

Abstract
Porous silicon nanoparticles (PSNPs) offer tunable pore structure and easily modified surface chemistry, enabling high loading capacity for drugs with diverse chemicophysical properties. While PSNPs are also cytocompatible and degradable, PSNP integration into composite structures can be a useful approach to enhance carrier colloidal stability, drug-cargo loading stability, and endosome escape. Here, we explored PSNP polymer composites formed by coating of oxidized PSNPs with a series of poly[ethylene glycol-block-(dimethylaminoethyl methacrylate-co-butyl methacrylate)] (PEG-DB) diblock copolymers with varied molar ratios of dimethylaminoethyl methacrylate (D) and butyl methacrylate (B) in the random copolymer block. We screened and developed PSNP composites specifically toward intracellular delivery of microRNA inhibitory peptide nucleic acids (PNA). While a copolymer with 50 mol % B (50B) is optimal for early endosome escape in free polymer form, its pH switch was suppressed when it was formed into 50B polymer-coated PSNP composites (50BCs). We demonstrate that a lower mol % B (30BC) is the ideal PEG-DB composition for PSNP/PEG-DB nanocomposites based on having both the highest endosome disruption potential and miR-122 inhibitory activity. At a 1 mM PNA dose, 30BCs facilitated more potent inhibition of miR-122 in comparison to 40BC (p = 0.0095), 50BC (p < 0.0001), or an anti-miR-122 oligonucleotide delivered with the commercial transfection reagent Fugene 6. Using a live cell galectin 8-based endosome disruption reporter, 30BCs had greater endosomal escape than 40BCs and 50BCs within 2 h after treatment, suggesting that rapid endosome escape correlates with higher intracellular bioactivity. This study provides new insight on the polymer structure-dependent effects on stability, endosome escape, and cargo intracellular bioavailability for endosomolytic polymer-coated PSNPs.
Keywords: porous silicon nanoparticles, endosomal escape, galectin-8, micro-RNA, peptide nucleic acid
Graphical Abstract
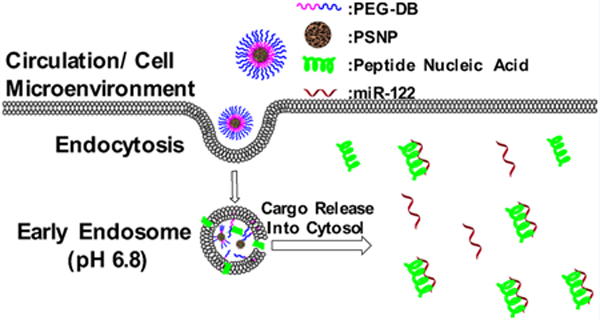
1. INTRODUCTION
Inorganic nanomaterials are attractive drug delivery vehicles due to their physical robustness compared to self-assembled carriers such as polymeric micelles and liposomes.1–3 Porous silicon nanoparticles (PSNPs) in particular benefit from high drug-loading capacity, biodegradability and cytocompatibility, tunability of size and shape,4,5 and applicability for loading of diverse types of small molecules, proteins, and nucleic acids.6–8 The large pore volume enables therapeutics to be loaded into PSNPs in a variety of solvents via capillary forces.9 Additionally, aqueous loading of biologics is possible with PSNPs, whereas encapsulation within most polymers [e.g. poly(lactic-co-glycolic acid)] requires use of harsh solvents that can denature biologics.10–13 Relative to most other inorganic materials, a major advantage of silicon is that it undergoes hydrolysis in the presence of water to break down into the safe byproduct silicic acid on a timescale of hours to weeks, which is desirable for drug delivery applications.14 Si–H bonds throughout the bulk of porous silicon are prone to spontaneous hydrolysis, which drives particle degradation. However, as the oxidation level of silicon is increased, through thermal or chemical methods, Si–O bonds are formed that slow the rate of hydrolysis.15 In addition to varying the degree of porous silicon (PSi) oxidation, surface functionalization with hydrophobic alkoxysilanes can provide another means to reduce water penetration and slow the hydrolytic degradation rate and drug release from PSNPs.16,17
The synthetic control of particle size and shape,8 pore size,18 and degradation behavior19 make PSNPs promising for clinical nanomedicines, although PSNPs inherently lack some critical characteristics for drug delivery.11,14 First, bare PSNPs lack colloidal stability and aggregate in the presence of physiologic saline and serum, which can cause accumulation within and blockage of lung or other tissue capillary beds after intravenous injection in vivo.11 Another shortcoming of PSNPs is that they enter cells via endocytosis mechanisms and become entrapped within endo-lysosomal vesicles, yielding limited intracellular bioavailability. This is especially limiting for biomolecular drugs such as proteins and nucleic acid therapies. PSNP modification with various silane derivatives is one means that can be used to vary surface characteristics, such as charge, which can be leveraged to promote PSNP surface interactions with drug cargo and increase colloidal stability.
Alternatively, PSNPs can be coated with polymers that increase colloidal stability, increase cell uptake, and impart pH-driven endosome escape mechanisms.7,11 For example, PSNPs have been combined with polymers and dendrimers such as polyethylenimine,20–22 polyamidoamine,23 and poly(propylene imine)21,24,25 to create hybrid materials with improved colloidal stability by imparting a combination of steric and cationic charge–charge repulsion to the particles. These hybrids can also provide biologic cargo escape from endosomes via the proton sponge effect.26 However, nanoparticles with cationic surface charge bind to serum proteins and can still have aggregation issues in complex biological solutions.27 Additionally, highly positive surface charge (approximately +20 mV or greater) can cause nanomaterials to have adverse interactions with red blood cells and cytotoxicity.21–23,28
Cationic nanoparticle cytotoxicity can be reduced by surface functionalization and shielding with polyethylene glycol (PEG).29,30 Here, we pursue polymer-coated PSNP hybrids comprising different compositions of the diblock copolymer poly[ethylene glycol-block-(dimethylaminoethyl methacrylate-co-butyl methacrylate)] (PEG-DB). Copolymerization of the D monomer provides a protonatable, cationic functionality that interacts with anionic, oxidized PSNPs for surface coating and also integrates tunable pH-responsive function. The B monomer adds pendant hydrophobic alkyl tails to the polymer to promote endosome membrane interactions and to contribute to porous silicon polymer-coating stability; the amount of B monomer also tunes the copolymer block solubility pKa of the copolymerized tertiary amine-containing D monomer and consequently the pH switch behavior of the copolymer.31,32
PEG-DB was previously optimized for the delivery of small interfering RNA (siRNA) in the form of electrostatically formed polyplexes (PEG-DB + siRNA).33 This prior study showed that a 50/50 molar ratio of D/B (50B) in the second block was optimal for intracellular gene silencing siRNA delivery, a concept which has been successfully applied in several settings.34–36 Here, we seek to deliver peptide nucleic acids (PNA) as microRNA inhibitors due to the favorable properties of PNA over more conventional 2′-OMe-modified RNA oligonucleotides (AMOs), including higher binding affinity to target nucleic acids and lower susceptibility to nuclease degradation.37 However, PNA, which has a peptide backbone, is not amenable to charge-based electrostatic complexation with 50B or other traditional cationic transfection reagents. This motivated our pursuit of formulation strategies for PNA loading into PSNPs followed by polymer coating to form stable and endosome-escaping anti-microRNA nanotherapeutics.
PSNPs coated with 50B polymer to form composites (50BCs) have a suppressed pH response in comparison to the free 50B polymer, motivating further exploration of the effect of PEG-DB polymer composition on the performance of PSNP and polymer composites in order to develop a formulation that activates in the early endosome pH environment. Specifically, free 50B polymer provides membrane disruption at early endosomal pH of 6.8, while our earlier studies showed that 50BC does not robustly activate until a lower pH of 6.2.11 Carrier activation within and escape from the early endosomal vesicles is critical, as acidity and enzymes that lead to cargo degradation are increased along the endo-lysosomal trafficking pathway.38–40 Furthermore, during the transition from early to late endosomes, cargo can be exocytosed back into the extracellular space via an endosome recycling mechanism, thus decreasing cargo intracellular retention.38 It has been posited that endosome escape is most efficient in early endosomal vesicles,41,42 which matches our own historical observations that robust membrane disruption at pH 6.8 is a good predictor of carrier ability to achieve intracellular bioactivity.33,34 Therefore, we sought to thoroughly explore how the composition of PEG-DB, specifically the D:B ratio, affects the performance of hybrid polymer-coated PSNPs for intracellular PNA delivery. We are targeting formulations that activate at an early endosomal pH and testing whether this feature correlates with intracellular PNA anti-microRNA bioactivity.
