

Abstract
T cells are vital for adaptive immune responses that protect against pathogens and cancers. The T cell receptor (TCR)–CD3 complex comprises a diverse αβ TCR heterodimer in noncovalent association with three invariant CD3 dimers. The TCR is responsible for recognizing antigenic peptides bound to MHC molecules (pMHC), while the CD3 dimers relay activation signals to the T cell. However, the mechanisms by which TCR engagement by pMHC is transmitted to CD3 remain mysterious, although there is growing evidence that mechanosensing and allostery both play a role. Here, we carried out NMR analysis of a human autoimmune TCR (MS2–3C8) that recognizes a self-peptide from myelin basic protein presented by the MHC class II molecule HLA-DR4. We observed pMHC-induced NMR signal perturbations in MS2–3C8 that indicate long-range effects on TCR β chain conformation and dynamics. Our results demonstrate that, in addition to expected changes in the NMR resonances of pMHC-contacting residues, perturbations extend to the Vβ/Vα, Vβ/Cβ, and Cβ/Cα interfacial regions. Moreover, the pattern of long-range perturbations is similar to that detected previously in the β chains of two MHC class I-restricted TCRs, thereby revealing a common allosteric pathway among three unrelated TCRs. Molecular dynamics (MD) simulations predict similar pMHC-induced effects. Taken together, our results demonstrate that pMHC binding induces long-range allosteric changes in the TCR β chain at conserved sites in both representative MHC class I- and class II-restricted TCRs, and that these sites may play a role in the transmission of signaling information.
Keywords: T cell receptor, peptide–MHC, allostery, NMR, T cell triggering
Graphical Abstract
Introduction
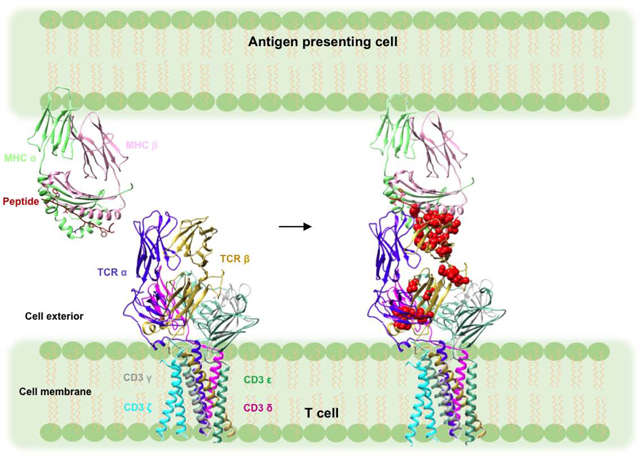
T cells are key players in host immune defense against microbial pathogens and tumors. The T cell receptor (TCR)–CD3 complex on the surface of T cells mediates this process. The TCR–CD3 complex consists of a genetically diverse αβ TCR heterodimer that is noncovalently associated with the invariant CD3 dimers CD3εγ, CD3εδ, and CD3ζζ [1]. The complete three-dimensional structure of the TCRαβ/CD3εγ/CD3εδ/CD3ζζ complex was recently determined by cryoEM (Fig. 1) [2]. The TCR is responsible for recognition of peptide fragments presented by major histocompatibility complex (MHC) molecules on antigen-presenting cells (APCs). These peptides arise from proteolysis of self or foreign proteins inside APCs expressing MHC class I or class II molecules [3]. After TCR ligation of peptide–MHC (pMHC) ligands, the CD3 subunits transmit activation signals to the T cell. The CD3 ε, γ, δ, and ζ chains possess immunoreceptor tyrosine-based activation motifs (ITAMs), which are located at the cytoplasmic side of the TCR–CD3 complex. These ITAMs are believed to dissociate from the lipid bilayer following TCR binding of pMHC, thus exposing the ITAMs to phosphorylation by the Src kinase Lck [4–7]. However, the exact mechanism whereby TCR engagement is relayed to CD3, a process known as early T cell activation, is poorly understood [1].
Figure 1:

Model of the TCR–CD3 complex with bound pMHC ligand. The model was constructed by superposing the crystal structure of the MS2–3C8–MBP–HLA-DR4 complex (PDB 3O6F) [23] onto the cryoEM structure of the unbound TCR–CD3 complex (PDB 6JXR) [2] through the shared Cα/Cβ domains. The inset shows a close-up view of the Cβ FG loop in the TCR–CD3 complex. The Cβ FG loop is positioned immediately above CD3εγ. The side chains of Cβ FG loop residues that undergo NMR signal perturbation in the MS2–3C8(wt)–MBP–HLA-DR4 complex (see Results) are drawn in stick representation.
Numerous models have been put forward to explain early T cell activation [1, 8–12]. These models involve one or a combination of several potential mechanisms: segregation, aggregation, mechanosensing and/or allostery. Binding of pMHC could induce segregation of the inhibitory phosphatase CD45 from TCR–CD3 complexes, allowing phosphorylation of CD3 ITAMs by Lck [13, 14]. TCR ligation could cause aggregation of TCR–CD3 complexes and a concomitant increase in the density of associated Lck molecules that results in enhanced phosphorylation of CD3 ITAMs [15]. According to the mechanosensor model, movement of the TCR and APC membranes over each other during cell–cell contacts produces a mechanical force that leads to dissociation of CD3 ITAMs from the T cell membrane and their exposure to Lck [16, 17]. In the allosteric model, pMHC binding induces long-range changes in TCR structure and/or dynamics that render CD3 ITAMs accessible to Lck [18, 19].
The first evidence for dynamically-driven TCR activation came from hydrogen/deuterium (H/D) exchange, which revealed ligand-induced TCR rigidification [20]. More recently, NMR spectroscopy has been used to address the possibility of long-range allosteric changes in TCR dynamics caused by pMHC binding. Two TCRs, both MHC class I-restricted, have been examined so far: 1) a mouse TCR (B4.2.3) specific for an HIV-1-derived peptide bound to H2-Dd [18] and 2) a human TCR (A6) specific for the Tax peptide of human T cell lymphocytic virus-1 (HTLV-1) bound to HLA-A2 [19]. These NMR studies, in conjunction with molecular dynamics (MD) simulations [19], have provided direct evidence for allosteric signaling from the V to the C domains. Three allosteric sites in the C domains have been identified: 1) the Cβ FG loop, 2) the Cβ αA (H3) helix, and 3) the Cα AB loop. These sites are positioned to communicate with CD3 subunits in the TCR–CD3 cryoEM structure [1, 2].
A special attraction of TCR allostery is that it, like mechanosensing [16, 17], can account for the remarkable ability of single pMHC molecule binding to an exclusively monomeric TCR to initiate T cell signaling with no need for pMHC-induced TCR dimerization or oligomerization [21]. However, the enormous clonal diversity of TCRs (105–108 unique sequences per individual) [22] presents a major intellectual challenge for allosteric models of early T cell activation, since it appears unlikely that multiple allosteric mechanisms could have arisen during evolution of the TCR–CD3 complex. To resolve this conundrum, we hypothesize that pMHC binding generates conserved dynamic or structural changes in Cα and Cβ, despite the diversity of TCR–pMHC interfaces and of Vα and Vβ sequences.
