

Abstract
Transient focal neurologic episodes (TFNEs) are brief disturbances in motor, somatosensory, visual, or language functions that can occur in patients with cerebral amyloid angiopathy (CAA) and may be difficult to distinguish from TIAs or other transient neurologic syndromes. They herald a high rate of future lobar intracerebral hemorrhage, making it imperative to differentiate them from TIAs to avoid potentially dangerous use of antithrombotic drugs. Cortical spreading depression or depolarization triggered by acute or chronic superficial brain bleeding, a contributor to brain injury in other neurologic diseases, may be the underlying mechanism. This review discusses diagnosis, pathophysiology, and management of CAA-related TFNEs.
Introduction
Cerebral amyloid angiopathy (CAA) is a disease of the small arteries and arterioles that predominantly affects the cortex and leptomeninges.1 Age-related sporadic CAA is caused by deposition of β-amyloid (Aβ), while rare genetic forms of the disease can be caused by deposition of Aβ or other amyloid proteins. This amyloid deposition damages the vessel wall, leading to thickening, hyalinization, and smooth muscle cell loss, ultimately leading to parenchymal brain injury due to both bleeding and ischemia.1
Originally recognized primarily as a cause of lobar intracerebral hemorrhage (ICH), recent research shows that CAA also causes convexity subarachnoid hemorrhage (cSAH) and transient neurologic symptoms.2,3 These consist of short (typically under 30 minutes), frequently recurrent stereotyped episodes of focal (usually sensory or motor) disturbances, often exhibiting a spreading progression, where the symptoms smoothly migrate over minutes to adjacent body parts as represented in the cerebral cortex (e.g., from the hand up the arm into the face).4,5 The difficulty in clinical diagnosis is that similar symptoms occur in other conditions such as TIA, migraine with aura, or seizures. Various terms have been used to describe this syndrome, including “amyloid spells.”6 We recommend the term CAA-related transient focal neurologic episodes (CAA-related TFNEs)5 to distinguish the transient neurologic symptoms seen in CAA from other causes of temporary neurologic disturbances and to avoid the nonspecific term spells.
Recognizing CAA-related TFNEs is important. Patients with CAA-related TFNE are at substantial risk of subsequent lobar ICH5 that would be exacerbated by inappropriate prescription of antithrombotics for a presumed TIA diagnosis. The high observed rates of ICH following TFNE raise the question whether the mechanisms underlying TFNE also predispose to future neuropathologic events such as vascular rupture or neuronal injury.
This review synthesizes information on the clinical and neuroimaging features, pathophysiology, diagnosis, prognosis, and management of CAA-related TFNE.
Methods
This narrative review was informed by a systematic search of the PubMed database on November 8, 2020, using the terms cerebral amyloid angiopathy or caa or “cerebral amyloid angiopathy” (MeSH) and transient or TFNE or transient ischemic attack or TIA or subarachnoid hemorrhage (text word) or siderosis (text word) or subarachnoid hemorrhage or “amyloid” (MeSH) or amyloid and spells or spell. The search returned 437 articles, of which 173 were related to the topic and reviewed in full. Hand-searching identified 4 additional articles.
Clinical Presentation
CAA-related TFNEs were sporadically reported as early as the 1980s.7 An early case series identified a characteristic spread of symptoms into contiguous body areas and evidence of small hemorrhagic lesions or subsequent large ICH in cortical locations corresponding to the neurologic symptoms.8
The increasing awareness of CAA as a clinical and pathologic entity, and the more widespread availability of blood-sensitive MRI sequences that allow diagnosis by application of the Boston criteria,9,10 have increased our understanding and recognition of CAA-related TFNEs.8,11,12 In a European multicenter retrospective cohort study of 172 patients with CAA based on the Boston criteria,5 TFNEs were the most common clinical presentation of CAA after lobar ICH, being present in 14.5%. TFNE clinical phenomenology was classified into 2 groups: predominantly positive or predominantly negative symptoms, each being equally common (52% vs 48%, respectively), with 25% having both positive and negative symptoms. The commonest positive symptom consisted of transient paresthesias in the mouth or hand (32%), often but not always with a gradual spread to contiguous body parts. The negative symptoms included focal weakness and dysphasia. A minority (<20%) had limb-jerking episodes or transient visual disturbances involving blurred vision or visual loss, flickering, or flashing lights and transient zig-zags (teichopsia). Most participants (68%) had multiple episodes, nearly always stereotyped (i.e., recurrent episodes similar or identical to the initial presentation). TFNEs lasted <6 minutes in 44% of patients, <30 minutes in 70%, and ≤3 hours in 96%. A subsequent systematic review13 confirmed a high frequency of positive spreading sensory symptoms (about 80%) but also predominantly negative symptoms (such as hemiparesis or nonfluent dysphasia) in around 40%.
Neuroimaging
CAA-related TFNEs are closely, but not exclusively, associated with convexity subarachnoid hemorrhage (cSAH) or cortical superficial siderosis (cSS) (figure 1), implicating superficial bleeding in the pathophysiology of this condition.14 Two cohort studies showed that the majority of patients with CAA-related TFNEs (first ever or recurrent) have 1 or both of cSAH or cSS (58%5 and 83%,13 respectively). Disseminated cSS is much more common in patients with CAA presenting with TFNEs than patients with CAA presenting with ICH.15,16
Figure 1. Convexity Sulcal Subarachnoid Hemorrhage (cSAH) and Cortical Superficial Siderosis (cSS).
