

Summary
Oxidative stress is a ubiquitous cellular challenge implicated in aging, neurodegeneration, and cancer. By studying pathogenic mutations in the tumor suppressor BRCA2, we identify a general mechanism by which oxidative stress restricts mitochondrial (mt)DNA replication. BRCA2 inactivation induces R-loop accumulation in the mtDNA regulatory region and diminishes mtDNA replication initiation. In BRCA2-deficient cells, intracellular reactive oxygen species (ROS) are elevated, and ROS scavengers suppress the mtDNA defects. Conversely, wild-type cells exposed to oxidative stress by pharmacologic or genetic manipulation phenocopy these defects. Mechanistically, we find that 8-oxoguanine accumulation in mtDNA caused by oxidative stress suffices to impair recruitment of the mitochondrial enzyme RNaseH1 to sites of R-loop accrual, restricting mtDNA replication initiation. Thus, oxidative stress impairs RNaseH1 function to cripple mtDNA maintenance. Our findings highlight a molecular mechanism that links oxidative stress to mitochondrial dysfunction and is elicited by the inactivation of genes implicated in neurodegeneration and cancer.
Keywords: BRCA2, oxidative stress, RNaseH1, mitochondrial DNA replication, R-loops, SETX, PRPF8, cancer, neurodegeneration
Graphical abstract
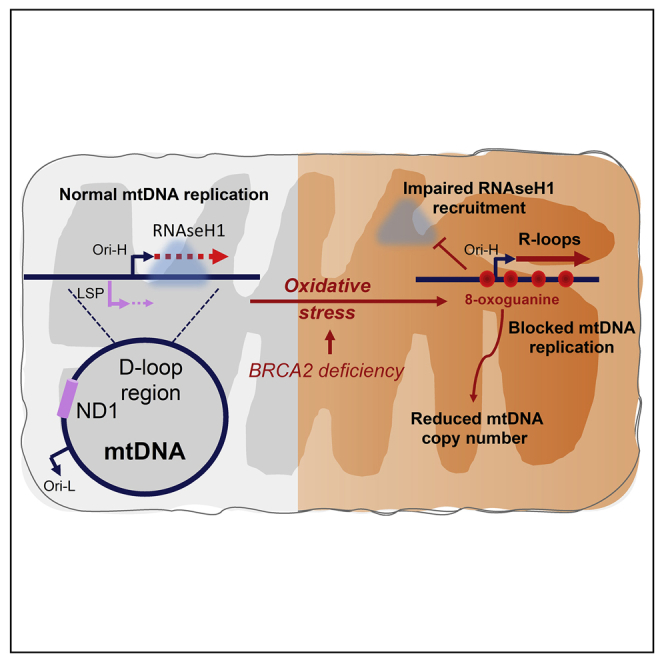
Highlights
-
•
BRCA2-deficient cells accumulate mtDNA R-loops due to oxidative stress
-
•
This stress creates 8-oxoguanine lesions impairing RNaseH1 recruitment to mtDNA
-
•
RNaseH1 impairment triggers R-loop formation and restricts mtDNA replication
-
•
Other sources of oxidative stress also cripple mtDNA maintenance via this mechanism
Oxidative stress ubiquitously challenges cellular homeostasis. Renaudin et al. show that oxidative stress impairs RNase H1 recruitment via 8-oxoguanine accumulation, eliciting unscheduled RNA-DNA hybrids (R-loops) that block mtDNA replication. These findings define the mechanism underlying a specific molecular consequence of oxidative stress, with links to aging-related human diseases.
Introduction
Inherited germline mutations affecting a single allele of the BRCA2 tumor suppressor predispose to cancers of the breast, ovaries, pancreas, prostate, and other tissues (Breast Cancer Linkage, 1999). BRCA2-deficient cells exhibit profound instability of the nuclear genome (reviewed in Venkitaraman, 2014), which has been ascribed to the loss of essential BRCA2 functions in the control of RAD51 activity during DNA repair by homologous recombination (Davies et al., 2001; Pellegrini et al., 2002), the stabilization of stalled DNA replication forks (Lomonosov et al., 2003; Schlacher et al., 2011), or the accurate segregation of chromosomes during mitosis (Choi et al., 2012; Daniels et al., 2004; Mondal et al., 2012). Recently, we and others have shown that BRCA2-deficient cells accumulate unscheduled RNA-DNA hybrids (R-loops) throughout the nuclear genome (Bhatia et al., 2014; Shivji et al., 2018) via the loss of a function for BRCA2 in promoting the release of RNA polymerase II from promoter-proximal pausing sites adjoining the transcription-start sequences of actively transcribed genes (Shivji et al., 2018). R-loop formation has not only been implicated in the regulation of transcription elongation and termination in the nuclear genome (Crossley et al., 2019) but has also been detected in the mitochondrial genome (Lee and Clayton, 1996; Wanrooij et al., 2012), where its functional role is incompletely understood.
Transcription and replication in mitochondrial DNA (mtDNA) (Anderson, 1981), an intron-free circle of 16.6 kb, are regulated via sequence motifs located in non-coding region (NCR) (or D-loop region). In this region, two heavy-strand promotors (HSP1 and HSP2) and a light-strand promotor (LSP) control mtDNA transcription (Gustafsson et al., 2016; Ojala et al., 1981), whereas mtDNA replication initiates at the replication origin of the heavy strand (OriH) adjacent to a guanine-rich, conserved sequence block (CSB-)II (Pham et al., 2006). mtDNA replication initiation at the OriH by polymerase gamma (POLG) (Holt et al., 2000; Yang et al., 2002; Yasukawa et al., 2006) is primed by a small 7S RNA generated by the POLRMT enzyme (Gustafsson et al., 2016), which may form an R-loop structure with cognate mtDNA to regulate this process (Holt, 2019). Collectively, these considerations prompted us to examine whether BRCA2 deficiency perturbs mitochondrial genome maintenance.
Here, we report that BRCA2 inactivation induces R-loops in the regulatory non-coding region of mtDNA, accompanied by diminished mtDNA replication and mtDNA deletions typical of human cancers. Strikingly, the exposure of wild-type cells to oxidative stress suffices to phenocopy these mitochondrial anomalies. Conversely, in BRCA2-deficient cells, intracellular reactive oxygen species (ROS) are elevated and ROS scavengers suppress mitochondrial defects. Our results identify a mechanism underlying these events. We show that BRCA2 deficiency as well as oxidative stress impair the recruitment of the mitochondrial R-loop processing enzyme, RNaseH1, to sites of R-loop formation in mtDNA. Together, our findings demonstrate that oxidative stress cripples mitochondrial genome maintenance via unscheduled R-loop accrual.
Besides cancer, oxidative stress and mitochondrial defects also accompany many human neurodegenerative disorders (Islam, 2017). Indeed, we find that depletion of the RNA processing factors SETX or PRPF8, which are mutated in amyotrophic lateral sclerosis, ataxia-oculomotor apraxia, and retinitis pigmentosa (Moreira et al., 2004; Skourti-Stathaki et al., 2011; Xu et al., 2018), is enough to induce mtDNA R-loop accumulation and endogenous oxidative stress. Thus, our findings not only reveal unrecognized cellular functions of BRCA2 relevant to carcinogenesis but also highlight a mechanism that may be implicated more widely in neurodegeneration and cancer.
Results
BRCA2 inactivation induces R-loop accumulation in the non-coding regulatory region of mtDNA
Recent evidence shows that BRCA2 deficiency causes unscheduled R-loop accumulation in the nuclear genome (Bhatia et al., 2014; Shivji et al., 2018). Because R-loops have also been implicated in mitochondrial genome maintenance, we tested in multiple experimental models whether BRCA2 deficiency caused their accumulation in mtDNA. R-loop accumulation detected by the S9.6 antibody specific to R-loop structures (Ginno et al., 2012) increases in cytosolic puncta reminiscent of mitochondria detected in patient-derived, BRCA2-deficient EUFA423 cells, when compared to their isogenic EUFA423-B2 counterparts reconstituted with full-length BRCA2 (Figure 1A). Cytosolic S9.6 staining co-localizes with the MitoTracker dye, confirming that it decorates mitochondria (Figures 1A and S1A), and is reduced by treatment with RNaseH, an enzyme that specifically cleaves RNA-DNA hybrids, but not with RNase III and T1 that cleaved double-stranded and single-stranded RNA, respectively, confirming its specificity (Figure S1B). DNA-RNA immunoprecipitation (DRIP) analyses using S9.6 antibody (Ginno et al., 2012) to survey R-loop formation across the mitochondrial genome demonstrate a specific increase at the D-loop region, which is not evident in mitochondrial protein-coding genes (ND1) or within the light-strand replication origin, OriL (Figure 1B). A similar increase in R-loop formation occurs selectively in the D-loop region of mtDNA derived from HeLa cells depleted of BRCA2 using short interfering (si)RNAs (Figures 1C–1E), when compared to isogenic controls. Three different Brca2-deficient cancer cell lines from a genetically engineered murine model (GEMM) for familial pancreatic ductal adenocarcinoma (Skoulidis et al., 2010) exhibit similar abnormalities (Figure S1C) when compared to pancreatic ductal adenocarcinoma cell lines from an otherwise isogenic GEMM bearing wild-type Brca2 (Skoulidis et al., 2010). Together, these results confirm that BRCA2 inactivation by RNA interference or gene targeting in human cells, as well as by cancer-causing mutations in human or murine cells, selectively induces R-loop accumulation in the non-coding D-loop region of mtDNA.
Figure 1.

