis (vascular calcification) Ageing |
CPPs were reported to bind not only to TLR4 but also to scavenger receptor-A. Modified from [8].
Abbreviations
- CKD
chronic kidney disease
- CPM
calciprotein monomer
- CPP
calciprotein particle
- HDL
high-density lipoprotein
- LDL
low-density lipoprotein
- RBA
receptor binding arm
Data Availability
All supporting data are included within the main article.
Competing Interests
The authors declare that there are no competing interests associated with the manuscript.
References
- 1.Wolfe-Simon F., Switzer Blum J., Kulp T.R., Gordon G.W., Hoeft S.E., Pett-Ridge J.et al. (2011) A bacterium that can grow by using arsenic instead of phosphorus. Science 332, 1163–1166 10.1126/science.1197258 [DOI] [PubMed] [Google Scholar]
- 2.Blair-West J.R., Denton D.A., McKinley M.J., Radden B.G., Ramshaw E.H. and Wark J.D. (1992) Behavioral and tissue responses to severe phosphorus depletion in cattle. Am. J. Physiol. 263, R656–R663 10.1152/ajpregu.1992.263.3.R656 [DOI] [PubMed] [Google Scholar]
- 3.Boaz M. and Smetana S. (1996) Regression equation predicts dietary phosphorus intake from estimate of dietary protein intake. J. Am. Diet. Assoc. 96, 1268–1270 10.1016/S0002-8223(96)00331-8 [DOI] [PubMed] [Google Scholar]
- 4.Cupisti A. and Kalantar-Zadeh K. (2013) Management of natural and added dietary phosphorus burden in kidney disease. Semin. Nephrol. 33, 180–190 10.1016/j.semnephrol.2012.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Uribarri J. (2009) Phosphorus additives in food and their effect in dialysis patients. Clin. J. Am. Soc. Nephrol.:CJASN 4, 1290–1292 10.2215/CJN.03950609 [DOI] [PubMed] [Google Scholar]
- 6.Akiyama K., Miura Y., Hayashi H., Sakata A., Matsumura Y., Kojima M.et al. (2019) Calciprotein particles regulate fibroblast growth factor-23 expression in osteoblasts. Kidney Int. 97, 702–712 10.1016/j.kint.2019.10.019 [DOI] [PubMed] [Google Scholar]
- 7.Brown E.M., Gamba G., Riccardi D., Lombardi M., Butters R., Kifor O.et al. (1993) Cloning and characterization of an extracellular Ca(2+)-sensing receptor from bovine parathyroid. Nature 366, 575–580 10.1038/366575a0 [DOI] [PubMed] [Google Scholar]
- 8.Kuro-o M. (2019) The Klotho proteins in health and disease. Nat. Rev. Nephrol. 15, 27–44 10.1038/s41581-018-0078-3 [DOI] [PubMed] [Google Scholar]
- 9.Shimada T., Hasegawa H., Yamazaki Y., Muto T., Hino R., Takeuchi Y.et al. (2004) FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J. Bone Miner. Res. 19, 429–435 10.1359/JBMR.0301264 [DOI] [PubMed] [Google Scholar]
- 10.Haut L.L., Alfrey A.C., Guggenheim S., Buddington B. and Schrier N. (1980) Renal toxicity of phosphate in rats. Kidney Int. 17, 722–731 10.1038/ki.1980.85 [DOI] [PubMed] [Google Scholar]
- 11.Hirano Y., Kurosu H., Shiizaki K., Iwazu Y., Tsuruoka S. and Kuro-o M. (2020) Interleukin-36alpha as a potential biomarker for renal tubular damage induced by dietary phosphate load. FEBS Open Bio. 10, 894–903 10.1002/2211-5463.12845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shiizaki K., Tsubouchi A., Miura Y., Seo K., Kuchimaru T., Hayashi H.et al. (2021) Calcium phosphate microcrystals in the renal tubular fluid accelerate chronic kidney disease progression. J. Clin. Invest. 10.1172/JCI145693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mathewson A.M., Fouque D. and Toft A.J. (2010) Dietary phosphate assessment in dialysis patients. J. Ren. Nutr. 20, 351–358 10.1053/j.jrn.2010.05.014 [DOI] [PubMed] [Google Scholar]
- 14.Denic A., Lieske J.C., Chakkera H.A., Poggio E.D., Alexander M.P., Singh P.et al. (2016) The substantial loss of nephrons in healthy human kidneys with aging. J. Am. Soc. Nephrol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou X.J., Rakheja D., Yu X., Saxena R., Vaziri N.D. and Silva F.G. (2008) The aging kidney. Kidney Int. 74, 710–720 10.1038/ki.2008.319 [DOI] [PubMed] [Google Scholar]