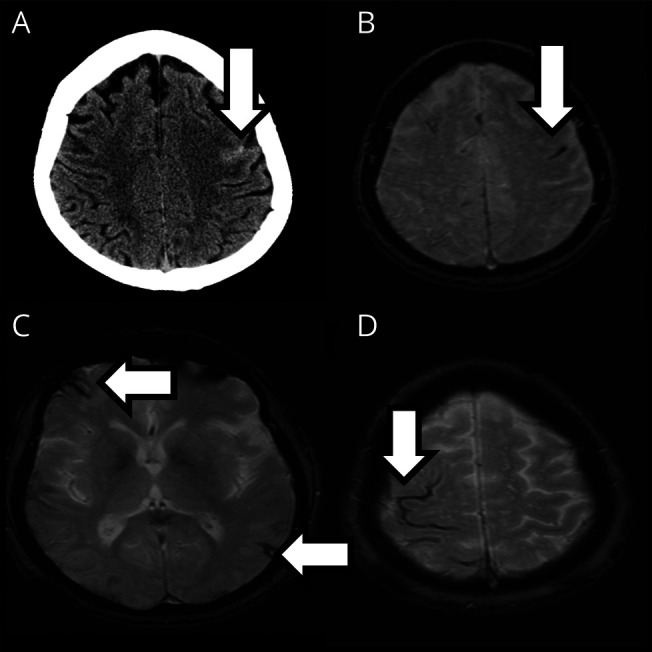
A 71-year-old woman presented with paresthesias and weakness of the right hand. (A) CT showed acute cSAH in a left frontal sulcus, visible as a linear hypointensity on T2*-weighted gradient-recalled echo (GRE) MRI (B). MRI GRE also showed 3 areas of cSS (arrows, C and D) in sulci without acute cSAH. One year later, the patient had a left parietal lobar intracerebral hemorrhage.
Pathophysiology
The slow spreading pattern of signs and symptoms congruent with cortical somatotopy, the preponderance of mixed positive and negative symptoms, and the transient and often stereotypic nature are the most conspicuous features of CAA-related TFNEs. These characteristics implicate the phenomenon of cortical spreading depolarization (CSD), also known as spreading depression, in response to superficial hemorrhagic lesions.
CSD is an electrophysiologic phenomenon associated with near complete depolarization of virtually all cell types in the brain tissue including neurons, glia, possibly perivascular nerves, and the vasculature.17 The sustained loss of neuronal membrane potentials and changes in neurochemical milieu preclude synaptic transmission and action potentials for minutes. As a result, there is complete electrophysiologic silence during CSD, which distinguishes CSD from seizures. The depolarization of CSD slowly propagates (a few millimeters per minute) by way of chemical contiguity, often for many centimeters across contiguous cortical regions, explaining the slow, smooth, somatotopic spread of neurologic deficits that is virtually pathognomonic for CSD. The cardinal features of CAA-related TFNEs, such as the marching pattern, positive and negative stereotypic symptomatology, and complete reversibility, strongly support CSD as the underlying mechanism, even though direct electrophysiologic evidence is lacking and may be impractical to acquire, as this would require invasive placement of subdural or depth electrodes.
The congruence between location of cSAH or cSS and the somatotopy of neurologic deficits (most often central sulcal cSAH and contralateral somatosensory migrating symptoms13) suggests that CSDs are triggered from these CAA lesions (figure 2). In the absence of direct mechanistic data, however, we can only speculate on what triggers CSD in CAA. CSDs can be triggered by hemorrhage, as described in the setting of ICH18 and aneurysmal subarachnoid hemorrhage (SAH).19 However, experimental data show that simple cortical exposure to whole blood does not trigger a CSD.20 In contrast, hemolyzed blood is a potent CSD trigger, presumably due to its high potassium (K+) content. This raises the interesting possibility that SAH trapped in a cortical sulcus acts as a persistent source of blood breakdown products, including K+, leading to recurrent, stereotyped CSD events that gradually diminish in frequency as the depolarizing substances are slowly cleared.21 Sulcal SAH could also facilitate CSD occurrence by exerting local mechanical pressure on adjacent cortex and by causing cortical arterial or venous thrombosis, both of which are known CSD triggers. Indeed, 2 case series show that sulcal SAH had adjacent cortical DWI lesions in about half of the cases.22,23 Mechanical pressure might be a CSD trigger in the setting of cortical microbleeds. However, the presence of locally elevated pressure adjacent to sulcal SAH or microbleeds has not been directly measured, and it would be technically difficult to do so. One final potential mechanism triggering CSD may be vasospasm caused by cSAH in the setting of CAA. However, this is less likely because vasospasm is a slower and more lasting event than TFNE; moreover, convexity and sulcal SAH are frequent after trauma, but do not lead to clinically relevant vasospasm or TFNEs.
Figure 2. Possible Mechanisms Triggering Spreading Depolarizations.