R-loops accumulate in the D-loop region of mtDNA in cells lacking BRCA2
(A) Immunofluorescence detection of R-loops with S9.6 antibody in BRCA2-deficient EUFA423 cells or EUFA423-B2 controls complemented with wild-type BRCA2. Plot shows the mean ± SD from three independent experiments. At least 70 cells were counted in each condition. The two-tailed Student’s t test was performed to determine statistical significance between the two groups. ∗p < 0.05. Scale bars, 10 μm; DAPI (4,6-diamidino-2-phenylindole) stains DNA, MitoTracker stains mitochondria. Magnification of one cell indicated by a yellow square is shown in Figure S1A.
(B) DRIP analysis with S9.6 antibody in EUFA423-B2 and EUFA423 cells. RNaseH1 treatment serves as a control for antibody specificity. Plots depict the mean ± SD from three independent experiments. The two-way ANOVA test was performed for all pairs to determine statistical significance. Statistically significant differences are indicated by ∗p < 0.05.
(C) Western blot showing BRCA2 depletion using specific (si)RNA (siBRCA2) for 72 h, compared to control (siCtrl). β-actin is the loading control.
(D) Immunofluorescence detection of R-loops with S9.6 antibody in HeLa Kyoto cells treated either with siCtrl or siBRCA2. Plot shows the mean ± SD from three independent experiments. At least 70 cells were counted in each condition. The two-tailed Student’s t test was performed to determine statistical significance between the two groups. ∗p < 0.05. Scale bars, 10 μm.
(E) DRIP analysis in HeLa Kyoto cells transfected either with siCtrl or siBRCA2. DRIP was performed 72 h after transfection and depicted as described in the preceding panels. Plots depict the mean ± SD from three independent experiments. Statistically significant differences are indicated ∗∗p < 0.01.
BRCA2 inactivation suppresses mtDNA replication
We detected a marked reduction in the incorporation of the nucleotide analog, EdU, into mtDNA following the depletion of BRCA2 from HeLa cells using siRNA (Figure 2A) or in CRISPR-Cas9-engineered BRCA2 nullizygous counterparts (Figures S2A and 2B) compared to parental HeLa Kyoto cells, speaking to a defect in mtDNA replication. We therefore tested the effect of BRCA2 deficiency on mtDNA recovery after chronic treatment with ethidium bromide (EtBr) for 4 days, which acutely diminishes mtDNA copy number in both parental HeLa Kyoto cells and their CRISPR-Cas9-engineered BRCA2 nullizygous counterparts (Figure 2C). In sharp contrast, however, the recovery of mtDNA copy number after EtBr washout was significantly slower in BRCA2-deficient cells than in wild-type controls (Figure 2C), confirming that BRCA2 inactivation suppresses mtDNA replication.
Figure 2.

BRCA2 deficiency diminishes mtDNA replication
(A and B) Measurement of mtDNA replication by EdU incorporation and “click-it” chemistry. Magnified views of the boxed regions 1,2 are shown below in separate panels. HeLa cells transfected with the indicated siRNA (A) or BRCA2-KO cells and their parental counterpart (B) were incubated with 6 μM aphidicolin for 4 h prior to incubation with EdU for 2 h. Plots show the mean ± SD from three independent experiments. At least 50 cells were counted in each experiment. The two-tailed Student’s t test was performed to determine statistical significance between the two groups. ∗∗p < 0.01. Scale bars, 10 μm.
(C) mtDNA content recovery measured by qPCR in HeLa Kyoto cells or their CRISPR-Cas9-engineered BRCA2 nullizygous counterparts (B2-KO), after ethidium bromide depletion for 4 days. The graph shows the mean ± SD from three independent experiments. The two-tailed Student’s t test was performed to determine statistical significance between the two groups at the latest time point. ∗p < 0.05.
(D) Fold change in the level of RNA transcripts emanating from the D-loop region measured by qPCR in HeLa Kyoto transfected with (si)RNA for BRCA2 or in EUFA423 cells compared with EUFA423 B2 controls expressing wild-type BRCA2. Plots show the relative mean ± SD from three independent experiments. The two- tailed Student’s t test was performed to determine statistical significance between the two groups. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
(E) mtDNA copy number variation in EUFA423 and its BRCA2-complemented counterpart, EUFA423_B2, in HeLa cells transfected with the indicated siRNA or in BRCA2 KO cells and the parental cell line. Copy number was assessed by qPCR and normalized using the nuclear gene GAPDH. Plots show the relative mean ± SD from three independent experiments. The two-tailed Student’s t test was performed to determine statistical significance between the two groups. ∗p < 0.05.
Nascent transcripts from the non-coding LSP motif usually serve as a primer for the initiation of mtDNA replication and accumulate in the D-loop region when replication initiation fails (Jiang et al., 2019). We therefore monitored their formation by a quantitative polymerase chain reaction (qPCR). In both HeLa cells depleted of BRCA2 using (si)RNA or in BRCA2-deficient EUFA423 cells, there was a marked increase in nascent transcripts in the mtDNA D-loop region when compared to controls (Figure 2D). By contrast, BRCA2 deficiency caused a decrease in the transcription of mitochondrially encoded genes, such as COX or ND1 (Figures S2B and S2C), indicating that the accumulation of nascent LSP-derived transcripts was not simply the result of increased mtDNA gene transcription.
These results prompted us to test whether the mitochondrial copy number was altered in BRCA2-deficient cells. Indeed, the relative amount of mtDNA measured by qPCR decreased in all three model systems, indicating a significant reduction in mtDNA copy number (Figure 2E). Taken together, our observations suggest that BRCA2 inactivation blocks the initiation of mtDNA replication to cripple mtDNA maintenance.
Oxidative stress in wild-type cells phenocopies the mitochondrial abnormalities provoked by BRCA2 deficiency
Oxidative stress has been implicated in both the genesis of R-loops in nuclear DNA (Teng et al., 2018), as well as in defective mtDNA replication (Pastukh et al., 2016). These connections prompted us to examine whether wild-type cells exposed to ROS accumulate mitochondrial R-loops. Indeed, treatment of wild-type HeLa cells with hydrogen peroxide (H2O2) increases mitochondrial R-loop formation detected by immunofluorescence (Figure 3A) or by DRIP (Figure 3B). H2O2-induced R-loop formation in wild-type cells is most marked in the D-loop region of mtDNA—just as it is in BRCA2-deficient cells (Figure 3B). Similarly, treatment of wild-type cells with etomoxir, a known inhibitor of carnitine palmitoyltransferase 1A (CPT1α) that at a high concentration increases intracellular ROS (Figure 3C), also triggers mtDNA R-loop accumulation (Figure 3D). Thus, oxidative stress in wild-type cells phenocopies the mitochondrial abnormalities provoked by BRCA2 deficiency, raising the possibility that defective mtDNA maintenance in BRCA2-deficient cells may arise from, or be exacerbated by, endogenous oxidative stress.
Figure 3.

Oxidative stress in wild-type cells phenocopies the mitochondrial abnormalities provoked by BRCA2 deficiency
(A) Immunofluorescence detection of R-loops with S9.6 antibody in HeLa Kyoto after treatment where indicated with 1mM H2O2 for 16 h. The plot shows the mean ± SD from three independent experiments. At least 80 cells were counted in each condition. Magnifications show details of selected cells as indicated. The two-tailed Student’s t test was performed to determine statistical significance between the two groups. ∗∗p < 0.01. Scale bars, 10 μm.
(B) DRIP analyses in HeLa Kyoto cells after treatment where indicated with 1 mM H2O2 for 16 h. DRIP was performed and depicted as described in Figure 1. The plot depicts the mean ± SD from three independent experiments. Statistically significant differences are indicated. ∗p < 0.05 and ∗∗∗p < 0.001.
(C) Bar graph showing relative ROS levels in HeLa Kyoto cells after 50 μM etomoxir treatment for 16 h. Intracellular ROS was measured using 5 μM of CM-H2DCFDA probe, incubated for 15 min. The plot shows the mean ± SD from three independent experiments. Magnifications show details of selected cells as indicated. The two-tailed Student’s t test was performed to determine statistical significance between the two groups. ∗∗∗p < 0.001.
(D) Immunofluorescence detection of R-loops with S9.6 antibody in HeLa Kyoto after treatment where indicated with 50 μM etomoxir for 16 h. Scale bars, 10 μm.
BRCA2 inactivation induces endogenous oxidative stress to promote R-loop accumulation and cripple mtDNA maintenance
Potential links between BRCA2 and the accumulation of ROS have previously been reported. Partner and localizer of BRCA2 (PALB2), a protein that binds directly to BRCA2 to control its intracellular localization (Xia et al., 2006), also engages Kelch-like ECH-associated protein 1 (KEAP1), an oxidative stress sensor for the master antioxidant transcription factor nuclear factor erythroid 2-related factor 2 (NRF2) (Ma et al., 2012). PALB2 depletion decreases the nuclear accumulation of NRF2, leading to an increase in cellular ROS (Ma et al., 2012). Mutant forms of PALB2 that lack binding to BRCA2 (or its partner, the breast cancer protein BRCA1) are active in promoting NRF2-dependent transcription (Ma et al., 2012). Nevertheless, BRCA1 is reported to regulate the expression of ROS-induced antioxidant genes via NRF2 (Bae et al., 2004; Gorrini et al., 2013), and BRCA1 deletion in murine or human cells induces ROS accumulation (Cao et al., 2007; Gorrini et al., 2013; Saha et al., 2009). By contrast, how BRCA2 deficiency affects cellular ROS accumulation remains unclear.