As noted above, both TCRs characterized so far by NMR are MHC class I-restricted [18, 19]. Here, we used NMR to analyze a human autoimmune TCR (MS2–3C8) that recognizes a self-peptide from myelin basic protein (MBP) presented by the multiple sclerosis-associated MHC class II molecule HLA-DR4 [23]. We show that, in addition to the expected residue-specific perturbations at the TCR–pMHC binding interface, there are numerous long-range effects on conformation and dynamics in the TCR β chain that are manifested as changes in chemical shifts and/or peak intensities. These changes propagate from the binding region to the TCR Vβ/Vα, Vβ/Cβ, and Cβ/Cα interfaces. Moreover, the pattern of the pMHC class II-induced changes in the TCR β chain is very similar to what was detected in NMR studies of the MHC class I-restricted TCRs B4.2.3 and A6 [18, 19], which are unrelated to each other or to MS2–3C8. Our results provide the first report of long-range effects in a TCR–pMHC class II interaction, adding to the growing body of data indicating that TCR β chains undergo conserved allosteric changes upon pMHC ligation.
Results
TCR system and NMR assignment of MS2–3C8 β chains
For a representative example of an MHC class II-restricted TCR, we used a human autoimmune TCR, MS2–3C8, which recognizes a self-peptide from MBP (MBP 111–129) and the MHC class II molecule HLA-DR4 (HLA-DRB1*0401). The crystal structure of the MS2–3C8 extracellular domain (VαCα/VβCβ) in complex with MBP–HLA-DR4 has been determined [23], providing a well-characterized binding interaction for comparison with the solution NMR results. Fig. S1 shows SDS-PAGE and size exclusion chromatograms of TCR MS2–3C8 and MBP–HLA-DR4 proteins used in this study.
Three variants of the MS2–3C8 ectodomain were investigated: the wild-type version MS2–3C8(wt), and two mutants, MS2–3C8(mut1) and MS2–3C8(mut2) (Fig. 2). MS2–3C8(mut1) has extensive NMR assignments that were obtained previously using 3D triple resonance NMR methods [24]. This mutant has an engineered interchain disulfide bond between C159α and C171β that is commonly employed in TCR structural biology to improve yields of the folded αβ heterodimer (Fig. 2). MS2–3C8(mut1) also contains a T29Aα mutation that increases the affinity for MBP–HLA-DR4 from a wild-type KD~5 μM to a KD~0.4 μM [23]. MS2–3C8(wt) lacks the engineered interchain disulfide, but instead retains the wild-type disulfide between C206α and C245β. The third variant, MS2–3C8(mut2), contains elements of both MS2–3C8(mut1) and MS2–3C8(wt), having the approximately 10-fold higher affinity of MS2–3C8(mut1) for MBP–HLA-DR4, as well as the native disulfide arrangement of MS2–3C8(wt). Together, these variants enable investigation into the possible roles of affinity and interchain disulfides on the transmission of long-range effects in an MHC class II-restricted TCR.
Figure 2:

Variants of the MS2–3C8 TCR ectodomain used in this study. (A) Crystal structure of MS2–3C8 (PBD 3O6F) [23] highlighting amino acid positions on the α (green) and β chains (wheat) where mutations were made. (B) Amino acid sequence comparisons between the three MS2–3C8 variants, MS2–3C8(mut1), MS2–3C8(mut2), and MS2–3C8(wt).
The focus of our study was on the effects of pMHC binding on the β chain conformation and dynamics of the TCR as this chain is thought to transmit most of the long-range allosteric changes from the pMHC ligation site to the T cell membrane surface [1, 18, 19]. We previously assigned 87% of the backbone amide resonances for the β chain of MS2–3C8(mut1) [24]. Most of these assignments were transferred to MS2–3C8(wt) and MS2–3C8(mut2) by inspection using a conservative approach (Fig. 3). We did not attempt to assign peaks corresponding to the mutated residues in the β chains of MS2–3C8(mut2) (S171, S189, C245) or MS2–3C8(wt) (T55, S171, S189, C245) due to significant peak shifts. In some cases, however, the neighboring residues could be assigned. Thus, 84.8% of the backbone amides were assigned for MS2–3C8(mut2) and 84.4% were assigned for MS2–3C8(wt). The backbone amide assignments for the β chains of MS2–3C8(mut1), MS2–3C8(mut2), and MS2–3C8(wt) are provided (Table S1) and a comparison of chemical shift differences between the variants is shown (Fig. S2). The differences are located near mutational sites that are mostly in the Cβ domain, around the interchain disulfide (Fig. S2A-F). Moreover, the chemical shift perturbations (CSPs) are similar between MS2–3C8(mut1) and MS2–3C8(mut2) when compared with MS2–3C8(mut1) and MS2–3C8(wt), where there are 5 and 7 amino acid differences, respectively (Fig. 2). In contrast, the CSPs between MS23C8(mut2) and MS2–3C8(wt) are relatively small, consistent with only 2 amino acid differences between these variants. The MS2–3C8(mut1) assignments were further compared with previous backbone resonance assignments made for the β chain of the human MHC class I-restricted TCR A6 in which the interchain disulfide between C172β and C159α is also present [19]. These comparisons showed that, while backbone amide chemical shifts differed widely in the variable Vβ domains of MS2–3C8(mut1) and A6 TCR ectodomains, their constant Cβ domains have similar assignments, as might be expected (Fig. S2G, H). The similarity of the Cβ domain chemical shifts determined independently from two different TCRs thus provides further confidence in their assignments.
Figure 3:

NMR backbone assignment of MS2–3C8 TCR β chains. (A) Overlaid two dimensional 1H-15N TROSY-HSQC spectra for MS2–3C8(mut1)α[β−2H, 15N] (black), MS2–3C8(mut2)α[β−2H, 15N] (green), and MS2–3C8(wt)α[β−2H, 15N] (red). Unassigned main chain signals are marked (x). (B) Expanded region highlighted (dashed box) in (A).
Interaction of MS2–3C8 TCR with MBP–HLA-DR4 in solution
The chemical shift perturbations (CSPs) and preferential loss of peak intensity of backbone amide resonances were monitored for specific residues using 2D 1H-15N TROSY-HSQC spectra of [2H, 15N]-labeled MS2–3C8 variants in the presence of unlabeled MBP–HLA-DR4. The NMR spectra indicate that bound and unbound states are in slow exchange, consistent with the observed binding constants. Assignments of backbone amide resonances for the bound state of the TCR were obtained by ascribing the CSP to the nearest shifted peak for a given residue when compared with the TROSY-HSQC spectrum of the unbound state. It is possible that some CSPs may be underestimated using this conservative assumption. The largest changes in CSPs and peak intensities occurred at the CDR loops of the MS2–3C8 ectodomains, which form the binding interface with the MBP–HLA-DR4 molecule [23]. However, additional changes were observed for residues well away from direct contacts with the pMHC ligand. These included perturbations throughout the Vβ domain and extending into the Cβ domain, indicating that MBP–HLA-DR4-binding leads to long-range effects on MS2–3C8 conformation and dynamics. Many of these remotely affected residues have backbone amide perturbations with comparable magnitude to some of the direct interactions. The results for each MS2–3C8 variant are described below, followed by a comparison of the NMR perturbations across the three mutants. Perturbations are referred to based on where the side chain is located for that residue (e.g. core, interface).
