

Abstract
Screening certain environmental chemicals for their ability to interact with endocrine targets, including the androgen receptor (AR), is an important global concern. We previously developed a model using a battery of eleven in vitro AR assays to predict in vivo AR activity. Here we describe a revised mathematical modeling approach that also incorporates data from newly available assays and demonstrate that subsets of assays can provide close to the same level of predictivity. These subset models are evaluated against the full model using 1820 chemicals, as well as in vitro and in vivo reference chemicals from the literature. Agonist batteries of as few as six assays and antagonist batteries of as few as five assays can yield balanced accuracies of 95% or better relative to the full model. Balanced accuracy for predicting reference chemicals is 100%. An approach is outlined for researchers to develop their own subset batteries to accurately detect AR activity using assays that map to the pathway of key molecular and cellular events involved in chemical-mediated AR activation and transcriptional activity. This work indicates in vitro bioactivity and in silico predictions that map to the AR pathway could be used in an integrated approach to testing and assessment for identifying chemicals that interact directly with the mammalian AR.
Keywords: Endocrine disruption, Androgen receptor, Alternative testing methods, In vitro
1. Introduction
Screening for chemicals that may affect endocrine signaling is of global concern, though the regulatory data requirements for endocrine bioactivity of chemicals vary by agency, intended chemical usage, and region (U.S. EPA, 2011, Government of Canada, 2016; EFSA, 2018). This task is complicated by the fact that there are too many chemicals requiring screening for potential endocrine activity (~10,000 in the US (U.S. EPA, 2012)) to evaluate using a combination of in vitro and in vivo assays. The US EPA’s Endocrine Disruptor Screening Program (EDSP) (U.S. EPA, 2007) has adopted high-throughput in vitro methods through the EDSP21 program (U.S. EPA, 2011) to help prioritize chemicals for additional testing in models of greater biological complexity. The most extensive progress has been evaluating chemicals’ interactions with the estrogen receptor (ER). A computational model was constructed using the output of screening 1811 chemicals in 18 distinct in vitro ER assays, which resulted in composite scores for agonist and antagonist activity (Browne et al., 2015; Judson et al., 2015; Judson et al., 2017; Watt and Judson, 2018). These 18 assays, and the corresponding cytotoxicity assay information, were obtained from the broader US EPA ToxCast and cross-agency Tox21 screening programs (Dix et al., 2007; Judson et al., 2010; Thomas et al., 2018; Thomas et al., 2019). The ER pathway model was validated against an internationally-developed set of in vitro ER reference chemicals (NICEATM/ICCVAM, 2011) and a corresponding set of in vivo reference chemicals identified using curated data from the in vivo rodent uterotrophic assay (Kleinstreuer et al., 2015). Based on the evaluation of this model against reference chemicals and recommendations of external Scientific Advisory Panels (SAPs) convened under the authority of the Federal Insecticide, Fungicide, and Rodenticide Act (FIFRA) (U.S. EPA, 2013; U.S. EPA, 2017), the US EPA is considering accepting the results of the ER pathway model (agonist mode) in lieu of three estrogen-related assays: the in vitro ER transactivation (OPPTS 890.1300, OECD TG455), in vitro ER binding (OPPTS 890.1250 OECD TG493), and in vivo uterotrophic EDSP Tier 1 screening assays (OPPTS 890.1600/OECD TG440). A large-scale QSAR modeling effort was also undertaken using the in vitro data from these 1811 chemicals to train multiple models and generate consensus ER binding, agonist, and antagonist predictions for tens of thousands of chemicals, including those subject to the EDSP (Mansouri et al., 2016; U.S. EPA, 2019).
A similar effort is being performed for the androgen receptor (AR). A corresponding AR pathway model was developed, using 11 in vitro AR assays with publicly available data from the ToxCast and Tox21 programs (Kleinstreuer et al., 2017). Because there was no existing internationally agreed upon in vitro reference chemical set for AR, one was developed for this project via a workgroup of the Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM). The results of the AR pathway model were also compared to the results of the Hershberger assay, a short term, in vivo screening assay measuring chemical effects on androgen-responsive accessory sex tissues in the male rat reproductive tract (Browne et al., 2018; Kleinstreuer et al., 2018). Notably, in vitro AR pathway model activity and results from the Hershberger assay were not as concordant as between the ER pathway in vitro model (agonist mode) and uterotrophic activity. In part, the Hershberger assay is more complicated, being sensitive to mechanisms other than interference of ligands interacting directly with the AR (Freyberger and Schladt, 2009; Browne et al., 2018). Further, for some chemicals, effects in the Hershberger were observed at doses where plasma concentrations would be expected to significantly exceeded corresponding media concentrations used for the in vitro tests (Browne et al., 2018; Kleinstreuer et al., 2018). The AR pathway model has been evaluated in an EDSP FIFRA SAP and its use in a regulatory context under the EDSP is under consideration (U.S. EPA, 2013; U.S. EPA, 2017).
The full ER pathway model presented a practical problem in that it is both expensive and logistically difficult to test a chemical across all 16 in vitro assays included in the published agonist-mode model. We addressed this problem by demonstrating that it is possible to achieve almost equivalent predictivity of ER agonist activity using as few as four assays (Judson et al., 2017). We focused on ER agonist activity because the expectation is that most xenobiotic chemicals that interact with the ER will act as agonists. The basic approach was to construct a series of simple assay subset models with all combinations of one to 16 agonist-mode assays and evaluate subset models against both the full model and the agonist in vitro and in vivo reference chemicals. The chemical-assay area under the curve (AUC) values for the assay subset models were estimated using simple linear combinations of individual assay AUC values. Panelists from the 2017 FIFRA SAP review of the AR pathway model suggested that a similar approach to reduce the number of assays required for the AR pathway model would be useful (U.S. EPA, 2017).
In this paper, we report the results of a similar procedure for the AR pathway model, using an updated, full AR model evaluated using AR in vitro and in vivo reference chemicals previously published (Kleinstreuer et al., 2017). Here, however, the full biologically based network structure is used for constructing the minimal assay models, rather than the linear data fitting strategy used with ER. The method used here is computationally intensive, and at the time the ER subset model was developed, the approach used here was not computationally tractable. Additionally, we developed both agonist and antagonist minimal assay models, the latter because AR antagonist activity is more prevalent among xenobiotic chemicals than is agonist activity, which has been previously observed for only a small set of steroid hormones and pharmaceutical chemicals.
2. Materials and methods
2.1. Reference chemicals
The in vitro reference chemicals were previously developed by an ICCVAM working group and reported by Kleinstreuer et al. (Kleinstreuer et al., 2017). To develop the list of in vitro reference chemicals, a targeted literature search was performed using chemical identifiers for 158 chemicals used in various validation efforts for AR assays (OECD, EPA, ECVAM, KoCVAM), and with activity in AR assays. Metadata included in this search included (among other fields) PubMed ID and year, hit response, potency metric and value (e.g. AC50), assay type, receptor type, species, reference agonist and antagonist, cytotoxicity information. Data was filtered to only include data from studies of the full-length receptor (e.g. not just ligand binding domain), and to include studies that reported concurrent cytotoxicity measurements for cell-based assays. Potency data for all results were converted to μM units, and the mean, standard deviation and 95% confidence intervals were calculated. To be called a positive agonist reference chemical, a chemical needed ≥3 experiments with ≥70% positive responses, plus at least one positive binding result. The same criteria were applied to antagonists, with the addition that the positive results could not have been confounded by cytotoxicity. To be called a negative agonist reference chemical, there had to be ≥ 3 negative calls with no positives. The same criteria were used for antagonists, except that there had to be ≥ 2 negative calls.
The in vivo reference chemicals were curated by Browne et al. (Browne et al., 2018; Kleinstreuer et al., 2018). To develop the in vivo reference chemical list, a systematic literature survey was carried out starting with a collection of 3200 chemicals and looking for results in the Hershberger assay. These chemicals included those that were run in EPA AR assays, chemicals used to validate the Hershberger assay at EPA and OECD, chemicals run in EPA EDSP Tier 1 assays, and chemicals identified as in vitro AR reference chemicals. Metadata for each study was extracted, including but not limited to: chemical identifiers, purity and source, strain and age of rat, intact or castrated, post-surgical recovery duration, number of animals per treatment group, route of administration, dose levels, control chemicals, ASTs measured and any significant effects. A complex set of decision criteria was used to include studies in the final database, which required studies to follow guideline protocols or be very close (“guideline-like”). The criteria for defining reference chemicals were: (1) a positive reference chemical was one with ≥2 Hershberger positive calls and one other related study positive result, with more positive results than negative results; (2) a negative reference chemical was one with ≥2 Hershberger negative calls with no positives OR 1 negative Hershberger and 1 other negative study with no positives. The class of other studies considered were in vivo studies that could assess the response to androgens/antiandrogens and that reported androgen-responsive endpoints, including: the male pubertal assay, 28 or 90 day repeated dose oral toxicity studies, and/or extended one generation studies in rats. All reference chemicals used in this work are listed in Supplemental Table S1. The in vitro reference chemicals are taken from Table 2 of (Kleinstreuer et al., 2017) and the in vivo reference chemicals are taken from Table 1 of (Kleinstreuer et al., 2018). A few of the reference chemicals from these sources were not used because data for all of the in vitro assays used here was not available.
Table 2.
Schema for antagonist activity confidence scoring.
| Source | Criteria | Confidence Score Contribution |
|---|---|---|
| AR pathway model | AUC (Antagonist) ≥ 0.1 | 2 |
| 0.1 > AUC (Antagonist) ≥ 0.001 | 1 | |
| Cell stress/cytotoxicity flag | average Z-score > 3 | 2 |
| average Z-score < 1 | −1 | |
| Confirmation assay dataa | true antagonist shift (hit/hit) | 3 |
| true antagonist shift (no hit/hit) | 2 | |
| FLAG: true antagonist shift but CI overlap | 1 | |
| FLAG: wrong direction shift (hit/hit) | −1 | |
| FLAG: wrong direction (hit/no hit) | −1 | |
| Maximum receptor value | AUC (antagonist) < AUC (any other) | −1 |
| Cell-free assay activity (A1-A3) | A1-A3 are all inactive | −1 |
See the original AR model paper for more details (Kleinstreuer et al., 2017).
Table 1.