Interestingly, we observe that intracellular ROS formation detected using the 2’,7’ –dichlorofluorescin diacetate (DCF-DA) probe, a fluorescent probe measuring hydroxyl or peroxyl radicals and other ROS, is elevated in patient-derived, BRCA2 mutant EUFA423 cells when compared to their isogenic EUFA423-B2 counterparts reconstituted with BRCA2 or in HeLa cells depleted of BRCA2 using siRNA (Figure 4A). Similar results are obtained when ROS are measured with the MitoSOX probe, which detects ROS produced by mitochondria (Figure 4B). Moreover, treatment of BRCA2 mutant EUFA423 cells with the ROS scavenger N-acetyl cysteine (NAC) not only reduces intracellular ROS but also causes a decrease in the selective accumulation of R-loops in the non-coding D-loop region of mtDNA detected by DRIP using the S9.6 antibody (Figure 4C). NAC treatment causes a similar decrease in mtDNA R-loop formation in HeLa cells depleted of BRCA2 using siRNA (Figures 4D and S3A). Thus, our findings confirm that R-loop accumulation in mtDNA following BRCA2 inactivation may arise from, or be exacerbated by, endogenous oxidative stress.
Figure 4.

BRCA2 inactivation induces endogenous oxidative stress to trigger R-loop accumulation and diminish mtDNA replication
(A and B) Bar graph showing relative ROS levels in HeLa Kyoto cells depleted of BRCA2 with (si)RNA for 72 h (left) or in BRCA2-deficient EUFA423 cells compared to EUFA423-B2 controls complemented with wild-type BRCA2 (right). Intracellular ROS was measured either using 5 μM of CM-H2DCFDA probe incubated for 15 min (A) or with 5 μM MitoSOX probe incubated for 10 min (B). The plots show the mean ± SD from three independent experiments. The two-tailed Student’s t test was performed to determine statistical significance between the two groups. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
(C and D) DRIP analysis of the D-loop region in EUFA423 cells compared to EUFA423-B2 controls (C) or in HeLa Kyoto treated with siBRCA2 (D). R-loop digestion by RNaseH1 serves as a control for antibody specificity. Cells were treated where indicated with 2 mM of NAC for 16 h. Error bars indicate the mean ± SD from three independent experiments. The two-way ANOVA test was performed for all pairwise comparisons to determine statistical significance. Statistically significant differences are indicated. ∗p < 0.05.
(E) mtDNA content recovery, in the presence or absence of NAC treatment for 4 days, measured by qPCR in HeLa Kyoto cells or their CRISPR-Cas9-engineered BRCA2 nullizygous counterparts (B2-KO), after ethidium bromide (EtBr) depletion for 4 days. The graph shows the mean ± SD from three independent experiments. The two-tailed Student’s t test was performed to determine statistical significance between the two groups at the latest time point. ∗p < 0.05.
Moreover, we find that endogenous oxidative stress also underlies defective mtDNA replication in BRCA2-deficient cells. Thus, NAC exposure almost completely reverses the observed impairment of mtDNA replication in BRCA2-deficient cells after EtBr treatment and wash-out (Figure 4E).
Oxidative stress is associated with a recurrent form of mitochondrial genome instability termed the “mitochondrial common deletion” (Nie et al., 2016). We compared the relative level of these deletions in BRCA2-deficient versus wild-type cells. It should be emphasized that the high sensitivity of the nested PCR method we used means that signals marking the deletion may arise from just a few copies of mtDNA. Nevertheless, such mtDNA deletions appear to occur more frequently in untreated BRCA2-deficient EUFA423 cells compared to isogenic controls (Figures S3B and S3C) and can be suppressed by NAC treatment (Figures S3B and S3C).
BRCA2 inactivation impairs the recruitment of RNaseH1 to sites of R-loop formation in mtDNA
Multiple proteins may be involved in the processing of aberrant R-loops in mtDNA (Silva et al., 2018). From among them, we focused on the mitochondrial enzyme RNaseH1, which cleaves R-loops formed as intermediates during transcription or replication in mtDNA (Suzuki et al., 2010). Interestingly, RNaseH1 recruitment to the D-loop region was diminished in HeLa cells depleted of BRCA2 using siRNA, relative to controls (Figure 5A), or in BRCA2 mutant EUFA423 cells compared to EUFA423-B2 controls (Figure 5B). Similar changes occur in CRISPR-Cas9-engineered BRCA2 nullizygous cells (Figure S2A) compared to parental HeLa Kyoto counterparts (Figure 5C), although total RNaseH1 levels are unaffected (Figure S4). Importantly, NAC treatment partially reverses the observed impairment in RNaseH1 recruitment to the D-loop region of mtDNA (Figure 5C), suggesting that it is caused by oxidative stress following BRCA2 deficiency. Indeed, consistent with this conclusion, exposure of wild-type cells either to H2O2 or etomoxir is also sufficient to diminish RNaseH1 recruitment to the D-loop region (Figure 5D). Thus, collectively, our results show that oxidative stress impairs recruitment of the mitochondrial enzyme RNaseH1 to the regulatory region of mtDNA, suggesting a mechanism underlying R-loop accumulation at these sites.
Figure 5.

Impaired recruitment of RNaseH1 to sites of R-loop formation in mtDNA
(A–D) Chromatin immunoprecipitation (ChIP) analysis with anti-RNaseH1 antibody of the mitochondrial D-loop region; OriL or ND1 gene shows similar ChIP analyses in BRCA2-KO cells compared to the parental HeLa Kyoto cells.
(B) Similar ChIP analyses in BRCA2-deficient EUFA423 cells compared to EUFA423-B2 controls expressing wild-type BRCA2.
(A and C) HeLa Kyoto cells treated either with (si)Ctrl (C) or (si)BRCA2 (A) and with 2 mM NAC for 16 h.
(D) Similar analysis of HeLa Kyto cells treated with 1 mM H2O2 or with 50 μM etomoxir for 16 h. The fold change in ChIP signal relative to an immunoglobulin G (IgG) isotype control is plotted. Plots show the mean ± SD from three independent experiments. The two-way ANOVA test was performed to determine statistical significance. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
8-oxoguanine accumulation in mtDNA diminishes RNaseH1 recruitment at sites of R-loop accrual
Oxidative stress induces DNA damage in the form of 8-oxoguanine base adducts (reviewed in Cadet et al., 2010), which are normally recognized by the 8-oxoguanine glycosylase (OGG1) for base excision repair (Baiken et al., 2021; German et al., 2013). 8-oxoguanine accumulation has previously been reported to inhibit the recruitment of DNA-binding proteins to the nuclear and mitochondrial genomes (Ba and Boldogh, 2018). Accordingly, we tested the hypothesis that oxidative stress might impair RNaseH1 recruitment to mtDNA via 8-oxoguanine accumulation.
Consistent with this hypothesis, 8-oxoguanine is elevated in the mitochondria of BRCA2-deficent cells, similar to cells lacking OGG1 (Figures 6A, S5A, and S5B). Moreover, OGG1 depletion leads to increased R-loop formation in mtDNA (Figures 6B and 6C), phenocopying similar defects in BRCA2-deficient cells, even without elevating ROS (Figure 6D). Notably, the recruitment of RNaseH1 to mtDNA regulatory regions is reduced after OGG1 depletion (Figure 6E), recapitulating what we observe in BRCA2-deficient cells or in wild-type cells exposed to oxidative stress. Finally, OGG1 depletion reduces mtDNA copy number (Figure 6F). Taken collectively, our findings provide multiple lines of evidence to suggest that oxidative stress restricts mtDNA maintenance via an unrecognized molecular mechanism wherein 8-oxoguanine accumulation impairs the recruitment of RNaseH1 to the regulatory region of mtDNA, causing R-loop accrual and inhibiting mtDNA replication.
Figure 6.

OGG1 deficiency induces mtDNA R-loops and impaired RNaseH1 recruitment
(A) Immunofluorescence staining for 8-oxoguanine (red) and MitoTracker (green) in HeLa Kyoto wild-type (WT) cells or in HeLa Kyoto cells engineered by CRISPR-Cas9 to generate BRCA2 nullizygosity (BRCA2-KO). A lower magnification view is shown in Figure S5A. Scale bars, 10 μm.
(B) Immunofluorescence detection of R-loops with S9.6 antibody in HeLa Kyoto transfected with the indicated siRNA. Magnifications show details of selected cells as indicated. Scale bars, 10 μm; DAPI, MitoTracker stains mitochondria.
(C) DRIP analysis in HeLa Kyoto cells transfected either with siCtrl or siOGG1. DRIP was performed 72 h after transfection and depicted as described in the preceding figures. Error bars indicate the mean ± SD from three independent experiments. Statistically significant differences are indicated. ∗∗p < 0.01.
(D) Bar graph showing relative ROS levels in HeLa Kyoto cells transfected for 72 h with (si)OGG1 (F) or with (si)Ctrl. Intracellular ROS was measured as above using 5 μM of CM-H2DCFDA probe. The plot shows the mean ± SD from three independent experiments. No statistical differences were detected by the two-tailed Student’s t test.
(E) ChIP analysis with anti-RNaseH1 antibody of the mitochondrial D-loop region, OriL, or ND1 gene in HeLa Kyoto cells treated either with (si)Ctrl or (si)OGG1. The fold change in ChIP signal relative to an IgG isotype control is plotted. Plots show the mean ± SD from three independent experiments. The two-way ANOVA test was performed to determine statistical significance. ∗p < 0.05.
(F) mtDNA copy number variation in HeLa cells transfected with the indicated siRNA. Copy number was assessed by qPCR and normalized using the nuclear gene GAPDH. Plots show the relative mean ± SD from three independent experiments. The two-tailed Student’s t test was performed to determine statistical significance between the two groups. ∗∗p < 0.01.
