

Abstract
Hepatocellular carcinoma (HCC) is the 5th most common and 2nd deadliest cancer worldwide. HCC risk factors include alcohol induced liver cirrhosis, which prompts hepatic inflammation, cell necrosis, and fibrosis deposition. As 25% of HCC cases are associated with alcohol induced liver disease, understanding the effects of the cirrhotic liver microenvironment on HCC tumor biology and therapeutic responses are critical. This study utilized the Oncopig Cancer Model—a transgenic pig model that recapitulates human HCC through induced expression of KRASG12D and TP53R167H driver mutations—to investigate the molecular mechanisms underlying alcohol induced liver disease. Oncopigs (n = 5) underwent fibrosis induction via infusion of ethanol and ethiodized oil (1:3 v/v dosed at 0.75 mL/kg) into the hepatic arterial circulation. Eight-weeks post induction, liver tissue samples from fibrotic and age-matched control (n = 5) Oncopigs were collected for histological evaluation and transcriptional profiling. Increased hepatic inflammation and fibrosis was observed in fibrotic Oncopigs via pathological assessment. Transcriptional profiling (RNA-seq) resulted in the identification of 4387 differentially expressed genes between Oncopig fibrotic and control livers. GO term enrichment analysis identified pathway alterations associated with cirrhosis progression in humans, including cell proliferation, angiogenesis, extracellular matrix deposition, and oxidation-reduction. Key alterations include activation of hepatic stellate cells, increased matrix metalloproteinase production, and altered expression of ABC and SLC transporter genes involved in transport of anticancer drugs.These results demonstrate Oncopig liver fibrosis recapitulates transcriptional hallmarks of human cirrhosis, making the Oncopig an ideal model for studying the effects of the cirrhotic liver microenvironment on HCC tumor biology and therapeutic response.
Keywords: Alcoholic liver disease, Fibrosis, Pig cancer model, Translational research, Matrix metalloproteinases
1. Introduction
Hepatocellular carcinoma (HCC), or primary liver cancer, is a deadly tumor that spans more than 780,000 new diagnoses and causes 750,000 annual deaths worldwide, accounting for more than 9% of yearly cancer mortality [1,2]. While global cancer incidence and mortality are generally decreasing, HCC incidence is not only rising but is projected to continually increase for the foreseeable future given the growing prevalence of chronic liver diseases [3]. Alcoholic liver disease represents a common chronic liver ailment which is progressive and incites liver cirrhosis—characterized by hepatic inflammation, cell necrosis, liver regeneration, and malignant transformation—with cancer developing at a 5-year incidence of 30% in at-risk cirrhotic populations [4]. The worldwide prevalence of liver cirrhosis approximates 4.5–9.5%, accounting for approximately 2% of all global mortality and affecting more than 600,000 people in the United States [5,6]. Alcohol-induced liver cirrhosis is the second most common cause of liver cirrhosis after hepatitis C viral infection in the United States, accounting for 20%–50% of the liver cirrhosis diagnoses. As the health status of the liver can have profound effects on HCC tumor biology and response to treatments [7], further insights into the molecular mechanisms underlying alcohol-induced liver cirrhosis and its effect on HCC tumor biology are critically needed.
Since alcohol consumption is a major cause of liver cirrhosis and increases the risk for HCC, development of animal models capable of exhibiting both HCC and alcohol induced liver cirrhosis is essential for investigating HCC detection, development and natural history, and response to treatment in its native comorbid cirrhotic background. A number of rodent animal models of HCC and alcohol induced cirrhosis are currently used in preclinical research; however, no rodent model exists that fully mirrors human alcoholic liver disease by developing liver fibrotic stages via alcohol consumption [8]. In order to overcome these limitations, new techniques have been introduced, such as the combination of ethanol administration with a second stimulus, such as LPS, leading to increased immune infiltrates, necrosis, and more severe liver injury [9,10]. However, their small size limits the ability to test novel imaging modalities and interventional approaches that show promise for improving HCC outcomes.
Pigs represent an ideal animal platform for development of a large animal HCC model for testing of interventional, immuno-, and combination therapeutic strategies for HCC due to their similar size, anatomy, physiology, immunology, metabolism, and genetics to humans. We previously developed an orthotopic porcine HCC model using the Oncopig Cancer Model, a transgenic pig model that recapitulates human cancer through development of site and cell specific tumors via induced expression of heterozygous KRASG12D and TP53R167H driver mutations [11–15]. Using the Oncopig, we have also demonstrated the ability to develop a pre-cirrhotic METAVIR F2–F3 model of alcoholic liver disease within 8 weeks via a single intravascular administration of an ethanol:ethiodized oil emulsion via the hepatic artery [16]. However, liver recovery consistent with the reversal of liver fibrosis in abstinent patients presenting with pre-cirrhotic alcoholic liver disease was observed [17,18], with fibrosis levels reducing to METAVIR grade F1–F2 within 20 weeks post induction [16]. These results indicate that a single induction procedure induces transient fibrosis, with prolonged or repeated alcohol exposure required to develop an irreversible METAVIR F4 porcine cirrhosis model.
Despite the need for additional model development, the Oncopig pre-cirrhotic model provides the opportunity to investigate the molecular mechanisms underlying alcoholic liver disease progression and recovery in a clinically relevant large animal HCC model. The aim of this study was to identify altered gene expression underlying previously reported increases in liver fibrosis and inflammation in response to alcohol exposure in Oncopigs [16]. The results presented here provide insights into the molecular mechanisms underlying fibrosis deposition and clearance in response to a single alcohol exposure, highlighting the translational relevance of the Oncopig alcohol induced liver disease model.
2. Methods
2.1. Study design
Complete details on animals, housing, and study design can be found in Gaba et al. [16]. Briefly, alcohol-induced liver fibrosis was induced in female Oncopigs (n = 5) at 60 days of age via infusion of absolute ethanol and ethiodized oil (1:3 v/v dosed at 0.75 mL/kg) into the hepatic arterial circulation [16]. This study was restricted to female pigs to control for sex differences between individuals. Female pigs were chosen over male pigs due to the need to castrate male subjects, which is not representative of the human population. Eight weeks post induction, Oncopigs were euthanized and liver samples were harvested for histological and transcriptomic analyses. Histological and transcriptomic results were compared to age and sex-matched healthy control Oncopigs (n = 5).
2.2. Sample collection
Eight weeks post fibrosis induction, fibrotic Oncopigs and age-matched controls were euthanized and fibrotic and normal liver tissues were harvested, respectively. The 8-week timepoint was chosen for histological and transcriptional based on previous studies demonstrating peak disease severity at 8 weeks post induction [16]. Liver segments were transected so that adjacent halves were fixed in formalin for histological evaluation and flash frozen in liquid nitrogen within 10 min of euthanasia for transcriptomic analysis. All samples were stored at −80 °C until processing.
2.3. Histological evaluation
Descriptive and semiquantitative histopathologic analyses were performed as part of our previous publication [16]. Briefly, formalin fixed samples were provided to the Research Histology and Tissue Imaging Core at the University of Illinois at Chicago for processing, embedding, sectioning, and staining. Slides were stained using H&E and Masson’s trichrome. Whole slides were scanned using a Hamamatsu Nanozoomer scanner (Hamamatsu Photonics, Hamamatsu, Japan), and digital images were visualized with NDP.view2 software (Hamamatsu) and blindly graded for fibrosis and inflammation by a board-certified veterinary pathologist according to a porcine-adapted METAVIR system [16]. For quantitative assessment of collagen/fibrosis, digital images of trichrome stained slides were also imported to ImageJ (NIH) [19] using BioFormats [20], and subjected to color deconvolution for quantification of trichrome positive collagen (fibrosis), expressed as a percentage of total liver tissue section area. The same board-certified veterinary pathologist oversaw and performed quality control assessments of thresholded image analysis masks, also in a blinded fashion, for the quantitative assessment of fibrosis.
2.4. RNA extraction
Total RNA was extracted from flash frozen Oncopig fibrotic (n = 5) and control (n = 5) liver samples using the AllPrep DNA/RNA Mini Kit (Qiagen, Valencia, CA, USA) following the manufacturer’s protocol. RNA integrity and the presence of genomic DNA was tested on an Agilent 2100 Bioanalyzer using an RNA Nano bio-analyzer chip by the Carver High-Throughput DNA Sequencing and Genotyping Unit (HTS lab, University of Illinois, Urbana, IL, USA). Samples with RNA integrity numbers greater than 7 were utilized for sequencing.
2.5. RNA sequencing
TruSeq stranded RNA-seq libraries (TruSeq Stranded RNA Sample Preparation Kit, Illumina, San Diego, CA, USA) were generated from high-quality RNA by the HTS lab. The resulting RNA-seq libraries underwent Illumina sequencing on an Illumina HiSeq4000. The libraries were paired-end sequenced to a total read length of 100 base-pairs. The data sets supporting the results of this article are available in the Sequence Read Archive under accession number PRJNA629513.
2.6. RNA-seq expression analysis
An average of 77.8 million raw reads were generated per sample, ranging from 61.9 to 89.5 million. Trim Galore v.0.4.4 [21] was used to trim raw reads for adapter contamination, A-tails, minimum quality score (20), and minimum length (20 bp), setting the –stringency option to 6. Trimmed reads were aligned to the swine reference genome (Sscrofa10.2) [22] using Tophat v.2.1.1 [23] using the –M, –G, and fr-firststrand options. In addition, the –r option was set to 20, the -p option to 10, the –read-realign-edit-dist option to 0, and the –mate-std-dev option to 180. The featureCounts function of the Subread v.1.5.2 [24] R package was used to extract raw counts from the aligned bam files using the -p and -B options. The produced count matrices were merged to generate a single counts matrix containing read counts for all the samples. The counts matrix was then imported into DESeq2 [25] for differential gene expression analysis. Normalized gene expression levels were also calculated for the quality assessment and visualization using DESeq2. The list of differentially expressed genes (DEGs) was subsequently extracted from the results generated by DESeq2 function. DESeq2 uses Benjamini-Hochberg adjustment method to calculate adjusted p-values for each gene. Genes with a adjusted p-values < 0.05 were considered differentially expressed.
2.7. GO-term analysis
GO term enrichment analysis was performed to identify GO terms enriched for DEGs using the Gene Ontology Consortium [26] with all Sus scrofa genes used as a background. Three domains (cellular component, molecular function, and biological process) were used to perform GO term enrichment analysis. The Bonferroni method was used to correct for multiple testing, and GO terms with a q-value < 0.05 were considered enriched.