MS2–3C8(mut1)–MBP–HLA-DR4 complex
For MS2–3C8(mut1)α[β−2H, 15N] in the presence of unlabeled MBP–HLA-DR4, experimentally significant CSP and/or preferential peak intensity losses are summarized in Fig. 4. Perturbations were detected at the TCR–pMHC binding interface in CDR1β (L24, A28, T29), CDR2β (N49, E50, G51), CDR3β (G96, G97, Y99, S101), and HV4β (S71, L72). Of these, the largest changes occurred in CDR1β and CDR3β, consistent with the observation that these CDR loops are at the center of the interaction with the pMHC in the crystal structure of the complex [23]. Additional CSP and peak intensity changes were detected for residues at the Vβ/Vα interface (T30, L42, R95, L103, F105) as well as in the Vβ core (S23, M45, A46, S48, V60, F65, I67, S75). Some of the perturbed Vβ core residues form close contacts (A46/I67, S48/S75, and M45/V60/F65). Residues in the VβCβ linker sequence (N119), the DE loop (R187), and the FG loop (G216, Q225, V231) also displayed backbone amide CSP and preferential peak intensity losses upon MBP–HLA-DR4 binding. These regions are all in close proximity at the interface between the Vβ and Cβ domains. Further perturbations extended even more remotely into the Cβ domain, mostly in the Cβ core (L143, A147, L190, L194, F208), and at the Cβ/Cα interface (F128, E134, T142, L146, G169, R195). As in the Vβ core, a number of the perturbed Cβ core residues are close to each other in the X-ray structure (e.g. A147/L190, and L143/L194/F208). At the Cβ/Cα interface, residues E134, T142, and R195 also form close contacts. Near the C-terminus, R242 and E132 both exhibited preferential peak intensity losses, and form a salt bridge in the crystal structure of the complex.
Figure 4:

Summary of backbone amide CSPs and differential peak intensity changes in the complex between MS2–3C8(mut1)α[β−2H, 15N] and MBP–HLA-DR4. (A) Expanded regions from the superimposed two dimensional 1H-15N TROSY-HSQC spectra of unbound (black) and MBP–HLA-DR4-bound (red) states of MS2–3C8(mut1)α[β−2H, 15N]. The spectrum of the bound state is scaled to match the peak intensities of most signals in the unbound state. This is done for visual purposes only to highlight the disproportional changes for specific residues upon binding. (B) Combined 1H and 15N chemical shift perturbations, Δδtotal (ppm), in the MS2–3C8(mut1) β chain versus residue number. The dashed line represents the mean value plus 1SD. Gray histogram bars indicate unassigned and proline residues. (C) Plot of percent loss of backbone amide peak intensity versus residue number. The dashed line corresponds with the mean plus 1SD. Hypervariable regions, αA, and the Cβ FG loop are highlighted in (B) and (C). (D) β chain residues in MS2–3C8(mut1) with experimentally significant changes (≥mean plus 1SD) upon binding to MBP–HLA-DR4: MS2–3C8(mut1) β chain (wheat); CSPs (blue); peak intensity changes (red); both CSP and peak intensity (purple); unassigned residues (gray); MS2–3C8(mut1) α chain (green); MBP peptide from pMHC (orange).
MS2–3C8(mut2)–MBP–HLA-DR4 complex
Binding experiments between MS2–3C8(mut2)α[β−2H, 15N] and unlabeled MBP–HLA-DR4 gave a similar pattern of perturbations in the 2D TROSY-HSQC spectra (Fig. 5). At the pMHC–TCR interface, CSP and/or peak intensity decreases were observed for CDR1β (L24, A28, T29), CDR2β (N49, E50, G51), CDR3β (G96, G97, Y99, S101), and HV4β (A70, S71, L72). Perturbed residues were also detected at the Vβ/Vα interface (T30, F32, Y34, Q36, L103, F105) and in the Vβ core (S23, M31, R35, S75, V79, A94). Perturbations in the Vβ domain were more extensive than for MS2–3C8(mut1) with an additional cluster of residues near the Vβ/Cβ region (S13, G14, Y56, V79, S81, H83, D86). A number of residues affected by pMHC binding in the Vβ core interact with each other (e.g. S23/A28/M31/A94, M31/S75, R35/V79/D86). Long-range changes were detected in the Vβ/Cβ interface for residues in the helical VβCβ linker region (K118, N119, F121), the end of the DE loop (Y188), and the FG loop (G216, L217, Q225, V231). Experimentally significant CSP and/or peak intensity changes were further detected at the Cβ/Cα interface (F128, S131, E134, T142, G169, L177) and in the Cβ core (L143, S192, F208), similar to the observations for MS2–3C8(mut1).
Figure 5:

Summary of backbone amide CSPs and differential peak intensity changes in the complex between MS2–3C8(mut2)α[β−2H, 15N] and MBP–HLA-DR4. (A) Expanded regions from the superimposed two dimensional 1H-15N TROSY-HSQC spectra of unbound (black) and MBP–HLA-DR4-bound (red) states of MS2–3C8(mut2)α[β−2H, 15N]. The spectrum of the bound state is scaled as in Fig. 4A. (B) Combined 1H and 15N chemical shift perturbations, Δδtotal (ppm), in the MS2–3C8(mut2) β chain versus residue number. The dashed line indicates the mean value plus 1SD. Gray histogram bars indicate unassigned and proline residues. (C) Plot of percent loss of backbone amide peak intensity versus residue number. The dashed line corresponds with the mean plus 1SD. Hypervariable regions, αA, and the Cβ FG loop are highlighted in (B) and (C). (D) β chain residues in MS2–3C8(mut2) with experimentally significant changes (≥mean plus 1SD) upon binding to MBP–HLA-DR4. Color coding as in Fig. 4D.
MS2–3C8(wt)–MBP–HLA-DR4 complex
As for the above cases, the 2D 1H-15N TROSY-HSQC spectrum of MS2–3C8(wt)α[β−2H, 15N] in the presence of unlabeled MBP–HLA-DR4 indicated a combination of local and long-range changes in the TCR β-chain (Fig. 6). CSP and/or peak intensity decreases were seen for CDR1β (T29), CDR2β (N49, E50, G51), CDR3β (G96, G97, Y99, S101), and HV4β (A70, S71, L72). Other more distant changes were found at the Vα/Vβ interface (T30, Y34, L42, T47, L103, F105) and in the Vβ core (I19, S23, A46, Y56, F65, V79, D86, A94, T109). As in the other two variants, the Vβ core residues with perturbations were generally close to each other (e.g. I19/T109, S23/A94, A46/Y56, F65/V79/D86). Moreover, perturbations extended into the Cβ domain, comparable with MS2–3C8(mut1) and MS2–3C8(mut2) samples. Residues in the VβCβ linker (K118), DE loop (Y188), and FG loop (Y215, G216, L217, Q225, V231) exhibited CSP and/or peak intensity loss effects, as did amino acids at the termini of the Cβ domain C-strand (E156), F-strand (Q213), and G-strand (Q233). A number of these perturbed residues are in close proximity (K118/Q225, E156/Q213/Y215, G216/L217/V231/Q233) similar to MS2–3C8(mut2). Perturbations also occurred at the Cβ/Cα interface (F128, S131, E134, H137, T142, G169, L177) and the Cβ core (V125, V127, C145, F200, F208, A237).
Figure 6:

Summary of backbone amide CSPs and differential peak intensity changes in the complex between MS2–3C8(wt)α[β−2H, 15N] and MBP–HLA-DR4. (A) Expanded regions from the superimposed two dimensional 1H-15N TROSY-HSQC spectra of unbound (black) and MBP–HLA-DR4-bound (red) states of MS2–3C8(wt)α[β−2H, 15N]. The spectrum of the bound state is scaled as in Figs. 4A and 5A. (B) Combined 1H and 15N chemical shift perturbations, Δδtotal (ppm), in the MS2–3C8(wt) β chain versus residue number. The dashed line corresponds with the mean value plus 1SD. Gray histogram bars indicate unassigned and proline residues. (C) Plot of percent loss of backbone amide peak intensity versus residue number. The dashed line represents the mean plus 1SD. Hypervariable regions, αA, and the Cβ FG loop are highlighted in (B) and (C). (D) β chain residues in MS2–3C8(wt) with experimentally significant changes (≥mean plus 1SD) upon binding to MBP–HLA-DR4. Color coding as in Figs. 4D and 5D.