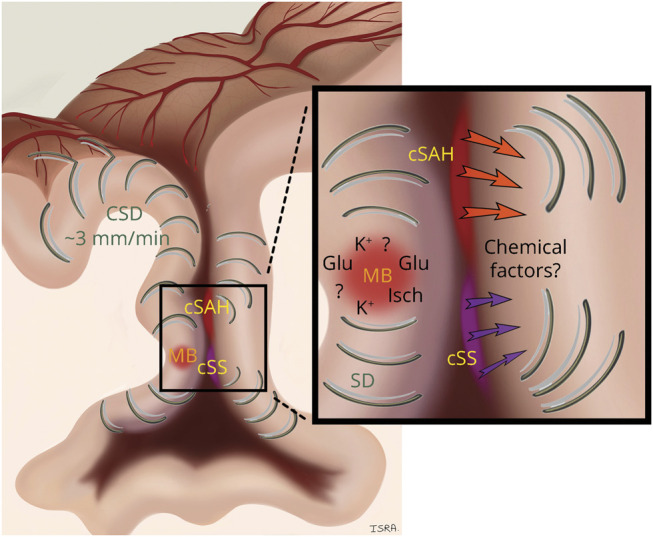
Schematic representation of hypotheses on the origin of spreading cortical depolarizations (CSDs) within a sulcus affected by cerebral amyloid angiopathy (CAA). Convexity subarachnoid hemorrhage (cSAH) and cortical superficial siderosis (cSS) could trigger CSD by releasing chemical factors that affect the brain tissue or pial vasculature. An acute cortical microbleed might also trigger CSD via ischemia (Isch) in the territory of the ruptured artery, via mechanical distortion of brain tissue by expanding microbleed, or by release of depolarizing factors from plasma leakage or hematoma lysis (e.g., potassium [K+] ions or glutamate [Glu]). Once initiated, CSDs propagate in cortical gray matter at a speed of ∼3 mm/min for many centimeters, creating a TFNE.
None of the abovementioned mechanisms directly related to SAH explains the apparent association between TFNEs and cSS without acute cSAH. Hemosiderin by itself has not been shown to trigger or facilitate CSDs, and any gliosis associated with cSS would be expected to suppress rather than facilitate CSD susceptibility.24 Seizure activity could trigger a CSD,25 which could help terminate the seizure but itself propagate in the tissue creating the TFNE. Whereas epileptic events have not been observed on routine electrophysiologic studies in patients with CAA with TFNEs,13,26,27 including in 1 patient with 2 episodes while undergoing continuous EEG,28 surface EEG is not highly sensitive for detecting focal seizures.
Another potential trigger for CSD is cerebral ischemia.17,29 Although CAA is primarily recognized as a disease characterized by hemorrhages, focal cerebral ischemia is common as well. Therefore, it is possible that recurrent ischemic events due to diseased cortical arterioles trigger recurrent stereotypic CSD events, and some eventually lead to cortical infarcts visible on DWI close to cSAH in CAA.22,23,30
CSDs dramatically increase oxygen and glucose consumption in brain tissue, disrupt the blood–brain barrier for more than 24 hours, induce vasoconstriction, and upregulate matrix metalloproteinase and proinflammatory cytokine expression.31-33 Despite all these changes, CSD is not by itself injurious to the brain tissue unless metabolically compromised. Indeed, numerous CSDs can occur in otherwise normal tissue without causing any cell death. However, in brains with CAA (and often coexisting Alzheimer disease pathology), these changes could have complex adverse effects on neurovascular structure and function, although it remains speculative whether TFNEs alter the disease course.
Diagnosis
The diagnosis relies on recognizing a compatible clinical syndrome accompanied by clinical, neuroimaging, or neuropathologic evidence of CAA and the absence of a more plausible alternative cause. TIA, migraine aura, and focal seizures are the most common alternative diagnoses to CAA-related TFNEs based on clinical symptoms prior to neuroimaging (table 1). Compared to TIA, CAA-related TFNEs are more likely to exhibit migratory spread, affect sensation, and recur in a stereotyped manner.34 Diagnostic characteristics of CAA-related TFNEs are shown in table 2.
Table 1.
Competing Causes of Transient Neurologic Symptoms

Table 2.