2.8. Statistical analysis
R v.3.4.1 was used for statistical analysis [27]. The Mann-Whitney-Wilcoxon test was used to assess statistically significant differences in fibrosis, inflammation scores, and percent fibrosis between fibrotic and control samples. ANOSIM was used to test for significant differences between fibrotic and control gene expression profiles.
3. Results
3.1. Increased hepatic fibrosis and inflammation in response to alcohol exposure
Our previous study identified significant histopathological differences between the Oncopig fibrotic and healthy control liver samples utilized in this study [16]. In this study, histological interpretations were performed on formalin fixed liver samples adjacent to flash frozen samples utilized for transcriptional profiling. Increased liver fibrosis and inflammation was observed in response to alcohol exposure 8 weeks post fibrosis induction (Fig. 1). No atypical histological changes were observed in the pigs that were considered to be purely a result of ethanol infusion (such as acute necrosuppurative inflammation/liquefactive necrosis, vasculitis, thrombosis, or infarction), or uniquely distinct from those histological changes described for human alcoholic liver disease. In addition, no evidence of tumor formation was observed in Oncopig livers 8-weeks post fibrosis induction based on macroscopic and histological examination. A previously developed porcine adapted METAVIR scheme [16] was used to quantify fibrosis and inflammation levels. METAVIR fibrosis and inflammation scores were significantly higher in Oncopig fibrotic compared to healthy liver samples (p-value = 0.01 and 0.009, respectively; Fig. 1C). In addition, quantitative image analysis of trichrome-stained collagen demonstrated an increase in mean percent fibrosis when comparing the fibrotic and control liver samples (average 14.96% vs 8.58%, respectively; Fig. 1C), although this increase was not statistically significant. Together, these results demonstrate successful induction of alcoholic liver disease via infusion of absolute ethanol and ethiodized oil into the Oncopig hepatic arterial circulation.
Fig. 1. Alcohol exposure results in fibrosis induction in Oncopig livers.
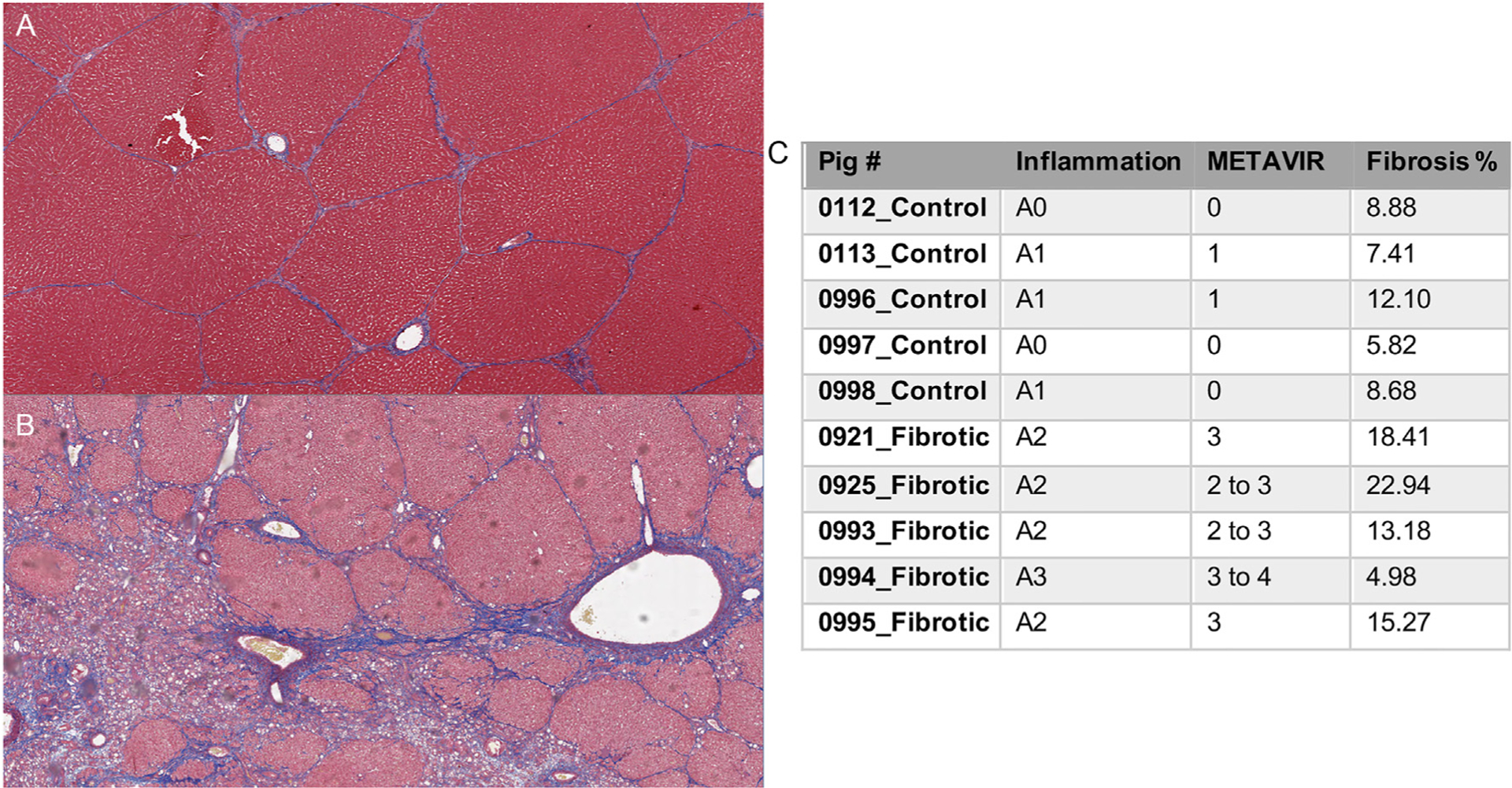
Representative images of Masson’s trichrome-stained Oncopig liver sections (2.5X) histologically graded for fibrosis using a porcine adapted METAVIR scheme. (A) Control, histologically normal Oncopig liver with normal pre-existing fibrous septa and associated classic hepatic-lobule architecture. (B) F3 fibrosis grade demonstrating numerous inter- and intra-lobular dissecting fibrous septa present at 8 weeks post-induction. (C) METAVIR inflammation, fibrosis scores, and percent fibrosis for each samples 8 weeks post-induction.
3.2. Reproducible effect of alcohol exposure on hepatic gene expression
In order to assess the impact of alcohol exposure on genome-wide hepatic gene expression profiles, PCA was performed for the 21,748 genes expressed in at least one sample. Fibrotic and control samples clustered by group (ANOSIM R = 0.248, P = 0.006; Fig. 2A), indicating significant and consistent genome-wide changes in gene expression induced by alcohol exposure in the fibrotic liver. In addition, a total of 4387 DEGs were detected between the fibrotic and control liver samples, with 2216 up-regulated and 2171 down-regulated in the fibrotic compared to the control samples (Fig. 2B and C, Table S1). As expected, samples also clustered by group when comparing the 4387 DEGs (ANOSIM R = 0.788, P = 0.011; Fig. 2C and D), providing further evidence that alcohol exposure results in reproducible changes in hepatic gene expression. In order to investigate the biological relevance of the identified DEGs, Gene Ontology (GO) term enrichment analysis was performed. In total, 239 GO terms were significantly enriched for DEGs, many of which are known to be associated with liver cirrhosis progression, including cell proliferation, angiogenesis, extracellular matrix remodeling, apoptosis, and oxidation-reduction processes (Fig. 3).
Fig. 2. Alcohol exposure results in reproducible alteration of hepatic gene expression profiles.
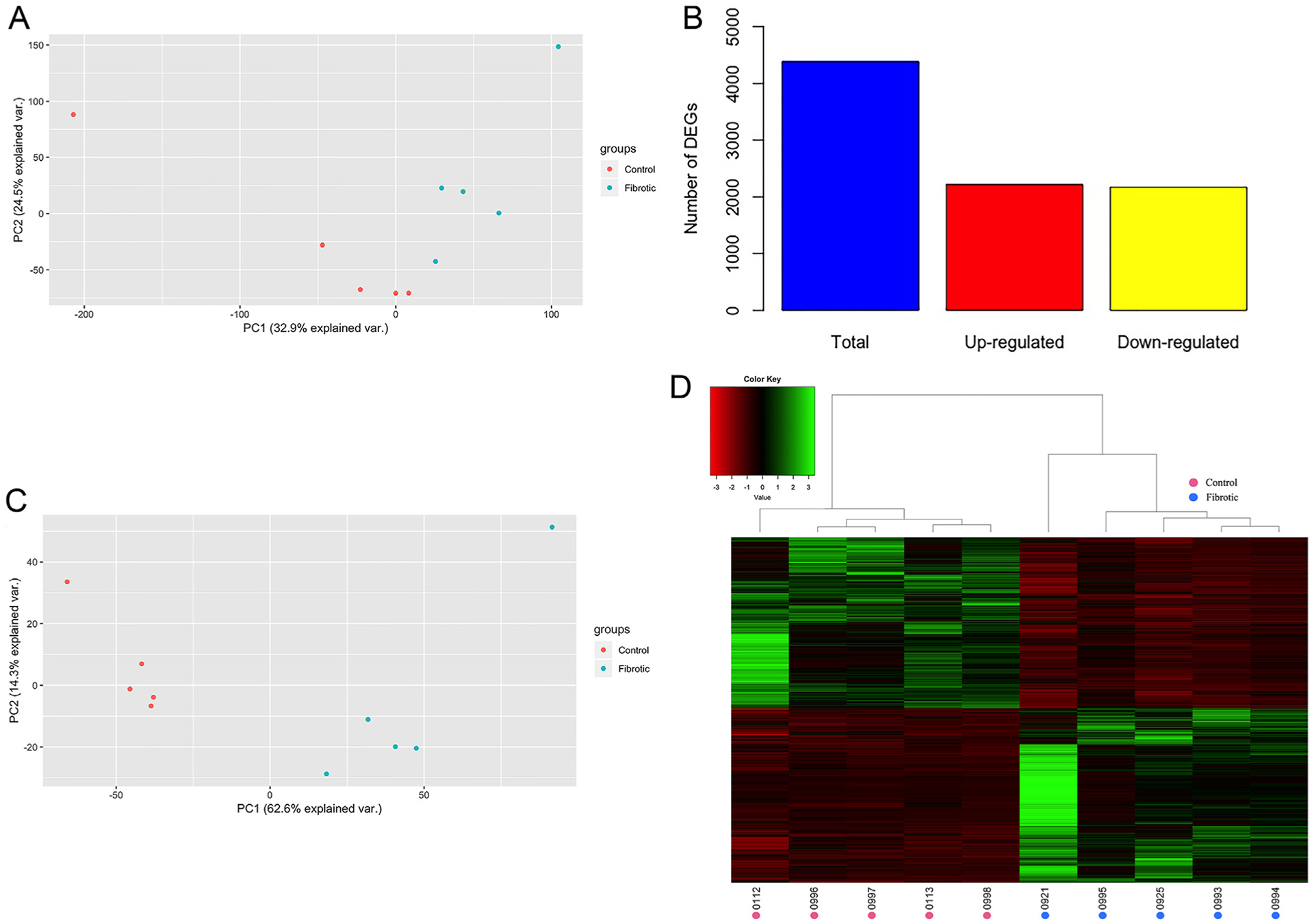
(A) PCA plot demonstrating clustering of samples by group based on the relative expression of 21,748 genes for which expression information was available for each sample. (B) Total number of DEGs, including those up- and down-regulated in the fibrotic compared to control group. (C) PCA plot demonstrating samples cluster by group based on the 4387 DEGs. (D) Heatmap of the normalized expression level of the 4387 DEGs for each sample, represented as z-scores. Dendrograms represent relationships between samples (columns) based on complete linkage clustering.