Conserved and consensus residues
Overall, the three MS2–3C8 TCR ectodomains exhibit similar CSP and peak intensity patterns when bound to MBP–HLA-DR4, with a significant number of the residue perturbations either present in at least two out of three TCRs (termed “consensus” here) or absolutely conserved in all three (Fig. 7A). The majority of the conserved (denoted in italics) and consensus amino acids are located in three regions of the MS2–3C8 β chain: 1) the MBP–HLA-DR4/MS2–3C8 binding interface (A28, T29, T30, N49, E50, G51, A70, S71, L72, G96, G97, Y99, S101); 2) the Vβ/Cβ interface (S13, K118, N119, G149, Y188, G216, L217, Q225, V231); and 3) the Cβ/Cα interface (F128, S131, E134, T142, G169, L177). Collectively, there are also extensive changes in the Vβ core, Vβ/Vα interface, and Cβ core across the three MS2–3C8 variants, but each of these regions has fewer residue perturbations present in more than one TCR (Vβ S23, A94, V79, D86; Vβ/Vα T30, L103, F105; Cβ A147, L143, F208).
Figure 7:

Comparison of changes in MHC class II-restricted TCR MS2–3C8 and MHC class I-restricted TCR A6 β chains upon pMHC-binding. (A-D) Summary of conserved and consensus MS2–3C8 residues perturbed by binding to the MBP–HLA-DR4. Color-coding for conserved residue perturbations present in all three MS2–3C8 variants: CSPs (blue); peak intensity changes (red); both CSP and peak intensity (purple). Color-coding for consensus residue perturbations: CSPs (cyan); peak intensity (orange); both CSP and peak intensity (light purple). (E-H) TCR A6 residues perturbed by binding to Tax–HLA-A2. Color-coding: CSPs (blue); peak intensity changes (red). Additional color coding: β chain (wheat); α chain (green); unassigned residues (gray); peptide from pMHC (pink); Cβ FG loop (yellow).
Conserved and consensus CSP and peak intensity changes in the CDR loops propagate into the connecting β-strands of the Vβ domain. The largest of these perturbations are in CDR3β and extend to adjacent residues L103 and F105 at the Vβ/Vα interface (Fig. 7B). Other consensus and conserved perturbed residues are also in close contact with each other spatially. Examples of such connected perturbations in the Vβ core are S23/A28/A94 and V79/D86 (Fig. 7B,C). At the Vβ/Cβ interface, K118 from the Vβ/Cβ linker region interacts with Q225 from the FG loop, while G216 and L217 are proximal to V231, also in the FG loop (Fig. 7C). At the Cβ/Cα interface, S131, E134, and T142 are all proximal (Fig. 7D), with the E134 and T142 side chains forming a stabilizing hydrogen bond, while L143 packs against F208 in the Cβ core. It should also be noted that a number of weaker unassigned peaks, corresponding to some of the unassigned regions in the CDR loops and interfacial regions, have significant CSP and peak intensity decreases upon binding to pMHC in all three MS2–3C8 variants.
Pairwise conservation
In terms of pairwise conserved patterns, MS2–3C8(mut2) and MS2–3C8(wt) have the highest number of perturbed residues in common (21 CSP + 19 PI = 40 total), MS2–3C8(mut2) and MS2–3C8(mut1) are intermediate (19 CSP + 14 PI = 33 total), while MS2–3C8(wt) and MS2–3C8(mut1) share the fewest (13 CSP + 15 PI = 28 total) (Fig. S3). The degree of similarity in perturbation patterns corresponds with the level of amino acid sequence similarity between the TCRs (Fig. 2). MS2–3C8(mut2) and MS2–3C8(wt) display the most similar response in binding to MBP–HLA-DR4, both having the same native disulfide arrangement, with no engineered interchain disulfide and only two amino acid differences. In contrast, MS2–3C8(mut1), which contains the interchain disulfide, has fewer perturbations in common with MS2–3C8(mut2) and MS2–3C8(wt). This may be due to the fact that MS2–3C8(mut1) has more amino acid differences with MS2–3C8(mut2) and MS2–3C8(wt), but it is also possible that the presence of the interchain disulfide plays a role in determining the extent of pMHC-induced changes. By comparison, the approximately 10-fold difference in the binding affinities of MS2–3C8(mut2) and MS2–3C8(wt) for MBP–HLA-A2 does not result in a significantly different perturbation pattern.
Effects of pMHC binding on the Cβ domain
We further note that, while the number of experimentally significant perturbations in Cβ is about the same in all three TCRs, MS2–3C8(mut2) has the most pronounced changes in the Cβ domain upon binding MBP–HLA-DR4 (Table S2). We hypothesize that this is because MS2–3C8(mut2) has the combined benefit of having the native disulfide arrangement as well as a 10-fold tighter binding affinity to MBP–HLA-DR4 than MS2–3C8(wt) [23]. In contrast, while MS2–3C8(mut1) has the same affinity for MBP–HLA-DR4 as MS2–3C8(mut2), it has an additional non-native interchain disulfide (Fig. 2) that may dampen the magnitude of long-range perturbations through restriction of conformational and dynamic changes.
Unique residue perturbations
While the conserved and consensus residues provide a framework for allosteric transmission of changes in conformation and dynamics, other residue perturbations are specific to a given TCR. The unique changes in the three MS2–3C8 TCRs upon pMHC binding are not localized to one region but are present over the entire β chain (Fig. S4). These pattern variations are presumably due to a combination of factors including modifications in disulfide architecture, the minor amino acid changes, differences in binding affinities to MBP–HLA-DR4, and experimental noise in the data.
Molecular dynamics simulations
MD simulations were employed on both unbound and pMHC-bound states of MS2–3C8 to measure in silico changes in MS2–3C8 β chain flexibility. Simulations were carried out to 1200 ns as described previously [19], employing the change in the root mean square fluctuation (RMSF) for the Cα atom of each residue as an indicator for effects of pMHC binding on backbone conformational dynamics. Overall, the results indicate that in silico binding of MBP–HLA-DR4 induces a net decrease in conformational fluctuations of the MS2–3C8 β chain (Fig. 8A, C;Fig. S5). All four hypervariable loops (CDR1β, CDR2β, CDR3β, HV4β) experience a decrease in conformational flexibility, reflected in positive ΔRMSF values, upon pMHC binding. Moreover, changes in RMSF propagate to the Cβ domain, resulting in significant rigidification of the FG loop and adjacent regions. Indeed, the FG loop exhibits the largest ΔRMSF values (up to 2.0–2.5 Å) in the β chain, exceeding even those observed for CDR3β. Estimation of the uncertainty in RMSF values was performed by analyzing the RMSF values of MS2–3C8 trajectories using 100 ns time regions in the last 1000 ns of simulations.