Diagnostic Characteristics of Cerebral Amyloid Angiopathy (CAA)–Related Transient Focal Neurologic Episodes (TFNEs)

Two cohort studies have defined operational criteria for identifying CAA-related TFNEs. The first of these articles defined CAA-related TFNE as “a clearly documented history of transient (≤24 hours), fully resolving, focal neurologic episodes accompanied by evidence of possible, probable, or definite CAA according to the Boston criteria, and no known alternative explanation other than CAA (e.g., structural brain lesion, atrial fibrillation, extracranial or intracranial stenosis).”5 Another article defined TFNEs as “a clinical episode of transient focal neurologic symptoms including numbness/tingling, weakness, dysarthria or aphasia lasting minutes to 1 hour with subsequent complete resolution.”13
The validated Boston criteria should be applied to infer the presence of moderate to severe CAA.9 In 2010, the criteria were modified to included cSS as equivalent to a lobar hemorrhage or microbleed.10 The Boston criteria have been validated pathologically in patients with lobar ICH9 but there are few pathology data available in patients with CAA-related TFNEs, because unlike ICH they are not fatal and not treated surgically. In 1 case, a 71-year-old woman with TFNEs related to a right cSAH and a concurrent contralateral lobar ICH had evidence of vascular Aβ at death, with more severe deposition in the leptomeningeal than the cortical vessels.35 Another pathology-confirmed CAA case consisted of a 58-year-old man with recurrent TFNEs followed by rapid cognitive decline and death.8 Indirect evidence of the validity of the Boston criteria to diagnose CAA-related TFNEs comes from prospective cohort studies showing that patients with CAA-related TFNE have a similarly high rate of subsequent lobar ICH as patients with lobar ICH and probable CAA.5,36
Investigations should at minimum consist of a history, physical and neurologic examination, bloodwork for metabolic causes of neurologic disturbances (electrolytes, creatinine, liver function tests, and complete blood count), coagulopathies (platelet count, prothrombin time, and partial thromboplastin time), and neuroimaging. CT will identify the cases with acute cSAH. MRI with susceptibility-weighted sequences (such as T2*-weighted gradient recalled echo or susceptibility-weighted imaging) has equivalent sensitivity to CT for SAH37 and will additionally identify cSS and lobar microbleeds that can point to the presence of CAA. MRI diffusion-weighted imaging (DWI) is useful because it may identify patterns of abnormalities consistent with infarction from thromboembolism rather than CAA; however, the clinician must be aware that small DWI abnormalities frequently occur in CAA as well,38 including in up to half of patients with CAA-related cSAH, usually adjacent to the acute hemorrhage22,23,30 (figure 3). In the subacute period, MRI is more useful than CT because blood products may have resolved on CT while MRI will still show susceptibility changes related to prior bleeding events.
Figure 3. MRI Diffusion-Weighted Imaging (DWI) in Cerebral Amyloid Angiopathy (CAA)–Related Transient Focal Neurologic Episodes (TFNEs) Compared With Ischemic Stroke.

(A–C) From the same patient, disseminated cortical superficial siderosis (cSS) on MRI susceptibility-weighted imaging (A) with a 3 mm focus of bright signal on DWI in the left superior frontal gyrus adjacent to cSS (B, circled) with hypointensity on apparent diffusion coefficient image (C, circled), indicating restricted diffusion. Small (≤10 mm) DWI-positive lesions are often seen in CAA with convexity subarachnoid hemorrhage and TFNEs, usually adjacent to regions of cSS and sometimes multiple.22 (D–F) DWI-positive lesion patterns seen in ischemic stroke but not CAA include single larger (>10 mm) DWI-positive infarcts (D), multiple DWI-positive infarcts restricted to a vascular perfusion territory (E), or DWI-positive small subcortical infarcts restricted to the territory of a single perforating artery (as in the thalamic recent subcortical infarct seen in F).
Patients with cSAH or cSS should at minimum undergo noninvasive angiography by CT or MRI to exclude distal aneurysms or vascular malformations. Invasive catheter angiopathy is probably not needed if there are radiologic markers meeting Boston criteria for CAA.
Although unlikely to be used in routine practice, Aβ markers are usually positive in patients with CAA-related TFNEs. Small cases series of patients with CAA-related TFNEs and cSAH have shown positive amyloid-PET39 and low CSF Aβ1-40 and Aβ1-42.23
Incidence of CAA-Related TFNEs
The incidence of CAA-related TFNEs in the general population is unknown, owing to general underrecognition and a lack of consensus diagnostic criteria. Among patients with acute nontraumatic cSAH, CAA was identified as the cause in a quarter to a third of patients of all ages11,40-47 including 76% of persons 60 years of age or older.40,46 Most of these patients with cSAH (≥75%) presented with TFNEs.15,16,22,36 Similarly, another study found that CAA accounted for the majority of cSS cases in a hospital-based radiology database.48 There are fewer data on the proportion of possible TIA that may instead be CAA-related TFNEs. In 1 prospective study of patients with possible ischemic symptoms who were consecutively consented to undergo MRI, 4/416 (1.0%) were retrospectively assessed as having CAA-related TFNEs.49 Thus, current evidence suggests that most nontraumatic cSAH in the elderly, 3-quarters of cSS, and 1% of suspected TIA cases may be related to CAA.
Prognosis
CAA-related TFNEs herald a high risk of future symptomatic hemorrhage, both ICH and acute cSAH.4 In a European multicenter study, 50% of patients with CAA-related TFNEs had symptomatic lobar ICH over a median period of 14 months.5 In the same study, a systematic review and meta-analysis of all relevant case reports and case series published showed a 24.5% (95% confidence interval [CI] 15.8–36.9) risk of symptomatic ICH at 8 weeks after TFNE, a risk uninfluenced by clinical features of the TFNE or previous symptomatic lobar ICH.5 In a meta-analysis of 9 patient cohorts with acute cSAH and probable CAA, the majority of which presented with TFNEs, the ICH rate was 19% per year (95% CI 13–27).36 These rates of ICH are higher than the risk of recurrent ICH after first CAA-related ICH (estimated at 7.4% per year50).