- 16.Schmitt R. and Melk A. (2017) Molecular mechanisms of renal aging. Kidney Int. 92, 569–579 10.1016/j.kint.2017.02.036 [DOI] [PubMed] [Google Scholar]
- 17.Cullen-McEwen L.A., Kett M.M., Dowling J., Anderson W.P. and Bertram J.F. (2003) Nephron number, renal function, and arterial pressure in aged GDNF heterozygous mice. Hypertension 41, 335–340 10.1161/01.HYP.0000050961.70182.56 [DOI] [PubMed] [Google Scholar]
- 18.Kunishige R., Mizoguchi M., Tsubouchi A., Hanaoka K., Miura Y., Kurosu H.et al. (2020) Calciprotein particle-induced cytotoxicity via lysosomal dysfunction and altered cholesterol distribution in renal epithelial HK-2 cells. Sci. Rep. 10, 20125 10.1038/s41598-020-77308-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blaine J., Chonchol M. and Levi M. (2015) Renal control of calcium, phosphate, and magnesium homeostasis. Clin. J. Am. Soc. Nephrol.: CJASN 10, 1257–1272 10.2215/CJN.09750913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baum M. and Quigley R. (2005) Proximal tubule water transport-lessons from aquaporin knockout mice. Am. J. Physiol. Renal. Physiol. 289, F1193–F1194 10.1152/ajprenal.00283.2005 [DOI] [PubMed] [Google Scholar]
- 21.Bank N., Su W.S. and Aynedjian H.S. (1978) A micropuncture study of renal phosphate transport in rats with chronic renal failure and secondary hyperparathyroidism. J. Clin. Invest. 61, 884–894 10.1172/JCI109014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Devaraj S., Duncan-Staley C. and Jialal I. (2010) Evaluation of a method for fibroblast growth factor-23: a novel biomarker of adverse outcomes in patients with renal disease. Metab. Syndr. Relat. Disord. 8, 477–482 10.1089/met.2010.0030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Isakova T., Wahl P., Vargas G.S., Gutierrez O.M., Scialla J., Xie H.et al. (2011) Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 79, 1370–1378 10.1038/ki.2011.47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Webster A.C., Nagler E.V., Morton R.L. and Masson P. (2017) Chronic kidney disease. Lancet 389, 1238–1252 10.1016/S0140-6736(16)32064-5 [DOI] [PubMed] [Google Scholar]
- 25.Hill N.R., Fatoba S.T., Oke J.L., Hirst J.A., O'Callaghan C.A., Lasserson D.S.et al. (2016) Global prevalence of chronic kidney disease - a systematic review and meta-analysis. PLoS ONE 11, e0158765 10.1371/journal.pone.0158765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Levey A.S., Eckardt K.U., Tsukamoto Y., Levin A., Coresh J., Rossert J.et al. (2005) Definition and classification of chronic kidney disease: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. 67, 2089–2100 10.1111/j.1523-1755.2005.00365.x [DOI] [PubMed] [Google Scholar]
- 27.Slatopolsky E., Brown A. and Dusso A. (1999) Pathogenesis of secondary hyperparathyroidism. Kidney Int. Suppl. 73, S14–S19 10.1046/j.1523-1755.1999.07304.x [DOI] [PubMed] [Google Scholar]
- 28.Stevens L.A., Li S., Wang C., Huang C., Becker B.N., Bomback A.S.et al. (2010) Prevalence of CKD and comorbid illness in elderly patients in the United States: results from the Kidney Early Evaluation Program (KEEP). Am. J. Kidney Dis. 55, S23–S33 10.1053/j.ajkd.2009.09.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pazianas M. and Miller P.D. (2020) Current Understanding of Mineral and Bone Disorders of Chronic Kidney Disease and the Scientific Grounds on the Use of Exogenous Parathyroid Hormone in Its Management. J. Bone Metab. 27, 1–13 10.11005/jbm.2020.27.1.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ketteler M. and Biggar P.H. (2013) Use of phosphate binders in chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 22, 413–420 10.1097/MNH.0b013e32836214d4 [DOI] [PubMed] [Google Scholar]
