

Abstract
In the first decade of targeted covalent inhibition, scientists have successfully reversed the previous trend that had impeded the use of covalent inhibition in drug development. Successes in the clinic, mainly in the field of kinase inhibitors, are existing proof that safe covalent inhibitors can be designed and employed to develop effective treatments. The case of KRASG12C covalent inhibitors entering clinical trials in 2019 has been among the hottest topics discussed in drug discovery, raising expectations for the future of the field. In this perspective, an overview of the milestones hit with targeted covalent inhibitors, as well as the promise and the needs of current research, are presented. While recent results have confirmed the potential that was foreseen, many questions remain unexplored in this branch of precision medicine.
Keywords: : covalent warhead, drug discovery, FDA-approved drugs, targeted covalent inhibitors
Lay abstract
If it is forever, it needs to be safe. Approved drugs on the market act with a multitude of different mechanisms, including both reversible and irreversible (permanent) inhibition of a target protein that is known to malfunction in a given disease. However, scientists are aware that increased side effects can potentially arise from irreversible drugs. Current research has focused on improving the safety of these drugs with the aim of developing more effective treatments. This perspective offers an overview of the current status of so-called ‘targeted covalent inhibition’ drug discovery.
Graphical abstract
It is common knowledge that a non-negligible number of approved drugs engage their target by covalent reaction. These compounds, which include aspirin and penicillin (to name the most well-known), were being used in the clinic long before their mechanism of action was elucidated [1,2]. A period of caution regarding covalent inhibitor promiscuity followed, in which many studies pointed out the risks associated with an irreversible reaction that can take place not only with the desired target but also with off-target proteins [3,4]. This resulted in drug discovery programs moving further and further away from reactive molecules. Filtering strategies for compound libraries have been devised against reactive motifs, some of which are recognized as pan-assay interfering compounds (PAINs) [5]. Until about 20 years ago this was considered the best practice in drug discovery and remains the gold standard for the development of reversible molecules. Eventually, a reverse trend appeared as researchers started to reconsider the potential of covalent inhibitors in addressing challenging targets, and this ultimately led to the resurgence of the field in the early 2010s [6]. As the historical development of covalent inhibitors is not the main focus here, interested readers are directed to a series of excellent reviews on the topic [3,6,7].
One of the major advantages of covalent inhibition is a prolonged residence time, which might at the same time cause safety concerns if the consequences of continued target inhibition are poorly understood [8]. Moreover, off-target activity could also be extended in the case of nonspecific reactivity, resulting in more serious side effects compared with noncovalent drugs [4]. Despite widespread efforts to avoid covalent mechanisms, the serendipitous discovery of such inhibitors in high-throughput screening has remained a concrete possibility even in libraries thoroughly filtered to exclude reactive moieties [9]. In fact, unexpected reactivity can be triggered or tremendously amplified by affinity-driven proximity and correct orientation toward a nucleophilic residue. However, these desirable properties can be challenging to predict for drug design, and tuning reactivity is not trivial. The concept of targeted reactivity has been slowly developed on the basis of novel experimental evidence and the advent of whole proteomic analytical techniques, showing that indeed covalent inhibition is not necessarily associated with promiscuity [10,11]. The increasing understanding and advancement of the field ultimately led from general skepticism to the concept of targeted covalent inhibitor (TCI) drug discovery, which triggered the current resurgence of covalent drugs. Both these now familiar expressions were minted in pivotal reviews to which the reader is directed for an amplified discussion [6,12]. Understanding the perils of covalent inhibition and combining it with careful design for selectivity can produce outstanding tools in the hands of a medicinal chemist and can also result in a more efficacious treatment. Thus a growing number of approaches for the rational design of covalent inhibitors have been devised due to their potential to expand the number of druggable targets [13,14]. A general fascination and curiosity surrounding the field is also demonstrated by the increasing blog discussions about TCIs (e.g., @CovalentMod on Twitter, Derek Lowe’s ‘In the PipeLine’ and Dennis Hu’s ‘Drug Hunter’).
The aim of the current perspective is to highlight the tangible advancement of TCIs in therapeutic application as well as the open questions of the field that have not been fully explored in translational settings. To support the reader’s understanding of the many aspects of this topic, detailed subtopic reviews will be indicated where available. The term TCI will be used to differentiate these advanced covalent inhibitors, which have been designed intentionally and with specific reactivity, as opposed to compounds with promiscuous reactivity and serendipitously discovered entities, which have long been used in the clinic. Successful examples of entities currently in the drug discovery pipelines will be used as case studies to guide the reader. Most importantly, common alerts for the discovery and development of TCIs will be discussed.
Targeted covalent inhibition mechanisms
As introduced earlier, TCIs are designed covalent inhibitors that contain an optimized affinity element and a relatively unreactive warhead (Figure 1A). Targeted covalent inhibition is most commonly described as a two-step mechanism (Figure 1B). First, a noncovalent interaction between the inhibitor and the target establishes a thermodynamic equilibrium. Second – driven by the proximity and correct orientation toward a specific residue – a covalent, usually irreversible, reaction follows favouring a shift to the right of the first equilibration step, and potentially driving the reaction to completion (i.e., depletion of either the protein target or the TCI). While this model is generally accepted to introduce the concept of TCI, different, more specific mechanisms can be used to describe such inhibitors on a case-by-case basis. An elegant compendium, coupled with unifying principles of mechanism nomenclature, has been written by Tuley and Fast and should be considered essential knowledge among medicinal chemists working with TCIs [15].
Figure 1. . Targeted covalent inhibition.

(A) Structural features of a targeted covalent inhibitor. (B) Targeted covalent inhibition entails a two-step mechanism: affinity encounter complex formation (determined by Kd = k–1/k1) and covalent bond formation (determined by rate constant k2). (C) Bar graph illustrating the timeline for the use of the terms ‘irreversible inhibition/inhibitors’ and ‘covalent inhibition/inhibitors’ in scientific literature. Data source: Pubmed, collected on 31 May 2020. The displayed data sets were obtained with the following search strings: Set A: ‘irreversible inhibitor’ OR ‘irreversible inhibition’; Set B: ‘covalent inhibitor’ OR ‘covalent inhibition’.
The two-step mechanism just described reveals that the potency of these compounds is time-dependent; thus if IC50 (inhibitory concentration that results in 50% target inhibition) is to be used for structure–activity relationship (SAR) comparison, the incubation time prior to measurement should be rigorously noted and kept consistent throughout analogue testing. While IC50 measurements are sometimes accepted in the literature, more appropriate parameters should be estimated for rigorous characterization of TCI, specifically kinact and KI (related to k2 and k−1.#x002F;k1, respectively; Figure 1B) [16]. The former parameter gives an estimation of the maximal labeling rate, while the latter is defined as the inhibitor concentration needed to reach 50% of kinact.#x00A0;and is an estimation of the potency of the compounds in the first affinity step, similar to Kd. For a thorough discussion of covalent inhibition kinetics and experimental determination thereof, a good starting point is provided by Strelow’s perspective [17]. In this work the author stresses that significant differences can be observed between kinetics determination in a biochemical assay (with purified protein) and within the cellular environment. When determining these properties in vitro, the kinetics of TCIs in the complex biological environment are not taken into consideration. For example, irreversible modification of a protein with high turnover could result in an apparent reversible effect, or even in ineffective inhibitors in cell experiments. Thus it might be more relevant to test TCIs directly in cells by exploiting established chemical biology techniques [18]. If sufficiently stable, the presence of a covalent bond with targeted proteins allows for easier manipulation of complex samples while preserving TCI adducts. Click chemistry can be used to append fluorescent or affinity tags to allow for visualization and enrichment of labeled proteins, which can be confidently assessed in proteomics searches [19]. These types of target engagement assays offer the advantage of simultaneously measuring improvements in potency, selectivity and further cellular pharmacokinetic and pharmacodynamic properties such as membrane permeability, toxicity and inhibitor-sequestering effects, for example by glutathione in the case of cysteine-reactive TCIs. However, dissecting the contribution of each aspect of inhibitor behavior is not trivial, and the support of biochemical experiments using simplified systems is often required.
In conclusion, while a covalent mechanism can offer a variety of advantages in terms of speeding up medicinal chemistry development, careful evaluation of assay results needs to be used to discriminate true TCIs from PAINs. As a general rule for drug discovery programs, an array of orthogonal assays should be sought to validate mechanistic hypotheses, and compound evaluation should be progressed toward the complex cell environment sooner rather than later after hit discovery.
The rise of targeted covalent drugs in the clinic
As proclaimed by the homonymous review by Singh et al. in 2011, a ‘resurgence of covalent drugs’ is currently taking place [6]. While the concept of ‘irreversible inhibition’ has been steadily discussed in the literature, a new connotation for TCIs, highlighted by the choice of the term ‘covalent inhibitor’, has seen a slowly increased interest that boomed from 2015 onwards (Figure 1C). The adoption of this terminology reflects scientists’ need to highlight the engineered design and specificity of such molecules and became absolutely necessary with the advent of covalent reversible inhibitors [20]. In general, TCI research has drifted away from the ‘irreversible inhibition’ term, which is more commonly associated with wide-ranging concerns around the intrinsic safety of this type of inhibitors.
Close examination of a comprehensive list of US FDA-approved drugs that are known to act with a covalent mechanism (see Supplementary Table 1) reveals some interesting trends in the evolutionary path of covalent drug discovery. The introduction of covalent drugs dates back to the beginning of the 20th century (and even earlier in Europe) with the debut of aspirin on the drug markets. Together with warfarin, alkylating agents, β-lactam antibiotics and proton pump inhibitors, these drugs are considered milestones in the discovery of covalent inhibitors and remain some of the most used therapies, thus implying their relatively tractable safety profile despite an underlying covalent mechanism of action. Yet most of these molecules were discovered serendipitously, with the elucidation of their chemical reactivity toward a specific target often following many years later. Their use in the clinic and their study in the laboratory have both been pivotal to the current advent of TCIs, despite the undeniable delays caused by a period of general concern and skepticism based on the advancement in toxicological studies [21]. For example, following the serendipitous discovery of β-lactam antibiotics, great advances have been reported in the antimicrobial field with the design of new inhibition strategies (e.g., the introduction of diazabicyclooctane β-lactamase inhibitors) [22]. These molecules were designed on the basis of a deep understanding of the serendipitously discovered natural substances, which act via an extremely selective covalent mechanism of action. An elegant review providing detailed insight into the mechanisms of approved covalent modifiers up to 2009 was compiled by Potashman and Duggan, who interestingly foresaw the advent of TCIs as a new starting point in drug discovery campaigns [23].
