

Abstract
Traumatic brain injury (TBI) triggers both central and peripheral inflammatory responses. Existing pharmacological drugs are unable to effectively and quickly target the brain inflamed regions, setting up a major roadblock towards effective brain trauma treatments. Nanoparticles (NPs) have been used in multiple diseases as drug delivery tools with remarkable success due to their rapid diffusion and specificity in the target organ. Here, leukocyte‐based biomimetic NPs are fabricated as a theranostic tool to directly access inflamed regions in a TBI mouse model. This NP systemic delivery is visualized using advanced in vivo imaging techniques, including intravital microscopy and in vivo imaging system. The results demonstrate selective targeting of NPs to the injured brain and increased NPs accumulation among the peripheral organs 24 h after TBI. Interestingly, increased microglial proliferation, decreased macrophage infiltration, and reduced brain lesion following the NPs treatments compared to sham vehicle‐treated mice are also found. In summary, the results suggest that NPs represent a promising future theranostic tool for TBI treatment.
Keywords: biomimicry, blood‐brain barrier, inflammation, leukosomes, liposomes
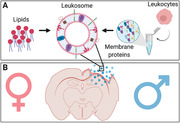
Leukosomes are fabricated using lipids mixed with membrane proteins extracted from cultivated leukocytes. Leukosomes are tested in vivo using a traumatic brain injury (TBI) mouse model and demonstrated increased targeting to inflamed organs following TBI in both sexes.
1. Introduction
Traumatic brain injury (TBI) is a serious and growing health problem in the United States, accounting for approximately 2.5 million emergency room visits, 56 000 deaths per year,[ 1 ] and more than $60 billion in direct medical expenses in palliative care and cognitive rehabilitation.[ 2 ] Immediately after injury, there is a breakdown of the blood‐brain barrier (BBB) and the neuroinflammatory cascade that triggers brain degeneration is activated. Common pathologic consequences of TBI include hematoma, subarachnoid hemorrhage, neuroinflammation, or axonal injury.[ 3 ] Most of these brain injuries also result in long‐term physical, emotional, and cognitive consequences. Yet, despite posing a significant threat to public health, TBI has no effective pharmacotherapy.[ 4 ]
Access to the brain from the periphery is generally regulated by the BBB, which presents a formidable obstacle for small and macromolecular cells to enter the brain. Neuroinflammation involves several molecular and cellular mediators, including the activation of resident microglia and the release of inflammatory cytokines and chemokines at the injury site.[ 5 ] Notably, not only does neuroinflammation occur after TBI, peripheral organs become inflamed due to the systemic inflammation post‐TBI[ 6 ] as we have previously demonstrated using the same TBI mouse model while assessing hepatic inflammation.[ 7 ] Previous studies have focused on central inflammation and have neglected the importance of immune cells that migrate from the periphery and play an important role in aggravating brain damage. After TBI, mechanical damage immediately ruptures the BBB, causing extravasation of plasma proteins, leukocytes,[ 8 ] T cells, monocytes, or macrophages to contribute to neuroinflammation,[ 8c,d ] neurotoxicity, neuronal death, and the extent of brain injury.[ 9 ] More specifically, the rapid invasion of inflammatory leukocytes (CD11b+/CD45+) at the site of damage leads to increased neuropathology[ 10 ] accompanied by activation of resident microglia and macrophages[ 11 ] after TBI.
As we and others have observed, activated peripheral immune cells and platelets are mobilized through the cerebral vasculature, penetrating the brain parenchyma after induced damage.[ 12 ] This peripheral inflammation affects many organs after TBI, causing inflammation and injury at the local level, such as the liver,[ 7 ] heart, lungs,[ 13 ] spleen,[ 14 ] intestines,[ 15 ] and gut microbiome.[ 16 ] The peripheral inflammatory effect should be studied in parallel with TBI. As such, this study explores the infiltration potential of nanoparticles (NPs) in peripheral organs that may be affected by TBI. A reduction in inflammation in the acute phase has been shown to reduce chronic inflammation in TBI models, and part of this inflammation comes from the periphery.[ 17 ] Therefore, reducing central and peripheral inflammation is crucial in restoring the injured brain. The transient BBB breakdown after TBI increases permeability within the injury pericontusional region and may offer a unique opportunity to deliver drugs. The use of NPs technology could bring novel opportunities to treat the damaged brain. NPs are used as a vehicle transporter of pharmacological treatments in various diseases. During the last few years, nanomedicine has also been used to treat brain disorders.[ 18 ] Several nanoformulations have been developed in recent decades to improve the solubility and pharmacokinetics of different drugs, improving drug targeting to reduce side effects.[ 19 ] However, brain‐targeted nanotherapeutics have numerous biological roadblocks, such as their inability to cross the BBB, uptake by the mononuclear phagocyte system, elimination by Kupffer cells in the liver, and glomerular filtration in the kidneys before reaching their target zone.[ 20 ] Biomimetic strategies represent a paradigm shift in NP design, enabling next‐generation platforms to interact and effectively affect the behavior of complex biological systems. Our group recently developed a novel biomimetic formulation that aims to mimic the composition of the cell membrane of leukocytes in their lipid (liposomes [Lipo]) and protein (leukosomes [Leuko]) compositions.[ 21 ] Specifically, Leuko target activated endothelia through CD11b and evade mononuclear phagocytic system (MPS) uptake through CD45, both of which are present on the surface of Leukos.[ 22 ] Moreover, Leuko bridge the gap between synthetic NPs (i.e., Lipo) and native cells (i.e., immune cells)[ 23 ] and they substantially advance the current paradigm for targeting and protection of NPs, which until now has been based solely on synthetic routes.[ 24 ] Also, we have previously elucidated also sex differences in the inflammatory response using the same TBI model.[ 5b ] Additional studies have highlighted the need to consider sex as a biological variable following TBI, specifically for any therapy that modifies the inflammatory response in females and males.[ 25 ] We explore in our study how sex affects nanotherapeutics outcomes after injury. These findings are essential to developing a more effective sex‐dependent TBI treatment.
In this study, we first characterized the physicochemical and biomimetic properties of Leuko and Lipo. Then, we assessed these two NPs groups’ capability to target the damaged brain tissue and peripheral organs in a more systemic approach in a sex‐dependent manner in a mouse model of TBI. Finally, we assessed changes in the inflammatory response and lesion size, along with macrophage infiltration following TBI. Thus, our results suggest that NPs may be a useful theranostic tool for targeting the damaged brain and inflamed organs in response to TBI.
2. Results and Discussion
2.1. Liposomes and Leukosomes Fabrication
During NP fabrication, one step distinguishes Lipo and Leuko: the addition of leukocytes’ membrane protein during the lipids thin‐film rehydration step (Figure 1a). Lipo and Leuko physicochemical and biomimetic properties were carefully characterized, as was reported in our previous work.[ 24, 26 ] Finally, NPs were systemically injected after TBI to analyze NP accumulation in different organs (Figure 1b) 24 h post‐TBI. We confirmed that adding 0.058 mg mL‐1 of leukocyte membrane proteins during the fabrication process did not affect the size or polydispersity index (PDI) of NPs (Figure 2a,b). However, we did notice that when membrane proteins were integrated into the Leuko membrane, the surface charge (i.e., zeta potential [ZP]) decreased by 1.7‐fold compared to the Lipo group (Figure 2c).
Figure 1.
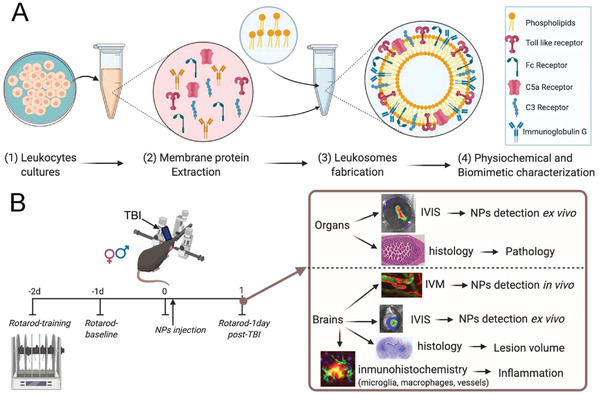
Schematic of nanoparticles’ (NPs) fabrication, characterization, and in vivo and ex vivo experiments. A1‐3) Leukosomes (Leuko) were fabricated using membrane protein extracted from cultivated leukocytes. A4) After fabrication, NPs were characterized for their physicochemical and biomimetic properties. B) NPs were tested in vivo using a traumatic brain injury (TBI) mouse model. They demonstrated increased targeting to inflamed organs following TBI, as assessed by two in vivo imaging techniques (i.e., in vivo imaging system [IVIS] and intravital microscopy [IVM]) followed by motor test (rotarod) and several histological and immunohistochemical techniques. This schematic was created using Biorender.com.
