

Abstract
Transfer RNAs (tRNAs) undergo extensive chemical modification within cells through the activity of tRNA methyltransferase enzymes (TRMs). Although tRNA modifications are dynamic, how they impact cell behavior after stress and during tumorigenesis is not well understood. This review discusses how tRNA modifications influence the translation of codon-biased transcripts involved in responses to oxidative stress. We further discuss emerging mechanistic details about how aberrant TRM activity in cancer cells can direct programs of codon-biased translation that drive cancer cell phenotypes. The studies reviewed here predict future preventative therapies aimed at augmenting TRM activity in individuals at risk for cancer due to exposure. They further predict that attenuating TRM-dependent translation in cancer cells may limit disease progression while leaving noncancerous cells unharmed.
Keywords: : anticodon, cancer, codon-bias, modification, oxidative stress, tRNA
Transfer RNAs (tRNAs) are fundamental biological molecules that complete the flow of genetic information from DNA to protein by reading cognate codons in mRNA. In humans, there are predicted to be 47 types of tRNAs for decoding 20 standard amino acids plus selenocysteine [1]. Across the three domains of life, it is typical for more than one copy of a gene for a particular species of tRNA to be present in the genome, although copy numbers vary between species [2]. These tRNAs are comprised of different isoacceptors – sequence variants charged with the same amino acid – that act together with the enzyme machinery of the ribosome to decode mRNA into protein during the process of translation. Notably, translation is a progressive process in which tRNAs charged with the amino acid corresponding to their anticodon decoding specificity enter the ribosome at the aminoacyl site (A-site). As the ribosome moves along the mRNA in the 5′ to 3′ direction, these aminoacylated tRNAs translocate to the peptidyl-site (P-site) where they are joined together by peptide bonds, one after the other, in the sequence dictated by the mRNA transcript. Finally, unacylated tRNAs translocate to the exit-site (E-site), recycling for the next round of charging and translation [3].
The degeneracy of the Universal Genetic Code lends more complexity to the process of translation: a total of 20 amino acids are represented by 61 codons, meaning that more than one codon, varying at the third base, also referred to as the wobble position, can be used to decode the same amino acid [4]. Further, different amino acids can share the same first two bases and vary only at the wobble position (i.e., Phe, Leu, His, Gln, Asn, Lys, Asp, Glu, Ser and Arg). In the case of these amino acids, residing in so-called split codon boxes (Figure 1), tRNA wobble base pairing is critical for discriminating the correct amino acid [5].
Figure 1. . The degeneracy of the Universal Genetic Code.

More than one codon, varying at the third base, or wobble position, can decode that same amino acid. Different amino acids that share the same first two bases and vary only at the wobble position reside in split-codon boxes (hatched lines); their specific decoding is influenced by wobble base tRNA modifications [5]. Codons found enriched in transcripts linked to the stress response or tumor promotion are in bold.
tRNAs can read their cognate codons using canonical Watson–Crick base pairing rules; however, tRNA–mRNA interactions are also influenced by post-transcriptional chemical modifications of the tRNA molecule, leading to noncanonical base-pairing interactions [6]. The most prominent chemical modification of tRNA is methylation, of the base or ribose sugar [7], which is catalyzed by a family of enzymes called tRNA methyltransferases (TRMs) [8]. These methyl-based modification chemistries play key roles in coordinating and regulating the process of translation at multiple levels. Modifications within the anticodon stem-loop at positions 37 and 34 (the wobble) are crucial for base pairing interactions that determine mRNA codon selection by tRNA, influencing both the accuracy and efficiency of translation [9–11]. Modifications outside of the anticodon stem-loop region contribute to and direct the folding of tRNA, altering the structure of the tRNA molecule in ways that can affect aminoacylation and interactions with the ribosome [5,12,13].
tRNA modifications influence other aspects of protein synthesis toward maintaining the fidelity of translation [14]. For example, modifications that alter tRNA stability can affect the abundance of a particular tRNA species within the cell [15–17]; RNA polymerase III-dependent transcriptional regulatory mechanisms will further contribute to tRNA populations within a cell and have been implicated as a downstream effect of oncogenic signaling pathways [18]. Regarding wobble base-pairing interactions, the codon context is an important consideration. Some sequences within mRNA transcripts make the ribosome more prone to frameshifting, and tRNA modifications can suppress this phenomenon and thus decrease the potential for translational errors [19,20]. Transfer RNA modifications can also influence the rate of translation of certain codon runs, particularly those that are present in duplicate and triplicate tandem or near-tandem repeats [21,22]. Finally, tRNA modifications can affect aminoacylation and represent another mechanism that serves to prevent translational errors by ensuring that the tRNA species is charged with the amino acid that corresponds to its specific anticodon [20].
Here, we review research that supports a role for tRNA modification in cancer, both at the initial stage of its development and as it advances to a malignant phenotype. First, we examine the connections between TRM activity and exposure to oxidative stress-inducing compounds. Given the well-established link between oxidative stress-induced DNA damage, mutation and carcinogenesis, this work implies that enhancing tRNA-dependent translational responses to oxidative stress could be an effective preventative strategy for cancer. Next, we will discuss tRNA modifications and altered tRNA modifying enzyme expression and activity associated with specific types of cancer, with the idea that the levels of ribonucleoside modification chemistries may be useful biomarkers of early disease or malignant progression. Last, we will highlight some of the exciting new mechanistic details that are beginning to emerge about how tRNA modification systems can translationally reprogram cells toward a cancerous phenotype. To facilitate this discussion, we consider a tRNA modification system to be the modifying enzymes, their catalyzed modifications and the codon-biased transcripts influenced by the modified tRNA at the level of translation.
Stress-induced tRNA methylation & methyltransferase activity
tRNAs are among the most chemically modified molecules within a cell, with 51 distinct modification chemistries identified in eukarya to date, many of which are conserved among all domains of life [23]. Although the number and type of modification will vary depending upon the individual tRNA, mammalian cytoplasmic tRNAs are estimated to carry, on average, 13–14 modifications [8,24]. Moreover, these modification chemistries are dynamic in nature and change in response to different environmental stresses, a strong indication that tRNA modifications play a regulatory role in the cell's response to stress [25]. Relevant to any discussion about cancer are the changes in tRNA modification chemistries that occur after exposure to oxidative stress. Using budding yeast (Saccharomyces cerevisiae) as a model eukaryote, Chan et al. (2010) report that exposure to hydrogen peroxide (H2O2) sodium hypochlorite (NaOCl) and sodium arsenite (NaAsO2) induces global changes in ribonucleoside modification chemistries, with significant increases in 2′-O-methylcytosine (Cm), 5-methylcytosine (m5C) and N2, N2-dimethylguanosine (m22G) in response to H2O2 and decreases in Cm in response to NaOCl and NaAsO2, with other modified nucleosides being either increased, or unchanged, with the latter two toxicants [25]. Oxidative stress describes a general cellular response that differs based upon the exposure toxicant and the corresponding free radicals that it produces within a cell. This aspect of oxidative stress is likely reflected in the distinct patterns of tRNA modifications generated upon exposure and suggests that each type of stress exposure will produce a distinct signature of tRNA modifications [25] that, in turn, could be used an indicator of potentially cancer-causing exposures.
If tRNA modifications act in the cell's response to stress, it follows that TRMs and certain mRNA transcripts (e.g., those enriched in codons that reside in split-codon boxes) must also be a part of this regulatory system. Indeed, exposure of budding yeast to H2O2 increases Trm4-dependent m5C modification of wobble position of tRNALEU(CAA) to enhance the translation of RPL22A, a stress-responsive ribosomal protein whose transcript is enriched with UUG codons (modified base is underlined) [26]. Studies in budding yeast have also linked Trm9 to a DNA damage-induced stress response [27]. Trm9 methylates the wobble uridine position of at least two tRNA species: 5-methoxycarbonylmethyluridine (mcm5U) on tRNAArg(UCU), and 5-methoxycarbonylmethyl-2-thiouridine (mcm5s2U) on tRNAGlu(UUC) [28,29]. Begley et al. (2007) found that in budding yeast exposed to the DNA alkylating agent methyl methanesulfonate, Trm9-dependent wobble modifications of tRNAArg(UCU) and tRNAGlu(UUC) enhanced the translation of transcripts enriched in AGA and GAA that are involved in DNA repair process (i.e., RNR1 and RNR3) and translational elongation (i.e., YEF3) [27].
While tRNA modification such as that catalyzed by Trm9 in budding yeast has been well documented to confer resistance to oxidative stress and DNA damage caused by alkylating agents, less is known about whether TRM9 confers resistance to DNA damage associated with cytochrome P450-activated carcinogens, such as aflatoxin B1 (AFB1). Aflatoxin B1 is a class I carcinogen and the most potent experimental liver carcinogen known [30]. Metabolic activation of AFB1 by CYP3A4 and CYP1A2 generates a highly reactive epoxide [31] that produces the DNA adduct 8,9-dihydro-8-(N7-guanyl)-9-hydroxyaflatoxin B1 (AFB1-N7-Gua) [32]. In addition, AFB1 exposure causes oxidative stress, as detected by 8-oxoguanine in DNA [33]. The importance of the genotoxicity of AFB1 is underscored by observations that a hotspot p53 mutation (i.e., a G to T transversion at codon 249 that recodes for serine instead of arginine) is often found in liver cancer patients from regions endemically contaminated with aflatoxin [34,35]. Thus, understanding how RNA modifications influence the DNA damage response to AFB1 will elucidate mechanisms by which AFB1 exerts its genotoxic effects.