On the other hand, the opposite trend can be delineated for nonspecific covalent modifiers. Despite still being extensively used in therapy, alkylating and cross-linking drugs such as cisplatin, nitrogen mustards and nucleic acid analogues saw a progressive decrease in approvals (from mechlorethamine in 1949 to bendamustine in 2008), mostly due to the high toxicity arising from the low selectivity of these drugs for their target, usually cancer cells. While a general, albeit slow, decay in the use of cytotoxic drugs as opposed to the more advanced targeted therapies is foreseeable, novel tactics are also being devised to exploit the power of such agents against cancer. For example, antibody–drug conjugates have proven successful in reducing chemotherapy side effects, combining the cytotoxic drug’s antitumor efficacy with the precise delivery offered by antibodies [24]. Similarly, more selective tumor treatments (e.g., natural compounds) can be cargoed with toxic drugs, including alkylating agents [25]. These strategies are in line with the novel concept of precision medicine and show the growing awareness of the side effects caused by these anticancer agents, most of which are inherently oncogenic.
The first TCIs in the clinic were kinase inhibitors, with afatinib and ibrutinib gaining FDA approval in 2013 [26–29]. About one TCI per year has been approved ever since (see Supplementary Table 1), thus confirming the efficacy of these drugs and the advantages in reduced toxicity brought by a careful design and balance of electrophilic properties. The majority of approved TCIs target protein kinases, a family of proteins previously considered challenging for drug design. More recently, the scope of TCIs in the clinic is clearly broadening, with the approval of selinexor (targeting XPO-1) [30,31] in 2019 and the many ongoing clinical trials against new targets; for example, KRAS, as discussed below. The field obviously maintains its challenges: TCIs can often fail in clinical trials due to unpredicted toxicity (e.g., CatK inhibitors balicatib and odonacatib) [32] and are obviously also susceptible to simple inefficacy (e.g., FAAH inhibitor PF-04457845) [33]. Although TCIs, bearing a reactive electrophile, might seem more prone to idiosyncratic side effects, a considerable number of reversible inhibitors have also been found to produce electrophilic metabolites [34]. In fact, the extremely low success rate of clinical trials is a widespread phenomenon, which is often independent from the inhibition mechanism. Increasing the thoroughness of metabolism and toxicology investigations in preclinical studies is key to reducing failures of both reversible inhibitors and TCIs at more advanced stages of clinical development [35].
Approachability of undruggable targets: KRAS as a case study
As introduced in the previous section, TCIs in the clinic have mostly tackled protein kinase targets. This is not surprising, as kinases have been one of the most studied protein families in the last decades of drug discovery, given their implication in different types of diseases [36]. Many reversible inhibitors have been designed and developed by medicinal chemists, with noteworthy success in the clinic [37]. Due to their high degree of homology, kinases have long been considered challenging targets. Although these proteins can play a quite specific function in physiology and thus disease, achieving selectivity toward a single member of this family with small molecules is an extremely demanding task. In fact, most approved kinase inhibitors target multiple members of the kinase family [37,38]. Perhaps counterintuitively, the introduction of a covalent warhead into potent yet nonselective reversible kinase inhibitors has proven successful in increasing the selectivity of these compounds, thus explaining the success of kinase TCIs in the clinic [39]. The identification of nonconserved nucleophilic residues in close proximity to the binding site is crucial for the success of this strategy and has so far been limited to Cys for FDA-approved drugs. On the other hand, relying on a nonconserved residue has the drawback that further mechanisms of resistance can arise. It is not uncommon for these nonfunctional residues to mutate in response to treatment, as has already been observed, for example, for the target of osimertinib [40]. A good review on approved covalent kinase inhibitors has been published recently; thus the success of TCIs in this field will not be addressed in further detail here [39].
More recently, a TCI with a non-kinase target was also approved, namely selinexor (Xpovio, 2019; Figure 2A), an inhibitor of protein nuclear export. Marketed by Karyopharm Therapeutics, this drug has proven efficacious in clinical trials against multiple myeloma [41]. Like most of the so-called selective inhibitors of nuclear export (SINEs) currently under study, selinexor was developed following the identification of XPO-1 as the target of a potent natural compound with antitumoral properties. Since the first reports of leptomycin B’s activity (Figure 2A) [42], tremendous efforts have led to the elucidation of this compound’s mechanism of action, which involves a Michael addition to Cys528 of XPO-1 [43]. This natural substance has helped in elucidating the role of XPO-1 both in the regulation of gene expression in malignancies and in HIV protein translocation to the cytoplasm [42,44]. The first reported PKF050-638, an ancestor of selinexor, was based on a phenotypic assay, rather than being structurally designed, likely due to the lack of structural information for XPO-1 at the time [30]. Although not specified, it is reasonable to guess that a library containing electrophiles might have been chosen due to the preceding understanding of leptomycin B’s covalent mechanism. In fact, once the crystal structure of XPO-1 was reported [45], scientists at Karyopharm followed up with computer-aided TCI discovery [46]. Finally, advances in genome editing techniques confirmed the covalent mechanism of action of this clinical candidate and identified the labeling site unequivocally as Cys528, a finding that was also supported by affinity pull-down experiments [47]. The drug was deemed safe in clinical trials and efficacious for the treatment of relapsed or refractory multiple myeloma [48]. Selinexor is being assessed further in several clinical trials for application alone or in combination against other malignancies [49] as well as for severe cases of COVID-19 [50]. While some reports claim the discovery of reversible inhibitors of XPO-1, the presence of obviously reactive moieties (e.g., α,β-unsaturated enones) and the insufficient experimental proof of reversibility make these claims dubious [51]. As a matter of fact, no reversible inhibitor against XPO-1 has reached clinical efficacy to date, thus making TCI an advantageous option for the druggability of this target. The example of selinexor is particularly suited to show both the slow progress in TCIs over the last 20 years outside of the kinase field, and the set of complementary techniques (e.g., gene editing, proteomics) that have developed in parallel and now allow for precise target identification and selectivity profiling of TCIs.
Figure 2. . Recent development of TCIs in the clinic.
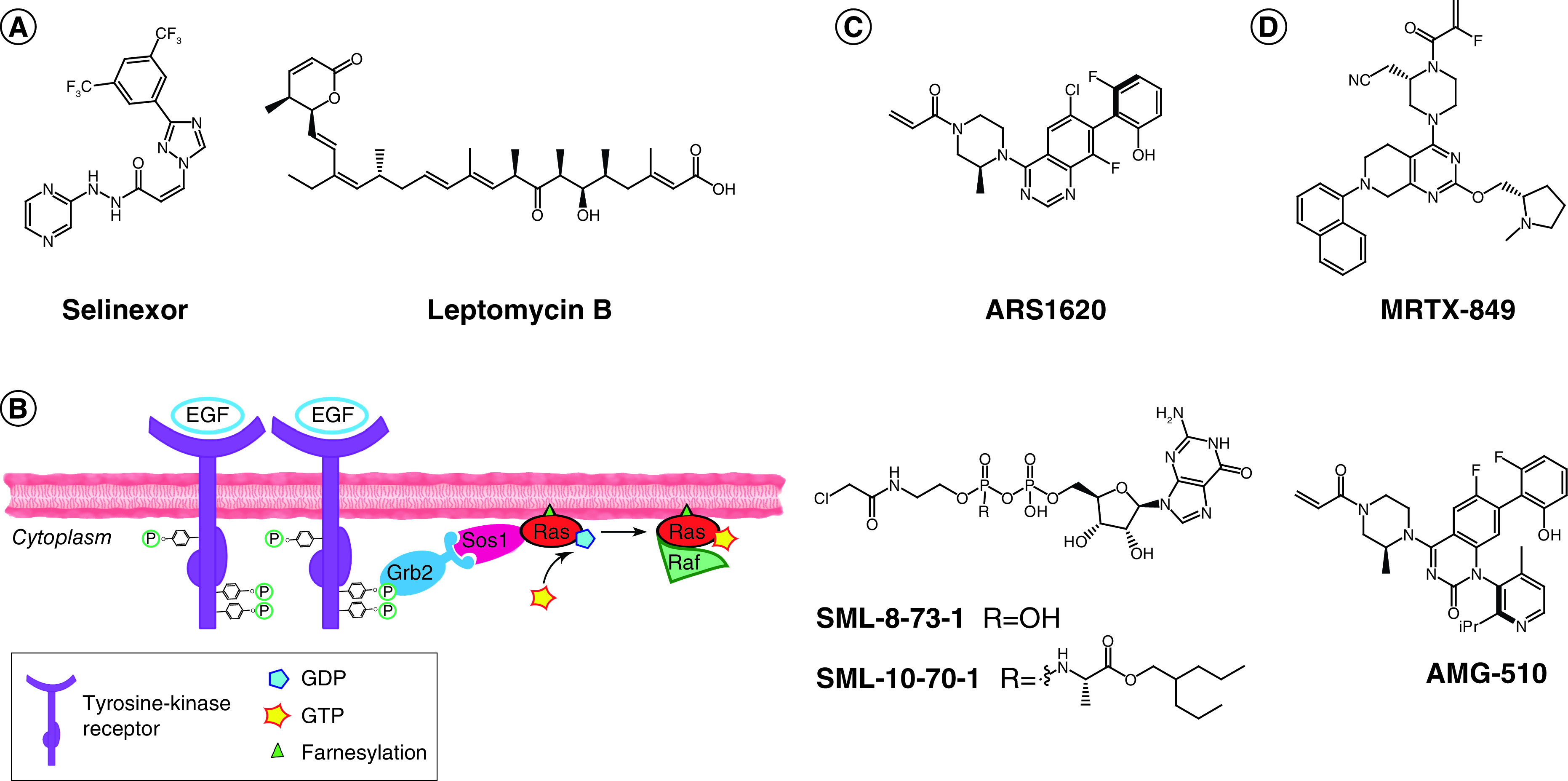
(A) Chemical structures of selinexor (left, FDA approved in 2019) and leptomycin B (right); both compounds bind covalently to Cys528 of XPO-1. (B) Ras activation cascade: upon detection of extracellular signal (e.g., EGF), tyrosine kinase receptors (e.g., EGFR) dimerize and cross-phosphorylate. Phosphate groups are recognized by an adaptor protein (e.g., GRB2), which activates a GEF protein (e.g., SOS1). SOS1 binds to GDP-RAS and induces nucleotide release. Cytoplasmic abundant GTP binds to RAS, resulting in the active conformation for signal transduction. Effector binding (e.g., Raf) occurs and the signal is further propagated to the cytoplasm, ultimately leading to regulation of specific gene expression. (C) Optimized covalent probes for KRASG12C. (D) Clinical candidates against KRASG12C for which the chemical structure is reported in the literature.