Figure 2.
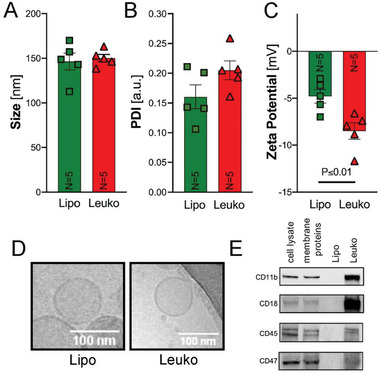
Nanoparticles’ (NPs) properties characterization. After fabrication, NPs were characterized for their physicochemical and biological properties using dynamic light scattering. No significant changes in A) size and B) polydispersity index (PDI) were observed. However, a significant decrease in C) zeta potential for leukosomes (Leuko) compared to liposomes (Lipo) was noticed. D) Representative cryo‐TEM images of Lipo and Leuko verified that no morphological changes occurred following membrane protein integration to the NPs. Scale bars = 100 nm. E) Western blots for leukocyte membrane protein markers: CD11b, CD18, CD47, and CD45 indicated their membrane integration in Leuko but absence in Lipo. Results are shown as mean ± SEM. Unpaired t‐test was used to determine statistical probabilities *p ≤ 0.05 among means considered statistically significant, n = 5.
Next, cryo‐transmission electron microscopy (TEM) images of both Lipo and Leuko were acquired. These images demonstrated a similar bilayer structure morphology in both NP groups (Figure 2d), implying that NP morphology was not affected by the protein integration. Then, we verified the presence of unique leukocyte proteins postfabrication. Four leukocyte membrane protein markers that are crucial for leukocyte inflammation targeting (e.g., CD11b, CD18, CD45, and CD47) were detected using western blot in the Leuko group, but not in the Lipo group (Figure 2e). Following this fabrication process and routine characterization protocol,[ 27 ] we demonstrated the fabrication of both Lipo and Leuko in a reproducible manner. Finally, we also determined that leukocyte membrane proteins were integrated into the NP bilayer and not absorbed outside or encapsulated inside for several reasons: a) the size and morphology of NPs would be affected if the proteins were absorbed on the NPs surface, b) the decreased ZP confirmed the presence of membrane protein on the NPs surface, and c) nonintegrated proteins would be separated during the purification step of dialysis. Subsequently, we also tested four leukocyte receptors to prove the presence of these proteins in the Leuko group and not in the Lipo group. These proteins, presence in the Leuko group is crucial in order to pass their unique natural biological activity, which is relevant for both inflammation and MPS evasion in the context of TBI. CD11b and CD18 are two subunits of the CD11 receptor, which is involved in numerous adhesion‐related associations between inflammatory cells such as monocytes, macrophages, natural killer cells, and granulocytes. However, it is also strongly displayed on activated microglia during the neurodegenerative process.[ 28 ] Thus, these receptors’ presence in the Leuko group should help enhance the targeting, interact with the trauma's immune niche, and, together with CD45 and CD47, avoid the phagocytosis from the MPS.[ 22 ]
2.2. In Vivo NPs Targeting to the Injured Brain
To assess NPs targeting to the injured brain upon systemic administration in vivo, the same amount of fluorescent Cy5.5‐1,2‐Distearoyl‐sn‐glycerol‐3‐phosphorylethanolamine (DSPE, Avanti) lipid (0.05 µL for 1 mL NPs formulation) was added to the NPs during the fabrication process for both Lipo and Leuko groups (see Experimental section for details). Although the same amount of fluorescent lipid was added to each NPs formulation, Leuko fluorescence was 1.1‐fold less than Lipo (Figure S1, Supporting Information). This factor was taken into consideration when we quantified the NPs biodistribution in vivo. The different fluorescence intensity can be explained by the fact that the Cy5.5 fluorophore is conjugated to a DSPE lipid that facilitates its integration into the hydrophobic region of the NP membrane.[ 29 ] Hence, leukocyte proteins in that same region might interfere with the fluorescent lipid integration. As a result, fewer fluorescent lipids were integrated into the Leuko membrane, which led to lower fluorescence intensity. For the TBI model, both mice sexes were injured, causing moderate‐to‐severe brain damage in the right somatosensory cortex (Figure 1b). Shortly after, either phosphate buffered saline (PBS, i.e., Vehicle [Vh]), Lipo, or Leuko were retro‐orbitally injected. After 24 h, animals under anesthesia were imaged, showing an accumulation of NPs only in the brains of the NPs treated TBI animals in both sexes (Figure 3a). Specifically, NPs can only reach the brain when the BBB is breached after TBI (i.e., mechanic impact), thus the usage of these fluorescent NPs can be potentially used as a diagnostics tool. Our TBI mouse model shows reproducible acute aspects of focal brain injury, including the BBB disruption and the activation of secondary injury processes due to inflammation.[ 5, 30 ] Moreover, TBI triggers leaky blood vasculature within the lesion area,[ 31 ] similar to a solid tumor's leaky vasculature.[ 32 ] Therefore, passive accumulation of both NPs formulations at the brain site was expected, as demonstrated in previous TBI works.[ 33 ] However, the significantly enhanced targeting of Leuko into the brain compared to Lipo may be due to different factors. For example, the inflamed endothelium surrounding the injury site may improve Leuko targeting, as demonstrated in local and tumor inflammation models.[ 26, 34 ]
Figure 3.
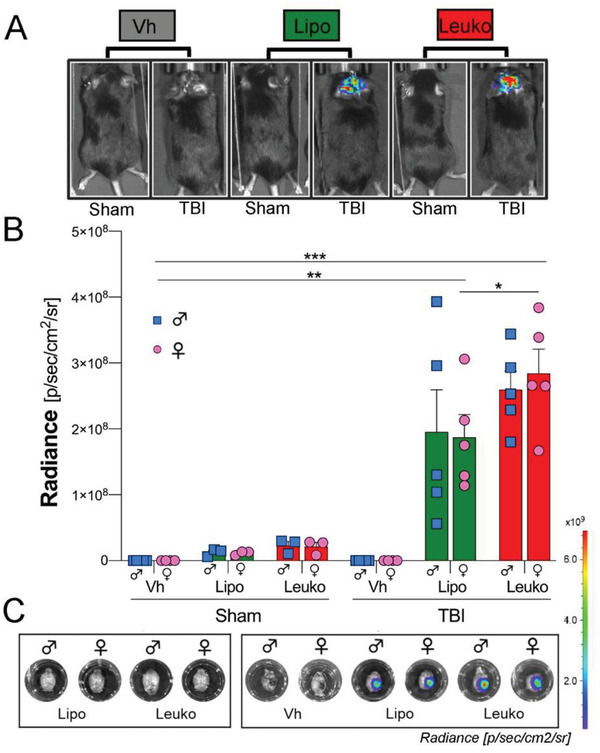
Nanoparticle (NPs) in vivo targeting of the injured brain following traumatic brain injury (TBI). A) Images show sham and TBI mice after being administrated with fluorescent NPs and vehicle (Vh). B,C) NPs groups demonstrated significantly higher brain targeting after TBI than Sham mice treated with the same group of NPs after 24 h following systemic administration, as verified using in vivo imaging system (IVIS). Notably, the female leukosomes (Leuko) group demonstrated higher brain vasculature targeting after TBI compared to the sham female liposomes (Lipo) group. Results are shown as mean ± SEM. Two‐way ANOVA followed by Tukey's multiple comparison test was used to determine statistical probabilities. *p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, n = 5.