2. EXPERIMENTAL METHODS
2.1. Polymer Library Synthesis.
2.1.1. Reversible Addition–Fragmentation Chain Transfer (RAFT) Chain Transfer Agent Synthesis.
The reversible addition–fragmentation chain transfer (RAFT) chain transfer agent (CTA) 4-cyano-4-(ethylsulfanylthiocarbonyl) sulfanylpentanoic acid (ECT) was synthesized as previously described, and the R-group of the CTA was subsequently conjugated to the hydroxy terminus of the PEG.31 Briefly, dicyclohexylcarbodimide (4 mmol, 0.82 g) was added to the stirring solution of mono methoxy-PEG (Mn = 5000, 2 mmol, 10 g), ECT (4 mmol, 1.045 g), and 4-dimethylaminopyridine (10 mg) in 50 mL of dichloromethane. The reaction mixture was stirred for 48 h. The precipitated cyclohexyl urea was removed by filtration, and the dichloromethane layer was concentrated and precipitated into diethyl ether twice. The precipitated PEG/ECT was washed three times with diethyl ether and dried under vacuum (yield ∼10 g). 1H nuclear magnetic resonance (NMR) spectroscopy (400 MHz CDCl3) revealed 85% substitution of the PEG with ECT.
2.1.2. PEG-DB RAFT Polymerization.
A library of rhodamine-containing PEG-DB polymers ranging from 20 mol % B to 70 mol % B (20B–70B) was synthesized via RAFT polymerization. Monomers were dissolved in a 10% N,N-dimethylformamide (DMF)/90% dioxane solution (mass of monomer + CTA equal to 20% of total mass) and reacted for 24 h under nitrogen atmosphere at 35 °C. The reaction was prepared such that 100% monomer conversion would achieve a degree of polymerization of 200. The radical initiator 2,2′-azobis(4-methoxy-2,4-dimethyl valero nitrile) (V-70) was added to the reaction at a 1:5 V-70 initiator to CTA molar ratio. Following 24 h, the resulting polymers were dialyzed against methanol in dialysis tubing with a 12k–14k molecular weight (MW) cutoff to remove unreacted monomers and residual solvent. Dialysis was then completed against deionized (DI) water to remove methanol, after which the polymers were frozen and lyophilized. Polymers were characterized for composition and MW by 1H NMR spectroscopy (Bruker 400 MHz spectrometer equipped with 9.4 T Oxford magnet). The absolute MW of the polymers was determined using DMF mobile phase gel permeation chromatography (GPC, Agilent Technologies, Santa Clara, CA, USA) with inline Agilent refractive index and Wyatt miniDAWN TREOS light scattering detectors (Wyatt Technology Corp., Santa Barbara, CA).
2.2. PSNP and PSNP/Polymer Composite Fabrication and Characterization.
2.2.1. PSNP Fabrication.
PSNPs were fabricated by electrochemical anodization of p-type silicon wafers (⟨100⟩, 0.01–0.02 Ω·cm) in an electrolyte solution containing 15% HF in ethanol using a procedure similar to what we previously reported.11 Briefly, alternating low (60 mA/cm2 for 8 s) and high (100 mA/cm2 for 2 s) current densities were applied to create alternating particle and sacrificial “lift-off” layers (400 cycles), respectively. Following anodization, films were removed from the wafer and fractured into nanoparticles via water-bath sonication. The PSNPs were subsequently sonicated by probe tip for 4 h in the presence of 35% H2O2 to yield oxidized PSNPs with an average zeta potential of −20 ± 2 mV, average nanoparticle diameter of 270 ± 30 nm, average pore diameter of 20 nm, and porosity near 86%.16
2.2.2. PSNP/Polymer Composite Fabrication and Characterization.
PSNPs were suspended into ethanol solutions of various PEG-DB polymers at either a 10:1, 5:1, or 2.5:1 weight ratio of PEG-DB/PSNP (PSNP concentration = 2 mg/mL). After mixing for 30 min, the PSNPs were washed twice with water and once with phosphate-buffered saline (PBS) to yield polymer-coated PSNPs.
2.2.3. Zeta Potential.
The pH-dependent zeta potential was measured at pH 7.4 (PBS) and pH 6.5. The pH 6.5 was created using a solution containing 1 mL of 10× PBS, 29 mL of DI water, and 2 μL of glacial acetic acid (17.5 M). The solution was vortex mixed at 900 rpm for 2 min and allowed to equilibrate for 30 s. A digital pH meter was used to confirm the pH following the 30 s equilibration and then again 24 h later. Zeta potential was determined using a Zetasizer NanoZS (Malvern Instruments, Worcestershire, UK). Particles were suspended at 0.1 mg/mL in either the pH 7.4 or pH 6.5 buffer, filtered with a 0.45 μm filter, and placed in a folded capillary cell.
2.2.4. Transmission Electron Microscopy.
Transmission electron microscopy (TEM) and scanning transmission electron microscopy (STEM) were performed using a Tecnai Osiris operating at 200 kV. Drift-corrected STEM–energy dispersive X-ray spectroscopy (EDS) maps were collected using the Bruker Esprit 1.9 software with a probe current on the order of 1 nA. Standardless quantification using the Cliff–Lorimer method was applied to determine the relative atomic percent of the elements of interest. PSNPs were suspended at a concentration of 1 mg/mL in biology grade water and allowed to evaporate overnight before imaging.
2.2.5. PNA Loading in PSNPs.
Anti-miR 122 PNA were purchased from PNA Bio (Newbury Park, CA). Sequence: ACAAACAC-CATTGTCACACTCCA-Cys. One mg of PNA was first dissolved in 100 μL of ethanol and then transferred to a vortexing 15 mL conical tube containing 1 mg of PSNPs suspended in 900 μL of PBS. The 1:1 mixture of PNA/PSNP was allowed to vortex for 4 h. The particles were then transferred to a 2 mL centrifuge tube and centrifuged at 21,000g for 20 min, rinsed once with DI water, resuspended in 1 mL of DI water, frozen at −20 °C, and lyophilized overnight. Loading was determined by dissolving silicon using 20 μL of a 0.5 M potassium hydroxide solution for 5 min and then diluting with ∼2 mL of DI water. To reduce interference from possible undissolved PSNP fragments, the solution was centrifuge for 5 min at 21,000g, then syringe filtered using a 0.25 μm syringe filter. A spectrophotometer was used to measure the absorbance of the solution at a wavelength of 260 nm, and comparison to a standard curve was used to determine the total mass of PNA loaded. It was determined that before polymer coating, 250 μg of PNA were loaded per 1 mg of PSNP (25 wt %), and that number was reduced to 15 wt % after polymer coating; PNA loading level was not affected by polymer composition.
2.3. In Vitro Assays.
2.3.1. Assaying Serum Stability of PSNP Polymer Coating.
The stability of the PSNP polymer coating as a function of varied PEG-DB compositions was assessed in the presence of fetal bovine serum (FBS). Briefly, 20BC–70BC composites were suspended in 1 mL of a 50% FBS/PBS solution at a concentration of 1 mg/mL and incubated at 37 °C in a water-bath for a predetermined length of time (0, 10, 40, and 60 min). Following each time point, particles were centrifuged at 21,000g, and 3 × 100 μL aliquots were removed for technical replicates and placed in a flat bottom black 96-well plate. The remaining supernatant was removed and replaced with a fresh 50% FBS solution, after which samples were incubated at 37 °C until the next time point. This repeated supernatant removal and serum exposure protocol was utilized to mimic in vivo infinite sink conditions where polymer exchange off and back on the PSNPs is unlikely to occur. Also, a 1 min time point was used to measure any initial rapid stripping of the polymer upon initial serum exposure. At the final time point, the remaining polymer was stripped from the PSNP composites by applying 1 mL of a 1% sodium dodecyl sulfate detergent solution. This suspension was shaken for 45 min and then centrifuged at 21,000g for 10 min to pellet the PSNPs. Subsequently, 3 × 100 μL aliquots were taken from the supernatant for technical replicates and placed in a black 96-well plate. A plate reader was used to measure the amount of rhodamine signal in the harvested supernatants at each time point and quantified relative to a standard curve for each of the PEG-DB polymers. The percent polymer stripped represented in the graph is the cumulative sum of the mass of polymer stripped at an individual time point plus the previous time points divided by the total mass of polymer coated [i.e., % stripped at T3 = (T1 + T2 + T3)/(mass of polymer coated)]. This experiment was repeated three times with three technical replicates per study; individual points on the graphs represent the average of the three technical replicates from each of the three independent studies.
2.3.2. pH-Dependent Polymer Shedding.
It is anticipated that the polymer coating should ideally be shed from the PSNP composites under pH conditions similar to an early stage endosome, facilitating endosomal membrane interaction and disruption. To assess the pH-dependent release of the polymers from the PSNPs, we followed a similar procedure to the serum stability assay except that the composite particles were incubated for a single time point of 60 min at 37 °C. The pH was adjusted using various ratios of monobasic and dibasic sodium phosphate to yield 50% FBS solutions with different pH values (7.4, 6.8, 6.2, and 5.6) representative of extracellular, early endosomal, late endosomal, and late endosomal/lysosomal environments, respectively.
