

Abstract
Background
Kiwifruit (Actinidia Lindl.) is considered an important fruit species worldwide. Due to its temperate origin, this species is highly vulnerable to freezing injury while under low-temperature stress. To obtain further knowledge of the mechanism underlying freezing tolerance, we carried out a hybrid transcriptome analysis of two A. arguta (Actinidi arguta) genotypes, KL and RB, whose freezing tolerance is high and low, respectively. Both genotypes were subjected to − 25 °C for 0 h, 1 h, and 4 h.
Results
SMRT (single-molecule real-time) RNA-seq data were assembled using the de novo method, producing 24,306 unigenes with an N50 value of 1834 bp. Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis of DEGs showed that they were involved in the ‘starch and sucrose metabolism’, the ‘mitogen-activated protein kinase (MAPK) signaling pathway’, the ‘phosphatidylinositol signaling system’, the ‘inositol phosphate metabolism’, and the ‘plant hormone signal transduction’. In particular, for ‘starch and sucrose metabolism’, we identified 3 key genes involved in cellulose degradation, trehalose synthesis, and starch degradation processes. Moreover, the activities of beta-GC (beta-glucosidase), TPS (trehalose-6-phosphate synthase), and BAM (beta-amylase), encoded by the abovementioned 3 key genes, were enhanced by cold stress. Three transcription factors (TFs) belonging to the AP2/ERF, bHLH (basic helix-loop-helix), and MYB families were involved in the low-temperature response. Furthermore, weighted gene coexpression network analysis (WGCNA) indicated that beta-GC, TPS5, and BAM3.1 were the key genes involved in the cold response and were highly coexpressed together with the CBF3, MYC2, and MYB44 genes.
Conclusions
Cold stress led various changes in kiwifruit, the ‘phosphatidylinositol signaling system’, ‘inositol phosphate metabolism’, ‘MAPK signaling pathway’, ‘plant hormone signal transduction’, and ‘starch and sucrose metabolism’ processes were significantly affected by low temperature. Moreover, starch and sucrose metabolism may be the key pathway for tolerant kiwifruit to resist low temperature damages. These results increase our understanding of the complex mechanisms involved in the freezing tolerance of kiwifruit under cold stress and reveal a series of candidate genes for use in breeding new cultivars with enhanced freezing tolerance.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12870-021-03152-w.
Keywords: Low temperature, Kiwifruit, Freezing tolerance, Full-length transcriptome, Cold stress
Background
Low temperature is one of the main abiotic stresses affecting horticultural plant development and growth and also restricts the geographical distribution of plants [1, 2]. Because plants are sessile organisms, low temperature can lead to decreased cell membrane fluidity and water potential and loss of protein function, resulting in enormous amounts of damage [3]. However, throughout their long-term evolutionary development, plants have evolved a set of complex mechanisms to adapt to low temperatures [4]. Freezing tolerance (FT) is considered an essential ability of plants to survive low-temperature stress and is an adaptive process during which the expression of proteins related to transcriptional regulation and functional materials is induced [5, 6]. In addition, complex metabolic regulatory activity is involved in FT, and metabolites such as antioxidant compounds (SOD, ascorbic acid), osmolytes (raffinose, trehalose) and nitrogenous compounds (glutathione and glycine betaine) have positive effects on increasing FT [7–9].
Genome data are basic information that can be used to facilitate research on FT mechanisms [10]. The A. chinensis (Actinidia chinensis) genome data were published in 2013; the genome has an ~ 140× sequencing depth, and the size of the current assembly is 616.1 Mb, with a total of 39,040 genes [11]. There are recently published kiwifruit genome assemblies for A. chinensis ‘Red 5’ and Actinidia eriantha ‘White’ as well as an improved assembly of ‘Hongyang’, all of which are diploid [12–14]. However, A. arguta is a tetraploid species, and its genome information is unknown due to genome complexity resulting from the polyploidy conditions. When genome data are absent, SMRT (single-molecule real-time) RNA-seq data can act as a useful tool to substitute for genomic [15]. SMRT long-read sequencing technology is currently becoming a popular method to obtain information on full-length (FL) mRNA molecules and has been extensively used for transcriptome profiling in various plant species [16–19]. FL transcript sequences that avoid incorrect assembly can show explicit information on each gene [20]. However, the SMRT method cannot calculate the mapped number of reads to quantify the expression level of genes, but this problem can be solved with NGS (next-generation sequencing) technology [21]. Furthermore, the SMRT sequencing method has a high error rate (up to 15%) for a single base, which can be corrected with NGS reads. Hence, hybrid RNA sequencing technology is a useful tool to obtain both qualitative and quantitative transcriptome results [22].
Transcriptome analysis has become an effective technology to characterize molecular regulatory activity and investigate the genes involved in response to various abiotic stresses, such as drought stress, waterlogging stress, and salt stress [23–25]. However, studies on transcriptional level changes under cold stress in kiwifruit are limited. In model plant species (Arabidopsis), previous studies have identified and confirmed several of the key genes and transcription factors (TFs) involved in the response to low-temperature stress [26]. Among these TFs, bHLH, MYB, and AP2/ERF TFs are considered important in terms of plant cold-responsive processes [27–29]. Many studies have shown the important roles of transcription factors, including CBFs (C-repeat binding factors), ICEs (inducers of CBF expression genes), and MYB15, in cold regulation and acclimation [30]. CBFs recognize and bind to cis-elements in the promoters of COR (cold-responsive) genes, thus triggering gene expression. At present, researchers have characterized several COR genes, including ZAT, COR15a, and KIN [31]. Moreover, metabolic pathways such as those of sugar metabolism, flavonoid metabolism, and amino acid metabolism are important in the response to cold stress [32–34]. Due to limited knowledge of the complex FT mechanism in kiwifruit, we planned to identify key pathways in kiwifruit through the transcriptome method.
A. arguta, unlike other Actinidia species, has an extensive geographic distribution varying from 25°N to 50°N. Moreover, these kiwifruit species have different FTs against cold stress. Especially in northern China, A. arguta has a remarkable FT, which implies that some changes in genetic mechanisms have occurred to enhance the FT of A. arguta [35]. However, studies on the FT mechanism in A. arguta are scarce. In this study, using SMRT sequencing and NGS analysis, we investigated two kiwifruit genotypes, KL and RB, with high and low FT, respectively. The physiological parameters of both genotypes were analyzed to strongly support the transcriptome analysis results. The present study aimed to identify differentially expressed genes and pathways involved in the FT of kiwifruit. Our results will reveal useful genes to develop new freezing-tolerant cultivars, which is important for coping with growing problems under low temperature for the kiwifruit industry in the near future.
Results
Physiochemical characteristics of kiwifruit under cold stress
The shoots of two kiwifruit genotypes with contrasting freezing tolerance (KL: freezing-tolerant; RB: freezing-sensitive) were subjected to − 25 °C for 0 h, 1 h and 4 h. The anatomical structural changes of the shoots were observed under the microscope. The anatomical structure of the shoots was not significantly different under cold stress in either genotype (Fig. 1a). Due to the relationship between anatomical structure and freezing tolerance (FT), we further investigated several indices of both untreated genotypes. There were several differences in both genotypes, including in the ratio of pith, vessel density, ratio of xylem, and ratio of phloem. The ratio of pith and the ratio of xylem in KL were higher than those in RB, whereas the vessel density and ratio of phloem in RB were higher than those in KL (Fig. 1b, c). These differences suggested that geographical distribution led to anatomical structural changes in both genotypes.
Fig. 1.

Microscopy observations of kiwifruit shoot anatomical structure. (a) Images of the anatomical structure of shoots after 0 h, 1 h, and 4 h of treatment (− 25 °C) for both genotypes. (b) Anatomical structure images of untreated shoots of both genotypes. I, the ratio of pith; II, the vessel density; III, the ratio of xylem; IV, the ratio of phloem. (c) Parameters of the anatomical structure of both genotypes. The bars represent the standard errors of the means (n = 3). The asterisks indicate that the values are significantly different (** for P < 0.01)
Two physiological indices, MDA content and H2O2 content, were measured to evaluate the FT of kiwifruit under cold stress. The plasma membrane has an important role in plants; however, low temperatures always lead to lipid peroxidation of the membrane. Membrane lipid peroxidation generates a set of metabolites, one of which is MDA. The MDA content in KL and RB showed a significant change from 0 h to 4 h (Fig. 2a). The MDA content in KL increased by approximately 13% from 0 h (15.8 μmol/g) to 4 h (17.9 μmol/g) at − 25 °C, whereas that in RB increased by approximately 26% from 0 h (17.9 μmol/g) to 4 h (22.5 μmol/g) at − 25 °C. Moreover, cold stress accelerated the accumulation of H2O2 in plants. The content of H2O2 in KL increased by 2-fold from 0 h (3.4 μmol/g) to 1 h (6.9 μmol/g) and then showed no significant change at 4 h (7.4 μmol/g); the content was consistent with that at 1 h. However, the H2O2 content in RB increased by 2.3-fold from 0 h (7.9 μmol/g) to 1 h (18.2 μmol/g) and then declined at 4 h (10.5 μmol/g) (Fig. 2b). Taken together, these results indicated that the membrane stability and level of oxidative stress in kiwifruit were affected by cold stress. Moreover, the damage that KL experienced from low-temperature stress was lower than that of RB.
Fig. 2.

Comparative physiological characterization of kiwifruit shoots of two different genotypes. (a) Malondialdehyde (MDA) content in the shoots. (b) Hydrogen peroxide (H2O2) content in the shoots. The experiments included three biological replicates. Multiple comparisons were performed for significant differences (P < 0.05, two-way ANOVA and Tukey’s test; the error bars represent the SDs)
Summary of NGS and SMRT transcriptome sequencing of kiwifruit
NGS transcriptome sequencing generated 906,672,934 raw reads and 866,664,476 clean reads (95.59%, 130.01 Gb) with an average Q30 value of 93.70% and an average GC content of 45.54% (Table 1). SMRT transcriptome sequencing generated 22,604,106 subreads (32.37 Gb in size, with an average length of 1388 bp and an N50 value of 1834 bp), that were corrected using the NGS data (Fig. 3a). A total of 350,711 circular consensus sequences (CCSs) were obtained from the filtration of the subreads. The number of full-length nonchimeric (FLNC) reads was 254,139, whose average length was 1602 bp.
Table 1.
Summary of the transcriptome data from PacBio RSII platform and Illumina Hiseq platform
| Platform | |
|---|---|
| PacBio RSII | |
| Total CCS | 350711 |
| Full length reads | 271699 |
| FLNC reads | 254139 |
| Average FLNC read length | 1602 bp |
| Subreads base (G) | 32.37 Gb |
| Subreads number | 22604106 |
| Average subreads length | 1388 |
| N50 | 1834 |
| Total unigenes | 24306 |
| TF number | 1953 |
| Illumina Hiseq | |
| Raw reads (average) | 50370718.56 |
| Clean reads (average) | 48148026.44 |
| Clean base (G) | 130.01 Gb |
| Base error rate (average) | 0.03% |
| Q30 (average) | 93.70% |
| GC content (average) | 45.54% |
Fig. 3.

