

Abstract
Background:
The PI3K/Akt/mTOR pathway is frequently activated in urothelial carcinoma (UC). We performed a phase II study evaluating the efficacy of the pan-isoform class I PI3 kinase (PI3K) inhibitor buparlisib in patients with platinum-refractory metastatic UC.
Methods:
Two cohorts were recruited: an initial genetically unselected cohort, and a subsequent expansion cohort of patients with PI3K/Akt/mTOR pathway-altered tumors. The primary endpoint was 2-month progression-free survival rate. A rate of 80% was considered promising using a minimax Simon 2-stage design. Secondary endpoints included safety and correlation of markers of PI3K pathway activation with outcome.
Results:
Six of 13 evaluable patients within the initial cohort exhibited stable disease (SD) and 1 a partial response (PR), below the cutoff of 9 patients required to proceed to stage 2. Three patients with SD and the patient with a PR harbored somatic TSC1 alterations. Four patients were subsequently recruited onto an expansion cohort, 3 with TSC1 alterations, and one with a PIK3CA activating mutation. No patient achieved disease control at 8 weeks and accrual was halted. Of 19 patients evaluable for toxicity, 17 exhibited treatment-related toxicities, two of whom had to discontinue therapy.
Conclusion:
Buparlisib exhibited modest activity in patients with metastatic UC whose tumors harbored TSC1 loss of function alterations; however, this was not a robust predictor of buparlisib response. The pattern of genetic co-alterations likely influences drug sensitivity. Given the modest clinical activity and substantial toxicity of buparlisib, future trials of PI3K inhibitors in UC should focus on isoform-selective PI3K inhibitors in genomically selected patients.
Keywords: Urothelial Carcinoma, Targeted Therapy, PI3K/Akt/mTOR pathway
Precis:
PI3K pathway inhibition with buparlisib exhibited modest activity in metastatic urothelial carcinoma with significant toxicity. Further studies of PI3K pathway inhibition should focus on patients with select genetic alterations and using isoform-selective inhibitors.
Introduction
Cisplatin-based chemotherapy remains the standard first-line option for patients with metastatic urothelial carcinoma (UC). While response rates are high, chemotherapy is rarely curative in the metastatic setting, with a 5-year overall survival (OS) rate of 13–15% with commonly used cisplatin-based regimens1. Since 2016, five immune checkpoint inhibitors have received FDA approval for locally advanced or metastatic UC. While dramatic long-term responses are observed with these agents, only a minority (13–29%2–8) of patients respond. Recently, the pan-FGFR inhibitor erdafitinib was FDA-approved for patients with metastatic UC with FGFR2/3 genomic alterations following platinum chemotherapy, based on an objective response rate of 40%9 and the antibody-drug conjugate enfortumab vedotin for patients who have progressed on chemotherapy and checkpoint blockade. Despite these recent advances, novel therapies are urgently needed for patients with pre-treated metastatic UC.
Activation of the phosphatidyl 3-inositol kinase (PI3K)/Akt/mechanistic target of rapamycin (mTOR) pathway is associated with urothelial cancer cell proliferation, migration, and invasion10, and inhibition of this pathway results in anti-tumor activity in preclinical models11, 12. Alterations of PIK3CA, the gene that encodes for the catalytic subunit of PI3K, were identified in 25% of bladder cancers in The Cancer Genome Atlas (TCGA), with 18% of patients having known oncogenic hotspot alleles13, 14. Phase II trials have examined everolimus, a selective inhibitor of mTOR15, 16, and the dual PI3K/mTOR inhibitor BEZ23517 in patients with metastatic UC. While these trials did not enrich for patients with PI3K pathway dysregulation, alterations in suppressors of the pathway, including TSC1 inactivating mutations18 and PTEN loss of expression by immunohistochemistry (IHC), were associated with mTOR inhibitor sensitivity and resistance, respectively12, 16. In the BEZ235 study, retrospective evaluation of tumor tissue identified 2/20 patients with pathway activation as defined by PTEN loss. Notably, there were no patients with PIK3CA activating mutations enrolled17. Taken together, these results indicate that a subset of patients with advanced UC may benefit from treatment with a PI3K/Akt/mTOR targeted therapy and that efficacy could be greater in patients whose tumors harbored specific molecular alterations.
Buparlisib (Novartis) is an oral pan-class I PI3K inhibitor which inhibits wild-type and mutant PI3K isoform signaling at nanomolar concentrations19. The drug was recently shown to improve PFS in hormone receptor positive, HER2-negative, advanced breast cancer20,21. We hypothesized that PI3K pathway signaling is upregulated in a subset of metastatic UC and that inhibition with BKM120 would result in inhibition of tumor growth and proliferation. We initiated a single-arm phase II study to define the safety and efficacy of buparlisib in patients with platinum-refractory, locally advanced or metastatic UC. We also correlated somatic genomic alterations in enrolled patients with response to therapy and investigated the mechanism of action of inhibitors of the PI3K/Akt/mTOR pathway using cell lines with PI3 kinase pathway alterations.
Methods:
The study was approved by the Memorial Sloan Kettering Cancer Center Institutional Review Board and registered with the National Cancer Institute (NCT01551030). It was performed under Good Clinical Practice guidelines and in accordance with the Declaration of Helsinki. All enrolled patients provided written informed consent.
Study Design and Treatment
This was an open-label, single arm phase II study of the pan-class I PI3K inhibitor buparlisib in patients with metastatic UC that had progressed on platinum-based chemotherapy. The primary endpoint was 2-month progression-free survival (PFS) rate. The trial employed a Simon two stage minimax design to discriminate between two-month PFS rates of <60% (considered not promising) and >80% (considered promising) with a type 1 error of 0.05 and power of 80%. In the Expansion Cohort, the planned sample size was 21 patients with somatic alterations in PIK3CA, AKT1, and/or TSC1. A one-stage study design was utilized to discriminate between an 8-week PFS rate of <60% versus ≥85%.
Secondary outcomes in the initial cohort included assessment of the safety and toxicity of buparlisib using Common Terminology Criteria for Adverse Events version 4.0 (CTCAE v4.0), the overall response rate (ORR) as assessed by RECIST v1.122, and assessment of genetic markers of an activated PI3K pathway in pretreatment samples and their correlation with subsequent treatment outcome.