Fig. 3. GO terms enriched for DEGs associated with liver cirrhosis progression.
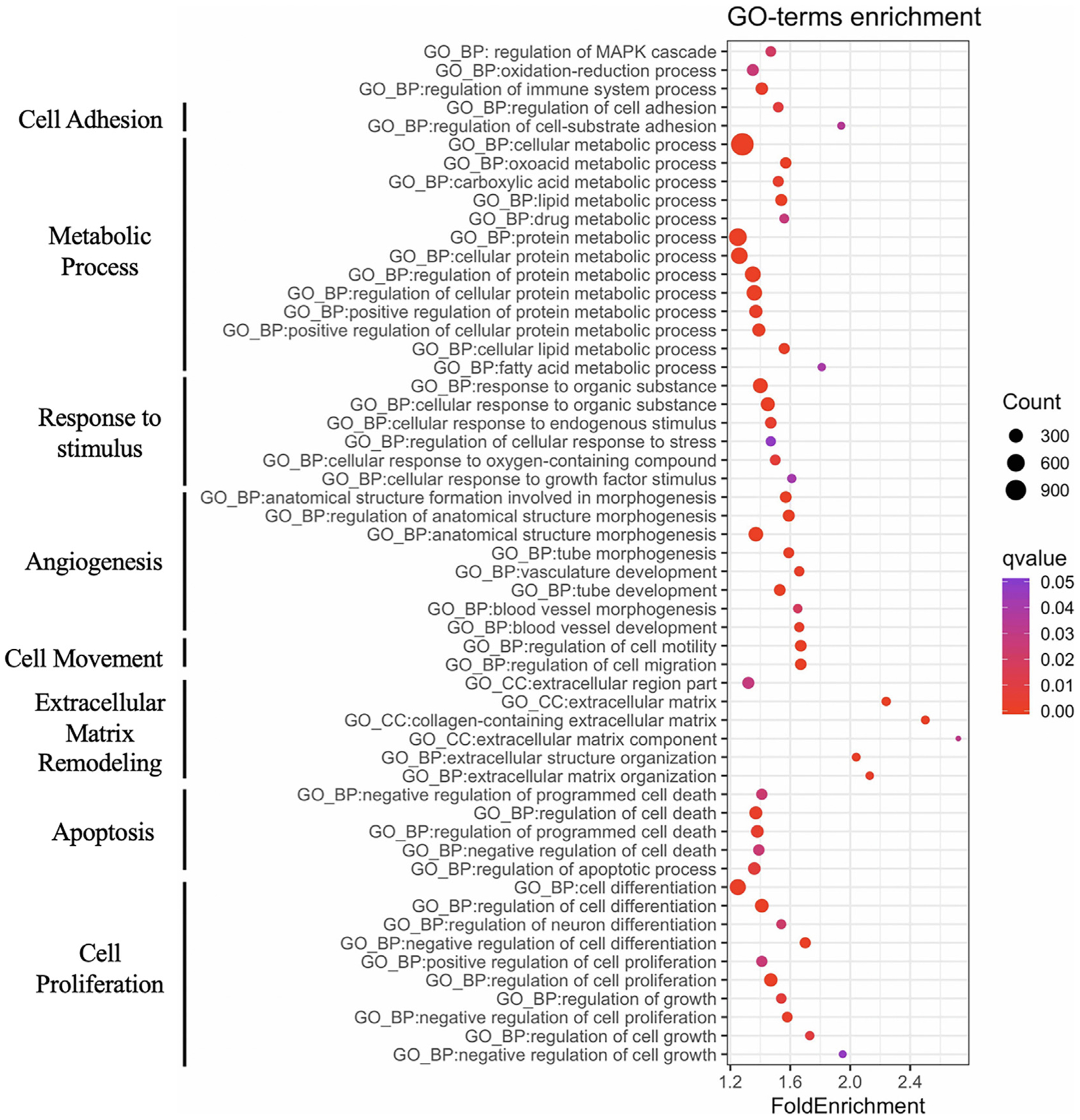
GO terms enriched for the 4387 DEGs are associated with biological processes involved in human liver cirrhosis progression.
3.3. Promotion of hepatic stellate cell activation and proliferation
As a result of chronic alcohol abuse, resident Kupffer cells and infiltrating macrophages, platelets, and sinusoidal endothelial cells secrete growth factors such as TGF-β and PDGF [28]. These growth factors trigger the proliferation of hepatic stellate cells (HSCs), which play a central role in the development of liver fibrosis through secretion of extracellular matrix (ECM) components. In this study, multiple growth factors from the TGF-β family (TGFB2 and TGFB3) were upregulated in Oncopig fibrotic liver samples (Log2 fold change 1.06 and 1.60, respectively). TGF-β plays an important role in the progression of liver disease by promoting hepatocyte cell death and HSC activation [29]. In addition, the PDGF receptor PDGFRA displayed increased expression in Oncopig fibrotic livers (Log2 fold change 0.69). PDGFRA is required for TGF-β signaling in human HSCs, with PDGFRA knockdown resulting in inhibition of SMAD-dependent TGF-β signaling [30]. PDGFRA also plays a role in cell survival and proliferation, with increased expression associated with liver injury [31]. Increased CD248 expression was also observed in Oncopig fibrotic livers (Log2 fold change 2.03), consistent with previous observations in human and mouse fibrotic livers where its expression is confined to activated HSCs [32]. CD248 is involved in PDGF signaling and promotes fibrosis deposition and proliferation of HSCs [32]. Finally, decreased expression of STAT1 was observed in the Oncopig fibrotic liver samples (Log2 fold change −0.69). STAT1 is a negative regulator of HSC proliferation whose downregulation accelerates liver fibrosis by enhancing PDGF-induced HSC proliferation [33]. Altered expression of these important mediators of HSC proliferation and activation in the Oncopig fibrotic liver indicates that, similar to humans, HSC proliferation and activation is responsible for progressing alcoholic liver disease in this model.
In addition to evidence of increased HSC activation and proliferation, evidence of reduced hepatocyte proliferation was observed in Oncopig fibrotic liver samples, including reduced HGF and STAT3 expression (log2 fold change −0.86 and −0.94, respectively). HGF stimulates hepatocyte proliferation and angiogenesis [34], partially through activation of STAT335. STAT3 is activated in hepatocytes in response to cytokine production, resulting in promotion of hepatocyte proliferation [35], with reduced compensatory hepatocyte proliferation observed in STAT3 deficient mice [36]. Reduced MYC expression was also observed in Oncopig fibrotic compared to control liver samples (log2 fold change −1.68; Fig. 4A). This is inconsistent with the elevated expression observed in human cirrhotic livers [37], where MYC overexpression by hepatocytes leads to hepatocyte proliferation, activation of HSCs, and induction of fibrogenesis (Fig. 4B). Together, these results suggest that while HSC activation and proliferation are primarily responsible for fibrosis deposition in this model, reduced MYC expression suggests that HSCs are no longer being activated 8 weeks post induction due to a lack of chronic alcohol exposure, leading to resolution of fibrosis and liver recovery. This is consistent with our previous report demonstrating that while alcohol exposure results in increased liver fibrosis and inflammation in Oncopigs, the lack of persistent alcohol exposure results in liver recovery within 20 weeks [16].
Fig. 4. Downregulation of MYC associated with inactivation of HSCs.