2.3.3. Red Blood Cell Hemolysis.
The pH-dependent hemolysis assay was used to gauge endosome disruption and escape potential of the various polymer-coated PSNP composites.43 Through a Vanderbilt IRB-approved protocol, blood was collected from anonymous donors into dipotassium ethylenediaminetetraacetic acid-coated Vacutainer tubes to prevent coagulation. Blood samples were centrifuged at 500g for 5 min to isolate cells, which were subsequently washed three times with 150 mM NaCl. The red blood cells were then split into four separate samples that were redispersed in phosphate buffers of varied pH (7.4, 6.8, 6.2, and 5.6). The buffered blood samples were placed in a 96-well plate and incubated with either free polymer at 40 μg/mL or polymer PSNP composites matched to this same polymer concentration. Triton X-100 was used as a positive control to determine 100% hemolysis. Samples were centrifuged, and supernatants were collected. The relative pH-dependent hemolysis of the polymers and PSNP polymer composites was determined by quantifying the supernatant absorbance at a wavelength of 541 nm of test samples relative to vehicle control and Triton X-100-treated samples.
2.3.4. Dual Luciferase Reporter Micro-RNA Inhibition Assay.
A Huh7-psiCHECK-2-miR122 cell line was cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% FBS, 2% penicillin/streptomycin, and 2 μg/mL ciprofloxacin. Huh7 cells were treated for 24 h with either an AMO [sequence: A*C*AAACAC-CAUUGUCACACU*C*C*A*-Chol-3′ (uppercase, 2′OMe; *, PS linkage)] delivered by the transfection reagent Fugene 6 or anti-miR122 PNA loaded into uncoated or PEG-DB-coated composite PSNPs. Initial studies investigated whether the ratio of PEG-DB to PSNPs utilized during composite fabrication affected the activity of the anti-miR122 PNA cargo. A bioactivity pre-screen was done on composites made at 10:1, 5:1, or 2.5:1 polymer/PSNP ratios for 30BC, 40BC, and 50BC, which motivated the use of the 10:1 ratio of PEG-DB/PSNP in the majority of the studies.
2.3.5. Live Cell Endosomal Escape.
Gal8-MDA-MB231 cells were plated in Corning 96-well Half-Area High-Content Imaging Glass-Bottom Microplates (Corning Inc.; product no. 4580) at a density of 6250 cells per cm2. Cells were left to adhere for 24 h before being treated with PNA formulations. After a 2 h treatment, media was aspirated and replaced with warm FluoroBrite DMEM supplemented with 25 mM HEPES, 10% FBS, and Hoechst 33342. Images were acquired with a Nikon C1si+ confocal microscope system on a Nikon Eclipse Ti-0E inverted microscope base, Plan Apo VC 20× objective, galvano scanner, and 408/488/543 dichroic mirror. Perfect Focus was used to maintain focus between wells. The Perfect Focus offset was set to the optimal focal plane of the Gal8-YFP in combination with the well-scanning mode to acquire images, removing microscopist bias. A software-controlled motorized stage moved the plate between images.
3. RESULTS AND DISCUSSION
The library of rhodamine acrylate (RhoA)-containing PEG-DB polymers was synthesized via RAFT polymerization with controlled MWs and low polydispersities (Figures 1, S1, and S2). All polymers had degrees of polymerization between 150 and 160 with targeted molar ratios of B/D closely matched to the polymerization reaction monomer feeds of 20:80, 30:70, 40:60, 50:50, and 60:40 (20B–70B). A low level of RhoA was also successfully integrated for polymer tracking and solution quantification purposes.
Figure 1.
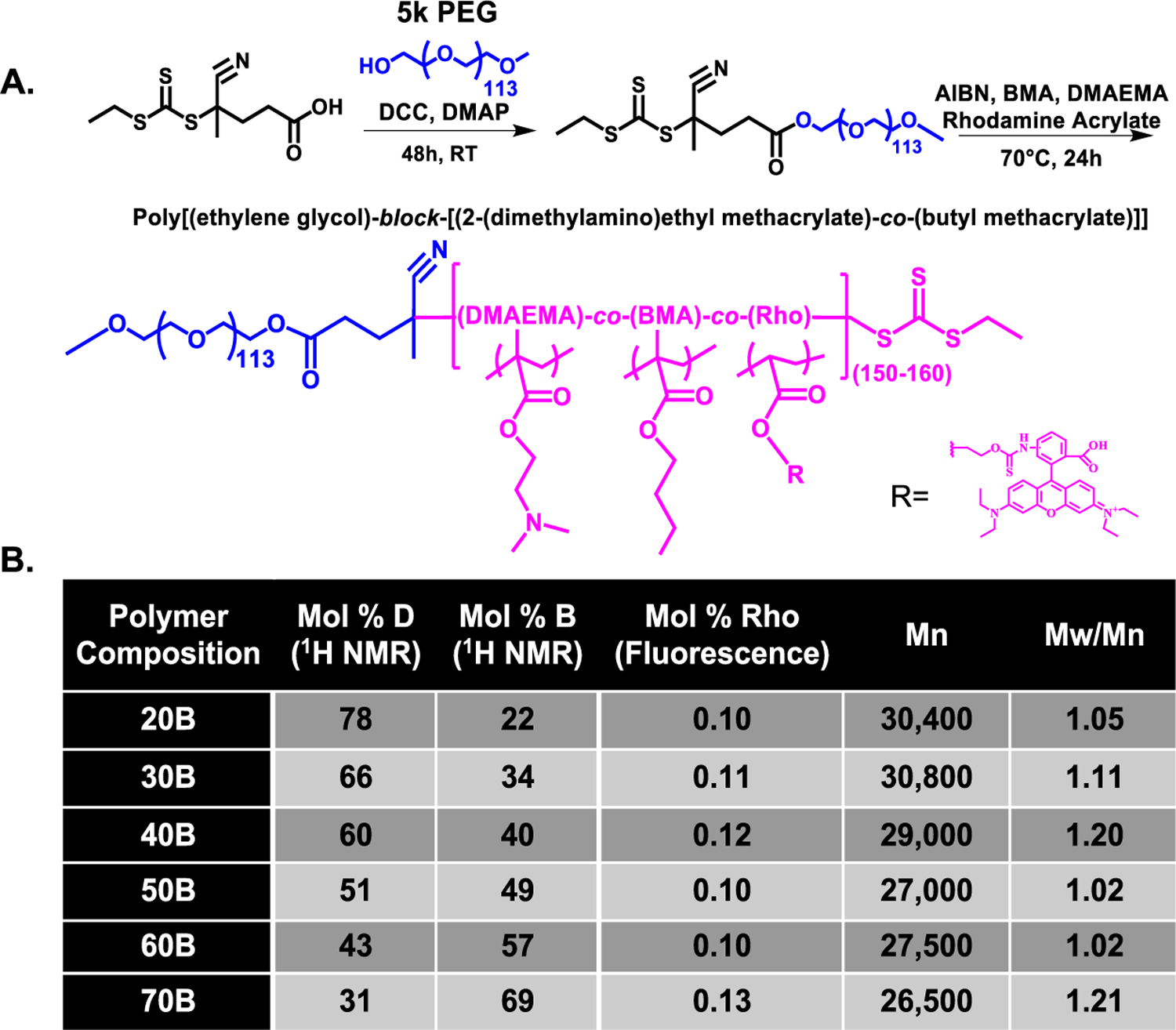
PEG-DB polymer library synthesis. (A) RAFT polymerization scheme for synthesis of PEG-DB library. (B) Experimentally determined monomer mole percentage, total diblock polymer MW (including 5000 g/mol PEG block), and polydispersity index of resultant polymers.
PSNPs were fabricated by electrochemical anodization of a p-type silicon wafer in the presence of hydrofluoric acid and subsequently sonicated in the presence of hydrogen peroxide to produce oxidized PSNPs. PSNPs were subsequently coated with various PEG-DB compositions to yield PSNP and polymer composites 20BC, 30BC, 40BC, 50BC, 60BC, and 70BC (Figure 2A). The final ratio of polymer to PSNP in the composite was measured using two methods. First, all polymer was stripped from the PSNP in ethanol, and the samples were measured relative to a rhodamine fluorescence standard curve generated with a dilution series of each polymer (Figure 2B). Formulations were also analyzed for polymer content via thermogravimetric analysis (TGA) (Figure 2C). Both measurements showed that all of the composites were approximately 20 wt % polymer by mass. The wt % of PEG-DB is critical, as a dense PEG coating contributes to colloidal stability of the particles, while the DB polymer block is the key component for mediating endosome escape and consequent PNA intracellular activity.11 We also measured the composite nanoparticle diameter by both dynamic light scattering (DLS) and SEM (Figure S3). Both methods indicated an average diameter of approximately 250 nm for bare PSNPs. DLS analysis of the polymer-coated PSNPs showed that the composite structures were approximately 20–50 nm larger in diameter compared to the bare particles (Figure 2D). A shift in zeta potential was also observed for the polymer-coated PSNPs. The bare PSNPs at pH 7.4 exhibited a zeta potential of −20 ± 3 mV, while all polymer-coated composites had an approximately neutral zeta potential, n = 3 (Figure 2E). A follow up study was done to gauge the composition-dependent change in zeta potential that would occur in the endosomal environment. At the early endosome-mimicking pH 6.5, the composites with low % B (20BC–40BC) had increased zeta potential relative to pH 7.4, while the higher % B composites (50BC–70BC) maintain approximately neutral zeta potential at the lower pH. These results are as anticipated, as the pKa of the PEG-DB polymers is composition dependent, with increasing B content correlating to a reduction in pKa (we previously measured pKa of 50B at ∼6.5 and derived the relationship that pKa = 7.36 – (0.019)x where x is mol % of B in the DB block).32
Figure 2.