Overview of SMRT sequencing results and annotation of unigenes. (a) Distribution of full-length reads. (b) Overlap between the number of all unigenes according to five databases. (c) Distribution of unigene annotations based on the NR database for the species distribution statistics. (d) KOG functional classification of all unigenes
Functional annotation of unigenes
To determine the possible functions of unigenes in kiwifruit, a total of 24,306 unigenes in the SMRT transcriptome were functionally annotated using five databases (the NR, GO, KEGG, KOG, and SwissProt databases). All the unigenes had an approximately 98% annotation rate in at least one database [NR (23,796, 97.90%), GO (15,833, 65.14%), KEGG (12,201, 50.20%), KOG (15,584, 64.12%), and SwissProt (20,715, 85.23%)] (Fig. 3b).
To determine the conservation level of the unigene sequences of kiwifruit in other plant species, the unigene sequences in kiwifruit were queried in the NCBI NR database (Fig. 3c). The unigene sequences in kiwifruit displayed the highest similarity to sequences from A. chinensis (86.57%), followed by those from Camellia sinensis (2.90%), Quercus suber (2.02%), Actinidia deliciosa (0.75%), Vitis vinifera (0.30%), Actinidia kolomikta (0.19%), and A. arguta (0.19%). The results suggested that the majority of unigenes were annotated in A. chinensis.
All the kiwifruit unigenes were subsequently queried against the KOG database (Fig. 3d). A total of 15,584 sequences were annotated, with 26 functional categories. ‘Posttranslational modification, protein turnover, and chaperones’ (2593, 16.64%); ‘general function prediction only’ (2593, 16.64%); and ‘signal transduction mechanisms’ (1543, 9.90%) were the top three categories. Furthermore, ‘cell motility’ (2, 0.012%) was the smallest category with the fewest unigenes.
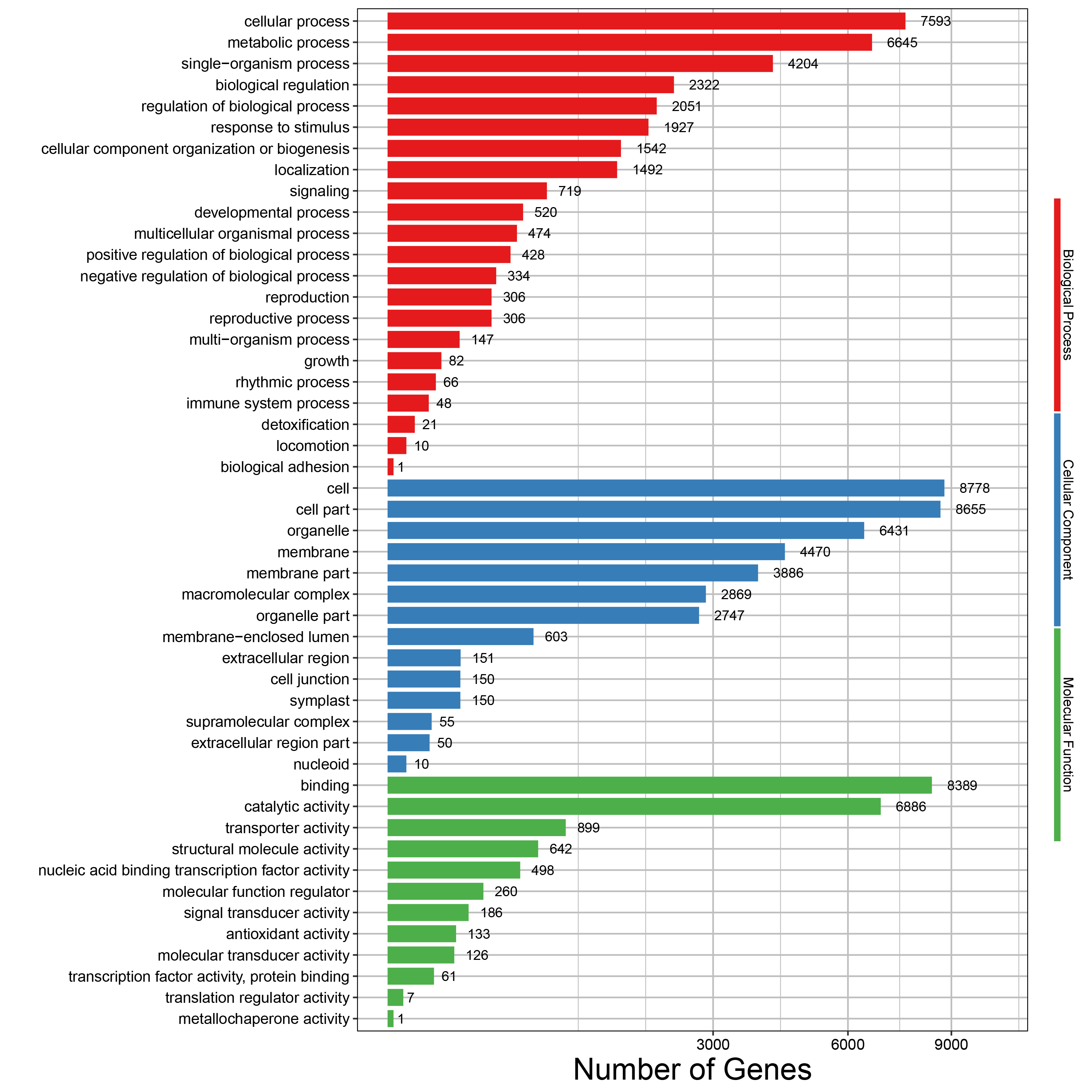
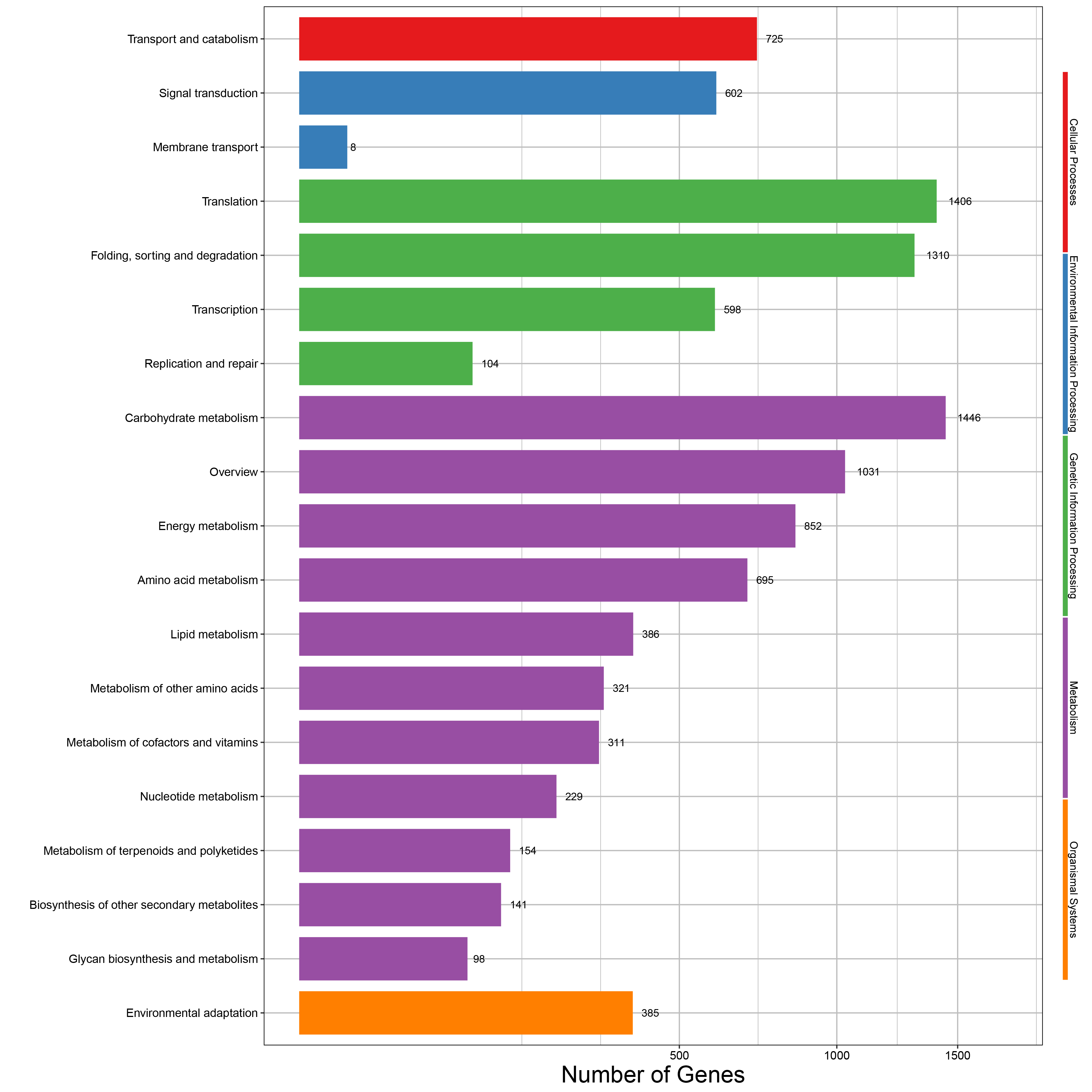
A total of 15,833 unigenes were annotated in the GO database (Fig. S1), which were successfully clustered into 48 functional groups, of which 14 categories belonged to BPs (biological processes), 22 belonged to CCs (cellular components), and 12 belonged to MFs (molecular functions). We also conducted an analysis based on KEGG pathways to obtain key information, including that on intracellular metabolic pathways and biological functions of genes in kiwifruit (Fig. S2). A total of 12,201 unigene sequences were clustered into 19 KEGG pathway categories. Moreover, the most significant category among these pathways was ‘carbohydrate metabolism’ (1446; 11.85%), followed by ‘translation’ (1406; 11.52%) and then ‘folding, sorting and degradation’ (1310; 10.73%).
Identification of DEGs
DEGs in the two contrasting genotypes were screened from a combination of NGS transcriptome data and SMRT transcriptome data. The expression levels of DEGs were compared between every two groups based on the FPKM values (Fig. 4). There were 473 DEGs (367 up- and 106 downregulated) between KL-0 h and KL-1 h, 579 DEGs (510 up- and 69 downregulated) between KL-0 h and KL-4 h, 866 DEGs (742 up- and 124 downregulated) between RB-0 h and RB-1 h, 925 DEGs (750 up- and 175 downregulated) between RB-0 h and RB-4 h, 10,300 DEGs (5028 up- and 5272 downregulated) between RB-0 h and KL-0 h, 10,539 DEGs (4980 up- and 5559 downregulated) between RB-1 h and KL-1 h, and 10,603 DEGs (5127 up- and 5476 downregulated) between RB-4 h and KL-4 h. These results indicated that the number of DEGs increased after low-temperature treatment in both genotypes. Moreover, RB exhibited more DEGs than did KL at 1 h and 4 h of treatment. For example, the number of DEGs in RB after 1 h of treatment was 866, which was almost twice that in KL DEGs (473). These differences in DEG numbers indicated that cold stress could induce more dynamic transcriptomic changes in RB than in KL and that KL had a stronger ability to maintain stable transcriptomes under cold stress. Furthermore, among the 3 time points, 4 h was the specific treatment time point that had the most abundant DEGs.
Fig. 4.