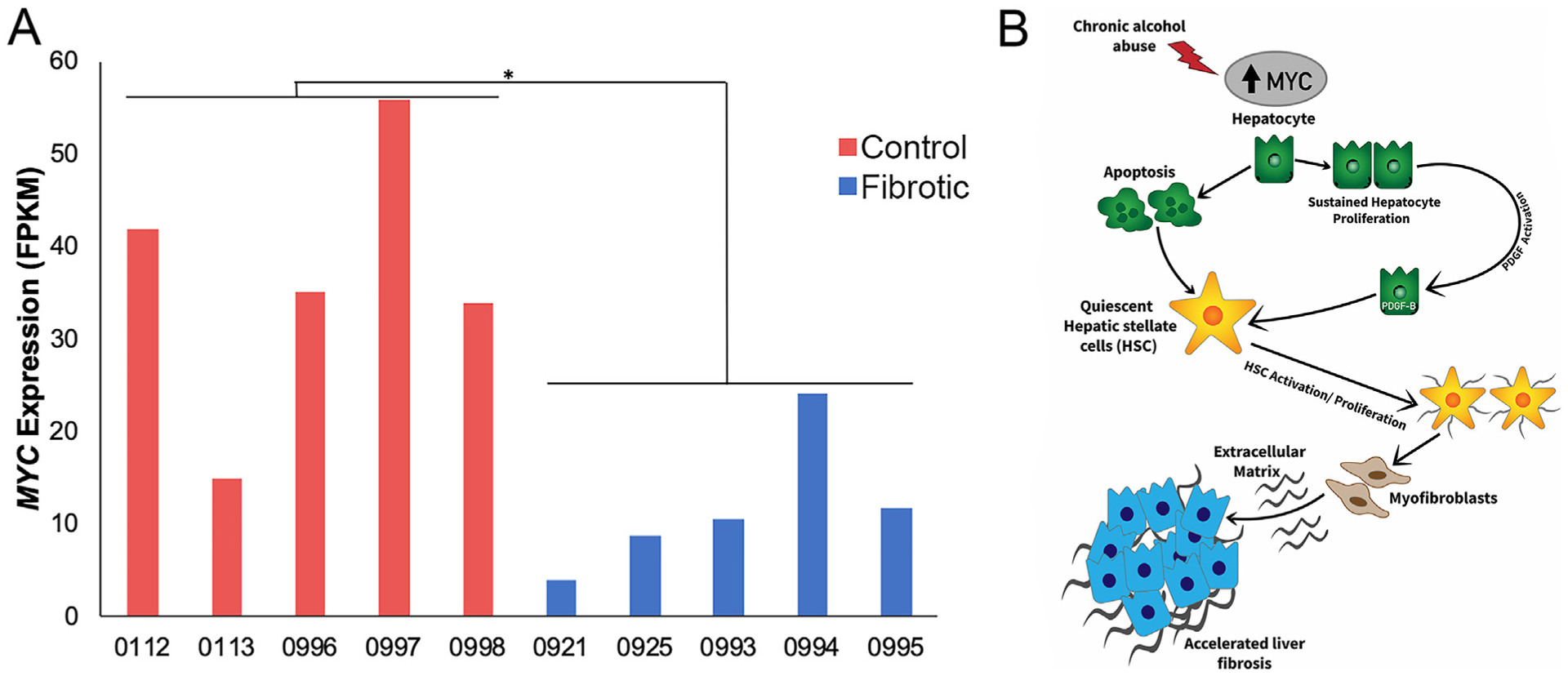
(A) Reduced expression of MYC was observed in the Oncopig fibrotic liver samples. (B) Diagram depicting upregulation of MYC due to chronic alcohol abuse resulting in hepatic apoptosis and activation and proliferation, which contributes to accelerated liver fibrosis. Adopted from Nevzorova et al. [37]. * denotes q-value = 9.5 × 10−4.
3.4. Increased matrix metalloproteinase and tissue inhibitors of metalloproteinase production as the main driver in ECM remodeling
In addition to secretion of ECM components, HSCs express matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs), which represent major regulators of liver fibrosis remodeling [38]. MMP overexpression generally results in fibrosis degradation, while increased TIMP expression results in MMP inhibition and increased fibrosis deposition. However, while MMPs are well known for their anti-fibrotic effects, they can also exert pro-fibrotic effects, with increased expression of a number of MMPs associated with liver disease progression [39,40]. The imbalance between MMP and TIMP expression therefore represents one of the main mechanisms leading to fibrosis deposition and resolution in human cirrhotic livers and other animal models of liver cirrhosis [41]. In this study, increased expression of TIMP1 and TIMP2 was observed in Oncopig fibrotic compared to control livers (Fig. 5). Both TIMP1 and TIMP2 are expressed at high levels in human cirrhosis patients and murine fibrotic liver models [42–44], with TIMP1 overexpression leading to increased fibrosis accumulation [45,46], while TIMP2 inhibition reduces fibrosis deposition by decreasing HSC activation and collagen accumulation [47].
Fig. 5. Differential MMP and TIMP expression in fibrotic Oncopig livers.
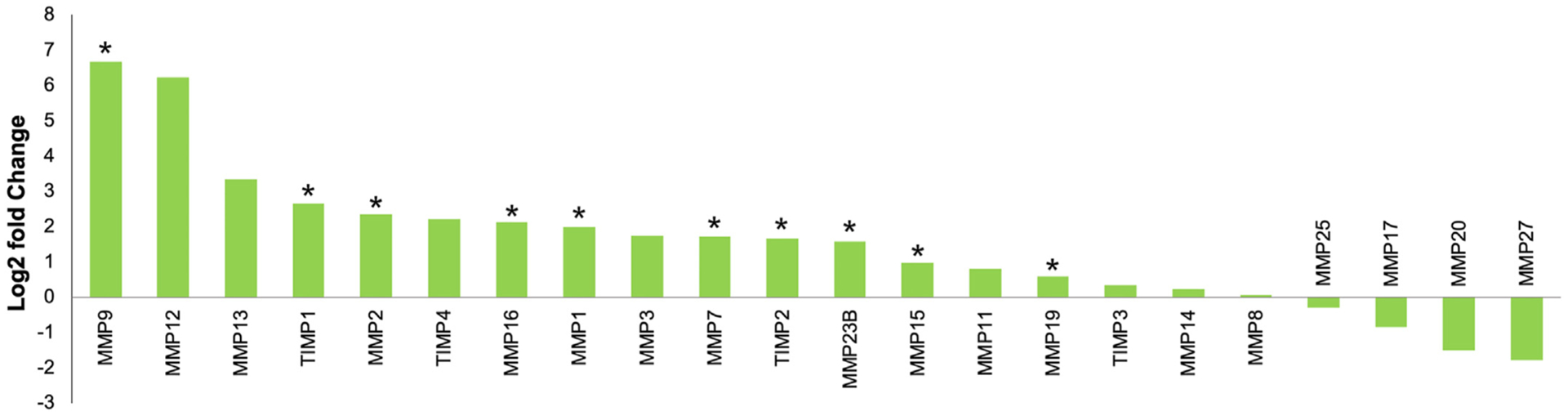
Altered expression of MMPs and TIMPs in Oncopig fibrotic compared to control livers. * denotes q-value < 0.05.
A number of MMPs were also significantly upregulated in Oncopig fibrotic compared to control livers (Fig. 5). This included several MMPs whose expression is increased in human liver cirrhosis and HCC. MMP19 is a pro-fibrotic MMP that promotes TGF-β signaling leading to liver fibrosis disease progression [40,48], while MMP16 and MMP7 expression is associated with HCC capsular invasion [49] and colorectal carcinoma liver metastasis, respectively [50]. Increased expression of MMPs involved in ECM clearance was also observed in Oncopig fibrotic livers. MMP9 is expressed by leukocytes in acutely damaged livers [51] and is responsible for the turnover and degradation of several ECM proteins, including fibronectin and type IV collagen [52]. MMP9 also promotes HSC apoptosis [53], and is associated with HCC metastasis and poor prognosis [54–57]. Another MMP displaying increased expression in Oncopig fibrotic livers was MMP2. MMP2 is expressed in human fibrotic livers [58] and contributes to reducing liver fibrosis severity by suppressing collagen type I expression [59,60], while MMP1 overexpression breaks down established fibrosis and induces hepatocyte proliferation [61]. The upregulation of MMPs involved in both fibrosis deposition and clearance in Oncopig fibrotic livers is consistent with our previous report demonstrating peak disease severity at 8 weeks post fibrosis induction followed by liver recovery [16].
3.5. Evidence of neoangiogenesis
Recent studies have identified vascular remodeling and abnormal angioarchitecture associated with the development and progression of liver fibrosis [62]. In this study, GO terms associated with angiogenesis were enriched for DEGs, including blood vessel development, blood vessel morphogenesis, and vasculature development (Fig. 3). Increased expression of several proangiogenic factors was observed in the Oncopig fibrotic liver samples (Fig. 6), including increased NRP1 expression, which has previously been shown to induce both angiogenesis and fibrosis deposition via activation of PDGF and TGF-β pathways in human and rat models [63]. Increased ANGPT2 expression was also observed in Oncopig fibrotic livers, consistent with human liver cirrhosis patients [64]. ANGPT2 inhibition in rat models results in reduced liver fibrosis through the induction of vessel normalization and attenuation of hepatic inflammatory infiltrates [64]. PECAM1 is a proangiogenic adhesion molecule whose expression in fibrotic livers has been shown to induce a profibrotic and premetastatic environment in response to alcohol-induced liver damage, and was overexpressed in this study [65]. Finally, increased SLIT2 expression, which promotes angiogenic [66] and fibrogenic activities in the liver [67], was observed in Oncopig fibrotic livers. Together, these results suggest neoangiogenesis was induced in response to alcohol exposure in Oncopigs through the same molecular mechanisms previously described in humans and rodent models.
Fig. 6. Increased expression of proangiogenic factors.
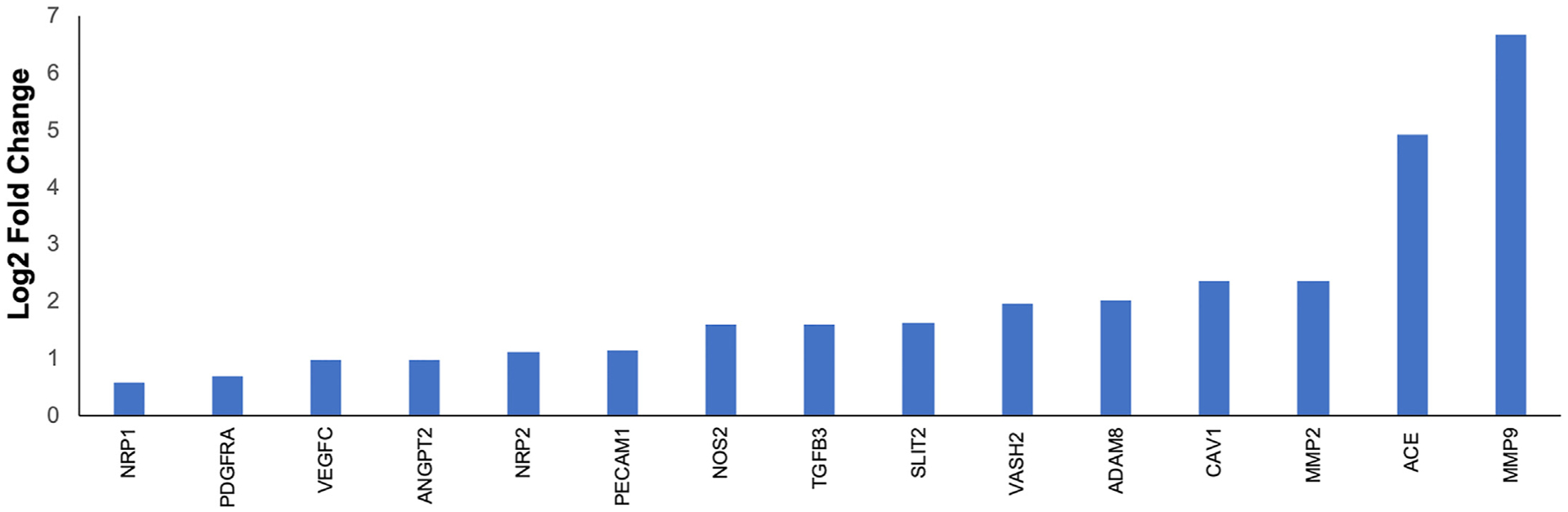
Increased expression of proangiogenic factors in Oncopig fibrotic compared to control livers.