Budding yeast has again been a useful tool for understanding the DNA damage response to AFB1. Exposure to AFB1 elicits transcriptional induction of DNA repair genes [36] and stalls replication in S phase [37]. St. John et al. (submitted) observed that trm9 diploid mutants are sensitive to AFB1. Considering that yeast mutants defective in DNA damage-associated ribonucleotide reductase (RNR) upregulation are also sensitive to AFB1 [38] and that tRNA modification contributes to the upregulation of RNR [27], it is possible that trm9-associated AFB1 sensitivity is associated with dNTP levels. Supporting the notion that RNR upregulation is important in genotoxic responses, Fasullo et al. (2007) observed that dun1 mutants defective in RNR transcription upregulation were also defective in postreplication repair. Further research would be necessary to determine whether trm9 mutants are also defective in postreplication repair mechanisms.
Mechanistic connections between tRNA modification systems and oxidative stress have been made in mammals for ALKBH8 (a human homolog of yeast Trm9) using mice to model Alkbh8 deficiency (i.e., Alkbh8-/-). One model defined the function of Alkbh8 as the enzyme required to catalyze the final step in the formation of mcm5U at the wobble position of tRNAs, including those charged with selenocysteine (tRNASec(UCA)), and that this Alkbh8-dependent catalytic event is a prerequisite for the subsequent formation of mcm5s2U and 5-methoxycarbonylmethyl-2′-O-methyluridine (mcm5Um) [39,40]. Uridine modifications at the wobble position of tRNASec(UCA) act together with cis-elements in seleno-protein decoding mRNAs to recode UGA as selenocysteine, which is an amino acid that lacks a dedicated codon [41,42]. The seleno-proteins derived from these UGA recoded transcripts include members of the glutathione peroxidase and thioredoxin reductase families, which play a critical role in ROS biology because selenocysteine is involved in active site redox reactions that detoxify intracellular ROS [41,43].
The capacity of ALKBH8 to act in biological processes linked to ROS detoxification was further examined in a distinct Alkbh8-/- mouse. Endres et al. (2015) investigated seleno-protein expression and ROS detoxification capacity in Alkbh8-/- murine embryonic fibroblasts and found decreased seleno-protein expression (linked to deficient UGA recoding), elevated intracellular ROS, and increased oxidative stress-associated DNA damage [44]. Since mouse and human ALKBH8 proteins are highly similar (94% within the methyltransferase domain, unpublished observations) and many TRMs share conserved functions among eukaryotes [8], it is likely that the observations of Alkbh8 function in mice will translate to humans.
Together, this body of work solidifies the role of tRNA modification systems in the oxidative stress response and suggests a broader phenomenon in which tRNA modifying enzymes are activated by oxidative stress, catalyze the modification of their target tRNA molecules, which go on to enhance the translation of stress-responsive transcripts (Figure 2) [21,45]. This model further predicts that TRMs are themselves components of stress-responsive signaling pathways, a prediction recently supported by the identification of hTRM9L as a relay target of phosphorylation by an oxidative stress-induced mitogen-activated protein kinase (MAPK) cascade that suppresses cell growth postexposure [46].
Figure 2. . A model of tRNA modifications in response to oxidative stress.
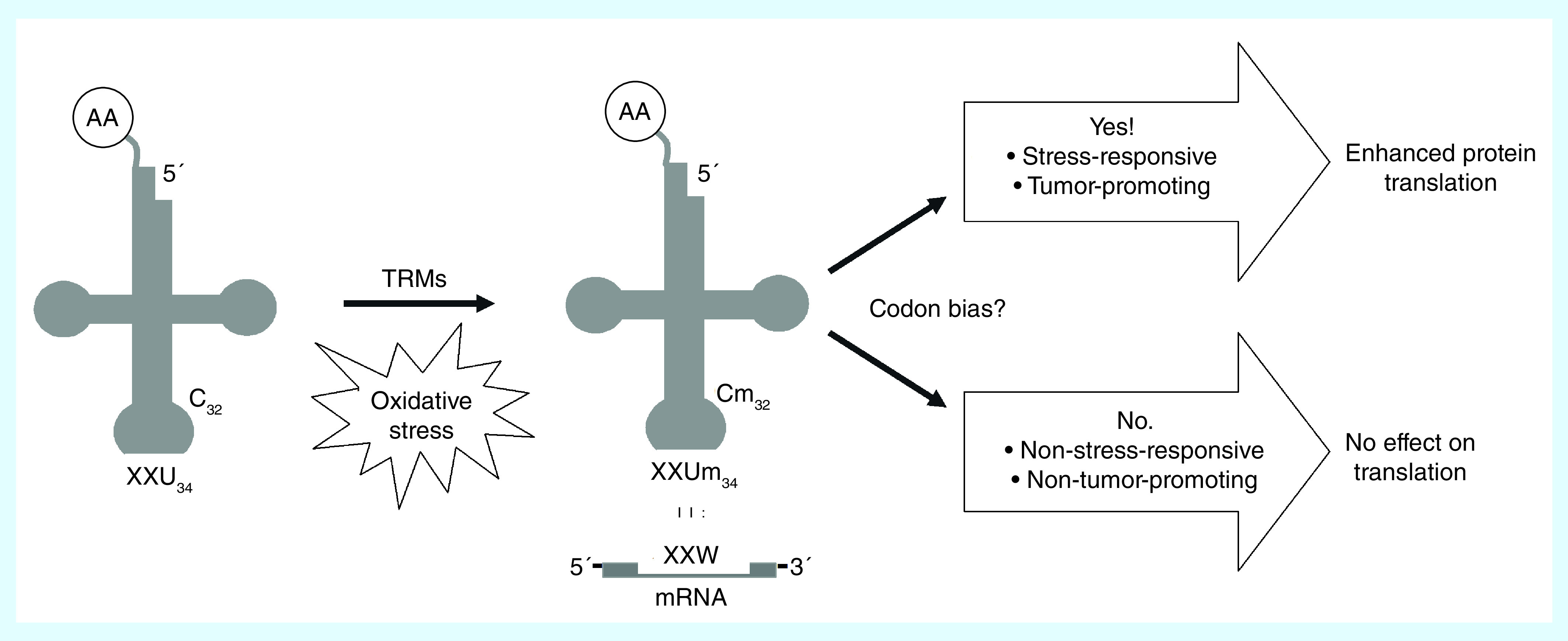
Oxidative stress either activates or augments the activity of TRMs that target specific tRNAs for modification (e.g., Um34) particularly at the wobble position. These modified tRNAs enhance the translation of transcripts with a codon bias that carry out various cellular activities including reactive oxygen species mitigation and promoting cancerous phenotypes.
TRM: tRNA methyltransferase enzyme.
Although ROS can act as critical redox-sensing molecules, signaling to pathways that regulate normal cell growth, hypoxic responses, immune cell recruitment and cell mobility [47–50], excessive ROS from toxicant exposure can cause DNA mutations, and in this capacity, ROS act in the process of cancer cell initiation. Thus, identifying a tRNA modification system like ALKBH8 -> tRNASec(UCA) -> seleno-protein translation has significant implications for cancer, as it suggests that enhancing tRNA-dependent translational responses to oxidative stress could suppress tumor-initiating mutations. Future studies will determine the feasibility of this type of cancer preventative strategy, requiring a full determination of whether the DNA damage in Alkbh8-/- cells (or other TRM deficient models with oxidative stress sensitivity) leads to wider spread genomic instability and exposure-induced cancer predisposition.
Links between tRNA modifications & cancer
In the previous section, we described how tRNA modifications play a role in the oxidative stress response, with implications for maintaining genome integrity and preventing cancer initiation. We now turn our attention to emerging evidence that supports the idea that tRNA modification systems are acting in cellular processes that directly support cancer cell phenotypes, including increased proliferation, metastatic potential and cancer stem cell survival. In fact, cancer cells have long been known to have higher rates of global protein synthesis compared with noncancerous cells [51]. Oncogenic activity across many types of cancer can be connected to various cellular processes that control protein translation, at both initiation and elongation stages, thus coupling increased cell proliferation to translational reprogramming of the cancer genome [52,53]. Here, we highlight tRNA modification systems that either suppress or stimulate cancer cell phenotypes, with a focus on work that supports that tRNA modification systems are critical components of oncogenic signaling pathways.
Nucleoside modifications associated with a cancer phenotype
Pseudouridine is an abundant and ubiquitous nucleoside modification [54] that is formed by the isomerization of uridine as catalyzed by pseudouridine synthases or PUSs [55]. Historically, pseudouridine (likely of tRNA origin [56]) was a frequently observed cancer-associated excreted nucleoside. Elevated levels of pseudouridine were found in the urine of patients with a variety of carcinomas including those of the gastrointestinal and respiratory tracts, breast, colon and bladder, as well as hematological malignancies, and also in the urine of tumor-bearing animals [20]. It is important to note that pseudouridine was not alone in these past reports, and awareness of aberrant nucleoside modifications, and modifying enzyme activity, within cancer dates back to the 1970s [57–60]. Many other modified nucleosides have since been detected as significantly altered in the urine of breast cancer patients and various cancer cell lines or tumor-bearing mice [61–65], and it is reasonable to suggest that nucleoside modifications on tRNA besides pseudouridine may act as cancer phenotypic markers. RNA nucleoside modification profiles can be easily obtained from patient urinary excretions, making them attractive as biomarkers of early disease or malignant progression [62]. These modification profiles could prove useful for cancer detection and intervention if developed to identify tRNA-specific modifications.
In pursuing this hypothesis, several groups have investigated the transition of uridine to pseudouridine (Ψ) on tRNAs as a potential cancer phenotypic marker. High-throughput Ψ-sequencing at the transcriptome level has shown that Ψ is widespread and incorporates at specific tRNA base sites within eukaryotic cells. Interestingly, this phenomenon of pseudouridinylation generates three classes of tRNA-derived noncoding RNA fragments that are upregulated in cancers: tRNA-derived small fragments (tRFs), tRNA-derived small RNAs (tsRNA) and tRNA halves [66]. Studies aimed at characterizing the function of Ψ-generated tsRNAs approximately 16–48 bases in length revealed that they accumulate in the nucleus and are exported into the cytoplasm, suggesting a potential role for these fragments in regulating gene expression [66,67]. Other links between tsRNAs and cancer come from studies with prostate cancer cell lines in which the expression of tsRNA-36 correlates with cell proliferation [68]; also, genes coding for specific tsRNAs-53 and -101 are located in a region of chromosome 17 that is deleted in chronic lymphocytic leukemia and lung cancer patients [69]. Further mechanistic details about how Ψ-generated tRNA fragments (via PUS7 enzyme activity) contribute to a cancer phenotype are described in the following section.