Depletion of SETX or PRPF8 suffices to trigger oxidative stress and R-loop accumulation in mtDNA
Besides cancer, oxidative stress and mitochondrial dysfunction have been implicated in human neurodegenerative disorders (Islam, 2017). Interestingly, it has been reported that inactivation of human SETX, which encodes an RNA helicase essential for genome-wide R-loop processing (Skourti-Stathaki et al., 2011) that is mutated in amyotrophic lateral sclerosis and ataxia-oculomotor apraxia (Moreira et al., 2004), causes a spontaneous increase in oxidative damage to DNA (Suraweera et al., 2007). Moreover, yeast strains bearing mutant forms of the SETX homolog, SEN1p, exhibit elevated intracellular ROS levels accompanied by mitochondrial depletion (Sariki et al., 2016). Strikingly, we find that SETX depletion using RNAi in HeLa cells was sufficient not only to induce R-loop accumulation in the regulatory region of mtDNA (Figure 7A), accompanied by excess nascent transcription in the mtDNA D-loop region reminiscent of impeded mtDNA replication (Figure 7B), but also to increase intracellular ROS levels (Figure 7C). Thus, these findings suggest that defective R-loop processing following SETX inactivation may be a cause of oxidative stress and mitochondrial anomalies.
Figure 7.

SETX or PRPF8 depletion triggers mtDNA R-loop accumulation and endogenous oxidative stress
(A) DRIP analyses in HeLa Kyoto cells after (si)RNA against SETX or PRPF8 for 48 h. Plots depict the mean ± SD from three independent experiments. The two-way ANOVA test was performed for all pairs to determine statistical significance. Statistically significant differences are indicated. ∗p < 0.05 and ∗∗∗p < 0.001.
(B) Fold change of the RNA level measure by qPCR with primers for D-loop region in HeLa Kyoto transfected with (si)RNA for (si)SETX or (si)PRPF8 compared with (si)Ctrl. Plots show the relative mean ± SD from three independent experiments. The two-tailed Student’s t test was performed to determine statistical significance between the two groups. ∗p < 0.05 and ∗∗p < 0.01.
(C and D) Bar graph showing relative ROS levels in HeLa Kyoto cells transfected for 72 h with (si)SETX (C), (si)PRPF8 (D), or (si)Ctrl. Intracellular ROS was measured as above using 5 μM of CM-H2DCFDA probe. The plot shows the mean ± SD from three independent experiments. The two-tailed Student’s t test was performed to determine statistical significance between groups. ∗∗p < 0.01.
Defects in RNA splicing induced by mutations affecting components of the pre-mRNA spliceosome have also been shown to induce genome-wide R-loop formation (Nguyen et al., 2017), which has been implicated in the pathogenesis of the pre-neoplastic disorder, myelodysplastic syndrome (Nguyen et al., 2018). We therefore tested the effects of depletion using (si)RNA from HeLa cells of PRPF8, which encodes a core component of the RNA spliceosome (Luo et al., 1999), whose inactivation engenders genome-wide aberrations in mRNA splicing (Wickramasinghe et al., 2015). Like SETX depletion, PRPF8 deficiency causes R-loop accumulation in mtDNA (Figure 7A), excess nascent transcription in the mtDNA D-loop region reminiscent of impeded mtDNA replication (Figure 7B), and increased intracellular ROS levels (Figure 7D). Collectively, our findings raise the possibility that R-loop accrual in mtDNA induced by oxidative stress may occur more widely in human diseases, like cancer or neurodegeneration.
Discussion
Pathogenic BRCA2 mutations impair mtDNA replication via R-loop accrual
Here, we identify a previously unrecognized mechanism through which pathogenic BRCA2 mutations trigger mitochondrial anomalies typical of cancer and other diseases (Figure S6). We find that BRCA2 inactivation causes unscheduled R-loop accumulation in the regulatory region of mtDNA proximal to mitochondrial replication origins (Figures 1B and 1E), to the greatest extent in patient-derived EUFA423 cells, compared to HeLa cells depleted of BRCA2 by RNAi or genetic engineering. In all cases, BRCA2 deficiency reduces the recruitment of RNaseH1, an R-loop processing factor essential for mtDNA replication, to the replication-initiating D-loop region (Figures 5A–5C). These events impede mtDNA replication (Figures 2A–2C), marked by the accumulation of sterile nascent transcripts that normally serve as replication initiation primers (Figure 2D), culminating in mitochondrial genomic deletions (Figure S3) typical of cancer and decreased mtDNA copy number (Figure 2E).
Endogenous oxidative stress and mitochondrial dysfunction
We present converging lines of evidence that endogenous oxidative stress triggers the mitochondrial defects that follow BRCA2 inactivation. Thus, the exposure of wild-type cells to oxidative stress is enough to recapitulate mtDNA R-loop accumulation and reduced RNaseH1 recruitment (Figure 3). Conversely, we confirm that BRCA2-deficient cells experience elevated intracellular ROS levels (Ma et al., 2012) and further demonstrate that ROS scavengers suppress mitochondrial anomalies (Figure 4), including mitochondrial genome deletions (Figure S3) and diminished mtDNA replication capacity (Figure 4).
BRCA2 has not been detected in the mitochondrial proteome (Calvo et al., 2016) and does not carry a mitochondrial localization signal. We therefore infer that excess ROS generation, or impaired ROS clearance, in BRCA2-deficient cells most likely originate from the loss of an extra-mitochondrial function of the protein, whose nature is presently uncertain. Its partner proteins BRCA1 and PALB2 are proposed to control ROS accumulation by regulating antioxidant gene expression (Gorrini et al., 2013; Ma et al., 2012), but the involvement of BRCA2 in this process is uncertain. Thus, how BRCA2 inactivation may increase intracellular ROS levels remains an open question.
A further clue to the link between BRCA2 deficiency and ROS accumulation comes from our observation that depletion of the R-loop helicase, SETX, or the spliceosome factor, PRPF8, also suffices to induce endogenous oxidative stress and mitochondrial defects (Figure 7). Like BRCA2, neither SETX nor PRPF8 is known to localize in mitochondria (Calvo et al., 2016). Instead, inactivation of these RNA processing factors (Skourti-Stathaki et al., 2011; Wickramasinghe et al., 2015)—like BRCA2 deficiency (Bhatia et al., 2014; Shivji et al., 2018)—causes the widespread formation of unscheduled R-loops in the nuclear genome, albeit by apparently distinct molecular mechanisms. These observations raise the possibility that genome-wide R-loop accumulation may in some way induce oxidative stress and, consequently, the defects in mitochondrial genome maintenance that we report in this paper. Moreover, the reported induction of nuclear genomic R-loops by ROS (Teng et al., 2018) suggests a mutually amplifying connection between nuclear R-loop accumulation and oxidative stress. But our work does not address either the relationship between nuclear and mitochondrial R-loop accumulation or the role of unscheduled R-loops formation in oxidative stress induction; both issues warrant further investigation.
Oxidative stress restricts mtDNA replication by impairing RNaseH1 recruitment
Notably, our observations suggest that oxidative stress provokes aberrations in mtDNA maintenance through a previously unrecognized molecular mechanism. We find that endogenous oxidative stress impairs RNaseH1 recruitment to the regulatory region of mtDNA in BRCA2-deficient cells, whereas exogenous oxidative stress can induce a similar phenotype in wild-type cells (Figure 5). Consistent with our conclusion, RNaseH1 has been implicated in mtDNA replication (Cerritelli et al., 2003), and inactivating mutations in human RNaseH1 increase mtDNA R-loop accumulation, diminish mtDNA replication, and cause adult-onset encephalomyopathy (Reyes et al., 2015). Notably, in RNaseH1-deficient cells, R-loops accumulate in the mtDNA D-loop region, accompanied by an increase in RNA transcripts from LSP-OriH (Akman et al., 2016; Holmes et al., 2015). These features phenocopy the abnormalities we observe in BRCA2-deficient cells (Figure 2B), supporting our conclusion that BRCA2 deficiency cripples mtDNA maintenance by impairing RNaseH1. Similar changes in RNA transcripts are also observed accompanying oxidative stress induced by SETX or PRPF8 inactivation (Figure 7B), consistent with a similar mechanism wherein oxidative stress restricts mtDNA maintenance via RNaseH1 impairment.
8-oxoguanine accumulation in mtDNA impairs RNaseH1 recruitment
Our findings provide several lines of evidence that oxidative stress diminishes RNaseH1 recruitment to mtDNA by inducing the accumulation of 8-oxoguanine base adducts. We find that cells depleted of the 8-oxoguanine excising enzyme, OGG1, increased mtDNA R-loop accumulation (Figures 6B and 6C) even without detectable changes in ROS levels (Figure 6D), accompanied by the accumulation of 8-oxoguanine in their mitochondria (Figure S4B). BRCA2 deficiency also increases mitochondrial 8-oxoguanine accumulation (Figures S4A and S4B). Finally, OGG1 depletion and the consequent increase in 8-oxoguanine accumulation are accompanied by diminished RNaseH1 recruitment to mtDNA (Figure 6E), phenocopying the alterations we detect in BRCA2-deficient cells. Overall, the findings we report in this paper suggest a previously unrecognized mechanism (Figure S6) in which oxidative stress—regardless of its origin—cripples mtDNA maintenance by increasing mtDNA oxidation, which in turn compromises RNaseH1 recruitment to the regulatory regions of mtDNA, impairing its essential functions in mtDNA replication initiation. The general significance of the proposed mechanism is enhanced by the ubiquity of oxidative stress as a cellular challenge and its implication in several human diseases. Whereas many deleterious effects of oxidative stress have been identified, our findings represent a rare example driven by a specific molecular mechanism.
Connections with inherited cancer mutations and neurodegenerative syndromes
Our results also provide a first link between mitochondrial anomalies and the cellular functions of the BRCA2 tumor suppressor, inherited mutations in which cause early-onset epithelial cancers. mtDNA instability and oxidative stress have both been implicated in carcinogenesis (Lee and Wei, 2009), raising the possibility that these anomalies also promote carcinogenesis in BRCA2 mutation carriers. If so, differences in oxidative stress between tissues lacking BRCA2 may contribute to the hitherto unexplained tissue selectivity of carcinogenesis in mutation carriers. Moreover, mtDNA instability has also been mooted as a marker of cancer progression (Lee and Wei, 2009), speaking to its potential value in cancer monitoring and early detection.