While the development of selinexor relied on a still-hybrid TCI approach, the eminent case of KRAS covalent inhibitors currently in clinical trials can be used as a case study to highlight the current advances in designed covalent inhibitors for tackling previously undruggable targets. The occurrence of malignant mutations of small RAS GTPases (specifically KRAS, HRAS and NRAS) has been well known to scientists for over 40 years [52]. In fact, almost 20% of all cancer patients are predicted to harbor a malignant mutation of one of these proteins [53]. Small monomeric RAS GTPases function as molecular switches, cycling from a GDP-bound (inactive) to a GTP-bound (active) state, in which they propagate signal by interacting with specific protein partners [54]. Posttranslational lipidation of RAS is catalyzed by FTases, after which farnesylated RAS localizes to the cellular membrane, where it detects and propagates activation of tyrosine kinase receptors downstream partners, ultimately leading to regulation of gene expression for cell growth and differentiation (Figure 2B) [54]. Despite numerous efforts to target RAS with different strategies – the most notable of which entailed indirect targeting with FTase inhibitors and failed at the clinical stage – RAS-targeted drugs have yet to appear in cancer treatment [55]. The lack of suitable pockets on the protein surface (with the exception of the nucleotide binding site) and the extremely high affinity to GTP, which is also abundant in cells, have prevented the success of conventional drug discovery approaches. Moreover, side effects due to affected collateral pathways are unavoidable when indirectly targeting RAS by inhibiting proteins involved in its functioning. Given the strong implication of RAS in cancer, medicinal chemistry efforts to tackle this target have never ceased. Thus, with the advent of TCIs, it did not take long before scientists started exploring this strategy to drug the undruggable RAS. KRASG12C is the most common RAS mutant in lung cancer [56]. The Cys12 mutation offers a desirable platform for this strategy, as the target nucleophile will only be found in malignant cells, while wild-type KRAS could potentially be left unaffected. The first report of a screen for KRASG12C inhibitors dates back to 2013, when Ostrem et al. performed a disulfide tethering assay using a library of 480 fragments and mass spectrometry to detect cross-disulfide linking under reducing conditions between KRAS and hit compounds [57]. Using KRASwt (which bears three native Cys residues) as a control, two hits showed almost complete and selective KRASG12C engagement; specifically, the inactive GDP-bound conformation was preferentially labeled by these compounds. Cocrystallization studies identified the binding site to be a previously reported allosteric shallow pocket [57,58]. This pocket is located in the proximity of the nucleotide binding site and is only available in GDP-bound KRASG12C and could further open to accommodate covalent binders [59]. Successive SAR optimization studies replaced the sulfide moiety with an acrylamide, and the affinity element of these compounds was tremendously improved in multiple steps of an intense medicinal chemistry campaign [57,60,61]. Indeed, the availability of precise structural information was pivotal for the rational and effective development of these initial hits into a potent lead substance, namely ARS1620 (Figure 2C). Compounds of this class act as trapping agents for the KRASG12C inactive conformation, thus rebalancing the altered equilibrium of overactivated KRAS found in cells expressing the malignant mutant. Interestingly, it was because of these molecules that the existence of a shifted yet cycling equilibrium in KRAS malignant isoforms could first be described, disproving previous theories of ‘constitutively active’ forms of RAS [62]. As already noted, the covalent handle was valuable for whole proteome selectivity analyses [60,61]. Another advantage in the discovery of KRASG12C covalent inhibitors was the availability of homozygous and heterozygous KRASG12C cell lines, as well as KRASwt and cell lines expressing further variants (e.g., KRASG12D) to confirm modification site identity and the selectivity of the therapeutic effects only in KRASG12C-expressing cells, without the need to resort to more laborious gene-editing techniques. ARS1620 was effective in reducing tumor growth and inducing tumor regression of in vivo xenograft mouse models harboring the relevant KRAS mutant, with no toxicity even at higher doses (1000 mg/kg), thus offering great promise for therapeutic application [61].
While the Shokat group was the first to publish covalent KRASG12C inhibitors, successively liaising with Wellspring Biosciences to develop improved molecules, the first candidate to win the race for clinical trials – AMG-510, which was also derived from ARS1620 – was reported by scientists at Amgen (Figure 2D) [59,63]. This drug has already shown promise in early Phase I studies [64]. In parallel, Mirati Therapeutics reported an improved compound, MRTX-849, that entered clinical trials shortly afterward (Figure 2D) [65]. Eventually, Wellspring Biosciences and Lilly joined this competition in the clinic (with drug candidates ARS-3248/JNJ-74699157 and LY3499446, respectively; both structures undisclosed), and there is increased hope that one of these four clinical candidates will succeed in the treatment of KRASG12C-harboring cancers [66].
Shortly after the first account of KRASG12C allosteric inhibitors, the Gray group also reported covalent probes that exploited the proximity of Cys12 to target the nucleotide binding site [67]. While these GDP-based probes (Figure 2C, compounds SML-8-73-1 and SML-10-70-1) are likely to lack important pharmacokinetic properties for application in the clinic (e.g., SML-8-73-1 is cell impermeable), they still remain valid as tool compounds. Moreover, they consolidate the concept that even extreme endogenous competition of a reversible binder, such as GTP, can be overcome by the introduction of a covalent modification, which over time can drive target occupancy kinetically, if allowed by pharmacokinetic and pharmacodynamic aspects. In fact, if a cysteine at this specific position is only found in the targeted GTPase, the cellular abundance of GTP is beneficial to prevent off-target activity that might arise from noncovalent interactions within homologous pockets.
The case of KRASG12C covalent inhibitors shows the importance of careful design and a thorough compound evaluation, which includes a deep understanding of the target protein’s lifecycle. While the accumulated knowledge concerning RAS in cancer may be a one of a kind, due to the high occurrence of oncogenic mutations of this protein, it still is encouraging for further TCI development. Other strategies to target KRAS are also currently in the clinic; for example, KRAS-SOS1 protein–protein interaction inhibitors from Boehringer Ingelheim (BI 1701963) [68] or anti-KRAS engineered T-cell receptor therapy from Gilead [69]. Nevertheless, the approval of a covalent inhibitor for KRAS, even if limited to malignancies specifically harboring a KRASG12C mutation, would be extremely significant for the advancement of TCIs. Furthermore, initial efforts are being made to expand activity toward KRASG12D [70]. Being able to target more abundant amino acids will be pivotal for increasing the potential of TCIs to specifically target malignant mutations of proteins. For this reason, research for specific warhead development has become more and more cutting-edge, as will be discussed in the following section.
Warheads: pushing new reactivities toward the clinic
Despite a steady success in the clinic, recent FDA-approved TCIs mostly rely on acrylamides as warheads (Figure 3A), recurrently targeting Cys residues (Figure 3B), except for the huge class of β-lactam antibiotics. This prevalence might derive from three main considerations:
-
■
Being the least common nucleophilic residue found in proteins, cysteines represent the ideal target for electrophilic drugs. Moreover, the environment can greatly influence the reactivity of nucleophilic residues; for example, Lys is usually protonated at endogenous pH, thus allowing a kinetic selection to occur in favor of Cys compared with other nucleophilic amino acids. This is also true among different Cys residues; for example, buried and disulfided Cys are unlikely to interfere, while pKa can vary greatly for solvent exposed Cys within different proteins [71].
-
■
Acrylamides are considered to bear the best balance of reactivity as Michael acceptors – not inherently promiscuous, yet sufficiently reactive when in proximity to a nucleophilic Cys – and thus offer a good starting point for a selective warhead in TCI design. However, this is a quite imperfect view, as the nature of acrylamide substituents can enormously affect reactivity [72,73].
-
■
There is a more consolidated understanding of α,β-unsaturated carbonyls as acceptors of hetero-Michael addition reactions, which has been built from the occurrence of such electrophiles in natural substances [74] and from the literature output of the first decades of TCIs [75].
Figure 3. . FDA-approved covalent inhibitors.
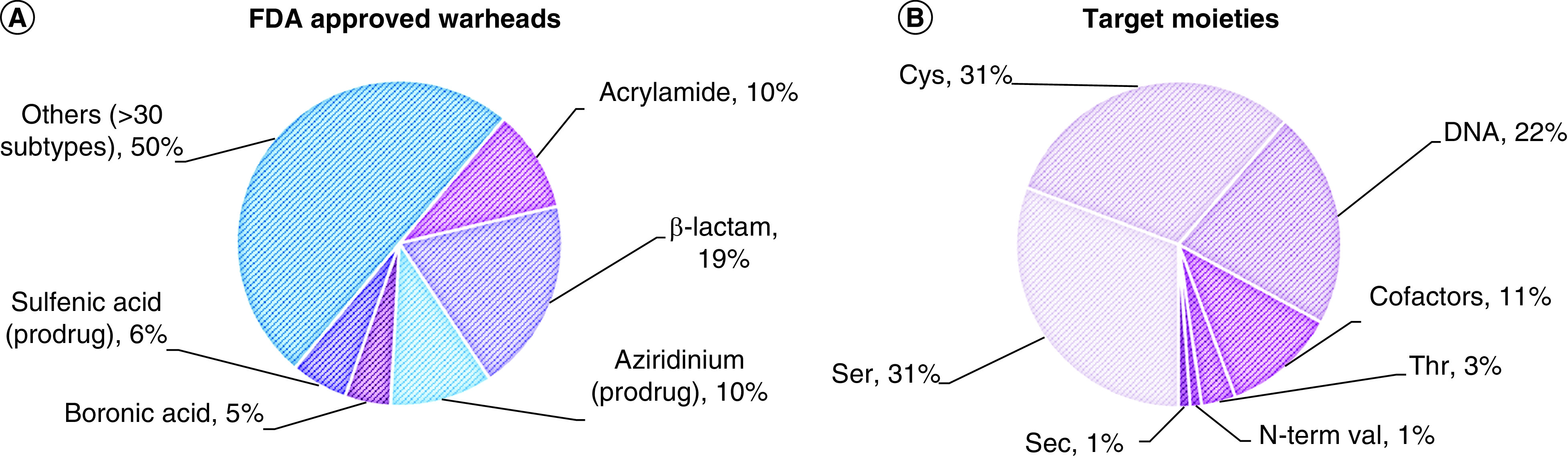
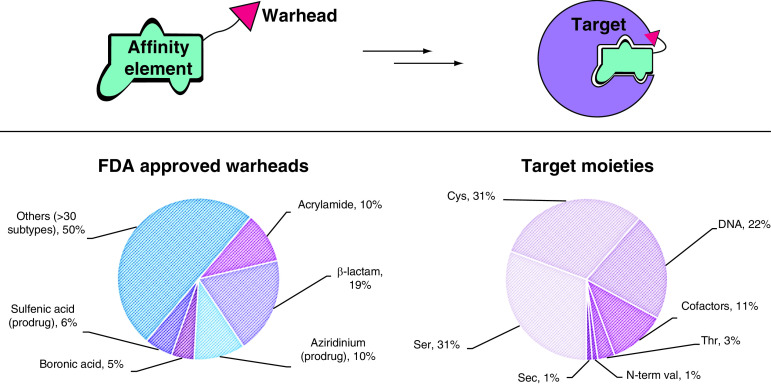
(A) Types of warheads found in FDA-approved drugs. In prodrugs, the warhead is masked. (B) Target moieties covalently bound by FDA-approved drugs. DNA includes all nonspecific alkylating agents (the most commonly modified site is N7 in purines). Cofactors include PLP, heme groups, FAD and NADH. Data source: Supplementary Table 1 (n = 88).
Sec: Selenocysteine.