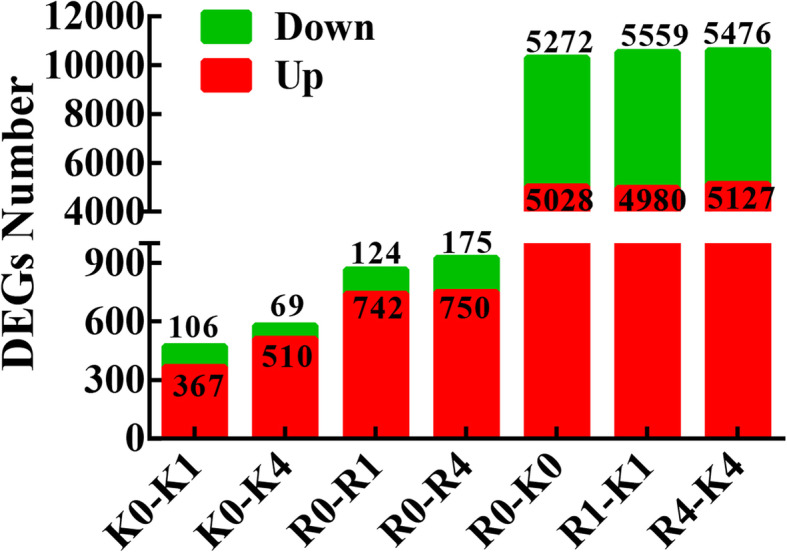
Summary of differential expression analysis of kiwifruit under cold stress. The numbers of upregulated and downregulated DEGs were identified in each comparison of different time intervals. K0, KL-0 h; K1, KL-1 h; K4, KL-4 h; R0, RB-0 h; R1, RB-1 h; R4, RB-4 h
KEGG classification of DEGs
The DEGs identified in KL between 0 h and 4 h were queried in the KEGG pathway database (Fig. 5a). KEGG pathway enrichment showed that the most significant subcategory among a series of pathways was ‘starch and sucrose metabolism’, followed by the ‘MAPK signaling pathway’, the ‘phosphatidylinositol signaling system’, ‘inositol phosphate metabolism’, and ‘arginine and proline metabolism’ in KL. However, KEGG analysis of RB between 0 h and 4 h showed that ‘starch and sucrose metabolism’ was involved in the response to cold stress (Fig. 5b). For the ‘starch and sucrose metabolism’ pathway, 11 transcripts were involved in this pathway, while most (9 out of 11) transcripts continuously had higher expression in KL than in RB. For the ‘MAPK signaling pathway’, 13 out of 18 transcripts involved in the MAPK signaling pathway had higher expression in KL than in RB. For the ‘phosphatidylinositol signaling system’ pathway, 5 of 8 transcripts had higher expression in KL than in RB. For the ‘inositol phosphate metabolism’ pathway, 2 transcripts of 4 transcripts had higher expression in KL, and for the ‘arginine and proline metabolism’ pathway, 4 of 5 transcripts had higher expression in KL (Table S1) (Fig. 5c). Taken together, these results highlighted the involvement of different pathways in the response to low-temperature stress.
Fig. 5.

KEGG enrichment of DEGs identified at 0 h vs. 4 h. (a) KEGG enrichment of DEGs between K0 (KL-0 h) and K4 (KL-4 h). The red boxes represent key pathways involved in the response to cold stress. (b) KEGG enrichment of DEGs between R0 (RB-0 h) and R4 (RB-4 h). The red boxes represent key pathways involved in the response to cold stress. (c) Heat map constructed based on the FPKM value of each gene of the key pathways
Identification of DEGs related to phytohormones and signaling
Among the DEGs, we identified several that were associated with phytohormones and calcium signaling. Seventeen DEGs involved in phytohormone pathways were selected, and their expression patterns are illustrated in Fig. 6a; these DEGs included 10 genes involved in the ethylene signaling process, 5 genes involved in the jasmonic acid signaling process, and 2 genes involved in the brassinosteroid signaling process (Table S2). For the ethylene process, 9 out of 10 genes were induced by cold stress in KL; these genes were expressed at levels higher than those in RB. The abovementioned 9 genes encoded ERF, EIN, and EBF proteins. For the jasmonic acid signaling process, 3 out of 5 genes had higher expression in KL than in RB; the above 3 genes encoded MYC2. For the brassinosteroid process, both genes had different expression levels in the two genotypes, and there were no changes in KL, whereas cold stress decreased the expression of 2 genes encoding TCH4 (xyloglucan endotransglucosylase) in RB. Moreover, a total of 96 transcripts encoded calcium signaling-related genes, including 1 CRLK (calcium/calmodulin-regulated receptor-like kinase), 14 CAMTAs (calmodulin-binding transcription activators), 22 CIPKs (CBL-interacting protein kinases), 46 CDPKs (calcium-dependent protein kinases), and 12 CNGCs (cyclic nucleotide/calmodulin-gated ion channels). Seventeen genes were significantly induced by cold stress in KL; however, 5 genes were significantly induced by low temperature in RB; the 12 genes with specific expression in KL included 1 CAMTA, 5 CIPK genes, 2 CDPK genes, and 4 CNGC genes (Fig. 6b).
Fig. 6.

Heat map of signal transduction-related genes in cold-stressed kiwifruit. (a) Heat map constructed based on hormone signal-related genes. (b) Heat map constructed based on calcium signal-related genes. Ethylene, ETH; jasmonic acid, JA; brassinolide, BR
Identification of DEGs related to starch and sucrose metabolism and measurement of sugar metabolites
A comparison between the KL-4 h and RB-4 h groups was performed. A KEGG enrichment scatterplot of the DEGs was constructed to provide a graphical presentation of the KEGG enrichment analysis results (Fig. 7a). The ‘starch and sucrose metabolism’ pathway, which was strongly enriched, was selected and presented. There were 3 significant metabolic processes in ‘starch and sucrose metabolism’: ‘cellulose degradation’, ‘trehalose synthesis’, and ‘starch degradation’. Nine DEGs were involved in the above 3 significant metabolic processes. Among these 9 functional genes, those encoding SS (sucrose synthase), CBH1 (cellobiohydrolase), beta-glucosidase (beta-GC), TPS, hexokinase, ADP-glucose pyrophosphorylase, SS (starch synthase), SBE (starch branching enzyme), and beta-amylase (BAM) were highly expressed in KL compared with RB (Fig. 7b). Therefore, we speculated that starch degradation, trehalose synthesis, and cellulose degradation played an important role in enhancing FT.
Fig. 7.

(a) KEGG enrichment of DEGs between KL-4 h and RB-4 h. The red box represents the key pathway involved in the response to cold stress. (b) DEGs involved in the starch and sucrose metabolism pathways. Gene expression is represented by the grids under the 4 h treatment, with FPKM values of both genotypes shown
The beta-GC, TPS, and BAM enzymes are key enzymes involved in ‘cellulose degradation’, ‘trehalose synthesis’, and ‘starch degradation’ processes, respectively. These enzymes were affected by cold stress but showed different changes in activity in both genotypes under cold stress (Fig. 8a, c, e). Under cold stress, beta-GC activity in KL showed an initial decrease and then an increase. However, in RB, beta-GC activity showed no significant change. Cold stress caused an increase in TPS activity. In KL, TPS activity remained relatively high; in contrast, cold stress suppressed TPS activity in the cold period in RB. BAM activity increased under cold stress in KL, whereas it decreased in RB. In addition, the BAM activity in KL was higher than that in RB throughout the measured period.
Fig. 8.

Changes in beta-GC (a), soluble sugar (b), TPS (c), trehalose (d), beta-amylase (e), and maltose (f) contents in the shoots of both genotypes under cold stress
The level of soluble sugar in kiwifruit shoots was also measured. Cold stress increased the soluble sugar content throughout the cold treatment in KL, and there was no significant difference in the soluble sugar content between the 4 h and 0 h treatments in RB (Fig. 8b). As shown in Fig. 8d, KL showed increased trehalose content at 4 h, whereas RB showed no significant difference throughout the cold treatment. Under cold stress, cold increased the maltose content in KL but decreased it in RB (Fig. 8f). These results indicated that kiwifruit actively stimulated a set of starch metabolic pathways to enhance FT to resist freezing damage during the low-temperature treatment periods. In addition, low temperature may promote soluble sugar, trehalose and maltose accumulation under cold stress to increase FT.
TFs in response to low-temperature stress
Transcription factors play a key role in cold stress and are involved in transcriptional regulatory networks. A total of 20 categories of TF families were identified (Fig. 9a). The DEGs encoding most of the TFs were involved in the low-temperature response, and the majority belonged to the ERF, bHLH, and MYB families. Members of 3 kinds of TF families have been previously identified to regulate FT. Consequently, DEGs derived from three transcription factor families (ERF, bHLH, and MYB) are shown in Fig. 9b, c, d. The 16 TFs (Table S3) were screened, 5 of which belonged to the ERF, 5 belonged to the bHLH, and 6 belonged to the MYB families. The 5 AaERF genes and 5 AaMYC genes had higher expression in KL than in RB, and 6 AaMYB genes had lower expression in KL than in RB. In addition to the abovementioned ERF, bHLH, and MYB family members, 4 genes belonging to the C2H2 family encode zinc finger proteins (ZAT6, ZAT10, and ZAT12), which had higher expression in KL than in RB; other members of TFs, including 1 HSF gene, 1 NAC gene, and 1 WRKY gene, were also significantly increased upon cold stress (Table 2).
Fig. 9.

Number and classification of TF-encoded genes among the unigenes (a). Heat map constructed based on FPKM expression values of ERFs (b), MYCs (c), and MYBs (d)
Table 2.
FPKM values of differentially expressed TF in both genotypes
| ID | Name | K0 | K1 | K4 | R0 | R1 | R4 |
|---|---|---|---|---|---|---|---|
| MWXS20249_01mi_transcript_23086 | ZAT6 | 27.10 | 127.74 | 169.98 | 6.75 | 15.77 | 17.10 |
| MWXS20249_01mi_transcript_23199 | ZAT10 | 11.69 | 97.55 | 162.67 | 3.30 | 68.27 | 62.87 |
| MWXS20249_01mi_transcript_24340 | ZAT10 | 61.80 | 259.50 | 362.35 | 44.81 | 181.13 | 163.13 |
| MWXS20249_01mi_transcript_25307 | ZAT12 | 0.81 | 14.02 | 30.38 | 0.00 | 0.00 | 0.00 |
| MWXS20249_01mi_transcript_2738 | HSF | 4.87 | 11.46 | 19.73 | 3.87 | 13.35 | 11.02 |
| MWXS20249_01mi_transcript_8860 | NAC | 2.97 | 14.45 | 15.23 | 1.27 | 3.78 | 3.33 |
| MWXS20249_01mi_transcript_4511 | WRKY | 9.24 | 19.70 | 29.89 | 6.20 | 12.51 | 14.13 |
Construction of WGCNA networks and identification of candidate genes in kiwifruit
To determine the correlation between samples and modules, we constructed a gene coexpression network using RNA-seq data. A network heatmap (interconnectivity plot) of the gene network is shown, which displays the resulting modules and corresponding hierarchical clustering dendrograms (Fig. 10a). Seven modules containing 60–251 DEGs each were identified. MDA, H2O2, beta-GC, soluble sugar, TPS, trehalose, BAM, and maltose contents were used as phenotypic data for calculating the relationships in the trait modules. The results of the correlation between the modules and starch-sucrose pathway indicated that the correlation values ranged from − 0.85 to 0.92 (Fig. 10b). The eigengenes of the blue modules showed significant correlations with the starch-sugar pathway. The heat map of the blue module showed that FPKM values were higher in KL than in RB (Fig. 10c). To further identify key candidate genes in the blue module, the hub genes in the blue module were calculated using Cytoscape software (Table S4). The results of the hub gene search showed that the connections of the sugar-related and transcription factor-regulated DEGs were in the blue module, and sugar-related genes were highly coexpressed with transcription factor-regulated genes (Fig. 10d).