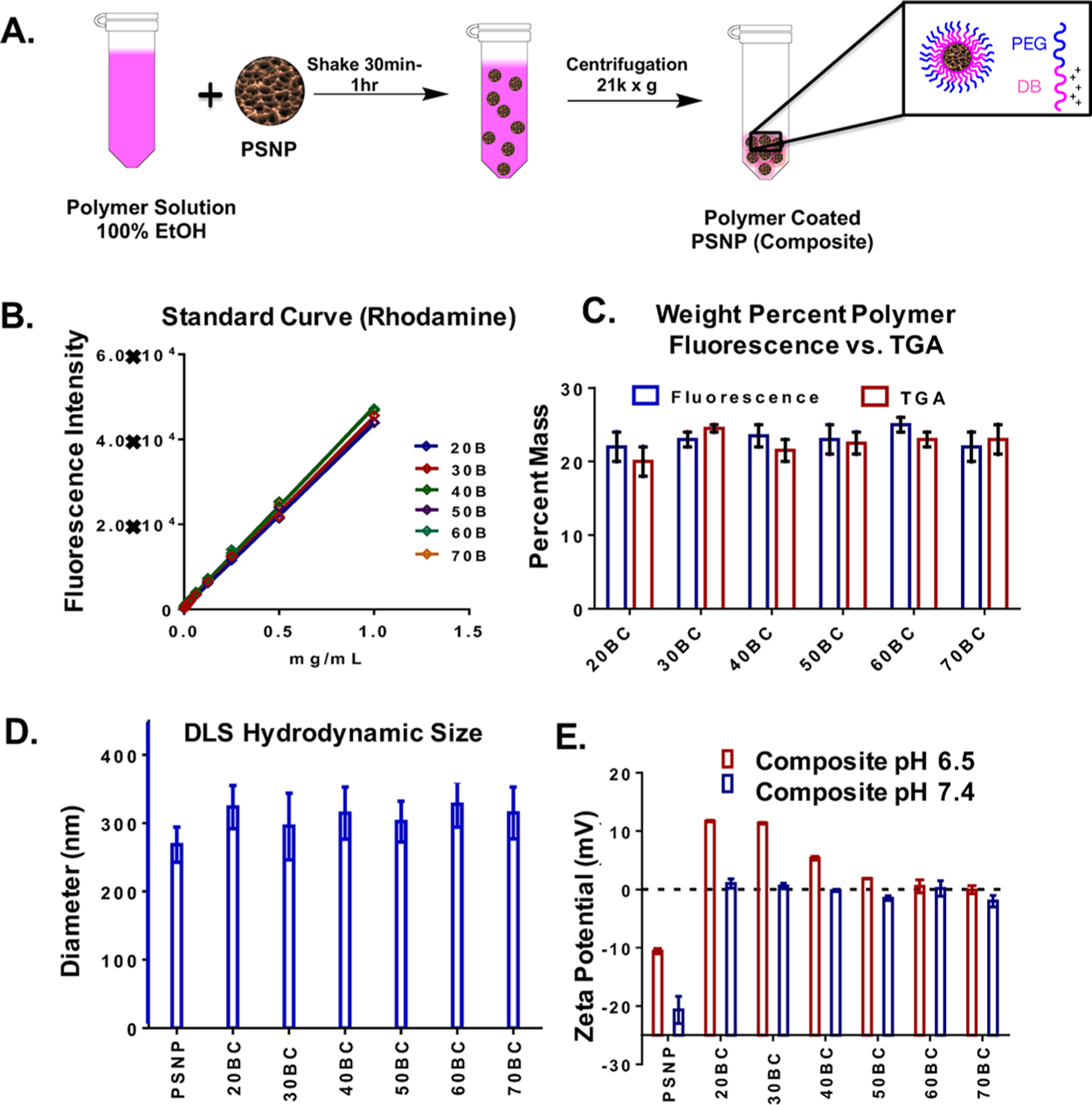
Production and characterization of PSNP polymer composites. (A) Fabrication scheme for PSNP/PEG-DB composites. (B) Standard curves for rhodamine-containing PEG-DB polymers. (C) Weight percent polymer per mg of PSNP measured by both TGA and rhodamine fluorescence. (D) Hydrodynamic diameter by DLS is similar for all composites and is 20–50 nm larger than bare PSNPs. (E) Zeta potential is approximately neutral for all composites at pH 7.4 vs −20 mV for bare, oxidized PSNPs. The zeta potential of the composites at pH 6.5 is increased in inverse proportion to the mol % B in the polymer. n = 3 unless indicated otherwise.
The bare PSNPs, PNA-loaded PSNPs, and polymer-coated composite PSNPs were further characterized by TEM imaging with EDS (Figure 3). For the bare PSNPs, carbon and nitrogen content is relatively low throughout the particle framework, while the PNA- and polymer-loaded PSNPs had higher nitrogen and carbon signals, respectively. For the polymer-coated group, a halo of higher density carbon signal could be visualized at the particle edge, indicative of the polymer coating on the PSNP. In the dehydrated state, the thickness of this outer layer appears to be in the 10–20 nm range. Note that the microscopy software used automatically scales each color in the image to a similar brightness; so, while distribution of the signal within a given image can be appreciated, the relative elemental composition between groups cannot be inferred from image visualization alone. Therefore, Figure 3 also includes a table with the respective atomic % for silicon, carbon, and nitrogen. Higher normalized atomic % N supports loading of PNA, which contains an amide backbone, into the PSNPs. PNA loading was further confirmed by UV/vis spectroscopy, which indicated 15 ± 2 wt % of PNA in the final, polymer-coated composites (Figure S4). The polymer coating on the PSNPs was also supported by the higher normalized atomic % C in the composite structures (62%). Although no apparent differences were present between the different polymer-coated composite PSNPs as a function of polymer composition, a full set of TEM images with EDS analysis is provided in Figure S5.
Figure 3.

TEM panel shows representative images for bare PSNPs, PNA-loaded PSNPs, and polymer-coated PSNPs. TEM and EDS data for all composites are provided in Figure S5A,B. From left to right, the images depict an individual particle, a high-resolution image of the particle edge, nitrogen map (PNA loading), and a silicon–carbon map (polymer coating). Scale bar = 100 nm for the TEM image on the left and 20 nm for the three images on the right. Note that the microscopy software used automatically scales each image to a similar brightness intensity, so the relative elemental composition between groups cannot be inferred from image visualization alone. The table at the bottom of the figure shows quantification of the normalized atomic % for each element from the EDS analysis.
Following characterization of PNA loading and polymer coating, we next analyzed polymer shedding from the PSNPs in serum and as a function of pH. We first exposed the composite particles to a 50% FBS/PBS solution at 37 °C and measured the amount of rhodamine-labeled polymer stripped from the particles over time under infinite sink-mimicking conditions (Figure 4A). This experiment allowed us to investigate the stability of all of the composites under conditions that model the circulation in vivo. In this study, we observed that there is a positive trend of increasing stability of the polymer coating with increasing hydrophobicity (% B). We next sought to understand how changes in pH affected the coating stability for the various composites while in the presence of serum, as it is anticipated that pH-triggered shedding at early endosome pH will be critical for polymer-mediated endo-lysosomal vesicle disruption. We incubated the particles for 60 min in a 50% FBS + buffered solutions with pH values representative of extracellular, early endosomal, late endosomal, and late endosomal/lysosomal pH (7.4, 6.8, 6.2, and 5.6, respectively) (Figure 4B). Similar to serum stability, an indirect relationship was observed between mol % B and pH required for polymer release. 20BC and 30BC show some stripping at physiological pH, which is indicative of premature release of the polymer and possible hemotoxicity, while the pH-driven polymer release profile from 40BC showed more idealized behavior with good stability at physiological pH and significant shedding at a pH consistent with early endosomal pH. Although 50BC-70BC polymers also show high stability at 7.4, their suppressed ability to rapidly shed the polymer at early endosomal pH is less desirable, as it decreases the ability of the polymer to mediate escape from early endosomes.
Figure 4.