The slow yet steady appearance of TCIs in the clinic in the last 7 years, albeit for a limited set of target moieties and warheads (Figure 3), is indicative of their promise and potential. The possibility of targeting more represented amino acids than Cys has opened new perspectives also in the field of kinase inhibition, allowing novel targets to be tackled (e.g., Tyr227 on SRPK1/2) [76]. The covalent inhibition field is still in its infancy and therefore the chemical armory is still expanding. Indeed, in 2017, a new warhead made it through FDA approval, with acalabrutinib relying on a 2-butynamide moiety to covalently target Cys481 on BTK. Like acrylamides, this electrophile reacts with Cys via Michael addition, but it offers a quite different geometry of both starting material and reaction product that can be crucial for precise targeting of a specific residue, not to mention that it masks a second Michael acceptor. To enhance the impact of TCIs in the clinic, broadening the number of tractable warheads is essential, expanding both on target scope and covalent mechanisms. Increasing the geometrical shapes available in the medicinal chemist’s toolbox is also fundamental to allow for more accurate structure-guided design.
A selection of covalent warheads, including some of those most commonly found in FDA-approved drugs, is shown in Figure 4. For a wider range of less common, emerging warheads the reader is directed to a compelling perspective from Gehringer and Laufercan [77].
Figure 4. . Nonexhaustive collection of warheads used for TCI design.
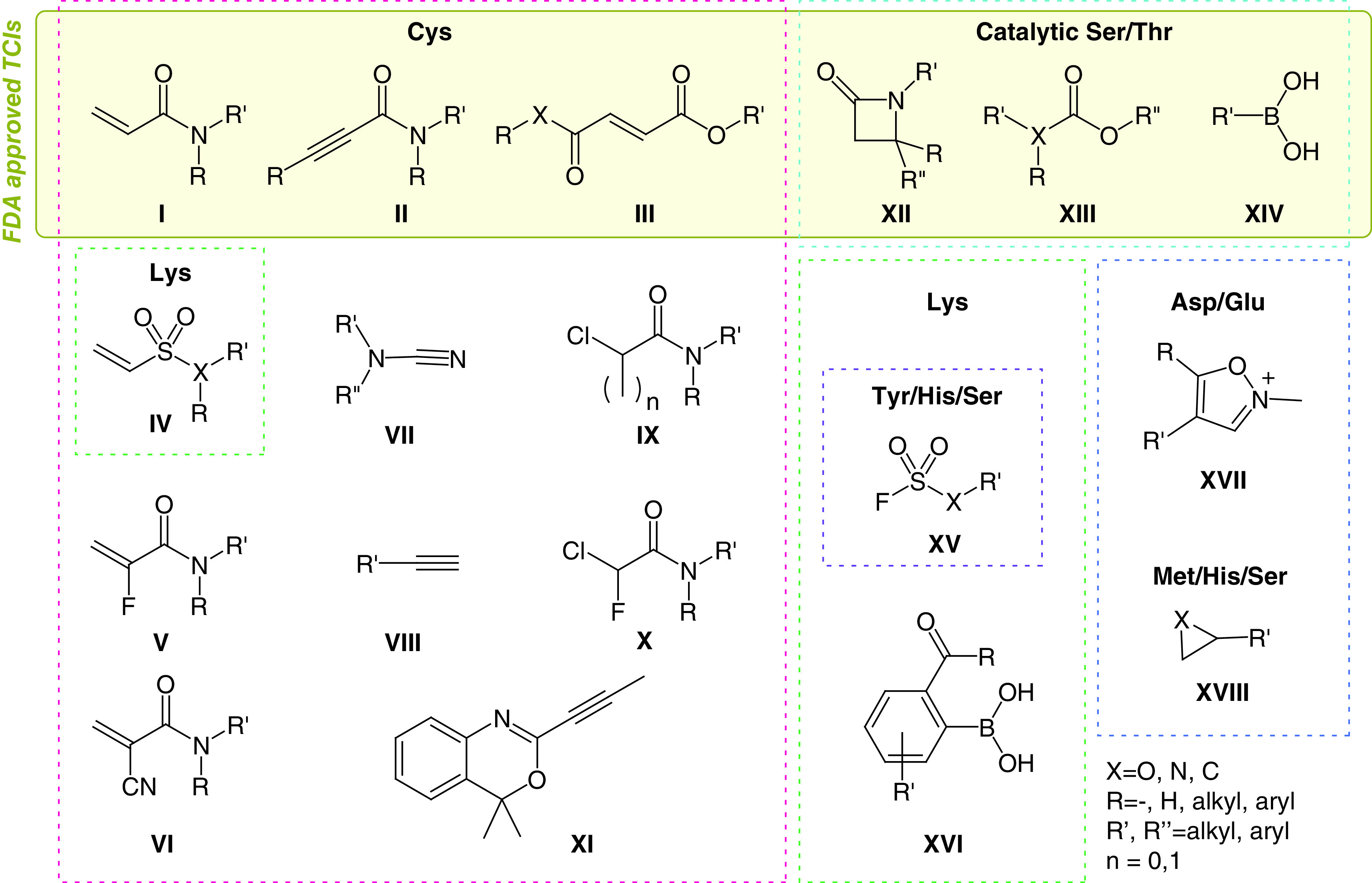
The warheads are grouped in panels by target residues (Cys, pink; catalytic Ser/Thr, cyan; Lys, green; Tyr/His/Ser, purple; Asp/Glu, blue; His/Ser, orange). Some warheads may be used to target multiple residues.
The list shown in Figure 4 is nonexhaustive and represents a biased focus on the most promising moieties currently in research. Each of these warheads has in fact shown some favorable properties that could be exploited in the clinic, such as stability and selectivity. The biggest set is represented by Cys-targeting warheads, as expected given that this is the most studied target residue for TCIs. As mentioned, the suitability and versatility of Michael acceptors is well known. The most represented ones are acrylamides (I), alkynylamides (II), fumarate derivatives (III) and vinylsulfones/sulfonamides (IV). Researchers are becoming aware of the significant effects of substituents over the reactivity of these functional groups [72,78]. While the selectivity of TCIs should mostly be driven by the specificity of noncovalent interactions and the correct orientation of the electrophilic warhead to target a nonconserved residue, reactivity tuning plays an important role not only in preventing promiscuity but also in enhancing on-target efficacy. Additionally, the importance of TCI kinetic selectivity should not be underestimated. In this regard, the introduction of strategic metabolic labilities to alter warhead reactivity, as pioneered by Zaro et al., opens a new and promising perspective [79]. Designed perturbation of warhead groups can be used in favor of desired reactivity tuning. For example, less reactive α-fluoroacrylamides (V) have recently become more common, as seen for the warhead of a KRASG12C clinical candidate (MRTX849, Figure 2D) [65,80]. The chemical basis for the reduced reactivity of this warhead is not yet clear and whether this effect can be associated with reversibility of the covalent bond has not been studied in detail. When a stronger electron-withdrawing group such as cyanate (VI) is introduced at the same position, the acidity of the adduct α-proton favors retro-Michael addition; thus covalent, yet reversible, inhibitors can be designed [81]. Pinner-type addition to cyanamides (VII) has been also used for the design of reversible TCIs [82–84]. Similarly, Cys can be added to activated heteroaryl alkynes (conjugate addition), while catalytic Cys residues have also recently been reported to react with nonactivated alkynes (VIII) [85]. The latter warhead offers extreme selectivity, the reactivity of alkynes in cells being so rare that they are considered bioorthogonal functional groups, highly exploited in click chemistry. Alternative mechanisms, such as nucleophilic substitution (SN2) of α-chloroacetamides (IX, n = 0), despite being in use, are often considered too promiscuous for drug design. Also in this case, introduction of further substituents on the α-carbon has been explored; for example, a sterically hindering methyl group (IX, n = 1) or an electronic-altering fluoride (X), both of which resulted in enhancement of desired properties [73,86]. Finally, efforts toward the expansion of the Cys-targeting armory recently led to the design of alkynyl benzoxazines (XI) [87]. However, further applications are needed to assess the tractability of this warhead.
Many successful covalent drugs target catalytic Ser and Thr, relying mostly on ּβ-lactams (XII) and carbamates/esters (XIII) for the former residue and boronic acids (XIV) for the latter. All these warheads, despite having shown good properties in terms of selectivity and pharmacokinetics, are too unreactive to be used to target these residues unless they are activated by their environment (e.g., an enzyme catalytic site, which favors deprotonation and consequent increased nucleophilicity of Ser/Thr). In their native state (i.e., not deprotonated), both Ser and Thr are significantly less nucleophilic than Cys; furthermore, they have a much higher pKa, so that even the correct orientation of a mildly reactive electrophile might be insufficient to trigger a covalent reaction. Thus only a few preliminary accounts have successfully targeted noncatalytic Ser [88], and specific warheads for these residues are needed, possibly catalyzing their deprotonation to favor reactivity.
The second most nucleophilic among noncatalytic residues is Lys, and increasing efforts are being pushed toward the design of selective electrophiles for this amino acid. Most Lys residues exposed on protein surfaces are protonated at physiological pH and contribute to enhanced protein solubility. However, Lys buried in protein pockets can have lower pKa, thus allowing for nucleophilic reactivity to be targeted. This phenomenon makes Lys residues attractive targets despite their abundance in proteins, as selectivity can be tuned by the site environment. For this reason, databases and prediction software for the pKa of specific residues are extremely relevant [89].
Currently, there are no FDA-approved TCIs targeting Lys despite increasing efforts in research [90]. While some of the Michael acceptors used against Cys can potentially (albeit less readily) react with Lys as well, protonation of the resulting amino-adduct favors retro-Michael addition, thus making this product less stable. Interestingly, vinyl sulfones/sulfonamide compounds (IV) tend to react faster with nitrogen nucleophiles, possibly due to a harder character of the conjugated acceptor in response to the higher delocalization of electrons toward the sulfur center. Thus these warheads have the potential to be optimized for lysine targeting [91,92].
Sulfonyl fluorides (XV, X = CH2) have been revived as click reagents for amines by Sharpless et al., and their intrinsic relative stability even in water has fostered application in biological systems [93]. However, in this complex environment many nucleophiles may be sufficiently reactive toward sulfonyl fluorides, and so far this functional group has been found to be able to react with Lys, Tyr, Ser, Thr, Cys and His [94,95]. On the other hand, the cognate fluorosulfate warhead (XV, X = O) is remarkably less reactive and thus offers much more promise in TCIs. In fact, this functional group has higher plasma stability and appears to be almost inert, so that only optimal proximity and orientation toward a sufficiently nucleophilic residue can trigger reactivity [94]. In addition to Lys, also Tyr and Ser have been targeted with this warhead, but its intrinsic reactivity is extremely low and promiscuous binding in the presence of an affinity element is rarely observed [88,96]. Recently, scientists at AstraZeneca have also demonstrated interest in covalently targeting Lys [97]. In an attempt to increase the effectiveness of a MCL-1 protein–protein interaction inhibitor, a reversible carbonyl warhead combined with a coordinating boronic acid (XVI) was used. This warhead proved effective in generating a long-lived covalent bond with a noncatalytic Lys that ultimately improved the cellular potency of the parent compound. Despite the absence of pharmacokinetic and selectivity data in this study, this warhead encompasses two functional groups already represented in the clinic, suggesting it might be viable in terms of safety and stability.