Assays used in the models. Assay ID and node correspond to Fig. 1. Detailed assay descriptions and protocols can be found at the EPA CompTox Chemicals Dashboard by searching the web page https://comptox.epa.gov/dashboard/assay_endpoints/ using the assay endpoint name. Detailed descriptions following the OECD harmonized template (http://www.oecd.org/ehs/templates/) are included in the supplemental information.
| Assay IDa | Node | Assay Endpoint Name | Assay endpoint ID | Source | Gene | Species | Type | Cell type and Detection Technology |
|---|---|---|---|---|---|---|---|---|
| A1 | N1 | NVS_NR_hAR | 711 | Novascreen | AR | Homo sapiens | Receptor Binding | Cell-free radioligand displacement |
| A2 | N1 | NVS_NR_cAR | 710 | Novascreen | AR | Pan troglodytes | Receptor Binding | Cell-free radioligand displacement |
| A3 | N1 | NVS_NR_rAR | 726 | Novascreen | AR | Rattus norvegicus | Receptor Binding | Cell-free radioligand displacement |
| A4 | N2/N6 | OT_AR_ARSRC1_0480 | 740 | Odyssey Thera | AR; SRC | Homo sapiens | Coregulator Recruitment | HEK293, Protein-fragment complementation |
| A5 | N2/N6 | OT_AR_ARSRC1_0960 | 741 | Odyssey Thera | AR; SRC | Homo sapiens | Coregulator Recruitment | HEK293, Protein-fragment complementation |
| A6 | N2/N6 | UPITT_HCI_U2OS_AR_TIF2_Nucleoli_Agonist | 2387 | University of Pittsburgh | AR | Homo sapiens | Nuclear Translocation/coactivator interaction | U2OS, Protein-fragment complementation |
| A7 | N3 | ATG_AR_TRANS_up | 115 | Attagene | AR | Homo sapiens | RNA Reporter Gene | HepG2, RT-PCR and Capillary electrophoresis |
| A8 | N4 | OT_AR_ARELUC_AG_1440 | 739 | Odyssey Thera | AR; ARE | Homo sapiens | Reporter Gene | HEK293, Protein-fragment complementation |
| A9 | N4 | TOX21_AR_BLA_Agonist_ratio | 761 | NCATS/NCGC | AR | Homo sapiens | Reporter Gene | HEK293, GAL4 b-lactamase reporter gene |
| A10 | N4 | TOX21_AR_LUC_MDAKB2_Agonist | 764 | NCATS/NCGC | AR | Homo sapiens | Reporter Gene | MDA-kb2, Luciferase-coupled ATP quantitation |
| A11 | N5 | ACEA_AR_agonist_80hr | 1855 | ACEA | AR | Homo sapiens | Real-time impedance | 22Rv1, Real-time cell electronic sensing |
| A12 | N6 | UPITT_HCI_U2OS_AR_TIF2_Nucleoli_Antagonist | 2386 | University of Pittsburgh | AR | Homo sapiens | Nuclear Translocation/coactivator interaction | U2OS, Protein-fragment complementation |
| A13 | N7 | TOX21_AR_BLA_Antagonist_ratio | 762 | NCATS/NCGC | AR | Homo sapiens | Reporter Gene | HEK293, GAL4 b-lactamase reporter gene |
| A14 | N7 | TOX21_AR_LUC_MDAKB2_Antagonist_0.5 nM_R1881 | 1816 | NCATS/NCGC | AR | Homo sapiens | Reporter Gene | MDA-kb2, Luciferase-coupled ATP quantitation |
Assays A6, A11 and A12 are new assays relative to those used in (Kleinstreuer et al., 2017).
2.2. Revised full model
The original AR pathway model is described in Kleinstreuer et al. (Kleinstreuer et al., 2017). For the full model described here, the original 11 assay AR pathway model was modified by including three additional assays. The first is an AR-dependent cell proliferation assay using a real-time cell impedance readout, run by ACEA Corporation (https://www.aceabio.com, San Diego CA). The other additions are nuclear translocation/coactivator interaction agonist and antagonist-mode assays developed by Johnston et al.(Hua et al., 2014; Fancher et al., 2018; Hua et al., 2018). These AR-TIF2 PPI (protein-protein interaction) assays incorporate both nuclear translocation and TIF2/SRC2 co-activator interactions. AR ligands stimulate nuclear translocation of the AR, and interaction with the nuclear TIF2 biosensor stabilizes the receptor in the nucleolus allowing quantitation by automated fluorescence imaging. For the original model, one of the antagonist assays (the TOX21 MDA-Kb2 antagonist assay) was run twice; once using a low concentration (0.5 nM) of the reference agonist, R1881, to initially induce AR pathway activity and then with a higher concentration (10 nM). In the original publication, the low concentration agonist version of the assay was used in the pathway model, and the high concentration version was used in combination with the low concentration version to inform an antagonist confirmation flag used in confidence scores considered in parallel with the pathway model results. Similarly, both versions of this assay are used here to test for an appropriate shift in the activity concentration in antagonist mode, indicative of true receptor-mediated activity. A true competitive antagonist should appear to be less potent (shifting the curve “right” to a larger AC50 value) when the assay is run with a higher concentration of the reference agonist, R1881. As before, this potency shift test was performed outside of the main model construction. The full set of assays used in the pathway model are given in Table 1.
Chemical and assay-wise concentration-response modeling was carried out using the ToxCast Pipeline (tcpl v 2.0.2) (Filer et al., 2017) to generate the summary values from curve-fitting used as inputs to the AR pathway model. Curve fitting uses a Hill or a Gain-Loss model; the latter is a rising Hill curve followed by a decrease in activity at higher concentrations. The concentration-response series is also fit to a constant model (response = 0). The model with the lowest Akaike Information Criteria (AIC) value is selected. The output parameters used here are AC50, Top and Hitcall, where AC50 is the concentration at half-maximum response, Top is the parameter defining the top of the Hill curve, and Hitcall equals 1 if Top exceeds an assay-dependent statistical threshold or is 0 otherwise. These values were extracted from invitrodb v3.2 (https://doi.org/10.23645/epacomptox.6062623.v4, available at ftp://newftp.epa.gov/Computational_Toxicology_Data/High_Throughput_Screening_Data/InVitroDB_v3.2/MySQL_Data/) (US EPA, 2019). A complete data set on 14 assays was available for 1820 chemicals. Assay data along with output of the full model are given in Supplemental Table S2, and assay targets within the AR signaling pathway are indicated in Fig. 1. The full model determines whether the pattern of activity is most consistent with agonist activity, antagonist activity or activity in one of multiple assay interference modes. Assay interference is a general term for a variety of processes that can cause false positive or false negative results in an assay due to a chemical interfering with the assay system or technology (Thorne et al., 2010; Bruns and Watson, 2012; Hsieh et al., 2015). For example, chemicals such as flavins are autofluorescent and therefore typically seen as “false positives” in assays that use fluorescence-based readouts. The chemical is assigned a mode (agonist, antagonist, other) based on which mode has the largest AUC value, where “other” corresponds to interference via a specific assay or a node in the pathway (Fig. 1). Two key results used from the full model are the AUC(agonist) and the AUC(antagonist). These pathway-level AUC values range from 0 to ~1 and monotonically increase with the log of the potency. Supplemental Fig. S1 shows the calibration curve between idealized AC50 values across all assays and the AUC values (see Kleinstreuer et al., 2017 for details). The AUC range is scaled so that a potent reference antagonist (Mifepristone) has an AUC (antagonist) score of 1.0. Note that in the original model, Hydroxyflutamide was the reference antagonist that was used to scale the model, but data for this chemical were not available for all of the new assays. Mifepristone was selected because it was the most potent AR antagonist in the new set of 14 assays tested. Chemicals more potent than Mifepristone can have AUC values greater than 1.
Fig. 1.
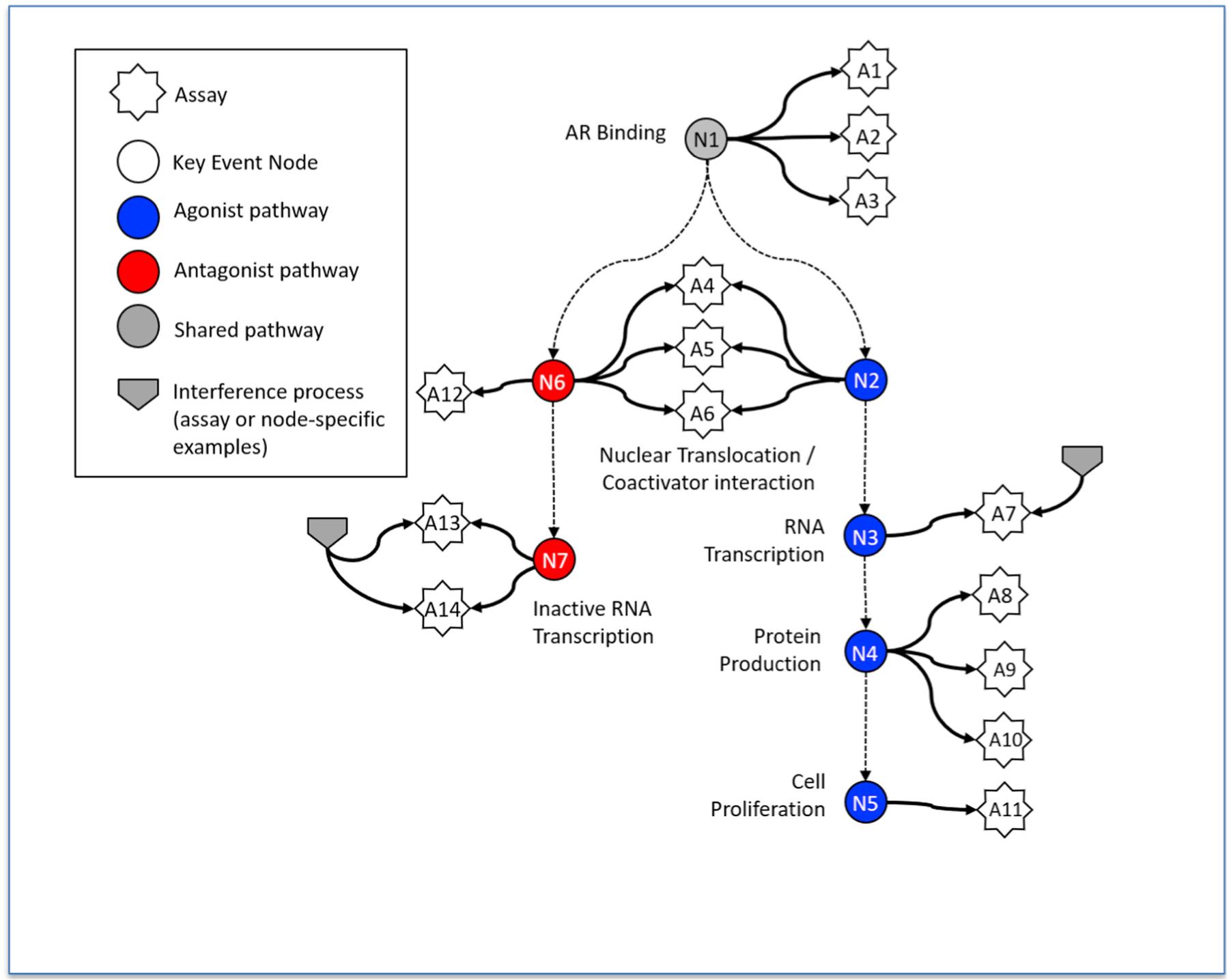
Diagram indicating the nodes in the AR signaling pathway associated with the assays. White stars indicate assays and correspond to the rows of Table 1. N1–N7 nodes correspond to processes in the underlying biological pathway, where blue is agonism, red is antagonism, and grey is common to both agonism and antagonism. Two representative assay interference processes are indicated, one affecting a single assay (A7) and another affecting a pair of related assays (A13 and A14). All single assays and groups of assays have their corresponding assay interference processes represented in the underlying mathematical model. See Kleinstreuer et al., 2017 for further details (Kleinstreuer et al., 2017). The strengths of these interference processes are indicated by the corresponding AUC values in Supplemental File S2.