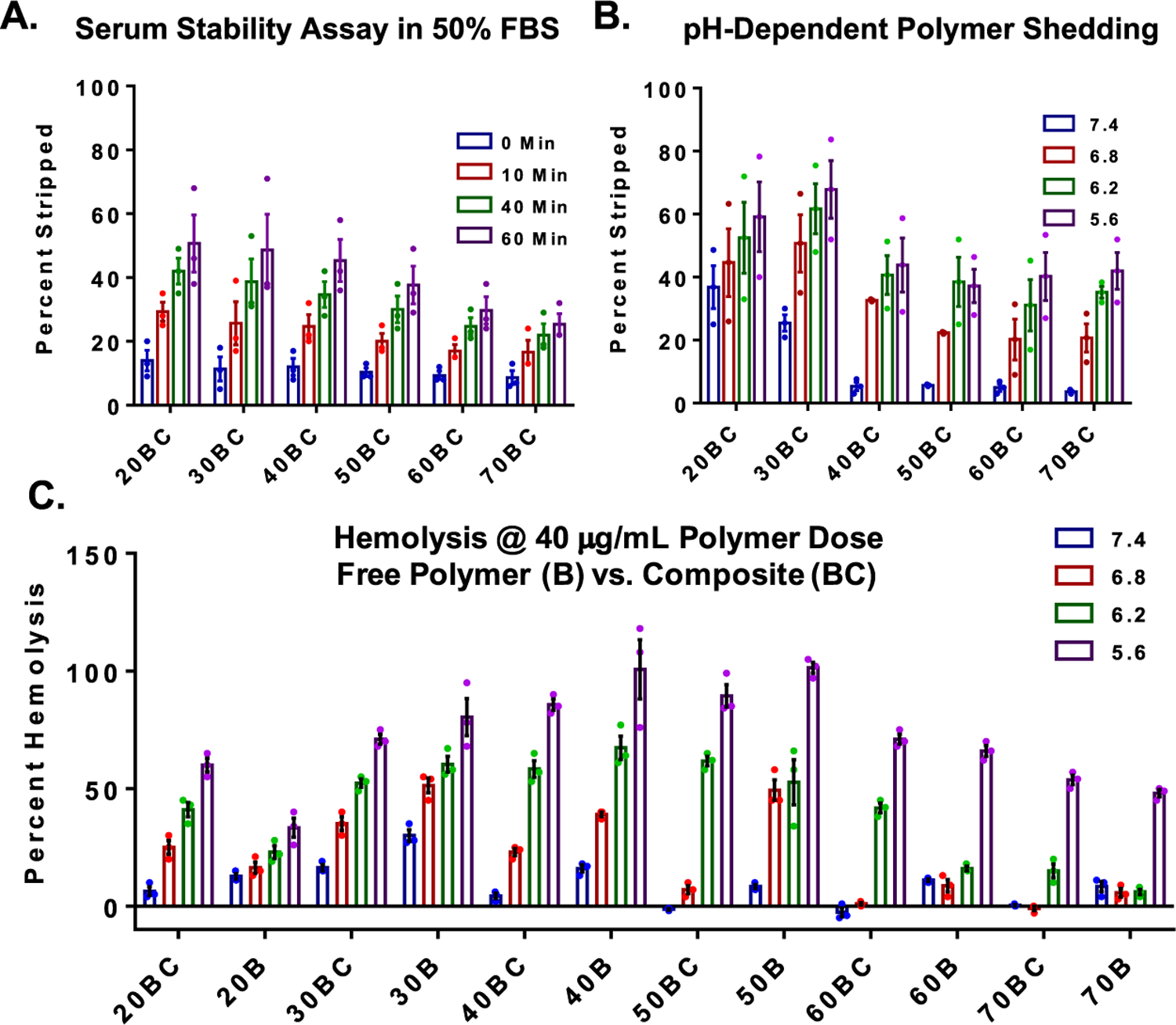
Serum stability of polymer coatings, pH-dependent polymer shedding, and membrane-disruptive behavior of nanocomposites. (A) Serum stability measurements of polymer coating in infinite sink conditions show a trend toward increasing stability with increasing hydrophobicity (% B). Significance was determined by two-way analysis of variance (ANOVA) (composition, p = 0.158; time, p < 0.0001; interaction, p = 0.0055) and post-hoc analysis using Tukey’s multiple comparison test (refer to Table S1). (B) pH-dependent polymer release from PSNPs. Significance was determined by two-way ANOVA (composition, p < 0.0015; pH, p < 0.0001; interaction, p = 0.9379) and post-hoc analysis using Tukey’s multiple comparison test (refer to Table S2). (C) pH-dependent red blood cell membrane disruption assay was used as an indicator for early endosome disruptive activity (hemolysis at pH 6.8 and below) and lack of hemotoxicity (no hemolysis at pH 7.4). The nanocomposites (labeled as BC) were compared to the polymers alone (labeled as B). Significance was determined by two-way ANOVA (composition, p < 0.0001; pH, p < 0.0001; interaction, p < 0.0001) and post-hoc analysis using Tukey’s multiple comparison (refer to Table S3). All experiments were n = 3, where individual points equal the average of technical replicates within independent experiments.
To further investigate the pH-dependent membrane disruptive behavior of the PSNP polymer composites, a red blood cell hemolysis assay was performed. The pH-dependent hemolysis assay is used as a surrogate measure for endosome disruption and toxicity for formulations developed to mediate intracellular bioavailability of biologics that are otherwise primarily sequestered within endo-lysosomal vesicles. Ideal candidate formulations demonstrate hemolysis (membrane disruptive activity) at acidic pH representative of the endosomes but will remain inactive at pH 7.4. Formulations that show significant hemolysis at pH 7.4 will typically cause cytotoxicity when screened in other cell types presumably due to outer cell membrane disruption and resultant necrotic cell death. For the current polymers, the DB polymer block mediates the membrane disruptive effect. This activity is dependent on three primary factors: sufficient hydrophobic content of the polymer to drive efficient lipid bilayer membrane insertion, pH-dependent stability of the polymer coating on the PSNPs (i.e., pH at which protonation of the D monomer causes sufficient electrostatic repulsion to destabilize the polymer coating on the PSNPs and expose the membrane interactive DB block), and polymer MW (not explored as a variable here). PEG-DB polymers showed altered pH-dependent hemolysis potential when coated onto PSNPs composites versus that in the free polymer form (BC vs B). In general, the composite BC formulations have reduced hemolysis magnitude relative to the free polymer B samples, and the pH at which the hemolysis is activated is lower in the BC than B format for most polymer compositions (Figure 4C).
Trends from these studies show that 50B free polymer has an ideal profile with minimal hemolysis at pH 7.4 and switch-like turn-on at pH 6.8, in agreement with our previous studies.33,43 However, for 50BC PSNP composites, a robust pH-dependent lysis “on switch” is not evident until a more acidic pH of 6.2. Although 30BC has a strong “on switch” at pH 6.8, being the only composite without statistically different membrane disruption activity from free 50B polymer at pH 6.8 (Table S3), the instability of the 30BC polymer coating at pH 7.4 generates some concern regarding its use for intravenous delivery in vivo. Conversely, 40BC composites create nearly 40% hemolysis at pH 6.8 and remain relatively inert at pH 7.4, suggesting that it may create the best balance between coating stability and pH-activated membrane disruption for ultimate intravenous use.
To test the polymer compositional effects on PSNP composite therapeutic cargo intracellular bioactivity, we used a reporter cell line that is capable of monitoring inhibition of miRNA-122, a microRNA with relevance to liver cholesterol processing and hepatitis C.44 A hepatoma carcinoma cell line (Huh-7) was modified to express both firefly and renilla luciferase based on Promega’s psiCHECK-2 system. Briefly, the complementary sequence for miR-122 was inserted downstream of the renilla gene such that miR-122 binding to this genomic site inhibits renilla expression. However, if miR-122 is inhibited, the expression of renilla is activated, permitting quantitative analysis of miR-122 inhibition. Conversely, firefly luciferase is independently and constitutively transcribed, allowing firefly luminescence to be used to normalize renilla luciferase activity and to gauge relative number of viable cells. A dual-luciferase reporter kit was used according to the manufacturing protocol (Promega) to quantify firefly and renilla luciferase activity via an in vivo imaging system. The 30BC–50BC composite groups generated significantly greater bioactivity than the AMO control. 30BC composites yielded the highest bioavailability of the PNA cargo at all doses, while the 40BC performed significantly better than 50BC at the highest 1 μM dose (Figure 5A). The cell viability fell in the range between the AMO treatment and the untreated cells for all PNSNPs at all doses tested, n = 3 (Figure 5B). We also completed a study with varying ratios of PEG-DB to PSNP for 30BC–50BC (10:1, 5:1, and 2.5:1) to determine if the degree of coating had an effect on bioavailability of PNA. It was observed that there is indeed a ratio-dependent effect on bioavailability of loaded cargo for the various composites; this supported the use of the 10:1 ratio in the studies shown here (Figure S6).
Figure 5.