Patients received buparlisib 100mg orally once daily until progression of disease or unacceptable toxicity with protocol-defined dose reductions included for toxicity. Baseline evaluations included physical examination, radiographic evaluation for RECIST-measurable metastatic disease, electrocardiogram, echocardiogram or multiple gated acquisition (MUGA) scan, chest x-ray, urinalysis, serum chemistry, hematology and coagulation profiles, and a serum pregnancy test in women of child-bearing age. Screening Generalized Anxiety Disorder 7-item (GAD-7) and Patient Health Questionnaire-9 (PHQ-9) neuro-psychiatric questionnaires were conducted to assess for pre-existing mood disorders on day 1 of each cycle due to the 26% rate of mood disorder observed in a phase I study of buparlisib19. Patients underwent clinical and toxicity assessments on days 1 and 15 of each treatment cycle. Radiographic evaluation was performed every 8 weeks from the start of treatment. Patients who withdrew consent or experienced rapid clinical progression prior to completing one treatment cycle (4 weeks) were not eligible for inclusion in the primary endpoint evaluation. Overall survival was calculated from date of treatment start to date of death, and progression-free survival was calculated from treatment start date to the date of progression. Kaplan Meier survival curves were generated using the R software package version 3.5.2.
Patient Eligibility
Key inclusion criteria included a histologically confirmed diagnosis of UC, a history of 1–4 prior cytotoxic chemotherapeutic agents in the perioperative and/or metastatic settings, including at least one platinum agent, gemcitabine, or a taxane, availability of archival tissue for next generation sequencing (NGS), adequate bone marrow, hepatic, and renal function, a Karnofsky Performance Status (KPS) of ≥ 60%, and a fasting plasma glucose of ≤120 mg/dL. Exclusion criteria included uncontrolled diabetes (fasting plasma glucose >120 mg/dL), acute or chronic pancreatitis, and kidney or liver disease. Patients with untreated brain metastases, significant gastrointestinal disorders potentially compromising drug absorption, or active cardiac disease were also excluded.
Specific exclusion criteria for mental illness included medically documented history of or active major depressive episode, bipolar disorder (I or II), obsessive-compulsive disorder, schizophrenia, a history of suicidal attempt or ideation, or homicidal ideation, ≥CTCAE grade 3 anxiety, and meeting the cut-off score of ≥10 in the PHQ-9 questionnaire or selecting a positive response regarding the potential for suicidal thoughts, or a score of ≥15 in the GAD-7 mood scale.
Tumor Sequencing
Archival formalin fixed paraffin embedded (FFPE) tissue from 18 tumors obtained from 11 of 13 patients evaluable for the primary endpoint underwent targeted next generation sequencing with a 279-gene version of the MSK-IMPACT (Integrated Molecular Profiling of Actionable Cancer Targets) assay23, with matched blood used for germline DNA. Sixteen tumors from 10 patients also underwent whole exome sequencing (WES) at the Broad Institute. The mean coverage by WES was 172x for tumor and 102x for matched normal. Tumor purity as estimated by the ABSOLUTE algorithm was 10–95%.24 For WES, MuTect25 was utilized to identify somatic single nucleotide variants (SNVs), ReCapSeg26 to define broad copy number alterations (CNAs) and ABSOLUTE24 to infer absolute allele specific CNAs. DeconstructSigs27 was utilized to identify mutation signatures. Artifacts introduced by DNA oxidation during sequencing or FFPE storage were removed using a pipeline developed by the Broad Institute28. For WES, all mutation calls were compared to a panel of germline samples and were removed if they appeared in more than three samples. Predicted functional impact of mutations was evaluated using the OncoKB database29. Schematic representations of the receptor tyrosine kinase (RTK) pathway were illustrated based on diagrams from pathway mapper30.
Cell Lines and Reagents
Human UC cell lines HCV29 (gift of M. Knowles) and MGH-U4 (gift of D. Theodorescu) were grown in RPMI1640 and MEM, respectively, with 10% fetal bovine serum at 37°C in 5% CO2. Antibodies were obtained from Cell Signaling: pAkt T308 (Catalog number; 2965), pAkt S473 (9271), Akt total (9272), pS6 S235/236 (2211), pS6 S240/244 (5364), S6 total (2317), p4E-BP1 T37/46 (9459), PARP (9542), and p70S6k (9205). Small molecule inhibitors were obtained from Selleck Chemical, including BYL719 (alpelisib), buparlisib, MK2206, and AZD8055. Cell lines were treated with vehicle control or one of the four drugs: BYL719 (2.5 μM), buparlisib (2.5 μM), MK2206 (2.5 μM), or AZD8055 (500nM). Cells were harvested at 0, 0.5, 4 and 8 hours, lysed in RIPA buffer and sonicated. Immunoblotting was conducted utilizing the antibodies described above.
Results
Patient Characteristics
Sixteen patients with metastatic UC were enrolled between May 2012 and June 2013 into the genetically unselected patient cohort. One patient was deemed ineligible for treatment due to elevations in serum amylase and lipase on screening bloodwork. Fifteen patients were evaluable for safety and toxicity, and 13 were evaluable for the primary endpoint (Figure 1). Baseline clinical demographics are displayed in Table 1 and are consistent with a typical population of patients with advanced UC. This was a heavily pre-treated patient cohort (median of 2 prior systemic regimens); moreover, 6 subjects had two adverse prognostic factors using the Bellmunt prognostic criteria31. The bladder was the primary disease site in 9 patients, and metastatic sites included non-regional lymph nodes (14), lung (6) and liver (5). Two of 15 patients who consented to treatment discontinued buparlisib prior to the 8-week evaluation, one from rapid progression of disease (development of malignant bowel obstruction) and one due to withdrawal of consent (patient preference). These patients were deemed as non-evaluable for the primary endpoint, as specified in the study protocol.
Figure 1.

CONSORT Diagram
Table 1.