- 31.Kuro-o M. (2013) Klotho, phosphate and FGF-23 in ageing and disturbed mineral metabolism. Nat. Rev. Nephrol. 9, 650–660 10.1038/nrneph.2013.111 [DOI] [PubMed] [Google Scholar]
- 32.Vervloet M.G., Sezer S., Massy Z.A., Johansson L., Cozzolino M. and Fouque D. (2017) The role of phosphate in kidney disease. Nat. Rev. Nephrol. 13, 27–38 10.1038/nrneph.2016.164 [DOI] [PubMed] [Google Scholar]
- 33.Fouque D., Horne R., Cozzolino M. and Kalantar-Zadeh K. (2014) Balancing nutrition and serum phosphorus in maintenance dialysis. Am. J. Kidney Dis. 64, 143–150 10.1053/j.ajkd.2014.01.429 [DOI] [PubMed] [Google Scholar]
- 34.Raun A., Cheng E. and Burroughs W. (1956) Ruminant nutrition, phytate phosphorus hydrolysis and availability to rumen microorganisms. J. Agric. Food Chem. 4, 869–871 10.1021/jf60068a006 [DOI] [Google Scholar]
- 35.Williams C., Ronco C. and Kotanko P. (2013) Whole grains in the renal diet–is it time to reevaluate their role? Blood. Purif. 36, 210–214 10.1159/000356683 [DOI] [PubMed] [Google Scholar]
- 36.Mullaney E.J., Daly C.B. and Ullah A.H. (2000) Advances in phytase research. Adv. Appl. Microbiol. 47, 157–199 10.1016/S0065-2164(00)47004-8 [DOI] [PubMed] [Google Scholar]
- 37.Moe S.M., Zidehsarai M.P., Chambers M.A., Jackman L.A., Radcliffe J.S., Trevino L.L.et al. (2011) Vegetarian compared with meat dietary protein source and phosphorus homeostasis in chronic kidney disease. Clin. J. Am. Soc. Nephrol.: CJASN 6, 257–264 10.2215/CJN.05040610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sullivan C., Sayre S.S., Leon J.B., Machekano R., Love T.E., Porter D.et al. (2009) Effect of food additives on hyperphosphatemia among patients with end-stage renal disease: a randomized controlled trial. JAMA 301, 629–635 10.1001/jama.2009.96 [DOI] [PubMed] [Google Scholar]
- 39.Gutiérrez O.M., Anderson C., Isakova T., Scialla J., Negrea L., Anderson A.H.et al. (2010) Low Socioeconomic Status Associates with Higher Serum Phosphate Irrespective of Race. J. Am. Soc. Nephrol. 21, 1953 10.1681/ASN.2010020221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McClelland R., Christensen K., Mohammed S., McGuinness D., Cooney J., Bakshi A.et al. (2016) Accelerated ageing and renal dysfunction links lower socioeconomic status and dietary phosphate intake. Aging (Albany NY) 8, 1135–1149 10.18632/aging.100948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuro-o M., Matsumura Y., Aizawa H., Kawaguchi H., Suga T., Utsugi T.et al. (1997) Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 390, 45–51 10.1038/36285 [DOI] [PubMed] [Google Scholar]
- 42.Hu M.C., Shi M., Zhang J., Pastor J., Nakatani T., Lanske B.et al. (2010) Klotho: a novel phosphaturic substance acting as an autocrine enzyme in the renal proximal tubule. FASEB J. 24, 3438–3450 10.1096/fj.10-154765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cha S.K., Ortega B., Kurosu H., Rosenblatt K.P., Kuro-o M. and Huang C.L. (2008) Removal of sialic acid involving Klotho causes cell-surface retention of TRPV5 channel via binding to galectin-1. Proc. Natl. Acad. Sci. U.S.A. 105, 9805–9810 10.1073/pnas.0803223105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chang Q., Hoefs S., van der Kemp A.W., Topala C.N., Bindels R.J. and Hoenderop J.G. (2005) The beta-glucuronidase klotho hydrolyzes and activates the TRPV5 channel. Science 310, 490–493 10.1126/science.1114245 [DOI] [PubMed] [Google Scholar]