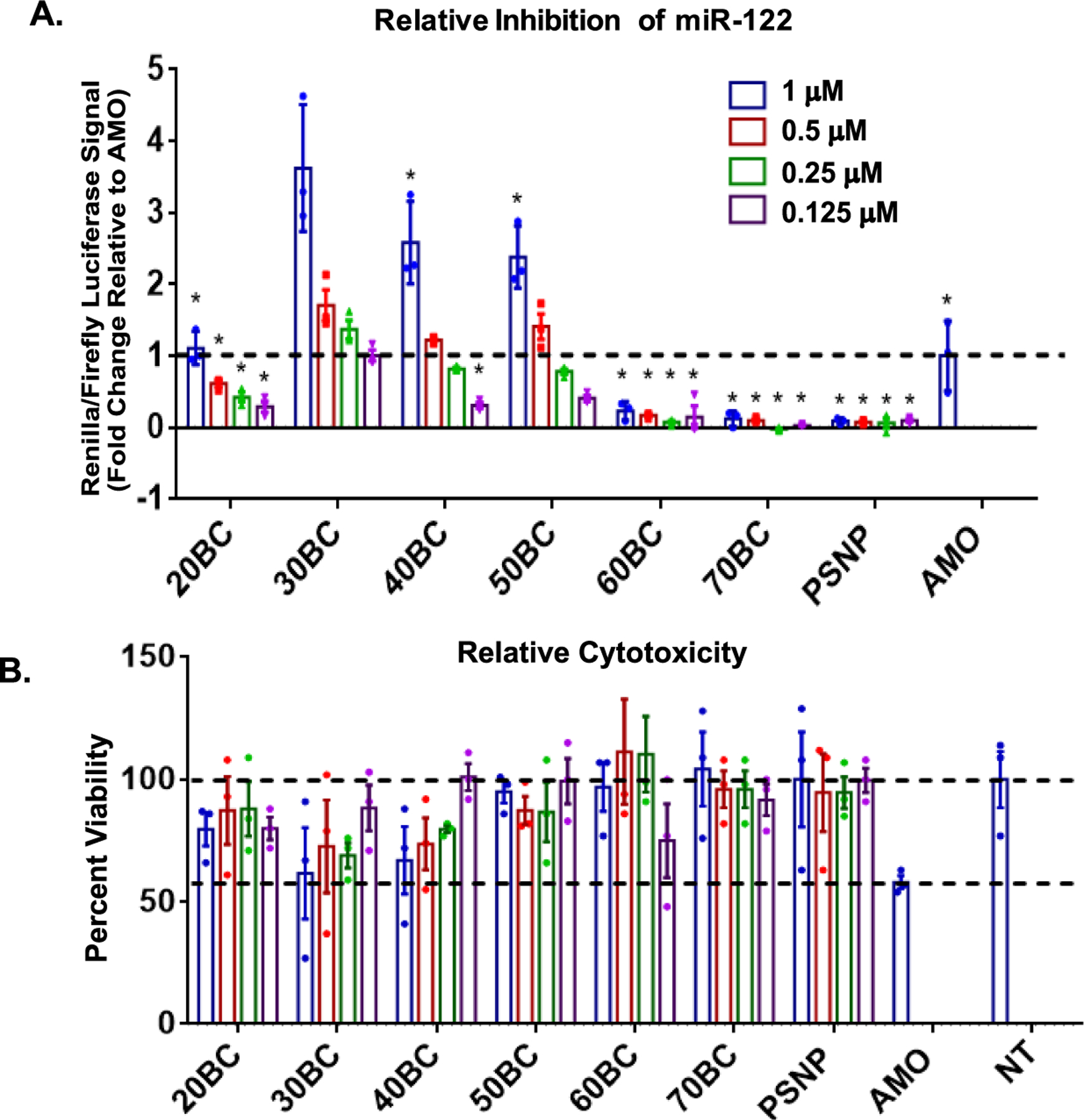
miR inhibition bioactivity and cytotoxicity for full polymer-coated PSNP composite library. (A) miR-122 Huh-7 dual-luciferase reporter cell line was used to quantify relative miR-122 inhibition levels. Statistical significance was determined by two-way ANOVA (p < 0.0001 for composition, concentration, and interaction) and Tukey’s post-hoc multiple comparisons test (stars indicate comparisons to 30BC with significantly less inhibition of miR-122 at the same dose, Table S4). (B) Cytotoxicity of the full library was measured normalized to not treated cells (NT). Significance was determined by two-way ANOVA (composition, p = 0.006; concentration, p = 0.9355; interaction, p = 0.769) and post-hoc comparisons using Tukey’s multiple comparisons test (Table S5). All experiments were done with technical replicates, and the data shown represent the average of technical replicates for three independent experiments. n = 3 unless otherwise indicated.
Finally, we utilized a recently described galectin-8 (Gal8) tracking method to observe and quantify endosomal disruption in live cells.32 Briefly, MDA-MB 231 cells were transfected with a retrovirus that encodes Gal8 fused with yellow fluorescent protein (YFP) for tracking purposes. At baseline, Gal8-YFP is evenly distributed within the cytosol of the cell, but when endosomal disruption occurs, Gal-8 redistributes and is concentrated onto specific glycans that are selectively located on the inner leaflet of the endosome. This causes a shift in fluorescent Gal8-YFP distribution into a more punctate organization (Figure 6A). This technique allows us, for the first time, to investigate how structure and composition determine the rate of endosomal disruption and how this correlates with a bioactivity measurement done in parallel. Here, we confirm that the level of rapid endosome disruption within 2 h post-treatment correlates, based on 30BC, 40BC, and 50BC data sets, n = 3, to the level of intracellular bioactivity. In the polymer-coated PSNP composites studied, 30BC had the strongest endosomolytic capacity and bioavailability (vesicle intensity = 1.8 × 105 ± 6.57 × 103) in an in vitro closed (non-sink) system.
Figure 6.
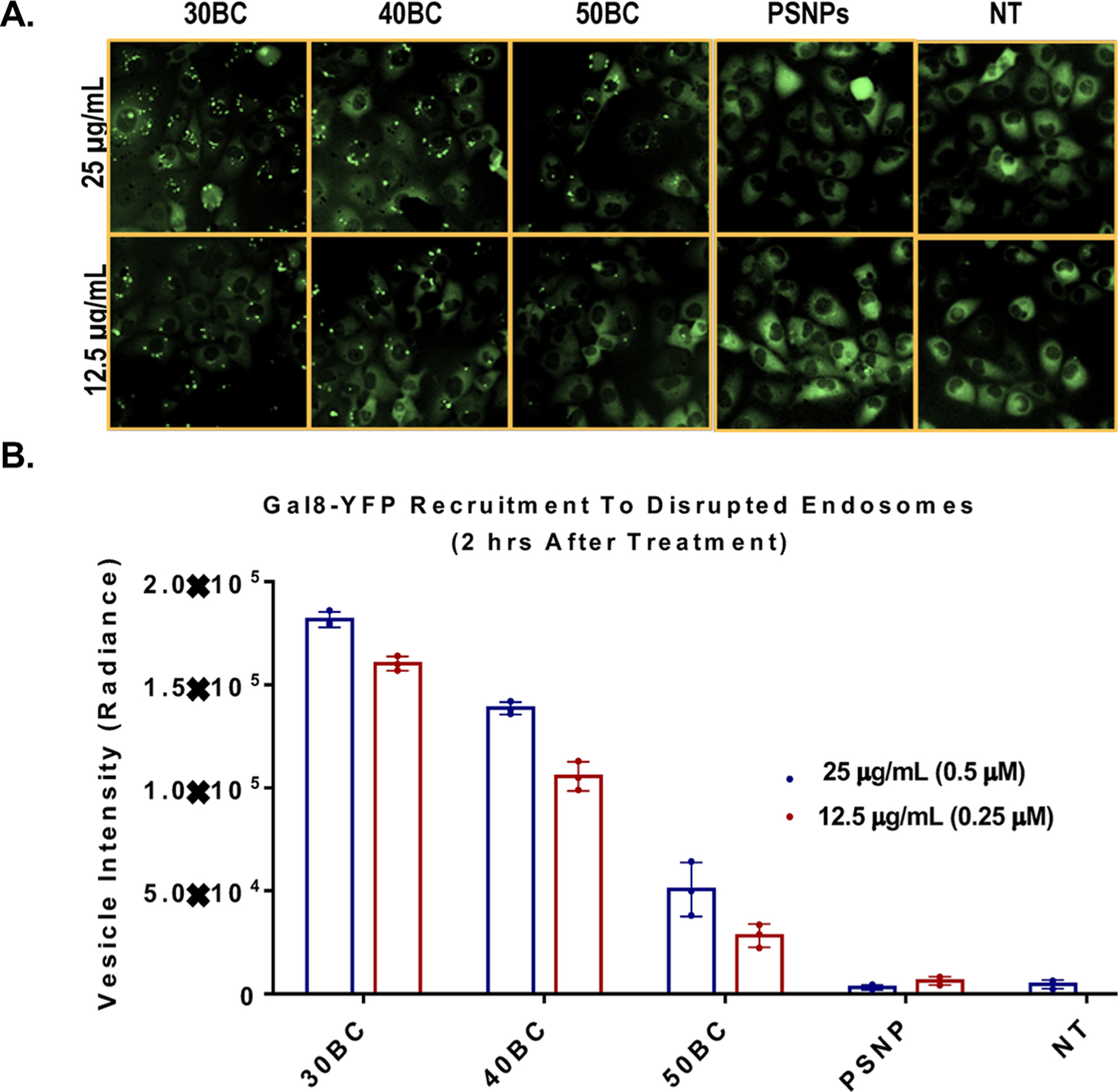
Gal8 recruitment assay demonstrates live cell endosome disruptive activity. (A) Microscopy showing dispersed Gal8 signal in the cytosol of the cell for samples that were treated with uncoated PSNPs or not treated (NT). Conversely, varied levels of Gal8 recruitment to disrupted endosomes was visualized as puncta in cells treated with the polymer-coated PSNP samples. (B) Graphical representation of the Gal-8 recruitment at a 2 h time point. Significance was determined by two-way ANOVA (p < 0.0001 for composition, concentration, and interaction) and a post-hoc comparison test using Sidak’s multiple comparisons test (Table S6). All dose-matched pairwise comparisons to 30BC were significant for all formulations. n = 3 unless otherwise indicated.