Moreover, mutations affecting SETX have been linked to neurodegenerative disorders, amyotrophic lateral sclerosis and ataxia-oculomotor apraxia (Moreira et al., 2004; Skourti-Stathaki et al., 2011). Similarly, PRPF8 mutations occur in an inherited form of the neurodegenerative syndrome, retinitis pigmentosa, in which the loss of retinal photoreceptor cells culminates in blindness (Liu and Zack, 2013; Xu et al., 2018), as well as in pre-neoplastic myelodysplasia (Nguyen et al., 2018). Our results implicate the mitochondrial anomalies actuated by the inactivation of these disease genes—however engendered—in disease pathogenesis.
Mitochondrial genomic deletions are frequently detected in human cancers (Yusoff et al., 2019), but their causation is unclear. Our findings raise the possibility that such mitochondrial genome alterations may be induced by endogenous oxidative stress triggered by defects in RNA processing or the inactivation of genes known to trigger R-loop accumulation in human cancers (Hatchi et al., 2015; Schwab et al., 2015). Moreover, defective R-loop processing also occurs in neurodegenerative disorders (Moreira et al., 2004; Skourti-Stathaki et al., 2011), in which mitochondrial dysfunction has also been implicated (Koopman et al., 2013). Thus, our work highlights a previously unrecognized mechanism that connects oxidative stress, mitochondrial genome maintenance, and human disease pathogenesis via defective R-loop processing through impaired RNaseH1 function.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| S9.6 | Home-made | N/A |
| BRCA2 | Merck Millipore | RRID:AB_2067762 |
| Beta-Actin | Sigma | RRID:AB_476744 |
| RNase H1 | Santa Cruz | Cat#: sc-292711 |
| 8-oxoguanine | Merck Millipore | RRID:AB_94925 |
| Chemicals, peptides, and recombinant proteins | ||
| CM-H2DCFDA | ThermoFisher | Cat#: C6827 |
| MitoSOX red | ThermoFisher | Cat#: M36008 |
| N-Acetyl cysteine | Sigma | Cat#: A9165 |
| H2O2 | Sigma | Cat#: H1009 |
| Etomoxir | Sigma | Cat#: E1905 |
| 37% formaldehyde solution | Sigma | Cat#: F8775 |
| RNase H | NEB | Cat#: M0297L |
| RNaseIII | NEB | Cat#: M0245S |
| RNaseT1 | ThermoScientific | Cat#: EN0541 |
| DAPI | Sigma | Cat#: D9542 |
| cOmplete, EDTA-free Protease Inhibitor Cocktail | Roche | Cat#: 11873580001 |
| Critical commercial assays | ||
| JetPRIME Transfection Reagent | Polyplus Transfections | Cat#: 114-07 |
| Click-iT EdU Alexa Fluor® 488 Imaging Kit | ThermoFisher Scientific | Cat#: C10086 |
| Experimental models: cell lines | ||
| HeLa Kyoto | From the laboratory of Jonathan Pines, Institute of Cancer Research, London | N/A |
| EUFA423 | From the European Fanconi Anemia Registry, VU University Medical Center, Amsterdam, the Netherlands | N/A |
| EUFA423 B2 | Hattori et al., 2011 | N/A |
| Pdx1Cre; KrasG12D/+; Trp53R270H/+; Brca2Wt/Wt or Brca2Tr/Δ11 | Skoulidis et al., 2010 | N/A |
| Oligonucleotides | ||
| Negative Control (si)RNA | QIAGEN | Cat#: 1027310 |
| BRCA2 (ON-TARGET plus SMARTpool) | Dharmacon | Cat#: L-003462-00 |
| PRPF8 (ON-TARGET plus SMARTpool) | Dharmacon | Cat#: L-012252-00 |
| SETX (si)RNA | QIAGEN | Cat#: SI05052236 and SI04183942 |
| OGG1 (ON-TARGET plus SMARTpool | Dharmacon | Cat#: L-005147-00 |
| D-loop_Fwd CTTTCATGGGGAAGCAGATTTG | Sigma | N/A |
| D-loop_Rev GCATGGGGAGGGGGTTTTG | Sigma | N/A |
| ND1_Fwd TCTCCACCCTTATCACAACA | Sigma | N/A |
| ND1_Rev GACTAGTTCGGACTCCCCTT | Sigma | N/A |
| OriL_Fwd CCCACAAACACTTAGTTAACAGCT | Sigma | N/A |
| OriL_Rev GGCCTCTTTTTACCAGCTCC | Sigma | N/A |
| Mt-CD1_Fwd AACCACAGTTTCATGCCCATC | Sigma | N/A |
| Mt-CD1_Rev TGTTAGTAAGGGTGGGGAAGC | Sigma | N/A |
| Mt-CD2_Fwd ACCCTATAGCACCCCCTCTAC | Sigma | N/A |
| Mt-CD2_Rev CTTGTCAGGGAGGTAGCGATG | Sigma | N/A |
| HPRT_Fwd TGACACTGGCAAAACAATGCA | Sigma | N/A |
| HPRT_Rev GGTCCTTTTCACCAGCAAGCT | Sigma | N/A |
| GAPDH_Fwd CTCCTGTTCGACAGTCAGC | Sigma | N/A |
| GAPDH_Rev TTCAGGCCGTCCCTAGC | Sigma | N/A |
| BRCA2 CRISPR/Cas9 KO (CTGTCTACCTG ACCAATCGA; ATGTAGCACGCATTCACATA; CGATTACCTGTGTACCCTTT) |
Santa Cruz | Cat#: sc-400700 |
| Software and algorithms | ||
| Prism version 5 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Fiji | (https://fiji.sc/) | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ashok R. Venkitaraman (arv22@nus.edu.sg).
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement as long as stocks remain available and reasonable compensation is provided by requestor to cover processing and shipment.
Experimental model and subject details
HeLa Kyoto (Female) (Adey et al., 2013), EUFA423 (Female) (Howlett et al., 2002) and EUFA423 B2 (Hattori et al., 2011) (stably transfected cell line with FLAG-BRCA2) and mouse PDAC cells (Skoulidis et al., 2010) (Pdx1Cre; KrasG12D/+; Trp53R270H/+; Brca2Wt/Wt or Brca2Tr/Δ11) were maintained in culture in DMEM 10% FBS, 1% Penicillin/streptomycin. All cell lines were authenticated using STR (Short Tandem Repeat) Profiling.
Method details
Treatments
N-Acetyl cysteine (Sigma) was added directly in the cell culture media at a final concentration of 2mM for 24hrs. H2O2 (Sigma) was diluted in PBS at a working concentration of 50mM and etomoxir (Sigma) was diluted in PBS at a working concentration of 60 mM and further diluted directly into the media at indicated concentrations.
Ethidium Bromide (Sigma) was added at the concentration of 50ng/mL and replace every day in fresh media.
Generation of BRCA2 knock-out
HeLa BRCA2-KO were made form HeLa Kyoto. Cells were plated in a 12 well plate and co-transfected with BRCA2 CRISPR/Cas9 KO Plasmid (h) (sc-400700, Santa Cruz Biotechnology) and BRCA2 HDR Plasmid (h) (sc-400700-HDR, Santa Cruz Biotechnology and Table S1) using Lipofectamine 2000 reagent. Two days after transfection, cells were re-plated in a 14 cm dish and selected with puromycin (1 ug/ml). Selected clones were expanded and screened by western blotting.
Plasmids (si)RNA transfection
JetPRIME transfection reagent (Polyplus) was used according to manufacturer’s instructions for transfecting (si)RNA or plasmid for CRISPR/Cas9 cells generation. (si)RNAs used were Negative Control (si)RNA (1027310 from QIAGEN), human BRCA2 (ON-TARGET plus SMARTpool, L-003462-00 from Dharmacon), human PRPF8 (ON-TARGET plus SMARTpool, L-012252-00 from Dharmacon), human SETX (SI05052236 and SI04183942 from QIAGEN), human OGG1 (ON-TARGET plus SMARTpool, L-005147-00 from Dharmacon.
ROS measurement and flow cytometry analysis
CM-H2DCFDA and MitoSOX red probes (ThermoFisher), dissolved in DMSO, was incorporated in DMEM without phenol red and incubated for 15 minutes at 37°C (final concentration of 5 μM). Cells were subsequently rinsed with PBS, trypsinised, collected and rinsed twice with PBS. Cells were analyzed using BD LSR-II cytometer with appropriate excitation and band pass filter.
Cell lysates, and western blotting
Whole cell extracts were prepared from HeLa Kyoto, EUFA423 and EUFA423 B2 cells using lysis buffer (50 mM Tris pH 7.5, 300 mM NaCl, 1 mM MgCl2, 0.1% SDS supplemented with protease and phosphatase inhibitors (Roche) and 350 u/ml benzonase (Millipore)). After 15 min of incubation at room temperature, the lysates were combined with 4 × LDS buffer containing DTT and denatured by boiling. The proteins were separated by SDS-PAGE.
Proteins of interest were identified by western blotting on 4%–12% Bis-tris/MES SDS-PAGE or 3%–8% Tris-acetate on PVDF Immobilon membranes. Blocking, 1° antibody (overnight 4°C) and 2° antibody in PBST-3% milk and detected with ECL or ECL prime chemiluminescence.
RNA extraction, cDNA synthesis
RNA extraction was performed using RNeasy mini Kit (QIAGEN) following manufacturer’s instructions. 3 ug of RNA was then converted to cDNA using FastGene Scriptase II cDNA Kit (Geneflow) according manufacturer’s instruction. cDNA was subsequently quantified and analyzed by qPCR with SYBR green mix (Roche).