- 45.Chen G., Liu Y., Goetz R., Fu L., Jayaraman S., Hu M.C.et al. (2018) alpha-Klotho is a non-enzymatic molecular scaffold for FGF23 hormone signalling. Nature 553, 461–466 10.1038/nature25451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhong X., Jagarlapudi S., Weng Y., Ly M., Rouse J.C., McClure K.et al. (2020) Structure-function relationships of the soluble form of the antiaging protein Klotho have therapeutic implications for managing kidney disease. J. Biol. Chem. 295, 3115–3133 10.1074/jbc.RA119.012144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shimada T., Kakitani M., Yamazaki Y., Hasegawa H., Takeuchi Y., Fujita T.et al. (2004) Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J. Clin. Invest. 113, 561–568 10.1172/JCI200419081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kurosu H., Ogawa Y., Miyoshi M., Yamamoto M., Nandi A., Rosenblatt K.P.et al. (2006) Regulation of fibroblast growth factor-23 signaling by klotho. J. Biol. Chem. 281, 6120–6123 10.1074/jbc.C500457200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu X., Ibrahimi O.A., Goetz R., Zhang F., Davis S.I., Garringer H.J.et al. (2005) Analysis of the biochemical mechanisms for the endocrine actions of fibroblast growth factor-23. Endocrinology 146, 4647–4656 10.1210/en.2005-0670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Urakawa I., Yamazaki Y., Shimada T., Iijima K., Hasegawa H., Okawa K.et al. (2006) Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 444, 770–774 10.1038/nature05315 [DOI] [PubMed] [Google Scholar]
- 51.Kuro-o M. (2018) Ageing-related receptors resolved. Nature 553, 409–410 10.1038/d41586-017-09032-4 [DOI] [PubMed] [Google Scholar]
- 52.Stubbs J.R., Liu S., Tang W., Zhou J., Wang Y., Yao X.et al. (2007) Role of hyperphosphatemia and 1,25-dihydroxyvitamin D in vascular calcification and mortality in fibroblastic growth factor 23 null mice. J. Am. Soc. Nephrol. 18, 2116–2124 10.1681/ASN.2006121385 [DOI] [PubMed] [Google Scholar]
- 53.Chen K., Wang S., Sun Q.W., Zhang B., Ullah M.F. and Sun Z. (2020) Klotho Deficiency Causes Heart Aging via Impairing the Nrf2-GR Pathway. Circ. Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kuro-o M. (2010) A potential link between phosphate and aging–lessons from Klotho-deficient mice. Mech. Ageing Dev. 131, 270–275 10.1016/j.mad.2010.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stenvinkel P., Painer J., Kuro-o M., Lanaspa M., Arnold W., Ruf T.et al. (2018) Novel treatment strategies for chronic kidney disease: insights from the animal kingdom. Nat. Rev. Nephrol. 14, 265–284 10.1038/nrneph.2017.169 [DOI] [PubMed] [Google Scholar]
- 56.Kuro-o M. and Moe O.W. (2016) FGF23-alphaKlotho as a paradigm for a kidney-bone network. Bone 100, 4–18 10.1016/j.bone.2016.11.013 [DOI] [PubMed] [Google Scholar]
- 57.Heiss A., DuChesne A., Denecke B., Grotzinger J., Yamamoto K., Renne T.et al. (2003) Structural basis of calcification inhibition by alpha 2-HS glycoprotein/fetuin-A. Formation of colloidal calciprotein particles. J. Biol. Chem. 278, 13333–13341 10.1074/jbc.M210868200 [DOI] [PubMed] [Google Scholar]
- 58.Jahnen-Dechent W., Büscher A., Köppert S., Heiss A., Kuro-o M. and Smith E.R. (2020) Mud in the blood the role of protein-mineral complexes and extracellular vesicles in biomineralisation and calcification. J. Struct. Biol. 212, 107577 10.1016/j.jsb.2020.107577 [DOI] [PubMed] [Google Scholar]
- 59.Jahnen-Dechent W., Heiss A., Schafer C. and Ketteler M. (2011) Fetuin-A regulation of calcified matrix metabolism. Circ. Res. 108, 1494–1509 10.1161/CIRCRESAHA.110.234260 [DOI] [PubMed] [Google Scholar]
- 60.Kuro-o M. (2018) Klotho and endocrine fibroblast growth factors: marker of chronic kidney disease progression and cardiovascular complications? Nephrol. Dial. Transplant. 34, 15–21 10.1093/ndt/gfy126 [DOI] [PubMed] [Google Scholar]