In these in vitro experiments, free polymer stripped off of the nanoparticles may potentially participate in endosome disruption and contribute to cargo bioavailability of 30BC. However, following systemic delivery in vivo, there are larger dilution and clearance effects, and therefore, 40BC may ultimately outperform 30BC in terms of safety and bioactivity due to higher stability of the polymer coating at pH 7.4 and in the presence of serum. These data further confirm that for polymer-coated PSNP composites, lower mol % B (specifically in the range of 30B–40B) achieves better delivery than 50B which was optimal in siRNA polyplex studies.
4. CONCLUSIONS
In this study, we investigated how tuning the ratio of cationic and hydrophobic content, specifically D/B monomer ratios, in the PEG-DB diblock copolymer can affect serum stability, membrane lysis potential, cytotoxicity, delivery of cargo to the cytosol, and endosomal escape of PSNP polymer composites. We observed that variation of polymer structure of endosomolytic polymer-coated PSNP composites had pronounced effects on stability, bioavailability, endosomal escape, and activity. In the series of polymer-coated PSNP composites studied, the 30 mol % B/70 mol % D (30B) proved to be the best for robust endosomal escape and activity when paired with PSNPs. The other most promising candidate was 40BC, which showed a slightly better balance of stability at physiological pH (7.4), membrane lysing potential at 6.8, and also more robust endosomal escape compared to the previously explored 50BC. In sum, this work illustrates the necessity to finely tune polymer-coated PSNP nanocarriers to maximize intracellular bioavailability and motivates further development and in vivo comparison of 30BC and 40BC for anti-miR therapies.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health R01 HL122347 and partially by National Science Foundation (DMR-120701). I.B.K. acknowledges funding from NIH NHLBI through a Research Diversity Supplement and Brock Fletcher from the NSF Graduate Research Fellowship Program. Experiments on imaging and quantification of Gal8 assay utilized the Vanderbilt High-Throughput Screening Core Facility which receives support from the Vanderbilt Institute of Chemical Biology and the Vanderbilt Ingram Cancer Center (P30 CA68485). The authors also acknowledge use of the Vanderbilt Institute of Nanoscale Science and Engineering (VINSE) for fabrication, characterization, and TEM and SEM imaging of PSNP composites.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsami.0c05827.
GPC of polymer library; polymer schematic and 1H NMR; SEM and TEM of bare PSNPs; PNA-loading quantification by absorbance; TEM and EDS of bare PSNPs and composite library; dose-dependent knockdown of miR-122 of 30BC–50BC; and statistical analysis table for Figures 4–6 (PDF)
The authors declare no competing financial interest.
Contributor Information
Isom B. Kelly, III, Department of Biomedical Engineering, Vanderbilt University, Nashville, Tennessee 37235, United States.
R. Brock Fletcher, Department of Biomedical Engineering, Vanderbilt University, Nashville, Tennessee 37235, United States.
James R. McBride, Vanderbilt Institute of Nanoscale Science and Engineering, Vanderbilt University, Nashville, Tennessee 37235, United States.
Sharon M. Weiss, Department of Electrical Engineering and Computer Science, Vanderbilt University, Nashville, Tennessee 37235, United States.
Craig L. Duvall, Department of Biomedical Engineering, Vanderbilt University, Nashville, Tennessee 37235, United States.
REFERENCES
- (1).Xu ZP; Zeng QH; Lu GQ; Yu AB Inorganic Nanoparticles as Carriers for Efficient Cellular Delivery. Chem. Eng. Sci 2006, 61, 1027–1040. [Google Scholar]
- (2).Mehrasa M; Asadollahi MA; Ghaedi K; Salehi H; Arpanaei A Electrospun Aligned PLGA and PLGA/gelatin Nanofibers Embedded with Silica Nanoparticles for Tissue Engineering. Int. J. Biol. Macromol 2015, 79, 687–695. [DOI] [PubMed] [Google Scholar]
- (3).Pawlik J; Łukowicz K; Cholewa-Kowalska K; Osyczka AM New Insights Into the PLGA and PCL Blending: Physico-Mechanical Properties and Cell Response. Mater. Res. Express 2019, 6, 085344. [Google Scholar]
- (4).Decuzzi P; Godin B; Tanaka T; Lee S-Y; Chiappini C; Liu X; Ferrari M Size and Shape Effects in the Biodistribution of Intravascularly Injected Particles. J. Controlled Release 2010, 141, 320–327. [DOI] [PubMed] [Google Scholar]
- (5).Mares JW; Fain JS; Beavers KR; Duvall CL; Weiss SM Shape-Engineered Multifunctional Porous Silicon Nanoparticles by Direct Imprinting. Nanotechnology 2015, 26, 271001. [DOI] [PubMed] [Google Scholar]
- (6).Watermann A; Brieger J Mesoporous Silica Nanoparticles as Drug Delivery Vehicles in Cancer. Nanomaterials 2017, 7, 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Xia B; Zhang W; Shi J; Xiao S.-j. Engineered Stealth Porous Silicon Nanoparticles via Surface Encapsulation of Bovine Serum Albumin for Prolonging Blood Circulation In Vivo. ACS Appl. Mater. Interfaces 2013, 5, 11718–11724. [DOI] [PubMed] [Google Scholar]
- (8).Li W; Liu Z; Fontana F; Ding Y; Liu D; Hirvonen JT; Santos HA Tailoring Porous Silicon for Biomedical Applications: From Drug Delivery to Cancer Immunotherapy. Adv. Mater 2018, 30, 1703740. [DOI] [PubMed] [Google Scholar]
- (9).Leonard F; Margulis K; Liu X; Srinivasan S; Magdassi S; Godin B Low Pressure Mediated Enhancement of Nanoparticle and Macromolecule Loading into Porous Silicon Structures. Open Mater. Sci 2014, 1, 44–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Liu D; Bimbo LM; Mäkilä E; Villanova F; Kaasalainen M; Herranz-Blanco B; Caramella CM; Lehto V-P; Salonen J; Herzig K-H; Hirvonen J; Santos HA Co-delivery of a Hydrophobic Small Molecule and a Hydrophilic Peptide by Porous Silicon Nanoparticles. J. Controlled Release 2013, 170, 268–278. [DOI] [PubMed] [Google Scholar]
- (11).Beavers KR; Werfel TA; Shen T; Kavanaugh TE; Kilchrist KV; Mares JW; Fain JS; Wiese CB; Vickers KC; Weiss SM; Duvall CL Porous Silicon and Polymer Nano-composites for Delivery of Peptide Nucleic Acids as Anti-MicroRNA Therapies. Adv. Mater 2016, 28, 7984–7992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Chaix A; Cueto-Diaz E; Delalande A; Knezevic N; Midoux P; Durand J-O; Pichon C; Cunin F Amino-acid Functionalized Porous Silicon Nanoparticles for the Delivery of pDNA. RSC Adv 2019, 9, 31895–31899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Bertucci A; Kim K-H; Kang J; Zuidema JM; Lee SH; Kwon EJ; Kim D; Howell SB; Ricci F; Ruoslahti E; Jang H-J; Sailor MJ Tumor-Targeting, MicroRNA-Silencing Porous Silicon Nanoparticles for Ovarian Cancer Therapy. ACS Appl. Mater. Interfaces 2019, 11, 23926–23937. [DOI] [PubMed] [Google Scholar]
- (14).Park J-H; Gu L; von Maltzahn G; Ruoslahti E; Bhatia SN; Sailor MJ Biodegradable Luminescent Porous Silicon Nanoparticles for In Vivo Applications. Nat. Mater 2009, 8, 331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Jarvis KL; Barnes TJ; Prestidge CA Surface Chemistry of Porous Silicon and Implications for Drug Encapsulation and Delivery Applications. Adv. Colloid Interface Sci 2012, 175, 25–38. [DOI] [PubMed] [Google Scholar]
- (16).Canham L Handbook of Porous Silicon; Springer, 2014. [Google Scholar]