The original antagonist mode model used an “antagonist confidence score” (CS) in addition to the AUC value (see Kleinstreuer et al., (2017) for details (Kleinstreuer et al., 2017). The CS accounted for factors beyond what is provided by the AUC value itself, including cytotoxicity, which can confound loss-of-signal antagonist assays, and the expected antagonist potency shift, described above. Here, we have slightly modified the calculation of the CS to add additional penalties for activity at concentrations well above where cytotoxicity is observed; to penalize chemicals for which one of the non-antagonist mode AUC values is greater than the antagonist mode AUC; and, to penalize chemicals that are active in multiple cell-based assays but in none of the cell-free assays (A1-A3). Here, cytotoxicity is measured using a large collection of assays from a variety of technologies, and not just from concurrent cytotoxicity assays corresponding to the current AR transactivation assays (Judson et al., 2016). Using invitrodb version 3.2, there were 86 possible cytotoxicity assays used in this calculation, and a minimum of 5 positive hitcalls were required to calculate a lower bound on a cytotoxicity concentration level. If the number of assays screened as positive was less than 5, a default value of 1000 μM was assigned due to lack of evidence to support a cytotoxic concentration. This last behavior is indicative of generalized cell stress-related assay interference. Cytotoxicity-based interference is characterized by a Z-score which measures where specific activity occurs relative to cytotoxic concentrations. A large Z-score (>3) indicates that specific activity occurs well below the cytotoxic level (Judson et al., 2016). The revised schema for the CS is given in Table 2. These weightings were somewhat arbitrary but were meant to emphasize what the authors felt were the most important factors for distinguishing true from false positive antagonist signals. The overall CS is the sum of the individual contributions, and the higher the score, the more confident one can be that the observed activity is due to antagonist activity rather than assay interference. Values can range from +7 to −4.
2.3. Subset models
The subset models are calculated using exactly the same method and software as the full model but using only a subset of the assays. We scanned through all combinations of 2–14 assays (a total of 214−15 = 16369 combinations) and calculated pathway AUC values for agonist, antagonist and other modes for all chemicals. As an example, when running the model with assays A1, A2, A4, A13 and A14 (refer to Fig. 1), only interference processes associated with N1, N7 and the individual assays will be included. For more details, refer to the original methodology of Kleinstreuer et al. (Kleinstreuer et al., 2017). This is significantly different than what was done for the ER minimal assay model, where we simply fit a linear model to the full assay AUC (agonist) values. The approach used here has the advantage that the subset models explicitly account for assay interference processes, just like with the full model, at the expense of increased computing time (~7 days on a 40-core Linux machine).
Subset models are named with the binary string corresponding to the assays used, prefixed with the letter A. For example, the model using the first two assays in Table 1 is named A11000000000000. We assume that the full 14-assay model provides the best estimate of the true AR activity of a chemical, but is still an estimate, and in particular still has uncertainties in determining weak antagonist effects (both false positives and false negatives). Several metrics were calculated comparing the AUC (agonist) and AUC (antagonist) values between each subset model and the full model, including the R2 (Pearson correlation coefficient) and root mean square error (RMSE) between the AUC(subset) and AUC (full). We also dichotomized the subset and full AUC values as follows to obtain a binary active/inactive call. All AUC values less than a cutoff of 0.1 are set to zero, and those at or above 0.1 are set to one. This cutoff corresponds to a potency of ~200 μM, which is the upper limit of testing in any of the in vitro assays (see Supplemental Fig. 1). (In the original full model, an intermediate group of chemicals with AUC in the range from 0.001 to 0.1 were classified as inconclusive, but that rule is not used in this calculation as it would artificially inflate the accuracy of this calculation.) It is possible for chemicals to be classified as active in both agonist and antagonist, and these could be selective androgen receptor modulators (SARMs). However, it is not possible to make a definitive call that a chemical is or is not a SARM based on the current data. Based on the dichotomized data, we calculate sensitivity, specificity, balanced accuracy and PPV and NPV (positive and negative predictive values) between the subset and full model, and between the subset model and the in vitro and in vivo reference chemical calls from the literature. Balanced accuracy (BA) is the average of sensitivity and specificity.
Uncertainty:
To understand the effect of assay uncertainty on the predictions, all chemical assay combinations were processed through the ToxBoot process (Watt and Judson, 2018) which produces 95% confidence intervals (CI) for the potency (AC50), and yields a probability from 0 to 1 that the chemical is active in the assay. The underlying algorithm uses a smooth nonparametric bootstrap method, selecting subsets of the concentration-response data and refitting the data 1000 times. Summary values from the 1000 fits of the bootstrap resampled data are available publicly in the ToxCast database, invitrodb (version 3.2), as level 7 data, including the width of the CI for the AC50 and the percent of refitted curves that were positive (hit percent). The full 14-assay AR model was run independently 100 times, where in each run, the assay-wise AC50 values were drawn from a normal distribution centered on the nominal value and having a standard deviation derived from the Toxboot AC50 distribution. The distribution of performance statistics for these models were used to judge the relative performance of the subset models.
2.4. Model application
Beyond the 1820 chemicals originally used to develop the full and subset models, we have data for up to 1000 additional chemicals in selected assays (A1, A2, A3, A7, A9, A10, A13 and A14; see Fig. 1). Using these additional data and the corresponding best available subset models, we can make forward predictions of AR agonist and antagonist activity. We combined the AUC values from the models with QSAR predictions from the Collaborative Modeling Project for Androgen Receptor Activity (CoMPARA) project, which developed 91 AR QSAR models in binding, agonist and antagonist modes that were then combined into consensus predictions (Mansouri, 2019; Mansouri et al., 2020). Of the 55,450 structures in the CoMPARA prediction set, the consensus models predicted that 19% would be active in binding mode, 3.9% would be active in agonist mode, and 21.9% would be active in antagonist mode. Subsequently, consensus predictions for ~750,000 chemicals have been calculated and are available from the EPA CompTox Chemicals Dashboard (https://comptox.epa.gov/dashboard).
2.5. Toxicokinetics
To facilitate model comparisons with the in vivo reference chemicals, lowest observed effect levels (LOELs) from the animal studies curated in Browne et al. (Browne et al., 2018; Kleinstreuer et al., 2018) were converted to internal concentrations using a standard in vitro toxicokinetics method (Rotroff et al., 2010; Wetmore et al., 2012; Wetmore et al., 2014; Wambaugh et al., 2015), implemented in the httk R package version 2.0.1 (Pearce et al., 2017) using the steady-state assumption. Briefly, fraction of chemical unbound to plasma protein (Fup) and intrinsic hepatic clearance (Clint) parameters are used to calculate the plasma concentrations relevant to specific dosing assumptions. Details are given below in the results section. To convert in vitro concentrations to in vivo doses, it is necessary to convert AUC values to concentrations, which is performed using the calibration curve of Supplemental File S1. Finasteride toxicokinetic parameters (Fup and Clint) were not available from the httk package so were taken from Zager and Barton, (2012). For all other chemicals described below, experimental toxicokinetic parameters Fup and Clint were used, i.e. no in silico predictions of these parameters were employed.
2.6. Software
All software used to run the calculations is written in R, version 3.5.2 (Ihaka and Gentleman, 1996) and is available from the author’s FTP site [ftp://newftp.epa.gov/COMPTOX/STAFF/rjudson/publications/Judson%20AR%202019/ (to be updated on final publication)]. This includes an R package vignette describing how to run all calculations used in this manuscript.
3. Results
There are 1239 of the 1820 chemicals that are active in at least one of the 14 assays. A heatmap of the AC50 values for the active chemicals and assays are shown in Supplemental Fig. S2. The variety of patterns of activity highlight the need for a model that accounts for both true activity and assay interference. All results for the new model are provided in Supplemental Table S2. The AUC values for active chemicals in the new model are slightly lower on average than in the original model because new model values were normalized using the reference antagonist, Mifepristone, which is more potent than the previous normalization reference chemical (Hydroxyflutamide), which was not tested in all of the new assays. In the new full model, the antagonist confidence score (CS) threshold of 2 was selected for comparing with the subset models (CS ≥ 2 high confidence, CS < 2 low confidence) because all but two of the in vitro reference chemical CS values were 2 or greater (Seven had a CS = 7, two had CS = 5, four had CS = 4, three had CS = 3, and one each had CS of 2, 1 and 0). The three lowest CS-value positive antagonist reference chemicals also had the lowest values of AUC (antagonist): o,p’-DDT (AUC = 0.15, CS = 1), Methoxychlor (AUC = 0.075, CS = 0), and Fenarimol (AUC = 0.065, CS = 2). The corresponding values for these chemicals in the original model were o,p’-DDT (AUC = 0.15, CS = 4), Methoxychlor (AUC = 0.04, CS = 2), and Fenarimol (AUC = 0.04, CS = 4). For these chemicals, the confidence scores have decreased because of the added penalties for cytotoxicity and other factors summarized in Table 2. The reference chemical activity from the original and new full models are shown in Supplemental Fig. S3. The main difference is that the positive antagonist reference chemical Zearalenone was inactive in the original full model but is now active (AUC (antagonist) = 0.125, CS = 3), due to strong activity in the UPitt nuclear translocation assays (A6 and A12), which reinforces the weak antagonist evidence seen without these assays. For reference, we list the most potent agonists and antagonists in the full model. The values in parentheses are the AUC values in the current full model vs. those in the original AR model of Kleinstreuer et al. The most potent agonists are 17beta-Trenbolone (1.29/1.59), 17-Methyltestosterone (1.29/1.55), 5alpha-Dihydrotestosterone (1.28/1.57), Testosterone propionate (1.23/1.53) and Norethindrone (1.26/1.52). The most potent antagonists in the full model are Mifepristone (1.0/1.21), Triphenyltin hydroxide (0.82/0.79), Tributyltin methacrylate (0.72/0.61), Nilutamide (0.71/0.85) and Dibutyltin dichloride (0.70/0.55). The cutoff of 0.1 for AUC (agonist) and AUC (antagonist) and an antagonist CS cutoff of 2 are used for calculating useful statistical correlations between the full and subset models. The quantitative AUC values for the original (11 assays) and new (14 assays) full models are highly correlated with R2 = 0.96 and RMSE = 0.022 for AUC (agonist) and 0.87 and 0.04 for AUC (antagonist). Plots showing the correlations are shown in Supplemental Fig. S4.