Overall, early observations of cancer-associated nucleoside modifications seeded the idea that deregulated tRNA modification could contribute to a cancer phenotype, which was predated further by ‘the hypothesis of aberrant methylation in neoplasms’ [20,59,70]. Fast forward to today, exciting new mechanistic details describing how defective or altered modification of tRNA (and other noncoding RNAs) can deregulate cell growth and differentiation leading to cancer have emerged. The following section is designed to highlight some of these significant findings.
tRNA modifying enzyme activity in cancer
Cellular transformation toward a cancerous phenotype requires gain-of-function mutations that activate oncogenes and loss-of-function mutations that inactivate tumor suppressor genes [71], and tRNA methyltransferases seem to be capable of taking on either role when driving a cancer phenotype. For instance, hTRM9L is capable of suppressing the growth of colon cancer cells, and its transcript is downregulated in several types of human carcinomas [72]. Moreover, the region harboring hTRM9L on chromosome 8 has been reported lost in cancers of the breast, bladder, prostate and colon [73–76] – a classic characteristic of a tumor suppressor.
Using colon cancer xenograft models, Begley et al. (2013) propose a two-pronged mechanism for hTRM9L-dependent tumor suppression that involves LIN9-induced cell-cycle arrest combined with attenuated HIF1-α-mediated gene expression that would otherwise support tumor growth under conditions of hypoxia. It is notable that HIF1-α mRNA is significantly enriched in AAA, GAA and CAA codons, which require U34 modifying enzymes, including the hTRM9L human homolog, ALKBH8, for efficient translation [77]. Taken together, these results suggest that hTRM9L may also effect the translation of HIF1-α mRNA and perhaps other transcripts that are sensitive to U34 modification. Another notable finding of this study was that hTRM9L deficient colon cancer cells were sensitive to the killing effects of paromomycin [72]. This result raises the possibility that aminoglycoside antibiotics could be used off-label as anticancer therapeutics aimed at further perturbing translation in cancers already harboring defective tRNA modification systems.
The hTRM9L enzyme has been further investigated in tumors and cell lines derived from lung and ovarian cancers. In lung cancer, evidence supports that hTRM9L attenuates the cell cycle by downregulating cyclin D1, and limits migratory and invasive potential by altering the expression of cadherin proteins (i.e., inhibiting N-cadherin expression while promoting E-cadherin expression) [78]. Immunohistochemical analysis of hTRM9L expression in malignant ovarian tumors shows that it is decreased relative to benign tumors, and overexpression of hTRM9L in ovarian cancer cells grown in culture inhibited growth and increased apoptosis via pRB and p53 proapoptotic signals, respectively [79]. Even though the cellular processes influenced by hTRM9L in colon, lung and ovarian cancers appear to be distinct, it is notable that they are all linked to cellular processes that inhibit cancer cell phenotypes (e.g., attenuating survival under hypoxic conditions; promoting cell cycle arrest or apoptosis; or limiting metastatic potential). Together, these reports support a tumor suppressive cellular role for hTRM9L.
Circling back to pseudouridine, we find that a new mechanism of translational control involving tRNA modification has been discovered in hematopoietic stem and progenitor cells, which sheds new light on the etiology of human myelodysplastic syndromes, particularly those at risk for transformation to acute myeloid leukemia [66]. Guzzi et al. (2018) found that a subclass of tRNA isoacceptors containing a 5′ terminal oligoguanine (TOG; e.g., tRNAAla, tRNACys and tRNAVal) undergo pseudouridylation by PUS7 at position eight, which leads to their formation into small mini tRNA fragments (mTOGs). These mTOGs are capable of repressing the initiation of protein synthesis; a mechanism believed to fine-tune protein translation during cell fate specification, producing just the right balance of proteins for stem cell self-renewal or differentiation. This balance may be upset in human myelodysplastic syndromes that have reduced PSU7 expression, and the resulting ‘over’ stimulation of protein synthesis may drive cellular transformation and progression to acute myeloid leukemia, which occurs in approximately one out of three cases [66,80].
Interestingly, tRNA modification by the methyltransferase NSUN2 (human homolog of yeast Trm4) has also been linked to the production of small 5′ tRNA fragments (tRFs), capable of regulating translation initiation [81,82]. The NSUN2 enzyme catalyzes the formation of 5-methylcytosine (m5C) at position 34 of tRNALec(CAA) [83], which, as previously mentioned for modifications at this wobble position, may stabilize anticodon-codon base pairing during translation. However, NSUN2-mediated m5C formation also prevents tRNA cleavage by angiogenin (a ribonuclease enzyme) into 5′ tRFs, which are also referred to as tRNA halves (tiRNAs) when the cleavage event occurs within the anticodon loop [81,84,85]. These tRFs are capable of repressing cap-dependent protein translation suggesting that NSUN2 regulates global rates of protein translation and points to additional roles for this tRNA methyltransferase in controlling translation beyond those that influence codon selection [86,87].
The implications of m5C protection against angiogenin cleavage for the stress response and cancer are further discussed below as new evidence requires that we bring DNMT2 into this discussion. However, at this point it is worth mentioning that loss-of-function NSUN2 mutations are implicated in human disease states that may be linked to reduced translation and stress that produces either hypo-proliferative or apoptotic effects in certain neuronal cell populations, resulting in growth retardation and impaired neurodevelopment [81,88–90]. Although premature, it is tempting to speculate that mutations in some tRNA modifying enzymes that are capable of influencing the production of tRFs leads to overprocessing of some tRNA fragments, attenuating proliferation or inducing cell death, whereas the oncogenic activity of others leads to an overproduction of tRFs that stimulate cell proliferation.
In fact, overexpression of NSUN2 has been documented in head and neck squamous carcinoma and breast cancer [91,92], and depletion of NSUN2 in breast cancer cell models diminishes certain characteristics associated with a malignant cancer phenotype, including proliferative capacity, and migratory and invasive potential, suggesting that it plays a protumorigenic, or oncogenic, role in cancer [93,94]. That stated, the role NSUN2 plays in cancer is likely complex and specific to the type of cancer cell that it acts within. Indeed, Blanco et al. (2016) observed an inverse correlation between NSUN2 expression and malignancy for human squamous cell carcinoma, and using a mouse model of Ras-induced squamous cell carcinoma define an intriguing new role for NSUN2 in maintaining a population of tumor-initiating stem cells through translational repression of stress-response transcripts [82].
Like NSUN2, DNMT2 (also known as TRDMT1) is an m5C tRNA methyltransferase that can target the anticodon stem loop (position 38) of specific tRNA species in mammals and Drosophila, including tRNAAsp(GUC), tRNAGly(GCC) and tRNAVal(AAC) [95–97]. Once designated as a DNA methyltransferase based on its close sequence homology to other enzymes in this family, DNMT2 is now considered to be tRNA methylation enzyme [95,97,98]. In fact, the tRNA methyltransferase activities of both NSUN2 and DNMT2 have been shown to protect tRNAs against oxidative stress-induced cleavage by angiogenin, an effect that could be cooperative in some cellular contexts [81,84,97]. Again, the fact that NSUN2 and DNMT2 catalyze tRNA modifications in the anticodon stem loop suggest a potential further role for these enzymes in translational regulation, perhaps at the level of codon selection, an idea that is supported in part by experiments in hematopoietic cells derived from Dnmt2-deficient mice, which show high levels of codon mistranslation due to an inability to recognize near-cognate codons (i.e., as decoded by tRNAAsp(GUC) and tRNAGlu(UUC)) [99]. In future, it will be interesting to determine the specific contributions of NSUN2 and DNMT2 to tumorigenesis regarding cell survival and proliferation or maintenance of a dedifferentiated state, and whether they make individual or coordinated contributions to a cancerous phenotype.
Additional links between wobble base tRNA modifications and cancer can be made through the Elongator complex subunit, ELP3. The ELP3-associated complex is part of an enzymatic sequence that includes the CTU1/2 complex and ALKBH8, which together catalyze the formation of mcm5U and mcm5s2U modifications at position 34 of tRNAs that decode AAA (LYS), GAA (GLU), CAA (GLN) and UGA as selenocysteine [39,100,101]. In recent years, exciting inroads have been made into understanding how ELP3-CTU1/2 acts as a critical component of different oncogenic signaling pathways that drive cancers of the intestine, breast and skin. In the Apc+/min mouse model of intestinal tumor development, Elp3 is induced by constitutive Wnt signaling where it is required for maintaining a population of tumor-initiating cells, with evidence to support that Elp3 does this via tRNA modifications that promote the translation of Sox9 [102].
In breast cancer, translational effects linked to ELP3-CTU1/2 have been implicated in WNT-dependent prometastatic processes, albeit through a different transcriptional effector, LEF1 [103,104]. In this model, ELP3-CTU1/2 enables U34 mcm5s2-tRNA modifications that promote the translation of proteins that act as transacting factors for the translation of transcripts with an internal ribosome entry site (IRES), so-called ITAFs (IRES-specific trans-acting factors). An analysis of codon content in ITAFs identified DEK as enriched in U34 mcm5s2 ‘sensitive’ codons (i.e., enriched in AAA/GAA/CAA codons), and through this bias ELP3-CTU1/2 enhances its translational elongation; DEK then goes on to promote the IRES-dependent translation of LEF1, which supports processes involved in the malignant progression of breast cancer [103].