Fig. 10.

Identification of coexpression network modules in kiwifruit. (a) Cluster dendrogram and heat map of genes subjected to coexpression module calculations. (b) Module-trait associations based on Pearson correlations. The color key from green to red represents r2 values from − 1 to 1, respectively. (c) Heat map constructed for the blue module genes. (d) Gene network of the blue module, which is positively correlated with beta-GC (r2 = 0.61), trehalose (r2 = 0.7), and BAM (r2 = 0.92) contents. The candidate genes within the blue module are highlighted in red due to the higher weight within the module and coded for gene descriptors based on annotations
Validation of candidate genes by qRT-PCR
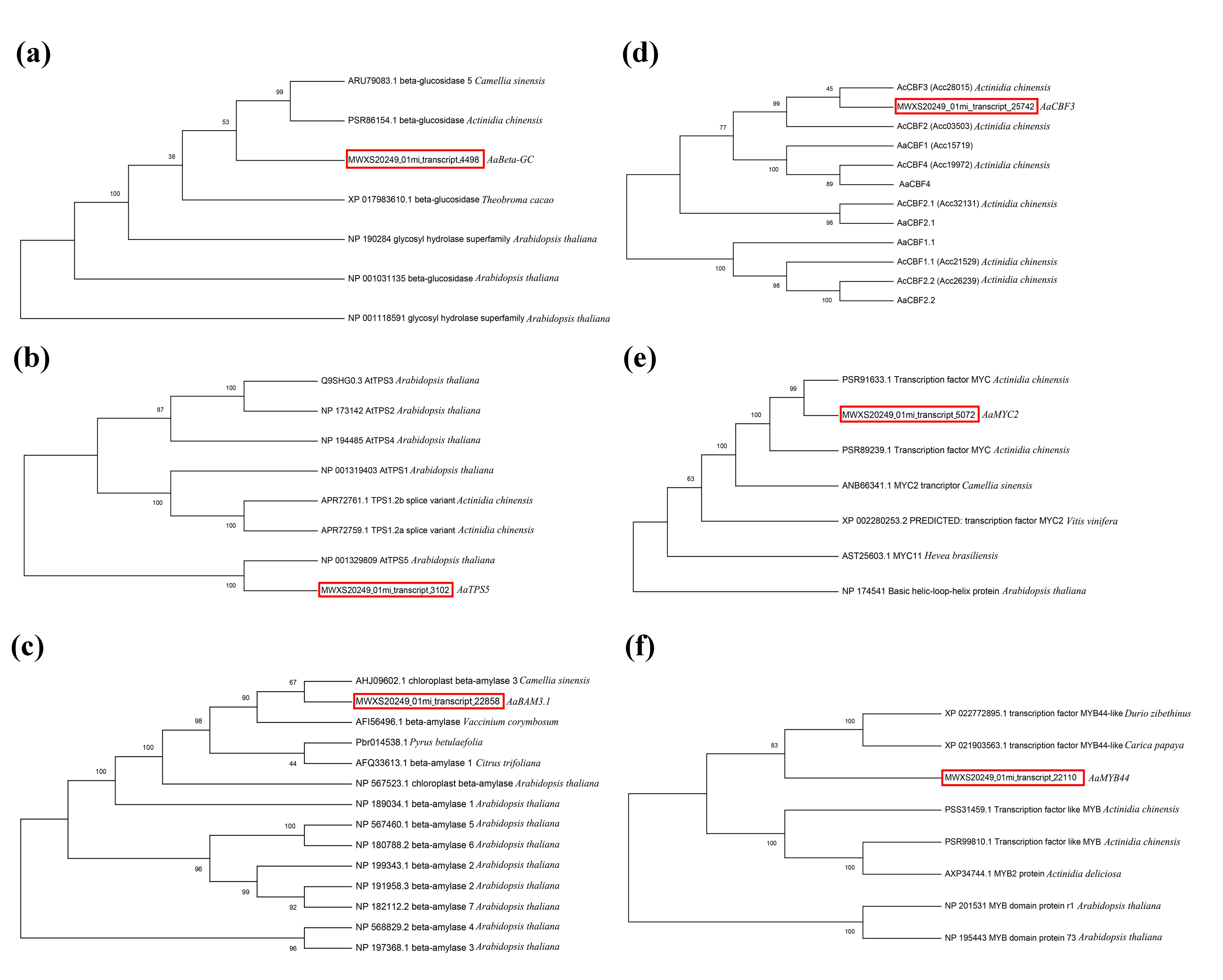
For further validation of the RNA-seq results and candidate genes linked to starch and sucrose metabolism, we selected 9 DEGs involved in ‘starch and sucrose metabolism’ and 3 DEGs identified as potential candidate TFs for qRT-PCR experiments. While the fold-change of gene expression levels measured by qRT-PCR and RNA-seq were different, the expression patterns of the majority of the 12 DEGs assessed by the two methods showed similar trends, which validated the reliability of the RNA-seq results (Fig. 11). Moreover, these results confirmed that beta-GC, TPS5, BAM3.1, CBF3, MYC2, and MYB44 were potential candidate genes involved in the regulation of FT (Fig. S3).
Fig. 11.

qRT-PCR verification of the expression pattern according to the RNA-seq data. The relative expression levels were calculated according to the 2-∆∆CT method, with the actin reference gene serving as a control
Discussion
Kiwifruit is not only an important fruit species but also an economically important plant species worldwide. Cold stress is an abiotic stress that harms the development of kiwifruit and limits its yield. To enhance FT, it is essential to explore the FT mechanism of kiwifruit. However, the ploidy of kiwifruit is comparatively complex and leads to complex genomic information, and the breeding of cultivars with high FT is obstructed by the absence of functional genomic data. We therefore used a hybrid sequencing method combining both SMRT and NGS sequencing to determine the regulatory network of gene expression and induction in shoots of kiwifruit under cold stress. Moreover, physiological and transcriptome analyses were carried out to determine the FT responses in kiwifruit in this study.
Involvement of starch and sucrose metabolism in high freezing tolerance
The enzymes and genes participating in starch degradation play an important role in the response to cold stress [36, 37]. In our study, a set of key genes associated with starch and sucrose metabolism were screened, and their expression levels were determined under cold stress. A majority of the identified genes were induced under cold stress, showing that cellulose degradation, trehalose accumulation and starch degradation were activated during this period, which may be important for the improvement of FT of kiwifruit (Fig. 8). Beta-glucosidase is a rate-limiting enzyme involved in the hydrolysis of cellulose [38]. Some beta-glucosidases can affect the structure of the cell wall, which might play a key role in cell adaptations to the physical deformations caused by cold stress [39]. After cold acclimation, A. thaliana plants with a mutation in sfr2–1, which encodes beta-glucosidase, are more sensitive to FT than the wild type is [40]. Under various abiotic stresses, beta-glucosidases hydrolyze inert precursors to release antioxidant substances [41]. A study on the transcriptional analyses of chickpea showed that the beta-glucosidase gene was screened to act as a differentially expressed gene under cold stress and that its expression was induced in response to low temperature [42]. Beta-glucosidase BGLU10 may be involved in cold, salt, and drought stress in A. thaliana [43]. In our study, the expression of the beta-glucosidase gene was upregulated under cold stress, and the expression of the beta-glucosidase gene in KL was higher than that in RB. Moreover, beta-glucosidase activity was activated, and the soluble sugar content also increased under cold stress.
Additionally, trehalose, a multifunctional disaccharide, exists extensively in various plant species [44, 45]. Trehalose forms a special protective structure on the surface of the cell membrane, protects the activity of functional protein molecules, and thus resists multiple abiotic stresses [46]. Both trehalose-6-phosphate and trehalose-phosphate-phosphatase play an important role in the synthesis of trehalose. Rice plants overexpressing OsTPS1 and OsTPP1 can acquire higher FT [47, 48]. TPS is the crucial controller, and there are 11 copies of the TPS gene in A. thaliana. Transgenic plants of A. thaliana overexpressing TPP and TPS increased the tolerance ability under low-temperature and salt stress [49]. Exogenous trehalose can induce the expression of stress-related genes in plants [50]. In this study, TPS genes were isolated and upregulated under cold stress. Moreover, OsICE1 can serve as a positively regulated TF to activate the expression of OsTPP [51]; likewise, we identified that both MYC2 and TPS were induced under cold stress in KL.
BAM is an important hydrolase that degrades starch to yield maltose and has a key effect on FT improvement [52]. The expression and activity of BAM were reported to be significantly induced after a 6 h treatment of cold stress in A. thaliana [53]. In kiwifruit, AaBAM3 was upregulated to increase the activity of beta-amylase from autumn to winter in China [54]. In pear, researchers have found the same results as those in kiwifruit [55]. Interestingly, in Poncirus trifoliata, there is a CBF-binding element in the promoter of PtrBAM1, and CBF can regulate the expression of PtrBAM1, which plays a role in FT by regulating the sugar metabolite content [56]. In our study, the results of kiwifruit were similar to those of other plant species, and BAM was upregulated under cold stress. In general, the abovementioned 3 genes may be key genes that enhance the FT of KL.
Pathways involved in cold signaling
The phosphatidylinositol signaling system and the inositol phosphate metabolic pathways have been reported to be associated with FT [57, 58]. The significant KEGG pathways during the 4 h treatment were enriched in both of these pathways in KL but not in RB (Fig. 5a, b). These results of the KEGG analyses showed that the gene expression levels of the different genotypes were different under low-temperature stress. Inositol 3-phosphate (IP3), which is generated by phospholipase C-mediated hydrolysis of phosphatidylinositol bisphosphate (PIP2), triggers a set of cellular processes by releasing calcium. These releases can increase calcium from low to high levels. Calcium also acts as a secondary messenger to transduce cold signals. In A. thaliana, the phosphatidylinositol-hydrolyzing phospholipase gene was upregulated under multiple abiotic stresses [59, 60]. Moreover, myo-inositol-3-phosphate synthase activity was shown to be activated during the ABA signaling response in Spirodela polyrhiza [61]. Overexpression of D-myo-inositol-3-phosphate synthase leads to elevated levels of inositol in Arabidopsis under salt stress [62]. In our study, three transcripts (MWXS20249_01mi_transcript_10951, MWXS20249_01mi_transcript_11392, MWXS20249_01mi_transcript_11458) encoding myotubularin phosphatases were identified; these proteins subject phosphatidylinositol to different phosphorylation patterns, resulting in the production of distinct phosphatidylinositol phosphates. The genes encoding myotubularin phosphatases were induced under cold stress to increase FT. Inositol-1,3,4-trisphosphate 5/6-kinase (MWXS20249_01mi_transcript_11353) is involved in inositol phosphate metabolism, which can also trigger signal transduction. Hence, KL might enhance its FT by activating genes related to both the phosphatidylinositol signaling system and the inositol phosphate metabolic pathways.