Subset models were evaluated against the revised full model using continuous statistics of R2 and RMSE for AUC(agonist) and AUC (antagonist), and discrete statistics of sensitivity, specificity and balanced accuracy after dichotomizing the chemicals with AUC≥0.1 set to positive (1) and those with AUC<0.1 set to negative (0). In reality, there is not a sharp distinction between chemicals active or inactive against AR based on the 0.1 AUC threshold. Instead, the larger the AUC values and the confidence score, the greater is the likelihood that there is true chemical-AR interaction. To judge the significance of the statistical differences between subset models and the full model by establishing a confidence interval for the full model, the full model was run 100 times adding the known experimental variability in chemical-assay potency. For each of these runs, the winning model (Hill, Gain-Loss or constant) was determined, along with its corresponding potency and efficacy parameters (AC50 and Top). The 95% CI for BA in agonist mode is [0.93,1.0] and for antagonist mode is [0.97,0.99].
The results of building subset models using increasing numbers of assays are summarized in Fig. 2. This shows the performance of the best-performing subset model as a function of the number of assays, as compared to the full model. First, performance based on reference chemicals was evaluated; except for the best 2-assay antagonist model and the best 4 assay agonist model, all of the highest performing subset models have 100% sensitivity, specificity and BA for the in vitro reference chemicals (red diamonds in Fig. 2). BA was also assessed for the larger chemical set. Beyond two assays, statistical agreement using all measures with the full model increases with the number of assays in each subset model. There are agonist subset models with as few as 6 assays and there are antagonist subset models with as few as 5 assays that have BA>0.95. Variability around the agonist full model is greater than around the antagonist model so that the best 5 assay agonist models are not statistically different from the full model once its variability is accounted for. In antagonist mode, only subset models with 6 assays have BA within the CI for the full model. Continuous measures of subset model performance as compared to the full model were also considered. For agonist mode (Fig. 2A), the R2 demonstrated improvement with addition of more assay data with the exception of a dip going from 2 to 3 assays. The best subset model with 6 assays had an R2 of 0.85. Adding more assays provided marginal improvement, with R2 approaching 1 at 10 or more assays. The antagonist mode models reach a plateau of an R2 between 0.85 and 0.9 using 4 assays and show little improvement until 10 or more assays are included.
Fig. 2.
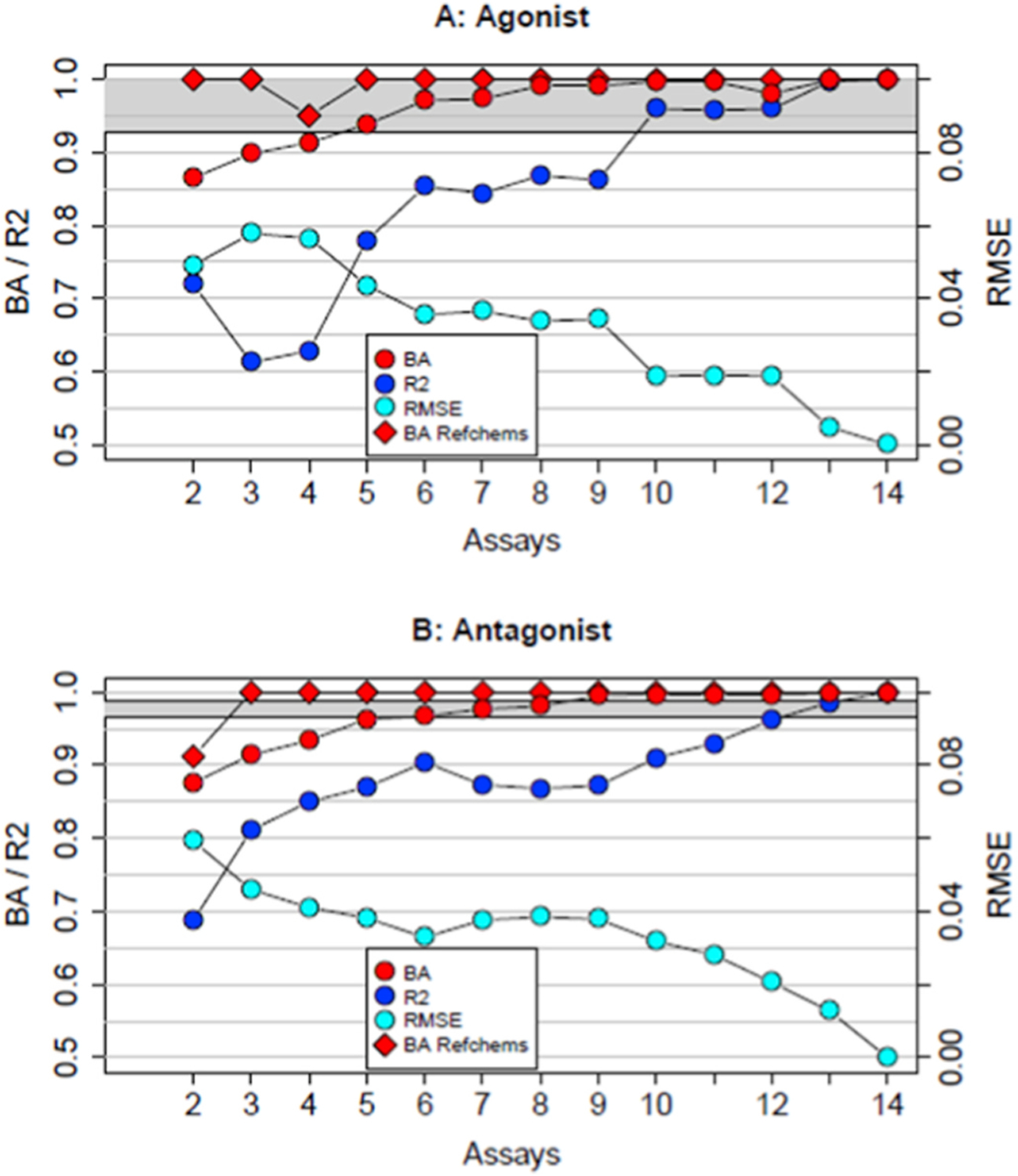
Summary of the best performing subset models for each number of assays. The model BA values are shown in red circles for all chemicals, and in red diamonds for the in vitro reference chemicals. The values of R2 (all chemicals) are shown in blue circles and for RMSE (all chemicals) in cyan circles. The y-scale for BA and R2 is on the left, and for RMSE is on the right. Panel A shows data for agonist mode and panel B shows data for antagonist mode. The grey bands show the 95% CI for the full model BA based on assay potency variability.
As an example, we show the results for the best performing 5-assay antagonist subset model (A01010000000111) in Fig. 3. This model uses assays A2, A4, A12, A13, and A14 from Table 1 and Fig. 1. These include one of the cell-free binding assays, one of the cofactor recruitment assays and all three antagonist assays. Note that these are all agonist/antagonist or antagonist-specific assays – no agonist-specific assays are included. The RMSE between the full and subset model AUC (antagonist) values is 0.038 and R2 is 0.87. As described in Methods, the continuous AUC values were dichotomized with values ≥ 0.1 set to active and those <0.1 set to inactive. This antagonist subset model has sensitivity of 0.97, specificity of 0.96 and BA of 0.96 for all chemicals with respect to the full model. For the in vitro reference chemicals, the classification statistics all equal 1. The all-chemical agonist mode statistics for this model are well below that for models optimized for agonist activity, which illustrates an important factor concerning these subset models with few assays, namely that they may perform well for one mode (agonist or antagonist) but not for both. The quantitative output for all subset models in agonist and antagonist modes are given in Supplemental Tables S3 and S4.
Fig. 3.
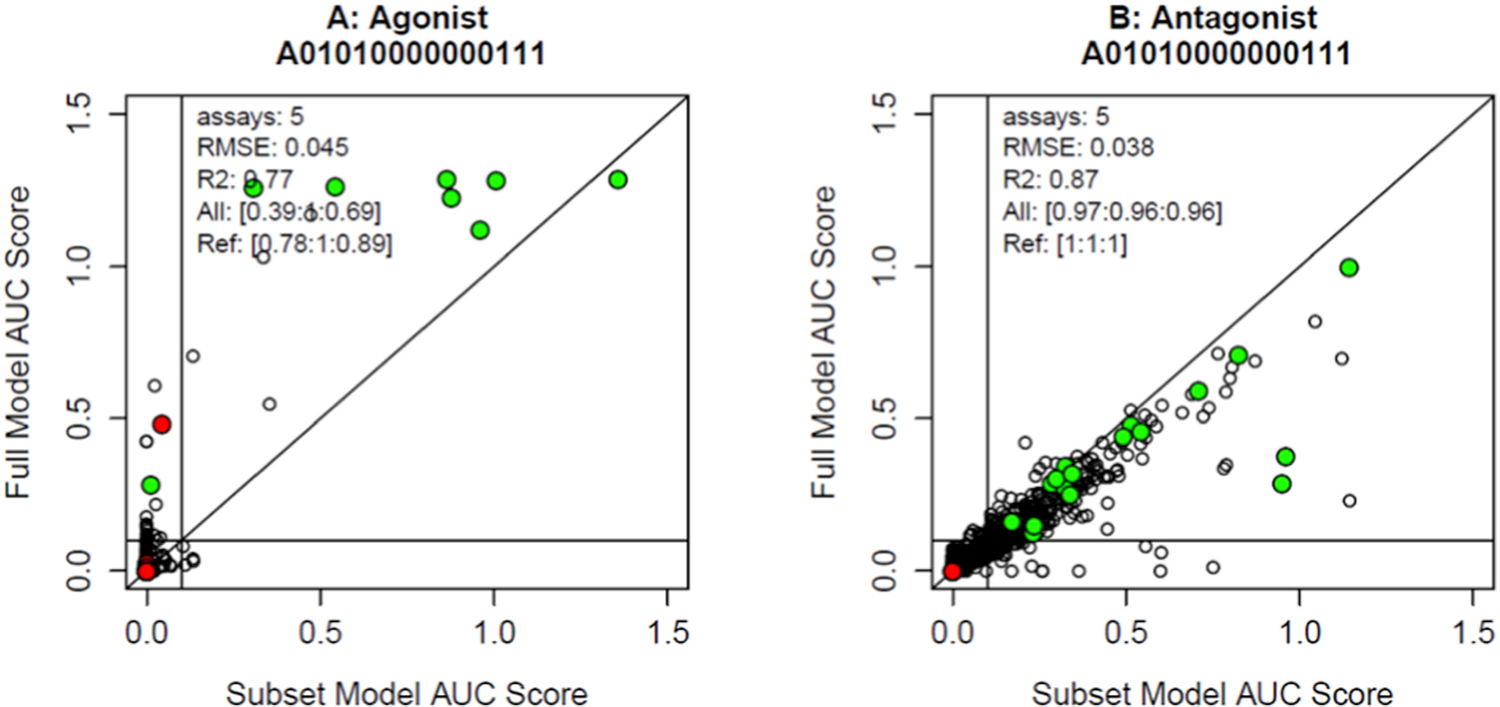
Results of the full model versus the subset model A01010000000111 across the 1820 chemicals. The x-axis is the subset model AUC and the y-axis is the full model AUC. Each point represents a chemical. Colored points are in vitro reference chemicals for the respective modes, where green are positive reference chemicals and red are negative. Panel A shows results for the agonist mode, and panel B shows results for the antagonist mode. In each mode, the legend provides the number of assays, the values of RMSE and R2, and in brackets the sensitivity, specificity and BA for all chemicals and the in vitro reference chemicals. There are horizonal and vertical lines at AUC = 0.1.