A further distinct mechanism of translational reprogramming by ELP3-CTU1/2 mediated U34 tRNA modification was identified in zebrafish and mouse models of melanoma, as well as in the human form of the disease [77]. Rapino et al. (2018) found that ELP3-CTU1/2 acts downstream of constitutive MAPK signaling, as driven by oncogenic BRAFV600E. This U34 tRNA modification system is responsible for maintaining high levels of HIF1-α protein in melanoma cells, which in turn drives a metabolic program that promotes cell survival and resistance to therapies that target aberrant MAPK signaling, such as vemurafenib and trametinib. Indeed, HIF1A mRNA is biased in codons requiring U34 tRNA modification for efficient translation, and translation-based assays confirm that HIF1A mRNA requires ELP3-CTU1/2 for its efficient translation into HIF1-α protein [77].
There are likely other codon-biased mRNA transcripts influenced by ELP3-CTU1/2 mediated tRNA modifications that decode proteins with prosurvival effects in melanoma, and perhaps other cancer cell types [77]. Nevertheless, therapeutic targeting of the ELP3-CTU1/2 tRNA modification system would be expected attenuate the translation of any transcript with a codon bias that requires U34 modification for optimal translational decoding, with potential antisurvival/proapoptotic consequences for tumor cells. Considering the treatment challenge that arises when patients acquire resistance to targeted MAPK inhibition in melanoma and other cancer types [105], the cumulative work described above has significant implications for combinatorial approaches to cancer treatment.
Additional studies of TRMs in diverse cancer cell line models
Although the above studies have shed exciting new light on many mechanisms by which tRNA methyltransferase activity can support a cancerous phenotype, they likely represent the tip-of-the-iceberg concerning our understanding of the role of deregulated tRNA modification systems in cancer. Indeed, studies involving human cancer cell models and tumor genome analysis have both substantiated a role for mutant TRM activity in cancer and continue to make new links between aberrant TRM activity and cancerous phenotypes, particularly for those previously discussed m5C tRNA methyltransferases, NSUN2 and DNMT2.
For example, Okamoto et al. (2012) document increases in NSUN2 gene copy number in multiple oral and colon cancer cell lines (i.e., HSC-2, HSC-4, Ca-9-22 and HT-29, LoVo, SW480, SW48, SW620, HCT116, DLD-1, respectively); notably, in these cell lines, gene amplification events correspond to increases in NSUN2 protein expression relative to noncancerous cell line models. Immunohistological analysis of human tumor tissue samples also shows elevated NSUN2 across many cancer types suggesting that the overproduction of NSUN2 may be a widespread tumorigenic event [106]. Future work will determine whether this tumor-associated NSUN2 expression is a result of gene amplification or other mechanisms that lead to the overexpression of this protein.
Across another independent panel of human cancer cell lines, DNMT2 shows variable levels of expression, with high levels in myeloid leukemia cells (i.e., HL-60, K562 and ML-1) and breast cancer cells (i.e., MCF-7), relative to lung (i.e., A549) or liver (i.e., HepG2) cancer cells [107]. Moreover, cancers cells that are ‘high expressers’ of DNMT2 can be targeted by 5-Azacytidine (5-AZA) as an RNA (rather than DNA) analog inhibitor of methylation [107,108]. COSMIC database analysis of a spectrum of human tumors identified somatic mutations that alter DNMT2 enzyme activity in vitro using tRNAAsp as a substrate [109,110]. Interestingly, the selected DNMT2 mutations (representing recurring tumor-associated amino acid substitutions) could either stimulate (i.e., E63K) or attenuate (e.g., G155S/C/V) DNMT2's methyltransferase activity, while others produced no difference in activity relative to wild-type DNMT2 (e.g., D226H/Y) [110]. It will be interesting for future studies to assign a respective oncogenic or tumor suppressive function to DNMT2 stemming from this initial assessment of stimulatory or inhibitory tumor-associated mutations.
Other genomic studies have identified gene amplification events for TRMT12 and ALKBH8 in breast and bladder cancer cell lines, respectively [111–113]. The ALKBH8 gene amplification in bladder cancer is an intriguing observation because of the established link between ALKBH8 and ROS detoxification (via tRNASec(UGA)) [44], and the fact that bladder cancer risk is strongly associated with exposure to toxicants that are potent inducers of oxidative stress [114]. At first, ALKBH8-dependent ROS detoxification seems inconsistent with the concept that gene amplification is a common oncogenic event that drives cancer formation. However, the role of ROS in cancer is complex, and emerging evidence in bladder cancer cell lines supports that in the later stages of this disease ROS may activate pathways that further stimulate cell growth and metastasis [115,116]. Thus, in the context of bladder cancer, ALKBH8 may play two possible roles. Initially, ALKBH8-dependent ROS detoxification could protect normal urothelial cells from oxidative stress-induced DNA damage, but in established urothelial carcinomas, ALKBH8 maintains ROS at sublethal levels that cancer cells exploit to further their growth and malignant progression. This phenomenon is thought to occur in cancers that upregulate antioxidant defense enzymes like superoxide dismutases [117,118].
Another piece of the puzzle: tRNA gene expression in cancer
Our discussion of tRNA modification systems thus far has not touched upon the availability, or abundance, of substrate tRNAs. As named, TRMs are methylating enzymes that target tRNAs, and without the availability of tRNA as a substrate, the tRNA modification system cannot act to either promote tumorigenesis or in response to stress. The level of tRNAs expressed (i.e., in response to different cellular conditions within specific cell types) can influence translation based on their ability to occupy the ribosome, which in turn affects the number of transcripts with complementary codons bound to the ribosome [119,120]. Factors affecting the abundance of substrate tRNAs include transcriptional regulation by RNA Pol III, as well as the relative number of tRNA isoacceptor genes within the genome, the latter of which in humans are known to be expressed in a tissue-specific manner [121,122]. Also, tRNA transcript levels are known to be downregulated in response to multiple forms of cellular stress (e.g., nutrient deprivation and DNA damage) through Maf1 (itself a target of the mTOR pathway), a negative regulator of the RNA Pol III transcriptional complex [14]. The idea that tRNA isoacceptor abundance may be a driving force for cancer, either alone, or in conjunction with aberrant tRNA modification systems is supported by recent studies that have performed global assessments of tRNA species across a variety of normal versus cancerous cells and tissues.
For example, genome-wide tRNA expression profiling has been used to determine the tRNA contents of various human cell lines and samples, which display distinct tRNA signatures correlating with their state of proliferation and differentiation [123,124], cellular programs that are deregulated in cancer. Goodzari et al. (2016) took a probe-based tRNA profiling approach to measure the content of each family of mature tRNAs in human breast cancer cell lines that model disease progression (i.e., nontumorigenic epithelial cells and those that are poorly or highly metastatic). They found that the upregulation of tRNAArg(CCG) and tRNAGlu(UUC) promoted breast cancer metastasis to the lungs of orthotopically injected mice and that the enhanced translation of GAA-containing transcripts, EXOSC2 and GRIPAP1, likely contributed to this gain in metastatic potential [119]. Since tRNAArg(CCG) is targeted for m5C methylation by NSUN2, the suggestion that upregulated tRNAArg(CCG) (at the transcript level) can promote breast cancer metastasis fits with other cancer models in which overexpression of NSUN2 increases metastatic potential [91–94].
The fact that tRNAGlu(UUC) decodes within a split-codon box and also undergoes U34 modification raises the question: does tRNAGlu(UUC) modification also affect the translational decoding of other GAA-enriched transcripts either driving breast cancer metastasis or acting at an earlier stage of disease? Although beyond the scope of Goodzari et al. (2016), it is interesting that Delaunay et al. (2016) similarly identified a tRNAGlu(UUC) influenced transcript (i.e., DEK) in their WNT-driven, prometastatic breast cancer model [103]. We may further ask, what is the relative abundance of these tRNA isoacceptors (i.e., tRNAArg(CCG) and tRNAGlu(UUC)) or others influenced by U34 modifications (i.e., tRNALys(UUU), tRNAGln(UUG), tRNASec(UCA)) in cancers with aberrant TRM activity [77,102,103,112]? If equal to or less abundant than other isoacceptors, it is possible that they undergo transcriptional upregulation (or other transcript stabilization) to participate in aberrant tRNA modification systems that drive cancer.
tRNA isoacceptor abundance may also play a critical role in the context of cancer-associated tRNA-derived fragments (tRFs). A recent study comparing tRFs in low- and high-grade prostate tumors versus normal adjacent prostate tissue found tRFs derived from tRNAPhe(GAA) and tRNALys(CUU) may be prognostic biomarkers for progression-free disease survival [125]. These findings are consistent with another study suggesting that tRFs may take on a tumor suppressive function in breast cancer [126]. Although the role of tRNA modification in generating cancer-associated tRFs has not been fully determined, the abundance of tRNA isoacceptors will surely be a contributing factor to their production. Notably, here we have described a tRNALys(CUU) isoacceptor associated with a potential tumor-suppressive function in prostate cancer, while its modified tRNALys(UUU) issoacceptor has been implicated in oncogenic pathways. Overall, any model of deregulated tRNA modification systems in cancer should consider the availability, or abundance, of specific tRNA species as a critical component.
Summary
This review aimed to highlight the dynamic nature of tRNA modifications and describe the emerging role that tRNA modification systems play in protecting cells against the potentially harmful effects of oxidative stress exposure. One of these effects is DNA mutation, which is a known initiator of cancer development and suggests that augmenting the activity of some TRM-dependent programs (e.g., as mediated by ALKBH8) could have antioxidant or other cytoprotective effects for cancers associated with exposure to ROS producing toxicants, including aflatoxin [44,127].
Further links to cancer come from a number of mechanistic studies demonstrating that aberrant TRM activity in cancer cells directs programs of codon-biased translation that directly contribute to cancer cell phenotypes. In this capacity, TRMs mediate cellular functions that can be either tumor suppressive (i.e., hTRM9L and PUS7) [66,72] or oncogenic (i.e., NSUN2, ELP3-CTU1/2, Figure 3) [77,93,94,102,103]. It is also possible that, in addition to activating mutations, oncogenic transcripts adopt a pattern of differential codon usage to enhance their translation [128]. It is clear that cancer phenotypes associated with malignant disease progression are supported by transcripts with a codon usage pattern that is distinct from noncancerous cells [77]. This makes cancer-associated tRNA modification systems an attractive future target for therapies that cripple cancer cells without harming normal tissue.