Plant hormones play an important role in regulating stress responses and activating downstream low-temperature pathways. ETH, JA, and BR hormones are vital stress hormones involved in the modulation of the responses to cold stress. We noticed that several ETH signaling-related genes, such as ERF, EIN, and EBF, were upregulated during cold stress in the freezing-tolerant genotype (KL). Both the stress-responsive JA signaling-related genes JAZ and MYC were induced under cold stress in KL (Fig. 6a); relatedly, PtrMYC2 of Poncirus trifoliata integrates JA signals to modulate cold-induced GB accumulation by directly regulating the expression of PtrBADH-l, a betaine aldehyde dehydrogenase (BADH)-like gene [9]. BR signaling is also the key pathway controlling the response to cold stress, and brassinazole-resistant 1 (BZR1) was shown to be upregulated under cold stress in Poncirus trifoliata to enhance its FT [63]. Furthermore, the TCH4 gene acts downstream of BZR1 [64]. In our study, we identified two TCH4 genes, and these genes were repressed under cold stress in RB; however, no change in their expression was found in KL. Both TCH4 genes might be the key reason for the decrease in FT of the sensitive genotype (RB).
The members of several kinase families are involved in cold signal transduction, including CDPK, CIPK, CRLK, CAMTA, and CNGC. The members of these gene families play an important role in regulating FT. In our study, we identified several DEGs, including 2 CDPK genes, 5 CIPK genes, 1 CAMTA gene, and 4 CNGC genes, indicating that the FT of kiwifruit might be enhanced through the Ca2+ signaling pathway. Interestingly, recent research in rice showed that cyclic nucleotide-gated channel (OsCNGC9) positively regulates chilling tolerance by mediating cytoplasmic calcium elevation [65]. In KL, 4 GNGC genes had higher expression than that in RB. These GNGC genes may be candidate genes for explaining the difference in FT between the genotypes.
The MAPK signaling pathway was also significantly induced, showing that this pathway is associated with cold stress responses [66]. Calcium/calmodulin binds to CRLK1 and activates it, after which CRLK1 interacts with MEKK1 (mitogen-activated protein kinase kinase kinase 1), leading to MAPK activation and freezing tolerance. MAPK cascades transduce cold signals into intracellular responses using various classes of protein kinases. With respect to kinases, some genes encoding MPKs (mitogen-activated protein kinases) were shown to be upregulated in response to low temperature in A. thaliana [67]. Some researchers identified another MAP3K that played an important role in cold stress signal transduction to enhance FT in Poncirus trifoliata [68]. In our study, a transcript (MWXS20249_01mi_transcript_12622) encoding MPK3/6 was identified, and its expression increased under cold stress. Cold-activated MPK3/6 phosphorylates ICE1 proteins to improve their stability and transcriptional activity, which consequently regulates CBF expression in and FT of plants.
Transcriptional regulatory pathways involved in the cold response
TFs, e.g., AP2/ERF, bHLH, and MYB, are differentially expressed at low temperature and have been reported to be associated with FT in A. thaliana. Moreover, the members of the AP2/ERF TF family were the most abundant TFs according to our data. The CBF gene belongs to the AP2/ERF family; this gene can induce the expression of cold-responsive genes (CORs) that are involved in multiple abiotic stress responses [69]. Previous studies have indicated that AtCBF1, 2, and 3 are responsive to cold stress in A. thaliana. Overexpression of the CBF gene was shown to activate the expression of COR genes and increase the FT of plants. In our study, the CBF1 gene was identified as a candidate gene for enhancing the FT of KL, and its upstream regulated genes, including CAMTA, EBF, EIN, ZAT10, and ZAT12, were also induced under cold stress in KL (Table 2). Members of the bHLH family also have a positive function in enhancing FT. bHLH TFs, which act upstream of CBF, can bind to the G/E-box motif of the CBF gene and subsequently improve FT by activating COR genes [70, 71]. Previous studies have shown that overexpression of ICE improves the FT of A. thaliana. According to our data, five MYC2 genes belonging to the bHLH family were upregulated in KL, whereas there were no significant changes in RB (Fig. 9c). MYB transcription factors may play an important role in the low-temperature response and changes in the expression levels of COR genes, affecting the physiological response to resist adverse environmental conditions. For example, AtMYB15 is a transcription regulatory gene that negatively regulates the FT of Arabidopsis [72]. MYBs were differentially expressed between KL and RB under cold stress, implying a key role of MYB genes in the low-temperature response under cold stress (Fig. 9d). Overall, TFs are crucial mediators in regulating the expression of COR genes to affect the FT of kiwifruit.
WGCNA of the gene network regulation of the FT mechanism
Freezing tolerance is the result of a complex biochemical pathway that is involved in the regulation of multiple genes; therefore, a single gene is not enough to explain FT. Genes involved in multiple metabolic pathways are usually regulated through coordinated expression, so correlation models are often used to identify gene coexpression networks [73, 74]. KEGG analysis and TF analysis revealed several candidate genes that respond to cold stress in kiwifruit in our study. We obtained 7 gene expression modules via WGCNA, of which one module was strongly correlated with starch-sucrose metabolism. The blue module from the WGCNA correlated significantly with beta-GC, TPS, and BAM. Through hub gene searching in the blue module, beta-GC, TPS5, BAM3.1, CBF3, MYC2, and MYB44 were screened as key genes. These results indicated that the above 6 candidate genes were key players in the cold response in kiwifruit. In general, the overview of these genes using coexpression networks would strengthen our understanding of coordinated regulation of multiple genes involved in the FT of kiwifruit.
Based on our results, we hypothesized that when kiwifruit are subjected to cold stress, various metabolites act as low-temperature sensors, after which cells release calcium through phosphatidylinositol signaling and inositol phosphate metabolism. Calcium interacts with CRLK to transduce cold signals through the MAPK signaling pathway, and then the MAPK signaling pathway triggers signal transduction via TFs such as ERF, bHLH, and MYB TFs. Moreover, plant hormones including ETH, JA, and BRs also serve as signaling molecules to regulate COR gene expression. Subsequently, the starch-sucrose pathway, which regulates osmolyte and redox balance, and the production of COR genes, including beta-GC, TPS, and beta-amylase, through activation of the CBF pathway and non-CBF pathway, in turn enhance FT. Overall, the accumulation of soluble sugars, trehalose, and maltose plays an important role in resisting adverse temperature conditions in kiwifruit. A diagram explaining the process involving kiwifruit FT is shown in Fig. 12.
Fig. 12.

Putative candidate model of gene involvement in the FT of kiwifruit. Phosphatidylinositol phosphate, PIP; phosphatidylinositol bisphosphate, PIP2; inositol 3-phosphate IP3; ethylene, ETH; jasmonic acid, JA; brassinolide, BR
Our results indicated that signaling transduction pathway and starch and sucrose pathway were key pathways to regulate kiwifruit freezing tolerance and that key genes involved in these pathways might lead to different FT between freezing tolerant genotype and sensitive genotype. However, one limitation in our study should also be considered: keeping in mind the limited number of samples (two samples) in current study, comparative transcriptome of two genotypes might lead to identify fake DEGs which were not related to freezing adaption but to different genetic background. Therefore, firstly, the two materials were compared with their respective untreatment materials (control) to sort out low temperature induced transcripts when we screened the differential transcripts. Secondly, transcriptome is a feasible method to identify key genes, the qRT-PCR results of key candidate genes validate authentic DEGs. Moreover, it is better method to find key genes through genome-wide association study (GWAS) or bulk-sequencing-analysis (BSA) after the genome of A. arguta is sequenced. In future studies, larger and more diverse samples should be investigated to further identify key DEGs. Additionally, the relationship of TFs and 3 sub-pathways including starch degradation, trehalose synthesis and cellulose degradation need to be clearly confirmed. Future studies also need to focus on how certain changes in the hub gene can affect other pathways or traits.
Conclusions
In the present study, an integrated transcriptome profile of contrasting kiwifruit genotypes under low temperature was analyzed via RNA-seq combined with SMRT and NGS technologies to reveal the FT mechanism. We identified the following genes in response to low temperature: (1) starch and sucrose metabolism-related genes (beta-GC, TPS5, BAM3.1) and (2) transcription factor genes (CBF3, MYC2, MYB44). Additionally, the ‘phosphatidylinositol signaling system’, ‘inositol phosphate metabolism’, ‘MAPK signaling pathway’, ‘plant hormone signal transduction’, and ‘starch and sucrose metabolism’ processes were involved in the cold response. These processes might play an important role in the responses to low temperature in kiwifruit. Our results provide novel viewpoints on a set of molecular mechanisms of kiwifruit FT. In future work, we will concentrate on the functional verification of the structural genes beta-GC, which is related to cellulose degradation, TPS, which is related to trehalose synthesis, and BAM, which is related to starch degradation, and explore how they work in response to cold stress.
Methods
Plant materials and cold treatment
Two A. arguta (Sieb. et Zucc.) Planch. et Miq. genotypes, ‘Kuilv male’ (KL) and ‘Ruby-3’ (RB), were collected in the national kiwifruit germplasm repository of Zhengzhou Fruit Research Institute, CAAS, Henan Province, China. KL originated from Jilin Province (125°E, 44°N; China), and RB originated from Henan Province (113°E, 34°N; China). Tissue culture-generated seedlings were planted in 3-L pots and then grown for 3 years. One-year-old shoots were collected in early January 2020, and then the detached shoots were put into a freezer in darkness. Three biological replicates of shoots for RNA sequencing (RNA-seq) were collected randomly from all plants of each genotype under − 25 °C treatment at 0 h, 1 h, and 4 h. At the same time, shoots were collected for measurement of physiological indices. The collected samples were frozen immediately in liquid nitrogen and stored in a − 80 °C freezer for further study.
Histological, physiological and biochemical analyses
The anatomical structure of the shoots was observed by the paraffin wax method for both genotypes [75]. In brief, the shoot samples were immediately submerged in FAA (5% formaldehyde:6% acetic acid:63% alcohol:26% H2O) fixative solution for 72 h. The shoot samples were then gradually dehydrated using an alcohol gradient. Later, the shoot sections were embedded in liquid paraffin wax (temperature 56–58 °C). The shoot sections (15 μm) for optical microscopy observations were sliced by a microtome. Each time point was represent by at least five images per sample.
The activities of beta-glucosidase (beta-GC), trehalose-6-phosphate synthase (TPS), and beta-amylase (BAM) and the contents of malondialdehyde (MDA), hydrogen peroxide (H2O2), soluble sugars, maltose, and trehalose were measured using corresponding assay kits according to the manufacturer’s instructions (Suzhou Comin Bioengineering Co., Ltd., Suzhou, China). The experiments involved three biological replicates.