3.6. Alcohol exposure results in increased oxidative stress
The liver is the major site for ethanol metabolism, and evidence has shown that oxidative stress is a significant pathogenic mechanism of alcohol induced fibrosis [68]. Oxidative stress results from overproduction of reactive oxygen species (ROS), and both acute and chronic alcohol consumption can induce oxidation-reduction processes and ROS production [69,70]. As expected, GO terms associated with oxidation-reduction were enriched for DEGs (Fig. 3). Upregulation of promotors of ROS production, including MICAL1, GPD2, NCF2, VCAM1, and AKR1B1 [71–75] was observed in Oncopig fibrotic liver samples (Fig. 7). ROS are also produced via ethanol metabolism by alcohol dehydrogenase (ADH) and aldehyde dehydrogenase (ALDH) enzymes [69]. Increased expression of several ADH and ALDH enzymes (ADH4, ALDH1B1, and ALDH1L2) was observed in the Oncopig fibrotic compared to control liver samples (Log2 fold change 1.32, 0.96, and 1.19, respectively). As expression of these genes contributes to ethanol oxidation and overproduction of ROS during alcohol consumption [76,77], the increased expression observed in this study suggests alcohol metabolism contributed to an overproduction of ROS and increased oxidative stress in the Oncopig fibrotic liver. However, it is important to note that reduced expression of RELSAT (promotor of ROS production) [78], in addition to increased expression of inhibitors of ROS production (GLUD1, MTHFR, and FASN) [79–81] was also observed in the Oncopig fibrotic liver samples (Fig. 7). The combined upregulation of promotors and inhibitors of ROS production is consistent with the above-mentioned transitionary phase between liver damage and recovery observed in this model, and suggests that while alcohol exposure results in increased oxidative stress and ROS production in Oncopig livers, the lack of chronic alcohol exposure may reduce oxidative stress and in part facilitate liver recovery.
Fig. 7. DEGs associated with oxidation-reduction processes.
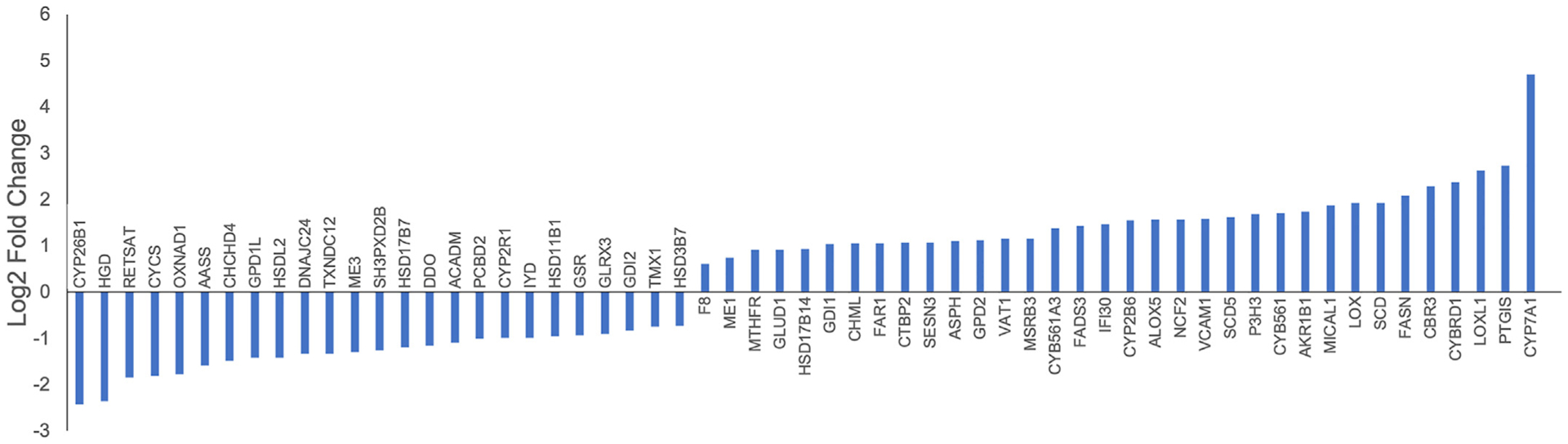
Up-regulated and down-regulated DEGs associated with the oxidation-reduction GO term in Oncopig fibrotic compared to control livers.
3.7. Altered drug metabolism in the oncopig fibrotic liver
As the liver plays a crucial role in metabolizing drugs, the functional status of the liver is especially important in patients undergoing chemotherapy or other therapeutic treatments [82]. We therefore assessed the impact of alcohol exposure on the expression of drug metabolizing enzyme gene families. Both increased (ABCC5 and ABCA7) and decreased (ABCB4, ABCE1, and ABCG8) expression of ABC transporters involved in chemotherapeutic efflux [83,84] was observed in Oncopig fibrotic livers (Fig. 8A). Increased expression of these ABC transporters is associated with cisplatin and doxorubicin resistance [85–90], both of which are commonly used to treat HCC clinically. In addition, differential expression of 64 SLC transporter genes was observed (38 downregulated, 26 upregulated; Fig. 8B), including transporters known to be involved in the transport of anticancer drugs: SL22A3 (Cisplatin, Oxaliplatin, irinotican, Vincristine, Melphane), SL22A7 (5-FU, Paclitaxel, Methotrexate), SLC31A2 (Cisplatin), and SLC7A2, SLC7A4, and SLC7A7 (Activicin) [91]. Finally, the drug metabolizing enzyme CBR3 was upregulated in Oncopig fibrotic livers (log2 fold change 2.29). Upregulation of CBR3 results in increased metabolism of doxorubicin leading to high doxorubicinol levels and cardiotoxicity in cancer patients [92]. Together, the increased and decreased expression of a variety of genes involved in drug metabolism and transport suggests Oncopigs with fibrotic livers may suffer from chemotherapeutic hepatotoxicity, providing a potential molecular mechanism for the reduced chemotherapeutic tolerance observed in people suffering from chronic liver diseases clinically.
Fig. 8. Altered expression of ABC and SLC transporters.
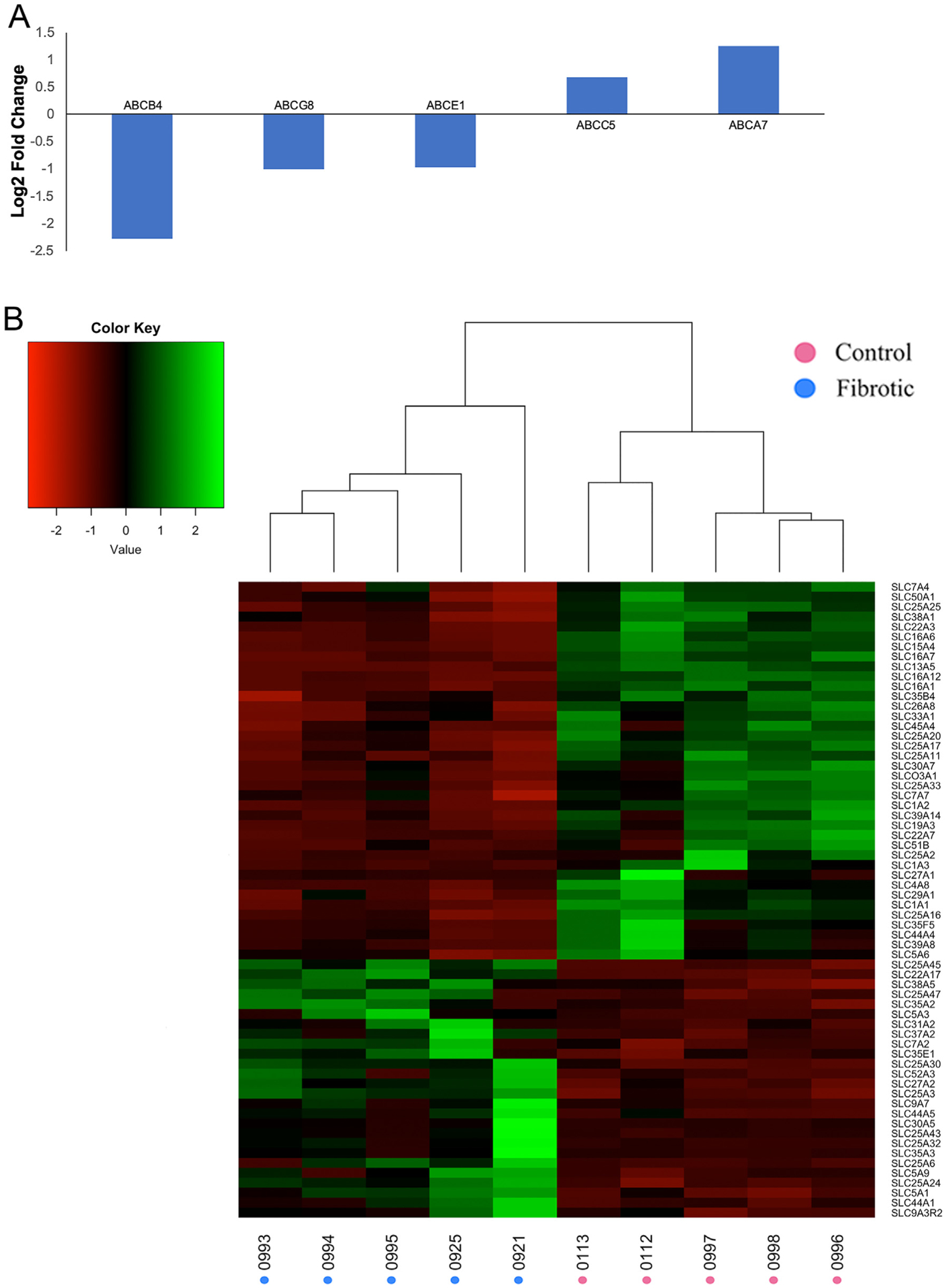
Altered expression of (A) ABC and (B) SLC transporter genes in Oncopig fibrotic compared to control liver samples. Dendrograms represent relationships between samples (columns) based on complete linkage clustering.