Figure 8:

Molecular dynamics simulations of pMHC binding to MHC class I- and class II-restricted TCR ectodomains. (A) ΔRMSF (unbound TCR – bound TCR) values for the MHC class II-restricted MS2–3C8 β chain. MS2–3C8 β chain residues that become more rigid upon binding to MBP-HLA-DR4 are positive, while residues with a gain in flexibility are negative. Residue positions of the CDR loops and the FG loop are highlighted. Error bars indicate ±1SD. (B) ΔRMSF values for the MHC class I-restricted TCR A6 β chain [19]. (C) Mapping of ΔRMSF values onto the β chain of MS2–3C8 in complex with MBP–HLA-DR4 (PDB 3O6F) [23]. Color coding: HLA-DR4 (gray); MBP peptide (pink); MS2–3C8 α chain (green); MS2–3C8 β chain (wheat); ΔRMSF > 0.2 Å (blue); 0.1 < ΔRMSF < 0.2 Å (light blue); −0.2 < ΔRMSF < −0.1 Å (orange); and ΔRMSF < −0.2 Å (red). (D) Mapping of ΔRMSF values onto the β chain of TCR A6 in complex with Tax–HLA-A2 (PDB 1QRN) [38]. Color coding: HLA-A2 (gray); Tax peptide (pink); A6 α chain (green); A6 β chain (wheat). ΔRMSF values are color coded as in (C).
The above simulations were compared with earlier in silico studies for the TCR A6 ectodomain in complex with its pMHC ligand, Tax–HLA-A2 [19]. Binding of Tax–HLA-A2 causes rigidification of the CDR1β and CDR3β loops in the A6 β chain, but an increase in flexibility for CDR2β and HV4β (Fig. 8B, D). This difference between A6 and MS2–3C8 may be due to the altered docking orientation of the pMHC with respect to the TCR in the two complexes, such that there are more contacts of the MBP peptide with the CDRβ loops in MS2–3C8 (Fig. 8C, D). For the most part, however, the A6 β chain has a similar pattern of ΔRMSF upon pMHC binding to what is observed for MS2–3C8, with effects extending into the Cβ domain. As in MS2–3C8, these changes are most pronounced in the Cβ FG loop, although there are also other decreases in Cβ chain mobility in A6 (Fig. 8B). The A6 and MS2–3C8 TCRs are representative examples from the MHC class I and class II-restricted TCRs, respectively. Overall, pMHC binding induces comparable long-range changes in conformational dynamics for both TCRs. Furthermore, these simulated allosteric affects are supported by the experimental NMR results presented here for MS2–3C8 as well as by the earlier data for A6 [19].
Network analyses of A6 and MS2–3C8 TCRs and their pMHC complexes were performed. A network is defined as a set of nodes with connecting edges. The nodes in this work represent the amino acid residues. The communities within a network were defined as the sub-structures of the network in which the nodes are more heavily interconnected to each other than to other nodes. It is interesting to see that both TCR A6 and MS2–3C8 share a similar network pattern before and after pMHC binding. For unbound TCRs, each of the Vα and Vβ domains were divided into two communities (Fig. S6A, C), unlike the Cα and Cβ domains, where each domain is a community. Notably, the FG loop is also a community, indicating its relatively independent motions. Upon pMHC binding, the two domains in Vα were merged into one community, with similar observations for the Vβ domain (Fig. S6B, D). We also calculated the betweenness of amino acid residues in the TCR-pMHC complexes for TCR A6 and MS2–3C8. The betweenness of a residue measures the importance of a node in signal transduction networks. We found that TCR A6 and MS2–3C8 have similar yet distinct betweenness patterns, such that TCR A6 utilizes both the α and β chains while MS2–3C8 relies mostly on the β chain (Fig. S7). However, there is considerable overlap of high betweenness residues in the β chain for both TCR A6 and MS2–3C8, consistent with our experimental NMR observations.
Discussion
The results described here are the first to report long-range NMR perturbations in a TCR β chain upon binding of an MHC class II molecule. Comparable studies have also been carried out between TCR ectodomains and MHC class I molecules [18, 19]. Recently, NMR and MD analyses were reported for a complex between the human antiviral TCR A6 and the MHC class I molecule HLA-A2 bound to the Tax antigen from HTLV-1 [19]. Experimentally significant chemical shift and peak intensity perturbations were observed for TCR A6 residues at the binding interface with pMHC as well as more remotely for residues at the Vβ/Vα, Vβ/Cβ, and Cβ/Cα interfaces, just as in MS2–3C8 (Fig. 7E-H). Thus, the overall patterns of pMHC-induced changes observed in the β chains of the MHC class I-restricted TCR A6 [19] and the MHC class II-restricted TCR MS2–3C8 reported here are similar. These patterns also resemble the pattern detected in NMR studies of the unrelated MHC class I-restricted TCR B4.2.3, which recognizes an HIV-1-derived peptide bound to H2-Dd [18].
TCR MS2–3C8, as well as TCRs B4.2.3 and A6 in previous NMR studies [18, 19], were produced by in vitro folding from bacterial inclusion bodies and were therefore not glycosylated, unlike native TCRs. The role, if any, of glycosylation in TCR function is unclear. Although there is no evidence that glycosylation affects TCR affinity for pMHC [25], glycosylation could potentially affect signaling. In one study [26], selective deletion of conserved N-glycosylation sites in Cα or Cβ increased the sensitivity of T cells to activation by tumor cells expressing cognate pMHC ligands. However, the mechanism underlying this enhancement is unknown, and might involve formation of non-physiological TCR multimers on the T cell surface.
A number of residue-specific long-range NMR perturbations are present in both TCRs MS2–3C8 and TCR A6. In MS2–3C8, K118 in the linker region is in close contact with Q225 in the FG loop and both exhibit NMR perturbations upon binding of MBP–HLA-DR4 (Fig. 7C). Similarly, the corresponding residues in TCR A6, K119 and Q226, are close in space and have preferential peak intensity changes when HLA-A2 is bound (Fig. 7G). Further, the perturbations observed at the Cβ/Cα interface are comparable for both TCRs MS2–3C8 and A6. Residues F128(129 in A6), S131(132), E134(135), and T142(143) exhibit NMR perturbations for both A6 and MS2–3C8 in the presence of pMHC (Fig. 7D, H). Residues E134(135) and T142(143) are within hydrogen bonding distance in both the MHC class I- and class II-restricted TCRs. Additionally, R195(196) is perturbed in MS2–3C8(mut1) and A6, but is not assigned in the other two MS2–3C8 variants. This residue forms a salt bridge with E134(135) and is in close contact with T142(143). Interestingly, NMR perturbations have also been observed at the Cβ/Cα interface for residues corresponding to E134 and T142 in the β chain of the mouse TCR B4.2.3 when bound to the mouse MHC class I molecule H2-Dd [18].
Together, these results point to a common set of residues involved in long-range NMR perturbations upon pMHC binding, providing a general framework for allosteric signaling between the pMHC and CD3 binding sites through conserved conformational and/or dynamic changes in Cβ (and possibly Cα), irrespective of the diversity of TCR–pMHC interfaces and of Vβ and Vα sequences. We propose that ligand-induced perturbations in the highly variable CDR loops are conveyed to the C domains through the highly conserved Vβ/Cβ and Vβ/Vα interfaces. This allosteric pathway is supported by NMR analysis of TCRs MS2–3C8 (this study), A6 [19] and B4.2.3 [18], in which many conserved residues in the Vβ/Cβ and Vβ/Vα interfaces undergo changes in chemical shifts and/or peak intensities. A unique structural feature of TCR β chains is the close juxtaposition of Vβ and Cβ domains, in striking contrast to Vα and Cα domains, and to antibody VH and CH1 or VL and CL domains [1]. Extensive interactions between Vβ and Cβ impose a rigid structure on the TCR that contrasts with the flexibility conferred on antibody Fab fragments by the elbow region between VH/CH1 and VL/CL modules. A rigid β chain conformation could enhance propagation of any allosteric changes in the TCR caused by pMHC engagement to CD3 subunits in the TCR–CD3 complex.