RNA extraction and RNA-seq
The samples were subjected to RNA extraction using an RNA Isolation Kit (Waryoung, China). Both the quality and concentration of the RNA samples were subsequently measured using an Agilent 2100 Bioanalyzer (Agilent Technologies, USA), a Qubit 3.0 Fluorometer (Invitrogen, USA), and a NanoDrop 2000 spectrophotometer (NanoDrop Products, USA). For Illumina libraries, the total RNA was subjected to mRNA purification using magnetic beads linked with oligo (dT) beads. The mRNA molecules were fragmented into short fragments, the short fragments were used as templates to synthesize cDNA, and the library was constructed. The library was subjected to 150 bp paired-end sequencing using the NGS tool of the Illumina HiSeq 2500 platform (Illumina Inc., USA). For PacBio libraries, cDNA was prepared using a SMRTer PCR cDNA Synthesis Kit (Clontech, USA), and PacBio libraries were constructed according to the PacBio protocol (http://www.pacb.com/wp-content).
Data processing
The short-read data were obtained via NGS, and raw reads were filtered by removing adapter reads, poly-N reads and low-quality reads to obtain clean reads [63]. The PacBio sequencing results were analyzed using the SMRT Pipe analysis workflow of the PacBio SMRT Analysis software suite (http://www.pacb.com/products-and-services/analytical-software/smrt-analysis). In brief, to obtain high-quality reads, the SMRT sequencing results were filtered by removing polymerase reads with a length of < 50 bp and a quality of < 0.75. The high-quality reads were subsequently chosen to obtain error-corrected circular consensus sequences (CCSs) for which the full passes were ≥ 0 and the quality was > 0.75. The CCSs were regarded as full-length (FL) transcripts when the 5′ primer sequence, 3′ primer sequence, and a poly-A tail were present. The artificial chimeric sequences were produced by direct linkages of two cDNA template strands due to the low concentrations of adapters or SMRTbell, whereas nonchimeric sequences in the FL sequence were considered to be FL-nonchimeric (FLNC) sequences. The FLNC transcripts were identified by searching for 5′ tail cDNA primers and poly-A tail signals in the CCSs. FLNC transcripts with a high-quality base were obtained for subsequent analysis.
Functional annotations of transcripts and identification of DEGs
Due to the absence of a reference genome for tetraploid kiwifruit, a reference transcriptome was assembled de novo from SMRT transcript data. Unigene sequences were annotated in various databases, and annotation information of the unigenes was obtained for those with an E-values of at least 1e-5. Five databases were used: the nonredundant protein sequence (NR) database, Gene Ontology (GO) database, Eukaryotic Ortholog Groups (KOG) database, Homologous Protein Family (Pfam) database, and Kyoto Encyclopedia of Genes and Genomes (KEGG) database. iTAK was used to predict plant transcription factors (TFs), and the TF family classification used by the database was adopted to identify TFs by hmmscan. The DESeq tool (R package version 1.10.1) was used to identify the DEGs (differentially expressed genes) between different comparison groups, and filtered standards were adjusted according to P-value < 0.05 and |log2 (fold-change)| ≥1. Via KEGG and GO databases, the enriched DEGs were used to identify significant metabolic pathways, and the filtered standard was an FDR-adjusted P-value of < 0.05.
Validation and expression analysis of DEGs by quantitative reverse transcription-PCR (qRT-PCR)
First-strand cDNA was synthesized from a total of 2 μg of template RNA using a ReverTra Ace qPCR RT Kit (Toyobo, Osaka, Japan) according to the manufacturer’s instructions. qRT-PCR was subsequently performed on a LightCycler 480 real-time PCR system (Roche, Basel, Switzerland) using 2x SYBR Green I Master Mix (Asbio Technology, Inc.) and the first-strand cDNA template. The qRT-PCR protocol was as follows: 95 °C for 5 min, followed by 45 cycles of denaturation at 95 °C for 10 s, annealing at 60 °C for 20 s, and extension at 72 °C for 20 s. The relative expression levels of each sample were calculated in relation to the expression of the reference gene β-actin using the 2-∆∆CT method. Three independent biological replications were included. All the primers used in this study are shown in Table S5.
Gene coexpression network analysis and network visualization
Weighted gene coexpression network analysis (WGCNA) was performed using the WGCNA package (R package version 1.69) with the default parameters. Using the FPKM values of the DEGs, an adjacency matrix was constructed. The resulting adjacency matrix was then converted into a topological overlap matrix (TOM). A dynamic hybrid tree-cut algorithm (via the R package dynamicTreeCut, v.1.63–1) was used to detect modules, with the default settings. The phenotypic data were used to identify the related modules, and a correlation matrix between phenotypes and gene modules was constructed using the WGCNA package. The genes from the related module were visualized using Cytoscape (v.3.8.2).
Statistical analyses
Two-way analysis of variance (ANOVA) accompanying Tukey’s test was performed for all data using SPSS 22 (IBM, Inc., Armonk, NY, USA) and Prism 7 (GraphPad Software, Inc., La Jolla, CA, USA). Each experiment was repeated at least three times independently, and Pearson’s one-tailed test was conducted at the P < 0.05 significance level.
Supplementary Information
{kind=link}
Additional file 1 Supplementary Fig. 1. GO functional classifications of all the unigenes.
{kind=link}
Additional file 2 Supplementary Fig. 2. KEGG functional classifications of all the unigenes.
{kind=link}
Additional file 3 Supplementary Fig. 3. Phylogenetic tree of candidate genes and homologous genes. (a) Beta-GC, (b) TPS5, (c) BAM3.1, (d) CBF3, (e) MYC2, and (f) MYB44.
Additional file 4 Table S1. DEGs related to Starch and sucrose metabolism, MAPK signaling pathway, Phosphatidylinositol signaling system, Inositol phosphate metabolism, Arginine and proline metabolism.
Additional file 5 Table S2. DEGs related to hormone and calcium signal.
Additional file 6 Table S3. DEGs related to TFs.
Additional file 7 Table S4. Top 100 hub genes in the blue module.
Additional file 8 Table S5. Primers used for quantitative real-time RT-PCR analysis.
Acknowledgements
Not applicable.
Authors’ contributions
SS, ML, XQ, JC, HG, YZ, LS, AM and DB conducted the experiments; JF organized and supervised the overall project; SS and ML performed the data analysis and wrote the manuscript writing; and CH edited the manuscript. All authors have read and approved the manuscript.
Funding
This study was funded by the National Key Research and Development Project of China (Grant No. 2019YFD1000200); the National Science Foundation of China (Grant No. 31801820); the Special Engineering Science and Technology Innovation, Chinese Academy of Agricultural Sciences (Grant No. CAAS-ASTIP-2015-ZFRI); and the Modern Agricultural Industrial Technology System of Henan Province (Grant No. S2014–11). The funders had no role in study design, collection, analysis, and interpretation of data and in writing the manuscript, but just provide the financial support.
Availability of data and materials
The raw data generated in this study have been deposited in the NCBI Short Read Archive (SRA) under the accession number PRJNA681641. All other relevant data contained within the paper are available in the paper and Supplementary Files.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflicts of interest concerning this work.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Shihang Sun and Miaomiao Lin contributed equally to this work.
Contributor Information
Chungen Hu, Email: chungen@mail.hzau.edu.cn.
Jinbao Fang, Email: fangjinbao@caas.cn.
References
- 1.Sun XM, Zhang LL, Wong DCJ, Wang Y, Zhu ZF, Xu GZ, Wang QF, Li SH, Liang ZC, Xin HP. The ethylene response factor VaERF092 from Amur grape regulates the transcription factor VaWRKY33, improving cold tolerance. Plant J. 2019;99(5):988–1002. doi: 10.1111/tpj.14378. [DOI] [PubMed] [Google Scholar]
- 2.Zhang LL, Zhao TT, Sun XM, Wang Y, Du C, Zhu ZF, Gichuki DK, Wang QF, Li SH, Xin HP. Overexpression of VaWRKY12, a transcription factor from Vitis amurensis with increased nuclear localization under low temperature, enhances cold tolerance of plants. Plant Mol Biol. 2019;100(1–2):95–110. doi: 10.1007/s11103-019-00846-6. [DOI] [PubMed] [Google Scholar]
- 3.Raju SKK, Barnes AC, Schnable JC, Roston RL. Low-temperature tolerance in land plants: are transcript and membrane responses conserved? Plant Sci. 2018;276:73–86. doi: 10.1016/j.plantsci.2018.08.002. [DOI] [PubMed] [Google Scholar]
- 4.Wisniewski M, Nassuth A, Arora R. Cold hardiness in trees: a Mini-review. Front Plant Sci. 2018;9:1394. doi: 10.3389/fpls.2018.01394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shen JZ, Zhang DY, Zhou L, Zhang XZ, Liao JR, Duan Y, Wen B, Ma YC, Wang YH, Fang WP, Zhu X. Transcriptomic and metabolomic profiling of Camellia sinensis L. cv. 'Suchazao' exposed to temperature stresses reveals modification in protein synthesis and photosynthetic and anthocyanin biosynthetic pathways. Tree Physiol. 2019;39(9):1583–1599. doi: 10.1093/treephys/tpz059. [DOI] [PubMed] [Google Scholar]
- 6.Singh AK, Dhanapal S, Yadav BS. The dynamic responses of plant physiology and metabolism during environmental stress progression. Mol Biol Rep. 2020;47(2):1459–1470. doi: 10.1007/s11033-019-05198-4. [DOI] [PubMed] [Google Scholar]
- 7.Yue C, Cao HL, Wang L, Zhou YH, Huang YT, Hao XY, Wang YC, Wang B, Yang YJ, Wang XC. Effects of cold acclimation on sugar metabolism and sugar-related gene expression in tea plant during the winter season. Plant Mol Biol. 2015;88(6):591–608. doi: 10.1007/s11103-015-0345-7. [DOI] [PubMed] [Google Scholar]
- 8.He Z, Zhao T, Yin Z, Liu J, Cheng Y, Xu J. The phytochrome-interacting transcription factor CsPIF8 contributes to cold tolerance in citrus by regulating superoxide dismutase expression. Plant Sci. 2020;298:110584. doi: 10.1016/j.plantsci.2020.110584. [DOI] [PubMed] [Google Scholar]