It is unclear whether this extremely high ICH risk is a direct consequence of TFNEs per se vs a consequence of the strong association of TFNEs with cSS and cSAH, which themselves predict future ICH.4,14 In a cohort of 236 patients with probable CAA presenting with non-ICH neurologic symptoms (22% with TFNEs, 68% with cognitive complaints, and 10% with other symptoms), presence of cSS, especially disseminated (i.e., affecting at least 4 cortical sulci), was an independent predictor of first-ever ICH.51 Additional studies52 and meta-analysis53 confirm that cSS is a risk factor for future ICH, independent of the number of microbleeds, in patients with CAA with or without prior history of ICH. A pooled meta-analysis of patients with cSAH found that patients fulfilling the modified Boston criteria for CAA have a high risk of future ICH: in those with probable CAA, the ICH rate per patient-year was 19% (95% CI 13–27) compared to 7% (95% CI 3–15) for those without probable CAA.36 Patients with CAA who present with cSAH have at least as high a risk of future ICH as the patients who present with ICH.54
Management
Like patients with TIA, those with TFNEs are most likely to present to a clinician after the resolution of 1 or more attacks of sensorimotor disturbance. Thus, hyperacute treatment is not usually possible or appropriate. Nevertheless, there is observational evidence that in acute cSAH there is an early risk (within 24 hours) of expansion of cSAH into the brain parenchyma as lobar ICH.35 Thus, despite a lack of direct clinical trial evidence, it might therefore be reasonable to lower blood pressure in TFNEs associated with acute cSAH presenting early (e.g., within 24 hours) to a similar level to that recommended for acute ICH (below 140 mm Hg systolic).55
About 30% of patients with cSAH and TFNEs are taking antithrombotic drugs.5,13 Where acute cSAH is demonstrated, it seems reasonable to withhold these drugs in the acute phase, for at least 24–48 hours given the potential for early expansion of cSAH to ICH, recurrent cSAH, or ICH. Depending on the strength of the ongoing indication for antithrombotic therapy, restarting can then be considered. As the risk of future ICH after cSAH and TFNE seems at least as high as that after ICH, it might be reasonable to avoid anticoagulant drugs for 7–8 weeks,56 but controlled trials are not available. Direct oral anticoagulants consistently show a ∼50% lower ICH risk than vitamin K antagonists,57 so are preferred after TFNEs associated with cSAH when there is a need for oral anticoagulation. In patients with atrial fibrillation and high ischemic stroke risk, left atrial appendage occlusion is a potential option in those at very high intracranial bleeding risk on oral anticoagulation,58 although the procedure does not obviate the need for postprocedure antithrombotics, at least in the short term, and randomized data are not available in patients with cSAH.
When associated with cSAH due to CAA, TFNEs often recur over a short time period,4 which can cause distress for patients. The TFNE attacks may respond to anticonvulsant drugs (e.g., levetiracetam), including those effective against migraine (e.g., topiramate),12 but there are no controlled trials and the episodes may be self-limited. Patients should be reassured that although these TFNE attacks may be distressing, each attack does not usually reflect new bleeding, and the natural history is usually of improvement and remission over days to weeks.
With regard to prevention of future intracranial hemorrhage (ICH or cSAH), there are no proven interventions in CAA-related TFNE. However, given that these patients are at high risk, we suggest it would be reasonable to follow ICH guidelines for prevention, including for blood pressure control to less than 130/80 mm Hg.55
Future Directions
CAA-related TFNEs should be suspected based on the typical clinical history of transient, often recurrent, usually spreading symptoms (suggesting CSDs), in conjunction with imaging evidence of cSAH, cSS, or microbleeds consistent with CAA. However, 2 major clinical questions surround CAA-related TFNEs. One is how to diagnose TFNEs reliably in clinical practice. A second question highlighted by the discussion of TFNE pathophysiology is whether CSD, the likely underlying mechanism for TFNEs, directly contributes to ICH risk or other types of tissue injury and thus represents a target for treatment.
One step towards improved diagnosis of CAA-related TFNEs will be to refine and validate the clinical-radiographic criteria for diagnosing CAA in patients without ICH. An international collaboration is currently underway to analyze cases with a range of CAA-like presentations (including TFNEs), available MRI scans, and neuropathology samples, with the goal of updating the Boston criteria and identifying more sensitive imaging biomarkers. Any improvements in biomarkers and diagnosis of CAA-related TFNEs would lead to reduced likelihood of misdiagnosis and mistreatment for these often complex clinical events.
The second more speculative question is whether clinical outcomes can be improved by preventing CSDs or other undefined mechanisms underlying TFNEs. One approach to this question would be to identify whether treatment with spreading depression–suppressing medications reduces risk for recurrent TFNEs as well as ICH and DWI-positive lesions on MRI. In principle, any treatment that reduces the susceptibility to CSD may suppress TFNEs, including migraine prophylactic interventions.59 Additional important information would come from observational studies that characterize whether TFNEs are associated with future tissue injury (such as ICH) independent of their association with cSAH, cSS, and other known radiologic predictors of ICH in CAA, potentially implicating isolated TFNEs as the cause of such injury. Studies in humans could be complemented by animal models of CAA-related spreading depolarization, allowing experimental testing of whether prevention of spreading depression affects the progression of other CAA-related processes such as hemorrhage, impaired vascular reactivity, and susceptibility to brain ischemia.
The minimum goal for future research on CAA-related TFNEs will be to improve the accuracy of the diagnosis and ensure correct treatment. The more ambitious goal will be to determine whether spreading depression is a rational therapeutic target in CAA, particularly in patients with TFNEs and potentially even those without clinically overt TFNEs.