4. Discussion
Alcohol consumption is a major cause of liver cirrhosis and increases the risk for HCC development. This, combined with high HCC morbidity and mortality rates and the increasing use of device-based treatments for HCC highlights the need for clinically relevant large animal models capable of exhibiting both HCC and alcohol induced liver cirrhosis to improve treatment responses. We have previously demonstrated our ability to induce liver fibrosis in the Oncopig cancer model through infusion of absolute ethanol and ethiodized oil into the hepatic arterial circulation, which results in increased liver fibrosis and inflammation within 8 weeks followed by liver recovery within 20 weeks [16]. This study investigated altered transcriptional profiles in fibrotic Oncopig liver samples at the peak of disease severity (8-weeks post alcohol exposure), resulting in the identification of altered gene expression consistent with pathway disruptions commonly observed in human cirrhosis, including those involved in extracellular matrix remodeling, cellular proliferation, angiogenesis, apoptosis, and oxidation-reduction processes. In addition, increased expression of a number of MMPs and TIMPs was observed, indicating that the molecular mechanisms underlying Oncopig fibrosis deposition and clearance are representative of those observed clinically in human cirrhotic patients.
The ECM is a major component of the cellular microenvironment with a highly dynamic structure, and ECM remodeling is a pivotal mechanism in the development of liver fibrosis [93]. ECM remodeling is characterized by a continuous two-way process consisting of fibrosis deposition and degradation, with liver cirrhosis resulting from environmental exposures leading to increased fibrosis deposition due to an imbalance between these two processes [94]. One of the main molecular mechanisms underlying liver fibrosis deposition is activation of HSCs leading to expression of MMPs, TIMPs, and ECM components [38]. Consistent with observations from human liver cirrhotic patients and preclinical rodent studies [41], transcriptional signals of HSC activation were observed in this study, including overexpression of TIMP1, TIMP2, and a number of MMPs. This result suggests that, consistent with human liver disease, HSC activation followed by production of MMPs and TIMPs represents the driving force responsible for Oncopig liver fibrosis deposition and progression of alcohol induced liver disease. In addition, increased expression of MMPs associated with liver metastases and HCC progression also suggest the Oncopig fibrotic liver microenvironment is ideal for tumor development.
While liver cirrhosis represents the irreversible end stage of alcoholic liver disease, liver fibrosis can be reversed back to a normal state if alcohol intake is stopped or greatly reduced [18,95]. After abstaining from alcohol consumption, activated HSCs are removed through apoptosis or reversal to their quiescent state [96,97], resulting in reduced TIMP and increased MMP expression [98]. This represents a transitional phase during which both ECM deposition and degradation are occurring simultaneously. The reduced MYC expression observed in this study—expressed at high levels as in human cirrhotic livers resulting in activation of HSCs and induction of fibrosis deposition [37]—is inconsistent with human cirrhosis, and suggests that HSCs are no longer being activated due to a lack of chronic alcohol exposure, resulting in resolution of fibrosis and liver recovery. This is further supported by the increased expression of a number of MMPs involved in fibrosis degradation and liver recovery, and represents a significant difference between the Oncopig fibrosis model and clinically observed human liver cirrhosis.
Previous studies have demonstrated that intrahepatic vascular remodeling is commonly observed during liver disease progression in response to chronic alcohol consumption. In fibrotic livers, increased venous pressure leads to reduced portal venous blood flow and oxygen delivery to the liver resulting in hypoxic conditions [99]. Hypoxia stimulates the formation of new blood vessels through activation of hypoxia inducible factors, upregulating expression of vascular endothelial growth factors and proangiogenic mechanisms. Although enrichment of GO terms associated with hypoxia was not observed in this study, increased expression of several proangiogenic factors was observed, including several whose expression is frequently upregulated in cirrhotic patients and rodent models. The increased expression of proangiogenic factors suggests hypoxic conditions may be present in Oncopig fibrotic liver samples following fibrosis induction, although further studies are required to confirm.
Although transcriptional signals demonstrating simultaneous fibrosis deposition and clearance was observed in this model at 8-weeks post fibrosis induction, transcriptional and immunohistochemical profiling of liver samples at multiple timepoints would have provided a clearer picture of the molecular mechanisms underlying disease onset and liver recover, and represents a limitation of the current study. While transcriptional alterations consistent with those observed in humans and rodent liver cirrhosis models were observed, the spontaneous liver recovery observed in our previous publication [16] and subsequent molecular dissimilarities observed in this study represent limitations of this model in its current form. The results of this study suggest that liver recovery is likely due to a lack of chronic alcohol exposure resulting in increased MMP production and subsequent ECM degradation. This result suggests that multiple transarterial administrations may be required to induce irreversible liver damage representative of METAVIR stage F4 cirrhosis. In addition, the lack of transcriptional alterations suggestive of increased inflammation in fibrotic liver samples is in contrast to the chronic liver inflammation observed in cirrhotic patients, representing a limitation of the current model. In addition to direct effects on the liver, excessive alcohol consumption disrupts the structural integrity of the gut, causing release of immunogenic compounds, primarily lipopolysaccharides, into the circulation resulting in activation of innate immune responses [100,101]. Growing evidence indicates inflammatory responses to the release of these immunogenic compounds plays a significant role in the pathogenesis of alcohol induced cirrhosis [102]. Therefore, the combination of cirrhosis induction via direct exposure of the liver to ethanol and chronic alcohol feeding may be required for development of a large animal model of alcohol induced liver cirrhosis, although further studies are required to confirm this. Additional improvement of the current Oncopig fibrosis model is required to more accurately model the molecular alterations and disease severity observed clinically in human cirrhotic liver patients.
As a large number of drugs are predominately metabolized by the liver, the pharmacokinetics of numerous drugs is altered in patients suffering from liver cirrhosis [82]. These changes are caused by altered expression of drug metabolizing enzymes and transporters, altered hepatic blood flow, and decreased plasma protein binding [103–105]. Because of this, the Food and Drug Administration recommends patients with varying levels of liver disease and hepatic function be enrolled in clinical studies investigating drugs predominately cleared by the liver [82]. However, the combination of competing clinical trials, stringent enrollment criteria, and lengthy accrual rates makes this logistically challenging, highlight the need for clinically relevant animal models to assess the impact of liver disease on drug metabolism and toxicity. The altered expression of 5 ABC and 64 SLC transporters observed in Oncopig fibrotic livers suggests Oncopigs may represent an ideal model to assess the impact of alcoholic liver disease on chemotherapeutic hepatotoxicity, especially for locoregional delivery-based drug trials where the size of the pig represents an advantage over currently available rodent models. However, further work is required to increase disease severity and assess chemotherapeutic hepatotoxicity in vivo to confirm the utility of the Oncopig liver fibrosis model for this purpose.
Supplementary Material
Acknowledgements
Histological services were provided by the Research Resources Center Research Histology and Tissue Imaging Core at the University of Illinois at Chicago, established with the support of the Vice Chancellor of Research. This work was supported by the United States Department of Defense (Translational Team Science Award CA150590), Guerbet USA LLC (Healthcare Professional Grant for ethiodized oil material transfer), the Illinois Health Disparities Initiative, and the University of Illinois Department of Radiology. DPR is supported by The National Institutes of Health, Office of the Director, award number K01ODO22982 and National Center for Advancing Translational Sciences, award numberL30 TR002126.
Declaration of conflict of interest
RCG receives research funding from the National Insitute of Health, the United States Department of Defense, Guerbet USA LLC, and Janssen Research & Development LLC. KMS receives research funding from the National Insitute of Health, Guerbet USA LLC, and Janssen Research & Development LLC. AY, DPR, and LBS have no conflict of interest.
Footnotes
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.biochi.2020.12.022.
References
- [1].GLOBOCAN 2012: Estimated Cancer Incidence, Mortality and Prevalence Worldwide in 2012.