Figure 3. . tRNA methyltransferase activity in cancer cells.

tRNA methyltransferase enzymes catalyze specific chemical modifications on tRNAs to direct translational programs that are either tumor suppressive (unfilled) or oncogeneic (filled) in cancer cells.
CBT: Codon-biased translation; tRF: Small tRNA-derived fragment.
Future perspective
Over the next decade, research in the field of tRNA modifications and cancer will determine the feasibility of targeting tRNA modification systems in cancer as a therapeutic. Considering the role of tRNA modifications in response to stress and cancer, there may be two strategies: cancer prevention and cancer intervention during disease progression, respectively. In either approach, critical gaps in our understanding of the fundamental biology of tRNA modifications must be filled. These gaps include but are not limited to, defining the chemical spectrum of tRNA modifications in response to stress and in normal versus cancerous cells; also, mapping modification chemistries to specific sites within specific tRNA isoacceptors. Advances in mass spectrometry combined with RNA sequencing techniques are already advancing the field toward this goal. At the same time, the cell signaling pathways that regulate tRNA modifications triggered by exposure or oncogenesis need to be elucidated. To get ahead of potential ‘off-target effects’ it will also be essential to identify cross-interactions between tRNA modification pathways and those that regulate other forms of noncoding RNA editing. Ultimately, targeting tRNA modification systems is an attractive therapeutic strategy for cancer because of the nature of codon-biased translation; that is, suppressing a single TRM-dependent modification would potentially attenuate the translation of a host of codon-biased transcripts driving multiple cancer cell phenotypes.
Executive summary.
Stress-induced tRNA methylation & methyltransferase activity
Transfer RNA modification chemistries are dynamic in nature, changing in response to oxidative stress-inducing toxicants.
Transcripts that encode proteins that act in the stress response (e.g., RPL22A and RNR1/3) have a codon bias that enhances their translation by tRNAs modified after exposure.
Yeast and mammalian cells deficient in tRNA methyltransferases (TRM) are sensitive to the cytotoxic effects of oxidative stress.
-
Mammalian homologs of yeast TRM9 have been linked to the oxidative stress-response in two ways:
ALKBH8 mitigates reactive oxygen species by modifying tRNASec(UCA) to recode the UGA stop codon as selenocysteine.
hTRM9L is a target of a stress-induced mitogen-activated protein kinase cascade to suppress cell growth post-exposure.
Links between tRNA modifications & cancer
Transfer RNA modification systems can support or suppress cancer cell phenotypes suggesting that they are capable of oncogenic and tumor suppressor activities.
Nucleoside modifications associated with a cancer phenotype
Thought to be of tRNA origin, pseudouridine is an abundant and ubiquitous nucleoside modification excreted in the urine of cancer patients.
Widespread cancer patient studies have now detected elevated levels of other modified nucleosides in urine.
If developed to identify tRNA-specific modifications, urinary excretions could be a useful biomarker of early-stage cancer.
tRNA modifying enzyme activity in cancer
Human TRM9L is capable of suppressing the growth of colon cancer; chromosomal regions harboring hTRM9L are lost in cancers of the breast, prostate and colon.
Loss of hTRM9L may suppress cancer cell phenotypes due to diminished translation of transcripts enriched in AAA, GAA and CAA codons, including LIN9 and HIF1-α.
Transfer RNA modifications regulate the production of small tRNA fragments capable of repressing translation to fine-tune protein synthesis for cell differentiation (i.e., PSU7) and during the oxidative stress response (i.e., NSUN2 and DNMT2).
Reduced PSU7 expression in myelodysplastic syndrome stimulates protein synthesis and is associated with progression to acute myeloid leukemia.
Oncogenic codon-biased translation has been implicated in cancers of the intestine, breast and skin through the ELP3-CTU1/2-ALKBH8 enzyme sequence that catalyzes U34 tRNA modifications.
ELP3-initiated U34 modifications enhance the translation of different transcripts depending upon the cancer type (i.e., Sox9 in intestinal tumors, DEK in breast cancer and HIFA in melanoma).
Additional studies of TRMs in diverse cancer cell line models
Multiple cancer cell line models and patient tumor studies have substantiated a role for deregulated TRM activity in cancer.
NSUN2 gene copy number is increased in oral and colon cancers and corresponds to increased protein expression.
DNMT2 is differentially expressed among cancers showing relatively high expression in cells derived from myeloid leukemia and breast cancer compared with those derived from lung or liver cancers.
Gene amplification events are known to occur for TRMT12 and ALKBH8 in breast and bladder cancer cells, respectively.
ALKBH8 overexpression in bladder cancer suggests an adaptive response to reactive oxygen species that may stimulate cell growth and metastasis.
Another piece of the puzzle: tRNA gene expression in cancer
Genome-wide tRNA expression profiling has identified distinct tRNA signatures that correlate with cell proliferation and differentiation.
The upregulation of some tRNA isoacceptors (i.e., tRNAArg(CCG) and tRNAGlu(UUC)) promote breast cancer metastasis; others (i.e., tRNAPhe(GAA) and tRNALys(CUU)) form tRFs associated with progression-free disease survival of prostate cancer.
Transfer RNA isoacceptor abundance likely plays a critical role in cancer development and thus, it should be considered in models of deregulated tRNA modification systems that drive cancer phenotypes.
Acknowledgments
This work serves to highlight some of the most recent findings in the fields of stress- and cancer-associated tRNA modifications. Although we have tried to be comprehensive in our review, we have undoubtedly missed the contributions of some scientists given the burgeoning nature of the field. For any missed work, please accept our sincere apologies. This paper was completed with support from the College of Arts and Sciences, SUNY Polytechnic Institute, and a SUNY Polytechnic Institutional Seed Grant. A Horner assisted with the composition and illustration of Figures 1 and 3.
Footnotes
Financial & competing interests disclosure
This work received support from a SUNY Polytechnic Institutional Seed Grant (LE), and the College of Arts and Sciences, SUNY Polytechnic Institute. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.Chan PP, Lowe TM. GtRNAdb 2.0: an expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res. 2016;44(D1):D184–D189. doi: 10.1093/nar/gkv1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marck C, Grosjean H. tRNomics: analysis of tRNA genes from 50 genomes of Eukarya, Archaea, and Bacteria reveals anticodon-sparing strategies and domain-specific features. RNA. 2002;8(10):1189–1232. doi: 10.1017/s1355838202022021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dever TE, Green R. The elongation, termination, and recycling phases of translation in eukaryotes. Cold Spring Harb. Perspect. Biol. 2012;4(7):a013706. doi: 10.1101/cshperspect.a013706. 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crick FH. Codon--anticodon pairing: the wobble hypothesis. J. Mol. Biol. 1966;19(2):548–555. doi: 10.1016/s0022-2836(66)80022-0. [DOI] [PubMed] [Google Scholar]
- 5.Agris PF, Narendran A, Sarachan K, Vare VYP, Eruysal E. The importance of being modified: the role of RNA modifications in translational fidelity. Enzymes. 2017;41:1–50. doi: 10.1016/bs.enz.2017.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Agris PF, Vendeix FA, Graham WD. tRNA's wobble decoding of the genome: 40 years of modification. J. Mol. Biol. 2007;366(1):1–13. doi: 10.1016/j.jmb.2006.11.046. [DOI] [PubMed] [Google Scholar]
- 7.Svensson I, Bjork G, Bjork W, Johansson KE, Johansson A. Evidence for enzymatic methylation in vitro of the ribose moiety of RNA. Biochem. Biophys. Res. Commun. 1968;31(2):216–221. doi: 10.1016/0006-291x(68)90733-x. [DOI] [PubMed] [Google Scholar]
- 8.Towns WL, Begley TJ. Transfer RNA methytransferases and their corresponding modifications in budding yeast and humans: activities, predications, and potential roles in human health. DNA Cell Biol. 2012;31(4):434–454. doi: 10.1089/dna.2011.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Agris PF. Bringing order to translation: the contributions of transfer RNA anticodon-domain modifications. EMBO Rep. 2008;9(7):629–635. doi: 10.1038/embor.2008.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Capra JD, Peterkofsky A. Effect on in vitro methylation on the chromatographic and coding properties of methyl-deficient leucine transfer RNA. J. Mol. Biol. 1968;33(3):591–607. doi: 10.1016/0022-2836(68)90308-2. [DOI] [PubMed] [Google Scholar]
- 11.Karlsborn T, Tukenmez H, Mahmud AK, Xu F, Xu H, Bystrom AS. Elongator, a conserved complex required for wobble uridine modifications in eukaryotes. RNA Biol. 2015;11(12):1519–1528. doi: 10.4161/15476286.2014.992276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shugart L, Chastain BH, Novelli GD, Stulberg MP. Restoration of aminoacylation activity of undermethylated transfer RNA by in vitro methylation. Biochem. Biophys. Res. Commun. 1968;31(3):404–409. doi: 10.1016/0006-291x(68)90490-7. [DOI] [PubMed] [Google Scholar]
- 13.Gefter ML, Russell RL. Role modifications in tyrosine transfer RNA: a modified base affecting ribosome binding. J. Mol. Biol. 1969;39(1):145–157. doi: 10.1016/0022-2836(69)90339-8. [DOI] [PubMed] [Google Scholar]
- 14.Phizicky EM, Hopper AK. tRNA biology charges to the front. Genes Dev. 2010;24(17):1832–1860. doi: 10.1101/gad.1956510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Motorin Y, Helm M. tRNA stabilization by modified nucleotides. Biochemistry. 2010;49(24):4934–4944. doi: 10.1021/bi100408z. [DOI] [PubMed] [Google Scholar]
- 16.Chernyakov I, Whipple JM, Kotelawala L, Grayhack EJ, Phizicky EM. Degradation of several hypomodified mature tRNA species in Saccharomyces cerevisiae is mediated by Met22 and the 5’-3’ exonucleases Rat1 and Xrn1. Genes Dev. 2008;22(10):1369–1380. doi: 10.1101/gad.1654308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alexandrov A, Chernyakov I, Gu W, et al. Rapid tRNA decay can result from lack of nonessential modifications. Mol. Cell. 2006;21(1):87–96. doi: 10.1016/j.molcel.2005.10.036. [DOI] [PubMed] [Google Scholar]
- 18.Gomez-Roman N, Grandori C, Eisenman RN, White RJ. Direct activation of RNA polymerase III transcription by c-Myc. Nature. 2003;421(6920):290–294. doi: 10.1038/nature01327. [DOI] [PubMed] [Google Scholar]
- 19.Farabaugh PJ. Programmed translational frameshifting. Microbiol. Rev. 1996;60(1):103–134. doi: 10.1128/mr.60.1.103-134.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bjork G, Rasmuson T. Links between tRNA modification and metabolism and modified nucleosides as tumor markers. In: Grosjean H, Benne R, editors. Modification and Editing of RNA. American Society for Microbiology; Washington, DC: 1998. pp. 471–491. [Google Scholar]
- 21.Endres L, Dedon PC, Begley TJ. Codon-biased translation can be regulated by wobble-base tRNA modification systems during cellular stress responses. RNA Biol. 2015;12(6):603–614. doi: 10.1080/15476286.2015.1031947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Doyle F, Leonardi A, Endres L, Tenenbaum SA, Dedon PC, Begley TJ. Gene- and genome-based analysis of significant codon patterns in yeast, rat and mice genomes with the CUT Codon UTilization tool. Methods. 2016;107:98–109. doi: 10.1016/j.ymeth.2016.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cantara WA, Crain PF, Rozenski J, et al. The RNA Modification Database, RNAMDB: 2011 update. Nucleic Acids Res. 2011;39:D195–D201. doi: 10.1093/nar/gkq1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sprinzl M, Horn C, Brown M, Ioudovitch A, Steinberg S. Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res. 1998;26(1):148–153. doi: 10.1093/nar/26.1.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chan CT, Dyavaiah M, Demott MS, Taghizadeh K, Dedon PC, Begley TJ. A quantitative systems approach reveals dynamic control of tRNA modifications during cellular stress. PLoS Genet. 2010;6(12):1–9. doi: 10.1371/journal.pgen.1001247. e1001247. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Identifies distinct signatures of tRNA modification chemistries for oxidative stress-inducing toxicants.