Demographics and Clinical Characteristics of Patients Consented to Study (n=20)
| Patient Characteristics | Cohort | |
|---|---|---|
|
| ||
| Initial | Expansion | |
| Median age (range) | 65.5 (53–82) | 64 (56–82) |
|
| ||
| Gender | ||
| Male | 11 | 4 |
| Female | 5 | 0 |
|
| ||
| Karnofsky Performance Status | ||
| 70 | 1 | 1 |
| 80 | 7 | 3 |
| 90 | 8 | 0 |
|
| ||
| Bellmunt Criteria Score* | ||
| 0 | 0 | 0 |
| 1 | 10 | 3 |
| 2 | 6 | 1 |
|
| ||
| Number of Previous Therapies | ||
| 1 | 5 | 1 |
| 2 | 5 | 2 |
| 3 | 5 | 0 |
| 4 | 1 | 0 |
| 5 | 0 | 1 |
| Previous Therapies | ||
| Platinum-based | 18 | 3 |
| (Gemcitabine/Cisplatin, Gemcitabine/Carboplatin) | ||
| Taxane | 2 | 1 |
| (Paclitaxel) | ||
| Antimetabolite | 8 | 2 |
| (Pemetrexed) | ||
| Targeted Agent | 4 | 0 |
| (Gemcitabine/Pazopanib, Gemcitabine/Cisplatin/Sunitinib, Gemcitabine/Cisplatin/Bevacizumab or placebo, Docetaxel/Glesatinib) | 0 | 1 |
| Immunotherapy | ||
| (Atezolizumab) | 3 | 2 |
| Other | ||
| (Gemcitabine, Gemcitabine/Docetaxel, Gemcitabine + Radiotherapy) | ||
|
| ||
| Primary Tumor Site | ||
| Bladder | 9 | 4 |
| Upper Tract | 7 | 0 |
| Renal Pelvis | 5 | |
| Ureter | 2 | |
|
| ||
| Histologic Subtype | ||
| UC NOS | 12 | 3 |
| Micropapillary/Glandular Mixed | 1 | 0 |
| Glandular | 0 | 1 |
| Squamous | 3 | 0 |
|
| ||
| Metastatic Sites at Screening | ||
| Bone | 0 | 3 |
| Lung | 6 | 2 |
| Liver | 5 | 1 |
| Nodes | 14 | 1 |
| Other⸸ | 8 | 0 |
1 point each for ECOG>0, Hemoglobin <10g/dL, Liver metastases33
spleen, ureteral stump, L psoas, adrenal (2), body wall, thigh, vagina, peritoneal
Seven (54%) of the thirteen evaluable patients in the unselected cohort were progression-free on 8-week follow-up CT scan, including six with stable disease (SD) as best response and one patient with a partial response (PR) of 16 months duration. The latter patient was a 63-year-old man with lung and lymph node metastases who demonstrated a 74% reduction in target lesions on buparlisib (Figure 2C, 3A). Retrospective analysis of this patient’s archival bladder tumor tissue from a transurethral resection revealed a nonsense mutation in TSC1 (R500*) with loss of heterozygosity (LOH) of the wild-type allele (Figure 3B). The median PFS was 3.2 months (Range, 1.4–15.8, Figure 2A), and the median OS was 8.7 months (2.7–63.8, Figure 2B). The median time on therapy for all 15 patients was 55 days (range, 3–483 days). When stratified by Bellmunt prognostic criteria, there was no significant difference in PFS (Supplemental Figure 1A); however, there was a worse OS in patients with a score of 2 compared to 1, although this was not statistically significant (p=0.06; Supplemental Figure 1B). The patient who achieved a partial response to buparlisib ultimately developed a new brain metastasis.
Figure 2.





Treatment Response. A) Progression Free Survival; B) Overall Survival C) Waterfall Plot of Radiographic Best Response to buparlisib by RECIST v1.1.
Figure 3.
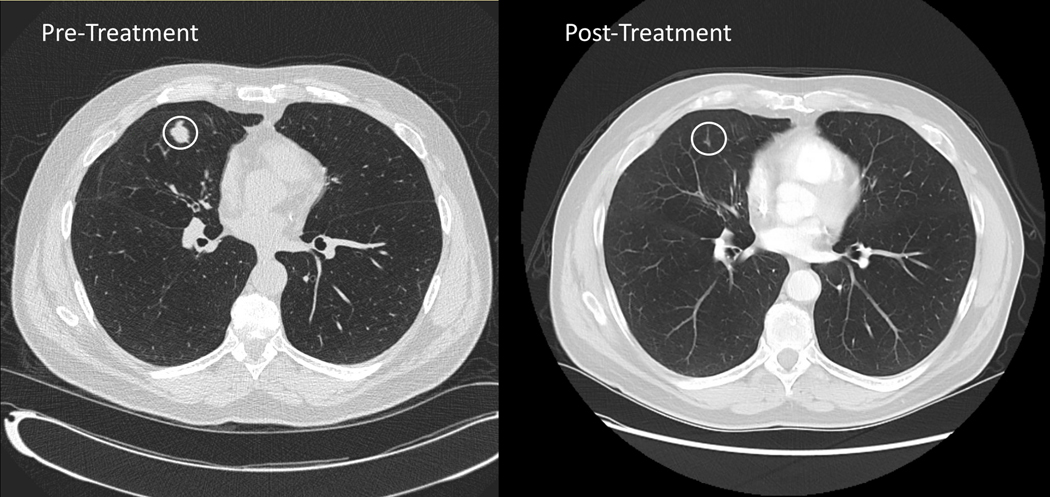
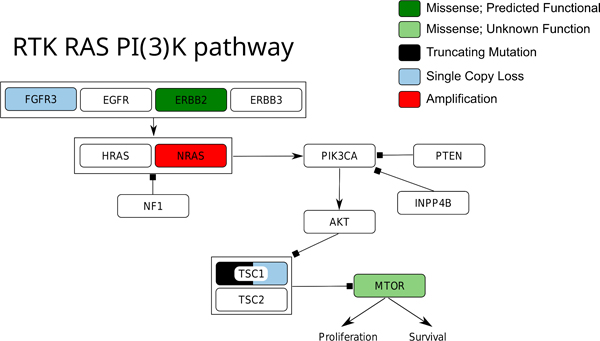
Partial Responder to BKM120 A) Best Radiographic Response in Target Lesion within the Right Lung. B) Receptor Tyrosine Kinase Pathway Map of Genetic Alterations in Archival Tissue
As the 2-month PFS rate of 54% in the unselected cohort was below the pre-specified threshold to proceed to the second stage of the study, and since a durable response was observed in a patient with a truncating TSC1 mutation, the study design was amended to include an expansion cohort restricted to patients with known or likely oncogenic somatic mutations in PIK3CA, AKT1/2/3, PTEN, or TSC1. Four of 21 planned patients were enrolled onto this expansion arm before the study was discontinued by the sponsor. Their clinical characteristics are summarized in Table 1 and their sequencing and clinical outcomes in Table 2. These patients had a similar median Karnofsky Performance Status (KPS) of 80% versus 85% for the patients enrolled onto the initial cohort. Three patients had TSC1 nonsense mutations and one an activating hotspot PIK3CA (E542K) mutation (Table 2). In the expansion cohort, one patient was removed from the study for grade 3 transaminitis and a grade 3 rash after 28 days of buparlisib therapy. The other 3 patients experienced disease progression within 8 weeks (Figure 2A; duration on therapy: 16, 53, and 55 days). Overall survival is shown in Figure 2B.
Table 2.