Interestingly, our in vivo imaging system (IVIS) analysis revealed NPs accumulation at the brain injury site in both groups 24 h post‐injury, without any evidence of NPs diffusion in other brain regions. Moreover, no differences in NPs brain targeting were observed when comparing females’ and males’ injured brains ex vivo in each of the NPs groups. Notably, a statistically significant higher (1.5‐fold) brain accumulation of Leuko compared to Lipo was noticed in female TBI mice. For male TBI mice a nonsubstantial 1.3‐fold uptake of Leuko compared to Lipo was observed (Figure 3b,c). This lack of significant difference among the male NPs treated groups can be explained by the low resolution of the IVIS measurements. The fluorescence measured by the IVIS was emitted by the whole organ (i.e., tissue and blood vessels) and not only from the inflamed endothelial sites.
2.3. NPs Filtering Organs Biodistribution after TBI
To further determine whether adding leukocyte membrane proteins to NPs impacts their biodistribution in vivo after TBI, the mice filtering organs (e.g., lungs, spleen, kidneys, and liver) were collected, and the NPs fluorescence in these organs was measured ex vivo using IVIS. The highest fluorescent signal among the collected organs was detected in the liver, followed by the spleen, kidneys, and lungs (Figure 4a,b). Subsequently, to evaluate if both Leuko and Lipo were tolerated in vivo, the mentioned organs were collected, washed, fixed, sectioned, and stained using hematoxylin and eosin (H&E) to evaluate tissue damage. When compared with tissue from a control PBS‐injected mouse, no abnormal or pathological morphology differences were observed in any of the two NPs groups’ organs. Additionally, there were no significant sex differences (Figure S2, Supporting Information). However, we did notice a lower accumulation of NPs in the lungs overall. This lower accumulation was expected due to the small size of NPs (<200 nm). Notably, the differential biodistribution between TBI and sham groups was unexplored in previous reports.[ 35 ] Nevertheless, it has been proposed that TBI also induces alterations in the bloodstream and peripheral organs.[ 36 ]
Figure 4.
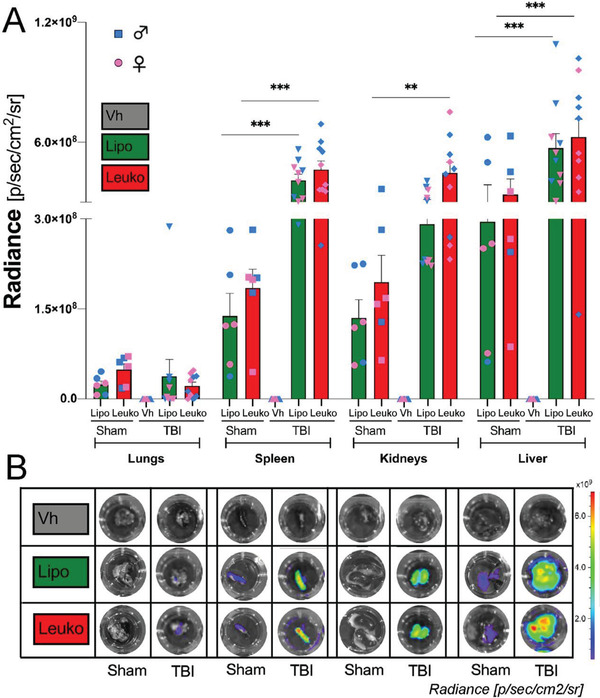
Nanoparticles (NPs) in vivo targeting of peripheral organs following traumatic brain injury (TBI). TBI mice were systemically administrated fluorescent NPs. A) NPs groups demonstrated significantly higher peripheral organ targeting 24 h after TBI and NPs administration compared to sham mice as verified B) in vivo and ex vivo using in vivo imaging system (IVIS). Interestingly, statistically significant higher levels of NPs accumulation were observed in the filtering organs (e.g., spleens, kidneys, and livers) of TBI mice. Results are shown as mean ± SEM. Two‐way ANOVA followed by Tukey's multiple comparison test was used to determine statistical probabilities. *p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, n = 5 (TBI), n = 3 (Sham).
Although this is an emerging area, it is interesting to note that NPs accumulate in filtering organs in the TBI group, but not in the sham group. Many questions arose after this observation. We assume, it could be due to the augmenting clearance, predominantly renal, after injury;[ 38 ] however, this phenomenon could explain the kidney accumulation and should be reported in other TBI models. Unfortunately, this information has mainly been considered secondary in previous studies.[ 39 ] There are many possibilities, but they are beyond the scope of the current study. Still, this secondary target could be exploited by future peripheral anti‐inflammatory approaches to treat TBI as a systemic condition, and not just as brain inflammation.[ 40 ]
2.4. NPs in the Damaged Brain and Their Distribution in the Cerebral Vessels after TBI
To further assess the specific targeting area in both NPs groups, we used Intravital Microscopy (IVM) analysis to focus on specific brain regions in vivo. Specifically, we focused on the activated brain vasculature immediately (Figure 5a,b) and 24 h after (Figure 5c,d) NPs administration. This analysis verified qualitatively higher Leuko inflamed endothelial targeting, similar to what was previously observed by our group in different disease models.[ 27 ] Interestingly, we also noticed that Leuko targeting is localized to the vasculature wall and engulfed by the surrounding brain tissue (Figure S3, Supporting Information). In contrast, we did not qualitatively observe NPs diffusion in uninjured brain tissue, suggesting that probably paracellular diffusion events, as the one detected from Leuko, are limited to the area surrounding the injury for at least 24 h (i.e., the time that passed from the NPs administration). This last observation was also verified using immunofluorescence (IF) staining (Figure 5e–j). Here, as well, we did not detect sex differences within the NPs groups. However, Leuko were more effective in targeting the female group's injury area than Lipo, possibly due to the better inflammatory response in females compared to males following brain injury as was reported by Villapol et al.[ 5b ]
Figure 5.
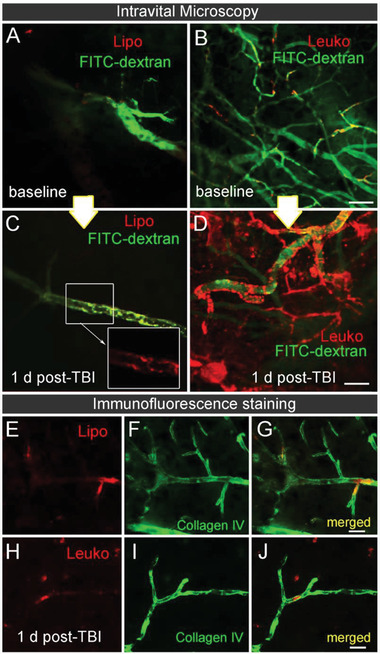
Cerebrovascular targeting of leukosomes in injured mice using intravital microscopy (IVM). TBI mice were systemically injected with NPs immediately after the injury. IVM images capturing the cerebrovascular targeting of A,B) NPs before (baseline) and C,D) 24 h after TBI and NPs administration were acquired. Higher leukosomes (Leuko) adherence to the lesion blood vessel walls was observed compared to the liposomes (Lipo) injected group. E–J) The presence of NPs inside the cerebral vasculature 24 h post‐injection is demonstrated using representative immunofluorescence images of blood vessels (Collagen IV, green) and NPs (red). Scale bar: A–D) 20 µm and E–J) 50 µm.