- 26.Chan CT, Pang YL, Deng W, et al. Reprogramming of tRNA modifications controls the oxidative stress response by codon-biased translation of proteins. Nat. Commun. 2013;3:937. doi: 10.1038/ncomms1938. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Describes an oxidative stress-induced tRNA modification system mediated by yeast Trm4 that enhances the translation of a stress-associated ribosomal protein.
- 27.Begley U, Dyavaiah M, Patil A, et al. Trm9-catalyzed tRNA modifications link translation to the DNA damage response. Mol. Cell. 2007;28(5):860–870. doi: 10.1016/j.molcel.2007.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Describes a tRNA modification system mediated by yeast Trm9 that responds to DNA damage and enhances the translation of proteins involved in DNA repair.
- 28.Kalhor HR, Clarke S. Novel methyltransferase for modified uridine residues at the wobble position of tRNA. Mol. Cell. Biol. 2003;23(24):9283–9292. doi: 10.1128/MCB.23.24.9283-9292.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deng W, Babu IR, Su D, Yin S, Begley TJ, Dedon PC. Trm9-catalyzed tRNA Modifications regulate global protein expression by codon-biased translation. PLoS Genet. 2015;11(12):1–23. doi: 10.1371/journal.pgen.1005706. e1005706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kew MC. Synergistic interaction between aflatoxin B1 and hepatitis B virus in hepatocarcinogenesis. Liver Int. 2003;23(6):405–409. doi: 10.1111/j.1478-3231.2003.00869.x. [DOI] [PubMed] [Google Scholar]
- 31.Baertschi SW, Raney KD, Shimada T, Harris TM, Guengerich FP. Comparison of rates of enzymatic oxidation of aflatoxin B1, aflatoxin G1, and sterigmatocystin and activities of the epoxides in forming guanyl-N7 adducts and inducing different genetic responses. Chem. Res. Toxicol. 1989;2(2):114–112. doi: 10.1021/tx00008a008. [DOI] [PubMed] [Google Scholar]
- 32.Eaton DL, Gallagher EP. Mechanisms of aflatoxin carcinogenesis. Annu. Rev. Pharmacol. Toxicol. 1994;34:135–172. doi: 10.1146/annurev.pa.34.040194.001031. [DOI] [PubMed] [Google Scholar]
- 33.Guindon-Kezis KA, Mulder JE, Massey TE. In vivo treatment with aflatoxin B1 increases DNA oxidation, base excision repair activity and 8-oxoguanine DNA glycosylase 1 levels in mouse lung. Toxicology. 2014;321:21–26. doi: 10.1016/j.tox.2014.03.004. [DOI] [PubMed] [Google Scholar]
- 34.Bressac B, Kew M, Wands J, Ozturk M. Selective G to T mutations of p53 gene in hepatocellular carcinoma from southern Africa. Nature. 1991;350(6317):429–431. doi: 10.1038/350429a0. [DOI] [PubMed] [Google Scholar]
- 35.Hsu IC, Metcalf RA, Sun T, Welsh JA, Wang NJ, Harris CC. Mutational hotspot in the p53 gene in human hepatocellular carcinomas. Nature. 1991;350(6317):427–428. doi: 10.1038/350427a0. [DOI] [PubMed] [Google Scholar]
- 36.Keller-Seitz MU, Certa U, Sengstag C, Wurgler FE, Sun M, Fasullo M. Transcriptional response of yeast to aflatoxin B1: recombinational repair involving RAD51 and RAD1. Mol. Biol. Cell. 2004;15(9):4321–4336. doi: 10.1091/mbc.E04-05-0375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fasullo M, Chen Y, Bortcosh W, Sun M, Egner PA. Aflatoxin B(1)-associated DNA adducts stall S phase and stimulate Rad51 foci in Saccharomyces cerevisiae . J. Nucleic Acids. 2010;2010:1–9. doi: 10.4061/2010/456487. 456487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fasullo M, Sun M, Egner P. Stimulation of sister chromatid exchanges and mutation by aflatoxin B1–DNA adducts in Saccharomyces cerevisiae requires MEC1 (ATR), RAD53, and DUN1. Mol. Carcinog. 2008;47(8):608–615. doi: 10.1002/mc.20417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Songe-Moller L, Van Den Born E, Leihne V, et al. Mammalian ALKBH8 possesses tRNA methyltransferase activity required for the biogenesis of multiple wobble uridine modifications implicated in translational decoding. Mol. Cell. Biol. 2010;30(7):1814–1827. doi: 10.1128/MCB.01602-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fu D, Brophy JA, Chan CT, et al. Human AlkB homolog ABH8 Is a tRNA methyltransferase required for wobble uridine modification and DNA damage survival. Mol. Cell. Biol. 2010;30(10):2449–2459. doi: 10.1128/MCB.01604-09. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Demonstrates a role for human ALKBH8 (yeast Trm9 homolog) and its associated mcm5U wobble base modification in the DNA damage response.
- 41.Driscoll DM, Copeland PR. Mechanism and regulation of selenoprotein synthesis. Annu. Rev. Nutr. 2003;23:17–40. doi: 10.1146/annurev.nutr.23.011702.073318. [DOI] [PubMed] [Google Scholar]
- 42.Carlson BA, Xu XM, Gladyshev VN, Hatfield DL. Selective rescue of selenoprotein expression in mice lacking a highly specialized methyl group in selenocysteine tRNA. J. Biol. Chem. 2005;280(7):5542–5548. doi: 10.1074/jbc.M411725200. [DOI] [PubMed] [Google Scholar]
- 43.Veal EA, Day AM, Morgan BA. Hydrogen peroxide sensing and signaling. Mol. Cell. 2007;26(1):1–14. doi: 10.1016/j.molcel.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 44.Endres L, Begley U, Clark R, et al. Alkbh8 regulates selenocysteine-protein expression to protect against reactive oxygen species damage. PLoS ONE. 2015;10(7):1–23. doi: 10.1371/journal.pone.0131335. e0131335. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Connects ALKBH8 to the oxidative stress response via ALKBH8-dependent tRNASec(UCA) modifications that enhance the translation of selenocysteine-containing proteins.