In antagonist mode, there are 30 subset models with 5 or 6 assays with BA for all chemicals of 0.95 or better and with BA = 1 for the in vitro reference chemicals. However, the performance of these subset models is generally not concordant with the in vivo antagonist reference chemical activity and is qualitatively the same as was seen in the original full model discussed previously (Kleinstreuer et al., 2018), and will be discussed in more detail below. Of the antagonist subset models with high performance against the in vitro reference chemicals, all 30 use the cell-based antagonist assays A12 (nuclear translocation) and A14 (reporter gene); 29 use the cell-based reporter gene assay A13; 21 use the cofactor recruitment assay A5; and all use one or two of the three receptor binding assays (A1-A3). The most commonly used cell-free binding assay is the chimpanzee version, used in 20 models. The complete scoring matrix for the antagonist subset models is given in Supplemental Table S4.
In agonist mode, there are 26 subset models with 6 or 7 assays that have BA for all chemicals ≥0.95 and BA = 1 for the in vitro and in vivo agonist reference chemicals. Of these agonist subset models, all 26 use assays A9 and A11, 23 use assay A7, 19 use A8 and all use at least one of the cell-free binding assays (18 rat, 10 chimpanzee, 5 human). Three of the models use the antagonist assay A14, which is likely just a result of noise in the data. The best 5-assay agonist subset model has BA = 0.94 for all chemicals, and the best 4-assay agonist subset model has BA = 0.91. These smaller models have high specificity but lower sensitivity (0.82–0.89). The complete scoring matrix for the agonist subset models is given in Supplemental Table S3.
We next demonstrate a direct use of the current assays and the subset models built from them. Beyond the 1820 chemicals originally used to develop the full and subset models, we have data for up to 1000 additional chemicals in selected assays. Therefore, for subset models using only these additional data, we can make forward predictions of AR agonist and antagonist activity. To illustrate this capability, we selected the best available agonist and antagonist subset models that only used assays with data for additional chemicals and applied them to the “forward testing” chemical set. To illustrate how one could build a weight-of-evidence approach to prioritizing chemicals for further testing, we combined the AUC values from the models with the consensus QSAR predictions from the Collaborative Modeling Project for Androgen Receptor Activity (CoMPARA) project, described in the Methods section (Mansouri, 2019; Mansouri et al., 2020).
The agonist subset model selected to illustrate this approach is A11000010100000, using the assays A1, A2, A7 and A9 (two binding assay and two agonist transactivation assay). This is not a particularly well-performing subset model because it contains neither the cell proliferation nor the nuclear translocation assays, both of which significantly increase agonist assay performance. These assays were excluded because no additional chemicals beyond the 1820 were tested in these assays. The performance of this model for all chemicals is sensitivity = 0.82 and specificity = 0.99. Because of the high specificity, positive results of this model should be accurate. However, with relatively low sensitivity, some true positive chemicals will be missed. There were a total of 884 chemicals with forward testing data for all of these assays (A1, A2, A7, A9), and 10 have AUC(agonist) > 0.1 (1.3%), listed in Table 3. The full data set for this model is given in Supplemental Table S5. Of these, 7 have steroid backbones and these are predicted to be 7 of the 8 most potent of the actives. The CoMPARA QSAR predictions indicate that all but two of these chemicals will be active against AR in one or both of agonist or antagonist mode (7-Diethylamino-4-methylcoumarin and Tripropylene glycol diacrylate are the exceptions). There is one chemical where the QSAR results predict it to be an antagonist rather than an agonist (Tetradonium bromide), but the antagonist AUC is much smaller than the agonist AUC (AUC (agonist) = 0.14, AUC (antagonist) = 0.01).
Table 3.
New chemicals predicted to have AR agonist activity using the agonist subset model A11000010100000.
| CASRN | Name | AUC (Agonist) | Steroid | CoMPARAa |
|---|---|---|---|---|
| 71–58–9 | Medroxyprogesterone acetate | 1.28 | Y | 111 |
| 17230–88–5 | Danazol | 1.02 | Y | 111 |
| 566–48–3 | 4-Hydroxyandrostenedione | 0.54 | Y | 110 |
| 434–03–7 | Ethisterone | 0.41 | Y | 110 |
| 6533–00–2 | dl-Norgestrel | 0.32 | Y | 110 |
| 52–76–6 | Lynestrenol | 0.31 | Y | 111 |
| 91–44–1 | 7-Diethylamino-4-methylcoumarin | 0.26 | N | 000 |
| 313–06–4 | Estradiol cypionate | 0.23 | Y | 111 |
| 42978–66–5 | Tripropylene glycol diacrylate | 0.16 | N | 000 |
| 1119–97–7 | Tetradonium bromide | 0.14 | N | 101 |
These binary values indicate activity in binding, agonist, antagonist modes. For example, 101 indicates an active prediction for antagonist and binding modes and inactive prediction for agonist mode.
The antagonist subset model selected is A01010000000111 (see Fig. 3), using assays A2, A4 and A12-A14 (one binding assay, one cofactor recruitment assay, the antagonist nuclear translocation assay and two antagonist transactivation assays). This is the best 5-assay subset model with all chemical BA = 0.96 and BA = 1 for the in vitro reference chemicals. A total of only 31 new chemicals had data for all five of these assays for use in forward prediction. Six had AUC (antagonist) > 0 and are listed in Table 4. All data for this set of 31 chemicals is given in Supplemental Table S6. Oryzalin was active in the original full AR model with AUC(antagonist) = 0.15. This chemical is not structurally similar to any other AR antagonists and the CoMPARA QSAR project predicts that it is inactive in both agonist and antagonist mode. 3,3′-Dimethoxybenzidine was borderline negative (AUC (antagonist) = 0.07) in the original full model, Triphenyl phosphate was inactive in the original full model with AUC(antagonist) = 0.04 and PharmaGSID_47315 (a failed pharmaceutical compound) was active in the original model with AUC (antagonist) = 0.22.
Table 4.
New chemicals predicted to have AR antagonist activity using the subset model A01010000000111.
| CASRN | Name | AUC (antagonist)a | CoMPARAb |
|---|---|---|---|
| 19044–88–3 | Oryzalin | 0.27 [0.15] | 000 |
| 119–90–4 | 3,3′-Dimethoxybenzidine | 0.24 [0.07] | 101 |
| 115–86–6 | Triphenyl phosphate | 0.09 [0.04] | 000 |
| 444610–91–7 | PharmaGSID_47315 | 0.08 [0.22] | 101 |
| 120–11–6 | 1-Benzyloxy-2-methoxy-4-(1-propenyl)benzene | 0.08 | 000 |
| 754–91–6 | Perfluorooctanesulfonamide | 0.05 | 000 |
Numbers are AUC (antagonist) values from the new subset model. Numbers in brackets are the corresponding values from the original full model of Kleinstreuer et al. Note that only a subset of the chemicals in the table were evaluated in that model.
The coding in this column indicates CoMPARA predicted activity in binding, agonist and antagonist modes.
The in vivo reference chemical set was identified from literature reviews and had reproducible in vivo activity in multiple Hershberger assays or one Hershberger assay and another in vivo assay with androgen responsive endpoints (Browne et al., 2018; Kleinstreuer et al., 2018). In vivo reference chemicals included 39 chemicals (3 active in the agonist mode, 20 active in the anti-androgenic mode and 16 negative). For the agonist mode in vivo reference chemicals, 99.5% of subset models with 5 or more assays had a BA of 1.0, meaning all in vivo reference chemicals were predicted correctly. However, there are only three in vivo reference agonists and all are highly potent pharmaceuticals (17beta-Trenbolone, 17-Methyltestosterone, Testosterone propionate).
For the antagonist in vivo reference chemicals (based on results of the Hershberger assay), neither the full model nor the subset models show good predictions. With the best subset model (A01010000000111), the comparison is shown in Table 5, there are 7 true positives, 13 false negatives (in vivo active, in vitro inactive), 5 false positives (in vivo inactive, in vitro active) and 10 true negatives. The performance for the full model was the same. For a safety screening method such as this, false negatives are of special concern. However, the analysis of the original full model relative to these antagonist Hershberger active chemicals found that most of the Hershberger positive, AR model negative, chemicals only showed activity at concentrations above the upper limit of testing in the in vitro assays (Browne et al., 2018; Kleinstreuer et al., 2018). We re-examined this issue with the current data set. LOEL values (in units of mg/kg/day) were taken from Table 1 of Kleinstreuer et al. (2018) (Kleinstreuer et al., 2018). These values were converted to internal concentrations using a standard in vitro toxicokinetics method (Rotroff et al., 2010; Wetmore et al., 2012; Wetmore et al., 2014; Wambaugh et al., 2015), implemented in the httk R package (v 2.0.1) (Pearce et al., 2017) using a time-dependent PBPK model using the option to model rat physiology. This model used the dosing schedule relevant to the Hershberger assay (one dose per day, for 10 days total). This model assumes 100% bioavailability. The result used was the average plasma concentration (Cavg). For some chemicals the PBPK model failed because the relevant experimental parameters were not available, so the analytical concentration at steady state (Css) was substituted. Hershberger LOEL values were converted to plasma concentrations by multiplying by Cavg. Cavg and Css are highly correlated with Cavg being larger on average by a factor of 1.4.
Table 5.