- 61.Kuro-o M. (2020) Phosphate as a pathogen of arteriosclerosis and aging. J. Atheroscler. Thromb. 28, 203–213 10.5551/jat.RV17045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pasch A., Farese S., Graber S., Wald J., Richtering W., Floege J.et al. (2012) Nanoparticle-based test measures overall propensity for calcification in serum. J. Am. Soc. Nephrol. 23, 1744–1752 10.1681/ASN.2012030240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rodriguez-Ortiz M.E., Lopez I., Munoz-Castaneda J.R., Martinez-Moreno J.M., Ramirez A.P., Pineda C.et al. (2012) Calcium deficiency reduces circulating levels of FGF23. J. Am. Soc. Nephrol. 23, 1190–1197 10.1681/ASN.2011101006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Quinn S.J., Thomsen A.R., Pang J.L., Kantham L., Brauner-Osborne H., Pollak M.et al. (2013) Interactions between calcium and phosphorus in the regulation of the production of fibroblast growth factor 23 in vivo. Am. J. Physiol. Endocrinol. Metab. 304, E310–E320 10.1152/ajpendo.00460.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ewence A.E., Bootman M., Roderick H.L., Skepper J.N., McCarthy G., Epple M.et al. (2008) Calcium phosphate crystals induce cell death in human vascular smooth muscle cells: a potential mechanism in atherosclerotic plaque destabilization. Circ. Res. 103, e28–e34 10.1161/CIRCRESAHA.108.181305 [DOI] [PubMed] [Google Scholar]
- 66.Sage A.P., Lu J., Tintut Y. and Demer L.L. (2011) Hyperphosphatemia-induced nanocrystals upregulate the expression of bone morphogenetic protein-2 and osteopontin genes in mouse smooth muscle cells in vitro. Kidney Int. 79, 414–422 10.1038/ki.2010.390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Reynolds J.L., Joannides A.J., Skepper J.N., McNair R., Schurgers L.J., Proudfoot D.et al. (2004) Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: a potential mechanism for accelerated vascular calcification in ESRD. J. Am. Soc. Nephrol. 15, 2857–2867 10.1097/01.ASN.0000141960.01035.28 [DOI] [PubMed] [Google Scholar]
- 68.Villa-Bellosta R., Bogaert Y.E., Levi M. and Sorribas V. (2007) Characterization of phosphate transport in rat vascular smooth muscle cells: implications for vascular calcification. Arterioscler. Thromb. Vasc. Biol. 27, 1030–1036 10.1161/ATVBAHA.106.132266 [DOI] [PubMed] [Google Scholar]
- 69.Villa-Bellosta R. and Sorribas V. (2009) Phosphonoformic acid prevents vascular smooth muscle cell calcification by inhibiting calcium-phosphate deposition. Arterioscler. Thromb. Vasc. Biol. 29, 761–766 10.1161/ATVBAHA.108.183384 [DOI] [PubMed] [Google Scholar]
- 70.Hamano T., Matsui I., Mikami S., Tomida K., Fujii N., Imai E.et al. (2010) Fetuin-mineral complex reflects extraosseous calcification stress in CKD. J. Am. Soc. Nephrol. 21, 1998–2007 10.1681/ASN.2009090944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Smith E.R., Ford M.L., Tomlinson L.A., Rajkumar C., McMahon L.P. and Holt S.G. (2012) Phosphorylated fetuin-A-containing calciprotein particles are associated with aortic stiffness and a procalcific milieu in patients with pre-dialysis CKD. Nephrol. Dial. Transplant. 27, 1957–1966 10.1093/ndt/gfr609 [DOI] [PubMed] [Google Scholar]
- 72.Nakazato J., Hoshide S., Wake M., Miura Y., Kuro-o M. and Kario K. (2019) Association of calciprotein particles measured by a new method with coronary artery plaque in patients with coronary artery disease: a cross-sectional study. J. Cardiol. 74, 428–435 10.1016/j.jjcc.2019.04.008 [DOI] [PubMed] [Google Scholar]
- 73.Miura Y., Iwazu Y., Shiizaki K., Akimoto T., Kotani K., Kurabayashi M.et al. (2018) Identification and quantification of plasma calciprotein particles with distinct physical properties in patients with chronic kidney disease. Sci. Rep. 8, 1256 10.1038/s41598-018-19677-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All supporting data are included within the main article.