Expansion Cohort Genomic Alterations and Treatment Response
| Patient | Mutation | Time on Therapy (Days) | PFS (Months) | OS (Months) | Reason for Study Discontinuation | 8-Week Imaging |
|---|---|---|---|---|---|---|
| 1 | TSC1 Y185* | 53 | 1.0 | 2.6 | Progression | N/A |
| 2 | TSC1 R424Nfs* | 55 | 1.7 | 9.2 | Progression | Y; PDǂ |
| 3 | PI3K E542K | 28 | 1.9 | 20.3 | Toxicity | N/A |
| 4 | TSC1 Q516* | 16 | 0.6ǂǂ | 1.7 | Progression | N/A |
New non-target lesions consistent with progression of disease
Patient ineligible for primary endpoint evaluation due to receiving <1 cycle of therapy
Safety and Tolerability
Treatment-related grades 1–2 and all grades 3–4 adverse events (AEs) for the 15 evaluable patients in the initial cohort are shown in Table 3. Six patients (40%) required dose reduction of buparlisib for treatment-related AEs, and treatment was discontinued in one patient for grade 3 hyperglycemia and another patient for grade 3 transaminitis and rash. The patient with hyperglycemia had preexisting diabetes and experienced recurrent hyperglycemic events mandating buparlisib discontinuation despite dose reduction and modification of anti-hyperglycemic agents. Fourteen patients (93%) experienced treatment-related AEs. The most common treatment-related grade 1–2 AEs included hyperglycemia (13), fatigue (12), and transaminitis (ALT elevation, 5; AST elevation, 4). Forty-nine grade 3 or 4 AEs were observed in 13 patients, with 11 occurring in 4 patients deemed to be treatment-related. The most frequent grades 3–4 events were lymphopenia (n=19), hyperglycemia (6), and anemia (5). There were no toxicity-related deaths on study. One patient manifested grade 3 depression within 2.5 weeks of starting buparlisib, with concomitant suicidal ideation. This AE subsided with temporary drug cessation, and upon dose reduction from 100mg to 60mg, the patient had only a single grade 1 depression-related adverse event recorded in the subsequent 2.5 months on study.
Table 3.
CTCAE v4.0 treatment-related Grade 1–2 and all Grade 3–4 adverse events, Unselected Cohort
| Adverse Event | Grade 1–2 (#Patients) | Grade 3–4 (#Events / #Patients [#Treatment-Related]) |
|---|---|---|
| Hyperglycemia | 13 | 6/2 (5) |
| Fatigue | 12 | 0 |
| Weight loss | 8 | 0 |
| Mucositis oral | 7 | 0 |
| Diarrhea | 6 | 0 |
| Alanine aminotransferase increased | 5 | 0 |
| Nausea | 5 | 0 |
| Rash maculo-papular | 5 | 1/1 (1) |
| Aspartate aminotransferase increased | 4 | 1/1 (0) |
| Depression | 3 | 1/1 (1) |
| Dry Skin | 4 | 0 |
| Hyperkalemia | 4 | 0 |
| Photosensitivity | 4 | 0 |
| Anorexia | 3 | 0 |
| Anxiety | 3 | 0 |
| Dyspepsia | 3 | 0 |
| Cholesterol high | 2 | 0 |
| Creatinine increased | 2 | 0 |
| Flatulence | 2 | 0 |
| Hypophosphatemia | 2 | 2/1 (0) |
| Pruritus | 2 | 2/1 (0) |
| Rash acneiform | 2 | 0 |
| Skin & subcutaneous tissue disorders Other; | ||
| Erythema | 2 | 1/1 (1) |
| Rosacea | 1 | 0 |
| Vomiting | 2 | 0 |
| Abdominal pain | 1 | 1/1 (0) |
| Alopecia | 1 | 0 |
| Alkaline phosphatase increased | 1 | 0 |
| Constipation | 1 | 0 |
| Cough | 1 | 0 |
| Endocrine disorders – Other; SIADH | 1 | 0 |
| Gastrointestinal disorders – other; Dry heaves | 1 | 0 |
| Hypertriglyceridemia | 1 | 0 |
| Hypocalcemia | 1 | 0 |
| Hypokalemia | 1 | 2/1 (2) |
| Hypernatremia | 1 | 0 |
| Hyponatremia | 1 | 1/1 (0) |
| Lymphocyte count decreased | 1 | 19/2 (0) |
| Muscle weakness | 1 | 0 |
| Platelet count decreased | 1 | 1/1 (0) |
| White blood cell decreased | 1 | 1/1 (0) |
| Anemia | 0 | 5/1 (0) |
| Hyperuricemia | 0 | 1/1 (0) |
| Infection | 0 | 1/1 (0) |
| Small intestinal obstruction | 0 | 1/1 (0) |
| Syncope | 0 | 2/2 (0) |
| Thromboembolic event | 0 | 1/1 (0) |
Treatment-related grades 1–2 and all grade 3–4 AEs for the molecularly selected cohort are presented in Table 4. Three patients experienced 29 treatment-related grade 1–2 AEs, and 2 experienced 11 grade 3–4 AEs, including transaminitis (ALT or AST increased, 4 events each), lymphopenia (2) and a maculo-papular rash (1). Nine of these grade 3–4 events in one patient were suspected to be treatment-related.
Table 4.
CTCAE v4.0 treatment-related Grade 1–2 and all Grade 3–4 adverse events, Expansion Cohort
| Adverse Event | Grade 1–2 (#Patients) | Grade 3–4 (#Events / #Patients [#Treatment-Related]) |
|---|---|---|
| Fatigue | 3 | 0 |
| Hypertriglyceridemia | 2 | 0 |
| Nausea | 2 | 0 |
| Activated partial thromboplastin time prolonged | 1 | 0 |
| Alanine aminotransferase increased | 1 | 4/1 (4) |
| Alkaline phosphatase increased | 1 | 0 |
| Anemia | 1 | 0 |
| Aspartate aminotransferase increased | 1 | 4/1 (4) |
| Bloating | 1 | 0 |
| Blood bilirubin increased | 1 | 0 |
| Cholesterol high | 1 | 0 |
| Concentration impairment | 1 | 0 |
| Constipation | 1 | 0 |
| Dyspnea | 1 | 0 |
| Dry Skin | 1 | 0 |
| Edema limbs | 1 | 0 |
| Hiccups | 1 | 0 |
| Hypoalbuminemia | 1 | 0 |
| Hyperglycemia | 1 | 0 |
| Palmar-plantar erythrodysesthesia syndrome | 1 | 0 |
| 1 | 0 | |
| Platelet count decreased | 1 | 0 |
| Peripheral motor neuropathy | ||
| Resp, thoracic & mediastinal disorder Other; | 1 | 0 |
| Rhinorrhea | 1 | 0 |
| Vomiting | 1 | 0 |
| Weight loss | 1 | 2/1 (0) |
| Lymphocyte count decreased | 0 | 1/1 (0) |
| Rash maculo-papular |
Next Generation Sequencing Analysis of Archival Tumor Tissue
Pre-treatment tumors from 11 patients from the unselected cohort were of sufficient quality for retrospective targeted NGS and 10 of 11 of these patients had adequate material for whole exome sequencing (WES). The median non-synonymous mutational burden within the tumor samples analyzed by WES was 3 mutations per megabase (muts/Mb), lower than the rate of 5.8 muts/Mb observed in urothelial TCGA cohort (p=0.4, Supplemental Figure 2.)32. Mutation rates for genes commonly altered in UC were similar to TCGA, including TP53, FGFR3, and ARID1A. The most common amplification events involved CCND1 (4 patients, 4 samples), ERBB2 (1 patient, 4 samples), and RAF1 (2 patients, 3 samples). Recurrent bi-allelic deletions were seen in CDKN2A (2 samples, 2 samples) and monoallelic deletions were observed in TP53 (5 patients, 6 samples), and in TSC1, RB1, RXRA, TLR4, AKAP11, and FOXO1 in 5 samples each.