2.5. NPs Modulate the Neuroinflammatory Response Following TBI
For quantitative analysis, we focused on the primary somatosensory cortex region, which is located immediately adjacent to the injury site (Figure 6a,c). We found that TBI in NP‐treated males caused a rapid increase in the number of Iba‐1 positive cells at 24 h posttrauma. Significant differences were observed in both the Lipo‐males and the Leuko‐females compared to their respective Vh counterparts (Figure 6b). The increase in the number of Iba‐1 positive cells may be associated with an increase in protective and repair microglia[ 41 ] that would facilitate the protective function of NPs, but further analysis is necessary to determine this process. It is possible that the accumulation of local microglia was directly induced by the presence of NPs at the injury site, although not investigated elsewhere. TBI induces the rapid infiltration of blood‐derived peripheral immune cells into the brain at the acute phase post‐trauma. F4/80 is a well‐characterized membrane protein, and it is highly expressed on macrophages to rapidly infiltrate the injured cortex following TBI.[ 42 ] NPs groups showed a decrease of F4/80 positive cells in the injured cortex compared to Vh groups 24 h post‐TBI (Figure 6c,d). The attenuated peripheral inflammation due to NPs administration can be attributed to NPs administration's effect at the systemic level and the possible effect of peripheral organs blocking the inflammatory response, thereby reducing infiltration of macrophages. Reduction of infiltrating inflammatory cells has been reported recently from another immunomodulatory approach to TBI.[ 43 ] Further, injections of miR‐146a regulating inflammation containing NPs into the brain immediately following an experimental TBI model significantly reduced hippocampal tumor necrosis factor (TNF) receptor‐associated factor 6 and interleukin‐1 receptor‐associated kinase 1 levels.[ 44 ] Thus, a potential mechanism for NPs to control the neuroinflammatory response seems independent from the physical‐chemical variables.
Figure 6.
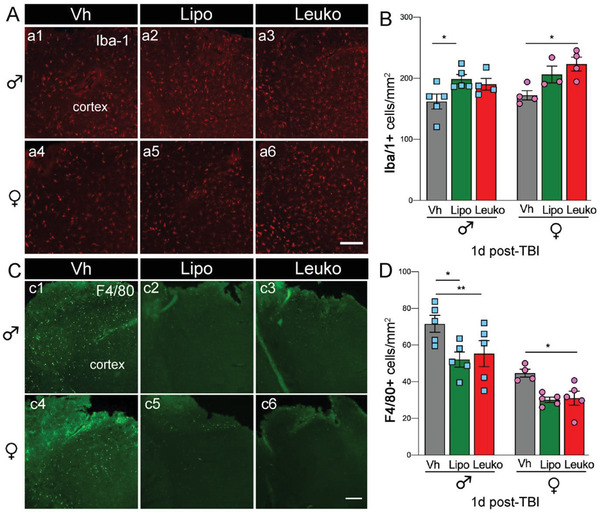
Microglia and macrophages cortical recruitment following nanoparticles (NPs) administration. A,B) An increased number of microglia (Iba‐1, red) positive cells a1–a6) was observed in the cerebral cortex of male liposomes (Lipo) and female leukosomes (Leuko) treated mice compared to their vehicle (Vh) treated counterparts at 24 h following TBI and systemic administration of NPs. Notably, no significant differences were seen between the Lipo and Leuko populations in the male and female groups. C,D) Macrophage (F4/80, green) population was increased in Vh treated mice compared to male Lipo, male Leuko, and female Leuko mice (c1–c6). No significant differences were detected between the Lipo and Leuko treated groups for either sex. Two‐way ANOVA followed by Tukey's multiple comparison test was used to determine statistical probabilities. *p ≤ 0.05, ** p ≤ 0.01, n = 5 (TBI), n = 3 (Sham). Scale bar: 50 µm.
2.6. NPs Reduce the Lesion Size and Improve Motor Ability
To evaluate the NPs therapeutic effect during the acute stage after TBI, we have assessed the TBI lesion size. We found that the lesion volume was reduced ≈28.6 % in NPs‐treated males and ≈21.6 % in NPs‐treated females compared to their Vh counterparts (n = 5 for all groups) (Figure 7a,b). Several TBI models have demonstrated functional improvements using NPs therapy. For example, dexamethasone encapsulating NPs were found as effective treatment after TBI. This NPs treatment not only increased survival, but also reduced anxiety, apoptosis, and BBB disruption among the test animal groups.[ 39c ] Additionally, in our study, we assessed TBI mice's motor ability using a rotarod after NPs treatments. Our results indicate that regardless of sex, the functional motor test was significantly improved among the Lipo group compared to their Vh treated counterparts (Figure 7c).
Figure 7.
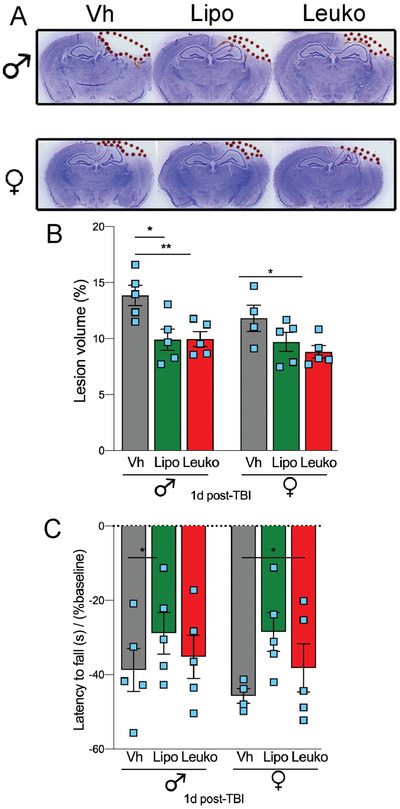
Nanoparticles (NPs) administration reduces lesion volume and improves motor recovery after TBI. The effect of NPs on the injured brain lesion was measured using histological techniques. A) Representative images of brain sections stained with cresyl‐violet 24 h after TBI followed by administration of liposomes (Lipo), leukosomes (Leuko), or vehicle (Vh). The dotted line indicates the lesion area composed of the cavity and edematous area. B) Statistically significant reduced lesion volume was found among NPs treated male groups and Leuko treated female groups compared to their Vh treated counterparts. C) Motor recovery was assessed using the rotarod. NPs treatment improved motor function, but this improvement was statistically different in injured mice treated with Lipo compared to those treated with Leuko. Two‐way ANOVA followed by Tukey's multiple comparison test was used to determine statistical probabilities. *p ≤ 0.05, ** p ≤ 0.01, n = 5 (TBI), n = 3 (Sham).
3. Conclusions
In this work, we have demonstrated the feasibility of using biomimetic NPs as a theranostic tool for TBI treatment. We have developed a novel delivery system that 1) penetrates the injured brain while carrying a fluorescent agent that targets the local brain inflammation; 2) can be used as a diagnostic tool for BBB breaching; and 3) reduces the lesion size. Notably, the NPs fluorescent marker can be replaced with a contrast agent 1,2‐distearoyl‐sn‐glycero‐3‐phosphoethanolamine‐N‐diethylenetriaminepentaacetic acid (gadolinium salt) to allow this formulation future clinical translation use, this time carrying magnetic resonance imagining (MRI) contrast agent for the TBI detection. Moreover, the fact that only empty NPs only, induced a therapeutic effect after TBI might pave the way for developing more efficient nanotherapeutics by encapsulating drugs inside the NPs.
Biomimetic NPs are a promising tool that can accelerate a therapy's translation more efficiently than cellular or exosome‐based techniques due to the ease of fabrication, purification, and similarity between the different NPs batches. Both of the NPs described in this study are biomimetic, mimicking either the mice cells’ lipidic composition only or the leukocytes’ protein composition along with the mice cells’ lipidic composition. The membrane's protein, presented on the Leuko NPs, favored brain lesion targeting and longer systemic circulation time (i.e., Leuko were still circulating systemically circulated 24 h after injury). Relevant differences in NPs biodistribution after brain trauma in filtering organs within the current study were also found, suggesting a potential local and systemic future targeting strategy. Our results demonstrated that biomimetic NPs could be strong candidates for acute pharmacologic treatment in TBI patients. By observing the infiltration of NPs into inflamed tissues, we can now design how to overcome the therapeutic challenges of TBI using pharmacological therapy targeting specific inflammatory mechanisms.