- [2].Flores A, Marrero JA, Emerging trends in hepatocellular carcinoma: focus on diagnosis and therapeutics, Clin. Med. Insights Oncol 8 (2014) 71–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM, Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States, Can. Res 74 (2014. June 1) 2913–2921. [DOI] [PubMed] [Google Scholar]
- [4].Singal AG, El-Serag HB, Hepatocellular carcinoma from epidemiology to prevention: translating knowledge into practice, Clin. Gastroenterol. Hepatol 13 (2015. November) 2140–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Scaglione S, Kliethermes S, Cao G, et al. , The epidemiology of cirrhosis in the United States: a population-based study, J. Clin. Gastroenterol 49 (2015. September) 690–696. [DOI] [PubMed] [Google Scholar]
- [6].Mokdad AA, Lopez AD, Shahraz S, et al. , Liver cirrhosis mortality in 187 countries between 1980 and 2010: a systematic analysis, BMC Med. 12 (2014. September 18) 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Tsai MJ, Chang WA, Huang MS, Kuo PL, Tumor microenvironment: a new treatment target for cancer, ISRN Biochem 2014 (2014) 351959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Yanguas SC, Cogliati B, Willebrords J, et al. , Experimental models of liver fibrosis, Arch. Toxicol 90 (2016. May) 1025–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Karaa A, Thompson KJ, McKillop IH, Clemens MG, Schrum LW, S-adenosyl-L-methionine attenuates oxidative stress and hepatic stellate cell activation in an ethanol-LPS-induced fibrotic rat model, Shock 30 (2008. August) 197–205. [DOI] [PubMed] [Google Scholar]
- [10].Apte UM, Banerjee A, McRee R, Wellberg E, Ramaiah SK, Role of osteopontin in hepatic neutrophil infiltration during alcoholic steatohepatitis, Toxicol. Appl. Pharmacol 207 (2005. August 22) 25–38. [DOI] [PubMed] [Google Scholar]
- [11].Schachtschneider K, Gaba R, Schwind R, et al. , Validation of the oncopig platform as a translational porcine model for human hepatocellular carcinoma, J. Vasc. Intervent. Radiol 28 (2017) S60. [Google Scholar]
- [12].Schachtschneider KM, Schwind RM, Darfour-Oduro KA, et al. , A validated, transitional and translational porcine model of hepatocellular carcinoma, Oncotarget 8 (2017. September 8) 63620–63634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Schachtschneider KM, Schwind RM, Newson J, et al. , The oncopig cancer model: an innovative large animal translational oncology platform, Front Oncol 7 (2017) 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Schook LB, Collares TV, Hu W, et al. , A genetic porcine model of cancer, PloS One 10 (2015), e0128864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Gaba R, Ray C, Schwind R, Darfour-Oduro K, Rund L, Schook L, The Oncopig cancer model: a novel genetically inducible porcine model of hepatocellular carcinoma for interventional radiology locoregional therapy testing, J. Vasc. Intervent. Radiol 27 (2016) S221. [Google Scholar]
- [16].Gaba RC, Mendoza-Elias N, Regan DP, et al. , Characterization of an inducible alcoholic liver fibrosis model for hepatocellular carcinoma investigation in a transgenic porcine tumorigenic platform, J. Vasc. Intervent. Radiol 29 (2018. August), 1194–202.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ellis EL, Mann DA, Clinical evidence for the regression of liver fibrosis, J. Hepatol 56 (2012) 1171–1180, 2012/May/01/. [DOI] [PubMed] [Google Scholar]
- [18].Ismail MH, Pinzani M, Reversal of liver fibrosis, Saudi J. Gastroenterol 15 (2009. January) 72–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Schneider CA, Rasband WS, Eliceiri KW, NIH Image to ImageJ: 25 years of image analysis, Nat. Methods 9 (2012. July) 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Linkert M, Rueden CT, Allan C, et al. , Metadata matters: access to image data in the real world, J. Cell Biol 189 (2010. May 31) 777–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Krueger F A wrapper tool around Cutadapt and FastQC to consistently apply quality and adapter trimming to FastQ files. Babraham Bioinformatics.Volume: 516, Pages: 517. [Google Scholar]
- [22].Groenen MAM, Archibald AL, Uenishi H, et al. , Analyses of pig genomes provide insight into porcine demography and evolution, Nature 491 (2012 2012/November/01) 393–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL, TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions, Genome Biol. 14 (2013 2013/April/25) R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Liao Y, Smyth GK, Shi W, featureCounts: an efficient general purpose program for assigning sequence reads to genomic features, Bioinformatics 30 (2014. April 1) 923–930. [DOI] [PubMed] [Google Scholar]
- [25].Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2, Genome Biol. 15 (2014) 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gene Ontology Consortium, Going forward, Nucleic Acids Res. 43 (2015. January) D1049–D1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].R.C.R. Team, A Language and Environment for Statistical Computing, R Foundation for Statistical Computing, Vienna, Austria, 2014. [Google Scholar]
- [28].Cheng K, Mahato RI, Gene modulation for treating liver fibrosis, Crit. Rev. Ther. Drug Carrier Syst 24 (2007) 93–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Dooley S, ten Dijke P, TGF-β in progression of liver disease, Cell Tissue Res. 347 (2012. January) 245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Liu C, Li J, Xiang X, et al. , PDGF receptor-α promotes TGF-β signaling in hepatic stellate cells via transcriptional and posttranscriptional regulation of TGF-β receptors, Am. J. Physiol. Gastrointest. Liver Physiol 307 (2014. October 1) G749–G759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kikuchi A, Monga S, Activation of platelet-derived growth factor receptor alpha in hepatic stellate cells during chronic liver injury, Faseb. J 30 (2016), 924.3–.3, 2016/April/01. [Google Scholar]
- [32].Wilhelm A, Aldridge V, Haldar D, et al. , CD248/endosialin critically regulates hepatic stellate cell proliferation during chronic liver injury via a PDGF-regulated mechanism, Gut 65 (2016. July) 1175–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Jeong W-I, Park O, Radaeva S, Gao B, STAT1 inhibits liver fibrosis in mice by inhibiting stellate cell proliferation and stimulating NK cell cytotoxicity, Hepatology 44 (2006) 1441–1451. [DOI] [PubMed] [Google Scholar]
- [34].Nakamura T, Mizuno S, The discovery of hepatocyte growth factor (HGF) and its significance for cell biology, life sciences and clinical medicine, Proc. Jpn. Acad. Ser. B Phys. Biol. Sci 86 (2010) 588–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Cong M, Iwaisako K, Jiang C, Kisseleva T, Cell signals influencing hepatic fibrosis, Int J Hepatol 2012 (2012) 158547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wang H, Lafdil F, Kong X, Gao B, Signal transducer and activator of transcription 3 in liver diseases: a novel therapeutic target, Int. J. Biol. Sci 7 (2011. April 27) 536–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Nevzorova YA, Hu W, Cubero FJ, et al. , Overexpression of c-myc in hepatocytes promotes activation of hepatic stellate cells and facilitates the onset of liver fibrosis, Biochim. Biophys. Acta 1832 (2013. October) 1765–1775. [DOI] [PubMed] [Google Scholar]
- [38].Benyon RC, Arthur MJ, Extracellular matrix degradation and the role of hepatic stellate cells, Semin. Liver Dis 21 (2001. August) 373–384. [DOI] [PubMed] [Google Scholar]
- [39].Duarte S, Baber J, Fujii T, Coito AJ, Matrix metalloproteinases in liver injury, repair and fibrosis, Matrix Biol. 44–46 (2015. May-Jul) 147–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Naim A, Pan Q, Baig MS, Matrix metalloproteinases (MMPs) in liver diseases, J Clin Exp Hepatol 7 (2017. December) 367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Herbst H, Wege T, Milani S, et al. , Tissue inhibitor of metalloproteinase-1 and −2 RNA expression in rat and human liver fibrosis, Am. J. Pathol 150 (1997. May) 1647–1659. [PMC free article] [PubMed] [Google Scholar]
- [42].Roeb E, Purucker E, Breuer B, et al. , TIMP expression in toxic and cholestatic liver injury in rat, J. Hepatol 27 (1997. September) 535–544. [DOI] [PubMed] [Google Scholar]
- [43].Busk TM, Bendtsen F, Nielsen HJ, Jensen V, Brunner N, Moller S, TIMP-1 in patients with cirrhosis: relation to liver dysfunction, portal hypertension, and hemodynamic changes, Scand. J. Gastroenterol 49 (2014. September) 1103–1110. [DOI] [PubMed] [Google Scholar]
- [44].Boker KH, Pehle B, Steinmetz C, Breitenstein K, Bahr M, Lichtinghagen R, Tissue inhibitors of metalloproteinases in liver and serum/plasma in chronic active hepatitis C and HCV-induced cirrhosis, Hepato-Gastroenterology 47 (2000. May-Jun) 812–819. [PubMed] [Google Scholar]
- [45].Yoshiji H, Kuriyama S, Miyamoto Y, et al. , Tissue inhibitor of metalloproteinases-1 promotes liver fibrosis development in a transgenic mouse model, Hepatology 32 (2000. December) 1248–1254. [DOI] [PubMed] [Google Scholar]
- [46].Yoshiji H, Kuriyama S, Yoshii J, et al. , Tissue inhibitor of metalloproteinases-1 attenuates spontaneous liver fibrosis resolution in the transgenic mouse, Hepatology 36 (2002. October) 850–860. [DOI] [PubMed] [Google Scholar]
- [47].Hu YB, Li DG, Lu HM, Modified synthetic siRNA targeting tissue inhibitor of metalloproteinase-2 inhibits hepatic fibrogenesis in rats, J. Gene Med 9 (2007. March) 217–229. [DOI] [PubMed] [Google Scholar]
- [48].Jirouskova M, Zbodakova O, Gregor M, et al. , Hepatoprotective effect of MMP-19 deficiency in a mouse model of chronic liver fibrosis, PloS One 7 (2012), e46271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Arai I, Nagano H, Kondo M, et al. , Overexpression of MT3-MMP in hepatocellular carcinoma correlates with capsular invasion, Hepato-Gastroenterology 54 (2007. Jan-Feb) 167–171. [PubMed] [Google Scholar]
- [50].Zeng ZS, Shu WP, Cohen AM, Guillem JG, Matrix metalloproteinase-7 expression in colorectal cancer liver metastases: evidence for involvement of MMP-7 activation in human cancer metastases, Clin. Canc. Res 8 (2002. January) 144–148. [PubMed] [Google Scholar]
- [51].Hamada T, Fondevila C, Busuttil RW, Coito AJ, Metalloproteinase-9 deficiency protects against hepatic ischemia/reperfusion injury, Hepatology 47 (2008. January) 186–198. [DOI] [PubMed] [Google Scholar]
- [52].Ram M, Sherer Y, Shoenfeld Y, Matrix metalloproteinase-9 and autoimmune diseases, J. Clin. Immunol 26 (2006. July) 299–307. [DOI] [PubMed] [Google Scholar]
- [53].Zhou X, Murphy FR, Gehdu N, Zhang J, Iredale JP, Benyon RC, Engagement of alphavbeta3 integrin regulates proliferation and apoptosis of hepatic stellate cells, J. Biol. Chem 279 (2004. June 4) 23996–24006. [DOI] [PubMed] [Google Scholar]
- [54].Arii S, Mise M, Harada T, et al. , Overexpression of matrix metalloproteinase 9 gene in hepatocellular carcinoma with invasive potential, Hepatology 24 (1996. August) 316–322. [DOI] [PubMed] [Google Scholar]
- [55].Sakamoto Y, Mafune K, Mori M, et al. , Overexpression of MMP-9 correlates with growth of small hepatocellular carcinoma, Int. J. Oncol 17 (2000. August) 237–243. [DOI] [PubMed] [Google Scholar]