DNA extraction and mitochondrial copy number assessment
DNA extraction was performed as described previously (Shivji et al., 2018). Briefly, cells were resuspended in gDNA extraction buffer (TE supplemented with 100mM NaCl, 0,5% SDS, and 5uL of proteinase K (stock at 20mg/mL)) and incubated over-night at 55°C. Then a phenol-chloroform purification followed by isopropanol precipitation was performed. The DNA pellet was washed with 70% ethanol and resuspended in an appropriate volume of TE buffer. MtDNA copy number was determined by amplification of a nuclear DNA (GAPDH) and a mtDNA (ND1) fragment in a real-time PCR for all samples. The DNA input was 10 ng. All samples were measured independently in triplicate. The PCR reaction was performed at 50°C for 2min, 95°C for 10min, followed by 40 cycles of 95°C for 15 s and 60°C for 1min.
Finally, mtDNA copy number was calculated as the ratio of Ct values for the GAPDH and D-loop fragments.
ChIP
Cells were crosslinked in DMEM containing 1% formaldehyde (Sigma, F8775) at room temperature for 10 minutes with rotation. The crosslinking reaction was stopped by adding glycine (Sigma, G8898) to a final concentration of 0.125 M for 5 minutes at room temperature. After two washes with cold PBS, cells were collected in 5 mL of cold PBS and centrifuged at 1500 rpm for 5 minutes. Cell pellets were lysed in 5 mM EDTA, 50 mM Tris-HCl (pH 8.0), 1% SDS supplemented with protease and phosphatase inhibitors. Total cell lysates were sonicated (Bioruptor, Diagenode) to obtain chromatin fragments of an average length of 200-600 bp and then centrifuged at 10,000 rpm for 10 minutes at 4°C. 10ug of chromatin was diluted 10-fold with 5 mM EDTA, 50 mM Tris-HCl (pH 8.0), 0.5% NP-40, 200 mM NaCl supplemented with protease and phosphatase inhibitors. Chromatin extracts were precleared with protein A/G Dynabeads (ThermoFisher) for 40 minutes. The precleared chromatin was incubated over-night with gentle rotation, with antibodies or control antibody. The following day, antibodies were captured by adding 20uL of beads and incubated at 4°C with rotation for 2 hours. Immunoprecipitates were washed with: 1X: 20 mM Tris-HCl pH 8, 2 mM EDTA, 0.1% SDS, 1% Triton X-100 and 165 mM NaCl; 1X: 20 mM Tris-HCl pH 8, 2 mM EDTA, 0.1% SDS, 1% Triton X-100 and 500 mM NaCl; 1X: 10 mM Tris-HCl pH 8, 1 mM EDTA, 1% NP-40, 1% Na-deoxycholate and 250 mM LiCl; 1X: 50 mM HEPES pH 7.6, 1 mM EDTA, 1% NP-40, 0.7% Na-deoxycholate and 500 mM LiCl; 2X with TE (10 mM Tris.HCl pH7.5, 1 mM EDTA). After the final wash, DNA was eluted with 150 uL of 0.1 M sodium carbonate, 1% SDS at 65C for 30 minutes with 1200 rpm agitation. To reverse crosslink, 6 uL of 5 M NaCl and 2uL proteinase K (20 mg/ml) were added and incubated at 65°C with gentle agitation for at least 3 h. DNA was purified using a PCR purification kit (QIAGEN) and analyzed by qPCR using SyBr green Mix (Roche) with the primers of interest.
DRIP
Genomic DNA, extracted with a previously described phenol/chloroform procedure, was digested at 37°C with a cocktail of restriction enzymes (20u/uL EcoRI, 20u/uL HindIII, 20u/uL XbaI, 25u/uL SSPI, 10u/uL BsrGI) in buffer 2.1 (NEB) with or without RNase H (5u/uL, NEB). Following a second DNA purification step with standard phenol/chloroform procedure, 10 ug of digested DNA was diluted in 900 uL of TE buffer (5 mM EDTA, 50 mM Tris HCl, pH 8) and 100uL of 10X DRIP buffer (100 mM NaH2PO4, 1.4 M NaCl, 0.5% Triton-X) was added. Chromatin was precleared with 20uL of protein G Dynabeads for 40 minutes at 4°C. Then 10ug of S9.6 antibody was added to the supernatant and incubated at 4C overnight with gentle rotation. Antibody capture was done with 20uL of beads and incubation at 4°C for 2h. Immunoprecipitates were washed twice with 1X DRIP buffer, and once in 1x DRIP buffer + 330 mM NaCl. After the last wash, DNA was eluted by adding DRIP elution buffer (50 mM Tris-HCl, pH 8, 10 mM EDTA, 0.5% SDS) and incubated at 65°C for 45 minutes. DNA was finally purified using PCR purification kit (QIAGEN) and analyzed by qPCR with SYBR green mix (Roche).
Immunofluorescence for R-loop detection
Appropriate number of cells were plated on coverslips the day before transfection and after 72 h ((si)RNA) or 24 h (plasmid) transfection were stained with Mitotracker green FM (ThermoFisher – M7514) with a final concentration of 300 nM for 20 minutes, then fixed in cold methanol for 10 minutes at −20°C, followed by incubation for 1 minute in cold acetone at room temperature. The coverslips were then quickly washed in SSC 4X buffer thrice then incubated for 30 minutes in SSC 4X, 3% BSA to prevent non-specific interactions. Primary antibody was incubated overnight at 4°C (S9.6 1/200, purified in house at concentration of 0.8 ug/uL). After three washes with PBS-tween 0.05%, coverslips were incubated at room temperature with secondary antibodies Alexa Fluor 488 or Alexa Fluor 568 (Molecular Probes, 1/500).
Enzymatic treatments with RNaseH1, RNaseIII (NEB) or RNaseT1 (Thermo) were performed after methanol/acetone fixation for 30 minutes at 37C in PBS and then rinsed in SCC 4X twice before blocking.
Immunofluorescence for 8-oxoguanine detection
Appropriate number of cells were plated on coverslips the day before transfection and after 72 h ((si)RNA) or 24 h (plasmid) transfection were stained with mitotracker green FM (ThermoFisher – M7514) with a final concentration of 300 nM for 20 minutes, then fixed in PFA 4% for 15 minutes at room temperature, followed by three washes in PBS. The coverslips were then permeabilized PBS-Triton-x 0,5% buffer and rinse thrice. And then incubated for 30 minutes in PBS-tween 0,05%, 3% BSA to prevent non-specific interactions. Primary antibody was incubated for 1 hour. After three washes with PBS-tween 0.05%, coverslips were incubated at room temperature with secondary antibodies Alexa Fluor 488 or Alexa Fluor 568 (Molecular Probes, 1/500).
EdU incorporation
In order to maximize EdU incorporation into the mtDNA, cells were incubated with 6 μM Aphidicolin (Sigma) for 4 hours followed by 2 hours incubation with the nucleotide analog 5-ethynyl-29-deoxyuridine (EdU). Click-iT chemistry (Life Technologies) was used to detect incorporated EdU into mtDNA according to instructions.
Coverslips were mounted with DAKO mounting medium (S3023) supplemented with DAPI (Sigma D9542) after three additional washes with PBS-tween 0.05%. Analysis. Stained cells were imaged on a Zeiss 880 confocal microscope. Maximum projections of the Z stacks of each field were analyzed using Fiji software (https://fiji.sc/). Quantification of immunofluorescence in the mitochondria compartment were done after removing the nuclear staining by using a DAPI mask. Then measurements in the mitochondria compartment were done using the Mitotracker staining as a mask.
Mitochondrial 4977 bp common deletion measurement
DNA was extracted by phenol/chloroform purification and 150 ng was amplified by PCR (COX gene) or nested PCR (common deletion, (PCR1 - 15 cycles with annealing temperature of 60°C; PCR2 - 30 cycles with annealing temperature of 62°C)) using AccuPrime Pfx DNA polymerase (Thermo Fisher). The common deletion of 4977 bp was indicated by the appearance of a 358 bp band on 1.5% agarose gel. Band intensity was quantified by densitometry analysis using Fiji software.
Primers
Primers used in this study including gRNA for CRISPR/Cas9 engineering are listed in Table S1.
Antibodies
Antibodies used in this study are listed in Table S2. S9.6 antibody, used to detect RNA-DNA hybrids, was purified as described previously (Shivji et al., 2018).
Quantification and statistical analysis
Statistical analysis and n number is indicated throughout in the figure legends. Data is represented as mean ± standard. Statistical significance was determined using a two-tailed Student’s t test for testing significance between groups. For ChIP and DRIP analysis, statistical significances were performed using two-way analysis of variance (2-way ANOVA) followed by a Bonferroni correction. All tests were performed using GraphPad Prism 5.0. Statistical details including p values can be found in the figure legends.
Acknowledgments
We thank an anonymous reviewer for sharing the insight that our observations reveal a general mechanism wherein oxidative stress—whatever its origin—restricts mtDNA replication by impairing RNAseH1 recruitment. We thank Dr. Michal Minczuk (MRC Mitochondrial Biology Unit, Cambridge), Dr. Serena Nik-Zainal, and Prof. Barry Halliwell (NUS) for critically reading our manuscript and Dr. Christian Frezza, his laboratory, and members of A.R.V.’s laboratory for helpful comments and discussion. We thank Dr. Feng Zhang (Broad Institute, Boston) for providing the Cas9 plasmid and Dr. Jan Karlseder (UCSD); the mClover LaminA plasmids, via Addgene; and the CIMR flow cytometry facility for assistance with cell sorting. This work was supported by a Krishnan-Ang Fellowship to M.S. and by Medical Research Council (MRC) Programme grants MC_UU_12022/1 and MC_UU_12022/8 to A.R.V.