Acknowledgment
The authors thank Isra Tamim for help in drafting figure 2.
Glossary
- Aβ
β-amyloid
- CAA
cerebral amyloid angiopathy
- CI
confidence interval
- cSAH
convexity subarachnoid hemorrhage
- CSD
cortical spreading depolarization
- cSS
cortical superficial siderosis
- DWI
diffusion-weighted imaging
- ICH
intracerebral hemorrhage
- SAH
subarachnoid hemorrhage
- TFNE
transient focal neurologic episode
Appendix. Authors
Study Funding
The authors report no targeted funding.
Disclosure
Dr. Smith has received grants from the Canadian Institutes of Health Research and Brain Canada, consulting fees from Bayer, Biogen, Cyclerion, and Javelin, royalties from UpToDate, and is an Associate Editor of Stroke. Dr. Charidimou and Dr. Ayata report no disclosures. Dr. Werring has received personal fees from Alnylam, Bayer, and Portola. Dr. Greenberg has served on data monitoring or advisory boards for Biogen, Roche, Washington University/IQVIA, and Bayer and receives royalties from UpToDate. Go to Neurology.org/N for full disclosures.
References
- 1.Vinters HV. Cerebral amyloid angiopathy: a critical review. Stroke. 1987;18(2):311-324. [DOI] [PubMed] [Google Scholar]
- 2.Greenberg SM, Al-Shahi Salman R, Biessels GJ, et al. Outcome markers for clinical trials in cerebral amyloid angiopathy. Lancet Neurol. 2014;13(4):419-428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Banerjee G, Carare R, Cordonnier C, et al. The increasing impact of cerebral amyloid angiopathy: essential new insights for clinical practice. J Neurol Neurosurg Psychiatry. 2017;88(11):982-994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Charidimou A, Baron JC, Werring DJ. Transient focal neurological episodes, cerebral amyloid angiopathy, and intracerebral hemorrhage risk: looking beyond TIAs. Int J Stroke. 2013;8(2):105-108. [DOI] [PubMed] [Google Scholar]
- 5.Charidimou A, Peeters A, Fox Z, et al. Spectrum of transient focal neurological episodes in cerebral amyloid angiopathy: multicentre magnetic resonance imaging cohort study and meta-analysis. Stroke. 2012;43(9):2324-2330. [DOI] [PubMed] [Google Scholar]
- 6.Coates R, Bell SM, Coley S, Blackburn DJ. Cerebral amyloid angiopathy: amyloid spells and cortical superficial siderosis. Pract Neurol. 2015;15(2):124-126. [DOI] [PubMed] [Google Scholar]
- 7.Smith DB, Hitchcock M, Philpott PJ. Cerebral amyloid angiopathy presenting as transient ischemic attacks. J Neurosurg. 1985;63(6):963-964. [DOI] [PubMed] [Google Scholar]
- 8.Greenberg SM, Vonsattel JP, Stakes JW, Gruber M, Finklestein SP. The clinical spectrum of cerebral amyloid angiopathy: presentations without lobar hemorrhage. Neurology. 1993;43(10):2073-2079. [DOI] [PubMed] [Google Scholar]
- 9.Knudsen KA, Rosand J, Karluk D, Greenberg SM. Clinical diagnosis of cerebral amyloid angiopathy: validation of the Boston criteria. Neurology. 2001;56(4):537-539. [DOI] [PubMed] [Google Scholar]
- 10.Linn J, Halpin A, Demaerel P, et al. Prevalence of superficial siderosis in patients with cerebral amyloid angiopathy. Neurology. 2010;74(17):1346-1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raposo N, Viguier A, Cuvinciuc V, et al. Cortical subarachnoid haemorrhage in the elderly: a recurrent event probably related to cerebral amyloid angiopathy. Eur J Neurol. 2011;18(4):597-603. [DOI] [PubMed] [Google Scholar]
- 12.Roch JA, Nighoghossian N, Hermier M, et al. Transient neurologic symptoms related to cerebral amyloid angiopathy: usefulness of T2*-weighted imaging. Cerebrovasc Dis. 2005;20(5):412-414. [DOI] [PubMed] [Google Scholar]
- 13.Ni J, Auriel E, Jindal J, et al. The characteristics of superficial siderosis and convexity subarachnoid hemorrhage and clinical relevance in suspected cerebral amyloid angiopathy. Cerebrovasc Dis. 2015;39(5-6):278-286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Charidimou A, Linn J, Vernooij MW, et al. Cortical superficial siderosis: detection and clinical significance in cerebral amyloid angiopathy and related conditions. Brain. 2015;138(pt 8):2126-2139. [DOI] [PubMed] [Google Scholar]
- 15.Charidimou A, Boulouis G, Fotiadis P, et al. Acute convexity subarachnoid haemorrhage and cortical superficial siderosis in probable cerebral amyloid angiopathy without lobar haemorrhage. J Neurol Neurosurg Psychiatry. 2018;89(4):397-403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raposo N, Calviere L, Cazzola V, et al. Cortical superficial siderosis and acute convexity subarachnoid hemorrhage in cerebral amyloid angiopathy. Eur J Neurol. 2017;25(2):253-259. [DOI] [PubMed] [Google Scholar]