- [56].Sun MH, Han XC, Jia MK, et al. , Expressions of inducible nitric oxide synthase and matrix metalloproteinase-9 and their effects on angiogenesis and progression of hepatocellular carcinoma, World J. Gastroenterol 11 (2005. October 14) 5931–5937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Nart D, Yaman B, Yilmaz F, Zeytunlu M, Karasu Z, Kilic M, Expression of matrix metalloproteinase-9 in predicting prognosis of hepatocellular carcinoma after liver transplantation, Liver Transplant. 16 (2010. May) 621–630. [DOI] [PubMed] [Google Scholar]
- [58].Takahara T, Furui K, Yata Y, et al. , Dual expression of matrix metalloproteinase-2 and membrane-type 1-matrix metalloproteinase in fibrotic human livers, Hepatology 26 (1997. December) 1521–1529. [DOI] [PubMed] [Google Scholar]
- [59].Onozuka I, Kakinuma S, Kamiya A, et al. , Cholestatic liver fibrosis and toxin-induced fibrosis are exacerbated in matrix metalloproteinase-2 deficient mice, Biochem. Biophys. Res. Commun 406 (2011. March 4) 134–140. [DOI] [PubMed] [Google Scholar]
- [60].Radbill BD, Gupta R, Ramirez MC, et al. , Loss of matrix metalloproteinase-2 amplifies murine toxin-induced liver fibrosis by upregulating collagen I expression, Dig. Dis. Sci 56 (2011. February) 406–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Iimuro Y, Nishio T, Morimoto T, et al. , Delivery of matrix metalloproteinase-1 attenuates established liver fibrosis in the rat, Gastroenterology 124 (2003. February) 445–458. [DOI] [PubMed] [Google Scholar]
- [62].Park S, Kim JW, Kim JH, Lim CW, Kim B, Differential roles of angiogenesis in the induction of fibrogenesis and the resolution of fibrosis in liver, Biol. Pharm. Bull 38 (2015) 980–985. [DOI] [PubMed] [Google Scholar]
- [63].Cao S, Yaqoob U, Das A, et al. , Neuropilin-1 promotes cirrhosis of the rodent and human liver by enhancing PDGF/TGF-beta signaling in hepatic stellate cells, J. Clin. Invest 120 (2010. July) 2379–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Pauta M, Ribera J, Melgar-Lesmes P, et al. , Overexpression of angiopoietin-2 in rats and patients with liver fibrosis. Therapeutic consequences of its inhibition, Liver Int. 35 (2015. April) 1383–1392. [DOI] [PubMed] [Google Scholar]
- [65].Raskopf E, Gonzalez Carmona MA, Van Cayzeele CJ, Strassburg C, Sauerbruch T, Schmitz V, Toxic damage increases angiogenesis and metastasis in fibrotic livers via PECAM-1, BioMed Res. Int 2014 (2014) 712893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Li S, Huang L, Sun Y, et al. , Slit2 promotes angiogenic activity via the robo1-VEGFR2-ERK1/2 pathway in both in vivo and in vitro studies, Invest. Ophthalmol. Vis. Sci 56 (2015. August) 5210–5217. [DOI] [PubMed] [Google Scholar]
- [67].Zeng Z, Wu Y, Cao Y, et al. , Slit2-Robo2 signaling modulates the fibrogenic activity and migration of hepatic stellate cells, Life Sci. 203 (2018. June 15) 39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Galicia-Moreno M, Gutierrez-Reyes G, The role of oxidative stress in the development of alcoholic liver disease, Rev. Gastroenterol. México 79 (2014. Apr-Jun) 135–144. [DOI] [PubMed] [Google Scholar]
- [69].Zakhari S, Overview: how is alcohol metabolized by the body? Alcohol Res. Health 29 (2006) 245–254. [PMC free article] [PubMed] [Google Scholar]
- [70].Bosoi CR, Rose CF, Oxidative stress: a systemic factor implicated in the pathogenesis of hepatic encephalopathy, Metab. Brain Dis 28 (2013. June) 175–178. [DOI] [PubMed] [Google Scholar]
- [71].Cook-Mills JM, Marchese ME, Abdala-Valencia H, Vascular cell adhesion molecule-1 expression and signaling during disease: regulation by reactive oxygen species and antioxidants, Antioxidants Redox Signal. 15 (2011. September 15) 1607–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Deng W, Wang Y, Gu L, et al. , MICAL1 controls cell invasive phenotype via regulating oxidative stress in breast cancer cells, BMC Canc. 16 (2016. July 18) 489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Italiano D, Lena AM, Melino G, Candi E, Identification of NCF2/p67phox as a novel p53 target gene, Cell Cycle 11 (2012. December 15) 4589–4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Singh G, Mitochondrial FAD-linked glycerol-3-phosphate dehydrogenase: a target for cancer therapeutics, Pharmaceuticals (Basel) 7 (2014. February 11) 192–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Yadav UC, Ramana KV, Srivastava SK, Aldose reductase inhibition suppresses airway inflammation, Chem. Biol. Interact 191 (2011. May 30) 339–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Singh S, Brocker C, Koppaka V, et al. , Aldehyde dehydrogenases in cellular responses to oxidative/electrophilic stress, Free Radic. Biol. Med 56 (2013. March) 89–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Luo X, Kranzler HR, Zuo L, Lappalainen J, B-z Yang J Gelernter, ADH4 gene variation is associated with alcohol dependence and drug dependence in European Americans: results from HWD tests and case–control association studies, Neuropsychopharmacology 31 (2006) 1085–1095, 2006/May/01. [DOI] [PubMed] [Google Scholar]
- [78].Pang XY, Wang S, Jurczak MJ, Shulman GI, Moise AR, Retinol saturase modulates lipid metabolism and the production of reactive oxygen species, Arch. Biochem. Biophys 633 (2017. November 1) 93–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Fried R, Chapter 10 - selected micronutrients and the metabolic basis for their support of endothelial health and erectile function, in: Fried R (Ed.), Erectile Dysfunction as a Cardiovascular Impairment, Academic Press, Boston, 2014, pp. 293–328. [Google Scholar]
- [80].Lorin S, Tol MJ, Bauvy C, et al. , Glutamate dehydrogenase contributes to leucine sensing in the regulation of autophagy, Autophagy 9 (2013. June 1) 850–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Li b, Espinoza I, Liu H, Lupu R, Inhibition of fatty acid synthase induces reactive oxygen species (ROS) to inhibit HER2 overexpressing breast cancer cell growth, Canc. Res 67 (2007) 4462. [Google Scholar]
- [82].Prasad B, Bhatt DK, Johnson K, et al. , Abundance of phase 1 and 2 drug-metabolizing enzymes in alcoholic and hepatitis C cirrhotic livers: a quantitative targeted proteomics study, Drug Metabol. Dispos 46 (2018) 943–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].El-Awady R, Saleh E, Hashim A, et al. , The role of eukaryotic and prokaryotic ABC transporter family in failure of chemotherapy, Front. Pharmacol 7 (2016) 535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Hlavata I, Mohelnikova-Duchonova B, Vaclavikova R, et al. , The role of ABC transporters in progression and clinical outcome of colorectal cancer, Mutagenesis 27 (2012. March) 187–196. [DOI] [PubMed] [Google Scholar]
- [85].Gotovdorj T, Lee E, Lim Y, et al. , 2,3,7,8-Tetrachlorodibenzo-p-dioxin induced cell-specific drug transporters with acquired cisplatin resistance in cisplatin sensitive cancer cells, J. Kor. Med. Sci 29 (2014. September) 1188–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Huang JF, Wen CJ, Zhao GZ, et al. , Overexpression of ABCB4 contributes to acquired doxorubicin resistance in breast cancer cells in vitro, Canc. Chemother. Pharmacol 82 (2018. August) 199–210. [DOI] [PubMed] [Google Scholar]
- [87].Pratt S, Shepard RL, Kandasamy RA, Johnston PA, Perry W 3rd, Dantzig AH, The multidrug resistance protein 5 (ABCC5) confers resistance to 5-fluorouracil and transports its monophosphorylated metabolites, Mol. Canc. Therapeut 4 (2005. May) 855–863. [DOI] [PubMed] [Google Scholar]
- [88].Oiso S, Takayama Y, Nakazaki R, et al. , Factors involved in the cisplatin resistance of KCP4 human epidermoid carcinoma cells, Oncol. Rep 31 (2014. February) 719–726. [DOI] [PubMed] [Google Scholar]
- [89].Zheng D, Dai Y, Wang S, Xing X, MicroRNA-299–3p promotes the sensibility of lung cancer to doxorubicin through directly targeting ABCE1, Int. J. Clin. Exp. Pathol 8 (2015) 10072–10081. [PMC free article] [PubMed] [Google Scholar]
- [90].Stokes E, Shuang T, Zhang Y, et al. , Efflux inhibition by H2S confers sensitivity to doxorubicin-induced cell death in liver cancer cells, Life Sci. 213 (2018. November 15) 116–125. [DOI] [PubMed] [Google Scholar]
- [91].Li Q, Shu Y, Role of solute carriers in response to anticancer drugs, Mol Cell Ther 2 (2014) 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Schaupp CM, White CC, Merrill GF, Kavanagh TJ, Metabolism of doxorubicin to the cardiotoxic metabolite doxorubicinol is increased in a mouse model of chronic glutathione deficiency: a potential role for carbonyl reductase 3, Chem. Biol. Interact 234 (2015. June 5) 154–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Lu P, Takai K, Weaver VM, Werb Z, Extracellular matrix degradation and remodeling in development and disease, Cold Spring Harb Perspect Biol (2011. December 1) 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Magdaleno F, Schierwagen R, Uschner FE, Trebicka J, Tipping” extracellular matrix remodeling towards regression of liver fibrosis: novel concepts, Minerva Gastroenterol. Dietol 64 (2018. March) 51–61. [DOI] [PubMed] [Google Scholar]
- [95].Jung YK, Yim HJ, Reversal of liver cirrhosis: current evidence and expectations, Korean J Intern Med 32 (2017. March) 213–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Puche JE, Saiman Y, Friedman SL, Hepatic stellate cells and liver fibrosis, Comp. Physiol 3 (2013. October) 1473–1492. [DOI] [PubMed] [Google Scholar]
- [97].Vaiphei K, Stellate cells in health and disease, Indian J. Med. Res 145 (2017) 407. [Google Scholar]
- [98].Campana L, Iredale JP, Extracellular matrix metabolism and fibrotic disease, Current Pathobiology Reports 2 (2014 2014/December/01) 217–224. [Google Scholar]
- [99].Park S, Kim JW, Kim JH, Lim CW, Kim B, Differential roles of angiogenesis in the induction of fibrogenesis and the resolution of fibrosis in liver, Biol. Pharm. Bull 38 (2015) 980–985. [DOI] [PubMed] [Google Scholar]
- [100].Rao R, Endotoxemia and gut barrier dysfunction in alcoholic liver disease, Hepatology 50 (2009. August) 638–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Seki E, Schnabl B, Role of innate immunity and the microbiota in liver fibrosis: crosstalk between the liver and gut, J. Physiol 590 (2012. February 1) 447–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Albano E, Vidali M, Immune mechanisms in alcoholic liver disease, Genes Nutr 5 (2010. June) 141–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Johnson TN, Boussery K, Rowland-Yeo K, Tucker GT, Rostami-Hodjegan A, A semi-mechanistic model to predict the effects of liver cirrhosis on drug clearance, Clin. Pharmacokinet 49 (2010. March) 189–206. [DOI] [PubMed] [Google Scholar]
- [104].Wang L, Collins C, Kelly EJ, et al. , Transporter expression in liver tissue from subjects with alcoholic or hepatitis C cirrhosis quantified by targeted quantitative proteomics, Drug Metab. Dispos 44 (2016. November) 1752–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Rasool MF, Khalil F, Laer S, Optimizing the clinical use of carvedilol in liver cirrhosis using a physiologically based pharmacokinetic modeling approach, Eur. J. Drug Metab. Pharmacokinet 42 (2017. June) 383–396. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.