One patient with amplification of PIK3CA, which encodes for the alpha catalytic subunit of PI3K, had SD lasting 98 days (0% best RECIST response; Table 5), whereas all three patients with activating mutations in PIK3CA had disease progression as best response. No mutations were identified in PTEN; however, monoallelic loss was present in 1 patient with SD (9% best RECIST response) who remained on therapy for 83 days before treatment discontinuation for excessive toxicity. Finally, TSC1 alterations were identified in 4 patients: 2 tumors had bi-allelic inactivation, and 2 single copy loss. Of the 2 patients with bi-allelic TSC1 inactivation, 1 achieved a PR (74% reduction in RECIST targets) and remained on buparlisib for 483 days, whereas the other patient had SD (−2% response) and remained on study drug for 110 days.
Table 5.
Association Between Genetic Alterations in PIK3CA, PTEN and TSC1 and Treatment Response
| 8-Week Treatment Response | Patient | Gene | Genetic Alteration | Functional Impact* | Best RECIST Response | Time on Therapy (days) |
|---|---|---|---|---|---|---|
| PR | TUR | TSC1 | R500* LOH | Inactivating | −74% | 483 |
|
| ||||||
| SD | RC | TSC1 | S331Efs*10 LOH | Inactivating | −2% | 110 |
| Lung Metastasis | TSC1 | S331Efs*10 | Inactivating | |||
|
| ||||||
| TUR | PIK3CA | Amplification | Activating | 0% | 86 | |
| TSC1 | LOH | Deletion | ||||
|
| ||||||
| TUR | 7% | 106 | ||||
|
| ||||||
| RC | PTEN | LOH | Deletion | 9% | 83 | |
| Sigmoid Colon | PTEN | LOH | Deletion | |||
| Invasion | ||||||
|
| ||||||
| RC Micropapillary | 11% | 167 | ||||
| RC UC, NOS | ||||||
|
| ||||||
| PD | Nephroureterectomy | 23% | 55 | |||
|
| ||||||
| Nephroureterectomy | PIK3CA | H1047R | Activating | 27% | 55 | |
|
| ||||||
| RC | PIK3CA | E542K | Activating | 36% | 48 | |
| RC LN | TSC1 | LOH | Deletion | |||
|
| ||||||
| RC | 45% | 41 | ||||
|
| ||||||
| CP | RC-1 | NA | 3 | |||
| RC-2 | ||||||
| RC-LN | ||||||
| Metastatic LN | PIK3CA | R108H | Activating | |||
CP= Clinical progression due to malignant bowel obstruction
LN= Lymph Node
NOS = Not Otherwise Specified
PD= Progressive Disease
RC=Radical Cystectomy
SD= Stable Disease
TUR= Transurethral Resection
= Analyzed via MSK IMPACT Only
Several additional potentially oncogenic alterations were identified by WES of the tumor from the patient who achieved a durable PR (Figure 3B). These included NRAS amplification and a ERBB2 (S310F) mutation. A mutation of unknown significance in MTOR (L133F) was also identified by both MSK-IMPACT and WES. Notably, the ERBB2 (S310F) mutation identified by WES was not detected by MSK-IMPACT despite robust sequencing coverage of the ERBB2 gene by both assays, which may suggest tumor heterogeneity. WES had been performed on a newly extracted DNA sample from the same tissue block, and thus this discordance in sequencing results may indicate intratumor heterogeneity. Additionally, analysis of the WES data indicated an amplification of chromosome 4 in the region of the AREG and EREG genes, which are ligands for the EGFR receptor that could induce both EGFR and downstream PI3K activation.
In Vitro Analysis of PI3K Pathway Inhibition
Since buparlisib’s anti-tumor effect was presumed to stem from inhibition of PI3K, the durable response of a patient with an inactivating mutation in TSC1 was unexpected. This was because inactivation of TSC1 has been hypothesized to induce mTOR activation independent of upstream PI3K/AKT signaling. We therefore sought to better characterize the effects of buparlisib on PI3K pathway activity using two UC cell lines, HCV29 (TSC1 Q55* mutant) and MGH-U4 (PIK3CA H1047R mutant). Both cell lines were exposed to a panel of inhibitors of PI3K pathway inhibitors, including buparlisib, alpelisib (BYL719, an alpha selective PI3K inhibitor), MK2206 (an allosteric pan-isoform Akt inhibitor), and AZD8055 (a dual mTORC1/2 kinase inhibitor; Supplemental Figure 3). As expected, treatment of the PIK3CA (H1047R) mutant MGH-U4 cell line with all four drugs resulted in inhibition of mTOR signaling as reflected by reduced expression of phosphorylated S6 and 4E-BP1, two downstream efforts of the mTORC1 complex. In the TSC1 Q55* mutant HCV29 cell line, the selective PI3K alpha inhibitor BYL719 and the AKT inhibitor MK2206 both inhibited AKT activation as assessed by a decrease in phosphorylation of AKT but had no effect on the phosphorylation of S6 and 4E-BP1. Treatment of HCV29 cells with AZD8055, an mTOR kinase inhibitor, resulted in reduced expression of phosphorylated AKT, S6 and 4E-BP1 consistent with its purported ability to inhibit both mTORC1 and mTORC2. In contrast to the PI3K alpha selective inhibitor BYL-719, buparlisib, like AZD8055, inhibited activation of kinases both upstream and downstream of TSC1 in HCV29 cells, as evidenced by reduced expression of phosphorylated Akt, S6, and 4E-BP1. Taken together, these data suggest that TSC1 mutant cell lines may be resistant to AKT and alpha selective PI3 kinase inhibitors but may retain sensitivity to buparlisib.