The perturbations in Cβ propagate to a region near the C-terminus of the TCR ectodomain that is involved in contacts with CD3 domains in solution [24] and in the cryoEM structure of the TCR–CD3 complex [1, 2]. Even relatively small rearrangements in the orientation of the individual Vβ/Vα, Vβ/Cβ, and Cβ/Cα interfaces, apparent from the NMR results presented, may be sufficient to induce changes in TCR interactions with CD3 and promote downstream T cell signaling events. Importantly, such an allosteric mechanism does not preclude other modes for transmission of signaling events such as mechanosensing [16, 17]. It may well be that these two mechanisms operate in concert to transfer binding at the pMHC interface to structural and dynamic changes near the T cell membrane [1]. According to this view, mechanical force arising from sliding of the T cell over the APC during immune surveillance is relayed to CD3 via conserved allosteric sites in the TCR C domains. In this unified model, force serves to reinforce allosteric communication between TCR and CD3.
A striking result of the MD simulations of pMHC binding to both MS2–3C8 and A6 [19] is ligand-induced rigidification of the Cβ FG loop (Fig. 8A, B). Indeed, in both TCRs, the largest RMSF differences occur in the Cβ FG loop, which protrudes from the interface between the Vβ and Cβ domains. Remarkably, these differences exceed those in any of the CDR loops, including CDR3β, which directly contact pMHC. In agreement with MD simulations, our NMR analysis of TCRs MS2–3C8 and A6 [19] also revealed ligand-induced changes in the Cβ FG loop. The 16-residue FG loop is a distinctive feature of Cβ domains and is not present in Cα or in antibody CH1 or CL domains. In the cryoEM structure of the TCR–CD3 complex [2], the Cβ FG loop is situated over CD3εγ (Fig. 1), but does not make direct contacts, at least not in unliganded TCR–CD3. A biological role for the Cβ FG loop in TCR signaling is supported by functional studies. Deletion of the Cβ FG loop dramatically reduced T cell sensitivity to activation by pMHC [27, 28]. Moreover, the Cβ FG loop has been shown to allosterically regulate the lifetime of TCR–pMHC bonds in single-molecule experiments [29].
Thermodynamic measurements of TCR–pMHC interactions have not revealed an enthalpic or entropic signature for TCR binding, either for MHC class I- or class II-restricted TCRs [30]. Some TCRs bind pMHC with favorable enthalpy changes and unfavorable entropy changes, possibly reflecting overall TCR rigidification, as suggested by MD simulations of TCRs A6 [19] and MS2–3C8 (this study). However, other TCRs bind pMHC with favorable entropy changes. Indeed, the interaction of TCR A6 with Tax–HLA-A2 is entropically favorable [31]. By contrast, another TCR, B7, binds Tax–HLA-A2 with an unfavorable entropy change, even though B7 and A6 dock similarly onto Tax–HLA-A2 and use the same Vβ segment [31]. These seemingly contradictory findings are not too surprising considering that the overall binding thermodynamics associated with molecular recognition reflects a complex interplay of multiple factors, such as conformational changes, burial of hydrophobic and hydrophilic surfaces, formation of hydrogen bonds and salt bridges, and incorporation or expulsion of water and ions [30].
In summary, the pMHC-induced NMR perturbation effects observed here are reproducible, with not only conserved changes among the three human MHC class II-restricted TCR variants in this study but also similar, and in some cases the same, changes in both human and mouse MHC class I-restricted TCRs. Our results therefore suggest that these allosteric affects may be general across all TCRs. This is also supported by extensive amino acid sequence analysis of TCRs, which indicates that while V domains have high sequence diversity, they have more sequence conservation at interfaces with other TCR domains [19]. We note that our experimental analysis focuses entirely on perturbations in the main chain. It is quite possible that larger effects will become apparent when analysis of side chains is considered. Further experimental studies on the effects of pMHC binding on TCR conformation and dynamics will require more detailed analyses using heteronuclear NMR relaxation and other spectroscopic methods, as well as approaches that are probably yet to be developed.
Materials and Methods
Production of TCR MS2–3C8 with isotope-labeled β chain
For binding experiments, the β chain of TCR MS2–3C8 (wild-type and mutants) with U-2H,15N labeling was obtained by inclusion body expression in Escherichia coli BL21(DE3) cells (ThermoFisher) transformed with the vector pET-26b (Novagen) as described previously [24]. D2O was purchased from Isotec. [15N]NH4Cl (Cambridge Isotopes) was the sole nitrogen source and [2H,12C]glucose (Cambridge Isotopes) was the sole carbon source. Unlabeled TCR MS2–3C8 α chain was produced as inclusion bodies in E. coli BL21(DE3) cells as described [24]. TCRs MS2–3C8(wt), MS2–3C8(mut1), and MS2–3C8(mut2) were prepared by in vitro folding of unlabeled α and labeled β chains as described [24]. Correctly folded TCR heterodimers were purified using sequential MonoQ and Superdex S-75 columns (GE Healthcare).
Production of MBP-HLA-DR4
A codon-optimized gene encoding the extracellular region of the HLA-DR4 α chain (residues 1–182) was synthesized (GenScript) and cloned into the mammalian expression vector pcDNA3.4-TOPO with a C-terminal Fos leucine zipper (GGGGGLTDTLQAETDQLEDEKSALQTEIANLLKEKEKLEFILAA) and His6 tag. A codon-optimized gene encoding the extracellular region of the HLA-DR4 β chain (residues 1–190) was cloned into pcDNA3.4-TOPO with a C-terminal Jun leucine zipper (GGGGRIARLEEKVKTLKAQNSELASTANMLREQVAQLKQKVMNH) and an N-terminal sequence comprising MBP 114–126 (FSWGAEGQRPGFG) followed by a 16-mer peptide linker (SGGGSLVPRGSGGGGS). The HLA-DR4 α and β chain constructs were mixed in a 1:1 molar ratio and transfected into Expi293F cells at 2.5 × 106 cells/ml using an ExpiFectamine 293 Transfection Kit (ThermoFisher). The culture was harvested after 96 h and MBP–HLA-DR4 purified from the supernatant using sequential Ni2+-NTA and Superdex 200HR columns (GE Healthcare).
NMR spectroscopy
The α[unlabeled]β[2H, 15N]-MS2–3C8 TCR samples employed in NMR experiments were 100 μM in 50 mM sodium phosphate buffer (pH 7.0) with 100 mM sodium chloride and 1% v/v protein inhibitor cocktail added (Roche). For each MS2–3C8 mutant, an NMR spectrum was first acquired on the unbound TCR at 25 °C using a Bruker Avance III 900 MHz spectrometer fitted with a [1H/13C/15N]-cryoprobe and Z-axis gradients. NMR spectra were processed utilizing NMRPipe [32] and analyzed with SPARKY [33]. Unlabeled MBP–HLA-DR4 (1.0–2.0 mol eq) in the above NMR buffer was then added to MS2–3C8 TCR samples and 2D 1H-15N TROSY-HSQC spectra were recorded. Sample addition was carried out as described previously [19] and spectra of pMHC-bound states were compared with control spectra of the unbound TCR state under the same conditions. Chemical shift perturbations were determined using Δδtotal=((WHΔδH)2 + (WNΔδN)2)1/2, where ΔδH and ΔδN are 1H and 15N shift changes between the bound and unbound states of MS2–3C8 TCR, and the weighting factors are WH=1 and WN=0.2. Peak intensity changes were measured utilizing SPARKY. The range of uncertainty in chemical shifts is Δδtotal≤0.005 ppm. This was based on measurement of amide chemical shifts in different samples of the same TCR and is consistent with our earlier observations [24]. The error in peak intensity is approximately ±2%. This was estimated from the ratio of the peak intensity for baseline noise and the average peak intensity of amide protons. The baseline noise level was determined from the median of 30 randomly sampled peak intensities using an automated routine in SPARKY. For both chemical shift and peak intensity analysis, mean values were determined and changes greater than the mean plus 1SD were mapped onto the MS2–3C8 structure as described previously [19]. Structures were analyzed and illustrated with Pymol (Schrodinger).