- 45.Dedon PC, Begley TJ. A system of RNA modifications and biased codon use controls cellular stress response at the level of translation. Chem. Res. Toxicol. 2014;27(3):330–337. doi: 10.1021/tx400438d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gu C, Ramos J, Begley U, Dedon PC, Fu D, Begley TJ. Phosphorylation of human TRM9L integrates multiple stress-signaling pathways for tumor growth suppression. Sci. Adv. 2018;4(7):1–12. doi: 10.1126/sciadv.aas9184. eaas9184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Diehn M, Cho RW, Lobo NA, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458(7239):780–783. doi: 10.1038/nature07733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012;24(5):981–990. doi: 10.1016/j.cellsig.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sies H. Role of metabolic H2O2 generation: redox signaling and oxidative stress. J. Biol. Chem. 2014;289(13):8735–8741. doi: 10.1074/jbc.R113.544635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000;279(6):L1005–1028. doi: 10.1152/ajplung.2000.279.6.L1005. [DOI] [PubMed] [Google Scholar]
- 51.Silvera D, Formenti SC, Schneider RJ. Translational control in cancer. Nat. Rev. Cancer. 2010;10(4):254–266. doi: 10.1038/nrc2824. [DOI] [PubMed] [Google Scholar]
- 52.Ruggero D. Translational control in cancer etiology. Cold Spring Harb. Perspect. Biol. 2013;5(2):1–27. doi: 10.1101/cshperspect.a012336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Truitt Ml, Ruggero D. New frontiers in translational control of the cancer genome. Nat. Rev. Cancer. 2016;16(5):288–304. doi: 10.1038/nrc.2016.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Charette M, Gray MW. Pseudouridine in RNA: what, where, how, and why. IUBMB Life. 2000;49(5):341–351. doi: 10.1080/152165400410182. [DOI] [PubMed] [Google Scholar]
- 55.Hamma T, Ferre-D'amare AR. Pseudouridine synthases. Chem. Biol. 2006;13(11):1125–1135. doi: 10.1016/j.chembiol.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 56.Borek E, Baliga BS, Gehrke CW, et al. High turnover rate of transfer RNA in tumor tissue. Cancer Res. 1977;37(9):3362–3366. [PubMed] [Google Scholar]
- 57.Craddock VM. Transfer RNA methylases and cancer. Nature. 1970;228(5278):1264–1268. doi: 10.1038/2281264a0. [DOI] [PubMed] [Google Scholar]
- 58.Borek E. Transfer RNA and transfer RNA modification in differentiation and neoplasia. Introduction. Cancer Res. 1971;31(5):596–597. [PubMed] [Google Scholar]
- 59.Viale GL. Transfer RNA and transfer RNA methylase in human brain tumors. Cancer Res. 1971;31(5):605–608. [PubMed] [Google Scholar]
- 60.Murphy TL, Cooper IA, Wray GW, Ironside PN, Matthews J. Transfer RNA and transfer RNA methylase activity in spleens of patients with Hodgkin's disease and histiocytic lymphoma. J. Natl Cancer Inst. 1976;56(2):215–219. doi: 10.1093/jnci/56.2.215. [DOI] [PubMed] [Google Scholar]
- 61.Bullinger D, Neubauer H, Fehm T, Laufer S, Gleiter CH, Kammerer B. Metabolic signature of breast cancer cell line MCF-7: profiling of modified nucleosides via LC-IT MS coupling. BMC Biochem. 2007;8:25. doi: 10.1186/1471-2091-8-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Frickenschmidt A, Frohlich H, Bullinger D, et al. Metabonomics in cancer diagnosis: mass spectrometry-based profiling of urinary nucleosides from breast cancer patients. Biomarkers. 2008;13(4):435–449. doi: 10.1080/13547500802012858. [DOI] [PubMed] [Google Scholar]
- 63.Willmann L, Schlimpert M, Halbach S, Erbes T, Stickeler E, Kammerer B. Metabolic profiling of breast cancer: differences in central metabolism between subtypes of breast cancer cell lines. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2015;1000:95–104. doi: 10.1016/j.jchromb.2015.07.021. [DOI] [PubMed] [Google Scholar]
- 64.Willmann L, Erbes T, Krieger S, Trafkowski J, Rodamer M, Kammerer B. Metabolome analysis via comprehensive two-dimensional liquid chromatography: identification of modified nucleosides from RNA metabolism. Anal. Bioanal. Chem. 2015;407(13):3555–3566. doi: 10.1007/s00216-015-8516-6. [DOI] [PubMed] [Google Scholar]
- 65.Dong C, Niu L, Song W, et al. tRNA modification profiles of the fast-proliferating cancer cells. Biochem. Biophys. Res. Commun. 2016;476(4):340–345. doi: 10.1016/j.bbrc.2016.05.124. [DOI] [PubMed] [Google Scholar]
- 66.Guzzi N, Ciesla M, Ngoc PCT, et al. Pseudouridylation of tRNA-derived fragments steers translational control in stem cells. Cell. 2018;173(5):1204–1216. doi: 10.1016/j.cell.2018.03.008. e1226. [DOI] [PubMed] [Google Scholar]
- 67.Zhang Y, Shi J, Chen Q. tsRNAs: new players in mammalian retrotransposon control. Cell Res. 2017;27(11):1307–1308. doi: 10.1038/cr.2017.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lee YS, Shibata Y, Malhotra A, Dutta A. A novel class of small RNAs: tRNA-derived RNA fragments (tRFs) Genes Dev. 2009;23(22):2639–2649. doi: 10.1101/gad.1837609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Balatti V, Nigita G, Veneziano D, et al. tsRNA signatures in cancer. Proc. Natl Acad. Sci. USA. 2017;114(30):8071–8076. doi: 10.1073/pnas.1706908114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Srinivasan PR, Borek E. Enzymatic alteration of nucleic acid structure. Science. 1964;145(3632):548–553. doi: 10.1126/science.145.3632.548. [DOI] [PubMed] [Google Scholar]
- 71.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 72.Begley U, Sosa MS, Avivar-Valderas A, et al. A human tRNA methyltransferase 9-like protein prevents tumour growth by regulating LIN9 and HIF1-alpha. EMBO Mol. Med. 2013;5(3):366–383. doi: 10.1002/emmm.201201161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kerangueven F, Essioux L, Dib A, et al. Loss of heterozygosity and linkage analysis in breast carcinoma: indication for a putative third susceptibility gene on the short arm of chromosome 8. Oncogene. 1995;10(5):1023–1026. [PubMed] [Google Scholar]
- 74.Knowles MA, Shaw ME, Proctor AJ. Deletion mapping of chromosome 8 in cancers of the urinary bladder using restriction fragment length polymorphisms and microsatellite polymorphisms. Oncogene. 1993;8(5):1357–1364. [PubMed] [Google Scholar]
- 75.Prasad MA, Trybus TM, Wojno KJ, Macoska JA. Homozygous and frequent deletion of proximal 8p sequences in human prostate cancers: identification of a potential tumor suppressor gene site. Genes Chromosomes Cancer. 1998;23(3):255–262. [PubMed] [Google Scholar]
- 76.Bardi G, Johansson B, Pandis N, et al. Cytogenetic analysis of 52 colorectal carcinomas – non-random aberration pattern and correlation with pathologic parameters. Int. J. Cancer. 1993;55(3):422–428. doi: 10.1002/ijc.2910550317. [DOI] [PubMed] [Google Scholar]
- 77.Rapino F, Delaunay S, Rambow F, et al. Codon-specific translation reprogramming promotes resistance to targeted therapy. Nature. 2018;558(7711):605–609. doi: 10.1038/s41586-018-0243-7. [DOI] [PubMed] [Google Scholar]; •• Connects the ELP3-initiated U34 tRNA modification system to oncogenic BRAF(V600E) in melanoma with implications for targeting drug-resistant cancers.
- 78.Wang S, Liu X, Huang J, et al. Expression of KIAA1456 in lung cancer tissue and its effects on proliferation, migration and invasion of lung cancer cells. Oncol. Lett. 2018;16(3):3791–3795. doi: 10.3892/ol.2018.9119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen HM, Wang J, Zhang YF, Gao YH. Ovarian cancer proliferation and apoptosis are regulated by human transfer RNA methyltransferase 9-like via LIN9. Oncol. Lett. 2017;14(4):4461–4466. doi: 10.3892/ol.2017.6750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dimitriou M, Woll PS, Mortera-Blanco T, et al. Perturbed hematopoietic stem and progenitor cell hierarchy in myelodysplastic syndromes patients with monosomy 7 as the sole cytogenetic abnormality. Oncotarget. 2016;7(45):72685–72698. doi: 10.18632/oncotarget.12234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Blanco S, Dietmann S, Flores JV, et al. Aberrant methylation of tRNAs links cellular stress to neuro-developmental disorders. EMBO J. 2014;33(18):2020–2039. doi: 10.15252/embj.201489282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Blanco S, Bandiera R, Popis M, et al. Stem cell function and stress response are controlled by protein synthesis. Nature. 2016;534(7607):335–340. doi: 10.1038/nature18282. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Provides details about how NSUN2 controls protein expression to regulate stem cell function and the role that the stress response plays in this process.