Comparison between the subset model A01010000000111and the Hershberger database results for antagonist mode.
| CASRN | Name | AUC Antagonista | AR Model AC50 (uM)b | AR Model POD (mg/kg/day)c | HB POD (mg/kg/day)d | HB In Vitro AC50 (uM)e | Cavg | AR Model Callf | HB DB Callg | Notes |
|---|---|---|---|---|---|---|---|---|---|---|
| 22248–79–9 | Z-Tetrachlorvinphos | 0.16 | 62.7 | 0.3 | >350 | >1000 | 324.27 | positive | negative | in vitro model active only near top of range tested |
| 133–07–3 | Folpet | 0.15 | 66.4 | 0.1 | >800 | >1000 | 13.86 | positive | negative | in vitro model active only near top of range tested |
| 113–48–4 | Octylbicycloheptenedicarboximide | 0.13 | 89.3 | 2.1 | >850 | >12.0 | 2.39 | positive | negative | in vitro model active only near top of range tested |
| 1897–45–6 | Chlorothalonil | 0.66 | 1.0 | 3.0 | >1000 | 0.1 | 0.84 | positive | negative | 1 negative HB study |
| 71751–41–2 | Abamectin | 0.45 | 5.0 | 7.7 | >5 | 4.8 | 0.48 | positive | negative | 1 negative HB study |
| 13311–84–7 | Flutamide | 0.54 | 2.5 | 37.7 | 0.1 | 1.6 | 0.32 | positive | antiandrogenic | agree positive |
| 50471–44–8 | Vinclozolin | 0.49 | 3.7 | 4.2 | 10 | 9.7 | 3.24 | positive | antiandrogenic | agree positive |
| 72–55–9 | p,p’-DDE | 0.34 | 12.1 | 37.8 | 5 | 3.6 | 0.36 | positive | antiandrogenic | agree positive |
| 32809–16–8 | Procymidone | 0.33 | 13.7 | 9.5 | 3 | 89.0 | 1.78 | positive | antiandrogenic | agree positive |
| 330–55–2 | Linuron | 0.33 | 13.8 | 0.4 | 10 | 1.0 | 125.00 | positive | antiandrogenic | agree positive |
| 67747–09–5 | Prochloraz | 0.30 | 17.0 | 20.5 | 50 | >240.3 | 20.03 | positive | antiandrogenic | agree positive |
| 98319–26–7 | Finasteride | 0.18 | 50.5 | 6.2 | 0.008 | >705.9 | 70.59 | positive | antiandrogenic | agree positive |
| 2921–88–2 | Chlorpyrifos | 0.04 | 409.8 | 144.1 | >12 | >2.1 | 6.94 | negative | negative | agree negative |
| 51–28–5 | 2,4-Dinitrophenol | 0.04 | 435.2 | 0.2 | >10 | >1000 | 6387.50 | negative | negative | agree negative |
| 61–82–5 | Amitrole | 0 | 1000.0 | 356.4 | >1000 | >1000 | 2.81 | negative | negative | agree negative |
| 1563–66–2 | Carbofuran | 0 | 1000.0 | 363.0 | >0.3 | >0.7 | 2.75 | negative | negative | agree negative |
| 66230–04–4 | Esfenvalerate | 0 | 1000.0 | 1339.2 | >9 | >123.4 | 0.75 | negative | negative | agree negative |
| 66332–96–5 | Flutolanil | 0 | 1000.0 | 648.2 | >1000 | >1.2 | 1.54 | negative | negative | agree negative |
| 57837–19–1 | Metalaxyl | 0 | 1000.0 | 408.4 | >375 | 60.1 | 2.45 | negative | negative | agree negative |
| 16752–77–5 | Methomyl | 0 | 1000.0 | 299.6 | >1 | 9.2 | 3.34 | negative | negative | agree negative |
| 21087–64–9 | Metribuzin | 0 | 1000.0 | 1089.4 | >80 | 41.9 | 0.92 | negative | negative | agree negative |
| 23135–22–0 | Oxamyl | 0 | 1000.0 | 120.0 | >0.5 | 13.3 | 2.79 | negative | negative | agree negative |
| 68359–37–5 | Cyfluthrin | 0 | 1000.0 | 1206.1 | 18 | 59.0 | 0.83 | negative | antiandrogenic | 2 negative HB studies, one positive |
| 52645–53–1 | Permethrin | 0 | 1000.0 | 1694.9 | 10 | 930.6 | 0.59 | negative | antiandrogenic | 2 negative HB studies, one positive |
| 2312–35–8 | Propargite | 0.005 | 335.0 | 33.1 | 15 | 162.5 | 4.65 | negative | antiandrogenic | 1 positive HB study |
| 13194–48–4 | Ethoprop | 0 | 1000.0 | 549.7 | 16 | 747.5 | 0.32 | negative | antiandrogenic | 1 positive HB study |
| 117–81–7 | Di (2-ethylhexyl) phthalate | 0.005 | 1000.0 | 1694.9 | 100 | 59.0 | 0.59 | negative | antiandrogenic | chemical does not act through AR |
| 60168–88–9 | Fenarimol | 0.08 | 154.2 | 33.1 | 200 | 930.6 | 4.65 | negative | antiandrogenic | in vitro HB AC50 > range tested |
| 51218–45–2 | Metolachlor | 0.08 | 178.6 | 549.7 | 500 | 162.5 | 0.32 | negative | antiandrogenic | in vitro HB AC50 > range tested |
| 84–74–2 | Dibutyl phthalate | 0 | 1000.0 | 668.9 | 500 | 747.5 | 1.49 | negative | antiandrogenic | in vitro HB AC50 > range tested |
| 1861–40–1 | Benfluralin | 0.003 | 1000.0 | 176.7 | 750 | 4245.0 | 5.66 | negative | antiandrogenic | in vitro HB AC50 > range tested |
| 36734–19–7 | Iprodione | 0 | 1000.0 | 104.9 | 200 | 1906.0 | 9.53 | negative | antiandrogenic | in vitro HB AC50 > range tested |
| 27314–13–2 | Norflurazon | 0 | 1000.0 | 2307.8 | 1000 | 433.3 | 0.43 | negative | antiandrogenic | in vitro HB AC50 > range tested |
| 23950–58–5 | Propyzamide | 0 | 1000.0 | 315.0 | 200 | 634.8 | 3.17 | negative | antiandrogenic | in vitro HB AC50 > range tested |
| 1582–09–8 | Trifluralin | 0.004 | 1000.0 | 1887.1 | 450 | 238.5 | 0.53 | negative | antiandrogenic | in vitro HB AC50 > range tested |
Notes:
AUC Antagonist from this subset model.
The nominal potency values (AC50) calculated using the AUC Antagonist value and the calibration curve.
The in vitro to in vivo extrapolation point of departure = AC50/Css.
The minimum LOEL for the chemical in the Hershberger database.
The in vitro equivalent potency for the Hershberger result = HB POD x Css.
The result is positive if AUC Antagonist ≥ 0.1 and negative otherwise.
Classification of Hershberger activity from (Kleinstreuer et al., 2018).
Of the 13 false negatives, 8 had predicted internal concentrations extrapolated from the in vivo LOEL range that were above the upper limit of in vitro testing. The remaining 5 of the 13 false negatives are as follows. Two chemicals have potentially ambiguous in vivo results with 2 positive studies and one negative apiece (Permethrin and Cyfluthrin). Another two have a single Hershberger study that was positive and were called positive due to a positive result in a related study type (Propargite and Ethoprop). Ethoprop’s single positive Hershberger assay had a LOEL of 16 mg/kg/day, equivalent to a predicted internal concentration of 13.3 μM. Ethoprop was inactive in all in vitro assays. Note that the Hershberger LOEL was 16 mg/kg/day, while the LD50 for Ethoprop is close to this, in the range of 26–34 mg/kg suggesting possible confounding general toxicity effects (see CompTox Chemicals Dashboard https://comptox.epa.gov/dashboard/dsstoxdb/results?search=DTXSID4032611#toxicity-values). The final false negative is Di (2-ethylhexyl) phthalate (DEHP). There is evidence that DEHP itself is not antiandrogenic but that its activity in the Hershberger assay is due to one or more metabolites and these may not be formed in vitro (Stroheker et al., 2005). Indeed, an alternative explanation for any of the false negatives is the lack of appropriate metabolic activation systems in the in vitro systems we have used.
Of the 5 false positive results (in vitro positive, in vivo negative), 3 have activity only near the top of the tested range (Folpet, Octylbicycloheptenedicarboximide and Z-Tetrachlorvinphos). It is always difficult to distinguish false from true in vitro activity in these cases, as activity can be confounded with cytotoxicity, and because there is usually only a single concentration with activity above the noise threshold, and this could just be an experimental artifact. The final 2 are Abamectin (single negative Hershberger study, only tested to 5 mg/kg/day) and Chlorothalonil (single negative study tested to 1000 mg/kg/day). These chemicals were classified as in vivo negative based on a combination of a single Hershberger result and a negative result in at least one other related in vivo assay. Both of these chemicals did not exhibit a statistically significant potency shift in the antagonist reporter gene confirmation assays. Further, all the false positive chemicals had relatively low overall confidence scores when evaluated in the full model (ranging from 0 to 2, with the exception of Chlorothalonil which had a score of 3). Another chemical of note based on mode of action is Finasteride, a 5α-reductase inhibitor. In this subset model, it is classified as an AR antagonist, with an AUC (antagonist) value of 0.18. In the full model, the AUC value is 0.09. It was weakly active (i.e., AC50 values > 10 μM) in 5 in vitro assays (A4, A5, A12, A13 and A14). While the pharmaceutical mode of action is through blocking formation of dihydrotestosterone from testosterone through 5α-reductase inhibition, the similarity of the finasteride steroid backbone to dihydrotestosterone may allow direct AR activity at higher concentrations. In summary, the majority of disagreements between the in vitro model and the in vivo antagonist reference chemicals may be due to the high doses/concentrations where activity was seen in vivo or confounding loss-of-signal effects due to generalized cell stress that precedes cytotoxicity. More detailed results for the full model are given in Supplemental Table S7.
It is important to note potential limitations of the toxicokinetic approach we have used. The underlying PBPK model is relatively simple (3 compartment) and generic. It assumes 100% oral bioavailability and only renal clearance. The accuracy can be limited in the cases of chemicals with low fraction unbound to plasma protein as the analytical method can be close to or below the level of detection. The model also assumes that the only route of metabolism is via liver. Comparisons with more detailed PBPK models and direct PK experiments show that in about 65% of chemicals, the value of Css is within a factor of 3.2 of the value obtained experimentally, an additional 28% were off by as much as 6-fold, and 8% of chemicals could be off by 2 orders of magnitude (Wambaugh et al., 2015).
A final issue to consider is the quantitative errors of prediction of Hershberger LOEL values using the IVIVE approach. There are 7 chemicals (the true positives) in the Hershberger reference set with Hershberger LOEL values (taken as the lowest of the LOELs across all studies for a given chemical). The root mean square error (RMSE) between the Hershberger LOEL and the IVIVE LOEL is 0.85 log (mg/kg/day). To calculate the IVIVE LOEL, we converted the antagonist AUC to a concentration using the calibration curve (Supplemental Fig. S1) and then divided by Cavg or Css. Values are given in Supplemental Table S7, column R. We compared this with the variation among Hershberger studies in the database of Browne et al.(Browne et al., 2018) The corresponding RMSE comparing one Hershberger LOEL to another for chemicals with replicate studies is 0.75 log (mg/kg/day), close to the value for Hershberger-to-IVIVE RMSE. A recent study [Pham et al. submitted] examining variability in vivo LOEL values across multiple study types reported RMSE values in the range of 0.47–0.63 log (mg/kg/day), which indicates that the Hershberger variability is at the high end of this spectrum.