Meanwhile, cutting-edge research is further advancing the field by targeting residues that are not conventionally considered nucleophilic, such as Asp and Glu. Although these studies are still in their infancy, it is noteworthy to mention the N-methyl isoxazonium warhead (XVII) that was successfully used to modify noncatalytic Asp and Glu residues [70,98]. Although its use is still limited for chemical biology research, due to instability and reactivity concerns, this warhead has been also reported in this context to highlight the existence of another powerful TCI tool. Substantial advances in reactivity, chemical stability and selectivity will be required for actual targeting of Asp/Glu in more translational applications. Sporadic examples have also appeared recently for targeting Met with epoxides (XVIII, X = O) [99]. Although this warhead, and the cognate aziridine (XVIII, X = N), does not usually present the optimal mild reactivity and can thus be promiscuous, it has been applied in approved drugs and could in principle be exploited to target less nucleophilic amino acids, provided that high selectivity can be obtained by the affinity element and reactivity can be tuned by warhead substituents.
Systematic studies on warhead reactivity, including the development of in silico predicting tools [100], have appeared in recent literature [72,73,78]. However, these studies remain sparse and difficult to compare as varying assay conditions have been employed. As a further advancement in the development of TCIs, it will be important to establish clear rules to predict warhead reactivity in order to minimize the need for case-by-case optimization.
Accessible resources for TCI discovery
Initial examples of TCIs – that is, kinase inhibitors – have adopted the late-stage introduction of an electrophilic warhead in optimized noncovalent lead compounds. This approach is preferable to limit warhead tuning efforts during SAR studies and has been more successful in the clinic to date [39]. However, as shown in some of the examples above (e.g., KRASG12C covalent inhibitors), the reverse approach can be significantly more straightforward, especially in the discovery of weak binders for challenging targets. Many pharmaceutical companies, as well as commercial vendors and academic laboratories, have developed proprietary electrophile libraries including subspecific selections of compounds for specific target residues or warheads. Both small molecule and fragment libraries are available for in silico and in vitro screening, with fragment-based drug discovery showing the highest promise in TCIs [11,101]. Covalent DNA-encoded libraries have also started appearing [102]. Guiding principles for the assembly of electrophilic fragment libraries have been described recently elsewhere [103]. In combination with increasing interest in TCI discovery, novel screening platforms have thus been developed with the aim of simplifying, accelerating and enhancing the accessibility of TCI drug discovery.
Mass spectrometry (MS) remains the leading detection method for protein covalent modification (e.g., via intact protein MS) as well as identification of peptide modification by tryptic digestion (Figure 5A). However, multiplexing strategies to reduce the times and costs associated with this technique are emerging [104]. One such example is the tethering assay used to discover KRASG12C covalent inhibitors (Figure 5B). In this method the target protein, usually containing a single available Cys, is incubated under reducing conditions with multiplexed hit compounds containing a disulfide warhead; thus the formation of covalent disulfide bonds will be reversible under the assay conditions. Compounds that have enhanced affinity to the proximity of the Cys will more favorably bind to the target, thus being prevalently detected in the covalent adducts by MS analyses. The tethering assay was initially developed to discover weak binders against challenging proteins, often by introduction of a mutant Cys at the desired site, but it is straightforward to see how this method was directly applicable for covalent fragment screens. However, less common disulfide libraries are usually required to exploit the equilibrium selection obtained with the reversible covalent reaction under reducing conditions. Thus the tethering assay concept has often diverted from its origin in TCI discovery to allow for employment of more convenient electrophile libraries [105].
Figure 5. . Overview of available screening platforms for the discovery of novel TCIs.
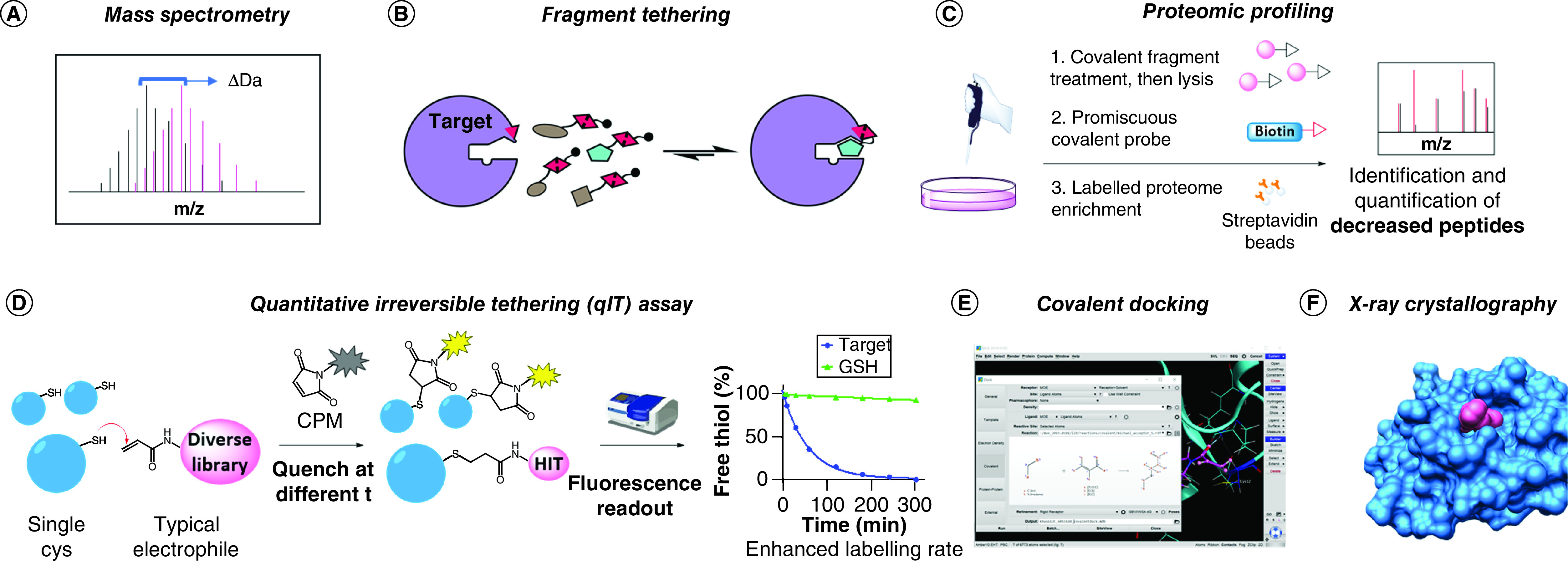
(A) Intact protein mass spectrometry: binding is shown by a shift in the measured protein MW. (B) Fragment tethering: hits are selected by favorable affinity under thermodynamic equilibrium conditions. (C) Proteomic profiling: targeted proteins are identified by the decrease in signal detected upon pull-down with broad spectrum covalent probes. (D) qIT assay: a fluorogenic substrate (CPM) is used to monitor the labeling reaction. (E) Covalent docking: hits are selected by in silico simulations. KRASG12C with ARS1620 in Molecular Operating Environment (MOE, Chemical Computing Group, Montreal, Canada) was used as an example to show the covalent docking interface (PDB: 5V9U). (F) X-ray crystallography: structural information of covalent fragment binding is generated. The structure shown derives from the Nir London lab’s recent effort for the COVID Moonshot campaign (fragment 1351 binding to COVID-19 Mpro catalytic Cys).
When large amounts of purified protein are difficult to access and selectivity issues have to be addressed as early as possible, proteomic techniques can provide a better platform (Figure 5C). Interestingly, this approach is largely unbiased, and both ligands for a specific target and targets that can be specifically liganded can be discovered [11,106,107]. Although profiling compound behavior in cells is highly advantageous in terms of selectivity and early assessment of pharmacokinetics, this technique requires more laborious sample preparation and specific instrumentation and thus is not accessible to every laboratory. Moreover, the technology is mostly incompatible with high-throughput screening. Nevertheless, studies on nucleophilic residues’ reactivity across the whole proteome have shown interesting results in terms of specific reactivities that have the potential to be targeted with carefully designed TCIs [108,109].
An orthogonal approach recently devised by Craven et al. offers a more accessible alternative to MS-based screens [110]. The quantitative irreversible tethering assay, or qIT, exploits a fluorogenic coumarinophenyl maleimide (CPM) that develops enhanced fluorescent signal upon Michael addition reaction (Figure 5D); thus the labeling rate of electrophilic compounds toward targets containing a single free Cys can be readily monitored by decrease in measured fluorescence over time. Despite requiring higher amounts of protein compared with MS, this assay offers the advantage of affordably screening large libraries of compounds in vitro. Furthermore, glutathione or other nucleophiles of choice can be used to discriminate between affinity-enhanced labeling rates and promiscuously highly reactive electrophiles that might be present in the library. However, orthogonal confirmation of hits by intact protein MS is required for assay result validation and to identify potential PAINs.
The most affordable approach may be represented by covalent docking (Figure 5E). Virtual screening technologies for the identification of new covalent ligands have been steadily improving with the implementation of new standalone software, protocols and web servers [111,112]. These include AutoDock [113], CovalentDock [114], CovDock [115], DOCKovalent [116], MOE (Figure 5E) and GOLD [117], most of which have been analyzed in comparative studies [118,119]. In addition to the obvious need for structural information concerning the target, the major limitation of covalent docking lies in the requirement for specific ligand and protein preparation, an often lengthy process that cannot always be automatized; results are thus inapplicable to large libraries [111]. Moreover, accounting for the energy gain associated with covalent bond formation can be tricky, and only warheads that share the same reaction mechanism can be productively ranked in a screen [118]. Nevertheless, selection of promising covalent hits from large libraries has been achieved with immense reduction in wet lab costs, as evidenced by numerous recent publications [120–122]. Moreover, in silico prediction tools represent a straightforward means to detect potential pseudopockets on a protein of interest; hence the increasing number of works relying on these calculations [123,124] and the appearance of free web servers such as the FTMap family [125]. Previously undruggable protein sites are now being reconsidered for TCI if they lie in the vicinity of a nucleophilic amino acid, and virtual interrogation is not uncommonly the first step in this re-evaluation process.
Actual structural information can instead be acquired by performing x-ray crystallography screening (Figure 5F). While this technique is usually associated with higher costs and presents a strict requirement for pre-existing and consolidated protein crystallization protocols, it offers the best starting point for rational TCI design. A remarkable example of this approach in covalent fragment-based drug discovery has been widely advertised among scientists and in social media for the structure-based discovery of inhibitors against COVID-19 Mpro. In particular, Nir London’s lab contributed to the COVID Moonshot campaign (@covid_moonshot) by screening a library of >1000 covalent fragments, following the elucidation of the structure of this virus main protease by Diamond Light Source [126,127].