- 17.Ayata C, Lauritzen M. Spreading depression, spreading depolarizations, and the cerebral vasculature. Physiol Rev. 2015;95(3):953-993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Helbok R, Schiefecker AJ, Friberg C, et al. Spreading depolarizations in patients with spontaneous intracerebral hemorrhage: association with perihematomal edema progression. J Cereb Blood Flow Metab. 2017;37(5):1871-1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dreier JP, Woitzik J, Fabricius M, et al. Delayed ischaemic neurological deficits after subarachnoid haemorrhage are associated with clusters of spreading depolarizations. Brain. 2006;129(pt 12):3224-3237. [DOI] [PubMed] [Google Scholar]
- 20.Oka F, Hoffmann U, Lee JH, et al. Requisite ischemia for spreading depolarization occurrence after subarachnoid hemorrhage in rodents. J Cereb Blood Flow Metab. 2017;37(5):1829-1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hartings JA, York J, Carroll CP, et al. Subarachnoid blood acutely induces spreading depolarizations and early cortical infarction. Brain. 2017;140(10):2673-2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beitzke M, Enzinger C, Pichler A, Wunsch G, Fazekas F. Acute diffusion-weighted imaging lesions in cerebral amyloid angiopathy-related convexal subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2018;38(2):225-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martinez-Lizana E, Carmona-Iragui M, Alcolea D, et al. Cerebral amyloid angiopathy-related atraumatic convexal subarachnoid hemorrhage: an ARIA before the tsunami. J Cereb Blood Flow Metab. 2015;35(5):710-717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sukhotinsky I, Dilekoz E, Wang Y, et al. Chronic daily cortical spreading depressions suppress spreading depression susceptibility. Cephalalgia. 2011;31(16):1601-1608. [DOI] [PubMed] [Google Scholar]
- 25.Dreier JP, Major S, Pannek HW, et al. Spreading convulsions, spreading depolarization and epileptogenesis in human cerebral cortex. Brain. 2012;135(pt 1):259-275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Charidimou A, Gang Q, Werring DJ. Sporadic cerebral amyloid angiopathy revisited: recent insights into pathophysiology and clinical spectrum. J Neurol Neurosurg Psychiatry. 2012;83(2):124-137. [DOI] [PubMed] [Google Scholar]
- 27.Calviere L, Cuvinciuc V, Raposo N, et al. Acute convexity subarachnoid hemorrhage related to cerebral amyloid angiopathy: clinicoradiological features and outcome. J Stroke Cerebrovasc Dis. 2016;25(5):1009-1016. [DOI] [PubMed] [Google Scholar]
- 28.Cooperman SS, Shah AK, Rajamani K. Amyloid spells. Neurol Clin Pract. 2019;9(2):e17-e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.von Bornstadt D, Houben T, Seidel JL, et al. Supply-demand mismatch transients in susceptible peri-infarct hot zones explain the origins of spreading injury depolarizations. Neuron. 2015;85(5):1117-1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Apoil M, Cogez J, Dubuc L, et al. Focal cortical subarachnoid hemorrhage revealed by recurrent paresthesias: a clinico-radiological syndrome strongly associated with cerebral amyloid angiopathy. Cerebrovasc Dis. 2013;36(2):139-144. [DOI] [PubMed] [Google Scholar]
- 31.Sadeghian H, Lacoste B, Qin T, et al. Spreading depolarizations trigger caveolin-1-dependent endothelial transcytosis. Ann Neurol. 2018;84(3):409-423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gursoy-Ozdemir Y, Qiu J, Matsuoka N, et al. Cortical spreading depression activates and upregulates MMP-9. J Clin Invest. 2004;113(10):1447-1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jander S, Schroeter M, Peters O, Witte OW, Stoll G. Cortical spreading depression induces proinflammatory cytokine gene expression in the rat brain. J Cereb Blood Flow Metab. 2001;21(3):218-225. [DOI] [PubMed] [Google Scholar]
- 34.Stanton JED, Chandratheva A, Wilson D, Hostettler IC, Islam S, Werring DJ. Clinical features distinguish cerebral amyloid angiopathy-associated convexity subarachnoid haemorrhage from suspected TIA. J Neurol. 2020;267(1):133-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beitzke M, Enzinger C, Wunsch G, Asslaber M, Gattringer T, Fazekas F. Contribution of convexal subarachnoid hemorrhage to disease progression in cerebral amyloid angiopathy. Stroke. 2015;46(6):1533-1540. [DOI] [PubMed] [Google Scholar]
- 36.Wilson D, Hostettler IC, Ambler G, Banerjee G, Jager HR, Werring DJ. Convexity subarachnoid haemorrhage has a high risk of intracerebral haemorrhage in suspected cerebral amyloid angiopathy. J Neurol. 2017;264(4):664-673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Verma RK, Kottke R, Andereggen L, et al. Detecting subarachnoid hemorrhage: comparison of combined FLAIR/SWI versus CT. Eur J Radiol. 2013;82(9):1539-1545. [DOI] [PubMed] [Google Scholar]