Discussion
This single-arm phase II clinical trial evaluated the efficacy of the pan-isoform class I PI3K inhibitor buparlisib in patients with platinum-refractory locally advanced or metastatic UC. Sixteen patients were recruited into an initial, genetically unselected cohort with an additional four patients enrolled onto an expansion cohort of patients known to have PI3K pathway alterations (as detected by targeted NGS of archival tumor tissue). Although this study did not meet its primary endpoint of an 80% 8-week PFS rate in the unselected cohort, one patient did achieve a durable PR lasting 15.9 months, while an additional 6 patients had stable disease as best response. The tumor from the partial responder had several alterations that may have contributed to the patients’ sensitivity to buparlisib, including a truncating mutation of TSC1, an activating ERBB2 mutation, and AREG/EREG gene amplification. In colorectal cancer, increased AREG and EREG expression has been shown to result in sensitization of tumors to anti-EGFR therapy,33 and EGFR activation can induce activation of the PI3K/Akt/mTOR pathway.34
While some anti-tumor effects of buparlisib were noted, the rate of buparlisib-related toxicity was significant, with 6 patients requiring dose reduction and two removed from the study due to grade 3 adverse events. The observed AE profile was similar to previous studies of buparlisib, including fatigue and hyperglycemia (a known on-target effect of PI3K pathway inhibition)35, 36. Buparlisib penetrates the blood brain barrier, a characteristic that has been viewed as advantageous for the treatment of glioblastoma multiforme,37, 38 but as a consequence, this drug can cause neuropsychiatric toxicities: two mood-related dose limiting toxicities and an overall 37% rate of treatment-related mood disorders were observed in the first-in-human buparlisib study, all of which were reversible with discontinuation of drug19. Notably, the rate of depression seen in this study was lower than that seen in previous trials35, 36.
We assessed the association between somatic alterations in the PI3K/Akt/mTOR pathway and response to buparlisib through targeted and whole exome sequencing of archival tumor tissue. In the unselected cohort, both patients with bi-allelic inactivation of TSC1 achieved disease control on 8-week imaging, including the partial responder whose tumor harbored a truncating mutation in one allele and LOH of the other allele. While buparlisib is considered a class I selective PI3 kinase, it exerts some inhibitory activity against classes III and IV PI3 kinases, including mTOR39. Bi-allelic loss of TSC1 may have resulted in dependence upon mTOR signaling for cell growth and proliferation in this patient’s tumor, subsequently resulting in heightened sensitivity to mTORC1 inhibition. Our cell line data confirmed that buparlisib, in contrast to the FDA approved alpha selective inhibitor alpelisib (BYL719), can inhibit mTORC1 signaling in TSC1 mutant lines as demonstrated by a reduction in the expression of phosphorylated S6, a downstream effector of the mTORC1 complex.
While the partial response observed in a TSC1 mutant UC patient in the genetically unselected cohort suggested that buparlisib may have had clinical activity in this molecularly defined subset of patients, none of the three patients with TSC1 mutations enrolled on the genetically selected cohort exhibited clinical benefit. These data suggest that TSC1 mutation is not a robust predictor of buparlisib response and that the pattern of co-mutational alterations likely influences drug sensitivity.
To date, trials of targeted therapy in metastatic UC have primarily focused on FGFR inhibition40, 41 with additional studies of HER2/342, 43 and mTOR inhibition15–17. The pan-FGFR inhibitors BGJ398 and erdafitinib displayed response rates of 25.4%44 and 40%9 in heavily pre-treated Patients with UC, and based on the latter, the FDA approved erdafitinib for the treatment of metastatic UC following progression on platinum-based chemotherapy40. A trial of afatinib in genetically unselected patients with platinum-refractory metastatic UC revealed a 3-month PFS rate of 21.7%42. Sequencing of archival tumors revealed that 5 of 6 patients with ERBB2 or ERBB3 alterations met the 3-month PFS primary endpoint, compared to 0 of 15 ERBB2/3 wild-type patients42. Finally, several case studies suggest that mTOR inhibition may be effective in patients with metastatic UC with TSC1/2 inactivating alterations and/or activating mutations in MTOR18, 45. These data indicate that patients with metastatic UC with the relevant sensitizing alteration can respond to targeted therapies, which underscores the importance of prospective selection. Indeed, a panel of UC experts recommended to the National Comprehensive Cancer Network that molecular profiling should be considered in all patients with advanced UC given the plethora of potentially targetable alterations in this disease46.
PI3K inhibition to date has not resulted in FDA approval of this class of agents as monotherapy in solid tumors. One reason for this may be that pan-isoform inhibition is associated with significant on- and off-target toxicity that limits administration of doses that adequately inhibit the target. Another possibility is that concomitant MAPK or other bypass signaling pathway activation reduces dependence on PI3K signaling for tumor growth and proliferation34, 39, 47, 48. Further complicating the application of targeted agents in UC are reports of significant intra49 and inter50-tumoral genetic heterogeneity in this disease as well as therapy-induced changes to the genomic profile of a tumor. Indeed, in our cohort, one patient in whom four separate tumors were sequenced harbored a PIK3CA activating mutation in only a single sample. The presence of a subclonal PIK3CA mutation may have resulted in reduced dependency on PI3K pathway signaling and thus reduced overall tumor response to buparlisib. Moreover, archival tumor tissue, which may not contain the most relevant genetic profile, was sequenced as opposed to metastatic biopsies for patients enrolled onto this study. Other limitations of the current study included the small number of patients treated with buparlisib, the lack of a control arm, and the absence of an intent-to-treat analysis.
Overall, while this study did not reach its intended therapeutic endpoint, buparlisib therapy did result in 8-week disease control in seven patients, including one durable PR. Consistent with other studies of buparlisib, the presence of a PIK3CA mutation was not predictive of drug response. This observation and the recent success of alpelisib in PIK3CA mutant breast cancer suggest that future studies in PIK3CA mutant patients with UC should focus on testing alpha-selective PI3K inhibitors in genetically defined patient populations. Given the genomic complexity of bladder cancer, such studies should also seek to test rational combination strategies.