Author contributions
A.R.V. and X.R. conceived the project; X.R. performed experiments and analyzed the data with A.R.V. M.L. generated the CRISPR cell lines and performed initial validation. M.S. helped with flow cytometry experiments. E.-M.S. undertook preliminary experiments that contributed to hypothesis development. A.R.V. wrote the manuscript with X.R.
Declaration of interests
The authors declare no competing interests.
Published: August 3, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109478.
Supplemental information
Data and code availability
This paper does not report original datasets. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Adey A., Burton J.N., Kitzman J.O., Hiatt J.B., Lewis A.P., Martin B.K., Qiu R., Lee C., Shendure J. The haplotype-resolved genome and epigenome of the aneuploid HeLa cancer cell line. Nature. 2013;500:207–211. doi: 10.1038/nature12064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akman G., Desai R., Bailey L.J., Yasukawa T., Dalla Rosa I., Durigon R., Holmes J.B., Moss C.F., Mennuni M., Houlden H. Pathological ribonuclease H1 causes R-loop depletion and aberrant DNA segregation in mitochondria. Proc. Natl. Acad. Sci. USA. 2016;113:E4276–E4285. doi: 10.1073/pnas.1600537113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson L. Identification of mitochondrial proteins and some of their precursors in two-dimensional electrophoretic maps of human cells. Proc. Natl. Acad. Sci. USA. 1981;78:2407–2411. doi: 10.1073/pnas.78.4.2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ba X., Boldogh I. 8-oxoguanine DNA glycosylase 1: beyond repair of the oxidatively modified base lesions. Redox Biol. 2018;14:669–678. doi: 10.1016/j.redox.2017.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae I., Fan S., Meng Q., Rih J.K., Kim H.J., Kang H.J., Xu J., Goldberg I.D., Jaiswal A.K., Rosen E.M. BRCA1 induces antioxidant gene expression and resistance to oxidative stress. Cancer Res. 2004;64:7893–7909. doi: 10.1158/0008-5472.CAN-04-1119. [DOI] [PubMed] [Google Scholar]
- Baiken Y., Kanayeva D., Taipakova S., Groisman R., Ishchenko A.A., Begimbetova D., Matkarimov B., Saparbaev M. Role of base excision repair pathway in the processing of complex DNA damage generated by oxidative stress and anticancer drugs. Front. Cell Dev. Biol. 2021;8:617884. doi: 10.3389/fcell.2020.617884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia V., Barroso S.I., García-Rubio M.L., Tumini E., Herrera-Moyano E., Aguilera A. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature. 2014;511:362–365. doi: 10.1038/nature13374. [DOI] [PubMed] [Google Scholar]
- Breast Cancer Linkage C., Breast Cancer Linkage Consortium Cancer risks in BRCA2 mutation carriers. J. Natl. Cancer Inst. 1999;91:1310–1316. doi: 10.1093/jnci/91.15.1310. [DOI] [PubMed] [Google Scholar]
- Cadet J., Douki T., Ravanat J.-L. Oxidatively generated base damage to cellular DNA. Free Radic. Biol. Med. 2010;49:9–21. doi: 10.1016/j.freeradbiomed.2010.03.025. [DOI] [PubMed] [Google Scholar]
- Calvo S.E., Clauser K.R., Mootha V.K. MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016;44(D1):D1251–D1257. doi: 10.1093/nar/gkv1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L., Xu X., Cao L.L., Wang R.-H., Coumoul X., Kim S.S., Deng C.-X. Absence of full-length Brca1 sensitizes mice to oxidative stress and carcinogen-induced tumorigenesis in the esophagus and forestomach. Carcinogenesis. 2007;28:1401–1407. doi: 10.1093/carcin/bgm060. [DOI] [PubMed] [Google Scholar]
- Cerritelli S.M., Frolova E.G., Feng C., Grinberg A., Love P.E., Crouch R.J. Failure to produce mitochondrial DNA results in embryonic lethality in Rnaseh1 null mice. Mol. Cell. 2003;11:807–815. doi: 10.1016/s1097-2765(03)00088-1. [DOI] [PubMed] [Google Scholar]
- Choi E., Park P.G., Lee H.O., Lee Y.K., Kang G.H., Lee J.W., Han W., Lee H.C., Noh D.Y., Lekomtsev S., Lee H. BRCA2 fine-tunes the spindle assembly checkpoint through reinforcement of BubR1 acetylation. Dev. Cell. 2012;22:295–308. doi: 10.1016/j.devcel.2012.01.009. [DOI] [PubMed] [Google Scholar]
- Crossley M.P., Bocek M., Cimprich K.A. R-loops as cellular regulators and genomic threats. Mol. Cell. 2019;73:398–411. doi: 10.1016/j.molcel.2019.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels M.J., Wang Y., Lee M., Venkitaraman A.R. Abnormal cytokinesis in cells deficient in the breast cancer susceptibility protein BRCA2. Science. 2004;306:876–879. doi: 10.1126/science.1102574. [DOI] [PubMed] [Google Scholar]
- Davies A.A., Masson J.Y., McIlwraith M.J., Stasiak A.Z., Stasiak A., Venkitaraman A.R., West S.C. Role of BRCA2 in control of the RAD51 recombination and DNA repair protein. Mol. Cell. 2001;7:273–282. doi: 10.1016/s1097-2765(01)00175-7. [DOI] [PubMed] [Google Scholar]
- German P., Szaniszlo P., Hajas G., Radak Z., Bacsi A., Hazra T.K., Hegde M.L., Ba X., Boldogh I. Activation of cellular signaling by 8-oxoguanine DNA glycosylase-1-initiated DNA base excision repair. DNA Repair (Amst.) 2013;12:856–863. doi: 10.1016/j.dnarep.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginno P.A., Lott P.L., Christensen H.C., Korf I., Chédin F. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol. Cell. 2012;45:814–825. doi: 10.1016/j.molcel.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorrini C., Baniasadi P.S., Harris I.S., Silvester J., Inoue S., Snow B., Joshi P.A., Wakeham A., Molyneux S.D., Martin B. BRCA1 interacts with Nrf2 to regulate antioxidant signaling and cell survival. J. Exp. Med. 2013;210:1529–1544. doi: 10.1084/jem.20121337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson C.M., Falkenberg M., Larsson N.G. Maintenance and expression of mammalian mitochondrial DNA. Annu. Rev. Biochem. 2016;85:133–160. doi: 10.1146/annurev-biochem-060815-014402. [DOI] [PubMed] [Google Scholar]
- Hatchi E., Skourti-Stathaki K., Ventz S., Pinello L., Yen A., Kamieniarz-Gdula K., Dimitrov S., Pathania S., McKinney K.M., Eaton M.L. BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol. Cell. 2015;57:636–647. doi: 10.1016/j.molcel.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori H., Skoulidis F., Russell P., Venkitaraman A.R. Context dependence of checkpoint kinase 1 as a therapeutic target for pancreatic cancers deficient in the BRCA2 tumor suppressor. Mol. Cancer Ther. 2011;10:670–678. doi: 10.1158/1535-7163.MCT-10-0781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes J.B., Akman G., Wood S.R., Sakhuja K., Cerritelli S.M., Moss C., Bowmaker M.R., Jacobs H.T., Crouch R.J., Holt I.J. Primer retention owing to the absence of RNase H1 is catastrophic for mitochondrial DNA replication. Proc. Natl. Acad. Sci. USA. 2015;112:9334–9339. doi: 10.1073/pnas.1503653112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt I.J. The mitochondrial R-loop. Nucleic Acids Res. 2019;47:5480–5489. doi: 10.1093/nar/gkz277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt I.J., Lorimer H.E., Jacobs H.T. Coupled leading- and lagging-strand synthesis of mammalian mitochondrial DNA. Cell. 2000;100:515–524. doi: 10.1016/s0092-8674(00)80688-1. [DOI] [PubMed] [Google Scholar]
- Howlett N.G., Taniguchi T., Olson S., Cox B., Waisfisz Q., De Die-Smulders C., Persky N., Grompe M., Joenje H., Pals G. Biallelic inactivation of BRCA2 in Fanconi anemia. Science. 2002;297:606–609. doi: 10.1126/science.1073834. [DOI] [PubMed] [Google Scholar]
- Islam M.T. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol. Res. 2017;39:73–82. doi: 10.1080/01616412.2016.1251711. [DOI] [PubMed] [Google Scholar]
- Jiang S., Koolmeister C., Misic J., Siira S., Kühl I., Silva Ramos E., Miranda M., Jiang M., Posse V., Lytovchenko O. TEFM regulates both transcription elongation and RNA processing in mitochondria. EMBO Rep. 2019;20:e48101. doi: 10.15252/embr.201948101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopman W.J.H., Distelmaier F., Smeitink J.A.M., Willems P.H.G.M. OXPHOS mutations and neurodegeneration. EMBO J. 2013;32:9–29. doi: 10.1038/emboj.2012.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D.Y., Clayton D.A. Properties of a primer RNA-DNA hybrid at the mouse mitochondrial DNA leading-strand origin of replication. J. Biol. Chem. 1996;271:24262–24269. doi: 10.1074/jbc.271.39.24262. [DOI] [PubMed] [Google Scholar]