4. Experimental Section
Reagents
Membrane protein extraction kit, chloroform, and 2‐Mercaptoethanol (Sigma Aldrich, Missouri, USA). PBS 10× solution, Syringe Filters 0.22 µm sterile PVDF, MilliporeSigma Milli‐Q Ultrapure Water Systems Accessory, Pierce Rapid Gold BCA Protein Assay Kit, and Pierce Bovine Serum Albumin (BSA) Standard Ampules 2 mg mL‐1 (Fisher Scientific, Pennsylvania, USA). Dipalmitoylphosphatidylcholine (DPPC), 1,2‐dioleoyl sn‐glycerol‐3 phosphocholine (DOPC), cholesterol (ovine wool, >98%) and 1,2‐dioleoyl‐sn‐glycerol‐3 phosphoethanolamine‐N‐(Cy5.5) (Avanti Polar Lipids, Inc, Alabaster, USA). Float‐A‐Lyzer G2 dialysis devices (Spectrum Labs, Massachusetts, USA). Dynamic light scattering (DLS) and disposable cuvettes primarily for measuring zeta potential (Malvern, Instruments, Worcestershire, UK). Semi microvolume disposable polystyrene cuvettes for size measurements, 10× tris buffered saline (TBS), 10× tris/glycine/SD, Precision Plus Protein Dual Color Standards, 10%Mini‐PROTEAN TGX Precast Protein Gels, Trans‐Blot Turbo Mini Nitrocellulose, 2× Laemmli Sample Buffer and Clarity Western ECL Substrate (Bio‐Rad Laboratories, California, USA). Polyester drain disc and Nuclepore Polycarbonate hydrophilic membranes (0.4, 0.2, and 0.08 µm) (Whatman, Buckinghamshire, UK). Antibodies for western blot rat anti‐CD11b (MAB11241), goat anti‐CD18 (AF2618), rabbit anti‐CD45 (EPR20033), goat anti‐CD47 (ab108415), anti‐rabbit IgG‐HRP, anti‐goat IgG‐HRP, and anti‐rat IgG (Bio‐Techne Corporation, Minnesota, USA).
Cell Culture
J774 murine macrophages were purchased from ATCC and cultured in Dulbecco's modified eagle's medium (DMEM) high glucose complete media supplemented with 1 % L‐glutamine and 1 % penicillin/streptomycin solution. They were maintained in humidified incubators under 5 % CO2, at 37 °C and passaged at 70–80% of confluence. Upon reception, cells were expanded and frozen as seed stocks of the first or second passage. All cells were passaged for a maximum of 3 or 4 weeks, after which new seed stocks were thawed for experimental use. All cells were grown at 37 °C in 5% CO2 and were maintained as a subconfluent monolayer using high glucose DMEM supplemented with 10 % fetal bovine serum and 1 % penicillin–streptomycin.
Membrane Protein Extraction and Quantification
Membrane proteins were extracted from the J774 cell line using the membrane extraction kit. Cells were first washed with 2 mL of the kit wash buffer followed by centrifugation at 4 °C, 300 g for 10 min, and the supernatant was discarded. Then, the pellet was resuspended with wash buffer, and the cells were split into 15 million of cells/tube marking up to 2 mL with wash buffer in each tube. Cells were rewashed with the same parameters, and the supernatant was discarded. Next, 2 mL of the kit extraction buffer were mixed with 10 µL protease inhibitor cocktail (PIC) and the supernatant was discarded. Afterward, the samples were incubated at 4 °C for 10 min, and they were then centrifuged at 4 °C, 16 000 g for 15 min. The supernatant was discarded, and the pellet was resuspended with 1 mL extraction buffer II and 5 µL PIC. Next, the sample was incubated at 4 °C for 30 min and centrifuged at 4 °C, 16 000 g for 15 min. Finally, the supernatant containing the MPS was collected and stored at ‐80 °C. MPS were quantified using the Pierce BCA Protein Assay Kit. Firstly, a standard calibration curve was prepared with the following concentrations—0, 25, 125, 250, 500, 750, 1000, and 1500 µg mL‐1—in triplicate using BSA and diluting it with 1× PBS. The samples and extraction buffer II were diluted with 1× PBS 1:5 (v:v) and 20 µL of all samples were loaded in triplicate in a 96‐well microplate and mixed with 200 µL of rapid Gold BCA reagent created by mixing reagents A and B 50:1 (v/v). The plate was covered with aluminum foil and incubated for 10 min at room temperature. The absorbance was measured at 480 nm with the plate reader.
Liposomes and Leukosomes and Fabrication
Lipo and Leuko were fabricated using DPPC, DOPC, and cholesterol (molar ratio 4:3:3 and initial lipid concentration 9 × 10‐3 m). DPPC, DOPC, and cholesterol were weighed and dissolved in chloroform at final concentrations of 20 mg mL‐1, mixing and sonicating at 45 °C for 5 min. Once all the lipids were dissolved, they were mixed (264 µL DPPC, 212 µL DPPC, 104 µL cholesterol, and 1420 µL chloroform) in a round‐bottom flask. The chloroform was evaporated using the rotary evaporator for 30 min, 45 °C, 0 psi, and 280 rpm, and the thin film was obtained. Next, the thin film was hydrated with 2 mL 1 × PBS for 30 min at 45 °C and 280 rpm, and it was then sonicated for 5 min at 45 °C for the Lipo. For the Leuko, one‐hundredth of the lipid weight was added and marked up to 2 mL with 1 × PBS. NPs were obtained using the extruder. Briefly, the heating system was set at 45 °C and connected to barrel. The samples were extruded using different cycles of polycarbonate membranes (400 nm for three times, 200 nm for three times, and 80 nm for five times). For the fluorescent NPs, the same procedure was followed, dissolving Cy5.5 in chloroform at final concentration of 1 mg mL‐1 and adding 264 µL DPPC, 212 µL DPPC, 104 µL cholesterol, 100 µL Cy5.5 and 1320 µL chloroform. For western blot, NPs were fabricated using NanoAssemblr, as we already described in the previous work with slight modifications.[ 45 ] Briefly, DPPC, DOPC, and cholesterol were weighed and dissolved in ethanol at final concentrations of 28, 22.5, and 11 mg mL‐1, respectively. Then, they were mixed and sonicated at 45 °C for 10 min. The solvent phase was obtained by mixing 94.3 µL DPPC, 94.3 µL DOPC and 94.3 µL cholesterol. The aqueous phase was made of 667 µL 1 × PBS for liposomes, while one‐hundredth of the lipid weight was added and marked up to 667 µL with 1 × PBS for Leuko. The solvent phase was warmed for 3 min at 45 °C, while the aqueous phase was warmed for 1 min at 45 °C before loading them into a NanoAssemblr cartridge. The following parameters were set: total volume 1 mL, flow ratio 2:1, flow rate 1 mL min‐1, start waste 0.15 mL, and end waste 0.05 mL.
Preparation and Characterization of NPs
Lipids and unbounded membranes were removed with the dialysis for in vivo studies. The samples were loaded in the float A‐Lyzer and put in 2 L 1× PBS. The buffer was changed two times (after 1 and 3 h) and the samples were collected after 19 h. The samples were sterilized by loading in a syringe and using a 0.22 µm PVDF filter. NPs size, PDI, and ZP were measured using the DLS. The size and PDI 5 µL of sample were mixed with 495 µL 1× PBS 1:100 (v/v) in the semimicrovolume disposable polystyrene cuvettes. For each sample, three measurements of 10 runs were acquired and averaged to get the size and PDI. For ZP, 10 µL of samples were mixed with 90 µL 1 × PBS and 900 µL MilliQ 1:9:90 (v/v/v) in the disposable cuvettes. For each sample, 3 measurements of 15 runs were acquired and the average was calculated to get the ZP. NPs fluorescence was determined using IVIS Spectrum with excitation 640 nm and emission 720 nm.
Western Blot Analysis
To prepare the samples, proteins were extracted and purified from NPs with chloroform and methanol. Briefly, NPs were centrifuged at 45 000 rpm for 1 h and the pellets were resuspended with 50 µL of MilliQ water, followed by gently mixing with 400 µL of methanol and 100 µL of chloroform. After centrifugation at 15 000 g for 2 min, 400 µL of methanol was added, then centrifuged again. Dried protein pellets were dissolved with 1× Laemmli sample buffer containing 2‐mercaptoethanol. 10 µg (CD11b, CD18) or 60 µg (CD45, CD47) of cell lysate with RIPA buffer or membrane protein was also prepared with 1× Laemmli sample buffer, and all samples were heated at 95 °C for 5 min. Samples were loaded onto 10% Mini‐PROTEAN TGX Precast Protein Gels and run for 2 h at 100 V. Proteins on the gel were transferred to the Trans‐Blot Turbo Mini Nitrocellulose membrane. 5% nonfat milk in tris buffered saline with 0.1% tween 20 was used to blocking and antibody dilution. Western blot was performed by incubating with rat anti‐CD11b (MAB11241, 1:3000 dilution), goat anti‐CD18 (AF2618, 1:3000 dilution), rabbit anti‐CD45 (EPR20033, 1:1000 dilution) and goat anti‐CD47 (ab108415, 1:1000 dilution). Their corresponding IgG conjugated with horseradish peroxidase (HRP) was used for the secondary detection. The membrane was developed with Clarity Western ECL Substrate. Protein bands were detected by ChemiDoc XRS+ System (Bio‐Rad).