- (17).Tsang CK; Kelly TL; Sailor MJ; Li YY Highly Stable Porous Silicon-Carbon Composites as Label-Free Optical Biosensors. ACS Nano 2012, 6, 10546–10554. [DOI] [PubMed] [Google Scholar]
- (18).Anglin E; Cheng L; Freeman W; Sailor M Porous Silicon in Drug Delivery Devices and Materials. Adv. Drug Delivery Rev 2008, 60, 1266–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Tsang CK; Kelly TL; Sailor MJ; Li YY Highly Stable Porous Silicon–Carbon Composites As Label-Free Optical Biosensors. ACS Nano 2012, 6, 10546–10554. [DOI] [PubMed] [Google Scholar]
- (20).Tong WY; Alnakhli M; Bhardwaj R; Apostolou S; Sinha S; Fraser C; Kuchel T; Kuss B; Voelcker NH Delivery of siRNA In Vitro and In Vivo Using PEI-Capped Porous Silicon Nanoparticles to Silence MRP1 and Inhibit Proliferation in Glioblastoma. J. Nanobiotechnol 2018, 16, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Ziemba B; Matuszko G; Appelhans D; Voit B; Bryszewska M; Klajnert B Genotoxicity of Poly(propylene imine) Dendrimers. Biopolymers 2012, 97, 642–648. [DOI] [PubMed] [Google Scholar]
- (22).Kafil V; Omidi Y Cytotoxic Impacts of Linear and Branched Polyethylenimine Nanostructures in a431 Cells. Bioimpacts 2011, 1, 23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Mukherjee SP; Byrne HJ Polyamidoamine Dendrimer Nanoparticle Cytotoxicity, Oxidative Stress, Caspase Activation and Inflammatory Response: Experimental Observation and Numerical Simulation. Nanomedicine 2013, 9, 202–211. [DOI] [PubMed] [Google Scholar]
- (24).Nadrah P; Porta F; Planinšek O; Kros A; Gaberšček M Poly(propylene imine) dendrimer caps on mesoporous silica nanoparticles for redox-responsive release: smaller is better. Phys. Chem. Chem. Phys 2013, 15, 10740–10748. [DOI] [PubMed] [Google Scholar]
- (25).Stasko NA; Johnson CB; Schoenfisch MH; Johnson TA; Holmuhamedov EL Cytotoxicity of Polypropylenimine Dendrimer Conjugates On Cultured Endothelial Cells. Biomacromolecules 2007, 8, 3853–3859. [DOI] [PubMed] [Google Scholar]
- (26).Prabhakar N; Zhang J; Desai D; Casals E; Gulin-Sarfraz T; Näreoja T; Westermarck J; Rosenholm J Stimuli-Responsive Hybrid Nanocarriers Developed by Controllable Integration of Hyperbranched PEI With Mesoporous Silica Nanoparticles for Sustained Intracellular siRNA Delivery. Int. J. Nanomed 2016, 11, 6591–6608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).McConnell KI; Shamsudeen S; Meraz IM; Mahadevan TS; Ziemys A; Rees P; Summers HD; Serda RE Reduced Cationic Nanoparticle Cytotoxicity Based on Serum Masking of Surface Potential. J. Biomed. Nanotechnol 2016, 12, 154–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Isaksson K; Åkerberg D; Posaric-Bauden M; Andersson R; Tingstedt B In vivo toxicity and biodistribution of intraperitoneal and intravenous poly-L-lysine and poly-L-lysine/poly-L-glutamate in rats. J. Mater. Sci.: Mater. Med 2014, 25, 1293–1299. [DOI] [PubMed] [Google Scholar]
- (29).Nimesh S; Chandra R Polyethylenimine Nanoparticles as an Efficient In Vitro siRNA Delivery System. Eur. J. Pharm. Biopharm 2009, 73, 43–49. [DOI] [PubMed] [Google Scholar]
- (30).Qi R; Gao Y; Tang Y; He R-R; Liu T-L; He Y; Sun S; Li B-Y; Li Y-B; Liu G PEG-conjugated PAMAM Dendrimers Mediate Efficient Intramuscular Gene Expression. AAPS J 2009, 11, 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Nelson CE; Kintzing JR; Hanna A; Shannon JM; Gupta MK; Duvall CL Balancing Cationic and Hydrophobic Content of PEGylated siRNA Polyplexes Enhances Endosome Escape, Stability, Blood Circulation Time, and Bioactivity In Vivo. ACS Nano 2013, 7, 8870–8880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Kilchrist KV; Dimobi SC; Jackson MA; Evans BC; Werfel TA; Dailing EA; Bedingfield SK; Kelly IB; Duvall CL Gal8 Visualization of Endosome Disruption Predicts Carrier-Mediated Biologic Drug Intracellular Bioavailability. ACS Nano 2019, 13, 1136–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Nelson CE; Kintzing JR; Hanna A; Shannon JM; Gupta MK; Duvall CL Balancing Cationic and Hydrophobic Content of PEGylated siRNA Polyplexes Enhances Endosome Escape, Stability, Blood Circulation Time, and Bioactivity In Vivo. ACS Nano 2013, 7, 8870–8880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Werfel TA; Jackson MA; Kavanaugh TE; Kirkbride KC; Miteva M; Giorgio TD; Duvall C Combinatorial Optimization of PEG Architecture and Hydrophobic Content Improves Ternary siRNA Polyplex Stability, Pharmacokinetics, and Potency In Vivo. J. Controlled Release 2017, 255, 12–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Werfel TA; Wang S; Jackson MA; Kavanaugh TE; Joly MM; Lee LH; Hicks DJ; Sanchez V; Ericsson PG; Kilchrist KV; Dimobi SC; Sarett SM; Brantley-Sieders DM; Cook RS; Duvall CL Selective mTORC2 Inhibitor Therapeutically Blocks Breast Cancer Cell Growth and Survival. Cancer Res 2018, 78, 1845–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Jackson MA; Werfel TA; Curvino EJ; Yu F; Kavanaugh TE; Sarett SM; Dockery MD; Kilchrist KV; Jackson AN; Giorgio TD; Duvall CL Zwitterionic Nanocarrier Surface Chemistry Improves siRNA Tumor Delivery and Silencing Activity Relative to Polyethylene Glycol. ACS Nano 2017, 11, 5680–5696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Menchise V; De Simone G; Tedeschi T; Corradini R; Sforza S; Marchelli R; Capasso D; Saviano M; Pedone C Insights into peptide nucleic acid (PNA) structural features: The crystal structure of a D-lysine-based chiral PNA-DNA duplex. Proc. Natl. Acad. Sci. U.S.A 2003, 100, 12021–12026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).LeCher JC; Nowak SJ; McMurry JL Breaking in and busting out: cell-penetrating peptides and the endosomal escape problem. Biomol. Concepts 2017, 8, 131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Cullen PJ; Steinberg F To Degrade or Not to Degrade: Mechanisms and Significance of Endocytic Recycling. Nat. Rev. Mol. Cell Biol 2018, 19, 679–696. [DOI] [PubMed] [Google Scholar]
- (40).Wittrup A; Ai A; Liu X; Hamar P; Trifonova R; Charisse K; Manoharan M; Kirchhausen T; Lieberman J Visualizing Lipid-Formulated siRNA Release from Endosomes and Target Gene Knockdown. Nat. Biotechnol 2015, 33, 870–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Evans BC; Fletcher RB; Kilchrist KV; Dailing EA; Mukalel AJ; Colazo JM; Oliver M; Cheung-Flynn J; Brophy CM; Tierney JW; Isenberg JS; Hankenson KD; Ghimire K; Lander C; Gersbach CA; Duvall CL An Anionic, Endosome-Escaping Polymer to Potentiate Intracellular Delivery of Cationic Peptides, Biomacromolecules, and Nanoparticles. Nat. Commun 2019, 10, 5012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Wang M; Li X; Ma Y; Gu H Endosomal Escape Kinetics of Mesoporous Silica-Based system for Efficient siRNA Delivery. Int. J. Pharm 2013, 448, 51–57. [DOI] [PubMed] [Google Scholar]
- (43).Evans BC; Nelson CE; Yu SS; Beavers KR; Kim AJ; Li H; Nelson HM; Giorgio TD; Duvall CL Ex Vivo Red Blood cell Hemolysis Assay for the Evaluation of pH-responsive Endosomolytic Agents for Cytosolic Delivery of Biomacromolecular Drugs. J. Visualized Exp 2013, 73, No. e50166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Connelly CM; Thomas M; Deiters A High-Throughput Luciferase Reporter Assay for Small-Molecule Inhibitors of Micro-RNA Function. J. Biomol. Screening 2012, 17, 822–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.