- Lee H.-C., Wei Y.-H. Mitochondrial DNA instability and metabolic shift in human cancers. Int. J. Mol. Sci. 2009;10:674–701. doi: 10.3390/ijms10020674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M.M., Zack D.J. Alternative splicing and retinal degeneration. Clin. Genet. 2013;84:142–149. doi: 10.1111/cge.12181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomonosov M., Anand S., Sangrithi M., Davies R., Venkitaraman A.R. Stabilization of stalled DNA replication forks by the BRCA2 breast cancer susceptibility protein. Genes Dev. 2003;17:3017–3022. doi: 10.1101/gad.279003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H.R., Moreau G.A., Levin N., Moore M.J. The human Prp8 protein is a component of both U2- and U12-dependent spliceosomes. RNA. 1999;5:893–908. doi: 10.1017/s1355838299990520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J., Cai H., Wu T., Sobhian B., Huo Y., Alcivar A., Mehta M., Cheung K.L., Ganesan S., Kong A.-N.T. PALB2 interacts with KEAP1 to promote NRF2 nuclear accumulation and function. Mol. Cell. Biol. 2012;32:1506–1517. doi: 10.1128/MCB.06271-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal G., Rowley M., Guidugli L., Wu J., Pankratz V.S., Couch F.J. BRCA2 localization to the midbody by filamin A regulates cep55 signaling and completion of cytokinesis. Dev. Cell. 2012;23:137–152. doi: 10.1016/j.devcel.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira M.-C., Klur S., Watanabe M., Németh A.H., Le Ber I., Moniz J.-C., Tranchant C., Aubourg P., Tazir M., Schöls L. Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat. Genet. 2004;36:225–227. doi: 10.1038/ng1303. [DOI] [PubMed] [Google Scholar]
- Nguyen H.D., Yadav T., Giri S., Saez B., Graubert T.A., Zou L. Functions of replication protein A as a sensor of R loops and a regulator of RNaseH1. Mol. Cell. 2017;65:832–847.e4. doi: 10.1016/j.molcel.2017.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen H.D., Leong W.Y., Li W., Reddy P.N.G., Sullivan J.D., Walter M.J., Zou L., Graubert T.A. Spliceosome mutations induce R loop-associated sensitivity to ATR inhibition in myelodysplastic syndromes. Cancer Res. 2018;78:5363–5374. doi: 10.1158/0008-5472.CAN-17-3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie H., Chen G., He J., Zhang F., Li M., Wang Q., Zhou H., Lyu J., Bai Y. Mitochondrial common deletion is elevated in blood of breast cancer patients mediated by oxidative stress. Mitochondrion. 2016;26:104–112. doi: 10.1016/j.mito.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojala D., Montoya J., Attardi G. tRNA punctuation model of RNA processing in human mitochondria. Nature. 1981;290:470–474. doi: 10.1038/290470a0. [DOI] [PubMed] [Google Scholar]
- Pastukh V.M., Gorodnya O.M., Gillespie M.N., Ruchko M.V. Regulation of mitochondrial genome replication by hypoxia: the role of DNA oxidation in D-loop region. Free Radic. Biol. Med. 2016;96:78–88. doi: 10.1016/j.freeradbiomed.2016.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini L., Yu D.S., Lo T., Anand S., Lee M., Blundell T.L., Venkitaraman A.R. Insights into DNA recombination from the structure of a RAD51-BRCA2 complex. Nature. 2002;420:287–293. doi: 10.1038/nature01230. [DOI] [PubMed] [Google Scholar]
- Pham X.H., Farge G., Shi Y., Gaspari M., Gustafsson C.M., Falkenberg M. Conserved sequence box II directs transcription termination and primer formation in mitochondria. J. Biol. Chem. 2006;281:24647–24652. doi: 10.1074/jbc.M602429200. [DOI] [PubMed] [Google Scholar]
- Reyes A., Melchionda L., Nasca A., Carrara F., Lamantea E., Zanolini A., Lamperti C., Fang M., Zhang J., Ronchi D. RNASEH1 mutations impair mtDNA replication and cause adult-onset mitochondrial encephalomyopathy. Am. J. Hum. Genet. 2015;97:186–193. doi: 10.1016/j.ajhg.2015.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha T., Rih J.K., Rosen E.M. BRCA1 down-regulates cellular levels of reactive oxygen species. FEBS Lett. 2009;583:1535–1543. doi: 10.1016/j.febslet.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sariki S.K., Sahu P.K., Golla U., Singh V., Azad G.K., Tomar R.S. Sen1, the homolog of human Senataxin, is critical for cell survival through regulation of redox homeostasis, mitochondrial function, and the TOR pathway in Saccharomyces cerevisiae. FEBS J. 2016;283:4056–4083. doi: 10.1111/febs.13917. [DOI] [PubMed] [Google Scholar]
- Schlacher K., Christ N., Siaud N., Egashira A., Wu H., Jasin M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011;145:529–542. doi: 10.1016/j.cell.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab R.A., Nieminuszczy J., Shah F., Langton J., Lopez Martinez D., Liang C.-C., Cohn M.A., Gibbons R.J., Deans A.J., Niedzwiedz W. The Fanconi anemia pathway maintains genome stability by coordinating replication and transcription. Mol. Cell. 2015;60:351–361. doi: 10.1016/j.molcel.2015.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivji M.K.K., Renaudin X., Williams C.H., Venkitaraman A.R. BRCA2 regulates transcription elongation by RNA polymerase II to prevent R-loop accumulation. Cell Rep. 2018;22:1031–1039. doi: 10.1016/j.celrep.2017.12.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva S., Camino L.P., Aguilera A. Human mitochondrial degradosome prevents harmful mitochondrial R loops and mitochondrial genome instability. Proc. Natl. Acad. Sci. USA. 2018;115:11024–11029. doi: 10.1073/pnas.1807258115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoulidis F., Cassidy L.D., Pisupati V., Jonasson J.G., Bjarnason H., Eyfjord J.E., Karreth F.A., Lim M., Barber L.M., Clatworthy S.A. Germline Brca2 heterozygosity promotes Kras(G12D) -driven carcinogenesis in a murine model of familial pancreatic cancer. Cancer Cell. 2010;18:499–509. doi: 10.1016/j.ccr.2010.10.015. [DOI] [PubMed] [Google Scholar]
- Skourti-Stathaki K., Proudfoot N.J., Gromak N. Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol. Cell. 2011;42:794–805. doi: 10.1016/j.molcel.2011.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suraweera A., Becherel O.J., Chen P., Rundle N., Woods R., Nakamura J., Gatei M., Criscuolo C., Filla A., Chessa L. Senataxin, defective in ataxia oculomotor apraxia type 2, is involved in the defense against oxidative DNA damage. J. Cell Biol. 2007;177:969–979. doi: 10.1083/jcb.200701042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y., Holmes J.B., Cerritelli S.M., Sakhuja K., Minczuk M., Holt I.J., Crouch R.J. An upstream open reading frame and the context of the two AUG codons affect the abundance of mitochondrial and nuclear RNase H1. Mol. Cell. Biol. 2010;30:5123–5134. doi: 10.1128/MCB.00619-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng Y., Yadav T., Duan M., Tan J., Xiang Y., Gao B., Xu J., Liang Z., Liu Y., Nakajima S. ROS-induced R loops trigger a transcription-coupled but BRCA1/2-independent homologous recombination pathway through CSB. Nat. Commun. 2018;9:4115. doi: 10.1038/s41467-018-06586-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkitaraman A.R. Cancer suppression by the chromosome custodians, BRCA1 and BRCA2. Science. 2014;343:1470–1475. doi: 10.1126/science.1252230. [DOI] [PubMed] [Google Scholar]
- Wanrooij P.H., Uhler J.P., Shi Y., Westerlund F., Falkenberg M., Gustafsson C.M. A hybrid G-quadruplex structure formed between RNA and DNA explains the extraordinary stability of the mitochondrial R-loop. Nucleic Acids Res. 2012;40:10334–10344. doi: 10.1093/nar/gks802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickramasinghe V.O., Gonzàlez-Porta M., Perera D., Bartolozzi A.R., Sibley C.R., Hallegger M., Ule J., Marioni J.C., Venkitaraman A.R. Regulation of constitutive and alternative mRNA splicing across the human transcriptome by PRPF8 is determined by 5′ splice site strength. Genome Biol. 2015;16:201. doi: 10.1186/s13059-015-0749-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia B., Sheng Q., Nakanishi K., Ohashi A., Wu J., Christ N., Liu X., Jasin M., Couch F.J., Livingston D.M. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol. Cell. 2006;22:719–729. doi: 10.1016/j.molcel.2006.05.022. [DOI] [PubMed] [Google Scholar]
- Xu G., Li T., Chen J., Li C., Zhao H., Yao C., Dong H., Wen K., Wang K., Zhao J. Autosomal dominant retinitis pigmentosa-associated gene PRPF8 is essential for hypoxia-induced mitophagy through regulating ULK1 mRNA splicing. Autophagy. 2018;14:1818–1830. doi: 10.1080/15548627.2018.1501251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M.Y., Bowmaker M., Reyes A., Vergani L., Angeli P., Gringeri E., Jacobs H.T., Holt I.J. Biased incorporation of ribonucleotides on the mitochondrial L-strand accounts for apparent strand-asymmetric DNA replication. Cell. 2002;111:495–505. doi: 10.1016/s0092-8674(02)01075-9. [DOI] [PubMed] [Google Scholar]
- Yasukawa T., Reyes A., Cluett T.J., Yang M.Y., Bowmaker M., Jacobs H.T., Holt I.J. Replication of vertebrate mitochondrial DNA entails transient ribonucleotide incorporation throughout the lagging strand. EMBO J. 2006;25:5358–5371. doi: 10.1038/sj.emboj.7601392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusoff A.A.M., Abdullah W.S.W., Khair S.Z.N.M., Radzak S.M.A. A comprehensive overview of mitochondrial DNA 4977-bp deletion in cancer studies. Oncol. Rev. 2019;13:409. doi: 10.4081/oncol.2019.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This paper does not report original datasets. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.