Cryo‐Transmission Electron Microscopy (Cryo‐TEM)
The morphology of liposomes and leukosomes and NPs solutions were determined and imaged at the Baylor College of Medicine Cryo‐Electron Microscopy Core Facility (BCM, Houston, TX). The Quantifoil R2/1, Cu 200 mesh Holey Carbon grids were pretreated with a 45 s air‐glow discharge to make the carbon surface hydrophilic. Alongside these grids, Quantifoil R2/1 200Cu +4 nm thin carbon grids were also glow discharged for 10 s to test the efficacy of the added layer of continuous carbon with binding of the NPs. Vitrification was performed using a Vitrobot Mark IV (FEI, Hillsboro, OR) operated at 18 °C and 100% humidity. Each grid had 3 µL of NPs sample applied to it and was subsequently blotted for 1–3 s before being immediately submerged in liquid ethane. The frozen grids were then transferred into a JEOL 3200FS microscope (JEOL) outfitted with a Gatan K2 Summit 4kx4k direct detector (Gatan, Pleasanton, CA) and a postcolumn energy filter set to 30 eV. Before imaging, the microscope was carefully aligned to prevent any beam‐induced aberrations or astigmatism that can negatively impact image quality. Images were collected at magnifications of 15 000× and 30 000× with respective pixel sizes of 2.392 and 1.232 angstroms. Images were collected using an exposure time of 1 s with an approximate dose rate of ≈20e‐/Å2/s per image.
Mice and Traumatic Brain Injury Model
Adult (3 months old) male and female C57BL/6J mice (Jackson Laboratories, Bar Harbor, ME) were kept under a 12:12 h light and dark cycle with access to food and water ad libitum. Mice were anesthetized with isoflurane (3% for induction, 1.5–2% for maintenance). A moderate TBI was performed on the left side of the brain at the primary motor and somatosensory cortices using an electromagnetically driven controlled cortical impact (CCI) device (Impact One stereotaxic impactor; Leica Microsystems, Buffalo Grove, IL).[ 16, 46 ] The impact site was located at 2 mm lateral and 2 mm posterior to Bregma, with a 3 mm diameter flat impact tip, impact velocity of 3.25 m s‐1, and impact depth of 2 mm. It has been previously determined that these parameters can induce a strong and acute inflammatory response.[ 5 ] Sham mice received identical anesthesia to serve as uninjured controls. All animal studies were approved by Institutional Animal Care and Use Committee (IACUC) at Houston Methodist Research Institute (IACUC: IS00004860). Thirty min after the impact, mice were administered Cy5.5‐labeled NPs via retro‐orbital injection under anesthesia and then were allowed to recover fully before being transferred to their home cages.
Intravital Confocal Microscopy
Intravital microscopy (IVM) was performed in accordance with the guidelines of the Animal Welfare Act and the Guide for the Care and Use of Laboratory Animals and The Houston Methodist IACUC. The IVM was equipped with an upright Nikon A1R laser scanning confocal microscope with a resonance scanner, motorized and heated stage, Nikon long‐working distance X4 and X20 dry plan‐apochromat objectives. It is housed within the Intravital Microscopy Core at the Houston Methodist Research Institute. Intravital imaging was performed on anesthetized mice before and after TBI. Anesthesia was maintained by inhalation of 2% isoflurane during surgical and imaging procedures. The animal was positioned on a heated stage with the head fixed in holders and a round coverslip on the exposed tissue. Each of them was injected with 50 µL of 0.1 mg mL‐1 of FITC‐dextran (70 kDa) via retro‐orbital injection to visualize the vessels and 100 µL of NPs (TEXAS‐Red labeled) while under the scope. The imaging lasted for about an hour since injection and was repeated on the same mice 24 h after the injection. Tracer was used to delineate the vasculature. All settings, including laser power, gain, offset, and pinhole diameter, were maintained throughout each acquisition. All images were analyzed with NIS‐Elements software. Data were obtained by averaging results on at least three images from three mice.
IVIS Imaging
Mice were euthanized 24 h postsurgery using CO2 (20%) and perfused with PBS. Brain, heart, lungs, liver, spleen, and kidneys were collected and IVIS imaging was assessed to target NPs biodistribution. IVIS image acquisition parameters were the following: Em = 720 nm, Ex = 640 nm, Epi‐illumination, Bin:(HR)4, FOV:18.4, f2, 0.5 s. Quantification of IVIS images was performed using the Living Image software.
Organs Paraffin Embedding
Lungs, liver, spleen, and kidney were sampled, fixed in 4 % paraformaldehyde for 48 h and transferred to 70 % ethanol. Tissues were processed in a Shandon Exelsion ES Tissue Processor and embedded in paraffin on a Shandon HistoCenter Embedding System, using the manufacturer's standard processing and embedding protocols. Slides were sectioned at 5 µm thickness.
Cresyl Violet Staining and Lesion Volume Measurements
Brains were removed, post‐fixed in 4% paraformaldehyde overnight, and stored at 4 °C in a 30% sucrose solution. A sliding microtome (Microm HM 430, Thermo Fisher Scientific, Waltham, MA) was used to section the brains at 20 µm thick in coronal orientation through the dorsal hippocampus. Brain sections were then cryoprotected in an antifreeze solution (30% glycerol + 30 % ethylene glycol + 40 % 0.01 m PBS) for storage at –20 °C. Cresyl‐violet (0.1%, Sigma‐Aldrich) was dissolved in distilled water and filtered. Every third brain section was mounted on poly‐d‐lysine‐coated slides and stained for 20 min with a cresyl‐violet solution. Sections were then dehydrated for 2 min sequentially with 100, 95, 70, then 50% ethanol, cleared in xylene for another 2 min, covered with DPX mounting media (Sigma‐Aldrich, CA) and coverslipped. Lesion area was assessed on 8 to 12 brain sections spaced equidistance (every 450 mm) apart, approximately between ‐1.70 to ‐2.70 mm from Bregma. Lesion volume was obtained by multiplying the sum of the lesion areas by the distance between sections. Percent of lesion volume was calculated by dividing each lesion volume by the total ipsilateral hemisphere volume (similarly obtained by multiplying the sum of the areas of the ipsilateral hemispheres by the distance between sections).
Hematoxylin and Eosin Staining
Tissues were dehydrated with 95% ethanol twice for 30 min, and then soaked in xylene for 1 h at 60–70 °C followed by paraffin for 12 h. For the mouse spleen, liver, and kidneys, 0.5 mL of 95% ethanol in dehydration was used. First, the tissues were stained with hematoxylin solution for 6 h at a temperature of 60–70 °C and were then rinsed in tap water until the water was colorless. Next, 10% acetic acid and 85% ethanol in water were used to differentiate the tissue 2 times for 2 min, and the tissues were rinsed with tap water.