- 83.Brzezicha B, Schmidt M, Makalowska I, Jarmolowski A, Pienkowska J, Szweykowska-Kulinska Z. Identification of human tRNA:m5C methyltransferase catalysing intron-dependent m5C formation in the first position of the anticodon of the pre-tRNA Leu (CAA) Nucleic Acids Res. 2006;34(20):6034–6043. doi: 10.1093/nar/gkl765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tuorto F, Liebers R, Musch T, et al. RNA cytosine methylation by Dnmt2 and NSun2 promotes tRNA stability and protein synthesis. Nat. Struct. Mol. Biol. 2012;19(9):900–905. doi: 10.1038/nsmb.2357. [DOI] [PubMed] [Google Scholar]
- 85.Anderson P, Ivanov P. tRNA fragments in human health and disease. FEBS Lett. 2014;588(23):4297–4304. doi: 10.1016/j.febslet.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sobala A, Hutvagner G. Small RNAs derived from the 5’ end of tRNA can inhibit protein translation in human cells. RNA Biol. 2013;10(4):553–563. doi: 10.4161/rna.24285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ivanov P, Emara MM, Villen J, Gygi SP, Anderson P. Angiogenin-induced tRNA fragments inhibit translation initiation. Mol. Cell. 2011;43(4):613–623. doi: 10.1016/j.molcel.2011.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Khan MA, Rafiq MA, Noor A, et al. Mutation in NSUN2, which encodes an RNA methyltransferase, causes autosomal-recessive intellectual disability. Am. J. Hum. Genet. 2012;90(5):856–863. doi: 10.1016/j.ajhg.2012.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Martinez FJ, Lee JH, Lee JE, et al. Whole exome sequencing identifies a splicing mutation in NSUN2 as a cause of a Dubowitz-like syndrome. J. Med. Genet. 2012;49(6):380–385. doi: 10.1136/jmedgenet-2011-100686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Abbasi-Moheb L, Mertel S, Gonsior M, et al. Mutations in NSUN2 cause autosomal-recessive intellectual disability. Am. J. Hum. Genet. 2012;90(5):847–855. doi: 10.1016/j.ajhg.2012.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lu L, Zhu G, Zeng H, Xu Q, Holzmann K. High tRNA transferase NSUN2 gene expression is associated with poor prognosis in head and neck squamous carcinoma. Cancer Invest. 2018;36(4):246–253. doi: 10.1080/07357907.2018.1466896. [DOI] [PubMed] [Google Scholar]
- 92.Frye M, Watt FM. The RNA methyltransferase Misu (NSun2) mediates Myc-induced proliferation and is upregulated in tumors. Curr. Biol. 2006;16(10):971–981. doi: 10.1016/j.cub.2006.04.027. [DOI] [PubMed] [Google Scholar]
- 93.Frye M, Dragoni I, Chin SF, et al. Genomic gain of 5p15 leads to over-expression of Misu (NSUN2) in breast cancer. Cancer Lett. 2010;289(1):71–80. doi: 10.1016/j.canlet.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 94.Yi J, Gao R, Chen Y, et al. Overexpression of NSUN2 by DNA hypomethylation is associated with metastatic progression in human breast cancer. Oncotarget. 2017;8(13):20751–20765. doi: 10.18632/oncotarget.10612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Goll MG, Kirpekar F, Maggert KA, et al. Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science. 2006;311(5759):395–398. doi: 10.1126/science.1120976. [DOI] [PubMed] [Google Scholar]
- 96.Khoddami V, Cairns BR. Identification of direct targets and modified bases of RNA cytosine methyltransferases. Nat. Biotechnol. 2013;31(5):458–464. doi: 10.1038/nbt.2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schaefer M, Pollex T, Hanna K, et al. RNA methylation by Dnmt2 protects transfer RNAs against stress-induced cleavage. Genes Dev. 2010;24(15):1590–1595. doi: 10.1101/gad.586710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jeltsch A, Ehrenhofer-Murray A, Jurkowski TP, et al. Mechanism and biological role of Dnmt2 in nucleic acid methylation. RNA Biol. 2017;14(9):1108–1123. doi: 10.1080/15476286.2016.1191737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tuorto F, Herbst F, Alerasool N, et al. The tRNA methyltransferase Dnmt2 is required for accurate polypeptide synthesis during haematopoiesis. EMBO J. 2015;34(18):2350–2362. doi: 10.15252/embj.201591382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Huang B, Johansson MJ, Bystrom AS. An early step in wobble uridine tRNA modification requires the Elongator complex. RNA. 2005;11(4):424–436. doi: 10.1261/rna.7247705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bauer F, Matsuyama A, Candiracci J, et al. Translational control of cell division by Elongator. Cell Rep. 2012;1(5):424–433. doi: 10.1016/j.celrep.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ladang A, Rapino F, Heukamp LC, et al. Elp3 drives Wnt-dependent tumor initiation and regeneration in the intestine. J. Exp. Med. 2015;212(12):2057–2075. doi: 10.1084/jem.20142288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Delaunay S, Rapino F, Tharun L, et al. Elp3 links tRNA modification to IRES-dependent translation of LEF1 to sustain metastasis in breast cancer. J. Exp. Med. 2016;213(11):2503–2523. doi: 10.1084/jem.20160397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nguyen A, Rosner A, Milovanovic T, et al. Wnt pathway component LEF1 mediates tumor cell invasion and is expressed in human and murine breast cancers lacking ErbB2 (her-2/neu) overexpression. Int. J. Oncol. 2005;27(4):949–956. [PubMed] [Google Scholar]
- 105.Karoulia Z, Gavathiotis E, Poulikakos PI. New perspectives for targeting RAF kinase in human cancer. Nat. Rev. Cancer. 2017;17(11):676–691. doi: 10.1038/nrc.2017.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Okamoto M, Hirata S, Sato S, et al. Frequent increased gene copy number and high protein expression of tRNA (cytosine-5-)-methyltransferase (NSUN2) in human cancers. DNA Cell Biol. 2012;31(5):660–671. doi: 10.1089/dna.2011.1446. [DOI] [PubMed] [Google Scholar]
- 107.Schaefer M, Hagemann S, Hanna K, Lyko F. Azacytidine inhibits RNA methylation at DNMT2 target sites in human cancer cell lines. Cancer Res. 2009;69(20):8127–8132. doi: 10.1158/0008-5472.CAN-09-0458. [DOI] [PubMed] [Google Scholar]
- 108.Stresemann C, Lyko F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int. J. Cancer. 2008;123(1):8–13. doi: 10.1002/ijc.23607. [DOI] [PubMed] [Google Scholar]
- 109.Forbes SA, Beare D, Gunasekaran P, et al. COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2014;43:D805–D811. doi: 10.1093/nar/gku1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Elhardt W, Shanmugam R, Jurkowski TP, Jeltsch A. Somatic cancer mutations in the DNMT2 tRNA methyltransferase alter its catalytic properties. Biochimie. 2015;112:66–72. doi: 10.1016/j.biochi.2015.02.022. [DOI] [PubMed] [Google Scholar]
- 111.Rodriguez V, Chen Y, Elkahloun A, Dutra A, Pak E, Chandrasekharappa S. Chromosome 8 BAC array comparative genomic hybridization and expression analysis identify amplification and overexpression of TRMT12 in breast cancer. Genes Chromosomes Cancer. 2007;46(7):694–707. doi: 10.1002/gcc.20454. [DOI] [PubMed] [Google Scholar]
- 112.Shimada K, Nakamura M, Anai S, et al. A novel human AlkB homologue, ALKBH8, contributes to human bladder cancer progression. Cancer Res. 2009;69(7):3157–3164. doi: 10.1158/0008-5472.CAN-08-3530. [DOI] [PubMed] [Google Scholar]
- 113.Ohshio I, Kawakami R, Tsukada Y, et al. ALKBH8 promotes bladder cancer growth and progression through regulating the expression of survivin. Biochem. Biophys. Res. Commun. 2016;477(3):413–418. doi: 10.1016/j.bbrc.2016.06.084. [DOI] [PubMed] [Google Scholar]
- 114.Burger M, Catto JW, Dalbagni G, et al. Epidemiology and risk factors of urothelial bladder cancer. Eur. Urol. 2013;63(2):234–241. doi: 10.1016/j.eururo.2012.07.033. [DOI] [PubMed] [Google Scholar]
- 115.Hempel N, Ye H, Abessi B, Mian B, Melendez JA. Altered redox status accompanies progression to metastatic human bladder cancer. Free Radic. Biol. Med. 2009;46(1):42–50. doi: 10.1016/j.freeradbiomed.2008.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hempel N, Melendez JA. Intracellular redox status controls membrane localization of pro- and anti-migratory signaling molecules. Redox Biol. 2014;2:245–250. doi: 10.1016/j.redox.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hempel N, Carrico PM, Melendez JA. Manganese superoxide dismutase (Sod2) and redox-control of signaling events that drive metastasis. Anticancer Agents Med. Chem. 2011;11(2):191–201. doi: 10.2174/187152011795255911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Pelicano H, Carney D, Huang P. ROS stress in cancer cells and therapeutic implications. Drug Resist. Updat. 2004;7(2):97–110. doi: 10.1016/j.drup.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 119.Goodarzi H, Nguyen HCB, Zhang S, Dill BD, Molina H, Tavazoie SF. Modulated expression of specific tRNAs drives gene expression and cancer progression. Cell. 2016;165(6):1416–1427. doi: 10.1016/j.cell.2016.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Demonstrates that the upregulation of tRNAArg(CCG) and tRNAGlu(UUC) can promote the metastatic behavior of breast cancer cells through enhanced codon-biased translation.
- 120.Novoa EM, Ribas DE, Pouplana L. Speeding with control: codon usage, tRNAs, and ribosomes. Trends Genet. 2012;28(11):574–581. doi: 10.1016/j.tig.2012.07.006. [DOI] [PubMed] [Google Scholar]
- 121.Dittmar KA, Goodenbour JM, Pan T. Tissue-specific differences in human transfer RNA expression. PLoS Genet. 2006;2(12):e221. doi: 10.1371/journal.pgen.0020221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tuller T, Carmi A, Vestsigian K, et al. An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell. 2012a;141(2):344–354. doi: 10.1016/j.cell.2010.03.031. [DOI] [PubMed] [Google Scholar]
- 123.Gingold H, Tehler D, Christoffersen NR, et al. A dual program for translation regulation in cellular proliferation and differentiation. Cell. 2014;158(6):1281–1292. doi: 10.1016/j.cell.2014.08.011. [DOI] [PubMed] [Google Scholar]
- 124.Pavon-Eternod M, Gomes S, Geslain R, Dai Q, Rosner MR, Pan T. tRNA over-expression in breast cancer and functional consequences. Nucleic Acids Res. 2009;37(21):7268–7280. doi: 10.1093/nar/gkp787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Olvedy M, Scaravilli M, Hoogstrate Y, Visakorpi T, Jenster G, Martens-Uzunova ES. A comprehensive repertoire of tRNA-derived fragments in prostate cancer. Oncotarget. 2016;7(17):24766–24777. doi: 10.18632/oncotarget.8293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Goodarzi H, Liu X, Nguyen HC, Zhang S, Fish L, Tavazoie SF. Endogenous tRNA-derived fragments suppress breast cancer progression via YBX1 displacement. Cell. 2015;161(4):790–802. doi: 10.1016/j.cell.2015.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Fasullo M, Freedland J, St John N, et al. An in vitro system for measuring genotoxicity mediated by human CYP3A4 in Saccharomyces cerevisiae . Environ. Mol. Mutagen. 2017;58(4):217–227. doi: 10.1002/em.22093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lampson BL, Pershing NL, Prinz JA, et al. Rare codons regulate KRas oncogenesis. Curr. Biol. 2013;23(1):70–75. doi: 10.1016/j.cub.2012.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]