- 9.Ming R, Zhang Y, Wang Y, Khan M, Dahro B, Liu J-H. The JA-responsive MYC2-BADH-like transcriptional regulatory module in Poncirus trifoliata contributes to cold tolerance by modulation of glycine betaine biosynthesis. New Phytol. 2021;229(5):2730–50. [DOI] [PubMed]
- 10.Wang Y, Xin H, Fan P, Zhang J, Liu Y, Dong Y, et al. The genome of Shanputao (Vitis amurensis) provides a new insight into cold tolerance of grapevine. Plant J. 2021;105(6):1495–506. [DOI] [PubMed]
- 11.Huang SX, Ding J, Deng DJ, Tang W, Sun HH, Liu DY, Zhang L, Niu XL, Zhang X, Meng M, Yu J, Liu J, Han Y, Shi W, Zhang D, Cao S, Wei Z, Cui Y, Xia Y, Zeng H, Bao K, Lin L, Min Y, Zhang H, Miao M, Tang X, Zhu Y, Sui Y, Li G, Sun H, Yue J, Sun J, Liu F, Zhou L, Lei L, Zheng X, Liu M, Huang L, Song J, Xu C, Li J, Ye K, Zhong S, Lu BR, He G, Xiao F, Wang HL, Zheng H, Fei Z, Liu Y. Draft genome of the kiwifruit Actinidia chinensis. Nat Commun. 2013;4(1):2640. doi: 10.1038/ncomms3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pilkington SM, Crowhurst R, Hilario E, Nardozza S, Fraser L, Peng YY, Gunaseelan K, Simpson R, Tahir J, Deroles SC, et al. A manually annotated Actinidia chinensis var. chinensis (kiwifruit) genome highlights the challenges associated with draft genomes and gene prediction in plants. BMC Genomics. 2018;19:19. doi: 10.1186/s12864-018-4656-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang W, Sun XP, Yue JY, Tang XF, Jiao C, Yang Y, Niu XL, Miao M, Zhang DF, Huang SX, et al. Chromosome-scale genome assembly of kiwifruit Actinidia eriantha with single-molecule sequencing and chromatin interaction mapping. GigaScience. 2019;8(4):10. doi: 10.1093/gigascience/giz027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu H, Ma T, Kang M, Ai F, Zhang J, Dong G, Liu J. A high-quality Actinidia chinensis (kiwifruit) genome. Horticulture Res. 2019;6(1):117. doi: 10.1038/s41438-019-0202-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu QY, Wang JW, Mao SX, Xu HR, Wu Q, Liang MT, Yuan YM, Liu MY, Huang K. Comparative transcriptome analyses of genes involved in sulforaphane metabolism at different treatment in Chinese kale using full-length transcriptome sequencing. BMC Genomics. 2019;20:377. doi: 10.1186/s12864-019-5758-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lou HQ, Ding MZ, Wu JS, Zhang FC, Chen WC, Yang Y, Suo JW, Yu WW, Xu CM, Song LL. Full-length transcriptome analysis of the genes involved in tocopherol biosynthesis in Torreya grandis. J Agric Food Chem. 2019;67(7):1877–1888. doi: 10.1021/acs.jafc.8b06138. [DOI] [PubMed] [Google Scholar]
- 17.Xu ZC, Peters RJ, Weirather J, Luo HM, Liao BS, Zhang X, Zhu YJ, Ji AJ, Zhang B, Hu SN, Au KF, Song J, Chen S. Full-length transcriptome sequences and splice variants obtained by a combination of sequencing platforms applied to different root tissues of Salvia miltiorrhiza and tanshinone biosynthesis. Plant J. 2015;82(6):951–961. doi: 10.1111/tpj.12865. [DOI] [PubMed] [Google Scholar]
- 18.Li YP, Dai C, Hu CG, Liu ZC, Kang CY. Global identification of alternative splicing via comparative analysis of SMRT- and Illumina-based RNA-seq in strawberry. Plant J. 2017;90(1):164–176. doi: 10.1111/tpj.13462. [DOI] [PubMed] [Google Scholar]
- 19.He L, Fu SH, Xu ZC, Yan J, Xu J, Zhou H, Zhou JG, Chen XL, Li Y, Au KF, Yao H. Hybrid sequencing of full-length cDNA transcripts of stems and leaves in Dendrobium officinale. Genes. 2017;8(10):257. doi: 10.3390/genes8100257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang XM, Chen SY, Shi X, Liu DN, Zhao P, Lu YZ, Cheng YB, Liu ZS, Nie XJ, Song WN, Sun Q, Xu S, Ma C. Hybrid sequencing reveals insight into heat sensing and signaling of bread wheat. Plant J. 2019;98(6):1015–1032. doi: 10.1111/tpj.14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Minio A, Massonnet M, Figueroa-Balderas R, Vondras AM, Blanco-Ulate B, Cantu D. Iso-Seq Allows Genome-Independent Transcriptome Profiling of Grape Berry Development. G3. 2019;9(3):755–767. doi: 10.1534/g3.118.201008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dong LL, Liu HF, Zhang JC, Yang SJ, Kong GY, Chu JSC, Chen NS, Wang DW. Single-molecule real-time transcript sequencing facilitates common wheat genome annotation and grain transcriptome research. BMC Genomics. 2015;16(1):1039. doi: 10.1186/s12864-015-2257-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma XS, Xia H, Liu YH, Wei HB, Zheng XG, Song CZ, Chen L, Liu HY, Luo LJ. Transcriptomic and Metabolomic studies disclose key metabolism pathways contributing to well-maintained photosynthesis under the drought and the consequent drought-tolerance in Rice. Front Plant Sci. 2016;7:1886. doi: 10.3389/fpls.2016.01886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luo HT, Zhang JY, Wang G, Jia ZH, Huang SN, Wang T, Guo ZR. Functional characterization of waterlogging and heat stresses tolerance gene pyruvate decarboxylase 2 from Actinidia deliciosa. Int J Mol Sci. 2017;18(11):2377. doi: 10.3390/ijms18112377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ho WWH, Hill CB, Doblin MS, Shelden MC, van de Meene A, Rupasinghe T, Bacic A, Roessner U. Integrative multi-omics analyses of barley Rootzones under salinity stress reveal two distinctive salt tolerance mechanisms. Plant Commun. 2020;1(3):100031. doi: 10.1016/j.xplc.2020.100031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Solanke AU. Signal transduction during cold stress in plants. Physiol Mol Biol Plants. 2008;14(1/2):69–79. doi: 10.1007/s12298-008-0006-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mizoi J, Shinozaki K, Yamaguchi-Shinozaki K. AP2/ERF family transcription factors in plant abiotic stress responses. BBA Gene Regul Mech. 2012;1819(2):86–96. doi: 10.1016/j.bbagrm.2011.08.004. [DOI] [PubMed] [Google Scholar]
- 28.Xie YP, Chen PX, Yan Y, Bao CN, Li XW, Wang LP, Shen XX, Li HY, Liu XF, Niu CD, Zhu C, Fang N, Shao Y, Zhao T, Yu J, Zhu J, Xu L, van Nocker S, Ma F, Guan Q. An atypical R2R3 MYB transcription factor increases cold hardiness by CBF-dependent and CBF-independent pathways in apple. New Phytol. 2018;218(1):201–218. doi: 10.1111/nph.14952. [DOI] [PubMed] [Google Scholar]
- 29.Li H, Ding YL, Shi YT, Zhang XY, Zhang SQ, Gong ZZ, Yang SH. MPK3-and MPK6-mediated ICE1 phosphorylation negatively regulates ICE1 stability and freezing tolerance in Arabidopsis. Dev Cell. 2017;43(5):630–642. doi: 10.1016/j.devcel.2017.09.025. [DOI] [PubMed] [Google Scholar]
- 30.Rubio S, Noriega X, Perez FJ. Abscisic acid (ABA) and low temperatures synergistically increase the expression of CBF/DREB1 transcription factors and cold-hardiness in grapevine dormant buds. Ann Bot. 2019;123(4):681–689. doi: 10.1093/aob/mcy201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tang K, Zhao L, Ren YY, Yang SH, Zhu JK, Zhao CZ. The transcription factor ICE1 functions in cold stress response by binding to the promoters of CBF and COR genes. J Integr Plant Biol. 2020;62(3):258–263. doi: 10.1111/jipb.12918. [DOI] [PubMed] [Google Scholar]
- 32.Kou S, Chen L, Tu W, Scossa F, Wang YM, Liu J, Fernie AR, Song BT, Xie CH. The arginine decarboxylase gene ADC1, associated to the putrescine pathway, plays an important role in potato cold-acclimated freezing tolerance as revealed by transcriptome and metabolome analyses. Plant J. 2018;96(6):1283–1298. doi: 10.1111/tpj.14126. [DOI] [PubMed] [Google Scholar]
- 33.Cheong BE, Ho WWH, Biddulph B, Wallace X, Rathjen T, Rupasinghe TWT, Roessner U, Dolferus R. Phenotyping reproductive stage chilling and frost tolerance in wheat using targeted metabolome and lipidome profiling. Metabolomics. 2019;15(11):144. doi: 10.1007/s11306-019-1606-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang M, Yang J, Su L, Sun K, Li DX, Liu YZ, Wang H, Chen ZQ, Guo T. Metabolic profile analysis and identification of key metabolites during rice seed germination under low-temperature stress. Plant Sci. 2019;289:110282. doi: 10.1016/j.plantsci.2019.110282. [DOI] [PubMed] [Google Scholar]
- 35.Sun S, Qi X, Wang R, Lin M, Fang J. Evaluation of freezing tolerance inActinidiagermplasm based on relative electrolyte leakage. Hortic Environ Biotechnol. 2020;61(4):755–765. doi: 10.1007/s13580-020-00272-4. [DOI] [Google Scholar]
- 36.Thalmann M, Santelia D. Starch as a determinant of plant fitness under abiotic stress. New Phytol. 2017;214(3):943–951. doi: 10.1111/nph.14491. [DOI] [PubMed] [Google Scholar]
- 37.Ouyang L, Leus L, De Keyser E, Van Labeke M-C. Seasonal changes in cold hardiness and carbohydrate metabolism in four garden rose cultivars. J Plant Physiol. 2019;232:188–199. doi: 10.1016/j.jplph.2018.12.001. [DOI] [PubMed] [Google Scholar]
- 38.Xu ZY, Lee KH, Dong T, Jeong JC, Jin JB, Kanno Y, Kim DH, Kim SY, Seo M, Bressan RA. A vacuolar β-glucosidase homolog that possesses glucose-conjugated abscisic acid hydrolyzing activity plays an important role in osmotic stress responses in Arabidopsis. Plant Cell. 2012;24(5):2184–2199. doi: 10.1105/tpc.112.095935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Akiyama T, Opassiri R, Cairns JK. A rice homolog of Arabidopsis sfr2 gene encoding beta-glucosidase is responsive to cold stress. Plant Cell Physiol. 2007;48:S237. [Google Scholar]
- 40.Thorlby G. The SENSITIVE TO FREEZING2 gene, required for FREEZING tolerance in Arabidopsis thaliana, encodes a beta-glucosidase. Plant Cell. 2004;16(8):2192–2203. doi: 10.1105/tpc.104.024018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Opassiri R, Pomthong B, Akiyama T, Nakphaichit M, Onkoksoong T, Cairns MK, Cairns JRK. A stress-induced rice (Oryza sativa L.) beta-glucosidase represents a new subfamily of glycosyl hydrolase family 5 containing a fascin-like domain. Biochem J. 2007;408(2):241–249. doi: 10.1042/BJ20070734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Khazaei M, Maali-Amiri R, Talei AR, Ramezanpour S. Differential transcript accumulation of Dhydrin and Beta-glucosidase genes to cold-induced oxidative stress in chickpea. J Agric Sci Technol. 2015;17(3):725–734. [Google Scholar]