- 38.Kimberly WT, Gilson A, Rost NS, et al. Silent ischemic infarcts are associated with hemorrhage burden in cerebral amyloid angiopathy. Neurology. 2009;72(14):1230-1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ly JV, Singhal S, Rowe CC, Kempster P, Bower S, Phan TG. Convexity subarachnoid hemorrhage with PiB positive PET scans: clinical features and prognosis. J Neuroimaging. 2015;25(3):420-429. [DOI] [PubMed] [Google Scholar]
- 40.Kumar S, Goddeau RP Jr., Selim MH, et al. Atraumatic convexal subarachnoid hemorrhage: clinical presentation, imaging patterns, and etiologies. Neurology. 2010;74(11):893-899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Graff-Radford J, Fugate JE, Klaas J, Flemming KD, Brown RD, Rabinstein AA. Distinguishing clinical and radiological features of non-traumatic convexal subarachnoid hemorrhage. Eur J Neurol. 2016;23(5):839-846. [DOI] [PubMed] [Google Scholar]
- 42.Rico M, Benavente L, Para M, Santamarta E, Pascual J, Calleja S. Headache as a crucial symptom in the etiology of convexal subarachnoid hemorrhage. Headache. 2014;54(3):545-550. [DOI] [PubMed] [Google Scholar]
- 43.Khurram A, Kleinig T, Leyden J. Clinical associations and causes of convexity subarachnoid hemorrhage. Stroke. 2014;45(4):1151-1153. [DOI] [PubMed] [Google Scholar]
- 44.Geraldes R, Sousa PR, Fonseca AC, Falcao F, Canhao P, Pinho e Melo T. Nontraumatic convexity subarachnoid hemorrhage: different etiologies and outcomes. J Stroke Cerebrovasc Dis. 2014;23(1):e23-e30. [DOI] [PubMed] [Google Scholar]
- 45.Mas J, Bouly S, Mourand I, Renard D, de Champfleur N, Labauge P. Focal convexal subarachnoid hemorrhage: clinical presentation, imaging patterns and etiologic findings in 23 patients. Rev Neurol. 2013;169(1):59-66. [DOI] [PubMed] [Google Scholar]
- 46.Beitzke M, Gattringer T, Enzinger C, Wagner G, Niederkorn K, Fazekas F. Clinical presentation, etiology, and long-term prognosis in patients with nontraumatic convexal subarachnoid hemorrhage. Stroke. 2011;42(11):3055-3060. [DOI] [PubMed] [Google Scholar]
- 47.Chertcoff A, Bandeo L, Pantiu F, et al. Convexity subarachnoid hemorrhage: clinical features and etiology of an Argentinian cohort. Arq Neuropsiquiatr. 2017;75(12):858-861. [DOI] [PubMed] [Google Scholar]
- 48.Linn J, Wollenweber FA, Lummel N, et al. Superficial siderosis is a warning sign for future intracranial hemorrhage. J Neurol. 2013;260(1):176-181. [DOI] [PubMed] [Google Scholar]
- 49.Zerna C, Modi J, Bilston L, Shoamanesh A, Coutts SB, Smith EE. Cerebral microbleeds and cortical superficial siderosis in patients presenting with minor cerebrovascular events. Stroke. 2016;47(9):2236-2241. [DOI] [PubMed] [Google Scholar]
- 50.Charidimou A, Imaizumi T, Moulin S, et al. Brain hemorrhage recurrence, small vessel disease type, and cerebral microbleeds: a meta-analysis. Neurology. 2017;89(8):820-829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Charidimou A, Boulouis G, Xiong L, et al. Cortical superficial siderosis and first-ever cerebral hemorrhage in cerebral amyloid angiopathy. Neurology. 2017;88(17):1607-1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wollenweber FA, Opherk C, Zedde M, et al. Prognostic relevance of cortical superficial siderosis in cerebral amyloid angiopathy. Neurology. 2019;92(8):e792-e801. [DOI] [PubMed] [Google Scholar]
- 53.Charidimou A, Boulouis G, Greenberg SM, Viswanathan A. Cortical superficial siderosis and bleeding risk in cerebral amyloid angiopathy: a meta-analysis. Neurology. 2019;93(24):e2192-e2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Calviere L, Viguier A, Patsoura S, et al. Risk of intracerebral hemorrhage and mortality after convexity subarachnoid hemorrhage in cerebral amyloid angiopathy. Stroke. 2019;50(9):2562-2564. [DOI] [PubMed] [Google Scholar]
- 55.Hemphill JC III, Greenberg SM, Anderson CS, et al. Guidelines for the management of spontaneous intracerebral hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2015;46(7):2032-2060. [DOI] [PubMed] [Google Scholar]
- 56.Klijn CJ, Schreuder FH. In patients with intracerebral haemorrhage and concomitant atrial fibrillation, optimal timing of reinitiating anticoagulants may be 7-8 weeks after ICH. Evid Based Med. 2017;22(3):108-109. [DOI] [PubMed] [Google Scholar]
- 57.Ruff CT, Giugliano RP, Braunwald E, et al. Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fibrillation: a meta-analysis of randomised trials. Lancet. 2014;383(9921):955-962. [DOI] [PubMed] [Google Scholar]
- 58.Schrag M, Mac Grory B, Nackenoff A, et al. Left atrial appendage closure for patients with cerebral amyloid angiopathy and atrial fibrillation: the LAA-CAA cohort. Transl Stroke Res. 2020;12(2):259-265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen SP, Ayata C. Novel therapeutic targets against spreading depression. Headache. 2017;57(9):1340-1358. [DOI] [PubMed] [Google Scholar]