Molecular dynamics simulations and analysis
The X-ray structure of the complex between TCR MS2–3C8 and MBP–MBP-HLA-DR4 (PDB 3O6F) [23] was utilized as a starting point for the MD simulations. The missing atoms and residues in the initial PDB structure were added using CHARMM. The C/D chains in the complex were used for the simulation of unbound TCR. The initial structure was solvated with TIP3P water molecules. The system was neutralized with sodium and chloride ions and the total ion concentration was 150 mM. The TCR–pMHC complex had a box size of 147×161×148 Å3 with 339,720 atoms and the TCR had a box size of 98×96×89 Å3 with 92,177 atoms. Energy minimization proceeded with 5000 conjugate gradient steps such that the protein was fixed whereas counterions and water molecules were movable. All atoms were then allowed to move in a further energy minimization of 5000 conjugate gradient steps. A series of dynamic cycles were performed to gradually relax each system in the equilibration stage. All simulations were carried out with the NPT ensemble at 300 K for 1200 ns in the production stage. The NAMD program [34] was employed for all MD simulations with a CHARMM27 force-field (CHARMM22 plus CMAP correction) [35]. The switching function was utilized to calculate short-range van der Waals interactions, with a twin range cut-off of 10.0 and 12.0 Å. The Particle Mesh Ewald method was used to calculate long-range electrostatic interactions, with a cut-off of 12.0 Å. The initial 200 ns of simulations were not included in RMSF calculations to reduce any effects due to crystal packing. Therefore, only conformations from the last 1000 ns of 1200 ns MD simulation trajectories were employed for analysis of RMSF. Each simulation consisted of 10000 frames. The Vα, Vβ, Cα and Cβ domains were superimposed separately to determine how interdomain motion effected RMSF values. As a reference, the averaged structures from the last 1000 ns were utilized.
Weighted network and community analysis
An edge between two nodes was defined if any heavy atoms from the two residues/water molecules are within 4.5 Å of each other in over 75% of the analyzed frames. Neighboring residues in sequence are not considered to be in contact because they will form numerous trivial suboptimal paths in the weighted network. The dynamical networks were constructed based on the MD trajectories. The community was identified by the Girvan-Newman algorithm [36]. The length of a path between two distant nodes is the sum of the edges weights between consecutive nodes along the path. The shortest path is obtained by optimization of this length. The betweenness of an edge is the number of pairwise optimal paths that cross that edge, and the betweenness of a node is a normalized percentage of optimal paths that cross the node. The network and community were plotted using the NetworkView [37] module in VMD.
Supplementary Material
Highlights.
How pMHC ligation triggers early T cell activation is not well understood.
Long-range pMHC-induced changes were detected in a MHC class II TCR by NMR and MD.
Perturbations in the TCR β chain extended to the Vβ/Vα, Vβ/Cβ, and Cβ/Cα regions.
The long-range effects are similar to those seen previously for two MHC class I TCRs.
Conserved allosteric sites in TCRs may play a role in the initiation of signaling.
Acknowledgements
This work was supported by National Institutes of Health Grant AI29893 (to R.A.M. and J.O.). The NMR facility is jointly supported by the University of Maryland, the National Institute of Standards and Technology, and a grant from the W. M. Keck Foundation. Molecular dynamics simulations were carried out utilizing the high-performance computational facilities of the Biowulf PC/Linux cluster at the National Institutes of Health, Bethesda, Maryland. It has also been funded in whole or in part with Federal funds from the Frederick National Laboratory for Cancer Research, National Institutes of Health, under contract HHSN261200800001E. This research was supported [in part] by the Intramural Research Program of NIH, Frederick National Lab, Center for Cancer Research.
Abbreviations used
- TCR
T cell receptor
- MHC
major histocompatibility complex
- pMHC
peptide–MHC
- APC
antigen-presenting cell
- ITAM
immunoreceptor tyrosine-based activation motif
- MBP
myelin basic protein
- H/D exchange
hydrogen/deuterium exchange
- MD
molecular dynamics
- HTLV-1
human T cell lymphotropic virus-1
- KD
dissociation constant
- TROSY
transverse relaxation optimized spectroscopy
- V
variable
- C
constant
- CDR
complementarity-determining region
- HV4
fourth hypervariable loop
- CSP
chemical shift perturbation
- HSQC
heteronuclear single quantum coherence
- RMSF
root mean square fluctuation
- RMSD
root mean square deviation
- PDB
Protein Data Bank
Footnotes
Declaration of interests
☒The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Mariuzza RA, Agnihotri P, & Orban J. (2020). The structural basis of T-cell receptor (TCR) activation: an enduring enigma. J. Biol. Chem 295, 914–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Dong D, Zheng L, Lin J, Zhang B, Zhu Y, Li N, Xie S, Wang Y, Gao N, & Huang Z. (2019). Structural basis of assembly of the human T cell receptor-CD3 complex. Nature 573, 546–552. [DOI] [PubMed] [Google Scholar]
- [3].Blum JS, Wearsch PA, & Cresswell P. (2013). Pathways of antigen processing. Annu. Rev. Immunol 31, 443–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Zhang H, Cordoba S-P, Dushek O, & van der Merwe PA (2011). Basic residues in the T-cell receptor ζ cytoplasmic domain mediate membrane association and modulate signaling. Proc. Natl. Acad. Sci. USA 108, 19323–19328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Guo X, Yan C, Li H, Huang W, Shi X, Huang M, Wang Y, Pan W, Cai M, Li L, Wu W, Bai Y, Zhang C, Liu Z, Wang X, Zhang XF, Tang C, Wang H, Liu W, Ouyang B, Wong CC, Cao Y, & Xu C. (2017). Lipid-dependent conformational dynamics underlie the functional versatility of T-cell receptor. Cell Res. 27, 505–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Gagnon E, Schubert DA, Gordo S, Chu HH, & Wucherpfennig KW (2012) Local changes in lipid environment of TCR microclusters regulate membrane binding by the CD3ε cytoplasmic domain. J. Exp. Med 209, 2423–2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Shi X, Bi Y, Yang W, Guo X, Jiang Y, Wan C, Li L, Bai Y, Guo J, Wang Y, Chen X, Wu B, Sun H, Liu W, Wang J, & Xu C. (2013). Ca2+ regulates T-cell receptor activation by modulating the charge property of lipids, Nature 493, 111–115. [DOI] [PubMed] [Google Scholar]