- 43.Wang PT, Liu H, Hua HJ, Wang L, Song CP. A vacuole localized beta-glucosidase contributes to drought tolerance in Arabidopsis. Chin Sci Bull. 2011;56(33):3538–3546. doi: 10.1007/s11434-011-4802-7. [DOI] [Google Scholar]
- 44.Benaroudj N, Lee DH, Goldberg AL. Trehalose accumulation during cellular stress protects cells and cellular proteins from damage by oxygen radicals. J Biol Chem. 2001;276(26):24261–24267. doi: 10.1074/jbc.M101487200. [DOI] [PubMed] [Google Scholar]
- 45.Fernandez O, Béthencourt L, Quero A, Sangwan RS, Clément C. Trehalose and plant stress responses: friend or foe? Trends Plant Sci. 2010;15(7):409–417. doi: 10.1016/j.tplants.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 46.Mostofa MG, Hossain MA, Fujita M, Tran LSP. Physiological and biochemical mechanisms associated with trehalose-induced copper-stress tolerance in rice. Sci Rep. 2015;5(1):11433. doi: 10.1038/srep11433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li HW, Zang BS, Deng XW, Wang XP. Overexpression of the trehalose-6-phosphate synthase gene OsTPS1 enhances abiotic stress tolerance in rice. Planta. 2011;234(5):1007–1018. doi: 10.1007/s00425-011-1458-0. [DOI] [PubMed] [Google Scholar]
- 48.Ge LF, Chao DY, Shi M, Zhu MZ, Gao JP, Lin HX. Overexpression of the trehalose-6-phosphate phosphatase gene OsTPP1 confers stress tolerance in rice and results in the activation of stress responsive genes. Planta. 2008;228(1):191–201. doi: 10.1007/s00425-008-0729-x. [DOI] [PubMed] [Google Scholar]
- 49.XL A, LF A, PQ A, YS A, JL B, XW A. Overexpression of the wheat trehalose 6 - phosphate synthase 11 gene enhances cold tolerance in Arabidopsis thaliana. Gene. 2019;710:210–217. doi: 10.1016/j.gene.2019.06.006. [DOI] [PubMed] [Google Scholar]
- 50.Bae H, Herman E, Bailey B, Ho B, Sicher R. Exogenous trehalose alters Arabidopsis transcripts involved in cell wall modification, abiotic stress, nitrogen metabolism, and plant defense. Physiol Plant. 2010;125(1):114–126. doi: 10.1111/j.1399-3054.2005.00537.x. [DOI] [Google Scholar]
- 51.Ding YL, Shi YT, Yang SH. Advances and challenges in uncovering cold tolerance regulatory mechanisms in plants. New Phytol. 2019;222(4):1690–1704. doi: 10.1111/nph.15696. [DOI] [PubMed] [Google Scholar]
- 52.Valerio C, Costa A, Marri L, Issakidis-Bourguet E, Pupillo P, Trost P, Sparla F. Thioredoxin-regulated -amylase (BAM1) triggers diurnal starch degradation in guard cells, and in mesophyll cells under osmotic stress. J Exp Bot. 2011;62(2):545–555. doi: 10.1093/jxb/erq288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Monroe JD, Storm AR. Review: the Arabidopsis β-amylase (BAM) gene family: diversity of form and function. Plant Sci. 2018;276:163–170. doi: 10.1016/j.plantsci.2018.08.016. [DOI] [PubMed] [Google Scholar]
- 54.Sun S, Fang J, Lin M, Qi X, Chen J, Wang R, Li Z, Li Y, Muhammad A. Freezing tolerance and expression of beta-amylase gene in two Actinidia arguta cultivars with seasonal changes. Plants. 2020;9(4):1–12. doi: 10.3390/plants9040515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhao LY, Yang TY, Xing CH, Dong HZ, Qi KJ, Gao JZ, Tao ST, Wu JY, Wu J, Zhang SL, Huang X. The beta-amylase PbrBAM3 from pear (Pyrus betulaefolia) regulates soluble sugar accumulation and ROS homeostasis in response to cold stress. Plant Sci. 2019;287:110184. doi: 10.1016/j.plantsci.2019.110184. [DOI] [PubMed] [Google Scholar]
- 56.Peng T, Zhu XF, Duan N, Liu JH. PtrBAM1, a beta-amylase-coding gene of Poncirus trifoliata, is a CBF regulon member with function in cold tolerance by modulating soluble sugar levels. Plant Cell Environ. 2014;37(12):2754–2767. doi: 10.1111/pce.12384. [DOI] [PubMed] [Google Scholar]
- 57.Zakharian E, Cao C, Rohacs T. Gating of transient receptor potential Melastatin 8 (TRPM8) channels activated by cold and chemical agonists in planar lipid bilayers. J Neurosci. 2010;30(37):12526–12534. doi: 10.1523/JNEUROSCI.3189-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu YN, Lu XX, Ren A, Shi L, Zhu J, Jiang AL, Yu HS, Zhao MW. Conversion of phosphatidylinositol (PI) to PI4-phosphate (PI4P) and then to PI (4,5) P-2 is essential for the cytosolic Ca2+ concentration under heat stress in Ganoderma lucidum. Environ Microbiol. 2018;20(7):2456–2468. doi: 10.1111/1462-2920.14254. [DOI] [PubMed] [Google Scholar]
- 59.Wimalasekera P, Holk M, Scherer GFE. Plant Phosphatidylcholine-Hydrolyzing Phospholipases C NPC3 and NPC4 with Roles in Root Development and Brassinolide Signaling in Arabidopsis thaliana RID A-7853-2010. Mol Plant. 2010;3(3):610–625. doi: 10.1093/mp/ssq005. [DOI] [PubMed] [Google Scholar]
- 60.Kravets VS, Kolesnikov YS, Kretynin SV, Kabachevskaya EM, Kukhar VP. Molecular and genetics approaches for investigation of phospholipase D role in plant cells. Biopolym Cell. 2010;26(3):175–186. doi: 10.7124/bc.000154. [DOI] [Google Scholar]
- 61.Smart CC, Fleming AJ. A plant gene with homology to D-myo-inositol-3-phosphate synthase is rapidly and spatially up-regulated during an abscisic-acid-induced morphogenic response in Spirodela polyrrhiza. Plant J. 2010;4(2):279–293. doi: 10.1046/j.1365-313X.1993.04020279.x. [DOI] [PubMed] [Google Scholar]
- 62.Smart CC, Flores S. Overexpression of D-myo-inositol-3-phosphate synthase leads to elevated levels of inositol in Arabidopsis. Plant Mol Biol. 1997;33(5):811–820. doi: 10.1023/A:1005754425440. [DOI] [PubMed] [Google Scholar]
- 63.Peng T, You XS, Guo L, Zhong BL, Mi LF, Chen JM, Xiao X. Transcriptome analysis of Chongyi wild mandarin, a wild species more cold-tolerant than Poncirus trifoliata, reveals key pathways in response to cold. Environ Exp Bot. 2021;184:104371. doi: 10.1016/j.envexpbot.2020.104371. [DOI] [Google Scholar]
- 64.Zhang CQ, Wang J, Gao X. Computational identification of transcriptional regulatory elements in Arabidopsis TCH4 promoter. Hereditas. 2008;30(5):620–626. doi: 10.3724/sp.j.1005.2008.00620. [DOI] [PubMed] [Google Scholar]
- 65.Wang J, Ren Y, Liu X, Luo S, Zhang X, Liu X, et al. Transcriptional activation and phosphorylation of OsCNGC9 confer enhanced chilling tolerance in rice. Mol Plant. 2021;14(2):315–29. [DOI] [PubMed]
- 66.Zhao CZ, Wang PC, Si T, Hsu CC, Wang L, Zayed O, Yu ZP, Zhu YF, Dong J, Tao WA, Zhu JK. MAP kinase cascades regulate the cold response by modulating ICE1 protein stability. Dev Cell. 2017;43(5):618–629. doi: 10.1016/j.devcel.2017.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tang W, Tang AY. Overexpression of Arabidopsis thaliana malonyl-CoA synthetase gene enhances cold stress tolerance by activating mitogen-activated protein kinases in plant cells. J Forestry Res. 2021;32(2):741–53.
- 68.Meng S, Dane F, Si Y, Ebel R, Zhang C. Gene expression analysis of cold treated versus cold acclimated Poncirus trifoliata. Euphytica. 2008;164(1):209–219. doi: 10.1007/s10681-008-9708-3. [DOI] [Google Scholar]
- 69.Shi YT, Ding YL, Yang SH. Molecular Reculation of CBF Sicnalinc in Colc acclimation. Trends Plant Sci. 2018;23(7):623–637. doi: 10.1016/j.tplants.2018.04.002. [DOI] [PubMed] [Google Scholar]
- 70.Wang YJ, Zhang ZG, He XJ, Zhou HL, Wen YX, Dai JX, Zhang JS, Chen SY. A rice transcription factor OsbHLH1 is involved in cold stress response. Theor Appl Genet. 2003;107(8):1402–1409. doi: 10.1007/s00122-003-1378-x. [DOI] [PubMed] [Google Scholar]
- 71.Feng XM, Zhao Q, Zhao LL, Qiao Y, Xie XB, Li HF, Yao YX, You CX, Hao YJ. The cold-induced basic helix-loop-helix transcription factor gene MdCIbHLH1 encodes an ICE-like protein in apple. BMC Plant Biol. 2012;12(1):22. doi: 10.1186/1471-2229-12-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang X, Ding Y, Li Z, Shi Y, Wang J, Hua J, Gong Z, Zhou JM, Yang S. PUB25 and PUB26 promote plant freezing tolerance by degrading the cold signaling negative regulator MYB15. Dev Cell. 2019;51(2):222–235. doi: 10.1016/j.devcel.2019.08.008. [DOI] [PubMed] [Google Scholar]
- 73.Sun MY, Li JY, Li D, Huang FJ, Wang D, Li H, Xing Q, Zhu HB, Shi L. Full-length transcriptome sequencing and modular organization analysis of the Naringin/Neoeriocitrin-related gene expression pattern in Drynaria roosii. Plant Cell Physiol. 2018;59(7):1398–1414. doi: 10.1093/pcp/pcy072. [DOI] [PubMed] [Google Scholar]
- 74.Umer MJ, Bin Safdar L, Gebremeskel H, Zhao S, Yuan P, Zhu H, Kaseb MO, Anees M, Lu X, He N, Gong C, Liu W. Identification of key gene networks controlling organic acid and sugar metabolism during watermelon fruit development by integrating metabolic phenotypes and gene expression profiles. Hortic Res. 2020;7(1):193. doi: 10.1038/s41438-020-00416-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nakata M, Sugiyama N, Pankasemsuk T. Morphological study of the structure and development of longan inflorescence. J Am Soc Hortic Sci. 2005;130(6):793–798. doi: 10.21273/JASHS.130.6.793. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1 Supplementary Fig. 1. GO functional classifications of all the unigenes.
Additional file 2 Supplementary Fig. 2. KEGG functional classifications of all the unigenes.
Additional file 3 Supplementary Fig. 3. Phylogenetic tree of candidate genes and homologous genes. (a) Beta-GC, (b) TPS5, (c) BAM3.1, (d) CBF3, (e) MYC2, and (f) MYB44.
Additional file 4 Table S1. DEGs related to Starch and sucrose metabolism, MAPK signaling pathway, Phosphatidylinositol signaling system, Inositol phosphate metabolism, Arginine and proline metabolism.
Additional file 5 Table S2. DEGs related to hormone and calcium signal.
Additional file 6 Table S3. DEGs related to TFs.
Additional file 7 Table S4. Top 100 hub genes in the blue module.
Additional file 8 Table S5. Primers used for quantitative real-time RT-PCR analysis.
Data Availability Statement
The raw data generated in this study have been deposited in the NCBI Short Read Archive (SRA) under the accession number PRJNA681641. All other relevant data contained within the paper are available in the paper and Supplementary Files.