Supplementary Material
Translational Relevance:
The PI3K/Akt/mTOR pathway is a critical cell growth and survival pathway that is frequently activated in many cancer types, including urothelial carcinoma (UC). This phase II study evaluated the safety and efficacy of the pan-isoform PI3K inhibitor buparlisib in patients with platinum-treated metastatic UC. Stable disease and one partial response were observed in patients with PI3K/Akt/mTOR pathway alterations, suggesting that more selective inhibitors of this pathway may be effective in molecularly selected subsets of patients with UC.
Acknowledgments
Funding: Bladder Cancer Advocacy Network Young Investigator Award (GC220702) and an institutional Core Grant (P30 CA008748).
VM reports personal fees from TerSera Therapeutics, outside the submitted work. HA reports personal fees from AstraZeneca, Bristol-Myers Squibb, outside the submitted work. MFB reports personal fees from Roche, grants from Grail, outside the submitted work. JER reports other from Novartis, during the conduct of the study, other from Illumina, grants and personal fees from AstraZeneca, Astellas Pharma, Bayer, QED Therapeutics, Seattle Genetics, personal fees and other from Bristol-Myers Squibb, personal fees from Chugai Pharma, Clinical Care Options, Clinical Mind, Intellisphere, Medscape, Peerview, Research To Practice, UpToDate, Vindico, Adicet Bio, Agensys, BioClin Therapeutics, EMD Serono, Fortress Biotech, GlaxoSmithKline, Inovio Pharmaceuticals, Janssen Oncology, Lilly, Merck, Pharmacyclics, Sensei Biotherapeutics, Western Oncolytics, grants, personal fees and other from Roche/Genentech, grants from Incyte, Jounce Therapeutics, Viralytics, Mirati Therapeutics, outside the submitted work. In addition, Dr. Rosenberg has a patent Predictor of platinum sensitivity pending. DBS reports personal fees from Pfizer, Loxo Oncology, Lilly Oncology, Vivideon Therapeutics, Q.E.D. Therapeutics, Illumina, outside the submitted work. EVA reports grants from Bristol-Myers Squibb, Novartis, personal fees from Illumina, Janssen, Ervaxx, Tango Therapeutics, InVitae, Genome Medical, Takeda, Third Rock Ventures, Roche, Syapse, outside the submitted work. In addition, Dr. Van Allen has a patent Discovery of retained intron as source of cancer neoantigens pending, a patent Discovery of chromatin regulators as biomarkers of response to cancer immunotherapy pending, and a patent Clinical interpretation algorithms using cancer molecular data pending. MIM reports personal fees from BioClin Therapeutics, grants from Merck, Acerta Pharma, Roche/Genentech, Bristol-Myers Squibb, Seattle Genetics, Astellas Pharma, Clovis Oncology, Inovio Pharmaceuticals, AstraZeneca, X4 Pharmaceuticals, Mirati Therapeutics, Boehringer Ingelheim, Constellation Pharmaceuticals, Jounce Therapeutics, Syndax, Innocrin Pharma, MedImmune, Cerulean Pharma, Incyte, other from Asieris, outside the submitted work. DFB reports other from Novartis, during the conduct of the study; personal fees from Merck Sharp & Dohme, Eisai, Fidia Farmaceutici S.p.A., Lilly, Urogen Pharma, grants and personal fees from Bristol-Myers Squibb, Roche/Genentech, Novartis, grants from Dendreon, outside the submitted work. GI reports personal fees from Mirati Therapeutics, Janssen, other from Novartis, outside the submitted work.
Footnotes
Conflicts of Interest:
BR, AB, SNS, MEB, IGG, AMR, ASM, PK, IO, AR, and AF declare that they have no conflicts of interest.
References
- 1.von der Maase H. et al. Long-Term Survival Results of a Randomized Trial Comparing Gemcitabine Plus Cisplatin, With Methotrexate, Vinblastine, Doxorubicin, Plus Cisplatin in Patients With Bladder Cancer. Journal of Clinical Oncology 23, 4602–4608 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Massard C. et al. Safety and Efficacy of Durvalumab (MEDI4736), an Anti–Programmed Cell Death Ligand-1 Immune Checkpoint Inhibitor, in Patients With Advanced Urothelial Bladder Cancer. Journal of Clinical Oncology 34, 3119–3125 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sharma P. et al. Nivolumab monotherapy in recurrent metastatic urothelial carcinoma (CheckMate 032): a multicentre, open-label, two-stage, multi-arm, phase 1/2 trial. The lancet oncology 17, 1590–1598 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sharma P. et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): a multicentre, single-arm, phase 2 trial. The lancet oncology 18, 312–322 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Bellmunt J. et al. Pembrolizumab as Second-Line Therapy for Advanced Urothelial Carcinoma. New England Journal of Medicine 376, 1015–1026 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Powles T. et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature 515, 558–562 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Balar AV et al. Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: a single-arm, multicentre, phase 2 trial. The Lancet 389, 67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Apolo AB et al. Avelumab, an Anti–Programmed Death-Ligand 1 Antibody, In Patients With Refractory Metastatic Urothelial Carcinoma: Results From a Multicenter, Phase Ib Study. Journal of Clinical Oncology 35, 2117–2124 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Loriot Y. et al. Erdafitinib in Locally Advanced or Metastatic Urothelial Carcinoma. New England Journal of Medicine 381, 338–348 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Knowles MA & Hurst CD Molecular biology of bladder cancer: new insights into pathogenesis and clinical diversity. Nat Rev Cancer 15, 25–41 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Seager CM et al. Intravesical Delivery of Rapamycin Suppresses Tumorigenesis in a Mouse Model of Progressive Bladder Cancer. Cancer Prevention Research 2, 1008–1014 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seront E. et al. PTEN deficiency is associated with reduced sensitivity to mTOR inhibitor in human bladder cancer through the unhampered feedback loop driving PI3K/Akt activation. British journal of cancer 109, 1586–1592 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao J. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling 6, pl1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cerami E. et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discovery 2, 401 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Milowsky MI et al. Phase II study of everolimus in metastatic urothelial cancer. BJU international 112, 462–470 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seront E. et al. Phase II study of everolimus in patients with locally advanced or metastatic transitional cell carcinoma of the urothelial tract: clinical activity, molecular response, and biomarkers. Annals of Oncology 23, 2663–2670 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Seront E. et al. Phase II study of dual phosphoinositol-3-kinase (PI3K) and mammalian target of rapamycin (mTOR) inhibitor BEZ235 in patients with locally advanced or metastatic transitional cell carcinoma. BJU international 118, 408–415 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Iyer G. et al. Genome Sequencing Identifies a Basis for Everolimus Sensitivity. Science (New York, N.Y 338, 221–221 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bendell JC et al. Phase I, Dose-Escalation Study of BKM120, an Oral Pan-Class I PI3K Inhibitor, in Patients With Advanced Solid Tumors. Journal of Clinical Oncology 30, 282–290 (2012). [DOI] [PubMed] [Google Scholar]