4. Discussion
We have described the construction of models of AR agonist and antagonist activity that use subsets of assays taken from a modified version of the full AR pathway model of Kleinstreuer et al. (2017). There is one agonist subset model with optimal performance with only 6 assays, but to achieve optimal performance for the antagonist mode, models can be built with as few as 5 assays. Optimal models for agonism are non-optimal for antagonism and vice versa (see Fig. 3). In both cases, the best subset of assays used multiple technologies and cell types, at different nodes in the AR signaling pathway. The best agonist subset models primarily used assays from the agonist nodes in Fig. 1 (N1–N5), while the best antagonist models used assays from the antagonist nodes (N1, N2, N6 and N7). This is all consistent with the results of the corresponding ER agonist subset modeling approach. As observed here, the sensitivity of a single assay for AR pathway activity (in agonist or antagonist mode) cannot achieve the same BA and sensitivity of a multi-assay model, emphasizing the need for an integrated approach to testing and assessment (IATA (OECD, 2019)) that employ multiple assays with different sources of potential interference, domains of applicability, and coverage of key biological events. We note here that the approach taken in this work is intended to provide flexibility in precisely which assays (potentially including in silico models) one would need to run to determine AR activity.
Not all of the AR assays are currently available through the original sources, so in order to implement subset models, one may need to find existing or develop new assays that have similar behavior to the ones selected for the optimal subsets. In general, the high-performing agonist models use a cell-free receptor binding assay, a cofactor recruitment assay, a transactivation assay, and a cell proliferation assay. These probe the main points of the pathway (binding, cofactor-recruitment, transcript/translation and proliferation) and use different and complementary readout technologies. In general, the high performing antagonist subset models use a binding assay, a cofactor recruitment assay and two or three complementary antagonist-mode transactivation assays. As with the agonist mode, the assays probe the main nodes in the antagonist pathway (binding, coregulator interaction, and inhibition of transactivation). The main difference is that multiple assays per node are needed, likely because of factors mentioned above: most antagonists have weak potency resulting in lower signal-to-noise ratios in the assays, which make it more difficult to distinguish true positives from noisy false positives. Additionally, antagonist assays are often confounded with cytotoxicity.
Given these points, we can make general recommendations for implementing new versions of these subset models. First, the currently used assays are either available commercially, or could be implemented using published protocols. With new assays, it would be necessary to calibrate the model performance against a set of reference chemicals. These might extend beyond the set used here. Fig. 2 shows that assay subset models with only one or two assays can have 100% predictivity of the in vitro reference chemicals but will not perform as well with a larger set of chemicals. Although the full model on the full set of chemicals is not necessarily the “truth”, it does provide a comprehensive benchmark against a large set of chemicals using a biologically based model of AR activity. One approach would be to supplement the initial reference chemicals with a set of borderline chemicals with which to characterize the performance of the new subset model against the full model. Here “borderline” means chemicals that are known to be active against AR but at concentrations near or above the typical in vitro testing range of ~100 μM. Given variable sensitivity of in vitro assays, some assays will detect these chemicals as active and some will not. For example, one could select a set of chemicals with AUC values (agonist or antagonist as required) in the range of 0.001–0.2 from Supplemental File S2. In agonist mode, there are 218 chemicals in this range, and in antagonist mode, there are 89 chemicals in this range with antagonist confidence score ≥2. Given a set of assay data (AC50, Top, Hitcall), our software could be used to calculate AUC(agonist) and AUC(antagonist). One feature of the full model that cannot in general be calculated for the subset models is the antagonist confidence score (CS). As shown in Table 2 and the accompanying description, this includes weights related to the AUC value, the presence of cytotoxicity in the same concentration range as apparent antagonist activity, the use of a confirmatory antagonist assay with a different concentration of the reference agonist, the comparison of AUC(antagonist) with assay interference AUC values and a confirmation that both cell free and cell-based assays show activity. Therefore, a further recommendation would be to include cytotoxicity assays and a confirmatory antagonist assay, at least for chemicals with an initial indication of antagonist activity. Note that the CS is mainly used to confirm true positives, and to exclude false positives, but not to protect against false negatives. Supplemental Fig. S5 show the relationship between the CS and AUC(Antagonist) showing that there is a strong relationship between these two values, but that there are cases where CS < 2 what AUC(Antagonist) well above 0.1.
To compare model predictions with in vivo effects, it is desirable to have a reliable set of in vivo reference chemicals. In this case, we compared subset models to reference chemicals identified in a literature review of Hershberger and other in vivo assays measuring androgen-responsive endpoints, systematically identified for this purpose. It should be noted that the aim of comparing the AR subset model with the in vivo reference chemicals identified from a review of Hershberger studies was not to predict the Hershberger response, but rather to predict chemicals that may interact with the AR in vivo. The Hershberger assay has been used historically to evaluate chemicals for their (anti) androgenic effects, but the assay has inherent variability and there are a variety of factors that may confound results (see Browne et al., 2018; Kleinstreuer et al., 2018 for more detailed discussion). Though the Hershberger assay and other in vivo tests that measure effects of test chemical on androgen responsive endpoints account for a more complete set of responses occurring in situ (e.g. effects of hepatic metabolism on parent chemical activity), results may be difficult to interpret due to the interaction of other targets and organ systems and a variety of compensatory mechanisms. Hence, results of the subset of multiple in vitro assays may better predict the effects of chemicals specifically on the AR pathway, and may provide high confidence in both the “true” positives acting through the steroid receptor and the “true” negatives due to the integration of multiple in vitro assays that account for different points in the signaling pathway using variable technologies.
Consideration should be given to the in vivo LOEL values of the reference chemicals as well. Using an in vitro to in vivo extrapolation approach could allow for adjustment of in vitro screening concentrations for more equitable comparison of results. Alternatively, one could use in vitro toxicokinetic calculations to aid in selecting the reference set of chemicals most appropriate for the in vitro testing data with respect to upper testing concentrations. Note that there may be some tradeoff in testing at higher concentrations in the form of a higher rate of assay artifacts generated. As demonstrated here, and discussed in more detail in (Kleinstreuer et al., 2018), much of the discordance between the in vitro model predictions and the in vivo results was likely due to relatively high-dose effects in the animal studies resulting in internal concentrations above the maximum tested concentrations in vitro.
A number of limitations of the AR pathway model are not addressed here. These limitations include thematic issues in high-throughput screening for bioactivity. First, the assays used in the current AR pathway model lack substantial xenobiotic metabolism and would require retrofitting with technology to confer this critical physiological capability (DeGroot et al., 2018). This would require additional screening, which would likely be resource-intensive. Further, there may be thousands of substances under the regulatory purview of multiple agencies internationally which require some level of information regarding AR pathway activity. This high number of substances may make in vitro screening, even in 5–6 assays, relatively impractical. Thus, use of quantitative structure activity response (QSAR) models may inform which substances should be prioritized for experimentally evaluating for AR pathway activity. Global crowdsourcing approaches have been leveraged in the CoMPARA work to build such QSAR models and screen the EDSP universe of chemicals (Mansouri, 2019). This set of QSAR models, and the overall CoMPARA consensus, were built using the complete data set we started with here. However, in forward validation of the CoMPARA models against an external test set from the literature, BA values were in the range of 0.70–0.75, which is significantly lower than what is seen with the in vitro subset models. Regardless, chemicals with evidence of AR activity from both the subset in vitro models and the QSAR models could be candidates for follow-up investigation. It is important to note that for AR antagonist activity in particular, the confidence scores and the AUC values must be considered in parallel.
The ER version of this approach was recently published by OECD as an IATA (OECD, 2019; U.S. EPA, 2019). We anticipate following a similar approach with the AR model and the minimal assay subsets. An IATA may be developed that includes other in silico/in vitro data and improves predictive performance and reduces limitation of the current model. An expected outcome of that process will be further definition of the requirements for validating specific implementations of the subset models, in particular the selection of evaluation chemicals and performance characteristics. The end result of this process will be internationally acceptable approaches to efficiently evaluate chemicals, with the current caveat of lacking metabolic activation, for their ability to interact with the ER and AR pathways.
Supplementary Material
Funding information
This work was wholly funded by the US Environmental Protection Agency.
Footnotes
Publisher's Disclaimer: Disclaimer
The views expressed in this article are those of the authors and do not necessarily reflect the views of policies of the U.S. Environmental Protection Agency, NICEATM or OECD. Mention of trade names or commercial products does not constitute endorsement or recommendation for use.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary data
For review, this material is available at ftp://newftp.epa.gov/COMPTOX/STAFF/rjudson/publications/Judson%20AR%202019/
Supplementary data to this article can be found online at https://doi.org/10.1016/j.yrtph.2020.104764.