In addition to the discussed screening approaches, other drug discovery methods that have not yet been extensively explored, such as NMR, could also be used for TCI. As already stressed, the need for structural information remains fundamental in TCI discovery, as the careful design of a correctly placed electrophile can be impossible without previous understanding of its binding mode. However, significant advances in structural biology have been made in recent years, including improvement of homology modeling software and the advent of CryoEM. The availability of these resources will be invaluable for the success of TCI approaches in drug discovery.
Afterword: be alert & learn from unpredicted reactivity
Some of the assays discussed above involve the use of end point measurements, which can be used throughout inhibitor development to rank derivatives. A full characterization of reaction mechanism and kinetics will often be conducted retrospectively and only on a handful of the most promising substances. The covalent KRASG12C inhibitors are a prominent example of this general trend: while the compounds are quickly progressing in clinical trials, only recently have full reaction mechanism studies been reported [128,129]. Interestingly, the role of Lys16 was found to be essential to trigger the highly favorable covalent reaction; we could technically say that KRAS catalyzes the reaction with its own inhibitor, by orienting the Michael acceptor and stabilizing the reactive intermediate [130]. Even though the ARS compounds were discovered serendipitously, it is becoming clear that the existence of such a specific mechanism is a major contributor to the success of these molecules. While the presence of this sweet spot for covalent inhibition via Michael acceptors on KRAS would have been hard to predict, we can retrospectively learn from this case and use the findings to produce better TCIs.
Another peculiar case was recently presented by Bashore and colleagues, who developed a series of cyanopyrrolidine-based covalent inhibitors of USP7 [131]. In this work, while investigating TCIs against the catalytic Cys of this protein, the authors came across an unusual desulfhydration mechanism, which was caused by the cyanopyrrolidine warhead and was specific for USP7 (compared with, say, USP30). This mechanism is perhaps an extreme example, but it shows that unpredictable products and reactivity, albeit uncommon, could result from TCIs. Thus investigation cannot stop after obtaining complete labeling of a protein; the mechanisms and kinetics of adduct hydrolysis should be always considered and excluded before considering a TCI truly irreversible. Surely further unpredicted mechanisms lie behind many of the TCIs currently studied, thus these cases are reported as a reminder against common traps of simplistic assumptions.
Conclusion
The current perspective was intended to provide an informative view of the current status of TCIs on the drug market and of the steps made toward the translation of research advances in the clinic. TCIs have fascinating potential to tackle challenging targets, including small GTPases, protein–protein interactions and nonprotein cell components, all of which represent cutting-edge quests in medicinal chemistry. Particularly, even if TCIs do not eventually revolutionize the clinical landscape, they serve as powerful tool compounds that are key to harnessing complex biological questions in basic and applied research. Undoubtedly, the last decade has seen a significant increase in academic research devoted to the investigation of covalent inhibitors, and great promise has been translated from industrial settings to the clinic, as seen for the KRASG12C inhibitors. While previous concerns regarding the safety of these molecules for human application were certainly justified, it is true that many physiological pathways are governed by covalent reactions, and many natural bioactive compounds were found to be safe despite being irreversible modifiers of proteins (e.g., β-lactam antimicrobial substances). It is difficult to predict how the future of TCI will look in 10 years’ time, but it is clear that these powerful molecules will accompany the field of medicinal chemistry for a long time and will be pivotal to significant chemical biology discoveries.
Future perspective
As anticipated by many medicinal chemists, the field of TCIs has shown a clear growth over the last decade. The efficacy of these inhibitors was shown in the clinic, with a rising number of new FDA-approved entities acting via a rationally designed covalent mechanism. While the advantages over reversible inhibition therapy need to be further investigated, TCIs will surely remain indispensable tools for chemical biology and a continuous expansion of this field is foreseeable for the near future. It is also possible that the development of novel TCIs will allow tackling more challenging therapeutic targets.
Executive summary.
A resurgence of covalent drugs is in place, with scientists showing a better grasp of safety issues and the potential of careful covalent inhibitor design.
At least eight targeted covalent inhibitors (TCIs) have been approved by the US FDA since 2013.
Research is exploring a variety of warheads beyond canonical acrylamides, and trying to target a wider range of nucleophilic residues beyond cysteine.
Several platforms for TCI discovery have been developed.
Overall progress is exemplified by well-known cases currently in the clinic; for example, KRASG12C inhibitors, used here as a case study.
Further efforts should be devoted to understanding and devising new mechanistic strategies for TCIs, with the aim of increasing the potential of these powerful tools.
Supplementary Material
Acknowledgments
The author would like to acknowledge M Maneiro Rey and D Lucy for the critical review of the manuscript, and E Tate for agreeing on revising the final manuscript and for the inspiring discussions that informed the author’s current view on the topic of covalent inhibition. The author also kindly appreciates H Wall for her support with manuscript submission to the journal.
Footnotes
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.future-science.com/doi/suppl/10.4155/fmc-2020-0236
Financial & competing interests disclosure
Molecular graphics (Figure 5) were performed with UCSF Chimera, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH P41-GM103311. The author has no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.Vane JR, Botting RM. The mechanism of action of aspirin. Thromb. Res. 110(5-6), 255–258 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Yocum RR, Rasmussen JR, Strominger JL. The mechanism of action of penicillin. Penicillin acylates the active site of Bacillus stearothermophilus D-alanine carboxypeptidase. J. Biol. Chem. 255(9), 3977–3986 (1980). [PubMed] [Google Scholar]
- 3.Bauer RA. Covalent inhibitors in drug discovery: from accidental discoveries to avoided liabilities and designed therapies. Drug Discov. Today 20, 1061–1073 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Baillie TA. Drug–protein adducts: past, present, and future. Med. Chem. Res. 29(7), 1093–1104 (2020). [Google Scholar]
- 5.Baell J, Walters MA. Chemistry: chemical con artists foil drug discovery. Nature 513(7519), 481–483 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Singh J, Petter RC, Baillie TA, Whitty A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 10(4), 307–317 (2011). [DOI] [PubMed] [Google Scholar]
- 7.Zhang T, Hatcher JM, Teng M, Gray NS, Kostic M. Recent advances in selective and irreversible covalent ligand development and validation. Cell Chem. Biol. 26(11), 1486–1500 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Copeland RA. The drug-target residence time model: a 10-year retrospective. Nat. Rev. Drug Discov. 15(2), 87–95 (2016). [DOI] [PubMed] [Google Scholar]
- 9.De Vita E, Schüler P, Lovell Set al. Depsipeptides featuring a neutral P1 are potent inhibitors of kallikrein-related peptidase 6 with on-target cellular activity. J. Med. Chem. 61(19), 8859–8874 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Mortenson DE, Brighty GJ, Plate Let al. ‘Inverse drug discovery’ strategy to identify proteins that are targeted by latent electrophiles as exemplified by aryl fluorosulfates. J. Am. Chem. Soc. 140(1), 200–210 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Backus KM, Correia BE, Lum KMet al. Proteome-wide covalent ligand discovery in native biological systems. Nature 534(7608), 570–574 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baillie TA. Targeted covalent inhibitors for drug design. Angew. Chemie Int. Ed. 55(43), 13408–13421 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Lagoutte R, Patouret R, Winssinger N. Covalent inhibitors: an opportunity for rational target selectivity. Curr. Opin. Chem. Biol. 39, 54–63 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Cheng SS, Yang GJ, Wang W, Leung CH, Ma DL. The design and development of covalent protein–protein interaction inhibitors for cancer treatment. J. Hematol. Oncol. 13(1), 1–14 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tuley A, Fast W. The taxonomy of covalent inhibitors. Biochemistry 57(24), 3326–3337 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Delineates guidelines for stratifying the mechanism of action of different covalent inhibitors.
- 16.Mah R, Thomas JR, Shafer CM. Drug discovery considerations in the development of covalent inhibitors. Bioorganic Med. Chem. Lett. 24(1), 33–39 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Strelow JM. A perspective on the kinetics of covalent and irreversible inhibition. SLAS Discov. 22(1), 3–20 (2017). [DOI] [PubMed] [Google Scholar]; •• Covers in depth the kinetics of covalent inhibition, with thoughtful consideration on translating in vitro results to cells.
- 18.Schürmann M, Janning P, Ziegler S, Waldmann H. Small-molecule target engagement in cells. Cell Chem. Biol. 23(4), 435–441 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Nomura DK, Maimone TJ. Target identification of bioactive covalently acting natural products. Curr. Top. Microbiol. Immunol. 420, 351–374 (2019). [DOI] [PubMed] [Google Scholar]
- 20.Lee CU, Grossmann TN. Reversible covalent inhibition of a protein target. Angew. Chemie Int. Ed. 51(35), 8699–8700 (2012). [DOI] [PubMed] [Google Scholar]
- 21.Kalgutkar AS, Dalvie DK. Drug discovery for a new generation of covalent drugs. Expert Opin. Drug Discov. 7(7), 561–581 (2012). [DOI] [PubMed] [Google Scholar]
- 22.Coleman K. Diazabicyclooctanes (DBOs): a potent new class of non-β-lactam β-lactamase inhibitors. Curr. Opin. Microbiol. 14(5), 550–555 (2011). [DOI] [PubMed] [Google Scholar]
- 23.Potashman MH, Duggan ME. Covalent modifiers: an orthogonal approach to drug design. J. Med. Chem. 52(5), 1231–1246 (2009). [DOI] [PubMed] [Google Scholar]; • Comprehensive review on covalent drugs mechanism of action (up to 2009).