- 20.Baselga J. et al. Buparlisib plus fulvestrant versus placebo plus fulvestrant in postmenopausal, hormone receptor-positive, HER2-negative, advanced breast cancer (BELLE-2): a randomised, double-blind, placebo-controlled, phase 3 trial. The lancet oncology 18, 904–916 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Di Leo A. et al. Buparlisib plus fulvestrant in postmenopausal women with hormone-receptor-positive, HER2-negative, advanced breast cancer progressing on or after mTOR inhibition (BELLE-3): a randomised, double-blind, placebo-controlled, phase 3 trial. The lancet oncology 19, 87–100 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Eisenhauer EA et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45, 228–247 (2009). [DOI] [PubMed] [Google Scholar]
- 23.Cheng DT et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. The Journal of Molecular Diagnostics 17, 251–264 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carter SL et al. Absolute quantification of somatic DNA alterations in human cancer. Nature Biotechnology 30, 413 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cibulskis K. et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nature Biotechnology 31, 213 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lichtenstein L, Woolf B, MacBeth A, Birsoy O. & Lennon N. Abstract 3641: ReCapSeg: Validation of somatic copy number alterations for CLIA whole exome sequencing. Cancer research 76, 3641–3641 (2016). [Google Scholar]
- 27.Rosenthal R, McGranahan N, Herrero J, Taylor BS & Swanton C. deconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biology 17, 31 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Costello M. et al. Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation. Nucleic Acids Research 41, e67–e67 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chakravarty D. et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precision Oncology, 1–16 (2017). [DOI] [PMC free article] [PubMed]
- 30.Bahceci I. et al. PathwayMapper: a collaborative visual web editor for cancer pathways and genomic data. Bioinformatics 33, 2238–2240 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bellmunt J. et al. Prognostic Factors in Patients With Advanced Transitional Cell Carcinoma of the Urothelial Tract Experiencing Treatment Failure With Platinum-Containing Regimens. Journal of Clinical Oncology 28, 1850–1855 (2010). [DOI] [PubMed] [Google Scholar]
- 32.Robertson AG et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 171, 540–556.e525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jing C, Jin YH, You Z, Qiong Q. & Jun Z. Prognostic value of amphiregulin and epiregulin mRNA expression in metastatic colorectal cancer patients. Oncotarget 7, 55890–55899 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tebbutt N, Pedersen MW & Johns TG Targeting the ERBB family in cancer: couples therapy. Nature Reviews Cancer 13, 663 (2013). [DOI] [PubMed] [Google Scholar]
- 35.Heudel PE et al. Phase II study of the PI3K inhibitor BKM120 in patients with advanced or recurrent endometrial carcinoma: a stratified type I–type II study from the GINECO group. British journal of cancer 116, 303 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bendell JC et al. Phase I, Dose-Escalation Study of BKM120, an Oral Pan-Class I PI3K Inhibitor, in Patients With Advanced Solid Tumors. Journal of Clinical Oncology 30, 282–290 (2012). [DOI] [PubMed] [Google Scholar]
- 37.Wen PY et al. Phase II trial of the phosphatidyinositol-3 kinase (PI3K) inhibitor buparlisib (BKM120) in recurrent glioblastoma. Journal of Clinical Oncology 32, 2019–2019 (2014). [Google Scholar]
- 38.Shih KC et al. A phase II study of the combination of BKM120 (buparlisib) and bevacizumab in patients with relapsed/refractory glioblastoma multiforme (GBM). Journal of Clinical Oncology 33, 2065–2065 (2015). [Google Scholar]
- 39.Maira S-M et al. Identification and Characterization of NVP-BKM120, an Orally Available Pan-Class I PI3-Kinase Inhibitor. Molecular cancer therapeutics 11, 317 (2012). [DOI] [PubMed] [Google Scholar]
- 40.Pal SK et al. Efficacy of BGJ398, a fibroblast growth factor receptor (FGFR) 1–3 inhibitor, in patients (pts) with previously treated advanced/metastatic urothelial carcinoma (mUC) with FGFR3 alterations. Journal of Clinical Oncology 34, 4517–4517 (2016). [Google Scholar]
- 41.Siefker-Radtke AO et al. A phase 2 study of JNJ-42756493, a pan-FGFR tyrosine kinase inhibitor, in patients (pts) with metastatic or unresectable urothelial cancer (UC) harboring FGFR gene alterations. Journal of Clinical Oncology 34, TPS4575-TPS4575 (2016). [Google Scholar]
- 42.Choudhury NJ et al. Afatinib Activity in Platinum-Refractory Metastatic Urothelial Carcinoma in Patients With ERBB Alterations. Journal of Clinical Oncology 34, 2165–2171 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Font Pous A. et al. Phase II trial of afatinib in patients with advanced/metastatic urothelial carcinoma (UC) with genetic alterations in ERBB receptors 1–3 who failed on platinum-based chemotherapy (CT). Journal of Clinical Oncology 36, TPS540-TPS540 (2018). [Google Scholar]
- 44.Pal SK et al. Efficacy of BGJ398, a Fibroblast Growth Factor Receptor 1–3 Inhibitor, in Patients with Previously Treated Advanced Urothelial Carcinoma with <em>FGFR3</em> Alterations. Cancer Discovery 8, 812 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kwiatkowski DJ et al. Mutations in TSC1, TSC2, and MTOR Are Associated with Response to Rapalogs in Patients with Metastatic Renal Cell Carcinoma. Clinical Cancer Research 22, 2445 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Network, N.C.C. NCCN Clinical Practice Guidelines in Oncology; Bladder Cancer (Version 3.2018). (2018).
- 47.Zhou W-Y, Zheng H, Du X-L & Yang J-L Characterization of FGFR signaling pathway as therapeutic targets for sarcoma patients. Cancer biology & medicine 13, 260–268 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang L. et al. A Functional Genetic Screen Identifies the Phosphoinositide 3-kinase Pathway as a Determinant of Resistance to Fibroblast Growth Factor Receptor Inhibitors in FGFR Mutant Urothelial Cell Carcinoma. European urology 71, 858–862. [DOI] [PubMed] [Google Scholar]
- 49.Thomsen MBH et al. Comprehensive multiregional analysis of molecular heterogeneity in bladder cancer. Scientific reports 7, 11702 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Faltas BM et al. Clonal evolution of chemotherapy-resistant urothelial carcinoma. Nature genetics 48, 1490 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.