Immunofluorescence Analysis
Free‐floating parallel brain sections were washed with PBS with 0.5% Triton X‐100 (PBS‐T) for 5 min and blocked with 5% normal goat serum (NGS, #S1000, Vector laboratories, Burlingame, CA) in PBS‐T for 1 h. Brain sections were incubated at 4 °C overnight in PBS‐T and 3% of NGS using the following primary antibodies: anti‐rabbit Iba‐1 (1:1000, Wako) for activated microglia/macrophages, anti‐rat F4/80 (1:200,R&D Systems) for infiltrated macrophages, or anti‐rabbit Collagen IV (1:3000, Chemicon) expressed on the surface vessels. After incubation, brain sections were washed in PBS‐T and incubated with the corresponding antirabbit or antimouse Alexa Fluor 568‐conjugated or Alexa Fluor 488‐conjugated IgG secondary antibodies (all 1:1000, Thermo Fisher Scientific, Waltham, MA) for 2 h at room temperature. Brain sections were rinsed with PBS ≈5 min and incubated in PBS with DAPI solution (1:50000, Sigma‐Aldrich, St. Louis, MO) for counterstained nuclei. The sections were rinsed with distilled water and coverslipped with Fluoro‐Gel with Tris Buffer mounting medium (Electron Microscopy Sciences, Hatfield, PA). For quantitative analysis of immunolabeled sections, unbiased standardized sampling techniques were implanted to measure tissue areas corresponding to the injured cortex showing positive immunoreactivity, as we have previously described.[ 5 ] To quantify the number of Iba‐1 and F4/80‐positive cells in the injured cortex, an average of four coronal sections from the lesion epicenter (‐1.34 to ‐2.30 mm from Bregma) were counted and imaged for each animal (n = 5 per group). Within each brain region, positive cells were counted in each of the three cortical fields (×20 objective, 151.894 mm2 per field) around the impact area, as we have previously described.[ 5b ] All the investigators were blinded for the experimental groups. Images were acquired on a Nikon motorized fluorescence microscope (Eclipse Ni‐U, Melville, NY) with a pco.Edge Scomos camera (4.2LT USB3) and analyzed using NIS‐Elements software.
Rotarod Test
The Rotarod, a motor behavior test, was selected to test motor coordination and dysfunction after TBI. Performance was assessed by the ability of mice to stay on a rotating rod apparatus (Ugo Basile Harvard Apparatus, PA, USA) as the speed gradually increased. Mice were tested 2 d before injury (baseline) and one day post‐injury. The rod was accelerated from 4 to 40 rpm in 2 min and the time the mice were able to stay on the rod was recorded as latency to fall in seconds. Latency to fall at one day after injury as percentage of baseline was calculated for each mouse.
Statistical Analysis
Unpaired t‐test was used to determine statistical probabilities comparing size, PDI, ZP, and NPs concentration during the formulation steps among the different NPs formulations. For biodistribution studies, two‐way analysis of variance (ANOVA) followed by Tukey's multiple comparison test was used to determine statistical probabilities. For rotarod, and histochemical analysis, a two‐way ANOVA was used with time after injury as one variable and sex as the other variable, then a post hoc test with Bonferroni multiple test correction. All data in this study are expressed as the mean, standard error of the mean (±SEM), and results were considered statistically significant at P‐values <0.05. The statistical analysis was processed with GraphPad Prism 8 Software (GraphPad; San Diego, CA, USA).
Conflict of Interest
The authors declare no conflict of interest.
Author Contributions
A.Z., F.T., and S.V. initiated, designed, planned, and oversaw all aspects of the study. A.Z., S.S., E.R., G.B., and S.V. performed the experimental work and data analysis, and they drafted the manuscript. All authors reviewed and edited the final version of the manuscript.
Supporting information
Supporting Information
Acknowledgements
This work was supported by grants from the National Institute for Neurological Disorders and Stroke (NINDS), R21NS106640 (S.V.), TIRR Foundation through Mission Connect grant (S.V.), and funds from Houston Methodist Research Institute (S.V.). The authors thank Ms. Manuela Sushnitha, Ms. Shashi Krishnamurthy, Dr. Tomoyuki Naoi, and Ms. Eliana Stetco for their technical help. The authors are indebted to Dr. Gillian Hamilton for editing the text. They thank the Baylor College of Medicine Cryo‐TEM Core Facility for electronic imaging support and the Pathology and Histology Core (HTAP). Figure 1 was created using Biorender.com.
Zinger A., Soriano S., Baudo G., De E. Rosa, Taraballi F., Villapol S., Biomimetic Nanoparticles as a Theranostic Tool for Traumatic Brain Injury. Adv. Funct. Mater. 2021, 31, 2100722. 10.1002/adfm.202100722
Contributor Information
Assaf Zinger, Email: ayzinger@houstonmethodist.org.
Francesca Taraballi, Email: ftaraballi2@houstonmethodist.org.
Sonia Villapol, Email: svillapol@houstonmethodist.org.
Data Availability Statement
Research data are not shared.
References
- 1.Taylor C. A., Bell J. M., Breiding M. J., Xu L., MMWR Surveill. Summ. 2017, 66, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ma V. Y., Chan L., Carruthers K. J., Arch. Phys. Med. Rehabil. 2014, 95, 986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Werner C., Engelhard K., Br. J. Anaesth. 2007, 99, 4. [DOI] [PubMed] [Google Scholar]
- 4.Diaz‐Arrastia R., Kochanek P. M., Bergold P., Kenney K., Marx C. E., Grimes C. J., Loh L. T., Adam L. T., Oskvig D., Curley K. C., Salzer W., J. Neurotrauma 2014, 31, 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Villapol S., Byrnes K. R., Symes A. J., Front. Neurol. 2014, 5, 82; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Villapol S., Loane D. J., Burns M. P., Glia 2017, 65, 1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Allen M., Ghosh S., Ahern G. P., Villapol S., Maguire‐Zeiss K. A., Conant K., Sci. Rep. 2016, 6, 35497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Villapol S., Kryndushkin D., Balarezo M. G., Campbell A. M., Saavedra J. M., Shewmaker F. P., Symes A. J., Am. J. Pathol. 2015, 185, 2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Aertker B. M., Kumar A., Prabhakara K. S., Smith P., Furman N. E. T., Hasen X., Cox C. S., Bedi S. S., J. Neurosci. Res. 2019, 97, 698; [DOI] [PubMed] [Google Scholar]; b) Lotocki G., de Rivero Vaccari J. P., Perez E. R., Sanchez‐Molano J., Furones‐Alonso O., Bramlett H. M., Dietrich W. D., J. Neurotrauma 2009, 26, 1123; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Whalen M. J., Carlos T. M., Kochanek P. M., Clark R. S., Heineman S., Schiding J. K., Franicola D., Memarzadeh F., Lo W., Marion D. W., Dekosky S. T., J. Neurotrauma 1999, 16, 583; [DOI] [PubMed] [Google Scholar]; d) Hartl R., Medary M., Ruge M., Arfors K. E., Ghajar J., Acta Neurochir. Suppl. 1997, 70, 240. [DOI] [PubMed] [Google Scholar]
- 9.a) Lee J., Costantini T. W., D'Mello R., Eliceiri B. P., Coimbra R., Bansal V., J Trauma Acute Care Surg. 2014, 77, 709; [DOI] [PubMed] [Google Scholar]; b) Woodcock T., Morganti‐Kossmann M. C., Front. Neurol. 2013, 4, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Jin X., Ishii H., Bai Z., Itokazu T., Yamashita T., PLoS One 2012, 7, e41892; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Castejon O. J., Castellano A., Arismendi G. J., Medina Z., J. Submicrosc. Cytol. Pathol. 2005, 37, 43; [PubMed] [Google Scholar]; c) Chodobski A., Chung I., Kozniewska E., Ivanenko T., Chang W., Harrington J. F., Duncan J. A., Szmydynger‐Chodobska J., Neuroscience 2003, 122, 853. [DOI] [PubMed] [Google Scholar]
- 11.Catania A., Lonati C., Sordi A., Gatti S., Brain, Behav., Immun. 2009, 23, 877. [DOI] [PubMed] [Google Scholar]