References
- Browne P, Judson RS, Casey WM, Kleinstreuer NC, Thomas RS, 2015. Screening chemicals for estrogen receptor bioactivity using a computational model. Environ. Sci. Technol 49 (14), 8804–8814. [DOI] [PubMed] [Google Scholar]
- Browne P, Kleinstreuer NC, Ceger P, Deisenroth C, Baker N, Markey K, Thomas RS, Judson RJ, Casey W, 2018. Development of a curated Hershberger database. Reprod. Toxicol 81, 259–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns RF, Watson IA, 2012. Rules for identifying potentially reactive or promiscuous compounds. J. Med. Chem 55 (22), 9763–9772. [DOI] [PubMed] [Google Scholar]
- DeGroot DE, Swank A, Thomas RS, Strynar M, Lee MY, Carmichael PL, Simmons SO, 2018. mRNA transfection retrofits cell-based assays with xenobiotic metabolism. J. Pharmacol. Toxicol. Methods 92, 77–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dix DJ, Houck KA, Martin MT, Richard AM, Setzer RW, Kavlock RJ, 2007. The ToxCast program for prioritizing toxicity testing of environmental chemicals. Toxicol. Sci 95 (1), 5–12. [DOI] [PubMed] [Google Scholar]
- EFSA, 2018. Guidance for the Identification of Endocrine Disruptors in the Context of Regulations (EU) No 528/2012 and (EC) No 1107/2009 [DOI] [PMC free article] [PubMed]
- Fancher AT, Hua Y, Camarco DP, Close DA, Strock CJ, Johnston PA, 2018. High-Content screening campaign to identify compounds that inhibit or disrupt androgen receptor-transcriptional intermediary factor 2 protein-protein interactions for the treatment of prostate cancer. Assay Drug Dev. Technol 16 (6), 297–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filer DL, Kothiya P, Setzer RW, Judson RS, Martin MT, 2017. tcpl: the ToxCast pipeline for high-throughput screening data. Bioinformatics 33 (4), 618–620. [DOI] [PubMed] [Google Scholar]
- Freyberger A, Schladt L, 2009. Evaluation of the rodent Hershberger bioassay on intact juvenile males–testing of coded chemicals and supplementary biochemical investigations. Toxicology 262 (2), 114–120. [DOI] [PubMed] [Google Scholar]
- ECCC/HC (2016) Government of Canada, 2016. Chemicals Management Plan (CMP) Science Committee Objectives Paper Meeting No. 5 - Integrating New Approach Methodologies within the CMP: Identifying Priorities for Risk Assessment Existing Substances Risk Assessment Program. [Google Scholar]
- Hsieh JH, Sedykh A, Huang R, Xia M, Tice RR, 2015. A data analysis pipeline accounting for artifacts in Tox21 quantitative high-throughput screening assays. J. Biomol. Screen 20 (7), 887–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Y, Camarco DP, Strock CJ, Johnston PA, 2018. High content positional biosensor assay to screen for compounds that prevent or disrupt androgen receptor and transcription intermediary factor 2 protein-protein interactions. Methods Mol. Biol 1683, 211–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Y, Shun TY, Strock CJ, Johnston PA, 2014. High-content positional biosensor screening assay for compounds to prevent or disrupt androgen receptor and transcriptional intermediary factor 2 protein-protein interactions. Assay Drug Dev. Technol 12 (7), 395–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihaka R, Gentleman R, 1996. R: a language for data analysis and graphics. J.Comput and Graphical Statistics 5, 299–314. [Google Scholar]
- Judson R, Houck K, Martin M, Richard AM, Knudsen TB, Shah I, Little S, Wambaugh J, Woodrow Setzer R, Kothya P, Phuong J, Filer D, Smith D, Reif D, Rotroff D, Kleinstreuer N, Sipes N, Xia M, Huang R, Crofton K, Thomas RS, 2016. Analysis of the effects of cell stress and cytotoxicity on in vitro assay activity across a diverse chemical and assay space. Toxicol. Sci 152 (2), 323–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judson RS, Houck KA, Kavlock RJ, Knudsen TB, Martin MT, Mortensen HM, Reif DM, Rotroff DM, Shah I, Richard AM, Dix DJ, 2010. In vitro screening of environmental chemicals for targeted testing prioritization: the ToxCast project. Environ. Health Perspect 118 (4), 485–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judson RS, Houck KA, Watt ED, Thomas RS, 2017. On selecting a minimal set of in vitro assays to reliably determine estrogen agonist activity. Regul. Toxicol. Pharmacol 91, 39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judson RS, Magpantay FM, Chickarmane V, Haskell C, Tania N, Taylor J, Xia M, Huang R, Rotroff DM, Filer DL, Houck KA, Martin MT, Sipes N, Richard AM, Mansouri K, Setzer RW, Knudsen TB, Crofton KM, Thomas RS, 2015. Integrated model of chemical perturbations of a biological pathway using 18 in vitro high-throughput screening assays for the estrogen receptor. Toxicol. Sci 148 (1), 137–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinstreuer NC, Browne P, Chang X, Judson R, Casey W, Ceger P, Deisenroth C, Baker N, Markey K, Thomas RS, 2018. Evaluation of androgen assay results using a curated Hershberger database. Reprod. Toxicol 81, 272–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinstreuer NC, Ceger P, Watt ED, Martin M, Houck K, Browne P, Thomas RS, Casey WM, Dix DJ, Allen D, Sakamuru S, Xia M, Huang R, Judson R, 2017. Development and validation of a computational model for androgen receptor activity. Chem. Res. Toxicol 30 (4), 946–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinstreuer NC, Ceger PC, Allen DG, Strickland J, Chang X, Hamm JT, Casey WM, 2015. A curated database of rodent uterotrophic bioactivity. Environ. Health Perspect 124 (5), 556–562. No 5 pp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansouri K, 2019. CoMPARA: Collaborative Modeling Project for Androgen Receptor Activity. Enhvironmental Health Perspectives; In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansouri K, Abdelaziz A, Rybacka A, Roncaglioni A, Tropsha A, Varnek A, Zakharov A, Worth A, Richard AM, Grulke CM, Trisciuzzi D, Fourches D, Horvath D, Benfenati E, Muratov E, Wedebye EB, Grisoni F, Mangiatordi GF, Incisivo GM, Hong H, Ng HW, Tetko IV, Balabin I, Kancherla J, Shen J, Burton J, Nicklaus M, Cassotti M, Nikolov NG, Nicolotti O, Andersson PL, Zang Q, Politi R, Beger RD, Todeschini R, Huang R, Farag S, Rosenberg SA, Slavov S, Hu X, Judson RS, 2016. CERAPP: collaborative estrogen receptor activity prediction project. Environ. Health Perspect 124 (7), 1023–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansouri K, Kleinstreuer N, Abdelaziz AM, Alberga D, Alves VM, Andersson PL, Andrade CH, Bai F, Balabin I, Ballabio D, Benfenati E, Bhhatarai B, Boyer S, Chen J, Consonni V, Farag S, Fourches D, Garcia-Sosa AT, Gramatica P, Grisoni F, Grulke CM, Hong H, Horvath D, Hu X, Huang R, Jeliazkova N, Li J, Li X, Liu H, Manganelli S, Mangiatordi GF, Maran U, Marcou G, Martin T, Muratov E, Nguyen DT, Nicolotti O, Nikolov NG, Norinder U, Papa E, Petitjean M, Piir G, Pogodin P, Poroikov V, Qiao X, Richard AM, Roncaglioni A, Ruiz P, Rupakheti C, Sakkiah S, Sangion A, Schramm KW, Selvaraj C, Shah I, Sild S, Sun L, Taboureau O, Tang Y, Tetko IV, Todeschini R, Tong W, Trisciuzzi D, Tropsha A, Van Den Driessche G, Varnek A, Wang Z, Wedebye EB, Williams AJ, Xie H, Zakharov AV, Zheng Z, Judson RS, 2020. CoMPARA: collaborative modeling project for androgen receptor activity. Environ. Health Perspect 128 (2), 27002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NICEATM/ICCVAM, 2011. Niceatm Draft ED BRD: BG1Luc ER TA Test Method – Section 3.0 Retrieved21 March, 2011, from. http://iccvam.niehs.nih.gov/methods/endocrine/BG1Luc/Section3-24Jan2011.pdf.
- OECD, 2019. Integrated Approaches to Testing and Assessment (IATA)
- Pearce R, Setzer R, Strope C, Sipes N, Wambaugh J, 2017. Httk: R package for high-throughput toxicokinetics. J. Stat. Software 79 (4), 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotroff DM, Wetmore BA, Dix DJ, Ferguson SS, Clewell HJ, Houck KA, Lecluyse EL, Andersen ME, Judson RS, Smith CM, Sochaski MA, Kavlock RJ, Boellmann F, Martin MT, Reif DM, Wambaugh JF, Thomas RS, 2010. Incorporating human dosimetry and exposure into high-throughput in vitro toxicity screening. Toxicol. Sci 117 (2), 348–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroheker T, Cabaton N, Nourdin G, Regnier JF, Lhuguenot JC, Chagnon MC, 2005. Evaluation of anti-androgenic activity of di-(2-ethylhexyl)phthalate. Toxicology 208 (1), 115–121. [DOI] [PubMed] [Google Scholar]
- Thomas RS, Bahadori T, Buckley TJ, Cowden J, Deisenroth C, Dionisio KL, Frithsen JB, Grulke CM, Gwinn MR, Harrill JA, Higuchi M, Houck KA, Hughes MF, Hunter ES, Isaacs KK, Judson RS, Knudsen TB, Lambert JC, Linnenbrink M, Martin TM, Newton SR, Padilla S, Patlewicz G, Paul-Friedman K, Phillips KA, Richard AM, Sams R, Shafer TJ, Setzer RW, Shah I, Simmons JE, Simmons SO, Singh A, Sobus JR, Strynar M, Swank A, Tornero-Valez R, Ulrich EM, Villeneuve DL, Wambaugh JF, Wetmore BA, Williams AJ, 2019. The next generation blueprint of computational toxicology at the U.S. Environmental Protection Agency. Toxicol. Sci 169 (2), 317–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas RS, Paules RS, Simeonov A, Fitzpatrick SC, Crofton KM, Casey WM, Mendrick DL, 2018. The US Federal Tox21 Program: a strategic and operational plan for continued leadership. ALTEX 35 (2), 163–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorne N, Auld DS, Inglese J, 2010. Apparent activity in high-throughput screening: origins of compound-dependent assay interference. Curr. Opin. Chem. Biol 14 (3), 315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- U.S. EPA, 2007. EPA Endocrine Disruptor Screening Program (EDSP) Retrieved8 August, 2008, from. http://www.epa.gov/endo/.
- U.S. EPA, 2011. EDSP21 Workplan Retrieved3 january, 2012, from. http://www.epa.gov/endo/pubs/edsp21_work_plan_summary%20_overview_final.pdf.
- U.S. EPA, 2012. Endocrine Disruptor Screening Program Universe of Chemicals and General Validation Principles U. S. E. P. Agency., Washington DC. [Google Scholar]
- U.S. EPA, 2013. FIFRA SAP Meeting held January 29–31, 2013 on the Scientific Issues Associated with “Prioritizing the Universe of Endocrine Disruptor Screening Program (EDSP) Chemicals using Computational Toxicology Tools” RetrievedOctober 28, 2013, from. http://ntp.niehs.nih.gov/NTP/About_NTP/SACATM/2013/September/SAPMtgRpt_Jan2013_508BE.pdf.
- U.S. EPA, 2017. FIFRA Scientific Advisory Panel; Notice of Public Meeting: Continuing Development of Alternative High-Throughput Screens to Determine Endocrine Disruption, Focusing on Androgen Receptor, Steroidogenesis, and Thyroid Pathways regulations.gov, Washington DC. [Google Scholar]
- U.S. EPA, 2019. Case Study on the Use of an Integrated Appraoch to Testing and Assessment for Estrogen Receptor Active Chemicals, Series on Testing and Assessment No. 309 E. Directorate, Paris (OECD). [Google Scholar]
- US EPA, 2019. ToxCast Database (invitroDB) Retrieved30 April, 2019, from. 10.23645/epacomptox.6062623.v3. [DOI]
- Wambaugh JF, Wetmore BA, Pearce R, Strope C, Goldsmith R, Sluka JP, Sedykh A, Tropsha A, Bosgra S, Shah I, Judson R, Thomas RS, Setzer RW, 2015. Toxicokinetic triage for environmental chemicals. Toxicol. Sci 147 (1), 55–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt ED, Judson RS, 2018. Uncertainty quantification in ToxCast high throughput screening. PLoS One 13 (7) e0196963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetmore BA, Allen B, Clewell HJ 3rd, Parker T, Wambaugh JF, Almond LM, Sochaski MA, Thomas RS, 2014. Incorporating population variability and susceptible subpopulations into dosimetry for high-throughput toxicity testing. Toxicol. Sci 142 (1), 210–224. [DOI] [PubMed] [Google Scholar]
- Wetmore BA, Wambaugh JF, Ferguson SS, Sochaski MA, Rotroff DM, Freeman K, Clewell HJ 3rd, Dix DJ, Andersen ME, Houck KA, Allen B, Judson RS, Singh R, Kavlock RJ, Richard AM, Thomas RS, 2012. Integration of dosimetry, exposure, and high-throughput screening data in chemical toxicity assessment. Toxicol. Sci 125 (1), 157–174. [DOI] [PubMed] [Google Scholar]
- Zager MG, Barton HA, 2012. A multiscale, mechanism-driven, dynamic model for the effects of 5alpha-reductase inhibition on prostate maintenance. PLoS One 7 (9) e44359. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.