- 24.Nejadmoghaddam M-R, Minai-Tehrani A, Ghahremanzadeh R, Mahmoudi M, Dinarvand R, Zarnani A-H. Antibody–drug conjugates: possibilities and challenges. Avicenna J. Med. Biotechnol. 11(1), 3–23 (2019). [PMC free article] [PubMed] [Google Scholar]
- 25.Chen Y, Jia Y, Song W, Zhang L. Therapeutic potential of nitrogen mustard based hybrid molecules. Front. Pharmacol. 9, 1453 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li D, Ambrogio L, Shimamura Tet al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene 27(34), 4702–4711 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dungo RT, Keating GM. Afatinib: first global approval. Drugs 73(13), 1503–1515 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Pan Z, Scheerens H, Li S-Jet al. Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase. ChemMedChem. 2(1), 58–61 (2007). [DOI] [PubMed] [Google Scholar]
- 29.De Claro RA, McGinn KM, Verdun Net al. FDA approval: ibrutinib for patients with previously treated mantle cell lymphoma and previously treated chronic lymphocytic leukemia. Clin. Cancer Res. 21(16), 3586–3590 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Daelemans D, Afonina E, Nilsson Jet al. A synthetic HIV-1 Rev inhibitor interfering with the CRM1-mediated nuclear export. Proc. Natl Acad. Sci. USA 99(22), 14440–14445 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van Neck T, Pannecouque C, Vanstreels E, Stevens M, Dehaen W, Daelemans D. Inhibition of the CRM1-mediated nucleocytoplasmic transport by N-azolylacrylates: structure–activity relationship and mechanism of action. Bioorganic Med. Chem. 16(21), 9487–9497 (2008). [DOI] [PubMed] [Google Scholar]
- 32.Brömme D, Panwar P, Turan S. Cathepsin K osteoporosis trials, pycnodysostosis and mouse deficiency models: commonalities and differences. Expert Opin. Drug Discov. 11(5), 457–472 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Huggins JP, Smart TS, Langman S, Taylor L, Young T. An efficient randomised, placebo-controlled clinical trial with the irreversible fatty acid amide hydrolase-1 inhibitor PF-04457845, which modulates endocannabinoids but fails to induce effective analgesia in patients with pain due to osteoarthritis of the knee. Pain 153(9), 1837–1846 (2012). [DOI] [PubMed] [Google Scholar]
- 34.Miller JA. The metabolism of xenobiotics to reactive electrophiles in chemical carcinogenesis and mutagenesis: a collaboration with Elizabeth Cavert Miller and our associates. Drug Metab. Rev. 30(4), 645–674 (1998). [DOI] [PubMed] [Google Scholar]
- 35.Schwöbel JAH, Koleva YK, Enoch SJet al. Measurement and estimation of electrophilic reactivity for predictive toxicology. Chem. Rev. 111(4), 2562–2596 (2011). [DOI] [PubMed] [Google Scholar]
- 36.Cohen P. Protein kinases – the major drug targets of the twenty-first century? Nat. Rev. Drug Discov. 1(4), 309–315 (2002). [DOI] [PubMed] [Google Scholar]
- 37.Ferguson FM, Gray NS. Kinase inhibitors: the road ahead. Nat. Rev. Drug Discov. 17(5), 353–376 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Roskoski R. Properties of FDA-approved small molecule protein kinase inhibitors. Pharmacol. Res. 144, 19–50 (2019). [DOI] [PubMed] [Google Scholar]
- 39.Abdeldayem A, Raouf YS, Constantinescu SN, Moriggl R, Gunning PT. Advances in covalent kinase inhibitors. Chem. Soc. Rev. 49(9), 2617–2687 (2020). [DOI] [PubMed] [Google Scholar]
- 40.Leonetti A, Sharma S, Minari R, Perego P, Giovannetti E, Tiseo M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br. J. Cancer 121(9), 725–737 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Drugs.com. Xpovio (selinexor): FDA Approval History. www.drugs.com/history/xpovio.html
- 42.Wolff B, Sanglier JJ, Wang Y. Leptomycin B is an inhibitor of nuclear export: inhibition of nucleo-cytoplasmic translocation of the human immunodeficiency virus type 1 (HIV-1) Rev protein and Rev-dependent mRNA. Chem. Biol. 4(2), 139–147 (1997). [DOI] [PubMed] [Google Scholar]
- 43.Sun Q, Carrasco YP, Hu Yet al. Nuclear export inhibition through covalent conjugation and hydrolysis of leptomycin B by CRM1. 110(4), 1303–1308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Komiyama K, Okada K, Tomisaka S, Beppu T, Hamamoto T, Umezawa I. Antitumor activity of leptomycin B. J. Antibiot. (Tokyo) 38(3), 427–429 (1985). [DOI] [PubMed] [Google Scholar]
- 45.Dong X, Biswas A, Süel KEet al. Structural basis for leucine-rich nuclear export signal recognition by CRM1. Nature 458(7242), 1136–1141 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kalid O, Toledo Warshaviak D, Shechter S, Sherman W, Shacham S. Consensus Induced Fit Docking (cIFD): methodology, validation, and application to the discovery of novel Crm1 inhibitors. J. Comput. Aided Mol. Des. 26(11), 1217–1228 (2012). [DOI] [PubMed] [Google Scholar]
- 47.Neggers JE, Vercruysse T, Jacquemyn Met al. Identifying drug-target selectivity of small-molecule CRM1/XPO1 inhibitors by CRISPR/Cas9 genome editing. Chem. Biol. 22(1), 107–116 (2015). [DOI] [PubMed] [Google Scholar]
- 48.US FDA (2019). FDA grants accelerated approval to selinexor for multiple myeloma. www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-selinexor-multiple-myeloma
- 49.Wang AY, Liu H. The past, present, and future of CRM1/XPO1 inhibitors. Stem Cell Investig. 6(6), 1–9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.ClinicalTrials.gov. Evaluation of activity and safety of oral selinexor in participants with severe COVID-19 infection. www.clinicaltrials.gov/ct2/show/NCT04349098?term=selinexor&draw=3&rank=14
- 51.Niu M, Chong Y, Han Y, Liu X. Novel reversible selective inhibitor of nuclear export shows that CRM1 is a target in colorectal cancer cells. Cancer Biol. Ther. 16(7), 1110–1118 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fernández-Medarde A, Santos E. Ras in cancer and developmental diseases. Genes Cancer 2(3), 344–358 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Prior IA, Hood FE, Hartley JL. The frequency of RAS mutations in cancer. Cancer Res. 80(14), 2969–2974 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Downward J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 3(1), 11–22 (2003). [DOI] [PubMed] [Google Scholar]
- 55.O'Bryan JP. Pharmacological targeting of RAS: recent success with direct inhibitors. Pharmacol. Res. 139, 503–511 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Prior IA, Lewis PD, Mattos C. A comprehensive survey of RAS mutations in cancer. Cancer Res. 72(10), 2457–2467 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503(7477), 548–551 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Taveras AG, Remiszewski SW, Doll RJet al. Ras oncoprotein inhibitors: the discovery of potent ras nucleotide exchange inhibitors and the structural determination of a drug-protein complex. Bioorganic Med. Chem. 5(1), 125–133 (1997). [DOI] [PubMed] [Google Scholar]
- 59.Lanman BA, Allen JR, Allen JGet al. Discovery of a covalent inhibitor of KRASG12C (AMG 510) for the treatment of solid tumors. J. Med. Chem. 63(1), 52–65 (2020). [DOI] [PubMed] [Google Scholar]
- 60.Patricelli MP, Janes MR, Li LSet al. Selective inhibition of oncogenic KRAS output with small molecules targeting the inactive state. Cancer Discov. 6(3), 316–329 (2016). [DOI] [PubMed] [Google Scholar]
- 61.Janes MR, Zhang J, Li LSet al. Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell 172(3), 578–589.e17 (2018). [DOI] [PubMed] [Google Scholar]
- 62.Lito P, Solomon M, Li L-S, Hansen R, Rosen N. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science 351(6273), 604–608 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Canon J, Rex K, Saiki AYet al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 575(7781), 217–223 (2019). [DOI] [PubMed] [Google Scholar]
- 64.Source: Amgen. Amgen announces first clinical data evaluating novel investigational KRASG12C inhibitor AMG 510 at ASCO 2019. Press release:www.amgen.com/media/news-releases/2019/06/amgen-announces-first-clinical-data-evaluating-novel-investigational-krasg12c-inhibitor-amg-510-at-asco-2019/
- 65.Fell JB, Fischer JP, Baer BRet al. Identification of the clinical development candidate MRTX849, a covalent KRASG12C inhibitor for the treatment of cancer. J. Med. Chem. 63(13), 6679–6693 (2020). [DOI] [PubMed] [Google Scholar]
- 66.Nagasaka M, Li Y, Sukari A, Ou S-HI, Al-Hallak MN, Azmi AS. KRAS G12C Game of Thrones, which direct KRAS inhibitor will claim the iron throne? Cancer Treat. Rev. 84, 101974 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lim SM, Westover KD, Ficarro SBet al. Therapeutic targeting of oncogenic K-ras by a covalent catalytic site inhibitor. Angew. Chemie Int. Ed. 53(1), 199–204 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.ClinicalTrials.gov. A study to test different doses of BI 1701963 alone and combined with trametinib in patients with different types of advanced cancer (solid tumours with KRAS Mutation). https://clinicaltrials.gov/ct2/show/NCT04111458
- 69.ClinicalTrials.gov. Administering peripheral blood lymphocytes transduced with a murine T-cell receptor recognizing the G12D variant of mutated RAS in HLA-A. https://clinicaltrials.gov/ct2/show/NCT03745326
- 70.Mcgregor LM, Jenkins ML, Kerwin C, Burke JE, Shokat KM. Expanding the scope of electrophiles capable of targeting K-Ras oncogenes. Biochemistry 56(25), 3178–3183 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Grimsley GR, Scholtz JM, Pace CN. A summary of the measured pK values of the ionizable groups in folded proteins. Protein Sci. 18(1), 247–251 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cee VJ, Volak LP, Chen Yet al. Systematic study of the glutathione (GSH) reactivity of N-arylacrylamides: 1. Effects of aryl substitution. J. Med. Chem. 58(23), 9171–9178 (2015). [DOI] [PubMed] [Google Scholar]
- 73.Flanagan ME, Abramite JA, Anderson DPet al. Chemical and computational methods for the characterization of covalent reactive groups for the prospective design of irreversible inhibitors. J. Med. Chem. 57(23), 10072–10079 (2014). [DOI] [PubMed] [Google Scholar]
- 74.Lagoutte R, Winssinger N. Following the lead from nature with covalent inhibitors. Chimia (Aarau). 71(10), 703–711 (2017). [DOI] [PubMed] [Google Scholar]
- 75.Jackson PA, Widen JC, Harki DA, Brummond KM. Covalent modifiers: a chemical perspective on the reactivity of α,β-unsaturated carbonyls with thiols via hetero-Michael addition reactions. J. Med. Chem. 60(3), 839–885 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hatcher JM, Wu G, Zeng Cet al. SRPKIN-1: A covalent SRPK1/2 inhibitor that potently converts VEGF from pro-angiogenic to anti-angiogenic isoform. Cell Chem. Biol. 25(4), 460–470.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gehringer M, Laufer SA. Emerging and re-emerging warheads for targeted covalent inhibitors: applications in medicinal chemistry and chemical biology. J. Med. Chem. 62(12), 5673–5724 (2019). [DOI] [PubMed] [Google Scholar]; • Review presenting an extensive analysis of TCI warheads from a clear chemistry perspective.