- [8].Natarajan K, Jiang J, May NA, Mage MG, Boyd LF, McShan AC, Sgourakis NG, Bax A, & Margulies DH (2018). The role of molecular flexibility in antigen presentation and T cell receptor-mediated signaling. Front. Immunol 9, 1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Buckle AM, & Borg NA (2018). Integrating experiment and theory to understand TCR-pMHC dynamics. Front. Immunol 9, 2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Schamel WW, Alarcon B, & Minguet S. (2019). The TCR is an allosterically regulated macromolecular machinery changing its conformation while working. Immunol. Rev 291, 8–25. [DOI] [PubMed] [Google Scholar]
- [11].Connolly A, & Gagnon E. (2019). Electrostatic interactions: From immune receptor assembly to signaling. Immunol. Rev 291, 26–43. [DOI] [PubMed] [Google Scholar]
- [12].Mallis RJ, Brazin KN, Duke-Cohan JS, Hwang W, Wang JH, Wagner G, Arthanari H, Lang MJ, & Reinherz EL (2019). NMR: an essential structural tool for integrative studies of T cell development, pMHC ligand recognition and TCR mechanobiology. J. Biomol. NMR 73, 319–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Davis SJ, & van der Merwe PA (2006). The kinetic-segregation model: TCR triggering and beyond. Nat. Immunol 7, 803–809. [DOI] [PubMed] [Google Scholar]
- [14].Chang VT, Fernandes RA, Ganzinger KA, Lee SF, Siebold C, McColl J, Jönsson P, Palayret M, Harlos K, Coles CH, Jones EY, Lui Y, Huang E, Gilbert RJC, Klenerman D, Aricescu AR, & Davis SJ (2016). Initiation of T cell signaling by CD45 segregation at ‘close contacts’. Nat. Immunol 17, 574–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].van der Merwe PA, & Dushek O. (2011). Mechanisms for T cell receptor triggering. Nat. Rev. Immunol 11, 47–55. [DOI] [PubMed] [Google Scholar]
- [16].Kim ST, Takeuchi K, Sun ZY, Touma M, Castro CE, Fahmy A, Lang MJ, Wagner G, & Reinherz EL (2009). The αβ T cell receptor is an anisotropic mechanosensor. J. Biol. Chem 284, 31028–31037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Brazin KN, Mallis RJ, Das DK, Feng Y, Hwang W, Wang JH, Wagner G, Lang MJ, & Reinherz EL (2015). Structural features of the αβTCR mechanotransduction apparatus that promote pMHC discrimination. Front. Immunol 6, 441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Natarajan K, McShan AC, Jiang J, Kumirov VK, Wang R, Zhao H, Schuck P, Tilahun ME, Boyd LF, Ying J, Bax A, Margulies DH, & Sgourakis NG (2017). An allosteric site in the T-cell receptor Cβ domain plays a critical signalling role. Nat. Comm 8, 15260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Rangarajan S, He Y, Chen Y, Kerzic MC, Ma B, Gowthaman R, Pierce BG, Nussinov R, Mariuzza RA, & Orban J. (2018). Peptide-MHC (pMHC) binding to a human antiviral T cell receptor induces long-range allosteric communication between pMHC- and CD3-binding sites. J. Biol. Chem 293, 15991–16005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hawse WF, Champion MM, Joyce MV, Hellman LM, Hossain M, Ryan V, Pierce BG, Weng Z, Baker BM (2012). Cutting edge: Evidence for a dynamically driven T cell signaling mechanism. J. Immunol 188, 5819–5823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Brameshuber M, Kellner F, Rossboth BK, Ta H, Alge K, Sevcsik E, Göhring J, Axmann M, Baumgart F, Gascoigne NRJ, Davis SJ, Stockinger H, Schütz GJ, & Huppa JB (2018) Monomeric TCRs drive T cell antigen recognition. Nat. Immunol 19, 487–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Qi Q, Liu Y, Cheng Y, Glanville J, Zhang D, Lee JY, Olshen RA, Weyand CM, Boyd SD, & Goronzy JJ (2014). Diversity and clonal selection in the human T-cell repertoire. Proc. Natl. Acad. Sci. USA 111, 13139–13144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Yin Y, Li Y, Kerzic MC, Martin R, & Mariuzza RA (2011). Structure of a TCR with high affinity for self-antigen reveals basis for escape from natural selection. EMBO J. 30,1137–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].He Y, Rangarajan S, Kerzic M, Luo M, Chen Y, Wang Q, Yin Y, Workman CJ, Vignali KM, Vignali DAA, Mariuzza RA, & Orban J. (2015). Identification of the docking site for CD3 on the T cell receptor β chain by solution NMR. J. Biol. Chem 290, 19796–19805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Rossjohn J, Gras S, Miles JJ, Turner SJ, Godfrey DI, McCluskey J. (2015). T cell antigen receptor recognition of antigen-presenting molecules. Annu. Rev. Immunol 33, 169–200. [DOI] [PubMed] [Google Scholar]
- [26].Kuball J, Hauptrock B, Malina V, Antunes E, Voss RH, Wolfl M, Strong R, Theobald M, Greenberg PD (2009). Increasing functional avidity of TCR-redirected T cells by removing defined N-glycosylation sites in the TCR constant domain. J. Exp. Med 206, 463–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sasada T, Touma M, Chang HC, Clayton LK, Wang JH, & Reinherz EL (2002). Involvement of the TCR Cβ FG loop in thymic selection and T cell function. J. Exp. Med 195, 1419–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Touma M, Chang HC, Sasada T, Handley M, Clayton LK, & Reinherz EL (2006). The TCR Cβ FG loop regulates αβ T cell development. J. Immunol 176, 6812–6823. [DOI] [PubMed] [Google Scholar]
- [29].Das DK, Feng Y, Mallis RJ, Li X, Keskin DB, Hussey RE, Brady SK, Wang JH, Wagner G, Reinherz EL, & Lang MJ (2015). Force-dependent transition in the T-cell receptor β-subunit allosterically regulates peptide discrimination and pMHC bond lifetime. Proc. Natl. Acad. Sci. USA 112, 1517–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Armstrong KM, Insaidoo FK, Baker BM (2008). Thermodynamics of T cell receptor–peptide/MHC interactions. J. Mol. Recognit 21, 275–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Davis-Harrison RL, Armstrong KM, Baker BM (2005). Two different T cell receptors use different thermodynamic strategies to recognize the same peptide/MHC ligand. J. Mol. Biol 346, 533–550. [DOI] [PubMed] [Google Scholar]
- [32].Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, & Bax A. (1995). NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- [33].Goddard TD, & Kneller DG (2004). SPARKY 3, University of California San Francisco. [Google Scholar]
- [34].Kale L, Skeel R, Bhandarkar M, Brunner R, Gursoy A, Krawetz N, Phillips J, Shinozaki A, Varadarajan K, & Schulten K. (1999). NAMD2: greater scalability for parallel molecular dynamics. J. Comp. Phys 151, 283–312. [Google Scholar]
- [35].Mackerell AD, Feig M, & Brooks CL (2004). Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem 25,1400–1415. [DOI] [PubMed] [Google Scholar]
- [36].Girvan M & Newman ME (2002). Community structure in social and biological networks. Proc. Natl. Acad. Sci. USA 99, 7821–7826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Eargle J & Luthey-Schulten Z. (2012). NetworkView: 3D display and analysis of protein·RNA interaction networks. Bioinformatics 28, 3000–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ding YH, Baker BM, Garboczi DN, Biddison WE, & Wiley DC (1999). Crystal structure of human A6 TCR complexed with HLA-A2 bound to altered HTLV-1 Tax peptide P6A. Immunity 11, 45–56. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.