- 12.Bortolotti P., Faure E., Kipnis E., Front. Immunol. 2018, 9, 1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qian Y., Gao C., Zhao X., Song Y., Luo H., An S., Huang J., Zhang J., Jiang R., J. Neurotrauma 2020, 37, 2131. [DOI] [PubMed] [Google Scholar]
- 14.Mader M. M., Lefering R., Westphal M., Maegele M., Czorlich P., Eur J Trauma Emerg. Surg. 2020, 1, 10.1007/s00068-020-01544-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sundman M. H., Chen N. K., Subbian V., Chou Y. H., Brain, Behav., Immun. 2017, 66, 31. [DOI] [PubMed] [Google Scholar]
- 16.Treangen T. J., Wagner J., Burns M. P., Villapol S., Front. Immunol. 2018, 9, 2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barrett J. P., Henry R. J., Shirey K. A., Doran S. J., Makarevich O. D., Ritzel R. M., Meadows V. A., Vogel S. N., Faden A. I., Stoica B. A., Loane D. J., J. Neurosci. 2020, 40, 2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Al‐Ahmady Z. S., Jasim D., Ahmad S. S., Wong R., Haley M., Coutts G., Schiessl I., Allan S. M., Kostarelos K., ACS Nano 2019, 13, 12470. [DOI] [PubMed] [Google Scholar]
- 19.Zinger A., Baudo G., Naoi T., Giordano F., Lenna S., Massaro M., Ewing A., Kim H. R., Tasciotti E., Yustein J. T. J. A. A. B. M., ACS Appl. Biomater. 2020, 3, 6737. [DOI] [PubMed] [Google Scholar]
- 20.Alexis F., Pridgen E., Molnar L. K., Farokhzad O. C., Mol. Pharm. 2008, 5, 505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zinger A., Brozovich A., Pasto A., Sushnitha M., Martinez J. O., Evangelopoulos M., Boada C., Tasciotti E., Taraballi F. J. N., Nanomater. 2020, 10, 2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martinez J. O., Molinaro R., Hartman K. A., Boada C., Sukhovershin R., De Rosa E., Kirui D., Zhang S., Evangelopoulos M., Carter A. M., Bibb J. A., Cooke J. P., Tasciotti E., Theranostics 2018, 8, 1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Csanyi G., Lucas R., Annex B. H., Circ. Res. 2020, 126, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Molinaro R., Evangelopoulos M., Hoffman J. R., Corbo C., Taraballi F., Martinez J. O., Hartman K. A., Cosco D., Costa G., Romeo I., Sherman M., Paolino D., Alcaro S., Tasciotti E., Adv. Mater. 2018, 30, 1702749. [DOI] [PubMed] [Google Scholar]
- 25.a) Biegon A., Front. Neurol. 2021, 12, 576366; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Stewart A. N., MacLean S. M., Stromberg A. J., Whelan J. P., Bailey W. M., Gensel J. C., Wilson M. E., Front. Neurol. 2020, 11, 802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.a) Zinger A., Sushnitha M., Naoi T., Baudo G., De Rosa E., Chang J., Tasciotti E., Taraballi F., ACS Nano, In press, 10.1021/acsnano.0c05792; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Corbo C., Molinaro R., Taraballi F., Toledano Furman N. E., Hartman K. A., Sherman M. B., De Rosa E., Kirui D. K., Salvatore F., Tasciotti E., ACS Nano 2017, 11, 3262. [DOI] [PubMed] [Google Scholar]
- 27.Zinger A., Brozovich A., Pasto A., Sushnitha M., Martinez J. O., Evangelopoulos M., Boada C., Tasciotti E., Taraballi F., Nanomaterials 2020, 10, 2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roy A., Fung Y. K., Liu X., Pahan K. J. J. O. B. C., J. Biol. Chem. 2006, 281, 14971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamamoto T., Teramura Y., Itagaki T., Arima Y., Iwata H., Sci. Technol. Adv. Mater. 2016, 17, 677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Acosta S. A., Tajiri N., Shinozuka K., Ishikawa H., Grimmig B., Diamond D., Sanberg P. R., Bickford P. C., Kaneko Y., Borlongan C. V. J. P. O., PloS One 2013, 8, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pham L. D. D., Hayakawa K., Seo J. H., Nguyen M. N., Som A. T., Lee B. J., Guo S., Kim K. W., Lo E. H., Arai K. J. G., Glia 2012, 60, 875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cabral H., Matsumoto Y., Mizuno K., Chen Q., Murakami M., Kimura M., Terada Y., Kano M., Miyazono K., Uesaka M. J. N. N., Nat. Nanotechnol. 2011, 6, 815. [DOI] [PubMed] [Google Scholar]
- 33.Bharadwaj V. N., Lifshitz J., Adelson P. D., Kodibagkar V. D., Stabenfeldt S. E. J. S. R., Sci. Rep. 2016, 6, 29988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lutton E. M., Farney S. K., Andrews A. M., Shuvaev V. V., Chuang G.‐Y., Muzykantov V. R., Ramirez S. H. J. F. I. N., Frontiers Neurology 2019, 10, 582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bharadwaj V. N., Nguyen D. T., Kodibagkar V. D., Stabenfeldt S. E. J. A. H. M., Adv. Healthcare Mater. 2018, 7, 1700668. [Google Scholar]
- 36.a) Villapol S., Neural Regen Res. 2016, 11, 226; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) McDonald S. J., Sharkey J. M., Sun M., Kaukas L. M., Shultz S. R., Turner R. J., Leonard A. V., Brady R. D., Corrigan F., J. Neurotrauma 2020, 37, 770. [DOI] [PubMed] [Google Scholar]
- 37.a) Weiss N., Thabut D. J. L. T., Liver Transplantation 2019, 25, 469; [DOI] [PubMed] [Google Scholar]; b) Wicker E., Benton L., George K., Furlow W., Villapol S., Biomed Res. Int. 2019, 2019, 5967816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Udy A. A., Jarrett P., Lassig‐Smith M., Stuart J., Starr T., Dunlop R., Deans R., Roberts J. A., Senthuran S., Boots R. J. J. O. N., J. Neurotrauma 2017, 34, 137. [DOI] [PubMed] [Google Scholar]
- 39.a) Shoffstall A. J., Atkins K. T., Groynom R. E., Varley M. E., Everhart L. M., Lashof‐Sullivan M. M., Martyn‐Dow B., Butler R. S., Ustin J. S., Lavik E. B. J. B., Biomacromolecules 2012, 13, 3850; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lashof‐Sullivan M. M., Shoffstall E., Atkins K. T., Keane N., Bir C., VandeVord P., Lavik E. B. J. P. O. T. N. A. O. S., Proc. Natl. Acad. Sci. 2014, 111, 10293; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Hubbard W. B., Lashof‐Sullivan M., Greenberg S., Norris C., Eck J., Lavik E., VandeVord P., Sci. Rep. 2018, 8, 10622; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Takahashi T., Marushima A., Nagasaki Y., Hirayama A., Muroi A., Puentes S., Mujagic A., Ishikawa E., Matsumura A. J. J. O. T., Surgery A. C., J. Trauma Acute Care Surg. 2020, 88, 677. [DOI] [PubMed] [Google Scholar]
- 40.Yates A. G., Anthony D. C., Ruitenberg M. J., Couch Y., Front. Immunol. 2019, 10, 2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yong H. Y. F., Rawji K. S., Ghorbani S., Xue M., Yong V. W., Cell Mol. Immunol. 2019, 16, 540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kumar A., Alvarez‐Croda D. M., Stoica B. A., Faden A. I., Loane D. J., J. Neurotrauma 2016, 33, 1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sharma S., Ifergan I., Kurz J. E., Linsenmeier R. A., Xu D., Cooper J. G., Miller S. D., Kessler J. A. J. A. O. N., Ann. Neurol. 2020, 87, 442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang W. X., Prajapati P., Vekaria H. J., Spry M., Cloud A. L., Sullivan P. G., Springer J. E., Neural Regener. Res. 2021, 16, 514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.a) Molinaro R., Pasto A., Corbo C., Taraballi F., Giordano F., Martinez J. O., Zhao P., Wang X., Zinger A., Boada C., Hartman K. A., Tasciotti E., Nanoscale 2019, 11, 13576; [DOI] [PubMed] [Google Scholar]; b) Zinger A., Adir O., Alper M., Simon A., Poley M., Tzror C., Yaari Z., Krayem M., Kasten S., Nawy G., Herman A., Nir Y., Akrish S., Klein T., Shainsky‐Roitman J., Hershkovitz D., Schroeder A., ACS Nano 2018, 12, 1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Villapol S., Balarezo M. G., Affram K., Saavedra J. M., Symes A. J., Brain 2015, 138, 3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
Research data are not shared.