- 78.Dahal UP, Gilbert AM, Obach RSet al. Intrinsic reactivity profile of electrophilic moieties to guide covalent drug design:: N-α-acetyl-l-lysine as an amine nucleophile. Medchemcomm. 7(5), 864–872 (2016). [Google Scholar]
- 79.Zaro BW, Whitby LR, Lum KM, Cravatt BF. Metabolically labile fumarate esters impart kinetic selectivity to irreversible inhibitors. J. Am. Chem. Soc. 138(49), 15841–15844 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Xia G, Chen W, Zhang Jet al. A chemical tuned strategy to develop novel irreversible EGFR-TK inhibitors with improved safety and pharmacokinetic profiles. J. Med. Chem. 57(23), 9889–9900 (2014). [DOI] [PubMed] [Google Scholar]
- 81.Serafimova IM, Pufall MA, Krishnan Set al. Reversible targeting of noncatalytic cysteines with chemically tuned electrophiles. Nat. Chem. Biol. 8(5), 471–476 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Oballa RM, Truchon JF, Bayly CIet al. A generally applicable method for assessing the electrophilicity and reactivity of diverse nitrile-containing compounds. Bioorganic Med. Chem. Lett. 17(4), 998–1002 (2007). [DOI] [PubMed] [Google Scholar]
- 83.Casimiro-Garcia A, Trujillo JI, Vajdos Fet al. Identification of cyanamide-based janus kinase 3 (JAK3) covalent inhibitors. J. Med. Chem. 61(23), 10665–10699 (2018). [DOI] [PubMed] [Google Scholar]
- 84.Conole D, Mondal M, Majmudar JD, Tate EW. Recent developments in cell permeable deubiquitinating enzyme activity-based probes. Front. Chem. 7, 876 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mons E, Jansen IDC, Loboda Jet al. The alkyne moiety as a latent electrophile in irreversible covalent small molecule inhibitors of cathepsin K. J. Am. Chem. Soc. 141(8), 3507–3514 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shindo N, Fuchida H, Sato Met al. Selective and reversible modification of kinase cysteines with chlorofluoroacetamides. Nat. Chem. Biol. 15(3), 250–258 (2019). [DOI] [PubMed] [Google Scholar]
- 87.McAulay K, Hoyt EA, Thomas Met al. Alkynyl benzoxazines and dihydroquinazolines as cysteine targeting covalent warheads and their application in identification of selective irreversible kinase inhibitors. J. Am. Chem. Soc. 142(23), 10358–10372 (2020). [DOI] [PubMed] [Google Scholar]
- 88.Fadeyi OO, Hoth LR, Choi Cet al. Covalent enzyme inhibition through fluorosulfate modification of a noncatalytic serine residue. ACS Chem. Biol. 12(8), 2015–2020 (2017). [DOI] [PubMed] [Google Scholar]
- 89.Pahari S, Sun L, Alexov E. PKAD: a database of experimentally measured pKa values of ionizable groups in proteins. Database (Oxford 2019, baz024 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pettinger J, Jones K, Cheeseman MD. Lysine-targeting covalent inhibitors. Angew. Chemie Int. Ed. 56(48), 15200–15209 (2017). [DOI] [PubMed] [Google Scholar]
- 91.Anscombe E, Meschini E, Mora-Vidal Ret al. Identification and characterization of an irreversible inhibitor of CDK2. Chem. Biol. 22(9), 1159–1164 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chen H, Huang R, Li Zet al. Selective lysine modification of native peptides: via aza-Michael addition. Org. Biomol. Chem. 15(35), 7339–7345 (2017). [DOI] [PubMed] [Google Scholar]
- 93.Dong J, Krasnova L, Finn MG, Sharpless KB. Sulfur(VI) fluoride exchange (SuFEx): another good reaction for click chemistry. Angew. Chemie Int. Ed. 53(36), 9430–9448 (2014). [DOI] [PubMed] [Google Scholar]
- 94.Gambini L, Baggio C, Udompholkul Pet al. Covalent inhibitors of protein–protein interactions targeting lysine, tyrosine, or histidine residues. J. Med. Chem. 62(11), 5616–5627 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jones LH, Kelly JW. Structure-based design and analysis of SuFEx chemical probes. RSC Med. Chem. 11(1), 10–17 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Baggio C, Udompholkul P, Gambini Let al. Aryl-fluorosulfate-based Lysine covalent pan-inhibitors of apoptosis protein (IAP) antagonists with cellular efficacy. J. Med. Chem. 62, 9188–91200 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Akçay G, Belmonte MA, Aquila Bet al. Inhibition of Mcl-1 through covalent modification of a noncatalytic lysine side chain. Nat. Chem. Biol. 12(11), 931–936 (2016). [DOI] [PubMed] [Google Scholar]
- 98.Martín-Gago P, Fansa EK, Winzker Met al. Covalent protein labeling at glutamic acids. Cell Chem. Biol. 24(5), 589–597.e5 (2017). [DOI] [PubMed] [Google Scholar]
- 99.Kharenko OA, Patel RG, Brown SDet al. Design and characterization of novel covalent bromodomain and extra-terminal domain (BET) inhibitors targeting a methionine. J. Med. Chem. 61(18), 8202–8211 (2018). [DOI] [PubMed] [Google Scholar]
- 100.Palazzesi F, Grundl MA, Pautsch A, Weber A, Tautermann CS. A fast ab initio predictor tool for covalent reactivity estimation of acrylamides. J. Chem. Inf. Model. 59(8), 3565–3571 (2019). [DOI] [PubMed] [Google Scholar]
- 101.Resnick E, Bradley A, Gan Jet al. Rapid covalent-probe discovery by electrophile-fragment screening. J. Am. Chem. Soc. 141(22), 8951–8968 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zambaldo C, Daguer JP, Saarbach J, Barluenga S, Winssinger N. Screening for covalent inhibitors using DNA-display of small molecule libraries functionalized with cysteine reactive moieties. Medchemcomm. 7(7), 1340–1351 (2016). [Google Scholar]
- 103.Keeley A, Petri L, Ábrányi-Balogh P, Keserű GM. Covalent fragment libraries in drug discovery. Drug Discov. Today 25(6), 983–996 (2020). [DOI] [PubMed] [Google Scholar]
- 104.Johansson H, Tsai YCI, Fantom Ket al. Fragment-based covalent ligand screening enables rapid discovery of inhibitors for the RBR E3 ubiquitin ligase HOIP. J. Am. Chem. Soc. 141(6), 2703–2712 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kathman SG, Statsyuk AV. Covalent tethering of fragments for covalent probe discovery. Medchemcomm. 7(4), 576–585 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Senkane K, Vinogradova E, Suciu Ret al. The proteome-wide potential for reversible covalency at cysteine. Angew. Chemie Int. Ed. 58(33), 11385–11389 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Counihan JL, Wiggenhorn AL, Anderson KE, Nomura DK. Chemoproteomics-enabled covalent ligand screening reveals ALDH3A1 as a lung cancer therapy target. ACS Chem. Biol. 13(8), 1970–1977 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Weerapana E, Simon GM, Cravatt BF. Disparate proteome reactivity profiles of carbon electrophiles. Nat. Chem. Biol. 4(7), 405–407 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Weerapana E, Wang C, Simon GMet al. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 468(7325), 790–797 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Craven GB, Affron DP, Allen CEet al. High-throughput kinetic analysis for target-directed covalent ligand discovery. Angew. Chemie Int. Ed. 57(19), 5257–5261 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kumalo H, Bhakat S, Soliman M. Theory and applications of covalent docking in drug discovery: merits and pitfalls. Molecules 20(2), 1984–2000 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sotriffer C. Docking of covalent ligands: challenges and approaches. Mol. Inform. 37(9), 1800062 (2018). [DOI] [PubMed] [Google Scholar]
- 113.Bianco G, Forli S, Goodsell DS, Olson AJ. Covalent docking using autodock: two-point attractor and flexible side chain methods. Protein Sci. 25(1), 295–301 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ouyang X, Zhou S, Su CTT, Ge Z, Li R, Kwoh CK. CovalentDock: automated covalent docking with parameterized covalent linkage energy estimation and molecular geometry constraints. J. Comput. Chem. 34(4), 326–336 (2013). [DOI] [PubMed] [Google Scholar]
- 115.Zhu K, Borrelli KW, Greenwood JRet al. Docking covalent inhibitors: a parameter free approach to pose prediction and scoring. J. Chem. Inf. Model. 54(7), 1932–1940 (2014). [DOI] [PubMed] [Google Scholar]
- 116.London N, Miller RM, Krishnan Set al. Covalent docking of large libraries for the discovery of chemical probes. Nat. Chem. Biol. 10(12), 1066–1072 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. Improved protein–ligand docking using GOLD. Proteins Struct. Funct. Bioinform. 52(4), 609–623 (2003). [DOI] [PubMed] [Google Scholar]
- 118.Scarpino A, Ferenczy GG, Keserü GM. Comparative evaluation of covalent docking tools. J. Chem. Inf. Model. 58(7), 1441–1458 (2018). [DOI] [PubMed] [Google Scholar]
- 119.Wen C, Yan X, Gu Qet al. Systematic studies on the protocol and criteria for selecting a covalent docking tool. Molecules 24(11), 1–20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chowdhury SR, Kennedy S, Zhu Ket al. Discovery of covalent enzyme inhibitors using virtual docking of covalent fragments. Bioorg. Med. Chem. Lett. 29(1), 36–39 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Shraga A, Olshvang E, Davidzohn Net al. Covalent docking identifies a potent and selective MKK7 inhibitor. Cell Chem. Biol. 26(1), 98–108.e5 (2019). [DOI] [PubMed] [Google Scholar]
- 122.Wan X, Yang T, Cuesta Aet al. Discovery of lysine-targeted eIF4E inhibitors through covalent docking. J. Am. Chem. Soc. 142(11), 4960–4964 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Olp MD, Sprague DJ, Goetz CJet al. Covalent-fragment screening of BRD4 identifies a ligandable site orthogonal to the acetyl-lysine binding sites. ACS Chem. Biol. 15(4), 1036–1049 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhang Y, Zhang D, Tian Het al. Identification of covalent binding sites targeting cysteines based on computational approaches. Mol. Pharm. 13(9), 3106–3118 (2016). [DOI] [PubMed] [Google Scholar]
- 125.Kozakov D, Grove LE, Hall DRet al. The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat. Protoc. 10(5), 733–755 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chodera J, Lee AA, London N, von Delft F. Crowdsourcing drug discovery for pandemics. Nat. Chem. 12(7), 581–581 (2020). [DOI] [PubMed] [Google Scholar]
- 127.PostEra. COVID-19. https://postera.ai/covid
- 128.Hansen R, Peters U, Babba Aet al. The reactivity-driven biochemical mechanism of covalent KRASG12C inhibitors. Nat. Struct. Mol. Biol. 25(6), 454–462 (2018). [DOI] [PubMed] [Google Scholar]; • Elegant elucidation of KRASG12C covalent inhibitors’ mechanism of action; a great example for the rigorous investigation of TCIs in general.
- 129.Khrenova MG, Kulakova AM, Nemukhin AV. Proof of concept for poor inhibitor binding and efficient formation of covalent adducts of KRASG12C and ARS compounds. Org. Biomol. Chem. 18(16), 3069–3081 (2020). [DOI] [PubMed] [Google Scholar]; • Elegant full elucidation of KRASG12C covalent inhibitors’ mechanism of action based on in silico modelling.
- 130.Statsyuk AV. Let K-Ras activate its own inhibitor. Nat. Struct. Mol. Biol. 25(6), 435–437 (2018). [DOI] [PubMed] [Google Scholar]
- 131.Bashore C, Jaishankar P, Skelton NJet al. Cyanopyrrolidine inhibitors of ubiquitin specific protease 7 mediate desulfhydration of the active-site cysteine. ACS Chem. Biol. 15(6), 1392–1400 (2020). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.